

Abstract
Background
We have shown previously that low‐density lipoprotein (LDL) can be oxidized in the lysosomes of macrophages, that this oxidation can be inhibited by cysteamine, an antioxidant that accumulates in lysosomes, and that this drug decreases atherosclerosis in LDL receptor–deficient mice fed a high‐fat diet. We have now performed a regression study with cysteamine, which is of more relevance to the treatment of human disease.
Methods and Results
LDL receptor–deficient mice were fed a high‐fat diet to induce atherosclerotic lesions. They were then reared on chow diet and drinking water containing cysteamine or plain drinking water. Aortic atherosclerosis was assessed, and samples of liver and skeletal muscle were analyzed. There was no regression of atherosclerosis in the control mice, but cysteamine caused regression of between 32% and 56% compared with the control group, depending on the site of the lesions. Cysteamine substantially increased markers of lesion stability, decreased ceroid, and greatly decreased oxidized phospholipids in the lesions. The liver lipid levels and expression of cluster of differentiation 68, acetyl–coenzyme A acetyltransferase 2, cytochromes P450 (CYP)27, and proinflammatory cytokines and chemokines were decreased by cysteamine. Skeletal muscle function and oxidative fibers were increased by cysteamine. There were no changes in the plasma total cholesterol, LDL cholesterol, high‐density lipoprotein cholesterol, or triacylglycerol concentrations attributable to cysteamine.
Conclusions
Inhibiting the lysosomal oxidation of LDL in atherosclerotic lesions by antioxidants targeted at lysosomes causes the regression of atherosclerosis and improves liver and muscle characteristics in mice and might be a promising novel therapy for atherosclerosis in patients.
Keywords: antioxidant, atherosclerosis, lipoprotein, low‐density lipoprotein
Subject Categories: Atherosclerosis, Aortic Dissection, Vascular Disease
Nonstandard Abbreviations and Acronyms
- NASH
nonalcoholic steatohepatitis
- TA
tibialis anterior
Clinical Perspective
What Is New?
The large clinical trials of antioxidants to treat cardiovascular disease have been disappointing; we have shown previously that low‐density lipoprotein can be oxidized in the lysosomes of macrophages and that this can be inhibited by cysteamine, an antioxidant that accumulates in lysosomes.
Cysteamine reduces atherosclerotic lesion formation in low‐density lipoprotein receptor–deficient mice fed a high‐fat diet.
We now show that cysteamine causes the regression of existing atherosclerotic lesions in low‐density lipoprotein receptor–deficient mice by 32% to 56% and substantially increases markers of stability in the lesions, and it also decreases lipid levels and inflammatory cytokines in liver and improves skeletal muscle function.
What Are the Clinical Implications?
Treatments for patients with atherosclerosis would ideally cause their existing lesions to regress and become more stable.
Cysteamine has these effects in mice and has beneficial effects on nonalcoholic steatohepatitis and skeletal muscle.
Antioxidants, such as cysteamine, that accumulate in lysosomes and inhibit low‐density lipoprotein oxidation in these organelles should be tested in clinical trials of cardiovascular disease and nonalcoholic steatohepatitis.
Cardiovascular disease is the leading cause of death in the world. Atherosclerosis is the underlying cause of most cardiovascular diseases, including coronary artery disease thrombotic strokes and peripheral arterial disease. Atherosclerotic lesions are sites of lipid deposition and chronic inflammation. 1 Inflammation participates in atherosclerosis during initiation and throughout all stages of lesion development. Atherogenesis involves accumulation of plasma‐derived lipoproteins and inflammatory cells (monocyte‐derived macrophages, T lymphocytes, and mast cells) into the subintimal space following endothelial activation. Lipids from retained apolipoprotein B–containing lipoproteins are taken up by activated macrophages by phagocytosis, pinocytosis, and through receptors (eg, cluster of differentiation [CD] 36 or LDL receptor‐related protein [LRP]), transforming them into macrophage‐derived foam cells. 2 The death of foam cells eventually leads to a lipid‐rich necrotic core under a collagen‐rich fibrous cap, which stabilizes the plaque and prevents access of the bloodstream to its thrombogenic core. The advanced lesions are vulnerable to rupture, leading to thrombosis, which can occlude the arteries supplying the heart, causing a myocardial infarction, or the brain, causing a thrombotic stroke. The likelihood of plaque rupture depends on a complex balance between the composition (stability) of the plaque and the presence of external stimuli, such as inflammation, mechanical or shear stresses, and hyperglycemic episodes.
Cells involved in the atherosclerotic process secrete and are activated by proinflammatory cytokines, such as tumor necrosis factor (TNF)‐α, interleukin‐1β, interleukin‐6, and chemokine ligand 2 (CCL2) (monocyte chemoattractant protein‐1 [MCP‐1]). Interleukin‐1β and TNF‐α can regulate the expression of adhesion molecules involved in early and late leukocyte recruitment. 3 The Canakinumab Anti‐Inflammatory Thrombosis Outcomes Study (CANTOS) trial recently showed that limiting the bioavailability of interleukin‐1β using a monoclonal antibody reduced clinical cardiovascular events by 15% in a trial of >10 000 patients who had recently experienced a heart attack and had high CRP (C‐reactive protein) levels. 4
Dietary cholesterol is also a trigger of hepatic inflammation and might be involved in the evolution of the inflammatory process during atherosclerosis. 5 The inflammatory processes related to the uptake of lipoproteins or modified lipoproteins 6 occur in both arteries and liver; and the production of inflammatory cytokines by macrophages in atherosclerotic lesions and by Kupffer cells, the resident macrophages of the liver, contributes to atherosclerosis and nonalcoholic steatohepatitis (NASH). 7 The activation of Kupffer cells increases the release of lipid peroxidation products and cytokines, which generate oxidative stress. 7 Recent investigations have indicated that lysosomal cholesterol accumulation in Kupffer cells contributes to the inflammatory response during NASH. 8 The presence of inflammation in a fatty liver is the key feature of NASH and precedes further disease progression. As macrophages play a pivotal role in both atherosclerosis and NASH, it has been suggested that pharmacological intervention should involve targeting macrophage activation directly. 9
The generation of proinflammatory molecules has paracrine actions and affects nonvascular tissues at numerous sites around the body, including skeletal muscle. The impact of atherosclerosis on skeletal muscle and vice versa has been extensively studied. 10 A large body of evidence has not only shown that skeletal muscle function can modify progression of atherosclerosis 11 , 12 but also that atherosclerosis has a negative impact on both the structure and function of skeletal muscle. 13 Indeed, epidemiological studies have shown that attenuation of muscle mass and function is associated with subclinical atherosclerosis. 14 Therefore, atherosclerosis treatment should bring about improvements not only to the primary site of the lesions but also to indirectly affected parts of the body, including the liver and skeletal muscle.
Multiple studies suggest that the presence of lysosomal cholesterol accumulation in macrophages, and not the total amount of intracellular lipids, is critical for the observed inflammatory response. 15 , 16 We have shown that lysosomes in macrophages are a site of low‐density lipoprotein (LDL) oxidation. 17 Seven days after taking up mechanically aggregated (vortexed) LDL or sphingomyelinase aggregated LDL, mouse J774 or human THP‐1 macrophage‐like cells and human monocyte‐derived macrophages generated ceroid in their lysosomes. 17 , 18 , 19 Ceroid (lipofuscin) is a polymerized product of lipid oxidation found within foam cells in atherosclerotic lesions. 20 , 21 The lysosomal oxidation of LDL is catalyzed by oxidation‐reduction active iron present in the lysosomes of macrophages through the generation of hydroperoxyl radicals at the lysosomal pH of 4.5. 18 , 22 , 23 This oxidation is inhibited by cysteamine (2‐aminoethanethiol), 18 an antioxidant that accumulates in lysosomes. 24 Cysteamine is used in patients for the lysosomal storage disease cystinosis, caused by the absence of the lysosomal cystine transporter cystinosin. 25 Recently, we have shown that cysteamine reduces atherogenic conditions caused by lysosomal LDL oxidation, such as lysosomal dysfunction, cellular senescence, and secretion of various proinflammatory cytokines, such as interleukin‐1β, TNF‐α, and interleukin‐6, and chemokines, such as CCL2, in human macrophages. 19
Preventing the progression of atherosclerosis or inducing regression of atherosclerotic plaques is central to strategies aimed at improving cardiovascular prognosis. 26 , 27 We have recently shown that cysteamine decreases the development of atherosclerosis in LDL receptor–deficient mice fed a high cholesterol diet after 8 weeks 19 at a dose equivalent to that given to patients with cystinosis. 25 However, treatments for atherosclerosis are given to patients who already have the disease, and we have therefore performed a regression study of cysteamine on existing atherosclerotic lesions in mice and assessed its effects on hepatic inflammation and skeletal muscle function.
