

Abstract
Background
Persistent activation of endoplasmic reticulum stress and the unfolded protein response (UPR) induces vascular cell apoptosis, contributing to atherogenesis. Aging and hypercholesterolemia are 2 independent proatherogenic factors. How they affect vascular UPR signaling remains unclear.
Methods and Results
Transcriptome analysis of aortic tissues from high fat diet–fed and aged ApoE −/− mice revealed 50 overlapping genes enriched for endoplasmic reticulum stress‐ and UPR‐related pathways. Aortae from control, Western diet (WD)–fed, and aged ApoE −/− mice were assayed for (1) 3 branches of UPR signaling (pancreatic ER eIF2‐alpha kinase /alpha subunit of the eukaryotic translation initiation factor 1/activating transcription factor 4, inositol‐requiring enzyme 1 alpha/XBP1s, activating transcription factor 6); (2) UPR‐mediated protective adaptation (upregulation of immunoglobulin heavy chain‐binding protein and protein disulfide isomerase); and (3) UPR‐mediated apoptosis (induction of C/EBP homologous transcription factor, p‐JNK, and cleaved caspase‐3). Aortic UPR signaling was differentially regulated in the aged and WD‐fed groups. Consumption of WD activated all 3 UPR branches; in the aged aorta, only the ATF6α arm was activated, but it was 10 times higher than that in the WD group. BiP and protein disulfide isomerase protein levels were significantly decreased only in the aged aorta despite a 5‐fold increase in their mRNA levels. Importantly, the aortae of aged mice exhibited a substantially enhanced proapoptotic UPR compared with that of WD‐fed mice. In lung tissues, UPR activation and the resultant adaptive/apoptotic responses were not significantly different between the 2 groups.
Conclusions
Using a mouse model of atherosclerosis, this study provides the first in vivo evidence that aging and an atherogenic diet activate differential aortic UPR pathways, leading to distinct vascular responses. Compared with dietary intervention, aging is associated with impaired endoplasmic reticulum protein folding and increased aortic apoptosis.
Keywords: aging, apoE deficient mice, hypercholesterolemia, unfolded protein response, vascular cells
Subject Categories: Basic Science Research, Vascular Biology, Animal Models of Human Disease
Nonstandard Abbreviations and Acronyms
- ATF6α
activating transcription factor 6 alpha
- BiP
immunoglobulin heavy chain‐binding protein
- CHOP
C/EBP homologous transcription factor
- eIF2α
alpha subunit of the eukaryotic translation initiation factor 2
- ER
endoplasmic reticulum
- IRE1α
inositol‐requiring enzyme 1 alpha
- ND
normal chow diet
- PDI
protein disulfide isomerase
- PERK
pancreatic ER eIF2‐alpha kinase
- UPR
unfolded protein response
- WD
Western diet
Clinical Perspective
What Is New?
Aging and hypercholesterolemia can both promote endoplasmic reticulum stress, but they activate differential unfolded protein response pathways in aorta, leading to distinct vascular responses.
Distinct from diet‐induced hypercholesterolemia, aging is associated with impaired endoplasmic reticulum protein folding machinery and exacerbated apoptosis in aortic vascular cells.
What Are the Clinical Implications?
Our study suggests that the unfolded protein response in arterial vessels can vary greatly according to different atherogenic risk factors, which provides new insights into the development of unfolded protein response–based treatment strategies.
Atherosclerosis is a progressive disease characterized by the accumulation of lipids and fibrous elements in large arteries. Despite considerable progress in the prevention and treatment of this condition, 1 , 2 atherosclerosis remains the leading cause of death and disability globally. 3 , 4 Hypercholesterolemia and vascular aging are the 2 strongest risk factors for atherosclerosis. However, whether differential molecular mechanisms induced by these 2 risk factors independently contribute to atherosclerosis remains unclear.
Numerous studies have demonstrated that excess lipid accumulation, a prerequisite for atherogenesis, can lead to endoplasmic reticulum (ER) stress, which is characterized by compromised protein folding and processing capabilities. 5 , 6 To cope with ER stress, cells mount an adaptive response—known as the unfolded protein response (UPR)—by activating 3 signal transduction pathways that are controlled by their respective ER‐resident transmembrane proteins: pancreatic ER kinase (PERK), inositol‐requiring enzyme 1 alpha (IRE1α), and activating transcription factor 6 (ATF6). 7 These pathways work cooperatively to rapidly lower the protein load in the ER through the phosphorylation of alpha subunit of the eukaryotic translation initiation factor 2 (eIF2α), as well as the upregulation of genes that enhance the ER protein folding machinery (such as immunoglobulin heavy chain‐binding protein [BiP] and protein disulfide isomerase [PDI]) and the ER‐associated degradation pathway via the induction of the transcriptional activators ATF6, ATF4, and XBP1. However, if these attempts fail to restore ER homeostasis, apoptosis will be executed through the transcriptional induction of CCAAT/enhancer‐binding protein homologous protein (CHOP), a transcription factor that mediates apoptosis, 8 as well as the activation of the c‐Jun NH2 terminal kinases (JNK), a downstream target of IRE1α. 9 UPR components, such as CHOP and phosphorylated PERK, are reported to be activated in human and murine atherosclerotic lesions. 10 , 11 , 12
Different from hypercholesterolemia, aging is thought to be related to dysregulation of the UPR, as evidenced by the reduced expression and activity of key ER chaperones and folding enzymes (eg, BiP and PDI), and the preferential activation of UPR‐mediated proapoptotic signaling, including increased CHOP expression and JNK phosphorylation. 13 , 14 Nevertheless, these findings have been largely obtained from research on age‐related neurodegenerative diseases, and it remains unclear how components of the UPR change in the vascular system of the elderly. In addition, compared with the ER stress resulting from high fat diet–induced hypercholesterolemia, the stress associated with vascular cell aging is more chronic, but milder. It remains to be determined whether different UPR signal transduction pathways are stimulated to adapt to aging‐associated chronic stress.
Given the causal relationship that exists between the UPR/ER stress and atherogenesis, targeting UPR components represents a promising therapeutic strategy for the treatment of cardiovascular diseases. However, the major challenge lies in how to selectively inhibit atherogenic UPR pathways without compromising the protective role of the UPR. 15 A better understanding of how different atherogenic factors (such as hypercholesterolemia and aging) trigger the UPR‐associated signaling networks may help identify the critical UPR components that contribute to atherogenesis, thus providing new insights into the development of effective treatments for atherosclerotic cardiovascular diseases.
In the present study, we compared the 3 arms of the UPR and their adaptive/apoptotic responses in aortic and lung tissues of ApoE −/− mice, the most frequently used animal model of atherosclerosis. With age, these mice can spontaneously develop aortic lesions, a process that can be greatly accelerated by Western diet (WD) feeding. 16 Herein, we showed that, different from WD‐induced hypercholesterolemia that triggers all 3 branches of the UPR‐related signaling network, aging selectively stimulated ATF6 activation and JNK phosphorylation, and greatly upregulated the transcription of CHOP and the ER chaperones BiP and PDI. Interestingly, the protein levels of BiP and PDI were reduced in the aorta, implying that these proteins are posttranslationally degraded. Our study provides the first in vivo evidence for the distinct impacts of hypercholesterolemia and aging on the vascular UPR in a murine model of atherosclerosis.
Methods
The authors declare that all supporting data are available within the article and its online supplementary files. Additional methods can be found in Data S1.
