

Abstract
BACKGROUND
Acute decompensated heart failure (ADHF) is associated with deterioration in renal function—an important risk factor for poor outcomes. Whether ADHF results in permanent kidney damage/dysfunction is unknown.
METHODS AND RESULTS
We investigated for the first time the renal responses to the development of, and recovery from, ADHF using an ovine model. ADHF development induced pronounced hemodynamic changes, neurohormonal activation, and decline in renal function, including decreased urine, sodium and urea excretion, and creatinine clearance. Following ADHF recovery (25 days), creatinine clearance reductions persisted. Kidney biopsies taken during ADHF and following recovery showed widespread mesangial cell prominence, early mild acute tubular injury, and medullary/interstitial fibrosis. Renal transcriptomes identified altered expression of 270 genes following ADHF development and 631 genes following recovery. A total of 47 genes remained altered post‐recovery. Pathway analysis suggested gene expression changes, driven by a network of inflammatory cytokines centered on IL‐1β (interleukin 1β), lead to repression of reno‐protective eNOS (endothelial nitric oxide synthase) signaling during ADHF development, and following recovery, activation of glomerulosclerosis and reno‐protective pathways and repression of proinflammatory/fibrotic pathways. A total of 31 dysregulated genes encoding proteins detectable in urine, serum, and plasma identified potential candidate markers for kidney repair (including CNGA3 [cyclic nucleotide gated channel subunit alpha 3] and OIT3 [oncoprotein induced transcript 3]) or long‐term renal impairment in ADHF (including ACTG2 [actin gamma 2, smooth muscle] and ANGPTL4 [angiopoietin like 4]).
CONCLUSIONS
In an ovine model, we provide the first direct evidence that an episode of ADHF leads to an immediate decline in kidney function that failed to fully resolve after ≈4 weeks and is associated with persistent functional/structural kidney injury. We identified molecular pathways underlying kidney injury and repair in ADHF and highlighted 31 novel candidate biomarkers for acute kidney injury in this setting.
Keywords: acute decompensated heart failure, acute kidney injury, kidney function, transcriptome
Subject Categories: Basic Science Research, Heart Failure, Nephrology and Kidney, Pathophysiology
Nonstandard Abbreviations and Acronyms
- ADHF
acute decompensated heart failure
- AKI
acute kidney injury
- AngII
angiotensin II
- AVP
arginine vasopressin
- CVP
central venous pressure
- eNOS
endothelial nitric oxide synthase
- GP6
glycoprotein VI
- IPA
Ingenuity Pathway Analysis
- MCs
mesangial cells
- PDGF
platelet‐derived growth factor
- PPAR
peroxisome proliferator‐activated receptor
- RNA‐seq
RNA sequencing
- TGF‐β
transforming growth factor‐β
- WRF
worsening renal function
Clinical Perspective
What Is New?
Characterization of serial functional and histological data during the development and resolution of acute decompensated heart failure (ADHF; using an ovine model) provides the first direct evidence that a single episode of ADHF can cause acute kidney injury (AKI), as demonstrated by cellular necrosis, tubular injury, and glomerular scarring, which is associated with a decline in kidney function that fails to fully resolve after ≈4 weeks.
Renal gene expression profiling of pathways associated with kidney injury and repair in ADHF suggest changes in expression may be driven by a network of inflammatory cytokines centered on IL‐1β (interleukin 1β).
What Are the Clinical Implications?
These data suggest that an episode of ADHF on a background of previously normal kidneys represents a significant event that can result in acute kidney injury and lead to permanent kidney injury and decrement in renal function.
Recognition and awareness of the significance of acute kidney injury is critical in clinical ADHF management to prevent unintended adverse renal effects and associated poor outcomes.
Inhibition of IL‐1β may have therapeutic potential in cardiorenal disease, including acute kidney injury in ADHF.
Heart failure (HF) is a leading cause of morbidity and mortality worldwide. Approximately 20% of currently middle‐aged people will develop HF in their remaining lifetime, 1 with ≈20% of patients dying in the first year after diagnosis and >50% after 5 years. 2 HF is a progressive syndrome characterized by recurrent episodes of acute worsening or decompensation and frequently accompanied by acute renal dysfunction—1 of the most important risk factors for poor clinical outcomes and death. 3 Indeed, the majority of individuals hospitalized with acute decompensated HF (ADHF) exhibit at least some degree of existing renal insufficiency, with perhaps as many as two thirds manifesting moderate to severe impairment. 4 Furthermore, a considerable number of patients incur acute worsening of kidney function, which if sustained, is associated with increased short‐term and long‐term mortality. 5
With both background impairment and additional decrements in renal function portending a worse prognosis in HF, 3 , 4 , 5 there is a need to better understand the pathophysiological mechanisms linking the failing heart and the kidney. Although kidney dysfunction in HF has traditionally been considered a result of impaired renal blood flow in the setting of depressed cardiac function, increasing evidence suggests contributory mechanisms are more complex and multifactorial. Putative contributors also include central venous congestion (and therefore renal venous hypertension), anemia, oxidative stress, inflammation, neurohormonal activation, and renal sympathetic overactivity. 6 , 7 However, despite considerable prior research, the pathophysiology of kidney dysfunction in HF remains incompletely understood. This is in a large part attributed to poor renal characterization. Importantly, it is not known whether acutely reduced kidney function secondary to ADHF leads to persistent kidney injury and repair by scarring—resulting in permanent kidney injury and dysfunction—or whether kidney structure and function recovers fully to predecompensated levels following successful HF treatment. Answers to these questions and a deeper understanding of molecular alterations in the kidney during development and resolution of ADHF may inform the management of patients with HF and point to therapeutic targets for renal preservation in HF.
We therefore investigated serial physiological (hemodynamic, neurohumoral, renal) and kidney histological and transcriptome (RNA sequencing [RNA‐seq]) responses during the development and resolution of ADHF using an ovine model of ADHF induced by rapid left ventricular (LV) pacing. 8
METHODS
RNA‐seq data have been deposited in the National Center for Biotechnology Information Gene Expression Omnibus database (www.ncbi.nlm.nih.gov/geo; Accession No. GSE180502). All other data and supporting materials/methods are either available within the article (and its online supplementary files) or from the corresponding author upon reasonable request.
Sheep Instrumentation
Studies were approved by the Animal Ethics Committee of the University of Otago–Christchurch (No. C23/10; in accordance with the New Zealand Animal Welfare Act 1999 and Amendment No. 2 [2015]) and conform to current National Institute of Health guidelines (8th edition).
A total of 9 adult Coopworth ewes (4–6 years; 47–69 kg; Lincoln University Farm, Canterbury, New Zealand) were instrumented via a left lateral thoracotomy as previously described 8 under general anesthesia (induced by intravenous diazepam [1×0.5 mg/kg]/intravenous ketamine [1×4 mg/kg]; maintained with 2% isoflurane/2 L per minute nitrous oxide/2 L per minute oxygen) and using approved perioperative/postoperative analgesia (intravenous carprofen [4 mg/kg once a day for ≥3 days], intravenous buprenorphine [0.005–0.01 mg/kg once a day for ≥3 days], intercostal bupivacaine [1×25 mg]/intercostal lignocaine [1×100 mg]), and antibiotics (intravenous cephazolin [2×20 mg/kg], intravenous enrofloxacin [1×2.5 mg/kg]). Briefly, catheters were placed via the jugular vein and in the left atrium for blood sampling and measurement of central venous pressure (CVP) and left atrial pressure; Konigsberg pressure‐tip transducers were inserted in the aorta to record mean arterial pressure (MAP) and into the apex of the left ventricle to obtain heart rate, LV pressures, and derivatives of pressure over time (dP/dt[max]); a Swan‐Ganz thermodilution catheter (American Edwards) was placed in the pulmonary artery for measurement of cardiac output; and a 7‐French His‐bundle electrode was stitched subepicardially to the left ventricle for subsequent cardiac pacing. A bladder catheter was inserted per urethra to allow timed urine collections. Adequacy of anesthesia was monitored via serial measurements of heart rate, blood pressure, capillary refill time, mucous membrane color, palpable blink response, jaw tone, anesthetic gas analysis, oxygen saturation, and end‐tidal CO2 levels.
Animals recovered for at least 14 days before commencing the study protocol, and throughout the study they were held in metabolic cages and received a diet of Lucerne chaff and food pellets (providing ≈75 mmol sodium/day, ≈150 mmol potassium/day) with free access to water.
Note that only ewes have been used in the current study because of practical issues with having an indwelling bladder catheter in a conscious male sheep within a metabolic crate. In addition, it is difficult to source male animals as male lambs are typically castrated within weeks of birth and sent off to the meat works before they are a year old.
Study Protocol
The study followed a longitudinal repeated‐measures design with sampling conducted during a 5‐day baseline period (days −4 to 0) followed by 14 days of rapid LV pacing at 220 bpm to induce ADHF (days 1 to 14, pace), and then termination of pacing and 25 days of recovery (days 1 to 25, recovery).
Hemodynamic measurements (CVP, left atrial pressure, MAP, heart rate, cardiac output, dP/dt[max]) were recorded using an online data acquisition system (PowerLab Systems; ADInstruments) and made before the commencement of pacing (baseline; days −4, −2, 0), during pacing (ADHF; days 1–4, 7–9, 10, 11, 14) and following the cessation of pacing (recovery; days 15–18, 21, 23, 25, 28, 30, 32, 35, 37, 39).
Blood samples were drawn following each hemodynamic measurement. Samples were taken into EDTA tubes on ice, centrifuged at 4 °C, and stored at either −20 °C or −80 °C before assay for ANP (atrial natriuretic peptide), BNP (B‐type natriuretic peptide), PRA (plasma renin activity), aldosterone, endothelin‐1, AVP (arginine vasopressin), and catecholamines. 8 For each analyte, all samples from individual animals were measured in the same assay to avoid interassay variability. Plasma sodium, potassium, creatinine, urea, albumin, and hematocrit were measured with every blood sample taken.
Water intake and urine collections for the measurement of volume and excretion of sodium, potassium, creatinine, and urea were made daily during the first 26 days of study and then every 2 to 3 days until protocol completion. Creatinine clearance was calculated as urine creatinine×volume/plasma creatinine.