METHODS
The data that support the findings of this study are available from the corresponding author on reasonable request.
Chemicals and Reagents
Chemicals and reagents were purchased from Sigma‐Aldrich (Dorset, UK) or Fisher Scientific Ltd (Loughborough, UK), unless otherwise stated. Solutions were prepared using ultrapure water generated from a Barnstead Nanopure system.
Animals and Diets
All procedures complied with the Animals (Scientific Procedures) Act 1986 and were approved by the Research Ethics Committee of the University of Reading. Nine‐week‐old female LDL receptor–deficient mice (Charles River, Margate, UK) were fed with normal laboratory chow for a week. Sixty animals were divided into 3 groups and fed cholate‐free high‐fat diet (Diet W; SDS, Horley, Surrey, UK) containing cocoa butter (15%, w/w) and cholesterol (0.25%, w/w) for 8 weeks to induce atherosclerosis. One group was euthanized for baseline measurements. The remaining 40 mice were switched to a normal chow diet and divided into 2 groups to receive no cysteamine or cysteamine (Cysteamine.HCl; Sigma) at 2.2 mmol/L (equivalent to that given to patients with cystinosis; ie, 42 mg of the free base mg/kg body weight/day 28 , 29 in purified water [of high electrical resistivity, 15 MΩ‐cm]) as drinking water, which was changed every day to ensure the availability of cysteamine. 19 At the end of 8 weeks, the mice were weighed and euthanized.
Blood and Tissue Collection
Blood was taken immediately by cardiac puncture with EDTA as anticoagulant. The chest cavity was cut open to expose the heart and 10 mL of PBS at pH 7.4 was slowly perfused through the circulatory system by placing the needle of the PBS‐loaded syringe into the left ventricle with a cut in the right atrium as the outlet. The PBS flush was continued until the perfusate coming from the vena cava was clear. The perfusion was then changed to 4% (w/v) paraformaldehyde to fix the heart. Blood samples were centrifuged, and plasma aliquots were stored at −80 °C for the measurement of lipid profile with an ILab600 chemical analyzer with kits supplied by Instrumentation Laboratory. 30
Quantification of Atherosclerosis
Atherosclerotic lesion severity was assessed by both en face analysis of the aorta and serial sections from the aortic root. The heart, aortic root, aortic arch, and descending aorta were dissected as described previously. 31 The dissected hearts were fixed with 4% (w/v) paraformaldehyde, cryopreserved in sucrose, and embedded in optimal cutting temperature (OCT) before sectioning. 31 The aortic root was serially sectioned into 10‐µm sections from where the aortic sinus appeared until the point where valve bases were shrunken, but still visible. Four sections per slide were saved, resulting in a total of 20 slides saved. Every other slide was stained with Oil Red O, Harris hematoxylin, and Light Green, for lesion analysis, as described previously. 32
For en face analysis of aorta area containing atherosclerotic plaques, aortas were perfused and postfixed in 4% (w/v) paraformaldehyde. The whole aortas (from the sinotubular junction to aortic bifurcation) were dissected out under a dissection microscope and rinsed with saline to remove the blood. All adventitial adipose tissue was removed by careful dissection. The aortas were opened longitudinally, pinned to a Petri dish filled with Sylgard 184 silicone elastomer (Dow Corning), and then stained with Oil Red O for neutral lipids using a previously published method. 33
The lesions were quantified by drawing around the digital images of the lesions using ImageJ and measuring their areas, using a scale with a known distance. The lesion areas were expressed as percentage area in the aortic root, and the en face was covered by lesions in the aortic arch and in the rest of the thoracic plus abdominal aorta.
Aortic Root Composition
To measure the percentage of infiltrating monocytes/macrophages, the aortic root sections were treated with the anti‐monocyte+macrophage antibody (Abcam) followed by Alexa Fluor 568 secondary antibody (Thermo, A‐11077). Smooth muscle cells were stained with α‐actin (Sigma‐Aldrich) followed by secondary Alexa Fluor 488 (Thermo, A‐11078), and collagen was detected by Picrosirius Red. 34 The sections were stained with anti‐mouse CD206 antibody (BioLegend, 141709) and anti‐mouse CD86 antibody (BioLegend, 105002) to quantify anti‐inflammatory and proinflammatory macrophage phenotypes, respectively. 35 The necrotic core was defined as the anuclear area (negative for hematoxylin and eosin staining). 36
Ceroid
The aortic root sections were processed for the quantity of ceroid present in the lesions. 20 , 21 , 37 The sections mounted on glass slides were treated with xylene for 10 minutes to extract the free lipids. The slides were then treated with 60% (v/v) isopropanol for 5 minutes, followed by Oil Red O for 10 minutes to stain for ceroid. Excess stain was removed by washing with 60% (v/v) isopropanol, and images were taken using a Zeiss AxioSkop system and analyzed by ImageJ software.
Oxidized Lipids
The sections of aortic roots were stained for oxidized phospholipids with the E06 mouse monoclonal antibody (IgM) (Avanti). 38 Endogenous peroxidase activity was blocked with 0.3% hydrogen peroxide in PBS for 15 minutes. After blocking nonspecific binding sites for 30 minutes (2% [w/v] BSA and 0.1% Triton X‐100 in PBS), slides were incubated with E06 dilution (1:100) overnight at 4 °C. The slides were washed 3 times with PBS and treated with a fluorescent secondary antibody (Alexa Fluor 488 goat anti‐mouse; 1:200) overnight at 4 °C. Slides were washed and images were taken using a Zeiss AxioSkop system. The percentage of positively stained areas was quantified by ImageJ. Two aortic root sections were scrapped from the glass slides, and lipids were extracted for high‐performance liquid chromatography analysis using methanol and hexane. 18 , 39 Lipid species were separated by reverse‐phase high‐performance liquid chromatography in a Waters C18 column (250×4.6 mm, 5 μmol/L particle size, 5‐μm guard column) with an Agilent 1100 high‐performance liquid chromatography system. Total cholesterol and 7‐ketocholesterol were detected at 234 nm using an acetonitrile/2‐propanol/water mobile phase (50/48.8/1.2, by volume) and a flow rate of 1 mL/min. The identities of the peaks were confirmed and quantified using commercially available standards.
Hepatic Gene Expression Analysis
Total RNA was isolated from mouse liver. Frozen liver was homogenized with 1 mL TRI Reagent (Sigma Aldrich, St. Louis, MO) and 1‐mm glass beads in a closed tube for 30 seconds at 5000 rpm. Chloroform (200 mL) was added; and after centrifugation, an aqueous phase was visible and transferred to a new tube. Then, isopropanol (0.5 mL) was added; and on centrifugation, an RNA pellet was formed. This was washed with 1 mL 70% (v/v) ethanol. After centrifugation, the supernatant was removed, and the pellet was dissolved in the appropriate volume of diethyl pyrocarbonate sterile water. All samples were always kept on ice, and RNAse‐free materials were used. Afterwards, the RNA concentration and RNA quality were determined with a NanoDrop ND‐1000 spectrophotometer. Total (RNA, 500 ng) from each subject was converted into first‐strand cDNA with the iScript cDNA synthesis kit (Bio‐Rad, Hercules, CA). Subsequently, changes in gene expression of inflammatory markers were determined using quantitative polymerase chain reaction (Applied Biosystems, 7900HT) with SensiMix SYBR with Fluorescein (Bioline) and 10 ng of cDNA. For each gene, a standard curve was generalized from a serial dilution of a cDNA pool. To normalize the amount of cDNA, 2 household genes were used (in our case, cyclophilin A and S12). Primer sets were developed with Primer Express 2.0 (Applied Biosystems, Foster City, CA) using default settings. Primer sequences can be found in Table S1. Data from quantitative polymerase chain reaction were analyzed according to the relative melting curve method.