Animals
All experimental animal procedures were approved by the Institutional Animal Care and Use Committee of the Boston University Medical Campus. ApoE‐deficient mice in a C57BL/6J background were acquired from the Jackson Laboratory (#002052, Bar Harbor, ME). Five‐week‐old ApoE −/− mice were divided into 3 groups and fed either a normal chow diet (ND; 4.5% fat, 0.02% cholesterol by weight) for 8 weeks (Young/ND group) or 40–52 weeks (Old/ND group); or a WD (purchased from Research Diets Inc., D12079B: 21% fat, 0.21% cholesterol by weight) for 8 weeks (Young/WD). Only male mice were used for these experiments.
Tissue Harvesting and Preparation
At the end of the feeding periods, blood was collected via the retro‐orbital sinus with the animals under anesthesia (intraperitoneal injection of ketamine [150 mg/kg] and xylazine [20 mg/kg]). Plasma was prepared by centrifugation of the collected blood samples at 2000g for 15 minutes and assayed for cholesterol and triglycerides using Infinity Cholesterol and Triglyceride Stable Reagents (Thermo Fisher Scientific, Middletown, VA). The mice were euthanized by cervical dislocation and immediately perfused with 30 mL of cold phosphate‐buffered saline via the right ventricle to remove blood components, following which the aortae were carefully dissected. The cleared aortae and lungs were flash‐frozen in liquid nitrogen for further biochemical analysis. Hearts (including aortic roots and ascending aorta) were removed, cut transversely, and embedded in optimum cutting temperature compound (Thermo Fisher Scientific). Serial sections (10‐µm thick) of ascending aortae were obtained for immunohistochemical analysis.
Immunofluorescence Analysis of Cross‐Sections of Ascending Aortae of ApoE −/− Mice
For immunofluorescence studies, fresh‐frozen aortic sections were fixed in ice‐cold acetone at −20°C for 10 minutes, followed by blocking with 2.5% horse serum for 1 hour. The sections were then incubated overnight at 4°C with primary antibodies targeting BiP (Cell Signaling Technology, #3183S, 1:500 dilution), CHOP (Thermo Fisher Scientific, #MA1‐250, 1:500 dilution), and activated caspase‐3 (Promega, #G748A, 1:250 dilution). Primary antibodies were detected using the VectaFluor Excel Amplified Anti‐rabbit IgG Dylight 594 Antibody Kit (Vector Laboratories, DK‐1594). Images were captured with a Nikon widefield deconvolution epifluorescence microscope equipped with a ×20 objective using filter sets for 4',6‐diamidino‐2‐phenylindole, fluorescein isothiocyanate conjugated, and Texas red.
Analysis of Proteins Associated With the UPR
To analyze the levels of UPR‐associated proteins, Western blotting was conducted, as previously described. 17 Briefly, frozen aortae and lungs were crushed to a fine powder in liquid nitrogen using a mortar and pestle, and then dissolved in radioimmunoprecipitation assay buffer containing protease and phosphatase inhibitors (Millipore Sigma) at 4°C. The cleared lysates were then suspended in SDS–PAGE sample buffer, separated by 4% to 12% NuPAGE, and blotted onto PVDF membranes. The membranes were then probed with primary antibodies against p‐PERK (Abclonal, #AP0886), p‐IRE1α (Thermo Fisher Scientific, #PA1‐16927), IRE1α (Cell Signaling Technology, #3294), ATF6α (Abcam, ab122897), p‐eIF2α (Cell Signaling Technology, #9721), p‐JNK (Cell Signaling Technology, #9255), CHOP (Cell Signaling Technology, #2895S), BiP (Cell Signaling Technology, #3183S), and cleaved caspase‐3 (Cell Signaling Technology, 9664), followed by incubation with the appropriate horseradish peroxidase–conjugated secondary antibodies (1: 5000 dilution). Membranes were developed with a chemiluminescent substrate and imaged using the KwikQuant Imager system (Kindle Biosciences, LLC). Target protein band densities were quantified and normalized to those of GAPDH, beta‐actin, or beta‐tubulin, as indicated. Band intensity data are expressed relative to the control group (Young/ND).
Quantitative Real‐Time Polymerase Chain Reaction
Total RNA was isolated from the aortae or lungs using TRIzol reagent (Invitrogen) following the manufacturer's instructions. One microgram of total RNA was reverse‐transcribed into cDNA using the HiFiScript gDNA Removal cDNA Synthesis Kit (CW Biosciences). ATF4, CHOP, total XBP1, spliced XBP1, BiP, EDEM, Ero1, and PDI transcripts were quantified in a CFX96 Real‐Time Polymerase Chain Reaction System (Bio‐Rad, USA) using a SYBR Green Polymerase Chain Reaction Master Mix (CW Biosciences) and the primers listed in Table. Gene expression levels were quantified using the ΔΔCt threshold cycle method with GAPDH as loading control, and are presented relative to the control group (Young/ND). The specificity of the amplicons was verified by melting curve analysis of the final products and a primer efficiency test.
Table 1.
Primers Used for qPCR
| Gene | Forward | Reverse |
|---|---|---|
| GAPDH | 5′‐ CATCACTGCCACCCAGAAGACTG‐3′ | 5′‐ ATGCCAGTGAGCTTCCCGTTCAG‐3′ |
| Atf4 | 5′‐ GGGTTCTGTCTTCCACTCCA‐3′ | 5′‐ AAGCAGCAGAGTCAGGCTTTC‐3′ |
| CHOP | 5′‐ CCACCACACCTGAAAGCAGAA‐3′ | 5′‐ AGGTGAAAGGCAGGGACTCA‐3′ |
| Xbp1 | 5′‐ TGGCCGGGTCTGCTGAGTCCG‐3′ | 5′‐ GTCCATGGGAAGATGTTCTGG‐3′ |
| X`bp1s | 5′‐ CTGAGTCCGCAGCAGGTG‐3′ | 5′‐ TAGCAGACTCTGGGGAAGGA‐3′ |
| Bip | 5′‐ TTCAGCCAATTATCAGCAAACTCT‐3′ | 5′‐ TTTTCTGATGTATCCTCTTCACCAGT‐3′ |
| Edem | 5′‐ CTACCTGCGAAGAGGCCG‐3′ | 5′‐ GTTCATGAGCTGCCCACTGA‐3′ |
| Ero1 | 5′‐ TTCTGCCAGGTTAGTGGTTACC‐3′ | 5′‐ GTTTGACGGCACAGTCTCTTC‐3′ |
| Pdi | 5′‐ TCTGAGATTCGACTAGCAAAGGT‐3′ | 5′‐ GCCACGGACACCATACTGC‐3′ |
qPCR indicates quantitative polymerase chain reaction.
Statistical Analysis
Data are presented as means±SEM. Statistical analysis was performed using GraphPad Prism 8.0 (GraphPad Software, La Jolla, CA). Differences between 2 groups were compared by the Mann–Whitney U test. Comparisons among multiple groups were conducted by 1‐way ANOVA followed by Dunnett's test. A P<0.05 was considered statistically significant. Combined with its statistical significance, the effect size that reflects the magnitude of effects was assessed by using . d=0.2, 0.5, and 0.8 are regarded as small, medium, and large effect sizes, respectively. The effect size of the difference between 2 groups is summarized in Table S1.