Serial kidney biopsies were performed in duplicate before pacing (baseline), on day 14 of pacing (HF), and after 25 days (nonpacing) recovery. For the first 2 procedures, the sheep were anesthetized (as described previously), and 2 adjacent percutaneous kidney biopsies were taken under ultrasonic guidance (Philips iE33; Philips Ultrasound) using a 14 g spring‐loaded automated biopsy gun (BARD Peripheral Vascular, Inc.). Final kidney samples were collected at study end immediately following euthanasia (intravenous Pentobarb300 100 mg/kg). On each occasion, 1 of the biopsies was fixed in 10% formalin (4 °C overnight) for histological analysis, and the other snap frozen in liquid nitrogen and stored at −80 °C for subsequent RNA extraction and RNA‐seq analysis.
Histological Analysis
Formalin‐fixed kidney biopsy samples were dehydrated in ethanol, embedded in paraffin, and cut into longitudinal 7‐µm sections before mounting. Sections were stained with hematoxylin–eosin or Masson’s trichrome and examined under light microscopy by a specialist pathologist. Specimens from 7×baseline, 9×HF, and 8×recovery biopsies were found to be acceptable for further evaluation. Presence of fibrosis (collagen deposition) was analyzed in digital images of Masson’s trichrome‐stained kidney sections (×10 objective magnification) by an image processing program (ImageJ) employing color‐based thresholding to determine percentage blue‐staining collagen.
RNA‐Seq and Bioinformatic Analysis
Full details of RNA extraction, library preparation, and bioinformatic analysis are provided in Data S1. Briefly, total RNA was extracted from frozen kidney biopsy samples and purified with RNeasy Midi Columns (Qiagen). To maximize statistical power, the 5×baseline, 6×HF, and 5×recovery serial samples were supplemented with kidney samples collected postmortem from animals euthanized before, during, or after the same pacing protocol. This provided a total of 14×baseline, 13×HF, and 8×recovery samples for RNA‐seq analysis. RNA‐seq libraries were prepared using total RNA and TruSeq stranded mRNA library prep kits (Illumina).
Statistical Analysis
Physiological Data Analysis
Changes during pacing and postpacing recovery were assessed by 1‐way repeated ANOVA (SPSS version 11.022). Where significant time effects were identified by ANOVA, level of significance at individual time points versus baseline (mean of samples taken before pacing) was determined by Fisher’s protected least‐significant difference tests. Results of baseline versus day 14 pacing and day 39 recovery are provided in the text. Significance was assumed at P<0.05.
Transcriptome Analysis
To confirm all biopsy samples were of kidney origin (ie, not contaminated with other tissue), the 1000 most abundant genes in each sample were tested for enrichment of tissue‐specific genes across 35 human tissues using the Tissue Enrich package 9 in R. 10 Kidney origin was confirmed for all but 3 samples (Figure S1), which were excluded, leaving a total of 11×baseline, 13×HF, and 8×recovery samples for differential expression analysis.
Gene expression was compared between time points (baseline to HF, HF to recovery, and baseline to recovery) using negative binomial generalized linear models with DESeq2 11 using a false discovery rate α=0.05. As not all animals underwent serial sampling, samples were treated as independent samples. Data for individual genes are presented as normalized read counts and corrected for library size using DESeq2 normalization. 11
Genes that differed between baseline and HF, HF and recovery, and baseline and recovery (P<0.01 after adjustment for multiple comparisons, fold change >1.2 or <0.83) were compared with 884 genes associated with acute kidney injury (AKI) as documented in 2 large databases: Harmonizome 12 and Ingenuity Pathway Analysis (IPA) knowledgebase (https://qiagenbioinformatics.com/products/ingenuity‐pathway‐analysis) 13 (Table S1). Enrichment for canonical pathways and nephrotoxicity networks was tested with Core and Comparative gene set enrichment analysis workflows using IPA software. The net effect of individual gene expression changes on the network/pathway (ie, whether the combined effect of individual gene expression changes predicted activation or inhibition of the network/pathway) was determined using Z scores (scores >2 indicate pathway activation, <−2 indicate pathway inhibition). Potential circulating or urinary biomarkers were identified using the IPA Biomarker Filter workflow and Protein Atlas secretome (www.proteinatlas.org). 14 A P value of P<0.01 after correction for multiple comparisons was used for all IPA analyses. Associations between candidate kidney biomarkers, selected neurohormones, and hemodynamic indexes were assessed with Pearson correlation (after ln‐transformation of data) and visualized on correlation matrices using the Corrplot package 15 in R. 10
RESULTS
Physiological Results
A total of 14 days of rapid LV pacing produced the hemodynamic deterioration, multifaceted hormonal activation, and sodium‐retaining hallmarks of human ADHF. Significant reductions were observed in cardiac output, dP/dt(max), and MAP together with increases in CVP, left atrial pressure, and calculated total peripheral resistance (calculated total peripheral resistance=MAP/CO), resulting in a halving of (calculated) renal perfusion pressure (MAP‐CVP; all P<0.001; Figure 1; Figure S2). Attendant neuroendocrine changes included marked rises in plasma ANP (P<0.001), BNP (P<0.01), PRA (P<0.05), aldosterone (P<0.01), endothelin‐1 (P<0.001), AVP (P<0.01), epinephrine (P<0.05), and norepinephrine (P<0.05; Figure S3). Renal function was adversely affected as evidenced by a halving in urine output and significant declines in urine sodium, potassium, urea, and creatinine excretion (all P<0.001; Figure 1; Figure S4). In addition, plasma creatinine levels rose (P<0.01; Table S2) and creatinine clearance was markedly reduced (P<0.001; Figure 1).
Figure 1. Mean±SEM serial physiological responses in 9 sheep before (baseline) and during development of acute decompensated heart failure (induced by left ventricular pacing at 220 bpm for 14 days) and during 25 days of recovery following termination of pacing.
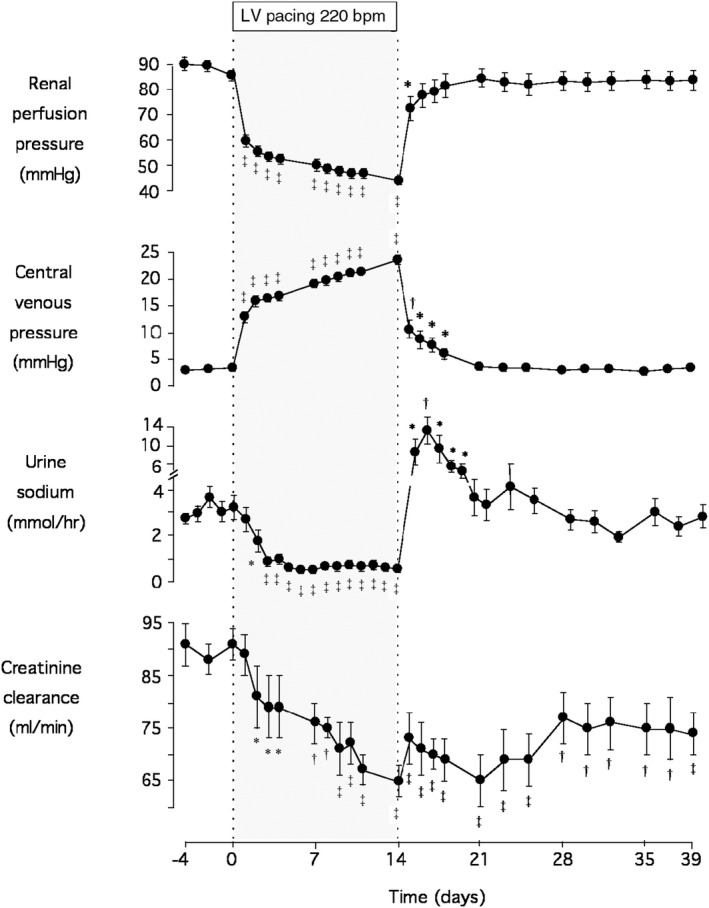
Significant differences from prepacing baseline levels are shown by the following: *P<0.05, † P<0.05, and ‡ P<0.001 by 1‐way ANOVA. bpm indicates beats per minute; and LV, left ventricular.
Following termination of rapid pacing and 25 days of recovery, most measurements returned to prepacing baseline levels. Exceptions included left atrial pressure (P<0.01) and the natriuretic peptides (ANP, P<0.01; BNP, P<0.05), which remained slightly but significantly elevated relative to baseline. Importantly, urine creatinine excretion (P<0.001) and creatinine clearance (P<0.001) remained significantly decreased compared with baseline even after 25 days of recovery, with plasma creatinine concentrations also tending to remain raised (P=0.075; Figure 1; Table S2; Figures S2 through S4).
With respect to the consistency of renal dysfunction evident with ADHF development, it should be noted that all sheep demonstrated rises in plasma creatinine (range, 6–28 µmol/L) and falls in creatinine excretion (range, −0.04 to −0.16 mmol/h) and clearance (range, −5.4 to −40.5 mL/min) during ADHF compared with baseline. Following 25 days of recovery, creatinine excretion remained reduced in all animals (range, −0.003 to −0.094 mmol/h) along with creatinine clearance (range, −5.1 to −28.5 mL/min).
Histological Results
The majority of kidney biopsies taken before pacing (baseline) had no significant histological abnormalities (5/7 samples [72%]; Figure 2A). In contrast, more than half of the samples taken during fully developed ADHF (day 14 of LV pacing) showed evidence of early mild acute tubular injury (5/9 samples [56%]), and most exhibited a prominence of glomerular mesangial cells (MCs; 7/9 samples [78%]). The same was true of biopsies taken after 25 days of ADHF recovery (63% showing both MC prominence and tubular injury). These also demonstrated some mild chronic inflammation (2/8 samples) as well as focal/patchy dystrophic calcification and medullary/interstitial fibrosis in nearly half of the specimens (3/8 samples for both; Figure 2A). Development of fibrosis is further supported by digital analysis (ImageJ) showing increased collagen deposition in all animals but 1 following development/recovery of ADHF, with levels increasing from 36±4.2% at baseline to 44.8±1.6% at HF (P<0.05) and 42.5±3.8% at recovery (1.0 > P>0.05) and can be visualized in representative serial kidney samples from the 3 states (baseline, HF, recovery) showing increased blue‐staining collagen (Figure 2B).