Skeletal Muscle Analysis
Dissection of the tibialis anterior (TA) was performed under oxygenated Krebs' solution (95% O2 and 5% CO2). Under circulating oxygenated Krebs' solution, one end of a silk suture was attached to the distal tendon of the TA and the other to a force transducer (FT03). The proximal tendon was secured in the experimental chamber by a silk suture. Silver electrodes were positioned on either side of the TA. A constant voltage stimulator was used to directly stimulate the TA, which was stretched to attain the optimal muscle length to produce maximum twitch tension. Tetanic contractions were invoked by stimulus trains of 500‐ms duration at 20, 50, 100, and 200 Hz. The maximum tetanic tension was determined from the plateau of the frequency‐tension curve. Following dissection, the muscle tissue was immediately frozen on liquid nitrogen–cooled isopentane and mounted in optimal cutting temperature compound (TAAB O023) cooled by dry ice/ethanol.
Histochemistry was performed on 10‐μm cryosections. Succinate dehydrogenase, dihydroethidium (DHE), and hematoxylin and eosin staining as well as morphometric analysis of muscle fiber size were performed as previously described. 40
Statistical Analysis
Data are presented as mean±SEM of at least 3 independent experiments of 17 to 20 mice within each group. Comparisons between control and treated samples were analyzed by 1‐way ANOVA with Dunnett or Tukey post hoc tests or, where appropriate, by t‐tests. Differences were considered significant at P<0.05.
RESULTS
Animal and Blood Analysis
In this regression study, the baseline mice were euthanized after 8 weeks on a high‐fat diet, whereas the other 2 groups were continued with or without cysteamine treatment for another 8 weeks on normal chow. The weight of the mice continued to increase a little for another 8 weeks compared with the baseline group mice; however, there was no significant difference in the weights of the cysteamine (2.2 mmol/L in drinking water) and noncysteamine (control) mice at the end of the experiment (Figure 1). After 8 weeks on the chow diet, there was a substantial decrease in the plasma total cholesterol and triacylglycerol, LDL cholesterol, and high‐density lipoprotein cholesterol concentrations compared with the baseline group. Cysteamine treatment did not have any effect on these concentrations (Figure 1).
Figure 1. Effect of cysteamine on body weights and plasma lipids in low‐density lipoprotein (LDL) receptor–knockout mice.
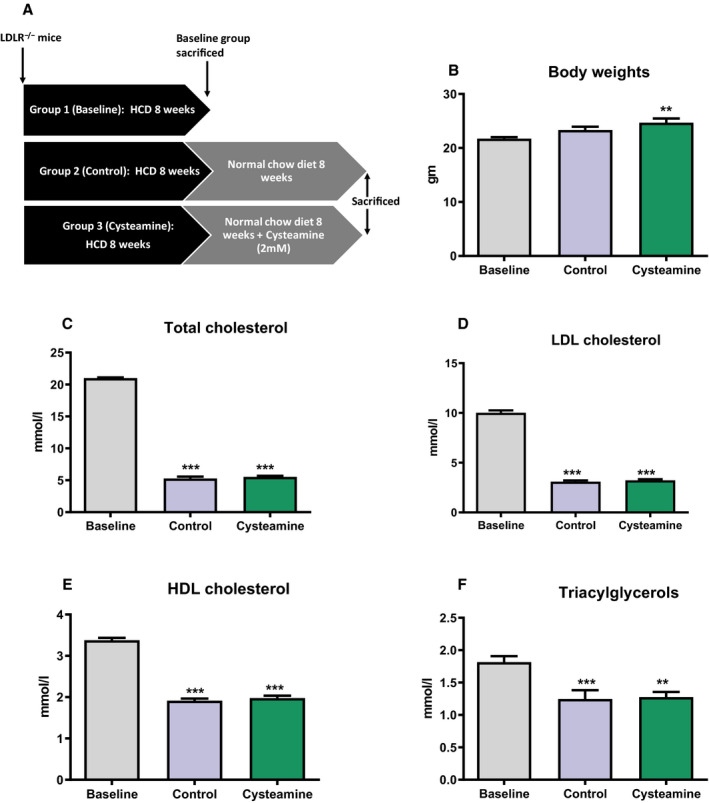
A, Plan of the study. Sixty female LDL receptor–deficient (LDLr−/−) mice were divided into 3 groups and fed a high‐cholesterol diet (HCD) for 8 weeks to induce atherosclerosis. One group was euthanized for baseline measurements. The remaining 40 mice were switched to a normal chow diet and divided into 2 groups to receive no cysteamine (control) or cysteamine at 2.2 mmol/L in drinking water for 8 weeks. Body weight of the mice and plasma lipids were measured at the end of the study. Cysteamine has no effect on the body weights (B); the levels of plasma lipids (C–F) were reduced after switching the mice to a normal chow diet, and cysteamine had no effect on them. There were 17 to 20 mice in each group, and the horizontal line shows the group mean±SEM. Data were analyzed by ANOVA, followed by the Tukey post hoc test. HDL indicates high‐density lipoprotein. **P<0.001 vs the baseline group, ***P<0.0001 vs baseline group.
Effects on Existing Atherosclerosis
Cysteamine Reduces Existing Atherosclerosis in Mice
We quantified atherosclerotic lesions in whole aortas by en face analysis and in aortic root sections by Oil Red O staining (Figure 2). There was no regression of atherosclerosis in the control group over the 8 weeks on a normal chow diet. The en face analysis of whole aorta showed that cysteamine treatment significantly reduced the preexisting atherosclerosis in the aortic arch and abdominal plus thoracic regions of the aortas (Figure 2A through 2C). The lesion area in the aortic arch was 14±6% of the total area in the cysteamine‐treated mice compared with 23±8% in the control mice (a reduction of 39%; P<0.05) and 27±9% in the baseline mice (a reduction of 48%; P<0.05). Cysteamine reduced lesions in the thoracic plus abdominal area of the aortas from 3.3±0.6% to 1.4±0.2% (a reduction of 56%; P<0.05) compared with the control mice and from 2.2±0.3% to 1.4±0.2% compared with the baseline mice (a reduction of 34%, although this was not statistically significant) (Figure 2C). The lesions in the root of the aorta were 31±6% in the cysteamine group compared with 46±7% in the control group (a reduction of 32%; P<0.05) and 47±6% in the baseline mice (a reduction of 33%; P<0.05) (Figure 2D and 2E).
Figure 2. Cysteamine reduced existing atherosclerosis in low‐density lipoprotein receptor–deficient mice.
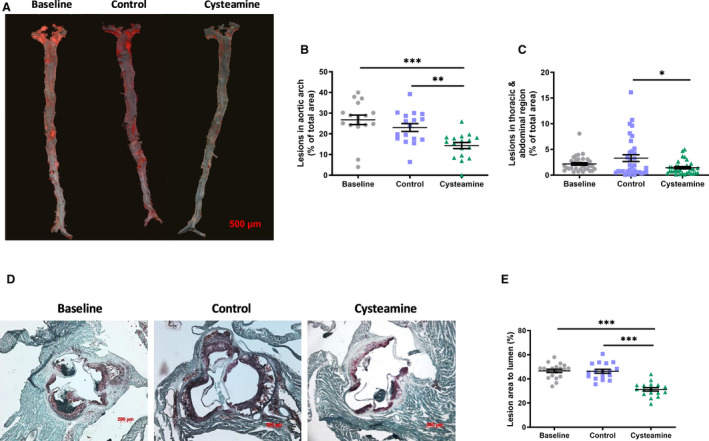
A, Representative images to show atherosclerotic lesions in the aorta of baseline, control, and mice treated with cysteamine (2.2 mmol/L in drinking water) stained with Oil Red O. Bar=500 μm. Data points show lesion areas in individual mice in each group in the aortic arch (B) and the rest of the thoracic plus abdominal aorta (C) of baseline, control, and cysteamine‐treated mice. Representative images to show atherosclerotic lesions in the aortic root (D) and lesion areas in individual mice (E). There were 17 to 20 mice in each group, and the horizontal line shows the group mean±SEM. Data were analyzed by ANOVA, followed by the Tukey post hoc test. *P<0.05, **P<0.01, ***P<0.001.
