

Abstract
Background
Cardiac fibrosis is the excessive deposition of extracellular matrix in the heart, triggered by a cardiac insult, aging, genetics, or environmental factors. Molecular imaging of the cardiac extracellular matrix with targeted probes could improve diagnosis and treatment of heart disease. However, although this technology has been used to demonstrate focal scarring arising from myocardial infarction, its capacity to demonstrate extracellular matrix expansion and diffuse cardiac fibrosis has not been assessed.
Methods and Results
Here, we report the use of collagen‐targeted peptides labeled with near‐infrared fluorophores for the detection of diffuse cardiac fibrosis in the β2‐AR (β‐2‐adrenergic receptor) overexpressing mouse model and in ischemic human hearts. Two approaches were evaluated, the first based on a T peptide that binds matrix metalloproteinase‐2‐proteolyzed collagen IV, and the second on the cyclic peptide EP‐3533, which targets collagen I. The systemic and cardiac uptakes of both peptides (intravenously administered) were quantified ex vivo by near‐infrared imaging of whole organs, tissue sections, and heart lysates. The peptide accumulation profiles corresponded to an immunohistochemically‐validated increase in collagen types I and IV in hearts of transgenic mice versus littermate controls. The T peptide could encouragingly demonstrate both the intermediate (7 months old) and severe (11 months old) cardiomyopathic phenotypes. Co‐immunostainings of fluorescent peptides and collagens, as well as reduced collagen binding of a control peptide, confirmed the collagen specificity of the tracers. Qualitative analysis of heart samples from patients with ischemic cardiomyopathy compared with nondiseased donors supported the collagen‐enhancement capabilities of these peptides also in the clinical settings.
Conclusions
Together, these observations demonstrate the feasibility and translation potential of molecular imaging with collagen‐binding peptides for noninvasive imaging of diffuse cardiac fibrosis.
Keywords: cardiomyopathy, collagen peptides, heart fibrosis, molecular imaging, molecular probes, preclinical imaging
Subject Categories: Cardiomyopathy, Imaging
Nonstandard Abbreviations and Acronyms
- α‐MHC
alpha‐myosin heavy chain
- β2‐AR
beta‐2‐adrenergic receptor
- Cy5.5
cyanine5.5
- ECM
extracellular matrix
- IHD
ischemic heart disease
- MMP‐2
matrix metalloproteinase‐2
- MTC
Masson’s trichrome
- NIR
near‐infrared
Clinical Perspective
What Is New?
A proof‐of‐concept usage of fluorescently‐labeled, collagen‐targeted peptides for direct molecular imaging of the heart’s extracellular matrix to precisely demonstrate interstitial cardiac fibrosis is presented in a mouse model of cardiomyopathy.
What Are the Clinical Implications?
Although cardiac magnetic resonance imaging allows direct detection of fibrotic patches or indirect viewing of interstitial fibrosis by T1 mapping, our proposed collagen tracer‐based technology tackles the ongoing challenge of direct imaging and accurate quantification of interstitial cardiac fibrosis.
Furthermore, the rapid clearance and nontoxicity of these collagen probes, together with their capacity to enhance fibrotic areas in human heart specimens, add critical value to the future translation efforts and ultimate clinical adoption of this molecular imaging technique.
Heart diseases diminish quality of life and cause severe economic burden worldwide. 1 Cardiac fibrosis, characterized by excessive deposition of collagen matrix within the myocardium, predicts the progression of heart disease and its associated pathologies, 2 including cardiomyopathies, ischemic heart disease, heart failure, systemic arterial disease, and stroke. Cardiac fibrosis is a major contributor to the cause of nearly all forms of cardiac disease, 3 resulting in chamber dilation, stiffening of the ventricular wall, abnormal electrical transmission, 4 poor cardiac nutrient and oxygen levels, 5 and a consequent decline in cardiac function. 6 The degree of the extracellular matrix (ECM) expansion as a reflection of adverse cardiac remodeling clinically correlates with mortality and worsening of composite end points, 7 such as systolic and diastolic heart functions. 8 , 9 Whereas in some cases the progression of disease and the level of cardiac fibrosis may be inconsistent, 10 a consensus surrounds the importance of the fibrotic process in heart pathology.
Promisingly, progress has been made in the understanding of molecular pathways that propel myocardial fibrosis, 11 , 12 and over 100 potential therapeutic targets have been identified. 13 Among them, several collagen types were shown to be upregulated during human heart disease, including the ECM collagens types I and III and the basement membrane collagen type IV. 14 , 15 Additionally, the enhanced activity of collagen‐modifying enzymes, such as matrix metalloproteinases, has been shown to be an indicator of active and progressive cardiomyopathic disease. 16 , 17
Endomyocardial biopsy is presently the gold‐standard method for evaluation and detection of cardiac fibrosis, but it is generally limited to the right ventricle. Furthermore, biopsy procedures are invasive, provide limited spatial information, and possess a risk of sampling error; biopsies are hence inadequate for assessment of cardiac fibrosis over time. 18 Current cardiac fibrosis imaging techniques are also insufficient. Nuclear imaging methodologies, such as singlephoton emission computed tomography and positron emission tomography, enable noninvasive and sensitive evaluation of macroscopic myocardial fibrosis, but cannot precisely identify diffuse fibrosis because of their limited resolution. An improved delineation is achieved on contrast‐enhanced cardiac magnetic resonance imaging using late gadolinium enhancement and T1 mapping of the myocardial wall, which progressively becomes the method of choice for precise evaluation of cardiac fibrosis. 19 , 20 , 21 Nevertheless, late gadolinium enhancement is considered indirect, as it relies on demonstration of the abnormal interstitial expansion but not the fibrotic matrix itself 19 , 20 , 21 . It is also limited by spatial resolution and image quality, especially in atrial disease because of the thinness of the atrial wall, 19 , 22 suffers from uncertain reproducibility, requires a high degree of operator dependence, 20 , 23 and has limited sensitivity in terms of payload of contrast agents. Taken together, a pressing need for new, more direct, and low‐cost diagnostic methodologies and treatment‐monitoring strategies becomes apparent.
Within this context, the potential benefits of an alternative, noninvasive, targeted molecular imaging technique of the cardiac ECM has been well‐recognized. 24 Conceptually, the use of specific tracers directed toward molecular targets within the fibrotic heart may overcome the present limitations by enabling enrichment of a tracer‐bound contrast agent in areas of fibrosis to improve signal‐to‐noise ratio and better demonstrate the cardiac condition. 19 In line with the above notions, one could further speculate that cardiac collagens and their digested forms could perfectly fit this purpose and represent excellent targets for direction of therapeutics and imaging agents into the diseased heart, bearing crucial diagnostic and therapeutic value.
Here, we report an innovative use of 2 peptide tracers for detection of diffuse cardiac fibrosis, the collagen‐targeting T peptide (TLTYTWS), which binds selectively to MMP‐2 (matrix metalloproteinase‐2)‐degraded collagen IV 25 , and the cyclic peptide EP‐3533, 26 , 27 which targets collagen I (Figure 1). We recently harnessed the T peptide for targeted delivery of drugs into MMP‐2–expressing cells, 28 whereas EP‐3533 was previously used for magnetic resonance imaging enhancement of type I collagen in murine models of myocardial infarction 26 , 27 , as well as liver 29 and pulmonary fibrosis. 30 However, the use of peptide probes for detection of diffuse cardiac fibrosis has never been described. For this purpose, we conjugated both tracers to the near‐infrared (NIR) fluorophore sulfo‐cyanine5.5 (Cy5.5) (excitation maximum 675 nm/emission maximum 694 nm) (Figure 1) to enable in vivo characterization of the widespread interstitial cardiac fibrosis that develops in the β2‐AR (beta‐2 adrenergic receptor) overexpressing mouse model. 31 , 32 , 33 , 34 Moreover, we also assessed our tracers in explanted, scar‐bearing human heart tissue from patients with ischemic heart disease (IHD). Our results demonstrate the capacity of these collagen peptide‐based approaches to detect diffuse interstitial fibrosis both experimentally and clinically, highlighting their potential as feasible, new molecular imaging strategies.
Figure 1. Chemical structures of sulfo‐cyanine5.5 (Cy5.5)‐conjugated T peptide, S peptide, and the cyclic peptide EP‐3533.
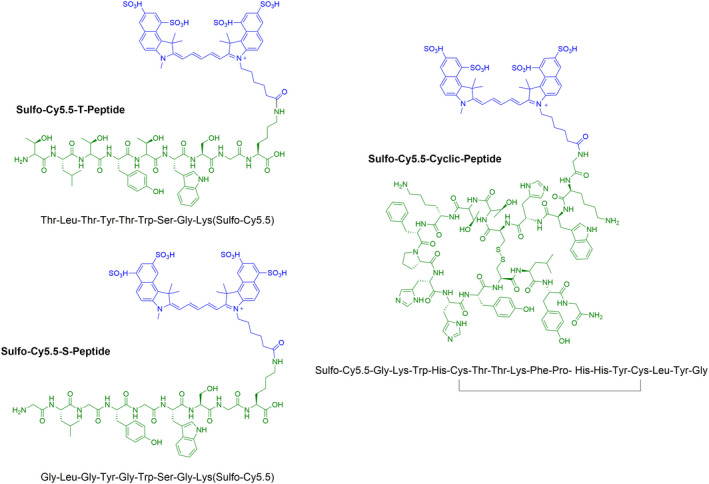
The primary peptide structure appears in green and the sulfo‐Cy5.5 conjugate in blue.
Methods
The data that support the findings of this study are either provided within the article or available from the corresponding author upon request.