Figure 2. Serial histological responses in sheep during development of, and recovery from, acute decompensated heart failure.
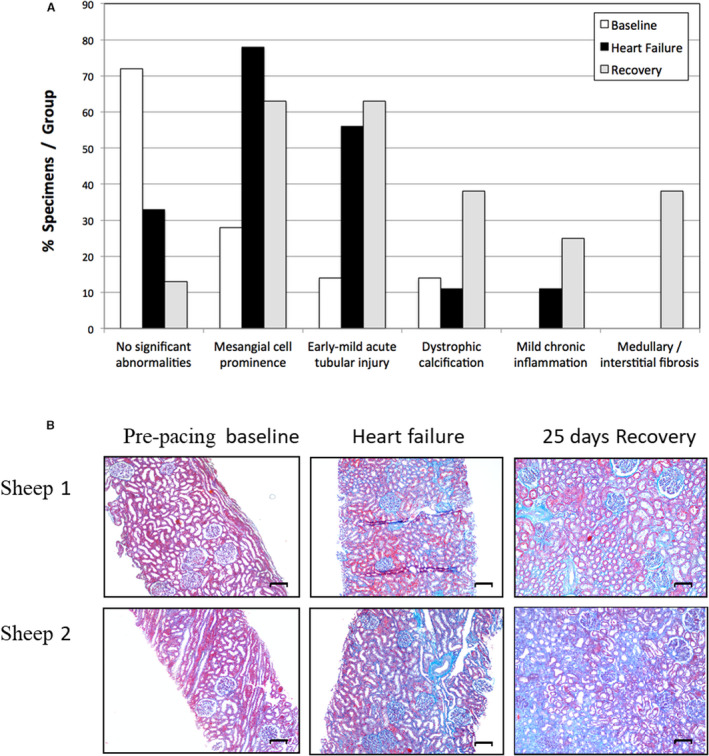
A, Principal histological findings in serial kidney biopsies taken in 9 sheep before (baseline, n=7), after development of acute decompensated heart failure (induced by left ventricular pacing at 220 bpm for 14 days; n=9), and after 25 days of recovery following termination of pacing (recovery, n=8). (Note that mesangial cell prominence is not included here as a significant histologic abnormality.). B, Masson’s trichrome staining of representative serial kidney biopsies taken before (baseline) and after development of acute decompensated heart failure (induced by left ventricular pacing at 220 bpm for 14 days) and after 25 days of recovery following termination of pacing (recovery). Blue staining indicates collagen deposition. Scale bar=100 μmol/L.
Transcriptome Results
RNA‐seq generated a mean±SD of 22.0±2.1×106 high‐quality read pairs per sample, which mapped to 31 553 unique genes in the sheep genome. Gene expression levels were compared between kidney biopsies collected at baseline (n=11), HF (n=13), and recovery (n=8). A total of 270 genes differed between baseline and HF (P<0.01 after adjustment for multiple comparisons, fold change >1.2 or <0.83, 47% upregulated in HF; Figure 3, Figure 4A, Table S3), 631 genes differed between HF and recovery (41% upregulated at recovery; Figure 3, Figure 4B, Table S3), and 47 genes differed between baseline and recovery (21% upregulated at recovery; Figure 3, Table S3). Of 827 unique genes that differed between time points, 152 (17%) had no known human homolog (most were in silico predicted transcripts) and were not analyzed further. The remaining 675 genes included 85 genes previously implicated in AKI (including mediators of fibrosis, inflammation and apoptosis, and components of the extracellular matrix), but not genes encoding early markers for AKI (including LCN2 [lipocalin 2]; IGFBP7 [insulin‐like growth factor‐binding protein 7] or TIMP2 [tissue inhibitor of metalloproteinases 2]) (Figure S5). The vast majority of differentially expressed genes (590 of 675 genes, 87%) had no prior reported association with AKI.
Figure 3. Genes altered in the kidney after development of, and following recovery from, acute decompensated HF; n=827; P<0.01 after adjustment for multiple comparisons, fold change >1.2 or <0.83).
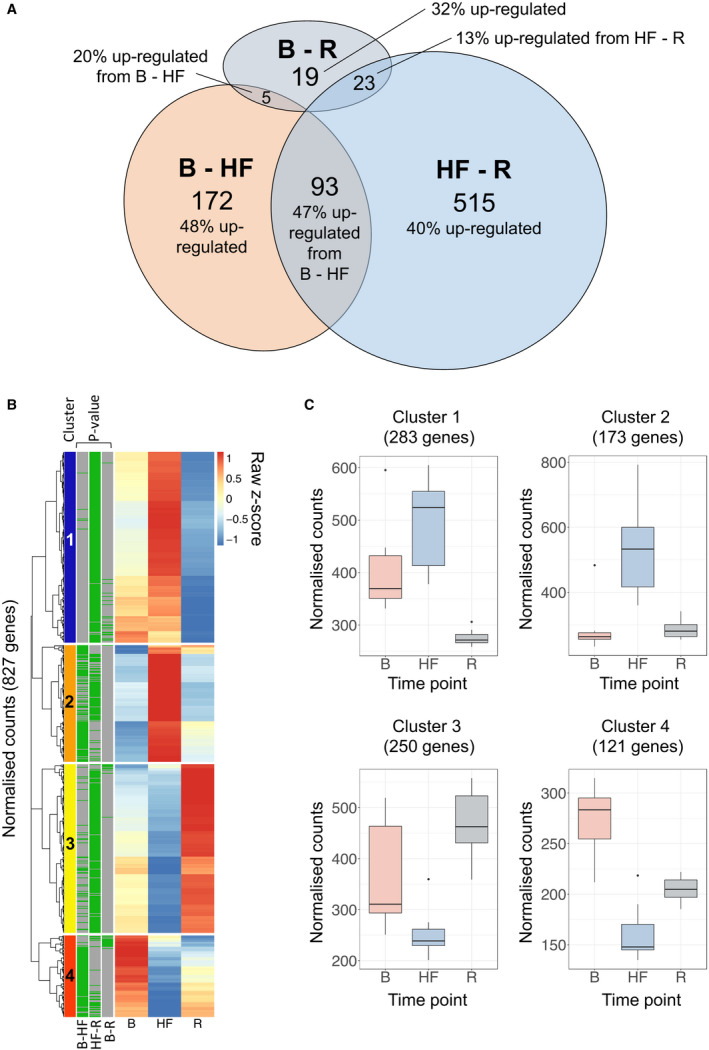
A, A total of 270 genes differed between B and HF and 631 genes differed between HF and R, of which 93 genes were altered across both data sets and were termed HF‐responsive genes. A total of 47 genes differed between B and R and were termed HF‐sustained genes. Percentages indicate the number of genes upregulated in the latter of the 2 time points. B, Heatmap showing median gene expression levels at B, HF, and R for genes differentially expressed between time points. Gene expression was scaled using Z scores to enable genes expressed at high and low abundance to be visualized on the same graph. For each gene, the color scale indicates high (red) or low (blue) expression relative to the other time points. Green bars (left) indicate differential expression between time points. Unsupervised hierarchical clustering identified four broad clusters of genes with similar patterns of expression across time points (clusters 1–4, far left). C, Box plots summarizing gene expressions for genes in each cluster in B (geometric mean) shown for all samples at each time point. B indicates baseline; HF, heart failure; and R, recovery.
Figure 4. Volcano plots showing genes altered from (A) baseline to heart failure, (B) heart failure to recovery, and (C) baseline to recovery.
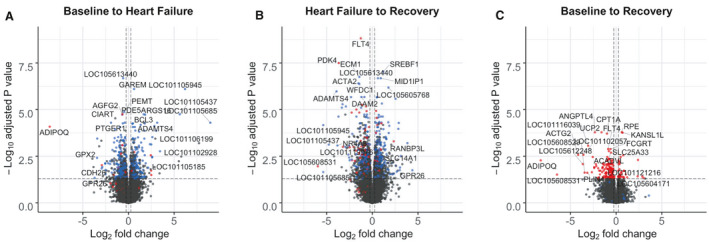
Genes to the right of the vertical dashed lines have a >1.2‐fold increase in expression; those to the left of the vertical dashed lines have a >1.2‐fold decrease in expression (ie, <0.83‐fold change) between time points. The horizontal dashed line indicates the threshold for statistical significance (P<0.01 after adjustment for multiple comparisons). Blue dots indicate HF‐responsive genes and red dots indicate HF‐sustained genes. Full gene names available from https://www.ncbi.nlm.nih.gov/gene/.
Cell Signaling Pathways
To investigate the molecular pathways altered in the kidney in response to ADHF development and recovery, comparative pathway analysis was performed for 675 genes altered between baseline and HF, HF and recovery, and baseline and recovery. Genes altered across these time points were enriched for a network of genes that regulate glomerulosclerosis and 8 molecular pathways (absolute Z score >2 and P<0.01 after adjustment for multiple comparisons; Figure 5A, Table S3). These were eNOS (endothelial nitric oxide synthase) signaling (repressed during ADHF development; Figure S6A); cholesterol biosynthesis and PPAR (peroxisome proliferator‐activated receptor) signaling (activated during recovery; Figure S6B and S6C); and GP6 (glycoprotein VI), PDGF (platelet‐derived growth factor), oncostatin M, acute phase response, and "hepatic" fibrosis signaling (all repressed during recovery; Figure S6D through S6H). The "hepatic" fibrosis pathway likely represents renal fibrosis given that the IPA knowledgebase lacks an annotated pathway for renal fibrosis and many genes/proteins are shared. All of these pathways have established roles in AKI. Among the 85 differentially expressed genes mapping to these pathways, 59 (69%) had not previously been implicated in AKI, and only 2 remained altered after recovery from ADHF. None of the identified pathways remained significantly activated or repressed after recovery.
Figure 5. Cell signaling pathways (A) and nephrotoxicity networks significantly enriched during HF and/or recovery (R) (B). (P<0.01 after correction for multiple comparisons.).