Cysteamine Increases Markers of Stability in Atherosclerotic Plaques
To assess markers of stability in the atherosclerotic lesions, we stained the aortic root sections to measure the percentage of lesions occupied by monocytes/macrophages (stained by anti‐monocyte+macrophage antibody), smooth muscle cells (α‐actin), and collagen (Picrosirius Red). Cysteamine substantially reduced the percentage area occupied by monocytes/macrophages by 38% compared with the non–drug‐treated control mice (P<0.05) and by 52% compared with the baseline mice (P<0.001) (Figure 3A). To determine the phenotype of the lesion macrophages, we stained aortic root lesions with CD86 (proinflammatory macrophage marker) and CD206 (anti‐inflammatory macrophage marker). Cysteamine reduced the area of the lesions occupied by proinflammatory macrophages by 61% compared with the area occupied by proinflammatory macrophages in the baseline mice (P<0.001) and by 55% compared with the control mice (P<0.001) (Figure 3B). The percentage area occupied by anti‐inflammatory macrophages increased significantly (P<0.001) in both untreated (by 146%) and cysteamine‐treated (195%) mice compared with the baseline mice (Figure 3C).
Figure 3. Effect of cysteamine on atherosclerotic plaque composition.
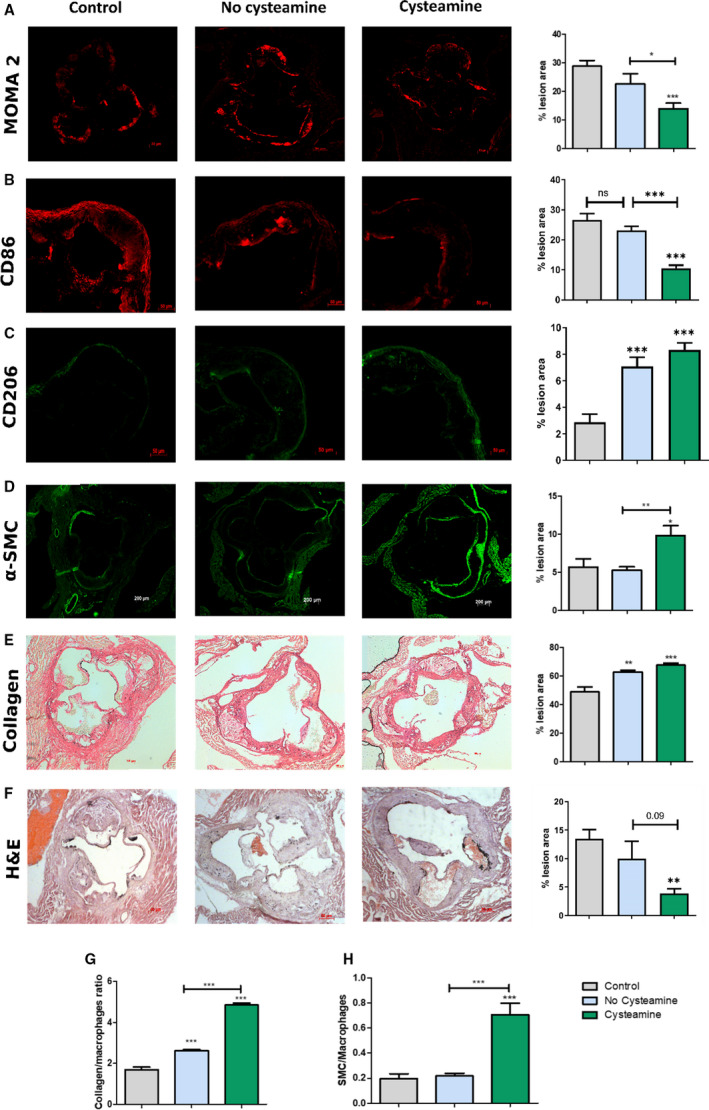
Sections from the aortic root were stained for monocytes/macrophages, cluster of differentiation (CD) 86, CD206, smooth muscle cells (SMCs), collagen, and hematoxylin and eosin (H&E), and the staining was quantified. Quantitation and representative photomicrographs of anti‐monocyte+macrophage antibody (MOMA‐2) (monocytes/macrophages) (A), CD86 (B), CD206 (C), actin (SMCs) (D), Picrosirius Red (collagen) staining (E) and H&E (F) in transverse sections from the aortic root of baseline, control, and cysteamine‐treated mice and percentage of acellular area. G and H, The ratio of collagen/macrophage areas and ratio of SMCs/macrophage areas were calculated as markers of lesion stability. Horizontal bars represent the group mean±SEM for 10 to 12 mice in each group. Data were analyzed by ANOVA, followed by the Tukey post hoc test. α‐SMC indicates α‐smooth muscle actin. *P<0.05, **P<0.01, ***P<0.001.
The percentage area occupied by smooth muscle cells was greatly increased by cysteamine by 85% compared with the non–cysteamine‐treated mice (P<0.01) (Figure 3D). Cysteamine increased the mean collagen content by 8% compared with the control group, although this was not statistically significant, and by 38% compared with the baseline mice (P<0.001) (Figure 3E). We measured the changes to the lesion necrotic cores by measuring the size of acellular areas in hematoxylin and eosin–stained sections (Figure 3F). The percentage area occupied by necrotic cores decreased greatly from 14±1.6% in the baseline mice to 4±0.8% in the cysteamine‐treated mice (P<0.01) and to 10±3.0% in the control mice (not significant) (Figure 3F).
A marker of lesion stability is the ratio of smooth muscle cell to monocyte/macrophage areas, and this was increased by cysteamine by 3‐fold compared with the control group (P<0.001) and by 3.6‐fold compared with the baseline group (P<0.001) (Figure 3I). The ratio of collagen to monocyte/macrophage areas, another marker for lesion stability, was increased 1.9‐fold by cysteamine compared with the controls (P<0.001) and by 2.9‐fold compared with the baseline group (P<0.001) (Figure 3H).
Cysteamine Reduces Levels of Oxidized Lipids in Aortic Root Lesions
We have previously shown that cysteamine prevents lysosomal oxidation of LDL in human macrophages. 41 To investigate the effect of cysteamine on lipoprotein oxidation in vivo, we measured the levels of ceroid, oxidized phospholipids, and 7‐ketocholesterol in the atherosclerotic lesions. Ceroid, a complex of protein associated with oxidized lipids, is commonly observed in human atherosclerotic lesions. 20 , 42 After washing the “soluble” lipids away with xylene, the aortic root sections were stained for ceroid with Oil Red O. The levels of ceroid in the aortic root continued to increase when the mice were switched to a normal chow diet (P<0.05), but this increase was prevented by cysteamine (P<0.05 compared with the control group) (Figure 4A and 4B). Cysteamine greatly decreased immunofluorescence staining with the E06 monoclonal antibody, which detects oxidized phospholipids, 38 , 43 by 73% compared with the control group (P<0.001) and by 77% compared with the baseline group (P<0.001) (Figure 4C and 4D). When adjusted for levels of total cholesterol in aortic root sections, cysteamine reduced 7‐ketocholesterol levels by 24% compared with the untreated control mice (P=0.05) (Figure 4G).
Figure 4. Effect of cysteamine on lipoprotein oxidation in aortic lesions.