Peptides Synthesis and Characterization
The linear heptapeptide TLTYTWS (termed in our study the “T peptide”) (Figure 1) was originally discovered by Mueller et al using phage display technology and described as a tumor‐homing peptide. 25 In principle, this peptide binds MMP‐2–degraded collagen IV and has dual affinity toward both the mouse and human proteins. 25 A control nonbinding peptide GLGYGWS (termed the “S peptide”) (Figure 1), was also generated to assess the T peptide specificity. Peptides were conjugated to sulfo‐Cy5.5 NIR fluorophore (below) (Figure 1).
Both the T and S peptides were synthesized using standard microwave‐assisted, solid‐phase peptide synthesis. Briefly, the coupling cycle of each Fmoc protected amino acid involved the use of 1‐[bis(dimethylamino)methylene]‐1H‐1,2,3‐triazolo[4,5‐b]pyridinium3‐oxide hexafluorophosphate and diisopropylethylamine as the base in dimethylformamide. Deprotection of Fmoc groups was achieved with 20% piperidine in dimethylformamide after each cycle, but no final N‐terminus Fmoc deprotection was performed. The global deprotection of protecting groups was achieved using a standard trifluoracetic acid mixture (Supplemental Material). The peptides were conjugated with a single molecule of the NIR‐soluble dye sulfo‐Cy5.5 (excitation/emission maximum 675/694; Lumiprobe, Hunt Valley, MD) by incubating a solution of peptide and dye in dimethylformamide at room temperature for 5 hours followed by removal of the Fmoc group from the N‐terminus using 20% piperidine in dimethylformamide, allowing the isolation of peptides at a 32% to 38% yield. Stock solutions were prepared in sterile PBS and further diluted in sterile saline for in vivo use.
For the cyclic peptide (Figure 1), a version of the peptide EP‐3533, 26 the linear sequence GKWHCTTKFPHHYCLYG was first synthesized, as mentioned above. Cyclization through the formation of an intramolecular disulfide bridge between the cysteine residues was achieved by in situ deprotection of the acetamido methyl group on cysteine residues followed by oxidation with iodine in dimethylformamide. After global deprotection, the cyclic peptide was labeled with sulfo‐Cy5.5, as described above. Fmoc groups were then removed from the N‐termini using 20% piperidine in dimethylformamide, whereas ivDDe was removed from lysine residues with 2% hydrazine in dimethylformamide, allowing the isolation of sulfo‐Cy5.5 cyclic peptide at 21% yield. This cyclic peptide was previously functionalized as a magnetic resonance imaging contrast probe and found to be effective for collagen type I targeting. 26 , 27 , 29 , 30 Furthermore, it associated in vitro with several sites on the mouse collagen I molecule, 26 making it an optimal probe for molecular imaging. Stock solutions were prepared in 10% DMSO and 10% ethanol (v/v) in sterile PBS, and further diluted in sterile saline for in vivo use. Refer to Data S1 for the detailed reagents and methods used for peptides synthesis and purification.
β2‐AR Transgenic Mouse Model
The well‐described β2‐AR transgenic mouse model, 32 in which cardiac‐specific expression of human β2‐AR occurs under the α‐MHC (α myosin heavy chain) promoter, was kindly provided by Xiao‐Jun Du (Baker Institute, Australia and Xi'an Medical University, China). This model produces classical phenotypes of heart disease, such as fibrosis and dilated cardiomyopathy, and has important clinical relevance. 31 , 32 , 33 , 34 Notably, our transgenic mice express constant levels of the β2‐AR transgene and therefore do not have variable cardiac phenotypes. 34
Human Heart Tissue
Human heart tissue was obtained from the Sydney Heart Bank, University of Sydney, as approved by the University of Sydney Human Research Ethics Committee (ethics approval number 2016/923). All subjects gave informed consent for usage of the heart tissue. The Sydney Heart Bank cardiac specimens are freshly cryopreserved within minutes of harvest (ie, it is not postmortem and not fixed), as previously described. 35 , 36 , 37 Five donor (nondiseased) hearts (aged 34–65 years, 1 man and 4 women) and 3 diffusely fibrotic heart specimens secondary to IHD 38 (aged 31–54 years, 3 men) were used. All samples were taken from the interventricular septum, a prevalent region for cardiac fibrosis. Tissues were kept at −80 °C until cryosectioning. Histological and immunohistochemical analyses were performed by standard procedures, as previously described, 39 , 40 and further detailed below.
Cardiac Tracer Uptake Studies in Whole Organs Using NIR Fluorescence Imaging
All animal experiments were approved by the Alfred Research Alliance Animal Ethics Committee, Monash University (approval numbers E/1941/2019/M and E/1625/2016/M). Twenty‐two heterozygote male β2‐AR transgenic mice and 26 littermate nontransgenic male controls were used (36 at 49 weeks old, 6 at 8–9 weeks old, and 6 at 7 weeks old). To assess peptide uptake in various mouse organs and particularly in the heart, fluorescently tagged peptides (0.15 mg/mL in saline) were injected intravenously into mice at 0.5 mg/kg of body weight via the tail vein (n=5 per group). Injections were performed by a person blinded to the administered substance. Four hours after injection, animals were humanely euthanized by an overdose of ketamine (300 mg/kg) and xylazine (30 mg/kg), and transcardially perfused with 20 mL of PBS. The liver, lung, spleen, kidney, brain, thigh muscle, and the heart were next harvested for analysis, washed with PBS, and kept on ice until imaging. The peptide fluorescence intensity in the various organs was then measured on the Odyssey CLx near‐infrared fluorescence (NIRF) 2‐dimensional scanner (Li‐Cor Biosciences, Lincoln, NE) at 169‐µm resolution using the manual L1 intensity and 0‐mm focus offset settings. Samples from an uninjected mouse served for background correction, whereas a 96‐well plate containing the tracer stock (0.15 mg/mL) was scanned as a reference for the highest possible value of each peptide. Analysis of peptide uptake in organs was done with Image Studio 5.2 software (Li‐Cor Biosciences) by defining a region of interest in each organ, followed by extraction of the mean fluorescence intensity value per pixel. Notably, pixels were used as a normalization method instead of tissue weight, because the Odyssey scanner generates only 2‐dimensional images. The biodistribution data were finally expressed as percentage of the maximal value (obtained from the injected stock) per tissue area in pixels (1 pixel=169×169 µm=0.0285 mm2).
Quantification of Peptide Accumulation in Mouse Heart Lysates
FastPrep‐96 high throughput bead beating grinder (MP Biomedicals, Santa Ana, CA) was used to obtain lysates from harvested hearts, separated into tissue chambers and homogenized based on wet weight/volume (milligrams per milliliter). In brief, atrial or ventricular tissues were weighed and placed in 0.5 mL tubes (attached cap, MP‐Bio catalog number 5076100) containing 200 mg of lysing matrix D (MP‐Bio catalog number 6983‐001). PBS was then added at 75% of the volume required to obtain 250 mg/mL of homogenate. The tissues next underwent 2 cycles of homogenization at 1800 oscillations/min, 45 seconds each. Lysates were supplemented thereafter with 4× RIPA buffer to make the remainder 25% of the volume (final concentrations: 0.022% β glycerophosphate, 1% 4‐nonylphenol, branched, ethoxylated, 0.018% sodium orthovanadate, 0.5% sodium deoxycholate, 0.038% EGTA, 1% sodium lauryl sulfate, 0.61% Tris, 0.029% EDTA, 0.88% NaCl, 0.112% sodium pyrophosphate decahydrate; v/v, pH 7.5) and placed on an orbital rocker for 1 hour at room temperature to complete the collagen solubilization process. Samples were kept frozen at −80 °C, thawed before use, and clarified by centrifugation at 1600g for 10 minutes at 4 °C. Fifty‐five microliters of each supernatant were finally sampled and scanned in black‐walled, clear bottom 96‐well plates (Corning Life Sciences, Kennebunk, ME) on the Odyssey CLx NIRF scanner (169‐µm resolution, manual L1 intensity, and 3‐mm focus offset settings). A standard curve of each peptide in PBS was prepared to convert fluorescence reads to peptide molarity values.
Blood Clearance and Peptide Toxicity Studies
Eight‐ to 9‐month‐old male, nontransgenic β2‐AR mice were intravenously injected with sulfo‐Cy5.5–labeled peptides (0.5 mg/kg), as described above. Blood samples (2.5 µL) were then collected from the tail vein opposing the injected vein at 2.5, ≈10, ≈15, and 240 minutes, and placed in 47.5 µL of citrated PBS (0.32% w/v sodium citrate). Samples (50 µL) were then transferred into black‐walled, clear bottom 96‐well plates and scanned on the Odyssey CLx NIRF scanner, as previously detailed. The total fluorescence per well (after subtraction of unlabeled blood background) was then plotted against time and the half‐life calculated from a one‐phase decay nonlinear regression.
Mice were humanely euthanized 24 hours after peptide administration and transcardially perfused with PBS (20 mL), followed by 10% (v/v) neutral‐buffered formalin (10 mL). The liver and kidneys were postfixed in neutral‐buffered formalin for 24 hours and embedded in paraffin. Peptide toxicity in hepatic and renal tissues was then evaluated from 4‐µm‐thick, hematoxylin and eosin–stained sections of treated versus untreated mice.
Ex Vivo Staining of Human Heart Sections With Collagen Peptide Tracers
Fresh‐frozen human IHD and nonfibrotic donor specimens were cryosectioned at 10 or 5 µm. Slides were washed twice for 7 minutes each in PBS at room temperature to remove the optimal cutting temperature material. Sections were then blocked in 10% v/v normal goat serum in PBS for 2 hours at room temperature. The blocking solution was then aspirated and the sections incubated for 72 hours at 4 °C with 0.188 and 0.0375 µg/mL of sulfo‐Cy5.5 cyclic peptide and T peptide, respectively, diluted in PBS. Sections were finally washed 3 times in PBS (5 minutes each) and mounted with ProLong Diamond Antifade mountant, followed by scanning on the 2‐dimensional Odyssey CLx NIRF scanner.