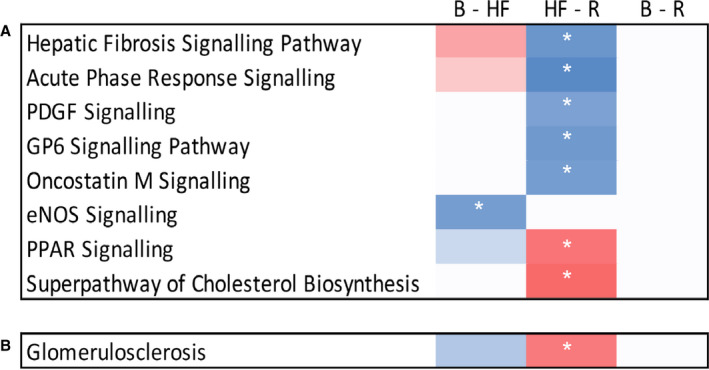
Pathways are clustered according to their pattern of activation at each time point. Pathways were considered activated (red boxes) if the activation Z score was >2 or repressed (blue boxes) if the activation Z score was <−2 (indicated by white asterisks). None of these pathways remained altered after recovery. Note that the pathway described as "hepatic" fibrosis likely represents renal fibrosis given that renal gene expression was measured, the Ingenuity Pathway Analysis knowledgebase lacks an annotated pathway for renal fibrosis, and many components of these pathways are shared. B indicates baseline; eNOS, endothelial nitric oxide synthase; GP6, glycoprotein VI; HF, heart failure; PDGF, platelet‐derived growth factor; PPAR, peroxisome proliferator‐activated receptor; and R, recovery.
Predicted Upstream Regulators
To identify potential upstream regulators, a statistical approach was used to identify regulator genes that may alter gene expression during ADHF development and recovery with a directionality consistent with the observed expression changes. The top‐predicted upstream regulator gene (ie, gene that promotes the development of ADHF and represses recovery) was the proinflammatory cytokine IL‐1β (interleukin 1β), a known regulator of kidney pathology (baseline to HF and HF to recovery, P<0.001 after adjustment for multiple comparisons; absolute Z score >3.5). A mechanistic network generated for IL‐1β indicated that the cytokine may activate a network of coregulators, including tumour necrosis factor, to alter the expression of 74 (27%) genes from baseline to HF and 263 (42%) genes from HF to recovery (Figure S7). Of these, 27 and 54 genes, respectively, may be regulated directly by IL‐1β (data not shown).
Candidate Plasma/Serum and Urinary Biomarkers for AKI in ADHF
To identify potential novel biomarkers of kidney injury and recovery in ADHF, the following 2 sets of genes were selected: (1) those that differed between baseline and recovery (termed HF‐sustained genes; n=47, 21% upregulated at recovery) and (2) those that differed from baseline to HF and from HF to recovery, but not from baseline to recovery (termed HF‐responsive genes; n=93, 47% upregulated at HF compared with baseline). From these, genes encoding proteins detectable in plasma, serum, or urine were selected as identifiers of potential candidate circulating or urinary markers and ranked based on their fold change in expression between time points. Of the 32 protein‐encoding genes identified (Figure 6), 13 were HF‐sustained genes, representing potential persistent markers of kidney damage or dysfunction in ADHF (Table 1), and 19 were HF‐responsive genes, representing potential markers for kidney repair in ADHF (Table 2). Although 5 HF‐sustained and 8 HF‐responsive genes had previously been implicated in AKI (42%; Tables 1 and 2, Figure 6), only 1 gene, REN (renin), had been investigated in patients with AKI and ADHF previously. 16 Consequently, the remaining 31 genes were prioritized as identifying potential novel markers for AKI in ADHF.
Figure 6. Transcriptome summary.
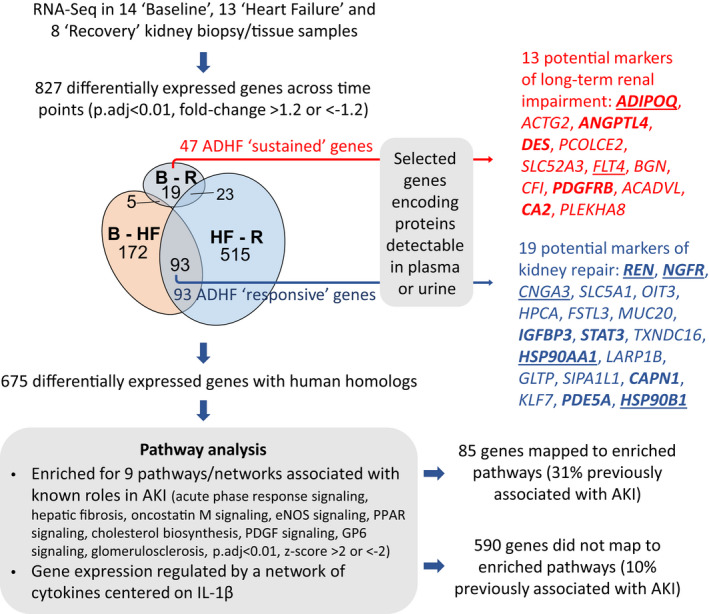
Kidney samples from animals that underwent serial sampling were supplemented with kidney samples collected postmortem from animals having undergone a similar pacing/recovery protocol, giving a total of 14×B, 13×HF, and 8×R samples for RNA sequencing analysis. Expression of 827 unique genes differed across the time course: B to HF, HF to R, and B to R. Of these, 675 genes had a human homolog and progressed to pathway analysis. Of 47 HF‐sustained genes (those that remained altered after 4 weeks of recovery), 13 genes encoded proteins detectable in plasma or urine and may represent markers of long‐term renal impairment in ADHF. Of 93 HF‐responsive genes (those that increased during ADHF development and decreased during recovery or vice versa), 19 genes encoded proteins detectable in plasma or urine and may represent markers of kidney repair in ADHF. Genes denoted in bold text indicate genes previously associated with AKI, genes that are underlined indicate genes that mapped to pathways shown to be enriched in pathway analysis. ACADVL indicates acyl‐CoA dehydrogenase very long chain; ACTG2, actin gamma 2, smooth muscle; ADHF, acute decompensated heart failure; ADIPOQ, adiponectin, C1Q, and collagen domain containing; AKI, acute kidney injury; ANGPTL4, angiopoietin like 4; B, baseline; BGN, biglycan CA2, carbonic anhydrase 2; CAPN1, calpain 1; CFI, complement factor I; CNGA3, cyclic nucleotide gated channel subunit alpha 3; DES, desmin; eNOS, endothelial nitric oxide synthase; FLT4, Fms related tyrosine kinase 4; FSTL3, follistatin like 3; GLTP, glycolipid transfer protein; GP6, glycoprotein VI; HF, heart failure; HPCA, hippocalcin; HSP90AA1, heat shock protein 90 alpha family class A member 1; HSP90B1, heat shock protein 90 beta family member 1; IGFBP3, insulin like growth factor binding protein 3; IL1β, interleukin 1β; KLF7, Kruppel like factor 7; LARP1B, La ribonucleoprotein domain family member 1B; MUC20, mucin 20; NGFR, nerve growth factor receptor; OIT3, oncoprotein induced transcript 3; p‐adj, p‐value adjusted for multiple comparisons; PCOLCE2, procollagen C‐endopeptidase enhancer 2; PDE5A, phosphodiesterase 5A; PDGF, platelet‐derived growth factor; PDGFRB, platelet derived growth factor receptor beta; PLEKHA8, pleckstrin homology domain containing A8; PPAR, peroxisome proliferator‐activated receptor; R, recovery; REN, renin; SIPA1L1, signal induced proliferation associated 1 like 1; SLC5A1, solute carrier family 5 member 1; SLC52A3, solute carrier family 52 member 3; STAT3, signal transducer and activator of transcription 3; and TXNDC16, thioredoxin domain containing 16.
Table 1.
Dysregulated Genes Encoding Candidate Responsive Biomarkers for AKI and Recovery in Acute Decompensated Heart Failure
| Symbol | Name | Cellular location | Plasma/serum | Urine | Known drug target* | Previous association with AKI† | Normalized read counts at B‡ | Fold change B‐HF§¶ | Fold change HF‐R§¶ | Fold change B‐R§¶ |
|---|---|---|---|---|---|---|---|---|---|---|
| REN ∥ | Renin | ES | Yes | No | Yes | Yes | 53 (48–102) | 7.1†† | −10.1†† | … |
| NGFR | Nerve growth factor receptor | PM | No | Yes | Yes | Yes | 86 (51–98) | −2.7** | 2.6** | … |
| CNGA3 | Cyclic nucleotide gated channel subunit alpha 3 | PM | Yes | No | No | No | 93 (85–126) | 2.6** | −3.3†† | … |
| SLC5A1 | Solute carrier family 5 member 1 | PM | Yes | No | No | No | 358 (197–574) | −2.4# | 2.5# | … |
| OIT3 | Oncoprotein induced transcript 3 | N | Yes | No | No | No | 29 (15–33) | 2.2** | 2.6# | … |
| HPCA | Hippocalcin | C | Yes | Yes | No | No | 50 (40–59) | −2.2** | 1.9# | … |
| FSTL3 | Follistatin like 3 | ES | Yes | No | No | No | 103 (91–126) | 2.1** | −1.9# | … |
| MUC20 | Mucin 20, cell surface associated | PM | No | Yes | No | No | 1280 (889–1441) | −2.0** | 1.9# | … |
| IGFBP3 | Insulin like growth factor binding protein 3 | ES | Yes | Yes | No | Yes | 495 (373–668) | −2.0# | 2.0# | … |
| STAT3 | Signal transducer and activator of transcription 3 | N | Yes | No | Yes | Yes | 1182 (1060–1274) | 2.0# | −1.9# | … |
| TXNDC16 | Thioredoxin domain containing 16 | ES | Yes | No | No | No | 255 (220–352) | −1.9†† | 1.7# | … |
| HSP90AA1 | Heat shock protein 90 alpha family class A member 1 | C | No | Yes | Yes | Yes | 10 647 (9601–11 011) | 1.7** | −1.5# | … |
| LARP1B | La ribonucleoprotein domain family member 1B | ES | Yes | No | No | No | 159 (153–195) | 1.6** | −1.5# | … |
| GLTP | Glycolipid transfer protein | C | No | Yes | No | No | 798 (734–914) | −1.5†† | 1.6†† | … |
| SIPA1L1 | Signal induced proliferation associated 1 like 1 | C | Yes | No | No | No | 820 (762–1283) | −1.5# | 1.7# | … |
| CAPN1 | Calpain 1 | C | No | Yes | No | Yes | 1448 (1204–1595) | −1.5** | 1.4# | … |
| KLF7 | Kruppel like factor 7 | N | Yes | No | No | No | 567 (524–625) | 1.4†† | −1.3# | … |
| PDE5A | Phosphodiesterase 5A | C | Yes | No | Yes | Yes | 2088 (1901–2231) | −1.4†† | 1.4†† | … |
| HSP90B1 | Heat shock protein 90 beta family member 1 | C | Yes | Yes | Yes | Yes | 8974 (8708–9585) | 1.4# | −1.4# | … |
AKI indicates acute kidney injury; B, baseline; C, cytoplasm; ES, extracellular space; HF, heart failure; N, nucleus; PM, plasma membrane; and R, recovery.