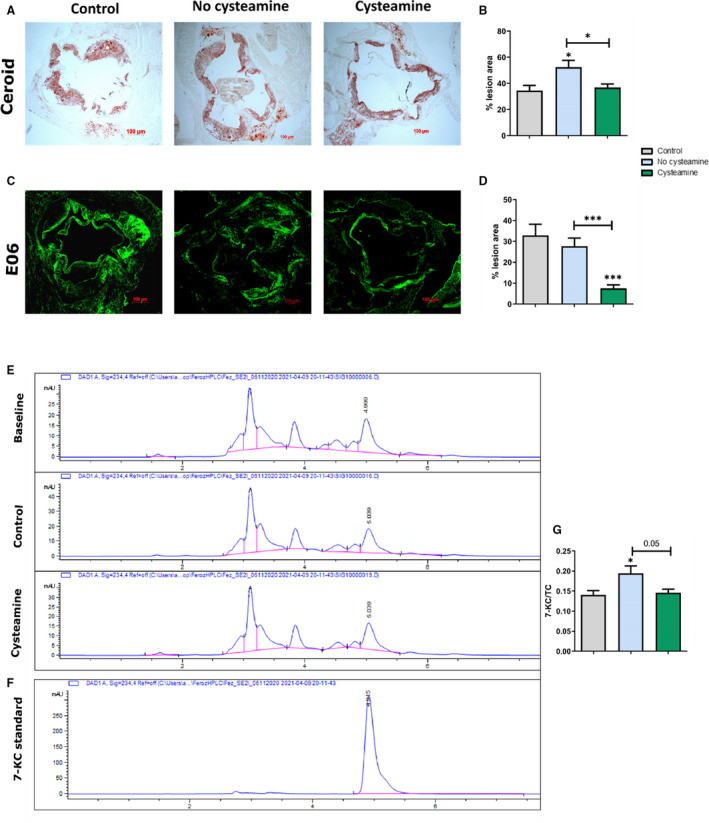
A and B, Quantitation and representative images to show ceroid levels in the atherosclerotic lesions. C and D, Quantitation and representative images to show immunofluorescence staining with the E06 monoclonal antibody against oxidized phospholipids. E, Representative high‐performance liquid chromatography chromatograms of aortic root lesions of baseline, control, and cysteamine‐treated mice. F, Chromatogram of 10 µmol/L 7‐ketocholesterol (7‐KC) standard at 234 nm. G, Ratio of 7‐KC/total cholesterol (TC) in the aortic lesions. There were 17 to 20 mice in each group, and the horizontal line shows the group mean±SEM. Data were analyzed by ANOVA, followed by the Tukey post hoc test. *P<0.05, ***P<0.001.
Cysteamine Reduces Lipids and Inflammation in the Liver
The mean total hepatic cholesterol in the cysteamine‐treated mice was much less than in the baseline mice (P<0.01) and the control mice (but of borderline significance when compared with the control mice, P=0.057) (Figure 5A). The triacylglycerol levels were reduced in both the cysteamine‐treated mice (P<0.001) and the untreated mice (P<0.05) compared with the baseline group, although the decrease appeared to be greater with cysteamine treatment (Figure 4B). The expression of CYP27 and ACAT2 (acetyl–coenzyme A acetyltransferase 2) was decreased in the mice treated with cysteamine compared with the control mice (Figure 5C and 5E). CYP7A1 expression was elevated during the regression phase but was not affected by cysteamine (Figure 5D).
Figure 5. Effect of cysteamine on hepatic cholesterol levels and inflammation.
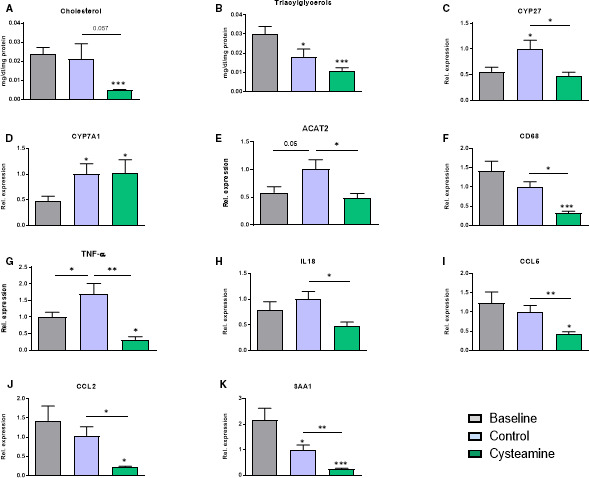
The mean total hepatic cholesterol (A) and triacylglycerol (B) levels were reduced by cysteamine compared with the baseline mice. Total RNA was isolated from mouse liver, and gene expression of various cholesterol‐metabolizing enzymes and proinflammatory proteins was measured. The increased expression of CYP 27 and ACAT2 (acetyl–coenzyme A acetyltransferase) in the control mice was reduced by cysteamine (C and E). CYP7A1 expression was elevated during the regression phase but was not affected by cysteamine (D). Cysteamine treatment reduced the expression of hepatic proinflammatory markers: cluster of differentiation (CD) 68, tumor necrosis factor (TNF)‐α, interleukin (IL)‐18, CCL5, CCL2, and serum amyloid A1 (SAA1) (F–K). Horizontal bars represent group the mean±SEM for 17 to 20 mice in each group. Data were analyzed by ANOVA, followed by the Tukey post hoc test. Rel. indicates relative. *P<0.05, **P<0.01, ***P<0.001.
Cysteamine treatment reduced the expression of CD68 (a marker for hepatic monocytes/macrophages) compared with both control and baseline mice (Figure 5F), suggesting that cysteamine reduced hepatic inflammation. We therefore measured the effect of cysteamine on various proinflammatory genes in liver (Figure 5G through 5K). There was a substantial reduction in the expression of proinflammatory cytokines, such as TNF‐α and interleukin‐18, by cysteamine compared with the untreated mice. Cysteamine also reduced the expression levels of chemokines, such as CCL2 (MCP‐1) and CCL5 (RANTES) compared with both untreated and baseline mice. The hepatic expression of serum amyloid A1, a short‐term phase protein released by liver cells in response to inflammation, was reduced during the regression phase (P<0.05) and was reduced further by cysteamine (P<0.01).
Effects on Skeletal Muscles
Skeletal muscle is sensitive to oxidative stress. Cysteamine appeared to have increased the skeletal muscle mass exemplified by the TA, but the increase was of borderline statistical significance compared with the control (P=0.06) and baseline (P=0.08) mouse groups (Figure 6D). We investigated the impact of cysteamine on muscle function by examining contraction parameters. There was a significant increase in twitch force in the cysteamine group compared with baseline values (Figure 6A). The tetanic force and specific force were increased in the cysteamine group when compared with either the baseline group or the control group (Figure 6B and 6C). We next examined cellular reasons that may underpin the improved function. We found that the percentage of aerobic fibers deploying oxidative metabolism, judged by quantifying the number of succinate dehydrogenase–positive fibers, was increased by cysteamine compared with the control group (Figure 6E and 6F). We examined the muscle for the presence of regenerating fibers based on the assumption that hypercholesterolemia or its sequelae will induce fiber death and then initiate a repair process. Indeed, we found a surprising high level of regenerated fibers, identified by centrally positioned nuclei after 8 weeks on the high‐fat diet. The number of regenerating fibers decreased following the withdrawal of the high‐fat diet but was not significantly affected by cysteamine (Figure 6G and 6H). We also determined the effect of cysteamine on muscle fibrosis. The degree of fibrosis was relatively high in the baseline group and remained so in both the cysteamine‐treated and control groups (Figure 6I and 6J). Finally, assessment of reactive oxygen species activity through DHE intensity showed that cysteamine significantly reduced the reactive oxygen species levels in the muscles by 52% compared with the control mice (P<0.001) (Figure 6K and 6L).
Figure 6. Effect of cysteamine on skeletal muscles.
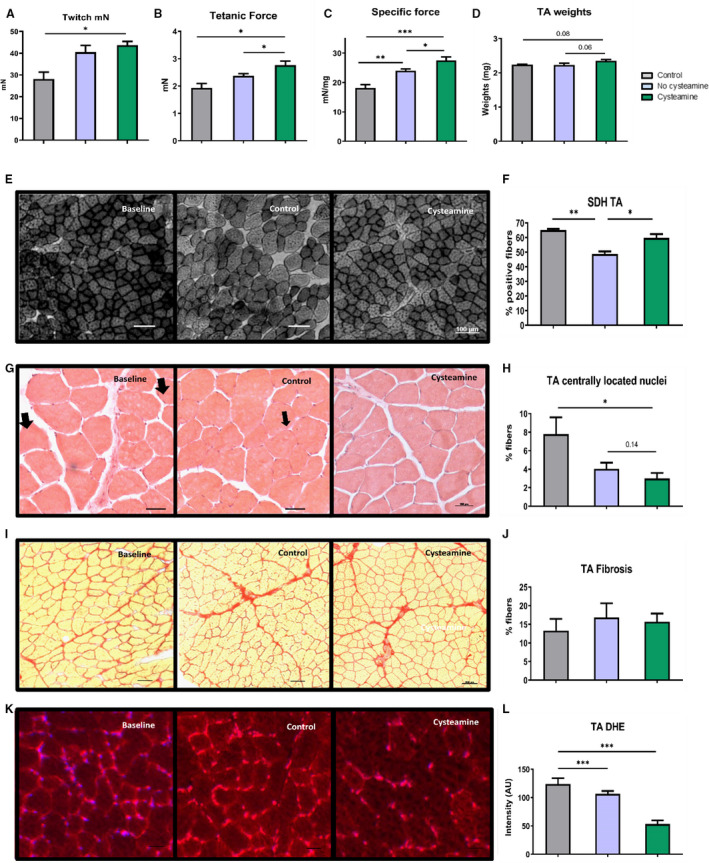
Muscle contraction was measured through ex vivo assessment of tibialis anterior (TA) twitch (A), tetanic force (B), and specific force (C). D, TA weights normalized to body weights. E, Qualitative changes in muscle function were evaluated by staining for succinate dehydrogenase (SDH). F, Quantification of stained SDH fibers. Hematoxylin and eosin–stained sections (G) were used to see muscle regeneration by calculating the percentage of fibers with centrally located nuclei (arrows) (H). I and J, Muscle fibrosis was calculated by staining for collagen deposition (Picrosirius Red) in TA. DHE‐stained sections (K) and quantification of DHE (L) are shown. Horizontal bars represent group mean±SEM for 17 to 20 mice in each group. Data were analyzed by ANOVA, followed by the Tukey post hoc test. *P<0.05, **P<0.01, ***P<0.001.