Immunohistochemistry and Immunofluorescence
A subgroup of fresh (unfixed, perfused) isolated mouse hearts were snap‐frozen on dry ice. Tissues were kept at −80 °C until cryosectioning at 10 μm in 5 identical series. One series was stained for Masson’s trichrome (MTC), whereas another underwent collagen immunohistochemistry. For the latter, sections were first incubated for 1 hour at room temperature in a blocking solution (5% goat serum in PBS). Anti‐collagen type I, anti‐collagen type III, and anti‐collagen type IV primary antibodies (SC‐59772, Santa Cruz, Dallas, TX; ab7778 and ab6586, Abcam, Melbourne, Australia, respectively; 10 µg/mL) were diluted in blocking solution and incubated with the sections overnight at 4 °C. The sections were then washed 3 times for 5 minutes each with PBS and incubated with 2 µg/mL of Alexa Fluor 647 donkey anti‐mouse (collagen I) or goat anti‐rabbit (collagen III and IV) secondary antibodies (Thermo Fisher Scientific, Melbourne, Australia) in blocking solution for 3 hours at room temperature. For costaining of collagen and the T peptide, we used perfused mouse hearts that were labeled in vivo with sulfo‐Cy5.5 T peptide for 4 hours, as described above. Sections then underwent the same collagen immunohistochemistry using anti‐collagen primary antibodies, but Alexa Fluor 488‐conjugated secondary antibodies (2 µg/mL; Thermo Fisher Scientific). In all experiments, nuclei were counter stained with Hoechst (5 µg/mL; Thermo Fisher Scientific) for 1 hour (added 2 hours after application of the secondary antibody). The sections were finally washed 3 times (5 minutes each) with PBS and mounted using ProLong Diamond Antifade mountant (Thermo Fisher Scientific). Images were acquired using the Nikon Imaging Software (NIS‐Elements; Nikon, Tokyo, Japan), on widefield fluorescence and confocal microscopes (Nikon Ti‐E and Nikon A1r, respectively), at ×20 magnification (Plan APO lens, Air, NA 0.75; Nikon) at identical settings for each collagen type. Quantification of the mean collagen signal intensity in each heart section was calculated from 4 random captures using Image J Fiji (available at: http://fiji.sc/) after thresholding.
Gelatin Zymography
MMP‐2 and ‐9 activity were evaluated in ventricular lysates (250 wet weight/mL) from perfused hearts of 8‐month‐old β2‐AR mice and their littermate controls, as previously described. 41 In brief, protein samples (60 µg/lane) were subjected to electrophoresis under nondenaturing conditions on SDS‐polyacrylamide gels containing 0.03% (w/v) gelatin. Following 3 washes of 30 minutes with Triton X‐100 solution (2.5% v/v) to remove SDS, gels were incubated overnight at 37 °C in developing buffer (10 mmol/L CaCl2, 50 mmol/L Tris‐HCl, pH 7.4, 150 mmol/L NaCl, and 1% (v/v) Triton X‐100) with gentle agitation. Bands of gelatinolytic activity were visualized as areas devoid of color after Coomassie blue staining.
Statistical Analysis
Statistical analyses were performed using GraphPad Prism version 9. Data sets with n=3 are presented as median±interquartile range, whereas those with n≥4 as mean±SEM. Pairwise analyses employed the Mann‐Whitney test (1‐ or 2‐tailed, as described in the figure legends), whereas ≥3 groups differing in 1 independent variable were analyzed by the Kruskal‐Wallis test with Dunn post hoc analysis. Data sets differing in 2 or more independent variables first passed both the Shapiro‐Wilk (n≥4) and the Kolmogorov‐Smirnov (n≥5) normality tests. A 2‐ or 3‐way ANOVA (with repeated measures when the right and left ventricles were separately sampled) was then performed, followed by Tukey or Sidak post hoc analysis (see figure legends). Each significantly‐different pair of columns by these parametric tests was also verified by a separate Mann‐Whitney test (not shown). A P≤0.05 was considered significant.
Results
Cardiac Collagens Profiles in the β2‐AR Mouse Model of Diffuse Cardiovascular Disease and in Human IHD
Using MTC staining and immunohistochemistry, we first evaluated, respectively, interstitial collagen expression and collagen subtypes in both clinical heart specimens (Figure S1) and in aged, cardiac fibrosis‐prone β2‐AR mice (10–11 months of age) 31 , 32 , 33 , 34 (Figure 2). As expected, distinct areas of fibrosis were observed by MTC staining in the human IHD tissues compared with healthy controls (Figure S1A), corresponding to a significant increase in the Ishak fibrosis scoring (0–6) of blue staining (P=0.05) (Figure S1B). Immunohistochemical analysis further showed a significant increase in collagen types I and IV (P=0.05) (Figure S1C, S1D, and S1F) and to a lesser extent of collagen type III (P=0.1) (Figure S1C and S1E) in the interventricular septum of IHD samples compared with the healthy donor specimens.
Figure 2. Fibrosis and collagen (Col) types in fibrotic β‐2‐adrenergic receptor (β2‐AR) transgenic mice (Tg) vs nontransgenic (Ntg) healthy controls.
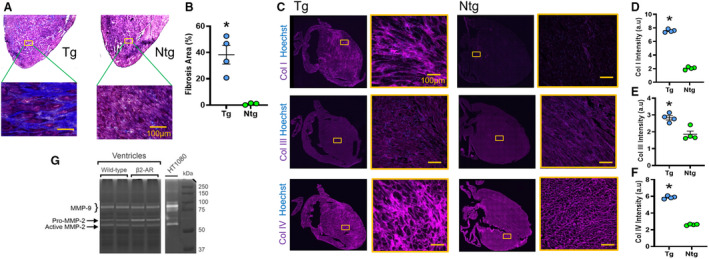
Representative images (A) and quantification (percentage of fibrotic area, B) of Masson’s trichrome–stained sections from Tg and Ntg mouse hearts. C through F, Representative heart images (C) and mean fluorescence intensities of Col I (D), Col III (E), and Col IV (F) in fibrotic (Tg) vs nonfibrotic (Ntg) control mice at 11 months of age. G, A gelatin zymogram of heart lysates from β2‐AR Tg and Ntg mice (8 months of age) demonstrating upregulation of MMP (matrix metalloproteinase)‐2 in this model, indicative of active disease. Mean±SEM. *P≤0.05 compared with the Ntg group by Mann‐Whitney test (1‐tailed). N=4 per group, except for the Ntg group in Figure 1B, where n=3. Scale bars=100 µm. a.u. indicates arbitrary unit; and HT1080 indicates conditioned medium from HT1080 human fibrosarcoma cell line stimulated with Phorbol 12‐myristate 13‐acetate (positive control).
Because the fibrotic areas in these clinical tissues represented mature (nonactive) scars after an ischemic heart attack, we also used the β2‐AR overexpressing mouse model. 32 This model is characterized by a nonischemic, diffuse, and active interstitial cardiac collagen deposition with a heart failure phenotype 32 developing in the heterozygous mouse from 6 months of age. 31 , 32 , 33 At 11 months of age, a striking, diffuse fibrosis covering about 40% of the myocardium was confirmed in our transgenic mice by MTC staining (P<0.01) (Figure 2A and 2B). Accordingly, the staining intensities of collagen types I, III, and IV were significantly elevated in the fibrotic β2‐AR transgenic mice compared with their nontransgenic controls (P<0.05) (Figure 2C through 2F). The β2‐AR mice also harbored an enhanced ventricular MMP‐2 activity on a gelatin zymogram (Figure 2G), as previously reported, 33 indicative of a collagen turnover and an active cardiomyopathic process. Together, the human IHD samples and mouse β2‐AR model provide us with excellent tools for assessment of collagen tracers against diffuse cardiac fibrosis. The β2‐AR transgenic mouse, in particular, manifests an active MMP‐2 and collagen IV–rich heart disease, ideal for studies using the unique T peptide.
The Collagen IV–Targeting T Peptide Detects Diffuse Myocardial Fibrosis in β2‐AR Mice
We next interrogated the potentials of type I and IV collagen‐targeted peptides (Figure 1) to visualize and quantify cardiac interstitial fibrosis in fibrotic β2‐AR mice. These mouse studies (Figure 3A) were designed to include in vivo tracer administration with ex vivo fluorescent imaging.
Figure 3. Collagen IV–targeted, T‐peptide enhancement of fibrotic mouse hearts by whole‐organ ex vivo imaging.