*Gene/protein product targeted by 1 or more pharmaceutical drugs.
†Genes associated with AKI in the Harmonizome database 12 and Ingenuity Pathway Analysis knowledgebase. 13
‡Normalized RNA sequencing read counts at B expressed as median (interquartile range).
§Expression fold changes between B, HF, and R.
||Genes enriched, or specifically expressed in, kidney (www.proteinatlas.org). 14
¶ P<0.05, # P<0.01, ** P<0.001, and †† P<0.0001 after adjustment for multiple comparisons.
Table 2.
Dysregulated Genes Encoding Candidate‐Sustained Biomarkers of Kidney Damage or Dysfunction in Acute Decompensated Heart Failure
| Symbol | Name | Cellular location | Plasma/serum | Urine | Known drug target* | Previous association with AKI† | Normalized read counts at baseline‡ | Fold change B‐HF§ ∥ | Fold change HF‐R§ ∥ | Fold change B‐R§ ∥ |
|---|---|---|---|---|---|---|---|---|---|---|
| ADIPOQ | Adiponectin, C1Q, and collagen domain containing | ES | Yes | No | No | Yes | 1 (0–182) | −394** | −307¶ | |
| ACTG2 | Actin gamma 2, smooth muscle | C | No | Yes | No | No | 45 (12–241) | −7.6¶ | −17.9¶ | |
| ANGPTL4 | Angiopoietin like 4 | ES | Yes | No | No | Yes | 193 (121–273) | −4.8** | −5.0# | |
| DES | Desmin | C | Yes | No | No | Yes | 378 (266–882) | −4.6¶ | ||
| PCOLCE2 | Procollagen C‐endopeptidase enhancer 2 | ES | No | Yes | No | No | 45 (26–87) | −2.7¶ | ||
| SLC52A3 | Solute carrier family 52 member 3 | PM | Yes | No | No | No | 260 (195–428) | −3.4** | −2.6¶ | |
| FLT4 | Fms related tyrosine kinase 4 | PM | Yes | No | Yes | No | 401 (280–510) | −2.4** | −1.8¶ | |
| BGN | Biglycan | ES | No | Yes | No | No | 1720 (1430–2272) | −1.8¶ | ||
| CFI | Complement factor I | ES | Yes | Yes | No | No | 1882 (1783–2191) | 1.5¶ | 1.7¶ | |
| PDGFRB | Platelet derived growth factor receptor beta | PM | No | Yes | Yes | Yes | 1324 (1105–1676) | −1.6¶ | ||
| ACADVL | Acyl‐CoA dehydrogenase very long chain | C | Yes | No | No | No | 5918 (5389–6703) | −1.5** | −1.5¶ | |
| CA2 | Carbonic anhydrase 2 | C | Yes | Yes | Yes | Yes | 12 771 (10 670–14 644) | −1.4¶ | ||
| PLEKHA8 | Pleckstrin homology domain containing A8 | ES | Yes | No | No | No | 499 (473–532) | 1.4¶ |
AKI indicates acute kidney injury; B, baseline; C, cytoplasm; ES, extracellular space; HF, heart failure; PM, plasma membrane; and R, recovery.
*Gene/protein product is targeted by 1 or more pharmaceutical drugs.
†Genes associated with AKI in the Harmonizome database 12 and Ingenuity Pathway Analysis knowledgebase. 13
‡Normalized RNA sequencing read counts at B expressed as median (interquartile range).
§Expression fold changes between B, HF, and R.
∥ P<0.05, ¶ P<0.01, # P<0.001, and ** P<0.0001 after adjustment for multiple comparisons.
Lastly, to explore the relationship between each of the genes that potentially translate to protein biomarkers with neurohormones and with hemodynamic measurements, correlation matrices were generated with data from 5 sheep for whom full serial measurements were available (Figures S8 and S9). Comparing changes from baseline to HF, HF‐responsive candidate biomarker generating genes clustered into 6 groups with similar profiles. These were (1) MUC20 (mucin 20) with urine urea and creatinine concentrations; (2) IGFBP3 (insulin like growth factor binding protein 3) and HPCA (hippocalcin) with fluid balance; (3) REN and LARP1B (La ribonucleoprotein domain family member 1B) with urine volume; (4) NGFR (nerve growth factor receptor), STAT3 (signal transducer and activator of transcription 3), PDE5A (phosphodiesterase 5A), CAPN1 (calpain 1), SIPA1L1 (signal induced proliferation associated 1 like 1), GLTP (glycolipid transfer protein), SLC5A1 (solute carrier family 5 member 1), and TXNDC16 (thioredoxin domain containing 16) with plasma urea and creatinine concentrations; (5) CNGA3 (cyclic nucleotide gated channel subunit alpha 3) and OIT3 (oncoprotein induced transcript 3) with MAP and renal perfusion pressure; and (6) HSP90AA1 (heat shock protein 90 alpha family class A member 1), KLF7 (Kruppel like factor 7), FSTL3 (follistatin like 3), and HSP90B1 (heat shock protein 90 beta family member 1) with PRA and plasma BNP concentrations (Figure S8A). In general, these relationships were not preserved from HF to recovery, suggesting a dynamic shift in relationships between gene expression, neurohormones, and hemodynamic responses as the model moves from developing ADHF through to recovery (Figure S8B). Similarly, comparing changes from baseline to recovery, HF‐sustained biomarkers clustered into 4 groups with similar profiles. These were (1) PDGFRB (platelet derived growth factor receptor beta) and SLC52A3 (solute carrier family 52 member 3) with urine sodium concentrations; (2) FLT4 (Fms related tyrosine kinase 4), PLEKHA8 (pleckstrin homology domain containing A8), CFI (complement factor I), and CA2 (carbonic anhydrase 2) with urine urea and creatinine concentrations; (3) ACADVL (acyl‐CoA dehydrogenase very long chain), PCOLCE2 (procollagen C‐endopeptidase enhancer 2), ACTG2 (actin gamma 2, smooth muscle), DES (desmin), ADIPOQ (adiponectin, C1Q, and collagen domain containing), and ANGPTL4 (angiopoietin like 4) with plasma creatinine concentrations; and (4) BGN (biglycan) with PRA and plasma BNP concentrations (Figure S9).
DISCUSSION
The present study exploits the unique advantages of a large, comprehensively instrumented, animal model in which ADHF can be reproducibly induced and then resolved. Serial physiological and kidney responses during ADHF induction and recovery showed that the pacing‐induced impairments in systemic/regional hemodynamics and widespread neurohumoral activation were associated with a decline in renal function as evidenced by avid sodium and volume retention, elevation of plasma creatinine concentrations, and reductions in creatinine excretion and clearance. Importantly, following pacing cessation and 25 days of recovery from ADHF while hemodynamic and hormonal indexes essentially returned to prepacing baseline levels, a degree of renal dysfunction persisted. These data suggest that an episode of ADHF on a background of previously normal kidneys can result in AKI that fails to fully resolve after ≈4 weeks recovery, leading to a permanent decrement in renal function.