DISCUSSION
We have previously shown that inhibiting lysosomal oxidation of LDL with cysteamine inhibits secretion of proinflammatory cytokines in human macrophages 41 and decreases the development of atherosclerosis in LDL receptor–deficient mice. 19
In the current study, we have shown that cysteamine causes the regression of atherosclerosis in LDL receptor–deficient mice, a model more appropriate for the treatment of human disease. Atherosclerosis in the aortic root, arch, and remaining thoracic plus abdominal aorta was significantly reduced by cysteamine (Figure 2), in the absence of any changes in plasma lipoprotein concentrations (Figure 1). At the end of the regression phase, there were reductions of 32% in the lesions in the aortic root, 39% in the aortic arch, and 56% in the rest of the aorta compared with the control group. Cysteamine reduced the area occupied by monocytes/macrophages in the aortic root by 38% compared with the control group (Figure 3A) and greatly reduced the levels of proinflammatory factors (Figure 3B).
Cysteamine also increased markers of stability in the atherosclerotic lesions. The areas occupied by smooth muscle cells and collagen were increased by 85% (P<0.001) and 8%, respectively, whereas acellular necrotic areas were reduced by 2.5‐fold compared with the control group (Figure 3D through 3F). The ratio of smooth muscle cell/monocyte/macrophage areas was increased by 3‐fold, and the ratio of collagen/monocyte/macrophage areas was increased by 3.5‐fold (Figure 3G and 3I). If similar effects were to occur in human atherosclerotic lesions, it should make them less liable to fissure, causing thrombosis, myocardial infarctions, and thrombotic strokes.
Foam cells in atherosclerotic lesions contain an advanced oxidation product of polymerized lipids and protein known as ceroid. 20 We have previously shown that macrophages generate ceroid in their lysosomes when they oxidize LDL in these organelles and that cysteamine inhibits its formation. 17 , 18 Herein, we have shown that cysteamine significantly reduced ceroid levels by 30% in the aortic root during the regression phase (Figure 4A and 4B). In addition, mice treated with cysteamine had only 8% of their lesion area stained with E06 for oxidized phospholipids as against 28% seen in the control group (Figure 4C and 4D). In addition, there were reduced levels of 7‐ketocholesterol in aortic lesions of mice treated with cysteamine compared with the control mice (Figure 4E and 4G). These results indicate that cysteamine prevents the lysosomal oxidation of lipoproteins to form oxidized lipids and advanced oxidation products, like ceroid. In line, it was demonstrated that, in patients with cystinosis, longer durations of cysteamine treatment were associated with decreased vascular calcification. 44
Cysteamine has been shown to sometimes increase the glutathione levels in cells, 45 , 46 and this might possibly be another mechanism by which cysteamine causes the regression of atherosclerotic lesions, but this remains to be proven.
Cysteamine had anti‐inflammatory effects in the liver. The expression of proinflammatory cytokines (TNF‐α and interleukin‐18), chemokines (CCL5 and CCL2), and serum amyloid A1 was reduced substantially by cysteamine (Figure 5). Cysteamine also reduced substantially the expression of CD68 (macrosialin), a marker for monocytes and macrophages, suggesting lower numbers of monocytes/macrophages in the liver. Unlike the plasma lipid levels, the hepatic cholesterol and triacylglycerol levels were significantly reduced by cysteamine. To investigate this further, we measured the expression of various cholesterol‐metabolizing enzymes. The expression of CYP27 and ACAT2 was reduced substantially by cysteamine, whereas there was no effect on CYP7A1. CYP27 encodes for sterol 27‐hydroxylase, mainly expressed by hepatic macrophages, which is responsible for the conversion of cholesterol into oxysterols (precursors of bile acids). 47 Hepatic ACAT2 encodes for acyl–coenzyme A/cholesterol transferase, which is responsible for esterification of cholesterol. 48 The hepatic expression levels of both CYP27 and ACAT2 might be lower in the cysteamine group as a consequence of the reduced hepatic cholesterol to be metabolized and esterified.
Cysteamine decreased the expression of CYP27, which is found mainly in hepatic macrophages, with no effect on CYP7A1 levels, which is found primarily in hepatocytes, suggesting that cysteamine has effects primarily on macrophages (Figure 5). CYP27 initiates the alternative bile acid synthesis pathway. 49 CYP7A1 encodes for the enzyme cholesterol 7α‐hydroxylase, which is the rate‐limiting enzyme in the synthesis of bile acids by the classic pathway. 49 , 50 Hepatocyte growth factor signaling pathway inhibits cholesterol 7α‐hydroxylase and bile acid synthesis in human hepatocytes, and increased expression has been shown to prevent atherosclerosis 51 and hepatic inflammation. 52 In our study, CYP7A1 expression was increased by switching the mice from a high‐fat to a normal chow diet.
Inflammation and oxidative stress both attenuate mitochondrial and skeletal muscle function in several pathological conditions, including diabetes mellitus and atherosclerosis. 53 A systemic inflammatory environment can lead to accelerated disease progression by interfering with mitochondrial function in skeletal muscle, which leads to increased production of reactive oxygen species. This has 2 major consequences: it leads to an attenuation of muscle function and promotes further rounds of inflammation. 54 Herein, we show that cysteamine increased the function of skeletal muscle. We propose that this effect might be attributable to changes in mitochondrial activity as there was an increase in the number of oxidative fibers following treatment. This might increase the capacity of the mice to undertake prolonged exercise, and if replicated in patients experiencing clinical symptoms attributable to atherosclerosis, it might help to increase their exercise capacity.
Conclusions
The finding that cysteamine caused the regression of atherosclerosis and substantially increased markers of stability in atherosclerotic lesions in LDL receptor–deficient mice adds to the evidence that the oxidation of LDL in the lysosomes of macrophages is important in atherosclerosis. Inhibiting lysosomal LDL oxidation by antioxidants directed to these organelles might be a novel therapy for atherosclerosis in patients. Cysteamine also had lipid‐lowering and anti‐inflammatory effects in the liver and might be a possible therapeutic approach for NASH. In addition, cysteamine increased skeletal muscle function and the proportion of aerobic fibers.
Sources of Funding
We thank the British Heart Foundation for a project grant (PG/15/98/31864).
Disclosures
None.
Supporting information
Table S1
Acknowledgments
We thank Dr Kim G. Jackson for analyzing lipid concentrations in the mouse plasma.
Supplementary Material for this article is available at https://www.ahajournals.org/doi/suppl/10.1161/JAHA.120.017524
For Sources of Funding and Disclosures, see page 13.