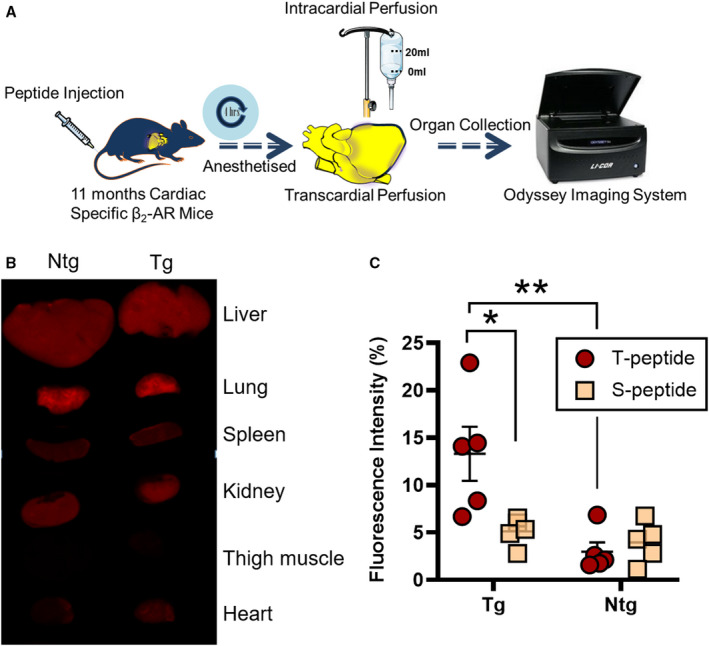
A, The study design. B, Representative ex vivo Odyssey images demonstrating the biodistribution of sulfo‐cyanine5.5 (Cy5.5) T peptide in perfused, transgenic (Tg) β2‐AR (β‐2‐adrenergic receptor) mice (right column) and nontransgenic (Ntg) controls (left column). Although cardiac uptake increases in Tg animals, the kidney, liver, and lung uptakes are consistently high irrespective of the genotype. C, Quantification of sulfo‐Cy5.5 T‐peptide–mediated cardiac enhancement of fibrotic Tg hearts compared with Ntg controls. The T‐peptide specificity is further observed against reduced accumulation of the control S peptide, but only in Tg mice (ie, at the level of disease). No differences between T and S peptides are observed in Ntg mice at the whole‐organ level. Mean±SEM. N=5 per group. *P<0.05. **P<0.01 by 2‐way ANOVA with Sidak post hoc.
We first examined the T peptide 25 and a nonbinding version, the S peptide (GLGYGWS), which served as control. Strikingly, when performing whole‐organ Odyssey scans (Figure 3B), no statistically different T‐peptide uptake was observed in any of the organs scanned except the heart, neither when comparing transgenic (fibrotic) mice versus nonfibrotic controls nor relating T versus S peptides (Figure S2). In the heart, however, we found a statistically significant difference in average peptide uptake, depending on both the genotype (F[1]=12.69, P<0.01) and the peptide type (F[1]=4.999, P<0.05), with a significant interaction between these terms (F[1]=8.061, P<0.05) (Figure 3C). A post hoc analysis then revealed that the fluorescence intensity of the T peptide (per unit area) in fibrotic β2‐AR mice was significantly higher compared with nonfibrotic animals (P<0.01) (Figure 3C), indicating a fundamental capacity of the T peptide to detect cardiac fibrosis. Furthermore, the fluorescence intensity of the S peptide compared with the T peptide was significantly lower in transgenic mice (P<0.05) (Figure 3C), but not in the nonfibrotic littermate controls (Figure 3C), suggesting specificity in the T‐peptide uptake but mainly at the level of disease. Similar results were notably obtained in 7‐month‐old β2‐AR mice harboring an earlier‐stage cardiomyopathy with an intermediate cardiac fibrosis (compared with the severe phenotype at 11 months of age) 33 , 42 , 43 , 44 (Figure S3). These important observations, in full correlation with the fibrotic phenotypes (Figure 2), laid exciting foundations for further development of the T peptide as an imaging tracer for diffuse cardiac fibrosis; nevertheless, because of the foundational role of these findings and potential differences between the T and S peptides (below), we sought to confirm these results by directly measuring peptide levels in tissue homogenates.
Because the right and left ventricles may pathologically defer in the β2‐AR model, 44 we first separated each heart to its compartments. The exact levels of fluorescent peptide trapped in each ventricular lysate (Figure 4A) were then extracted from standard curves of the T and S peptides at equimolar concentrations (accounting for inherent differences in molecular weight and brightness [fluorescence per mole] between them). Cardiac peptide concentrations significantly relied on the genotype (F[1]=236.1, P<0.0001), the peptide used (F[1]=64.05, P<0.0001), and as postulated, also on the side of the heart (F[1]=342.4, P<0.0001), with significant interactions between every combination of these variables (P<0.001) (Figure 4B). Consistent with the peptide accumulation profiles seen at the whole‐organ level (Figure 3), a post hoc analysis demonstrated significantly higher T‐peptide concentrations in fibrotic β2‐AR transgenic mice compared with nonfibrotic, nontransgenic controls in both the right (P<0.05) and left ventricles (P<0.0001), with the left chamber accumulating 4.2‐fold more T peptide than the right under disease conditions (P<0.0001) (Figure 4B).
Figure 4. Specific accumulation of T peptide in cardiac lysates from fibrotic β2‐AR (β‐2‐adrenergic receptor) mice.
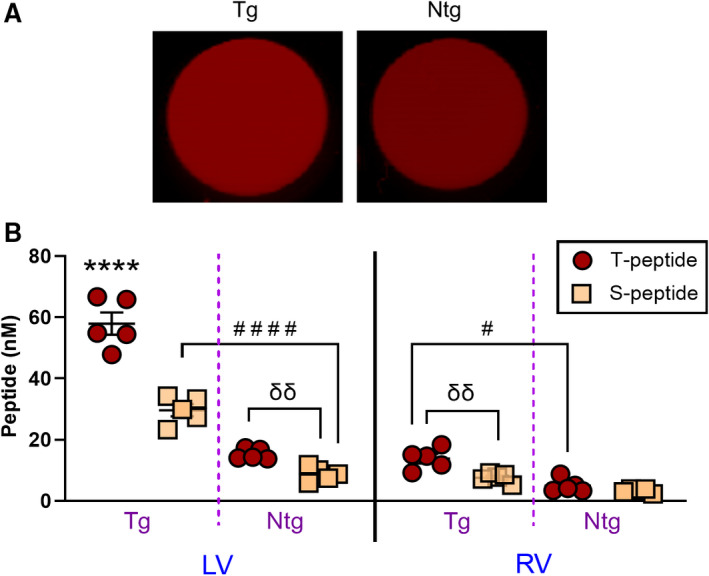
A, Representative Odyssey scans of 96 wells containing ventricular lysates from perfused hearts of fibrotic β2‐AR (Tg) and nonfibrotic (Ntg) control mice treated with sulfo‐cyanine5.5 (Cy5.5) T peptide. Fluorescent signal enrichment in the Tg sample can be observed. B, Quantification of sulfo‐Cy5.5 T‐ and S‐peptide levels in lysates from the left ventricle (LV) and right ventricle (RV) showing enhanced concentrations of the T peptide in fibrotic hearts (Tg) compared with nonfibrotic controls (Ntg). Reduced levels of the control S peptide compared with the T peptide is further observed in both chambers at the disease level (Tg), evidence of T‐peptide specificity. Enhanced T‐ vs S‐peptide uptake at the basal (Ntg) levels is also detected in the left but not in the right ventricle. Mean±SEM. N=5 per group. ****P<0.0001 against all other groups, #### P<0.0001. # P<0.05 by repeated‐measures 3‐way ANOVA with Tukey post hoc. δδ P<0.01 by an unpaired t test.
Importantly, from the peptide’s standard curves, it was revealed that the S peptide was 2.169‐fold brighter than the T peptide on an equimolar basis, as shown by the slope ratio of curves (Figure S4). Taking this factor into account and in line with our previous results in whole organs (Figure 3C), the post hoc analysis of the homogenate method also confirmed that the S‐peptide uptake was significantly reduced in transgenic mouse hearts compared with the T peptide, in both the left and right ventricles (P<0.0001 and P<0.01, respectively) (Figure 4B). Surprisingly, in contrast to the whole organ scans (Figure 3C), S‐peptide concentrations were also reduced in the left ventricles of nontransgenic hearts (P<0.01) (Figure 4B). Although the latter may be explained by a better accuracy of the homogenate‐based quantification method, these results overall reiterate that the T‐peptide uptake was largely specific.
To highlight additional aspects of the use of the T peptide in vivo, we gauged its blood clearance rate and possible toxicity (Figure S5). As anticipated, the T peptide was rapidly cleared with a half‐life of 8.96±0.19 minutes (Figure S5A) and induced no obvious toxic effects on hepatic and renal tissues (being the most highly exposed) when histologically evaluated 24 hours after administration (Figure S5B). These characteristics add to the attractiveness of the T peptide as a novel molecular imaging agent.
Imaging of Diffuse Cardiac Fibrosis in β2‐AR Mice Using a Collagen I–Targeted Cyclic Peptide
We next targeted type I collagen with a cyclic peptide named EP‐3533. 26 , 27 , 29 , 30 Encouragingly, whole organs imaging on the Odyssey NIRF scanner 4 hours after cyclic peptide administration (as performed with the T peptide) (Figure 3A) showed a significant increase in the uptake of this collagen I tracer in fibrotic ventricles of β2‐AR transgenic mice compared with their littermate nontransgenic controls (Figure 5A and 5B) (P<0.05). It was also interesting to note that the atrial tissue qualitatively showed a much stronger fluorescent peptide uptake than the ventricles under both heathy (nontransgenic) and diseased conditions (Figure 5A).
Figure 5. Collagen I‐targeted cyclic peptide enhancement of fibrotic mouse hearts and major organs in β2‐AR (β‐2‐adrenergic receptor) mice.