Results
ER Stress‐ and UPR‐Related Genes Were Enriched in the Aortae of Aged or High Fat Diet–Fed ApoE−/− Mice
To test whether ER stress and UPR‐related signaling in the aortae of ApoE−/− mice is modulated by both high fat diet–induced hypercholesterolemia and aging, we analyzed the following 2 publicly available transcriptome data sets obtained from the Gene Expression Omnibus database: GSE10000, which compares young and aged ApoE −/− mice, and GSE18443, which compares normal diet– and high fat diet–fed ApoE −/− mice. We identified 3115 genes that were differentially expressed (with an adjusted P value of <0.05) in both high fat diet–fed and aged ApoE −/−mice. Of these overlapping genes, 50 were enriched for those related to ER stress and the UPR (Figure 1A). Specifically, the expression of 9 UPR‐related genes, including Atf6α and Xbp1—key UPR signaling components—was significantly changed in both groups, while 2 mitochondrial pro‐apoptotic genes (Bax, Bok) that are known to promote apoptosis in response to ER stress were significantly upregulated (Figure 1B). We further analyzed these 2 data sets to identify the overlapping upregulated and downregulated genes, respectively (Figure 1A). The gene set enrichment and pathway analyses strongly supported that ER stress and the UPR in the aorta are likely affected by both hypercholesterolemia and aging, thus providing a rationale for further investigation.
Figure 1. Bioinformatic analysis of transcriptome data sets for aortae obtained from aged ApoE −/− mice fed a high‐fat diet revealed 50 overlapping genes that are enriched for ER stress‐ and UPR‐related pathways.
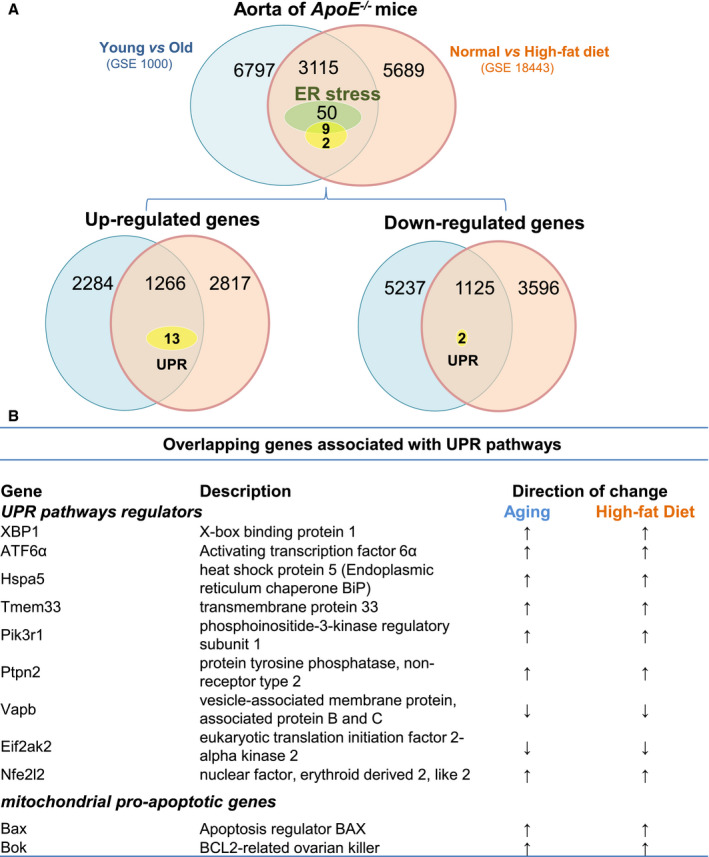
A, Two transcriptome data sets derived from studies on atherosclerosis were retrieved from the NCBI Gene Expression Omnibus database. One compares the gene expression profile of aortae from normal diet‐ and high fat diet–fed ApoE −/− mice (GSE18443), and the other (GSE10000) compares the gene expression profile of aortae between young and aged ApoE −/− mice. DEGs (adjusted P<0.05) in these 2 data sets were screened and compared with the overlapping DEGs, which were then subjected to Gene Ontology pathway analysis. The Venn diagram illustrates the overlay of DEGs (3115) between hypercholesterolemia (high‐fat diet vs normal diet) and aging (young vs old); 50 DEGs were enriched for ER stress, while 9 were associated with UPR‐related pathways. The overlapping upregulated or downregulated DEGs in these 2 groups are illustrated in the Venn diagram. B, List of overlapping UPR‐related genes in both aging and high fat diet–fed mice. DEGs indicates differentially expressed genes; ER, endoplasmic reticulum; and UPR, unfolded protein response.
Aging and Hypercholesterolemia Trigger Distinct UPR Signaling Events in the Aorta
As depicted in Figure 2A, 3 groups of ApoE −/− mice were used in this study: a control (ND) group (13 weeks old and fed a ND), a WD group (13 weeks old and fed a WD for 8 weeks), and an aged group (40–52 weeks old and fed a ND). We first assessed the effects of a WD and aging on plasma lipid levels in ApoE −/− mice. As shown in Figure 2B, compared with the control group, the plasma levels of total cholesterol were significantly increased (2.9‐fold) in the WD group, while the aged group showed slight, but not significant, increases in plasma triglyceride levels. No significant differences were observed in cholesterol levels. These results were consistent with those previously reported. 16 , 18 Of note, the basal blood cholesterol levels in young (control) ApoE −/− mice were 451.6±34.5 mg/dL, which was ≈4.5‐fold higher than those for C57BL/6 mice (<100 mg/dL).
Figure 2. Assessment of UPR‐related signaling in descending aortae of ApoE −/− mice from the control, WD‐fed, and aged groups.
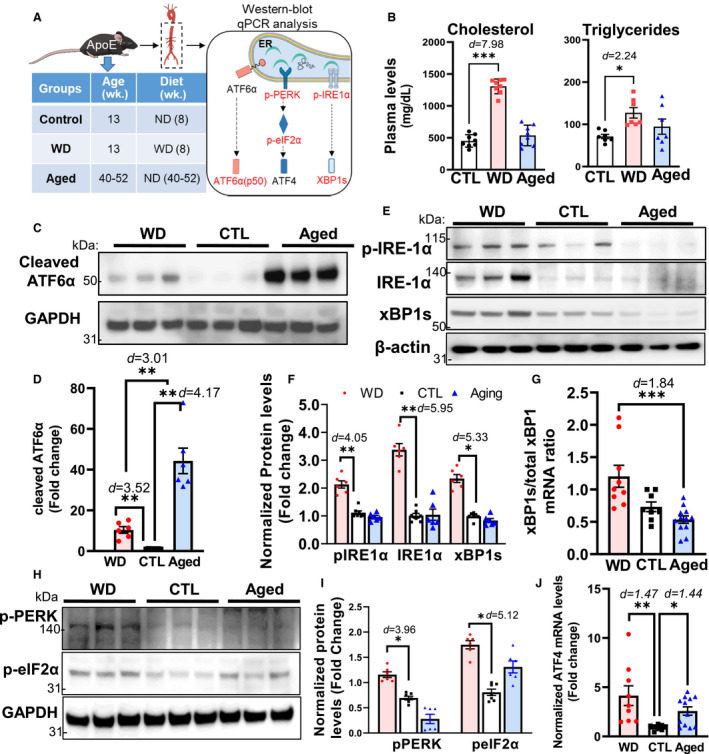
A, Schematic detailing the experimental groups and analysis. Descending aortae from ApoE −/− mice from the control (CTL, 13 weeks old and fed a ND, n=8), WD‐fed (13 weeks old and fed a WD for 8 weeks, n=9), and aged (40–52 weeks old and fed a ND, n=12) groups were assayed for the 3 arms of the UPR (initiated by the ER stress sensors ATF6α [C and D], IRE1α [E through G], and PERK [H through J]) using Western blotting and qPCR. B, The plasma levels of total cholesterol and triglycerides did not differ significantly between the aging and WD groups. C and D, In aged mice, ATF6α was activated to a greater extent than that in mice of the WD group as evidenced by the increased levels of cleaved ATF6α. E through G, In the aged group, the levels of phosphorylated IRE1α and spliced XBP1, as well as the ratio of spliced to total Xbp1 mRNA, were lower than those in the WD group. H through J, The levels of PERK/eIF2α signaling activation and Atf4 transcription were lower in the aged group than in the WD group. Data are presented as means±SE. *P<0.05, **P<0.01, ***P<0.001 compared with the control group, unless otherwise indicated. The effect size is assessed by cohen's d value. ATF6α indicates activating transcription factor 6 alpha; CTL, control; ER, endoplasmic reticulum; IRE1α, inositol‐requiring enzyme 1 alpha; ND, normal chow diet; PERK, pancreatic ER eIF2‐alpha kinase; qPCR, quantitative polymerase chain reaction; UPR, unfolded protein response; and WD, Western diet.