References
- 1. Libby P, Hansson GK. From focal lipid storage to systemic inflammation: JACC review topic of the week. J Am Coll Cardiol. 2019;74:1594–1607. DOI: 10.1016/j.jacc.2019.07.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Remmerie A, Scott CL. Macrophages and lipid metabolism. Cell Immunol. 2018;330:27–42. DOI: 10.1016/j.cellimm.2018.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Fatkhullina AR, Peshkova IO, Koltsova EK. The role of cytokines in the development of atherosclerosis. Biochem (Mosc). 2016;81:1358–1370. DOI: 10.1134/S0006297916110134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ridker PM, Everett BM, Thuren T, MacFadyen JG, Chang WH, Ballantyne C, Fonseca F, Nicolau J, Koenig W, Anker SD, et al. Antiinflammatory therapy with canakinumab for atherosclerotic disease. N Engl J Med. 2017;377:1119–1131. DOI: 10.1056/NEJMoa1707914. [DOI] [PubMed] [Google Scholar]
- 5. Kleemann R, Verschuren L, van Erk MJ, Nikolsky Y, Cnubben NHP, Verheij ER, Smilde AK, Hendriks HFJ, Zadelaar S, Smith GJ, et al. Atherosclerosis and liver inflammation induced by increased dietary cholesterol intake: a combined transcriptomics and metabolomics analysis. Genome Biol. 2007;8:R200. DOI: 10.1186/gb-2007-8-9-r200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wolf D, Ley K. Immunity and inflammation in atherosclerosis. Circ Res. 2019;124:315–327. DOI: 10.1161/CIRCRESAHA.118.313591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Nassir F, Ibdah JA. Role of mitochondria in nonalcoholic fatty liver disease. Int J Mol Sci. 2014;15:8713–8742. DOI: 10.3390/ijms15058713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bieghs V, Hendrikx T, van Gorp PJ, Verheyen F, Guichot YD, Walenbergh SMA, Jeurissen MLJ, Gijbels M, Rensen SS, Bast A, et al. The cholesterol derivative 27‐hydroxycholesterol reduces steatohepatitis in mice. Gastroenterology. 2013;144:167–178.e1. DOI: 10.1053/j.gastro.2012.09.062. [DOI] [PubMed] [Google Scholar]
- 9. Bieghs V, Rensen PCN, Hofker MH, Shiri‐Sverdlov R. NASH and atherosclerosis are two aspects of a shared disease: central role for macrophages. Atherosclerosis. 2012;220:287–293. DOI: 10.1016/j.atherosclerosis.2011.08.041. [DOI] [PubMed] [Google Scholar]
- 10. Kalanuria AA, Nyquist P, Ling G. The prevention and regression of atherosclerotic plaques: emerging treatments. Vasc Health Risk Manag. 2012;8:549–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Diep L, Kwagyan J, Kurantsin‐Mills J, Weir R, Jayam‐Trouth A. Association of physical activity level and stroke outcomes in men and women: a meta‐analysis. J Women's Health. 2002;2010:1815–1822. DOI: 10.1089/jwh.2009.1708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Shimba Y, Togawa H, Senoo N, Ikeda M, Miyoshi N, Morita A, Miura S. Skeletal muscle‐specific PGC‐1α overexpression suppresses atherosclerosis in apolipoprotein e‐knockout mice. Sci Rep. 2019;9:4077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sfyri P, Matsakas A. Crossroads between peripheral atherosclerosis, western‐type diet and skeletal muscle pathophysiology: emphasis on apolipoprotein E deficiency and peripheral arterial disease. J Biomed Sci. 2017;24:42. DOI: 10.1186/s12929-017-0346-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Campos AM, Moura FA, Santos SN, Freitas WM, Sposito AC. Sarcopenia, but not excess weight or increased caloric intake, is associated with coronary subclinical atherosclerosis in the very elderly. Atherosclerosis. 2017;258:138–144. DOI: 10.1016/j.atherosclerosis.2017.01.005. [DOI] [PubMed] [Google Scholar]
- 15. Razani B, Feng C, Coleman T, Emanuel R, Wen H, Hwang S, Ting JP, Virgin HW, Kastan MB, Semenkovich CF. Autophagy links inflammasomes to atherosclerotic progression. Cell Metab. 2012;15:534–544. DOI: 10.1016/j.cmet.2012.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hendrikx T, Walenbergh SM, Hofker MH, Shiri‐Sverdlov R. Lysosomal cholesterol accumulation: driver on the road to inflammation during atherosclerosis and non‐alcoholic steatohepatitis. Obes Rev. 2014;15:424–433. DOI: 10.1111/obr.12159. [DOI] [PubMed] [Google Scholar]
- 17. Wen Y, Leake DS. Low density lipoprotein undergoes oxidation within lysosomes in cells. Circ Res. 2007;100:1337–1343. DOI: 10.1161/CIRCRESAHA.107.151704. [DOI] [PubMed] [Google Scholar]
- 18. Ahmad F, Leake DS. Antioxidants inhibit low density lipoprotein oxidation less at lysosomal pH: a possible explanation as to why the clinical trials of antioxidants might have failed. Chem Phys Lipid. 2018;213:13–24. DOI: 10.1016/j.chemphyslip.2018.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wen Y, Ahmad F, Mohri Z, Weinberg PD, Leake DS. Cysteamine inhibits lysosomal oxidation of low density lipoprotein in human macrophages and reduces atherosclerosis in mice. Atherosclerosis. 2019;291:9–18. DOI: 10.1016/j.atherosclerosis.2019.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Mitchinson MJ. Insoluble lipids in human atherosclerotic plaques. Atherosclerosis. 1982;45:11–15. DOI: 10.1016/0021-9150(82)90167-8. [DOI] [PubMed] [Google Scholar]
- 21. Haka AS, Kramer JR, Dasari RR, Fitzmaurice M. Mechanism of ceroid formation in atherosclerotic plaque: in situ studies using a combination of Raman and fluorescence spectroscopy. J Biomed Opt. 2011;16:011011. DOI: 10.1117/1.3524304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Yuan XM, Li W, Olsson AG, Brunk UT. Iron in human atheroma and LDL oxidation by macrophages following erythrophagocytosis. Atherosclerosis. 1996;124:61–73. DOI: 10.1016/0021-9150(96)05817-0. [DOI] [PubMed] [Google Scholar]
- 23. Kurz T, Terman A, Gustafsson B, Brunk UT. Lysosomes in iron metabolism, ageing and apoptosis. Histochem Cell Biol. 2008;129:389–406. DOI: 10.1007/s00418-008-0394-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Pisoni RL, Park GY, Velilla VQ, Thoene JG. Detection and characterization of a transport system mediating cysteamine entry into human fibroblast lysosomes: specificity for aminoethylthiol and aminoethylsulfide derivatives. J Biol Chem. 1995;270:1179–1184. [DOI] [PubMed] [Google Scholar]
- 25. Dohil R, Gangoiti JA, Cabrera BL, Fidler M, Schneider JA, Barshop BA. Long‐term treatment of cystinosis in children with twice‐daily cysteamine. J Pediatr. 2010;156:823–827. DOI: 10.1016/j.jpeds.2009.11.059. [DOI] [PubMed] [Google Scholar]
- 26. De Backer G, Ambrosioni E, Broch‐Johnsen K, Brotons C, Cifkova R, Dallongeville J, Ebrahim S, Faergeman O, Graham I, Mancia G, et al. European guidelines on cardiovascular disease prevention in clinical practice: third joint task force of European and other societies on cardiovascular disease prevention in clinical practice (constituted by representatives of eight societies and by invited experts). Eur J Prev Cardiol. 2003;10:S1–S10. DOI: 10.1097/00149831-200308000-00004. [DOI] [PubMed] [Google Scholar]
- 27. Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults . Executive summary of the third report of the National Cholesterol Education Program (NCEP) expert panel on detection, evaluation, and treatment of high blood cholesterol in adults (adult treatment panel III). JAMA. 2001;285:2486–2497. DOI: 10.1001/jama.285.19.2486. [DOI] [PubMed] [Google Scholar]