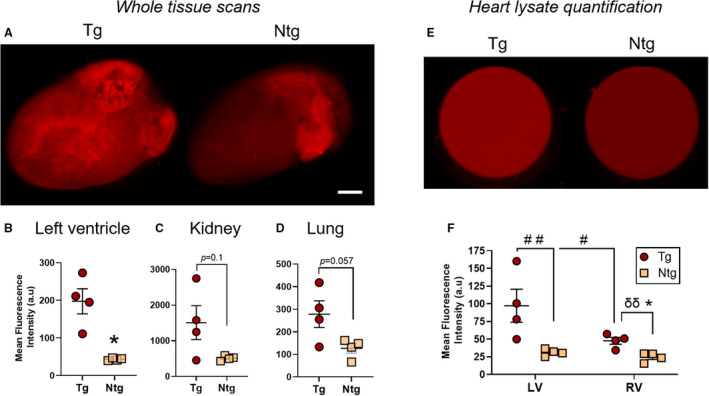
A through D, Representative ex vivo whole heart Odyssey images (A) and quantitation of cyclic peptide accumulation in the left ventricle (B), kidney (C), and lung (D) demonstrating cyclic peptide enhancement of perfused hearts and major organs in transgenic (Tg) vs nontransgenic (Ntg) β2‐AR mice 4 hours after peptide administration (0.5 mg/kg). E and F, Representative Odyssey scans (E) and quantification (F) of 96 wells containing left and right ventricular lysates from the hearts of these cyclic peptide–treated β2‐AR mice. Fluorescent signal enrichment in both chambers of the Tg mouse heart can be observed. Mean±SEM. N=4 per group. In (B through D and F), *P<0.05 by Mann‐Whitney test (1‐tailed). In (F), # P<0.05, ## P<0.01 by repeated‐measures 2‐way ANOVA with Sidak post hoc; δδ P<0.01 by an unpaired t test. Scale bar=1.8 mm. a.u. indicates arbitrary unit.
Furthermore, a near‐significant enhancement of peptide accumulation was detected also in the kidneys and lungs of fibrotic mice (Figure 5C and 5D, respectively) (P=0.057–0.1). These findings could indicate a wider‐spread fibrotic phenotype in this heart‐specific model, which is characterized by a left‐heart failure with pulmonary congestion 44 that could lead to pulmonary fibrosis following chronic lung overload and to a cardiorenal pathology. Nevertheless, when assessing both lungs and kidneys by MTC staining (Figure S6A and S6B), we identified only an age‐related circumferential peribronchial idiopathic fibrosis (Figure S6C), with no significant differences in circumferential peribronchial idiopathic fibrosis and in interstitial fibrosis between transgenic and nontransgenic mice (Figure S6A and S6C). These findings raise the possibility that an additional molecular target may exist for the cyclic peptide. Interestingly, no differences in elastin fiber deposition between the genotypes was identified in the lungs (Figure S6D), excluding this other important ECM protein as a possible off‐target and prompting the need for additional investigation into the subject.
To confirm the robust observations with the cyclic peptide in whole‐heart scans (Figure 5A and 5B), we tested our results once again in cardiac lysates (250 wet weight/mL) from the right and left lower‐heart chambers (Figure 5E). The cardiac concentrations of the cyclic peptide were significantly affected by both the genotype (F[1]=10.03, P<0.05) and the side of the heart (F[1]=8.368, P<0.05), but there was no significant interaction between these factors (F[1]=4.88, P=0.07). A post hoc analysis importantly revealed that the mean fluorescence intensity of sulfo‐Cy5.5 cyclic peptide in cardiac lysates from fibrotic transgenic animals was ≈2‐ to 3‐fold higher than in lysates from nontransgenic littermate controls (3.13‐ and 1.95‐fold for the left and right ventricles, respectively; P<0.01) (Figure 5F). Notably, the left transgenic ventricle attracted on average twice the amount of cyclic peptide (per gram tissue) than the right, evidence of a different fibrotic state of the ventricles in the β2‐AR model (P<0.05) (Figure 5F). Clearance and toxicity wise, the cyclic peptide resembled the T peptide in its rapid clearance rate (half‐life of 10.12±0.38 minutes) (Figure S5A) and lack of obvious toxicity to the liver and kidney 24 hours after exposure (Figure S5B). Taken as a whole, collagen I tracing by the cyclic peptide emerges as another promising and powerful strategy for molecular imaging of diffuse cardiac fibrosis.
Confirmation of the T Peptide Collagen‐Binding Specificity by Confocal Microscopy in Mouse Tissue
To prove the collagen‐binding specificity of the T peptide in the mouse, we conducted coimmunohistochemical analysis of the collagen subtypes previously characterized in the β2‐AR strain (Figure 2) together with the T peptide, labeled in vivo by intravenous injection. As shown in Figure 6, the sulfo‐Cy5.5 T peptide strongly colocalized with microscars of basement membrane type IV collagen (Figure 6A and Video S1), but less with fibrillar collagen types I and III (not shown), an observation further reflected by the degree of overlap and fine spatial staining patterns within the collagen deposits (Video S1yet a binding dependency on the collagen‐degrading activity of MMP‐2 in vivo remains to be confirmed.
Figure 6. Confocal microscopy of collagen (Col) IV binding by sulfo‐cyanine5.5 (Cy5.5)‐conjugated T peptide.
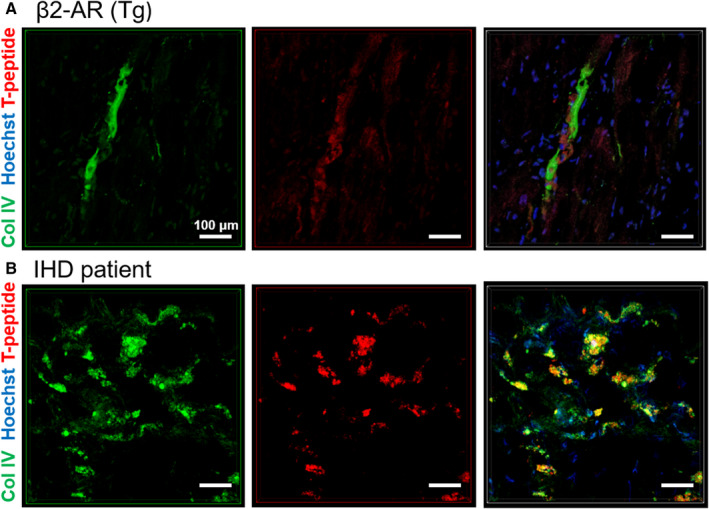
Colocalization of sulfo‐Cy5.5‐labeled T peptide (red; excitation/emission 647/703) with Col IV (green; excitation/emission 488/519) in heart sections from a fibrotic β2‐AR (β‐2‐adrenergic receptor) mouse (in vivo labeling with Cy5.5 T peptide for 4 hours) (A) and from a patient with ischemic heart disease (IHD) (ex vivo costaining) (B). Cell nuclei are depicted in blue (Hoechst). Scale bars=100 µm. Tg indicates transgenic.
Fibrosis Detection in Clinical IHD Specimens by the Collagen Peptide Tracers
Finally, we sought to validate these findings ex vivo in human tissue sections from heart specimens of patients with IHD and healthy donors. Both the T peptide and cyclic peptide conjugates enhanced the areas of clinical fibrosis, as could be inferred from their colocalization with collagen‐rich areas exposed by MTC staining in successive heart sections (Figure 7). Whereas the cyclic peptide staining pattern was generally characterized by a prominent retention in fibrotic areas throughout the sections, the colocalization of the T peptide with collagen deposits was more limited to the section periphery (Figure 7A and 7B). Nevertheless, these varying staining patterns likely represent the particular distribution of the different collagen‐type deposits within each specimen. Overall, the fibrotic regions of the IHD samples consistently showed enhanced peptide uptake compared with donor (nondiseased) tissue, irrespective of staining durations (from 30 minutes to 72 hours, not shown), indicative of an affinity of our tracers also to human heart collagens. Coimmunostaining of collagen IV and the T peptide revealed a high degree of overlap between the epitope and its tracer, confirming a prominent specificity of the T‐peptide probe also in human cardiac tissue (Figure 6B and Video S2).
Figure 7. Collagen binding by T and cyclic peptides in human heart tissue.
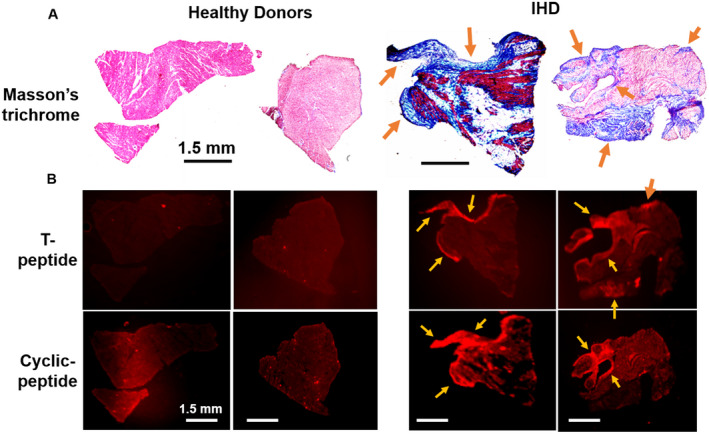
Representative corresponding images of Masson’s trichrome (MTC)‐stained (A) and ex vivo sulfo‐cyanine5.5 T‐ and cyclic peptide–labeled (B) human heart sections from patients with ischemic heart disease (IHD) (right) and from healthy donors (left). Colocalization of tracer accumulation in areas of cardiac fibrosis (blue patches on MTC staining, matching arrows) can be observed. Images represent 3 different specimens per tracer. Scale bars=1.5 mm.
Several limitations could have accompanied this clinical investigation, mainly for the use of T peptide, because these human scars are conceptually more established and less active, with minimal matrix metalloproteinase activity and collagen turnover. In addition, in vivo labeling of myocardial fibrosis in the mouse could have been more robust and less washable than ex vivo labeling of human tissue. However, although further studies are required, both the T peptide and the cyclic peptide appear to preferentially bind fibrotic human cardiac tissue, opening exciting possibilities for future translation of these tracers.