This suggestion is supported by serial kidney histology. Whereas prepacing kidney biopsies were generally noted as having no significant abnormalities, this was not the case for specimens taken either during ADHF or after 25 days of recovery. More than half of the ADHF samples showed evidence of early mild acute tubular injury, as did recovery specimens. Furthermore, nearly half of the recovery biopsies exhibited focal or scattered dystrophic calcification—a deposition of calcium salts that commonly occurs in degenerated or necrotic cells/tissue as a result of damage or injury 17 and can also indicate the presence of ongoing organ dysfunction. A conspicuous histopathological feature of the majority of biopsies taken following ADHF development was the prominence of glomerular MCs, an anomaly that generally still presents after 4 weeks of ADHF recovery. MC proliferation is a nonspecific response to assorted forms of glomerular insult and is a feature of many human and experimental kidney diseases. 18 Stimulation of MC replication is reported to occur via multiple mechanisms including hypoxia, inflammation, podocyte injury 19 and a large number of chemical mediators that encompass cytokines, growth factors, and hormones, with the latter including AngII (angiotensin II), aldosterone, endothelin‐1, and AVP 20 —all of which were elevated during ADHF in the current study (with increments in PRA implying concurrent elevation of AngII). Although a transient increase in MCs is thought to occur in response to glomerular injury as part of the process of tissue repair or “wound healing,” persistent MC proliferation caused by repetitive or more sustained glomerular injury is believed to precede, as well as promote, irreversible glomerular scarring and subsequent loss of renal function. 18 Indeed, abnormally proliferative MCs are themselves able to release mediators of proliferation, inflammation, and fibrosis (eg, AngII) 21 and to increase mesangial extracellular matrix accumulation, leading to progressive glomerulosclerosis and interstitial fibrosis. 22 Of particular significance, this process of tissue remodeling, mesangial expansion, and glomerular scarring progresses even after termination of the initial noxious stimulus, regardless of the type of glomerular injury. 23
Consistent with a process of progressive reparative glomerular fibrosis/scarring, the present study observed evidence of renal fibrosis subsequent to ADHF‐induced AKI, with histological signs of interstitial medullary fibrosis (mild focal or patchy) noted only in recovery biopsies. The continued renal impairment (ie, persistent reductions in creatinine excretion and clearance) seen in recovery is congruent with the loss of function typically resulting from the excess accumulation of the extracellular matrix and fibrosis as normal tissue architecture is replaced with scar tissue. In agreement with our findings, work from experimental studies of AKI in mice support the notion that fibrosis is ongoing after the initial AKI, with activated fibroblasts failing to return to their resting state. 24 Our results are also in keeping with the clinical observation that renal interstitial fibrosis is the strongest indicator of progression of chronic kidney disease (even if the primary disease is glomerular). 25
Although findings from multiple experimental and clinical studies have demonstrated that extended periods of chronic HF result in progressive renal pathology, 26 conclusions regarding the impact of ADHF on kidney structure/function are far less definitive. Observational studies in several patient groups, including those hospitalized for acute myocardial infarction and/or percutaneous coronary revascularization, have found that those patients diagnosed with an episode of AKI were more likely to develop end‐stage renal disease or chronic kidney disease, 27 with the severity, duration, and frequency of AKI appearing to be important predictors of outcomes. Furthermore, the development of chronic kidney disease in patients with idiopathic dilated cardiomyopathy without renal insufficiency at initial diagnosis was independently related to the frequency of HF admissions, 28 and the likelihood of patients hospitalized for ADHF developing worsening renal function increased in those presenting with renal dysfunction at baseline. 29 In other studies looking at acute worsening of kidney function in patients hospitalized with HF, a prospective, multicenter cohort registry 30 reported that of a total of 5625 patients, worsening renal function (WRF) occurred in 55.1% of patients and was an independent predictor of in‐hospital, short‐term (3‐month) and long‐term (1‐year) mortality. Among those patients with WRF, 61.9% showed transient WRF (recovery of serum creatinine level at discharge; mean hospitalization, 14.0±17.6 days) and 38.1% showed persistent WRF (nonrecovered creatinine level at discharge). Importantly, even transient WRF was shown to be a risk factor for 1‐year mortality compared with no WRF, with persistent WRF according no additive risk compared with transient WRF (despite this patient group being overall “sicker”). In another cohort of 27 309 patients from ADHERE (Acute Decompensated Heart Failure National Registry), 31 both transient (11.7%) and persistent (20.3%) WRF were associated with higher risks of 90‐day postadmission mortality compared with those without WRF (68.0%), although in this study persistent WRF was associated with a higher risk relative to transient WRF. These data suggest that WRF, whether transient or persistent, represents a significant event during acute HF that, even despite measurable recovery, has significant consequences beyond the initial episode. Although all of these works provide anecdotal evidence that episodes of acute exacerbation of HF are associated with the occurrence of AKI and development/progression of renal impairment, the current study presents direct serial functional and histological data showing that a single episode of ADHF can cause AKI, which has consequences and implications beyond the acute event—with tubular injury, cellular necrosis (as indicated by dystrophic calcifications), and glomerular scarring accompanied by a permanent loss of renal function. Of note, this loss of function, although relatively minor, was consistently demonstrated across all animals studied.
Many of the triggers/processes believed to contribute to the initial renal injury following an episode of HF decompensation, as well as promote subsequent progressive renal pathology and functional decline, were evident in the current study. These include reductions in renal perfusion pressure (resulting in low‐grade ischemia, hypoxia, and loss of kidney autoregulation), 32 increases in CVP (causing renal congestion and leading to increased renal interstitial pressure, tubular compression, and stimulation of inflammatory mediators), 33 and maladaptive neurohormonal activation. Notably, this includes increased sympathetic nervous traffic (which increases renal vascular resistance, augments renal renin release, and induces oxidative stress 34 ), endothelin‐1 (a potent renal vasoconstrictor that also possesses inflammatory and mitogenic properties and stimulates MC contraction and extracellular matrix production 35 ), AVP (stimulates MC proliferation and renin secretion and alters renal hemodynamics 36 ), and the renin‐angiotensin‐aldosterone system. Activation of the renin‐angiotensin‐aldosterone system axis and its main effector AngII, in particular, is considered pivotal to the pathogenesis of much renal disease, 37 with the peptide shown to exacerbate the initial injury and promote fibrosis both directly and indirectly via stimulation of aldosterone (promotes renal inflammatory and fibrotic processes, podocyte injury, and MC proliferation 20 ) and upregulation of numerous reactive oxygen species, chemokines, growth factors, and cytokines, most especially TGF‐β (transforming growth factor β)—a central regulator/mediator of fibrosis following AKI. 38
In an effort to elucidate the pathways involved in the development and recovery of AKI in ADHF, we performed RNA‐seq in sequential kidney biopsies (baseline, HF, recovery), identifying altered expression of >800 genes. Changes in gene expression were consistent with repression of the reno‐protective eNOS signaling pathway during ADHF development and activation of glomerulosclerosis and reno‐protective pathways and repression of proinflammatory and profibrotic pathways during ADHF recovery. Three observations can be made from these data. First, renal inflammation and ischemia (as a consequence of ADHF) markedly altered renal gene expression, potentially leading to a reduction in local generation of nitric oxide—an established process in the "injury phase" of AKI that can promote further tissue damage and initiate an adaptive or maladaptive repair response. 39 Second, recovery from ADHF was associated with a further substantial shift in renal gene expression involving more than twice the number of genes associated with ADHF development. These gene expression changes may simultaneously serve to (1) restore the expression of many genes altered during ADHF development to baseline levels, (2) promote glomerulosclerosis (consistent with histological analysis), and (3) mediate the switch toward healing and repair (consistent with the partial improvement in renal function during recovery), as evidenced by activation of reno‐protective pathways (cholesterol biosynthesis 40 and PPAR 41 signaling) and repression of proinflammatory (acute phase response 42 and oncostatin M 43 signaling), platelet activation (GP6 signaling 44 ), and profibrotic pathways (PDGF signaling 45 and "hepatic" fibrosis 39 ). Third, although most gene expression changes were transient, the expression of 47 genes remained altered (relative to baseline) after 4 weeks of recovery, suggesting that ADHF promotes a persistent shift in renal gene expression. These long‐term alterations may be associated with incomplete resolution of injury or maladaptive changes in kidney structure and potentially predispose to long‐term renal impairment. 39 Although all pathways have established roles in AKI, this study is the first to suggest their direct involvement in kidney injury and repair in ADHF.
Consistent with previous studies of AKI, 46 our data suggest that changes in renal gene expression during ADHF development and recovery may be driven by a network of inflammatory cytokines centred on IL‐1β. A recent post hoc analysis of CANTOS (Canakinumab Anti‐inflammatory Thrombosis Outcome Study) 47 suggested that selective targeting of IL‐1β improves cardiovascular outcomes in patients with chronic kidney disease and a history of myocardial infarction and systemic inflammation over a median of 3.7 years of follow‐up. 48 Although the precise mechanism of action was not described and there was no overall benefit on renal outcomes in this trial, these findings suggest that inhibition of IL‐1β may have therapeutic potential in cardiorenal disease, including AKI in ADHF.
Our data provide the first mechanistic insights into the molecular changes associated with AKI during the development of, and recovery from, ADHF. These data add to work by Pleasant et al, 49 who used RNA sequencing to investigate renal gene expression in murine pre‐symptomatic HF. In kidney tissue collected at 24 weeks, Pleasant et al identified 6 genes that differed by >1.5‐fold between transgenic mice that develop cardiomyopathy and later progress to HF (after 24 weeks) and matched wild‐type controls. We confirmed the involvement of 3 of the 6 genes (renin, arginase 2, α‐1 antitrypsin), supporting the idea that a small number of genes associated with AKI in established ADHF may also be altered before the onset of overt symptomatic HF and promote susceptibility to AKI. Beyond ADHF, we observed a number of genes in common with studies investigating early transcriptional responses to kidney injury and repair (<24 hours) in rodent models of renal ischemia/reperfusion. 50 , 51 , 52 Together these data suggest that AKI is associated with activation and repression of hundreds of renal genes, each with its own temporal profile, of which a small proportion may represent common pathways shared by multiple AKI etiologies.
To prioritize candidate markers for AKI in ADHF, we identified 32 genes encoding proteins detectable in urine or blood that were strongly responsive to ADHF (n=19) or showed a persistent change in expression (n=13). Of these, 13 have previously been implicated in AKI, but only 1, REN, was previously investigated in clinical studies of AKI in ADHF. 16 The HF‐responsive biomarkers showed a very similar magnitude and opposite direction of fold change between baseline and HF and HF and recovery, indicating a strong relationship with kidney injury/repair and highlighting their potential as markers for kidney repair in ADHF. Among these, CNGA3 (required for normal vision and olfactory signal transduction 14 ) and OIT3 (potentially involved in liver development and function 14 ) had large fold changes in expression and were correlated with renal perfusion pressure, an important determinant of renal function. Similarly, 2 of the HF‐sustained biomarkers, ACTG2 (a component of the cytoskeleton that mediates cell motility 14 ) and ANGPTL4 (a multifunctional protein that regulates lipid metabolism and apoptosis 14 ), showed consistent large decreases in expression across more than 1 time point. These sustained changes in gene expression may reflect incomplete resolution of injury or maladaptive changes in kidney structure that persist beyond other indexes of kidney injury, suggesting that they may be markers of long‐term renal impairment in ADHF.
Our study has several limitations. First, we were unable to obtain serial kidney biopsies at >3 time points owing to the invasive and highly technically demanding nature of sampling. Second, we did not detect altered expression of genes encoding emerging markers of AKI, such as LCN2 (which encodes NGAL [neutrophil gelatinase‐associated lipocalin]), IGFBP7 (insulin‐like growth factor‐binding protein 7), and TIMP2 (tissue inhibitor of metalloproteinases 2). Thus, although our data provide exciting novel mechanistic insights into the molecular changes associated with AKI in ADHF, we may have missed early events. Third, pathway analysis could only be performed for sheep genes for which there is an established human or rodent homologue. Consequently, several predicted protein‐coding genes displaying the greatest differences in expression between time points (potentially representing novel pathways and biomarkers of AKI in ADHF) were excluded from our analysis. Future work will investigate whether changes in renal gene expression can be detected at the protein level, and if these changes are detectable in circulation.