- 28. Bachmanov AA, Reed DR, Beauchamp GK, Tordoff MG. Food intake, water intake, and drinking spout side preference of 28 mouse strains. Behav Genet. 2002;32:435–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bouazza N, Tréluyer J‐M, Ottolenghi C, Urien S, Deschenes G, Ricquier D, Niaudet P, Chadefaux‐Vekemans B. Population pharmacokinetics and pharmacodynamics of cysteamine in nephropathic cystinosis patients. Orphanet J Rare Dis. 2011;6:86. DOI: 10.1186/1750-1172-6-86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Tricon S, Burdge GC, Jones EL, Russell JJ, El‐Khazen S, Moretti E, Hall WL, Gerry AB, Leake DS, Grimble RF, et al. Effects of dairy products naturally enriched with cis‐9, trans‐11 conjugated linoleic acid on the blood lipid profile in healthy middle‐aged men. Am J Clin Nutr. 2006;83:744–753. DOI: 10.1093/ajcn/83.4.744. [DOI] [PubMed] [Google Scholar]
- 31. Mohanta S, Yin C, Weber C, Hu D, Habenicht AJ. Aorta atherosclerosis lesion analysis in hyperlipidemic mice. Bio‐protocol. 2016;6:e1833. DOI: 10.21769/BioProtoc.1833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Baglione J, Smith JD. Quantitative assay for mouse atherosclerosis in the aortic root. Methods Mol Med. 2006;129:83–95. [DOI] [PubMed] [Google Scholar]
- 33. Baglione J, Smith JD. Quantitative assay for mouse atherosclerosis in the aortic root. In: Wang QK, ed. Cardiovascular Disease: Methods and Protocols Volume 2: Molecular Medicine. Humana Press; 2007:83–95. [DOI] [PubMed] [Google Scholar]
- 34. Bhatia VK, Yun S, Leung V, Grimsditch DC, Benson GM, Botto MB, Boyle JJ, Haskard DO. Complement C1q reduces early atherosclerosis in low‐density lipoprotein receptor‐deficient mice. Am J Pathol. 2007;170:416–426. DOI: 10.2353/ajpath.2007.060406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kadl A, Meher AK, Sharma PR, Lee MY, Doran AC, Johnstone SR, Elliott MR, Gruber F, Han J, Chen W, et al. Identification of a novel macrophage phenotype that develops in response to atherogenic phospholipids via Nrf2. Circ Res. 2010;107:737–746. DOI: 10.1161/CIRCRESAHA.109.215715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Yu P, Qian AS, Chathely KM, Trigatti BL. PDZK1 in leukocytes protects against cellular apoptosis and necrotic core development in atherosclerotic plaques in high fat diet fed ldl receptor deficient mice. Atherosclerosis. 2018;276:171–181. DOI: 10.1016/j.atherosclerosis.2018.05.009. [DOI] [PubMed] [Google Scholar]
- 37. Burt RC. The incidence of acid‐fast pigment (ceroid) in aortic atherosclerosis. Am J Clin Pathol. 1952;22:135–139. DOI: 10.1093/ajcp/22.2.135. [DOI] [PubMed] [Google Scholar]
- 38. Palinski W, Horkko S, Miller E, Steinbrecher UP, Powell HC, Curtiss LK, Witztum JL. Cloning of monoclonal autoantibodies to epitopes of oxidized lipoproteins from apolipoprotein E‐deficient mice: demonstration of epitopes of oxidized low density lipoprotein in human plasma. J Clin Investig. 1996;98:800–814. DOI: 10.1172/JCI118853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kritharides L, Jessup W, Gifford J, Dean RT. A method for defining the stages of low‐density lipoprotein oxidation by the separation of cholesterol‐ and cholesteryl ester‐oxidation products using HPLC. Anal Biochem. 1993;213:79–89. DOI: 10.1006/abio.1993.1389. [DOI] [PubMed] [Google Scholar]
- 40. Matsakas A, Macharia R, Otto A, Elashry MI, Mouisel E, Romanello V, Sartori R, Amthor H, Sandri M, Narkar V, et al. Exercise training attenuates the hypermuscular phenotype and restores skeletal muscle function in the myostatin null mouse. Exp Physiol. 2012;97:125–140. DOI: 10.1113/expphysiol.2011.063008. [DOI] [PubMed] [Google Scholar]
- 41. Ahmad F, Leake DS. Lysosomal oxidation of LDL alters lysosomal pH, induces senescence, and increases secretion of pro‐inflammatory cytokines in human macrophages. J Lipid Res. 2019;60:98–110. DOI: 10.1194/jlr.M088245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lee F‐Y, Lee T‐S, Pan C‐C, Huang A‐L, Chau L‐Y. Colocalization of iron and ceroid in human atherosclerotic lesions. Atherosclerosis. 1998;138:281–288. DOI: 10.1016/S0021-9150(98)00033-1. [DOI] [PubMed] [Google Scholar]
- 43. Hörkkö S, Bird DA, Miller E, Itabe H, Leitinger N, Subbanagounder G, Berliner JA, Friedman P, Dennis EA, Curtiss LK, et al. Monoclonal autoantibodies specific for oxidized phospholipids or oxidized phospholipid‐protein adducts inhibit macrophage uptake of oxidized low‐density lipoproteins. J Clin Investig. 1999;103:117–128. DOI: 10.1172/JCI4533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ueda M, O’Brien K, Rosing DR, Ling A, Kleta R, McAreavey D, Bernardini I, Gahl WA. Coronary artery and other vascular calcifications in patients with cystinosis after kidney transplantation. Clin J Am Soc Nephrol. 2006;1:555. DOI: 10.2215/CJN.01431005. [DOI] [PubMed] [Google Scholar]
- 45. Djurhuus R, Svardal AM, Mansoor MA, Ueland PM. Modulation of glutathione content and the effect on methionine auxotrophy and cellular distribution of homocysteine and cysteine in mouse cell lines. Carcinogenesis. 1991;12:241–247. DOI: 10.1093/carcin/12.2.241. [DOI] [PubMed] [Google Scholar]
- 46. Wilmer MJ, Kluijtmans LAJ, van der Velden TJ, Willems PH, Scheffer PG, Masereeuw R, Monnens LA, van den Heuvel LP, Levtchenko EN, Monnens LA, et al. Cysteamine restores glutathione redox status in cultured cystinotic proximal tubular epithelial cells. Biochim Biophys Acta. 1812;2011:643–651. DOI: 10.1016/j.bbadis.2011.02.010. [DOI] [PubMed] [Google Scholar]
- 47. Chiang JY. Bile acid regulation of gene expression: roles of nuclear hormone receptors. Endocr Rev. 2002;23:443–463. DOI: 10.1210/er.2000-0035. [DOI] [PubMed] [Google Scholar]
- 48. Parini P, Davis M, Lada AT, Erickson SK, Wright TL, Gustafsson U, Sahlin S, Einarsson C, Eriksson M, Angelin BO, et al. ACAT2 is localized to hepatocytes and is the major cholesterol‐esterifying enzyme in human liver. Circulation. 2004;110:2017–2023. DOI: 10.1161/01.CIR.0000143163.76212.0B. [DOI] [PubMed] [Google Scholar]
- 49. Chiang JYL, Ferrell JM. Bile acids as metabolic regulators and nutrient sensors. Annu Rev Nutr. 2019;39:175–200. DOI: 10.1146/annurev-nutr-082018-124344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Song KH, Ellis E, Strom S, Chiang JY. Hepatocyte growth factor signaling pathway inhibits cholesterol 7alpha‐hydroxylase and bile acid synthesis in human hepatocytes. Hepatol. 2007;46:1993–2002. [DOI] [PubMed] [Google Scholar]
- 51. Miyake JH, Duong‐Polk XT, Taylor JM, Du EZ, Castellani LW, Lusis AJ, Davis RA. Transgenic expression of cholesterol‐7‐alpha‐hydroxylase prevents atherosclerosis in C57BL/6J mice. Arterioscler Thromb Vasc Biol. 2002;22:121–126. [DOI] [PubMed] [Google Scholar]
- 52. Liu H, Pathak P, Boehme S, Chiang JY. Cholesterol 7alpha‐hydroxylase protects the liver from inflammation and fibrosis by maintaining cholesterol homeostasis. J Lipid Res. 2016;57:1831–1844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Marchio P, Guerra‐Ojeda S, Vila JM, Aldasoro M, Victor VM, Mauricio MD. Targeting early atherosclerosis: a focus on oxidative stress and inflammation. Oxid Med Cell Longev. 2019;2019:8563845. DOI: 10.1155/2019/8563845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Petrillo S, Pelosi L, Piemonte F, Travaglini L, Forcina L, Catteruccia M, Petrini S, Verardo M, D’Amico A, Musarò A, et al. Oxidative stress in Duchenne muscular dystrophy: focus on the NRF2 redox pathway. Hum Mol Genet. 2017;26:2781–2790. DOI: 10.1093/hmg/ddx173. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1