Discussion
Myocardial fibrosis characterizes the evolution of nearly all forms of myocardial diseases as it adversely affects the heart muscle. At the macro level, it alters ventricular geometry such as chamber volume and configuration. 45 On biochemical, histological, and functional characterization, the alterations are accompanied by cardiomyocytes apoptosis, 46 loss of molecular signaling, 47 hypertrophy, ECM synthesis and expansion, and progressive interstitial diffuse fibrosis. The severity of ECM expansion, commonly referred to as scaring, results in adverse cardiac remodeling, both in animal models and in patients. Importantly, major components participating in cardiac ECM expansion include fibrillar collagens, like type I and III, but also type IV basement membrane collagen.
As often performed by others using various models, 33 , 34 , 39 our traditional MTC staining demonstrated extensive fibrosis and excessive accumulation of collagen in the myocardium of aging β2‐AR transgenic mice. These mice were not only highly fibrotic, but also enriched in various collagen types, particularly collagen type I and IV. Because of the widespread abundance of these collagen types all through the β2‐AR mouse heart, it was logical to select this model and these molecular targets for the development of collagen‐avid probes for noninvasive imaging of diffuse cardiac disease. Success in this task could ultimately enable early detection, diagnosis, and monitoring of cardiac fibrosis‐associated heart disease. Accordingly, the T peptide, an MMP‐2–digested collagen IV tracer, 25 and a cyclic peptide that targets type I collagen with high affinity 26 , 29 , 30 were chosen, attempting to harness key molecular characteristics of cardiac ECM expansion (ie, collagen deposition) for diagnosis. We showed that diffuse collagen expression can be visualized using sulfo‐Cy5.5 cyclic‐ and T‐peptide–enhanced NIRF imaging, and that interstitial fibrosis is a suitable site for molecular imaging, that may be directly targeted for diagnostic and therapeutic purposes. This is contrary to current magnetic resonance imaging–based indirect methods for visualization and quantification of dispersed disease such as late gadolinium enhancement. 20 , 48
When factoring in further the affinity of the T peptide to MMP‐2–degraded collagen IV, which marks active cardiac disease, 16 , 17 and the reduced uptake of the control S peptide at both disease and baseline levels, the T peptide in particular represents a smart, specific, and sensitive probe that could be most suitable for disease stratification. Within this context, it will be also vital to unveil whether the activity of our probes can encompass the presence of cross‐linked collagens, shown to be clinically important for cardiac disease prognostication and outcome. 49 The cross‐linking of collagen type I by lysyl oxidase marks an acute cardiac fibrotic process and worsening fibrogenesis, because it promotes the build‐up of thicker and insoluble collagen fibers within the heart that are also resistant to matrix metalloproteinase degradation. 49 Consequently, several dedicated molecular imaging probes that target allysines (lysyl oxidase–generated, oxidized lysine residues that mediate collagen crosslinking) are being developed. 50 Retention of the T and/or cyclic peptide’s binding capacity also to cross‐linked collagen will add much value for future clinical adoption of these probes.
Another finding worth noting was an enhanced accumulation of the cyclic peptide in atria compared with the lower chambers of the heart (Figure 5A). This observation, also obtained with the T peptide (not shown), could stem from a well‐known increase (about 4‐fold) in the collagen content in normal atrial tissue compared with ventricles, a chamber difference that is maintained under diseased conditions 51 , 52 (possibly because of a more prominent profibrotic atrial signaling of TGF‐β1 [transforming growth factor‐β1]). 53 The capacity to strongly enhance the atrial tissue via collagen‐targeting probes opens exciting possibilities to use these strategies also for imaging and therapy of atrial conditions, a key clinical challenge.
Favorably, although this is a proof‐of‐concept study, both the T and cyclic peptides were administered intravenously at a well‐tolerated and nontoxic micromolar dose (estimated 2.5–4 µmol/L in the blood), extravasated into the rodent heart within a clinically relevant 4‐hour time frame (and most likely quicker), were rapidly cleared from the circulation (t1/2=≈10 minutes) (Figure S5), and demonstrated a binding capacity toward human fibrotic heart tissue, making their use consistent with clinical demands. Overall, the peptides’ ability to robustly and rapidly demonstrate interstitial fibrosis, albeit by ex vivo detection at this stage, arms these tracers with a promising translation potential as probes that enable early diagnosis, staging, and monitoring of heart disease, especially if configured as a background‐free contrast agent for use on a more advanced imaging modality such as positron emission tomography/computed tomography.
From an experimental aspect, our NIRF imaging approach represents a superior imaging technique for in vitro, ex vivo, and possibly also in vivo applications. NIRF imaging offers a simple‐to‐operate, nontoxic (ionizing radiation‐free), and low‐cost alternative to complex, expensive positron emission tomography and magnetic resonance imaging modalities. Additional advantages of ex vivo NIRF imaging include the ultralow autofluorescence emitted by biological tissues at these wavelengths and the extreme sensitivity of some scanners, such as the Odyssey used here; however, for extension of the NIRF imaging capacity for in vivo use, some issues remain to be overcome. For example, a limited sensitivity because of poor light penetration through tissue exists, as well as light absorption and scattering issues, affecting fluorophore excitation, emission, and signal collection capabilities above background. 54 Implementation of NIRF imaging at the second near‐infrared (NIR‐II) window (1000–1700 nm, above the NIR‐I range of 400–900 nm used here) offers a true breakthrough in this regard. 54 Evidently, despite the challenges, investigators are increasingly applying NIRF imaging in preclinical and even clinical settings. 55 , 56 , 57 allowing the vision of diffuse cardiac fibrosis imaging by NIRF‐conjugated peptide tracers in the foreseeable future.
Some limitations accompany our present study. First, as mentioned above, a possible off‐target binding partner may exist, especially for the cyclic collagen I peptide probe EP‐3533 (Figure 5 and Figure S6), raising the need to explore this possibility further for complete validation of our observation with this agent. Second, we did not assess whether the peptides can demonstrate myocardial fibrosis at early subclinical disease stage, though the ability of the T peptide to distinguish intermediate cardiac fibrosis in 7‐month‐old transgenic mice (Figure S3) and its specific uptake in aged, nontransgenic healthy mice (Figure 4) was encouraging in this context. Third, cardiac remodeling is a dynamic process, therefore age‐dependent, time‐dependent, 44 and disease‐specific quantitative changes in ECM expansion (eg, in the collagen content, type, typical proportions, and distinctive ECM morphologies such as cross‐linked collagen 49 ) may affect imaging resolution, create detection limits or, on the contrary, present opportunities for more accurate disease stratification. Additional studies are required to assess the optimal time window of visualization, as well as the potential to discriminate between different stages of cardiac disease via collagen deposition and tailored molecular probes.
In summary, we demonstrated here the feasibility of a targeted molecular imaging approach for detection of diffuse myocardial fibrosis using specific collagen probes. Experimentally, the NIR fluorophore‐conjugated, collagen‐binding peptide approach, combined with a NIRF imaging capability, directly detects myocardial interstitial fibrosis and enables both visualization and quantification of myocardial disease ex vivo with great sensitivity. Importantly, the affinity of these peptide tracers to fibrotic human cardiac tissue lays the foundation for future development of these methods also for clinical imaging of diffuse cardiac fibrosis and its associated heart disease.
Sources of Funding
This work has been funded by grant number GNT1120129, awarded to C.E.H. by the National Health and Medical Research Council of Australia (NHMRC). M. Ezeani is supported by a Government of Australia Training Research Program Scholarship and the Monash University, Faculty of Medicine, Nursing and Health Sciences Postgraduate Excellence Award. Dr Alt is a NHMRC Medical Research Future Fund (MRFF) Career Development Fellow (award number GNT1140465). C. Hagemeyer is a Senior Research Fellow of the NHMRC of Australia (award number GNT1154270). Dr Niego is supported by NHMRC grant number GNT1120129.
Disclosures
None.
Supporting information
Data S1
Figures S1–S6
Video S1
Video S2
Acknowledgments
The authors wish to thank Monash University’s Micro‐Imaging Platform (S. Cody, Dr Carmichael, and C. Johnson) and Histology Platform (C. Cohen and A. Shad), as well as the Mass Spectrometry and Proteomics Facility at the Bio21 Institute, University of Melbourne, for their professional support. We thank X‐J. Du (the Baker Institute, Australia and Xi'an Medical University, China) for kindly providing the β2‐AR transgenic mouse strain and for his generous advice, and the dedicated staff of the Precinct Animal Centre for their professional support and colony care during this project. The authors further thank Drs Harris and van Dam from Clarity Pharmaceuticals, Sydney, Australia, for their support. The authors thank C. dos Remedios and the late Dr Chang from the Sydney Heart Bank for the human heart tissues, and finally, the patients and staff of St. Vincent’s Hospital, Sydney, and the Australian Red Cross Blood Service.
Supplementary Material for this article is available at https://www.ahajournals.org/doi/suppl/10.1161/JAHA.121.022139
For Sources of Funding and Disclosures, see page 13.