In summary (see Figure 7), we have characterized the events leading to kidney injury and repair during the development of, and recovery from, ADHF (using an ovine model). By assessing changes in hormonal, hemodynamic, biochemical, and urine measures of cardio‐renal injury and repair, we provide the first direct evidence that an episode of ADHF leads to a decline in kidney function that fails to fully resolve after ≈4 weeks and is associated with important, potentially lasting, kidney injury. Renal gene expression profiling provides the first insight into the pathways associated with kidney injury and repair in ADHF, highlighting the potential importance of inflammatory mediators in regulating these processes. We have prioritized 31 novel candidates as biomarkers for AKI in the setting of ADHF.
Figure 7. Summary of physiological and kidney histological and transcriptomic responses to development of, and recovery from, ADHF.
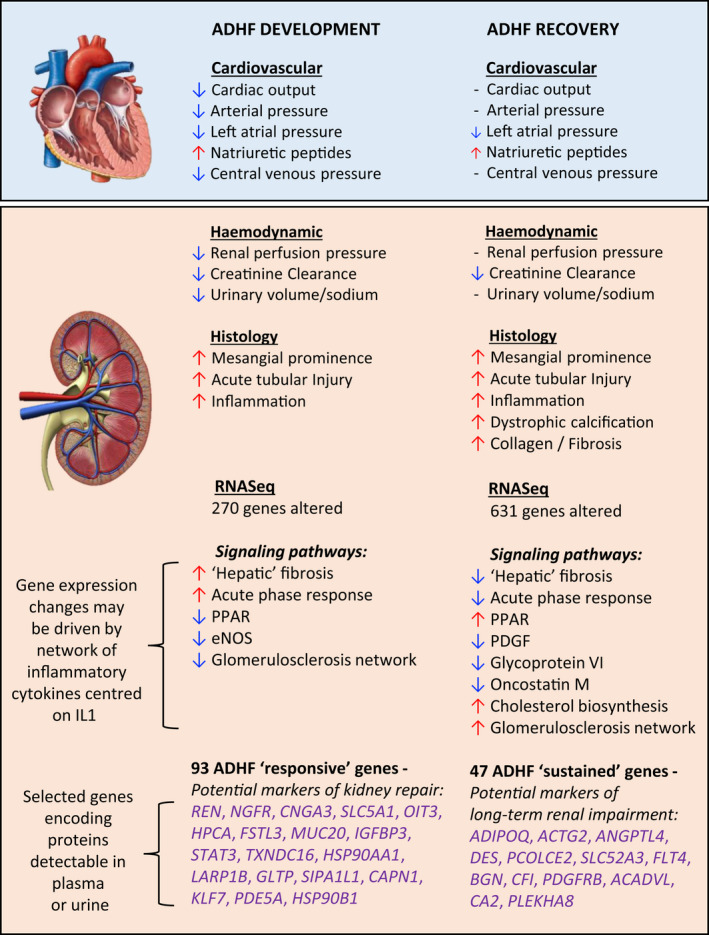
ACADVL indicates acyl‐CoA dehydrogenase very long chain; ACTG2, actin gamma 2, smooth muscle; ADHF, acute decompensated heart failure; ADIPOQ, adiponectin, C1Q, and collagen domain containing; ANGPTL4, angiopoietin like 4; BGN, biglycan CA2, carbonic anhydrase 2; CAPN1, calpain 1; CFI, complement factor I; CNGA3, cyclic nucleotide gated channel subunit alpha 3; DES, desmin; eNOS, endothelial nitric oxide synthase; FLT4, Fms related tyrosine kinase 4; FSTL3, follistatin like 3; GLTP, glycolipid transfer protein; HPCA, hippocalcin; HSP90AA1, heat shock protein 90 alpha family class A member 1; HSP90B1, heat shock protein 90 beta family member 1; IGFBP3, insulin like growth factor binding protein 3; IL1β, interleukin 1β; KLF7, Kruppel like factor 7; LARP1B, La ribonucleoprotein domain family member 1B; MUC20, mucin 20; NGFR, nerve growth factor receptor; OIT3, oncoprotein induced transcript 3; PCOLCE2, procollagen C‐endopeptidase enhancer 2; PDE5A, phosphodiesterase 5A; PDGF, platelet‐derived growth factor; PDGFRB, platelet derived growth factor receptor beta; PLEKHA8, pleckstrin homology domain containing A8; PPAR, peroxisome proliferator‐activated receptor; REN, renin; SIPA1L1, signal induced proliferation associated 1 like 1; SLC5A1, solute carrier family 5 member 1; SLC52A3, solute carrier family 52 member 3; STAT3, signal transducer and activator of transcription 3; and TXNDC16, thioredoxin domain containing 16.
Sources of Funding
This work was supported by grants from the Heart Foundation of New Zealand (Grant 1562) and Health Research Council of New Zealand (Grant 14/521).
Disclosures
None.
Supporting information
Data S1
Tables S1–S3
Figures S1–S9
Acknowledgments
We are grateful to the staff of the University of Otago–Christchurch Animal Laboratory for animal care, the staff of our Translational BioDiscovery Laboratory for performing hormone assays, and the Otago Genomics and Bioinformatics Facility (University of Otago–Dunedin, New Zealand) for performing RNA sequencing and assisting with bioinformatics analysis.
Supplementary Material for this article is available at https://www.ahajournals.org/doi/suppl/10.1161/JAHA.121.021312
For Sources of Funding and Disclosures, see page 16.
REFERENCES
- 1. Lloyd‐Jones DM, Larson MG, Leip EP, Beiser A, D'Agostino RB, Kannel WB, Murabito JM, Vasan RS, Benjamin EJ, Levy D, et al. Lifetime risk for developing congestive heart failure: the Framingham Heart Study. Circulation. 2002;106:3068–3072. doi: 10.1161/01.CIR.0000039105.49749.6F [DOI] [PubMed] [Google Scholar]
- 2. Taylor CJ, Ordonez‐Mena JM, Roalfe AK, Lay‐Flurrie S, Jones NR, Marshall T, Hobbs FDR. Trends in survival after a diagnosis of heart failure in the United Kingdom 2000–2017: population based cohort study. BMJ. 2019;364:l223. doi: 10.1136/bmj.l223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Fonarow GC, Adams KF Jr, Abraham WT, Yancy CW, Boscardin WJ; Adhere Scientific Advisory Committee SG and Investigators . Risk stratification for in‐hospital mortality in acutely decompensated heart failure: classification and regression tree analysis. JAMA. 2005;293:572–580. doi: 10.1001/jama.293.5.572 [DOI] [PubMed] [Google Scholar]
- 4. Heywood JT, Fonarow GC, Costanzo MR, Mathur VS, Wigneswaran JR, Wynne J; Committee ASA and Investigators . High prevalence of renal dysfunction and its impact on outcome in 118,465 patients hospitalized with acute decompensated heart failure: a report from the ADHERE database. J Card Fail. 2007;13:422–430. doi: 10.1016/j.cardfail.2007.03.011 [DOI] [PubMed] [Google Scholar]
- 5. Gottlieb SS, Abraham W, Butler J, Forman DE, Loh E, Massie BM, O'Connor CM, Rich MW, Stevenson LW, Young J, et al. The prognostic importance of different definitions of worsening renal function in congestive heart failure. J Card Fail. 2002;8:136–141. doi: 10.1054/jcaf.2002.125289 [DOI] [PubMed] [Google Scholar]
- 6. Bock JS, Gottlieb SS. Cardiorenal syndrome: new perspectives. Circulation. 2010;121:2592–2600. doi: 10.1161/CIRCULATIONAHA.109.886473 [DOI] [PubMed] [Google Scholar]
- 7. Sarraf M, Masoumi A, Schrier RW. Cardiorenal syndrome in acute decompensated heart failure. Clin J Am Soc Nephrol. 2009;4:2013–2026. doi: 10.2215/CJN.03150509 [DOI] [PubMed] [Google Scholar]
- 8. Rademaker MT, Charles CJ, Richards AM. Urocortin 1 administration from the onset of rapid left ventricular pacing represses progression to overt heart failure. Am J Physiol. 2007;293:H1536–H1544. doi: 10.1152/ajpheart.00377.2007 [DOI] [PubMed] [Google Scholar]
- 9. Jain A, Tuteja G. TissueEnrich: tissue‐specific gene enrichment analysis. Bioinformatics. 2019;35:1966–1967. doi: 10.1093/bioinformatics/bty890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. R Core Team . R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing; 2019. Available at: http://www.R‐project.org/. Accessed February 1, 2019. [Google Scholar]
- 11. Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA‐seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Rouillard AD, Gundersen GW, Fernandez NF, Wang Z, Monteiro CD, McDermott MG, Ma'ayan A. The harmonizome: a collection of processed datasets gathered to serve and mine knowledge about genes and proteins. Database (Oxford). 2016;2016:baw100. doi: 10.1093/database/baw100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kramer A, Green J, Pollard J Jr, Tugendreich S. Causal analysis approaches in Ingenuity Pathway Analysis. Bioinformatics. 2014;30:523–530. doi: 10.1093/bioinformatics/btt703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Uhlen M, Fagerberg L, Hallstrom BM, Lindskog C, Oksvold P, Mardinoglu A, Sivertsson A, Kampf C, Sjostedt E, Asplund A, et al. Proteomics. Tissue‐based map of the human proteome. Science. 2015;347:1260419. doi: 10.1126/science.1260419 [DOI] [PubMed] [Google Scholar]
- 15. Wei T, Simko V. R package "corrplot": visualization of a correlation matrix (version 0.84). 2017. Available at: https://github.com/taiyun/corrplot. Accessed July 17, 2017.