Next, we further defined the impact of hypercholesterolemia and aging on UPR‐related signaling pathways in the aorta. As shown in Figure 2A, descending aortae collected from control, WD, and aged ApoE −/− mice were subjected to Western blot and quantitative polymerase chain reaction analyses of the 3 branches (ATF6α, PERK, and IRE1α) of the UPR. As measured by the protein levels of its cleaved form (p50), ATF6 activation was significantly stimulated in both the aged and WD groups, but considerably more so in aged mice (44.3±6.29‐ and 10.4±1.61‐fold increases in the aged and WD groups, respectively; Figure 2C and 2D). In contrast, the other 2 arms of the UPR (activated by IRE1α and PERK) were only significantly activated in the WD group, but not in the aged group (Figure 2E through 2I). Regarding the IRE1α‐activated pathway, compared with the control group, p‐IRE1α and total IRE1α levels were increased by 2.13±0.13‐ and 3.38±0.22‐fold, respectively, in WD‐fed mice (Figure 2E and 2F). The mRNA encoding the transcription factor XBP1 is spliced by IRE1α, and is its only known substrate. This splicing event alters the XBP1 reading frame, resulting in an activated XBP1 protein capable of inducing the expression of UPR‐related genes. 19 Coincident with the IRE1α results, consuming a WD, but not aging, stimulated XBP1 activation, as evidenced by the increase in the levels of truncated XBP1 protein, as well as the ratio of spliced to total Xbp1 mRNA (Figure 2E through 2G). For the PERK/eIF2α/ATF4 signal transduction pathway, compared with the control group, PERK and its downstream target, eIF2α, were both activated in the WD group, which was supported by the significant increase observed in the levels of phosphorylated PERK and eIF2α (1.67±0.06‐ and 2.16±0.08‐fold, respectively; Figure 2H and 2I). Moreover, the mRNA levels of Atf4, a target gene of eIF2α, were also significantly increased in the WD group (4.14±1.0‐fold; Figure 2J). Collectively, these results indicated that UPR‐related signaling in arterial vessels is differentially regulated under aging and hypercholesterolemia. Specifically, the ATF6‐initiated UPR pathway, required for the transcriptional induction of ER chaperones and the ER‐associated degradation machinery, is selectively activated under aging, whereas hypercholesterolemia activates all 3 branches of the UPR.
Activity of the ER Protein Folding Machinery Is Reduced in Aged Aortae
UPR‐associated signaling counteracts ER stress primarily by improving the protein‐folding capacity of the ER, thereby maintaining its protein processing ability. 20 Herein, we evaluated the effects of aging and hypercholesterolemia on the key components of the ER protein‐folding machinery (BiP and PDI) in the aorta. As shown in Figure 3A and 3B, the protein levels of BiP and PDI in aortic lysates were reduced by ≈51%, and ≈47%, respectively, in aged ApoE −/− mice compared with those in their young counterparts. In contrast, Bip and Pdi mRNA levels were significantly higher in the aged group than in the control group (6.73±1.40‐ and 4.72±0.95‐fold, respectively), whereas only Bip was transcriptionally upregulated in mice of the WD group (Figure 3C and 3D). Similarly, immunostaining analysis of aortic cross‐sections supported that vascular BiP was differentially regulated following the consumption of a WD and aging (Figure 3E and 3F). The immunofluorescence signal intensity of BiP was significantly lower (≈34%) in aged aortae than in WD aortae. Taken together, our data demonstrated that hypercholesterolemia promoted the expression of ER chaperones, which is essential for the UPR‐mediated restoration of ER homeostasis. In contrast, in aged aortae, BiP and PDI were transcriptionally upregulated, whereas their protein levels were diminished, implying the involvement of posttranslational modulation. For instance, the oxidation of BiP and PDI has been reported to be increased in the liver of aging rats, and is related to a decrease in their expression and activity. 21
Figure 3. In aged ApoE −/− mice, the protein levels of the ER chaperones BiP and PDI are reduced, whereas their mRNA levels are significantly upregulated.
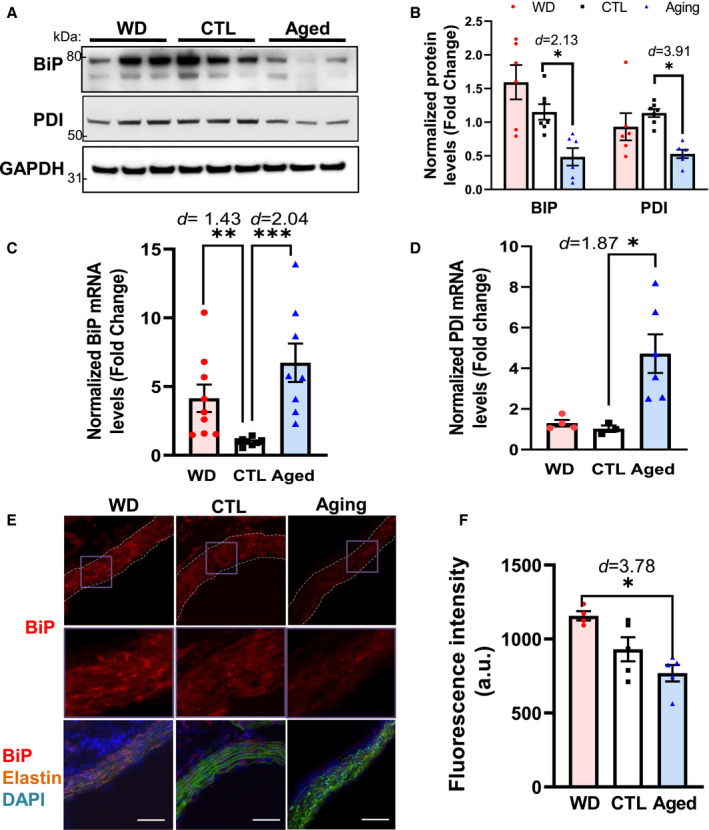
A and B, The protein levels of BiP and PDI were decreased in the aged group. C and D, The mRNA levels of Bip and Pdi were increased in the aortae of mice from the aged group. C and D, Immunofluorescence analysis of BiP expression in the aortae from each group of ApoE −/− mice. Fresh and frozen cross‐sections of aortae without evident lesions were immunostained for BiP (E) and fluorescence (red) was then quantified using ImageJ (F). Nuclei were counterstained with DAPI (blue), and elastin was visualized via its autofluorescence (green). The vessels are outlined in white dashed lines; purple boxes indicate the areas shown in higher magnification below the corresponding images. For each aortic cross‐section, the red fluorescence intensity was measured in 3 to 5 randomly selected image fields, averaged, and expressed as arbitrary units (a.u.; n=4–6). Data are presented as means±SE. *P<0.05, **P<0.01, ***P<0.001 vs the control group, unless otherwise indicated. The effect size is assessed by cohen's d value. BiP indicates immunoglobulin heavy chain‐binding protein; CTL, control; DAPI, 4',6‐diamidino‐2‐phenylindole; ER, endoplasmic reticulum; PDI, protein disulfide isomerase; and WD, Western diet. Magnification=200×, Scale bar=50 μm.