References
- 1. Dunbar SB, Khavjou OA, Bakas T, Hunt G, Kirch RA, Leib AR, Morrison RS, Poehler DC, Roger VL, Whitsel LP, et al. Projected costs of informal caregiving for cardiovascular disease: 2015 to 2035: a policy statement from the American Heart Association. Circulation. 2018;137:e558–e577. doi: 10.1161/CIR.0000000000000570 [DOI] [PubMed] [Google Scholar]
- 2. Senra T, Ianni BM, Costa ACP, Mady C, Martinelli‐Filho M, Kalil‐Filho R, Rochitte CE. Long‐term prognostic value of myocardial fibrosis in patients with Chagas cardiomyopathy. J Am Coll Cardiol. 2018;72:2577–2587. doi: 10.1016/j.jacc.2018.08.2195 [DOI] [PubMed] [Google Scholar]
- 3. Travers JG, Kamal FA, Robbins J, Yutzey KE, Blaxall BC. Cardiac fibrosis: the fibroblast awakens. Circ Res. 2016;118:1021–1040. doi: 10.1161/CIRCRESAHA.115.306565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Nattel S. Molecular and cellular mechanisms of atrial fibrosis in atrial fibrillation. JACC Clin Electrophysiol. 2017;3:425–435. doi: 10.1016/j.jacep.2017.03.002 [DOI] [PubMed] [Google Scholar]
- 5. Ugolini GS, Pavesi A, Rasponi M, Fiore GB, Kamm R, Soncini M. Human cardiac fibroblasts adaptive responses to controlled combined mechanical strain and oxygen changes in vitro. Elife. 2017;6:e22847. doi: 10.7554/eLife.22847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Burke MA, Chang S, Wakimoto H, Gorham JM, Conner DA, Christodoulou DC, Parfenov MG, DePalma SR, Eminaga S, Konno T, et al. Molecular profiling of dilated cardiomyopathy that progresses to heart failure. JCI Insight. 2016;1:e86898. doi: 10.1172/jci.insight.86898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wong TC, Piehler K, Meier CG, Testa SM, Klock AM, Aneizi AA, Shakesprere J, Kellman P, Shroff SG, Schwartzman DS, et al. Association between extracellular matrix expansion quantified by cardiovascular magnetic resonance and short‐term mortality. Circulation. 2012;126:1206–1216. doi: 10.1161/CIRCULATIONAHA.111.089409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Almehmadi F, Joncas SX, Nevis I, Zahrani M, Bokhari M, Stirrat J, Fine NM, Yee R, White JA. Prevalence of myocardial fibrosis patterns in patients with systolic dysfunction: prognostic significance for the prediction of sudden cardiac arrest or appropriate implantable cardiac defibrillator therapy. Circ Cardiovasc Imaging. 2014;7:593–600. doi: 10.1161/CIRCIMAGING.113.001768 [DOI] [PubMed] [Google Scholar]
- 9. Moreo A, Ambrosio G, De Chiara B, Pu M, Tran T, Mauri F, Raman SV. Influence of myocardial fibrosis on left ventricular diastolic function: noninvasive assessment by cardiac magnetic resonance and echo. Circ Cardiovasc Imaging. 2009;2:437–443. doi: 10.1161/CIRCIMAGING.108.838367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ellims AH, Shaw JA, Stub D, Iles LM, Hare JL, Slavin GS, Kaye DM, Taylor AJ. Diffuse myocardial fibrosis evaluated by post‐contrast T1 mapping correlates with left ventricular stiffness. J Am Coll Cardiol. 2014;63:1112–1118. doi: 10.1016/j.jacc.2013.10.084 [DOI] [PubMed] [Google Scholar]
- 11. Heymans S, Gonzalez A, Pizard A, Papageorgiou AP, Lopez‐Andres N, Jaisser F, Thum T, Zannad F, Diez J. Searching for new mechanisms of myocardial fibrosis with diagnostic and/or therapeutic potential. Eur J Heart Fail. 2015;17:764–771. doi: 10.1002/ejhf.312 [DOI] [PubMed] [Google Scholar]
- 12. Humeres C, Frangogiannis NG. Fibroblasts in the infarcted, remodeling, and failing heart. JACC Basic Transl Sci. 2019;4:449–467. doi: 10.1016/j.jacbts.2019.02.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Braunwald E. Heart failure. JACC Heart Fail. 2013;1:1–20. doi: 10.1016/j.jchf.2012.10.002 [DOI] [PubMed] [Google Scholar]
- 14. Nogami K, Kusachi S, Nunoyama H, Kondo J, Endo C, Yamamoto K, Murakami T, Tsuji T. Extracellular matrix components in dilated cardiomyopathy. Immunohistochemical study of endomyocardial biopsy specimens. Jpn Heart J. 1996;37:483–494. doi: 10.1536/ihj.37.483 [DOI] [PubMed] [Google Scholar]
- 15. Watanabe T, Kusachi S, Yamanishi A, Kumashiro H, Nunoyama H, Sano I, Nakahama M, Murakami T, Naito I, Ninomiya Y, et al. Localization of type IV collagen alpha chain in the myocardium of dilated and hypertrophic cardiomyopathy. Jpn Heart J. 1998;39:753–762. doi: 10.1536/ihj.39.753 [DOI] [PubMed] [Google Scholar]
- 16. Chen CL, Huang SK, Lin JL, Lai LP, Lai SC, Liu CW, Chen WC, Wen CH, Lin CS. Upregulation of matrix metalloproteinase‐9 and tissue inhibitors of metalloproteinases in rapid atrial pacing‐induced atrial fibrillation. J Mol Cell Cardiol. 2008;45:742–753. doi: 10.1016/j.yjmcc.2008.07.007 [DOI] [PubMed] [Google Scholar]
- 17. Wang GY, Bergman MR, Nguyen AP, Turcato S, Swigart PM, Rodrigo MC, Simpson PC, Karliner JS, Lovett DH, Baker AJ. Cardiac transgenic matrix metalloproteinase‐2 expression directly induces impaired contractility. Cardiovasc Res. 2006;69:688–696. doi: 10.1016/j.cardiores.2005.08.023 [DOI] [PubMed] [Google Scholar]
- 18. Hogan JJ, Mocanu M, Berns JS. The native kidney biopsy: update and evidence for best practice. Clin J Am Soc Nephrol. 2016;11:354–362. doi: 10.2215/CJN.05750515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ezeani M, Hagemeyer CE, Lal S, Niego B. Molecular imaging of atrial myopathy: towards early af detection and non‐invasive disease management. Trends Cardiovasc Med. 2020;S1050‐1738(20)30155‐9. doi: 10.1016/j.tcm.2020.12.002 [DOI] [PubMed] [Google Scholar]
- 20. Mewton N, Liu CY, Croisille P, Bluemke D, Lima JA. Assessment of myocardial fibrosis with cardiovascular magnetic resonance. J Am Coll Cardiol. 2011;57:891–903. doi: 10.1016/j.jacc.2010.11.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Stirrat J, White JA. The prognostic role of late gadolinium enhancement magnetic resonance imaging in patients with cardiomyopathy. Can J Cardiol. 2013;29:329–336. doi: 10.1016/j.cjca.2012.11.033 [DOI] [PubMed] [Google Scholar]
- 22. Bois JP, Glockner J, Sheldon S, Newman D, Lin G, Packer D, Brady P. Left atrial delayed enhancement by MRI is not associated with occurrence or type of atrial fibrillation: a single‐center experience. J Am Coll Cardiol. 2013;61:E403–E403. doi: 10.1016/S0735-1097(13)60403-9 [DOI] [Google Scholar]
- 23. Marrouche NF, Wilber D, Hindricks G, Jais P, Akoum N, Marchlinski F, Kholmovski E, Burgon N, Hu N, Mont L, et al. Association of atrial tissue fibrosis identified by delayed enhancement MRI and atrial fibrillation catheter ablation: the DECAAF study. JAMA. 2014;311:498–506. doi: 10.1001/jama.2014.3 [DOI] [PubMed] [Google Scholar]
- 24. de Haas HJ, Arbustini E, Fuster V, Kramer CM, Narula J. Molecular imaging of the cardiac extracellular matrix. Circ Res. 2014;114:903–915. doi: 10.1161/CIRCRESAHA.113.302680 [DOI] [PubMed] [Google Scholar]
- 25. Mueller J, Gaertner FC, Blechert B, Janssen KP, Essler M. Targeting of tumor blood vessels: a phage‐displayed tumor‐homing peptide specifically binds to matrix metalloproteinase‐2‐processed collagen iv and blocks angiogenesis in vivo. Mol Cancer Res. 2009;7:1078–1085. doi: 10.1158/1541-7786.MCR-08-0538 [DOI] [PubMed] [Google Scholar]
- 26. Caravan P, Das B, Dumas S, Epstein F, Helm P, Jacques V, Koerner S, Kolodziej A, Shen L, Sun W‐C, et al. Collagen‐targeted MRI contrast agent for molecular imaging of fibrosis. Angew Chem Int Ed Engl. 2007;46:8171–8173. doi: 10.1002/anie.200700700 [DOI] [PubMed] [Google Scholar]
- 27. Helm PA, Caravan P, French BA, Jacques V, Shen L, Xu Y, Beyers RJ, Roy RJ, Kramer CM, Epstein FH. Postinfarction myocardial scarring in mice: molecular MR imaging with use of a collagen‐targeting contrast agent. Radiology. 2008;247:788–796. doi: 10.1148/radiol.2473070975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wang T‐Y, Kendrick‐Williams LL, Choy MY, Gilmore KA, Bonnard T, Pearce HA, Law LS, Carmichael I, Cody SH, Alt K, et al. Collagen‐targeted theranostic nanosponges for delivery of the matrix metalloproteinase 14 inhibitor naphthofluorescein. Chem Mater. 2020;32:3707–3714. doi: 10.1021/acs.chemmater.9b02840 [DOI] [Google Scholar]