- 16. Yogasundaram H, Chappell MC, Braam B, Oudit GY. Cardiorenal syndrome and heart failure‐challenges and opportunities. Can J Cardiol. 2019;35:1208–1219. doi: 10.1016/j.cjca.2019.04.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Giachelli CM. Ectopic calcification: gathering hard facts about soft tissue mineralization. Am J Pathol. 1999;154:671–675. doi: 10.1016/S0002-9440(10)65313-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Schocklmann HO, Lang S, Sterzel RB. Regulation of mesangial cell proliferation. Kidney Int. 1999;56:1199–1207. doi: 10.1046/j.1523-1755.1999.00710.x [DOI] [PubMed] [Google Scholar]
- 19. Schlondorff D, Banas B. The mesangial cell revisited: no cell is an island. J Am Soc Nephrol. 2009;20:1179–1187. doi: 10.1681/ASN.2008050549 [DOI] [PubMed] [Google Scholar]
- 20. Briet M, Schiffrin EL. Aldosterone: effects on the kidney and cardiovascular system. Nat Rev Nephrol. 2010;6:261–273. doi: 10.1038/nrneph.2010.30 [DOI] [PubMed] [Google Scholar]
- 21. Vidotti DB, Casarini DE, Cristovam PC, Leite CA, Schor N, Boim MA. High glucose concentration stimulates intracellular renin activity and angiotensin II generation in rat mesangial cells. Am J Physiol Renal Physiol. 2004;286:F1039–F1045. doi: 10.1152/ajprenal.00371.2003 [DOI] [PubMed] [Google Scholar]
- 22. Kriz W, Gretz N, Lemley KV. Progression of glomerular diseases: is the podocyte the culprit? Kidney Int. 1998;54:687–697. doi: 10.1046/j.1523-1755.1998.00044.x [DOI] [PubMed] [Google Scholar]
- 23. Kashgarian M, Sterzel RB. The pathobiology of the mesangium. Kidney Int. 1992;41:524–529. doi: 10.1038/ki.1992.74 [DOI] [PubMed] [Google Scholar]
- 24. Bechtel W, McGoohan S, Zeisberg EM, Muller GA, Kalbacher H, Salant DJ, Muller CA, Kalluri R, Zeisberg M. Methylation determines fibroblast activation and fibrogenesis in the kidney. Nat Med. 2010;16:544–550. doi: 10.1038/nm.2135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Barnes JL, Glass WF. Renal interstitial fibrosis: a critical evaluation of the origin of myofibroblasts. Contrib Nephrol. 2011;169:73–93. [DOI] [PubMed] [Google Scholar]
- 26. Cruz DN, Schmidt‐Ott KM, Vescovo G, House AA, Kellum JA, Ronco C, McCullough PA. Pathophysiology of cardiorenal syndrome type 2 in stable chronic heart failure: workgroup statements from the eleventh consensus conference of the Acute Dialysis Quality Initiative (ADQI). Contrib Nephrol. 2013;182:117–136. doi: 10.1159/000349968 [DOI] [PubMed] [Google Scholar]
- 27. Chawla LS, Kimmel PL. Acute kidney injury and chronic kidney disease: an integrated clinical syndrome. Kidney Int. 2012;82:516–524. doi: 10.1038/ki.2012.208 [DOI] [PubMed] [Google Scholar]
- 28. Tanaka T, Nangaku M. The role of hypoxia, increased oxygen consumption, and hypoxia‐inducible factor‐1 alpha in progression of chronic kidney disease. Curr Opin Nephrol Hypertens. 2010;19:43–50. doi: 10.1097/MNH.0b013e3283328eed [DOI] [PubMed] [Google Scholar]
- 29. Han SW, Ryu KH. Renal dysfunction in acute heart failure. Korean Circ J. 2011;41:565–574. doi: 10.4070/kcj.2011.41.10.565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kang J, Park JJ, Cho Y‐J, Oh I‐Y, Park H‐A, Lee SE, Kim M‐S, Cho H‐J, Lee H‐Y, Choi JO, et al. Predictors and prognostic value of worsening renal function during admission in HFpEF Versus HFrEF: data from the KorAHF (Korean Acute Heart Failure) Registry. J Am Heart Assoc. 2018;7:e007910. doi: 10.1161/JAHA.117.007910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Krishnamoorthy A, Greiner MA, Sharma PP, DeVore AD, Johnson KW, Fonarow GC, Curtis LH, Hernandez AF. Transient and persistent worsening renal function during hospitalization for acute heart failure. Am Heart J. 2014;168:891–900. doi: 10.1016/j.ahj.2014.08.016 [DOI] [PubMed] [Google Scholar]
- 32. Basile DP, Anderson MD, Sutton TA. Pathophysiology of acute kidney injury. Compr Physiol. 2012;2:1303–1353. doi: 10.1002/cphy.c110041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Afsar B, Ortiz A, Covic A, Solak Y, Goldsmith D, Kanbay M. Focus on renal congestion in heart failure. Clin Kidney J. 2016;9:39–47. doi: 10.1093/ckj/sfv124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Rafiq K, Noma T, Fujisawa Y, Ishihara Y, Arai Y, Nabi AHMN, Suzuki F, Nagai Y, Nakano D, Hitomi H, et al. Renal sympathetic denervation suppresses de novo podocyte injury and albuminuria in rats with aortic regurgitation. Circulation. 2012;125:1402–1413. doi: 10.1161/CIRCULATIONAHA.111.064097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Neuhofer W, Pittrow D. Role of endothelin and endothelin receptor antagonists in renal disease. Eur J Clin Invest. 2006;36(suppl 3):78–88. doi: 10.1111/j.1365-2362.2006.01689.x [DOI] [PubMed] [Google Scholar]
- 36. Meijer E, Boertien WE, Zietse R, Gansevoort RT. Potential deleterious effects of vasopressin in chronic kidney disease and particularly autosomal dominant polycystic kidney disease. Kidney Blood Press Res. 2011;34:235–244. doi: 10.1159/000326902 [DOI] [PubMed] [Google Scholar]
- 37. Ruster C, Wolf G. Renin‐angiotensin‐aldosterone system and progression of renal disease. J Am Soc Nephrol. 2006;17:2985–2991. doi: 10.1681/ASN.2006040356 [DOI] [PubMed] [Google Scholar]
- 38. Efstratiadis G, Divani M, Katsioulis E, Vergoulas G. Renal fibrosis. Hippokratia. 2009;13:224–229. [PMC free article] [PubMed] [Google Scholar]
- 39. Ferenbach DA, Bonventre JV. Mechanisms of maladaptive repair after AKI leading to accelerated kidney ageing and CKD. Nat Rev Nephrol. 2015;11:264–276. doi: 10.1038/nrneph.2015.3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Naito M, Bomsztyk K, Zager RA. Renal ischemia‐induced cholesterol loading: transcription factor recruitment and chromatin remodeling along the HMG CoA reductase gene. Am J Pathol. 2009;174:54–62. doi: 10.2353/ajpath.2009.080602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Cheng CF, Chen HH, Lin H. Role of PPARalpha and its agonist in renal diseases. PPAR Res. 2010;2010:345098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Rabb H, Griffin MD, McKay DB, Swaminathan S, Pickkers P, Rosner MH, Kellum JA, Ronco C; Acute Dialysis Quality Initiative Consensus XWG . Inflammation in AKI: current understanding, key questions, and knowledge gaps. J Am Soc Nephrol. 2016;27:371–379. doi: 10.1681/ASN.2015030261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Luyckx VA, Cairo LV, Compston CA, Phan WL, Mueller TF. Oncostatin M pathway plays a major role in the renal acute phase response. Am J Physiol Renal Physiol. 2009;296:F875–F883. doi: 10.1152/ajprenal.90633.2008 [DOI] [PubMed] [Google Scholar]
- 44. Jansen MPB, Florquin S, Roelofs J. The role of platelets in acute kidney injury. Nat Rev Nephrol. 2018;14:457–471. doi: 10.1038/s41581-018-0015-5 [DOI] [PubMed] [Google Scholar]
- 45. Ostendorf T, Boor P, van Roeyen CR, Floege J. Platelet‐derived growth factors (PDGFs) in glomerular and tubulointerstitial fibrosis. Kidney Int Suppl (2011). 2014;4:65–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Anders HJ. Of inflammasomes, alarmins: IL‐1beta and IL‐1alpha in kidney disease. J Am Soc Nephrol. 2016;27:2564–2575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ridker PM, Everett BM, Thuren T, MacFadyen JG, Chang WH, Ballantyne C, Fonseca F, Nicolau J, Koenig W, Anker SD, et al. Antiinflammatory therapy with canakinumab for atherosclerotic disease. N Engl J Med. 2017;377:1119–1131. doi: 10.1056/NEJMoa1707914 [DOI] [PubMed] [Google Scholar]
- 48. Ridker PM, MacFadyen JG, Glynn RJ, Koenig W, Libby P, Everett BM, Lefkowitz M, Thuren T, Cornel JH. Inhibition of interleukin‐1beta by canakinumab and cardiovascular outcomes in patients with chronic kidney disease. J Am Coll Cardiol. 2018;71:2405–2414. [DOI] [PubMed] [Google Scholar]
- 49. Pleasant L, Ma Q, Devarajan M, Parameswaran P, Drake K, Siroky B, Shay‐Winkler K, Robbins J, Devarajan P. Increased susceptibility to structural acute kidney injury in a mouse model of presymptomatic cardiomyopathy. Am J Physiol Renal Physiol. 2017;313:F699–F705. doi: 10.1152/ajprenal.00505.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Liu J, Krautzberger AM, Sui SH, Hofmann OM, Chen Y, Baetscher M, Grgic I, Kumar S, Humphreys B, Hide WA, et al. Cell‐specific translational profiling in acute kidney injury. J Clin Invest. 2014;124:1242–1254. doi: 10.1172/JCI72126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Sharifian R, Okamura DM, Denisenko O, Zager RA, Johnson A, Gharib SA, Bomsztyk K. Distinct patterns of transcriptional and epigenetic alterations characterize acute and chronic kidney injury. Sci Rep. 2018;8:17870. doi: 10.1038/s41598-018-35943-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Xu K, Rosenstiel P, Paragas N, Hinze C, Gao X, Huai Shen T, Werth M, Forster C, Deng R, Bruck E, et al. Unique transcriptional programs identify subtypes of AKI. J Am Soc Nephrol. 2017;28:1729–1740. doi: 10.1681/ASN.2016090974 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1
Tables S1–S3
Figures S1–S9