UPR‐Mediated Apoptosis Is Highly Activated in Aged Aortae
In response to ER stress, the UPR helps maintain the functionality of the ER protein‐folding machinery. Under persistent stress, however, the UPR can instead facilitate apoptosis, mainly through the activation of CHOP‐ and JNK‐mediated apoptotic pathways. Therefore, we compared the effects of aging and hypercholesterolemia on UPR‐mediated proapoptotic signaling in ApoE −/− mice (Figure 4). Compared with the control group, the protein levels of CHOP and p‐JNK were significantly upregulated in aortic tissue of both WD‐fed (7.43±0.72‐ and 7.22±0.88‐fold, respectively) and aged ApoE −/− mice (22.8±3.0‐ and 11.6±1.82‐fold, respectively) (Figure 4A and 4B). Consistent with the protein levels, CHOP transcript levels were highly upregulated in these 2 groups (2.68±0.57‐fold and 5.37±1.40‐fold, respectively; Figure 4C). Fluorescence immunostaining analysis of cross‐sections of ascending aortae confirmed that CHOP expression was upregulated in aged mice relative to that in the control group (Figure 4D and 4E).
Figure 4. Aging preferentially activates UPR‐mediated proapoptotic signaling in the aortae of ApoE −/− mice.
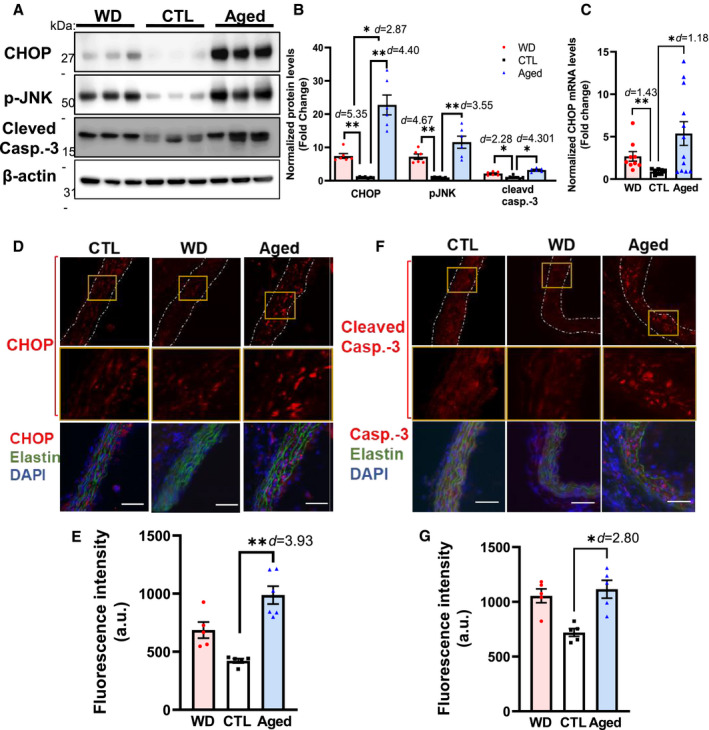
A through C, Aortae derived from aged ApoE −/− mice displayed a significant increase in the levels of the apoptosis executor CHOP (protein and mRNA), phosphorylated JNK, and cleaved Casp‐3 relative to WD‐fed mice. D through G, Immunofluorescence analysis of the aortic expression of CHOP (D and E), and cleaved Casp‐3 (F and G) in each group. Fresh and frozen cross‐sections of aortae without evident lesions were immunostained for CHOP (D) or cleaved Casp‐3 (F), and their fluorescence signals (red) were quantified using ImageJ (E and G, respectively). Nuclei were counterstained with DAPI (blue), and elastin was visualized via its autofluorescence (green). The yellow boxes indicate the areas shown in higher magnification below the corresponding images. From each aortic cross‐section, the red fluorescence intensity was quantified in 3 to 5 randomly selected image fields, averaged, and expressed as arbitrary units (a.u.; n=3–5). Data are presented as means±SE. *P<0.05, **P<0.01 vs the control group, unless otherwise indicated. The effect size is assessed by cohen's d value. Casp‐3 indicates caspase‐3; CHOP, C/EBP homologous transcription factor; CTL, control; DAPI, 4',6‐diamidino‐2‐phenylindole; UPR, unfolded protein response; and WD, Western diet. Scale bar=50 μm.
We further assessed the extent of cellular apoptosis in the aorta by measuring the levels of cleaved caspase‐3, a surrogate marker for apoptosis and a downstream target of the CHOP and JNK pathways. 22 Consistent with our CHOP and p‐JNK findings, the levels of cleaved caspase‐3 in the aortae of WD‐fed and aged ApoE−/− mice were significantly induced compared with those in the control group (2.14±0.22‐ and 3.20±0.23‐fold, respectively; Figure 4A and 4B); however, no significant difference was seen between the WD and aged groups (P>0.05). Similarly, immunofluorescence analysis indicated that cleaved caspase‐3 levels were significantly higher in the aged group (1.78±0.28‐fold; Figure 4F and 4G). Collectively, these data demonstrated that both aging and hypercholesterolemia can induce UPR‐mediated proapoptotic signaling pathways and cellular apoptosis in the aorta. Nevertheless, this effect was more pronounced with aging.
Difference Between Aging‐ and WD‐Triggered UPR Signaling Is Diminished in the Lungs of ApoE −/− Mice
We next examined whether aging and hypercholesterolemia have different effects on UPR‐related signaling in the lungs of ApoE −/− mice that are rich in microvascular vessels. As summarized in Figure 5, compared with the control group, all 3 UPR signaling pathways were activated in WD‐fed mice, and to a lesser extent also in aged mice. Specifically, ATF6α activation was increased in both the WD and aged groups. However, in contrast to the result observed in the aorta (Figure 2), aging induced less ATF6α activation than hypercholesterolemia (2.78±0.31‐ versus 12.7±1.06‐fold, respectively; Figure 5A and 5B). Although the IRE1α and PERK pathways were activated in the WD group, the mRNA levels of Atf4 and the ratio of spliced to total XBP1—downstream effectors of these 2 arms of the UPR—were not significantly upregulated. Combined, these data suggested that aging and hypercholesterolemia exert limited influence on UPR‐related signaling in the lungs of ApoE −/− mice, in contrast to that observed with the aorta.
Figure 5. Assessment of UPR‐related signaling in lung tissues obtained from control, WD‐fed, and aged ApoE −/− mice.