- 29. Fuchs BC, Wang H, Yang Y, Wei L, Polasek M, Schühle DT, Lauwers GY, Parkar A, Sinskey AJ, Tanabe KK, et al. Molecular MRI of collagen to diagnose and stage liver fibrosis. J Hepatol. 2013;59:992–998. doi: 10.1016/j.jhep.2013.06.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Caravan P, Yang Y, Zachariah R, Schmitt A, Mino‐Kenudson M, Chen HH, Sosnovik DE, Dai G, Fuchs BC, Lanuti M. Molecular magnetic resonance imaging of pulmonary fibrosis in mice. Am J Respir Cell Mol Biol. 2013;49:1120–1126. doi: 10.1165/rcmb.2013-0039OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Du XJ, Autelitano DJ, Dilley RJ, Wang B, Dart AM, Woodcock EA. Beta(2)‐adrenergic receptor overexpression exacerbates development of heart failure after aortic stenosis. Circulation. 2000;101:71–77. [DOI] [PubMed] [Google Scholar]
- 32. Milano CA, Allen LF, Rockman HA, Dolber PC, McMinn TR, Chien KR, Johnson TD, Bond RA, Lefkowitz RJ. Enhanced myocardial function in transgenic mice overexpressing the beta 2‐adrenergic receptor. Science. 1994;264:582–586. doi: 10.1126/science.8160017 [DOI] [PubMed] [Google Scholar]
- 33. Xu Q, Dalic A, Fang L, Kiriazis H, Ritchie RH, Sim K, Gao XM, Drummond G, Sarwar M, Zhang YY, et al. Myocardial oxidative stress contributes to transgenic beta(2)‐adrenoceptor activation‐induced cardiomyopathy and heart failure. Br J Pharmacol. 2011;162:1012–1028. doi: 10.1111/j.1476-5381.2010.01043.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Liggett SB, Tepe NM, Lorenz JN, Canning AM, Jantz TD, Mitarai S, Yatani A, Dorn GW II. Early and delayed consequences of beta(2)‐adrenergic receptor overexpression in mouse hearts: critical role for expression level. Circulation. 2000;101:1707–1714. [DOI] [PubMed] [Google Scholar]
- 35. Lal S, Li A, Allen D, Allen PD, Bannon P, Cartmill T, Cooke R, Farnsworth A, Keogh A, Dos Remedios C. Best practice biobanking of human heart tissue. Biophys Rev. 2015;7:399–406. doi: 10.1007/s12551-015-0182-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Mamidi R, Li J, Gresham KS, Verma S, Doh CY, Li A, Lal S, Dos Remedios CG, Stelzer JE. Dose‐dependent effects of the myosin activator omecamtiv mecarbil on cross‐bridge behavior and force generation in failing human myocardium. Circ Heart Fail. 2017;10:e004257. doi: 10.1161/CIRCHEARTFAILURE.117.004257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Polizzotti BD, Ganapathy B, Walsh S, Choudhury S, Ammanamanchi N, Bennett DG, dos Remedios CG, Haubner BJ, Penninger JM, Kuhn B. Neuregulin stimulation of cardiomyocyte regeneration in mice and human myocardium reveals a therapeutic window. Sci Transl Med. 2015;7:281ra245. doi: 10.1126/scitranslmed.aaa5171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Li M, Parker BL, Pearson E, Hunter B, Cao J, Koay YC, Guneratne O, James DE, Yang J, Lal S, et al. Core functional nodes and sex‐specific pathways in human ischaemic and dilated cardiomyopathy. Nat Commun. 2020;11:2843. doi: 10.1038/s41467-020-16584-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Aghajanian H, Kimura T, Rurik JG, Hancock AS, Leibowitz MS, Li LI, Scholler J, Monslow J, Lo A, Han W, et al. Targeting cardiac fibrosis with engineered T cells. Nature. 2019;573:430–433. doi: 10.1038/s41586-019-1546-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Khalil H, Kanisicak O, Vagnozzi RJ, Johansen AK, Maliken BD, Prasad V, Boyer JG, Brody MJ, Schips T, Kilian KK, et al. Cell‐specific ablation of Hsp47 defines the collagen‐producing cells in the injured heart. JCI Insight. 2019;4:e128722. doi: 10.1172/jci.insight.128722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Niego B, Lee N, Larsson P, De Silva TM, Au AE, McCutcheon F, Medcalf RL. Selective inhibition of brain endothelial Rho‐kinase‐2 provides optimal protection of an in vitro blood‐brain barrier from tissue‐type plasminogen activator and plasmin. PLoS One. 2017;12:e0177332. doi: 10.1371/journal.pone.0177332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Nguyen MN, Kiriazis H, Ruggiero D, Gao XM, Su Y, Jian A, Han LP, McMullen JR, Du XJ. Spontaneous ventricular tachyarrhythmias in beta2‐adrenoceptor transgenic mice in relation to cardiac interstitial fibrosis. Am J Physiol Heart Circ Physiol. 2015;309:H946–H957. doi: 10.1152/ajpheart.00405.2015 [DOI] [PubMed] [Google Scholar]
- 43. Nguyen MN, Su Y, Kiriazis H, Yang Y, Gao XM, McMullen JR, Dart AM, Du XJ. Upregulated galectin‐3 is not a critical disease mediator of cardiomyopathy induced by beta2‐adrenoceptor overexpression. Am J Physiol Heart Circ Physiol. 2018;314:H1169–H1178. doi: 10.1152/ajpheart.00405.2015 [DOI] [PubMed] [Google Scholar]
- 44. Du XJ, Gao XM, Wang B, Jennings GL, Woodcock EA, Dart AM. Age‐dependent cardiomyopathy and heart failure phenotype in mice overexpressing beta(2)‐adrenergic receptors in the heart. Cardiovasc Res. 2000;48:448–454. [DOI] [PubMed] [Google Scholar]
- 45. Masci PG, Schuurman R, Andrea B, Ripoli A, Coceani M, Chiappino S, Todiere G, Srebot V, Passino C, Aquaro GD, et al. Myocardial fibrosis as a key determinant of left ventricular remodeling in idiopathic dilated cardiomyopathy: a contrast‐enhanced cardiovascular magnetic study. Circ Cardiovasc Imaging. 2013;6:790–799. doi: 10.1161/CIRCIMAGING.113.000438 [DOI] [PubMed] [Google Scholar]
- 46. Piek A, de Boer RA, Sillje HH. The fibrosis‐cell death axis in heart failure. Heart Fail Rev. 2016;21:199–211. doi: 10.1007/s10741-016-9536-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Lohse MJ, Engelhardt S, Eschenhagen T. What is the role of beta‐adrenergic signaling in heart failure? Circ Res. 2003;93:896–906. doi: 10.1161/01.RES.0000102042.83024.CA [DOI] [PubMed] [Google Scholar]
- 48. Salerno M, Janardhanan R, Jiji RS, Brooks J, Adenaw N, Mehta B, Yang Y, Antkowiak P, Kramer CM, Epstein FH. Comparison of methods for determining the partition coefficient of gadolinium in the myocardium using T1 mapping. J Magn Reson Imaging. 2013;38:217–224. doi: 10.1002/jmri.23875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Lopez B, Ravassa S, Gonzalez A, Zubillaga E, Bonavila C, Berges M, Echegaray K, Beaumont J, Moreno MU, San Jose G, et al. Myocardial collagen cross‐linking is associated with heart failure hospitalization in patients with hypertensive heart failure. J Am Coll Cardiol. 2016;67:251–260. doi: 10.1016/j.jacc.2015.10.063 [DOI] [PubMed] [Google Scholar]
- 50. Chen HH, Waghorn PA, Wei L, Tapias LF, Schühle DT, Rotile NJ, Jones CM, Looby RJ, Zhao G, Elliott JM, et al. Molecular imaging of oxidized collagen quantifies pulmonary and hepatic fibrogenesis. JCI Insight. 2017;2:e91506. doi: 10.1172/jci.insight.91506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Hanna N, Cardin S, Leung TK, Nattel S. Differences in atrial versus ventricular remodeling in dogs with ventricular tachypacing‐induced congestive heart failure. Cardiovasc Res. 2004;63:236–244. doi: 10.1016/j.cardiores.2004.03.026 [DOI] [PubMed] [Google Scholar]
- 52. Nguyen M‐N, Ziemann M, Kiriazis H, Su Y, Thomas Z, Lu Q, Donner DG, Zhao W‐B, Rafehi H, Sadoshima J, et al. Galectin‐3 deficiency ameliorates fibrosis and remodeling in dilated cardiomyopathy mice with enhanced Mst1 signaling. Am J Physiol Heart Circ Physiol. 2019;316:H45–H60. doi: 10.1152/ajpheart.00609.2018 [DOI] [PubMed] [Google Scholar]
- 53. Rahmutula D, Marcus GM, Wilson EE, Ding CH, Xiao Y, Paquet AC, Barbeau R, Barczak AJ, Erle DJ, Olgin JE. Molecular basis of selective atrial fibrosis due to overexpression of transforming growth factor‐beta1. Cardiovasc Res. 2013;99:769–779. doi: 10.1093/cvr/cvt074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Wang S, Fan Y, Li D, Sun C, Lei Z, Lu L, Wang T, Zhang F. Anti‐quenching NIR‐II molecular fluorophores for in vivo high‐contrast imaging and pH sensing. Nat Commun. 2019;10:1058. doi: 10.1038/s41467-019-09043-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Choy G, O'Connor S, Diehn FE, Costouros N, Alexander HR, Choyke P, Libutti SK. Comparison of noninvasive fluorescent and bioluminescent small animal optical imaging. Biotechniques. 2003;35:1022–1026, 1028–1030. doi: 10.2144/03355rr02 [DOI] [PubMed] [Google Scholar]
- 56. Low AF, Tearney GJ, Bouma BE, Jang IK. Technology insight: optical coherence tomography—current status and future development. Nat Clin Pract Cardiovasc Med. 2006;3:154–162; quiz 172. doi: 10.1038/ncpcardio0482 [DOI] [PubMed] [Google Scholar]
- 57. Schols RM, Connell NJ, Stassen LP. Near‐infrared fluorescence imaging for real‐time intraoperative anatomical guidance in minimally invasive surgery: a systematic review of the literature. World J Surg. 2015;39:1069–1079. doi: 10.1007/s00268-014-2911-6 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1
Figures S1–S6
Video S1
Video S2