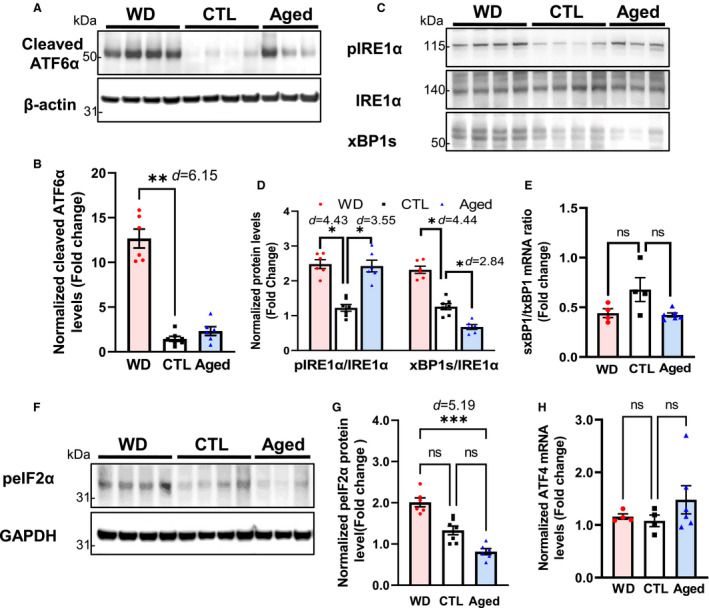
Lung tissues from ApoE −/− mice in the 3 groups were assessed for the activation of the 3 arms of the UPR (initiated by the endoplasmic reticulum stress sensors ATF6α (A and B), IRE1α (C through E), and PERK (F through H) using Western blotting and qPCR. In contrast to that observed in the aorta, the levels of ATF6α and p‐IRE1α/XBP1 signal activation were lower in the aged group than in the WD group; the PERK/peIF2α/ATF4 pathway was not significantly activated by either aging or a WD, but compared with the WD group, p‐IRE1α level was significantly lower, ***P<0.001. Data are presented as means±SE (n=6–8); *P<0.05, **P<0.01 vs the control group, unless otherwise indicated. The effect size is assessed by cohen's d value. ATF6α indicates activating transcription factor 6 alpha; CTL, control; IRE1α, inositol‐requiring enzyme 1 alpha; peIF2α, alpha subunit of the eukaryotic translation initiation factor 2; PERK, pancreatic ER eIF2‐alpha kinase; qPCR, quantitative polymerase chain reaction; UPR, unfolded protein response; and WD, Western diet.
Hypercholesterolemia and Aging Do Not Significantly Affect Adaptive and Apoptotic UPR Signaling in the Lungs of ApoE −/− Mice
In line with the findings that hypercholesterolemia and aging have a limited impact on the 3 branches of the UPR in the lungs, no significant changes were observed in ER chaperone levels or proapoptotic signaling in the lungs of mice from the 3 experimental groups (Figure 6). The protein and mRNA levels of the key ER protein‐folding components (ie, BiP and PDI) did not differ significantly among the 3 groups, even though the protein levels of BiP and PDI in the aged group showed a tendency to decrease (Figure 6A through 6C). Moreover, CHOP expression levels were not altered either in the WD or in the aged group, while JNK phosphorylation was stimulated in these 2 groups to an extent similar to that of the control group (7.55±0.57‐ and 7.08±0.52‐fold, respectively). Cleaved caspase‐3, a marker of apoptosis, was not induced in the lungs of mice from the WD and aged groups (Figure 6D and 6E).
Figure 6. In lung tissues, ER chaperone and CHOP levels were not significantly altered by either the consumption of a WD or aging when compared with the control group.
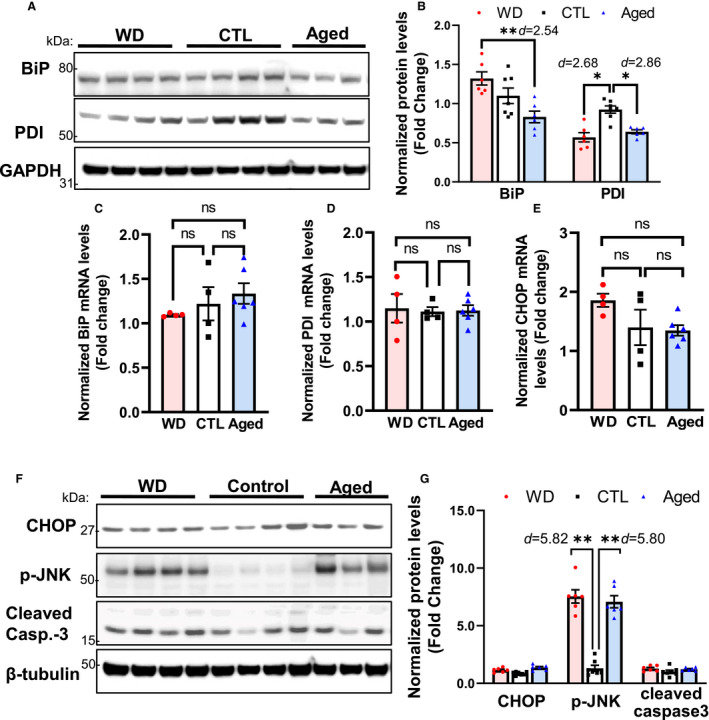
Lung tissues from the 3 groups of ApoE −/− mice were assessed for the activation of the unfolded protein response–mediated adaptive response (upregulation of the levels of the ER chaperones BiP and PDI; A through D) and apoptosis (increased expression of CHOP, p‐JNK, and cleaved caspase‐3; E through G). Only p‐JNK was markedly induced in the WD and aged groups. Data are presented as means±SE (n=6–8); *P<0.05, **P<0.01 vs the control group, unless otherwise indicated. The effect size is assessed by cohen's d value. BiP indicates immunoglobulin heavy chain‐binding protein; Casp‐3, caspase‐3; CHOP, C/EBP homologous transcription factor; CTL, control; ER, endoplasmic reticulum; ns, no significance; PDI, protein disulfide isomerase; and WD, Western diet.
Discussion
Our study demonstrated that hypercholesterolemia and aging, 2 strong risk factors for atherosclerosis, elicit distinct UPR signaling responses in the vascular system of ApoE −/− mice, as summarized in Figure 7. Specifically, aging can selectively activate ATF6 signaling in the aorta, but without significantly affecting the IRE1α/XBP1 and PERK/eIF1α axes; in contrast, hypercholesterolemia can stimulate all 3 UPR activation pathways. In aged aortae, the protein levels of the ER chaperones BiP and PDI were diminished, despite their marked upregulation at the mRNA level, which is usually indicative of adaptive UPR activation. In contrast, compared with WD‐fed mice, the expression of CHOP, p‐JNK, and cleaved caspase‐3 was greatly increased in the aged mice, indicating that UPR‐mediated apoptosis is enhanced with aging. This study provides the first in vivo evidence that vascular cells respond differently to aging and hypercholesterolemia with respect to UPR activation, which may contribute to the differential effects of these 2 risk factors on atherosclerosis.
Figure 7. Schematic summary detailing the distinct UPR pathways regulated by hypercholesterolemia and aging within aortic and lung tissues in a murine model of atherosclerosis.
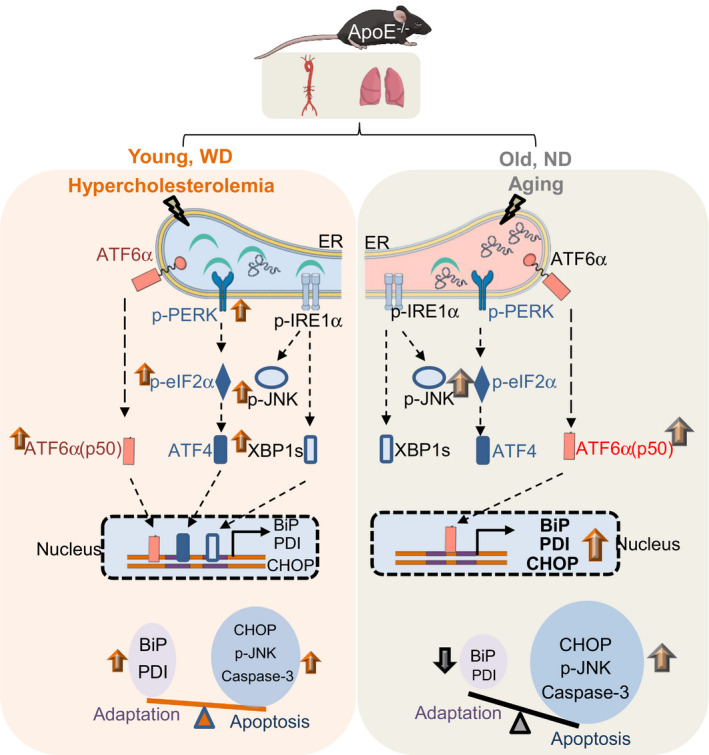
In ApoE −/− mice, the consumption of a WD induces hypercholesterolemia, and activates the 3 branches of the unfolded protein response (UPR) that are initiated by the activation of ATF6α (orange), PERK (blue), and IRE1α (gray) in the endoplasmic reticulum (ER). Key activated signaling components (red) were assessed by Western blotting and are indicated by green arrows. The activation of these 3 pathways results in the transcriptional upregulation of genes that facilitate adaptation (Bip and Pdi) and apoptosis (Chop) through ATF6α, ATF4, and XBP1s. Collectively, UPR‐related signaling cascades and the downstream transcription activation steps determine the balance between adaptation and apoptosis. Aging selectively activates the ATF6α pathway, which tips the balance toward apoptosis. ATF6α indicates activating transcription factor 6 alpha; BiP, immunoglobulin heavy chain‐binding protein; CHOP, C/EBP homologous transcription factor; ER, endoplasmic reticulum; IRE1α, inositol‐requiring enzyme 1 alpha; ND, normal chow diet; PDI, protein disulfide isomerase; p‐eIF2α, phosphorylated alpha subunit of the eukaryotic translation initiation factor 2; p‐PERK, phosphorylated pancreatic ER eIF2‐alpha kinase; UPR, unfolded protein response; and WD, Western diet.
The main finding of this study was that distinct UPR‐related pathways are activated by hypercholesterolemia and aging, even though these proatherogenic factors are both well‐recognized ER stressors. Several human and animal studies have indicated that elevated concentrations of free cholesterol or oxidized polyunsaturated fatty acids can directly induce ER stress and activate the UPR in endothelial cells, smooth muscle cells, and macrophages in the arterial wall. 10 , 23 , 24 , 25 , 26 , 27 In line with these reports, our results demonstrated that hypercholesterolemia because of the consumption of a WD can stimulate all 3 branches of the UPR, leading to the activation of both adaptive and apoptotic responses (Figures 2 and 4). The UPR is thought to be impaired with aging, as manifested by the decreased expression and activity of the ER‐resident chaperones BiP and PDI, 28 , 29 as well as the insufficient activation of UPR signaling pathways, such as the PERK/XBP1 pathway 30 ; this impaired ER quality control system, which aggravates ER stress and induces apoptosis, is implicated in the pathogenesis of age‐onset diseases. 31 Interestingly, our study showed that the ATF6α branch is selectively activated in the aged aorta, suggesting that ATF6α may play an important role in regulating aging‐related ER stress and arterial vascular responses. Additionally, the age‐related hyperactivation of ATF6α seems to be tissue‐specific, because no significant activation was observed in the lung tissues of ApoE −/− mice (Figures 4 and 5). Because ATF6α is required to induce the expression of genes that maintain the protein‐processing ability of the ER, including those coding for chaperones and elements of the ER degradation machinery, Atf6α‐null cells are particularly sensitive to long‐term or repeated stress. 32 , 33 This suggests that ATF6α hyperactivation represents a mechanism that allows vascular cells to specifically adapt to aging‐associated chronic stress. In support of this, we found that the transcription of the key ER chaperones—BiP and PDI—was enhanced in the aged aorta, and that their induction was likely to be primarily mediated by ATF6α activation as the PERK and IRE1α pathways, which also contribute to the upregulation of genes encoding ER chaperones, were suppressed. Similarly, the selective suppression of the PERK and IRE1α branches of the UPR may also explain how vascular cells adapt to aging‐induced chronic stress. PERK/eIF2α signaling is an acute stress response that can rapidly inhibit protein translation and relieve ER stress by immediately reducing the protein load. 34 The most likely cause for this rapid downregulation is that this pathway activates CHOP‐mediated apoptosis. 35 Prolonged IRE1α signaling may also be anti‐adaptive because IRE1α signaling is related to the activation of the proapoptotic JNK pathway. 35
Another major finding of this study was that, in the aging aorta, the protein levels of the ER chaperones BiP and PDI were markedly downregulated despite their significant transcriptional induction, whereas CHOP and apoptosis‐related signaling was highly stimulated (Figures 4 and 5). These results suggest that the role of the adaptive UPR (ie, the activation of ATF6 and the inhibition of the PERK and IRE1α pathways) may be to facilitate the adaptation to aging‐related chronic stress, but that the upregulated ER chaperones may subsequently be degraded in aging cells, for example, through oxidation, as has been observed in the aging liver and brain tissues. 28 , 29 The resultant defective protein folding and processing in the ER may aggravate ER stress, thus triggering a shift in the UPR from an adaptive response to apoptosis, as evidenced by the increased activation of CHOP and JNK. There are several lines of evidence to support that the inhibition of BiP can promote CHOP expression and apoptosis during prolonged ER stress, whereas increased expression of BiP allows cells to escape apoptosis. 36 , 37 , 38 Accordingly, restoring ER oxidative homeostasis and preserving the ER protein‐folding machinery may reshape the balance between adaptation and apoptosis, and may represent a new and effective therapeutic target for the treatment of atherosclerosis or other aging‐related diseases.
We used the well‐established ApoE −/− mouse model of atherosclerosis to study the effects of aging and WD‐induced hypercholesterolemia on UPR activation in the vascular system. APOE is a major antiatherogenic lipoprotein synthesized in the liver, neurons, and macrophages, and mediates the transport and clearance of circulating cholesterol. 39 Therefore, its deficiency disrupts cholesterol metabolism, and the plasma cholesterol levels of ApoE −/− mice are several‐fold higher than those of the background C57BL6J strain. 40 In these mice, as they age, lesions develop spontaneously in the major large arteries, while feeding them a WD greatly increases their plasma cholesterol levels and accelerates the formation of lesions. 16 , 18 That the plasma cholesterol levels do not change significantly with age helps to shed some light on the impact of aging on the vascular UPR. However, it is not possible to completely differentiate between the effects of age and the elevated basal cholesterol levels in ApoE −/− mice fed a chow diet. The higher plasma cholesterol concentrations may lead to additional ER stress and UPR activation in addition to that normally induced by aging and may not fully reflect the vascular ER stress and UPR of aged C57BL6 mice. This represents a major limitation of our study. The elevated basal cholesterol level may cause mild and chronic ER stress in arterial vascular cells, possibly resulting in the desensitization of the UPR. This may help explain the decreased activation of the PERK and IRE pathways observed in aged aorta. To test whether the aortic UPR signaling in young ApoE −/− mice differed from that of C57BL6 mice, we performed a comparative analysis of UPR‐related gene expression in aortae from ApoE −/− and C57BL6 mice at 14 and 30 weeks of age using a publicly available transcriptome data set (GSE163657). Principal component analysis indicated that the expression profile of UPR‐related genes was similar between young (14 weeks of age) C57BL6 and ApoE −/− mice, but showed significant differences at 30 weeks of age (Figure S1). This suggests that, at a young age, ApoE −/− mice can cope well with the ER stress imposed by the elevated basal cholesterol levels; with aging, however, the age‐related ER functional decline and the high cholesterol levels synergically promote UPR dysfunction.
In addition, given the antioxidant properties of APOE proteins, ApoE deficiency is likely to cause oxidative stress, 41 which may accelerate UPR failure. Notably, in candidate gene and genome‐wide association studies, ApoE was the first gene identified as being consistently associated with longevity. Compared with C57BL6 mice at the same age, ApoE −/− mice display cell senescence markers in the aorta, indicative of aggravated vascular aging. 42 , 43 Accordingly, ApoE −/− mice could be used as an animal model to investigate the in vivo UPR response to chronic stress, as well as the UPR‐related mechanisms that drive vascular aging and atherosclerosis.
Conclusions
Although both age and hypercholesterolemia (the strongest risk factors for atherosclerosis) can cause ER stress, they activate distinct UPR signaling networks and trigger differential responses in the aortae of atherosclerotic mice. Importantly, we found that while hypercholesterolemia activates all 3 arms of the UPR, aging selectively stimulates the ATF6α pathway, and aged mice exhibit an impaired ER protein‐folding capacity and increased levels of apoptosis. This study improves our understanding of vascular ER stress and the UPR triggered by different risk factors, and provides new insights into the development of effective UPR‐targeted therapeutics for the treatment of atherosclerotic cardiovascular diseases.
Sources of Funding
This work was supported by the National Institutes of Health (R01HL137771 to Han, R21AG058983 to Han, R21AA026922 to Han, R01HL13631 to Seta, and 1UL1TR001430 to BU CTSI); the Evans Medical Foundation (to Han); and the Boston University Undergraduate Research Opportunity Program (to Zhang, Zhou).
Disclosures
None.
Supporting information
Data S1
Table S1
Figure S1
Acknowledgments
The authors wish to acknowledge Boston University Cellular Imaging Core for the use of their instruments and their technical assistance.
(J Am Heart Assoc. 2021;10:e020441. DOI: 10.1161/JAHA.120.020441.)
For Sources of Funding and Disclosures, see page 14.
References
- 1. Virani SS, Alonso A, Benjamin EJ, Bittencourt MS, Callaway CW, Carson AP, Chamberlain AM, Chang AR, Cheng S, Delling FN, et al. Heart disease and stroke statistics‐2020 update: a report From the American Heart Association. Circulation. 2020;141:E139–E596. DOI: 10.1161/CIR.0000000000000757. [DOI] [PubMed] [Google Scholar]
- 2. Herrington W, Lacey B, Sherliker P, Armitage J, Lewington S. Epidemiology of atherosclerosis and the potential to reduce the global burden of atherothrombotic disease. Circ Res. 2016;118:535–546. DOI: 10.1161/CIRCRESAHA.115.307611. [DOI] [PubMed] [Google Scholar]
- 3. Wang JC, Bennett M. Aging and atherosclerosis: mechanisms, functional consequences, and potential therapeutics for cellular senescence. Circ Res. 2012;111:245–259. DOI: 10.1161/CIRCRESAHA.111.261388. [DOI] [PubMed] [Google Scholar]
- 4. Tietge UJ. Hyperlipidemia and cardiovascular disease: inflammation, dyslipidemia, and atherosclerosis. Curr Opin Lipidol. 2014;25:94–95. DOI: 10.1097/MOL.0000000000000051. [DOI] [PubMed] [Google Scholar]
- 5. Tabas I. The role of endoplasmic reticulum stress in the progression of atherosclerosis. Circ Res. 2010;107:839–850. DOI: 10.1161/CIRCRESAHA.110.224766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Erbay E, Babaev VR, Mayers JR, Makowski L, Charles KN, Snitow ME, Fazio S, Wiest MM, Watkins SM, Linton MF, et al. Reducing endoplasmic reticulum stress through a macrophage lipid chaperone alleviates atherosclerosis. Nat Med. 2009;15:1383–1391. DOI: 10.1038/nm.2067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Walter P, Ron D. The unfolded protein response: from stress pathway to homeostatic regulation. Science. 2011;334:1081–1086. DOI: 10.1126/science.1209038. [DOI] [PubMed] [Google Scholar]
- 8. Feng BO, Yao PM, Li Y, Devlin CM, Zhang D, Harding HP, Sweeney M, Rong JX, Kuriakose G, Fisher EA, et al. The endoplasmic reticulum is the site of cholesterol‐induced cytotoxicity in macrophages. Nat Cell Biol. 2003;5:781–792. DOI: 10.1038/ncb1035. [DOI] [PubMed] [Google Scholar]
- 9. Urano F, Wang XZ, Bertolotti A, Zhang Y, Chung P, Harding HP, Ron D. Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science. 2000;287:664–666. DOI: 10.1126/science.287.5453.664. [DOI] [PubMed] [Google Scholar]
- 10. Myoishi M, Hao H, Minamino T, Watanabe K, Nishihira K, Hatakeyama K, Asada Y, Okada K‐I, Ishibashi‐Ueda H, Gabbiani G, et al. Increased endoplasmic reticulum stress in atherosclerotic plaques associated with acute coronary syndrome. Circulation. 2007;116:107. DOI: 10.1161/CIRCULATIONAHA.106.682054. [DOI] [PubMed] [Google Scholar]
- 11. Zhou J, Lhoták S, Hilditch BA, Austin RC. Activation of the unfolded protein response occurs at all stages of atherosclerotic lesion development in apolipoprotein E‐deficient mice. Circulation. 2005;111:1814–1821. DOI: 10.1161/01.CIR.0000160864.31351.C1. [DOI] [PubMed] [Google Scholar]
- 12. Thorp E, Li G, Seimon TA, Kuriakose G, Ron D, Tabas I. Reduced apoptosis and plaque necrosis in advanced atherosclerotic lesions of Apoe‐/‐ and Ldlr‐/‐ mice lacking CHOP. Cell Metab. 2009;9:474–481. DOI: 10.1016/j.cmet.2009.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Naidoo N, Ferber M, Master M, Zhu Y, Pack AI. Aging impairs the unfolded protein response to sleep deprivation and leads to proapoptotic signaling. J Neurosci. 2008;28:6539–6548. DOI: 10.1523/JNEUROSCI.5685-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Paz Gavilán M, Vela J, Castaño A, Ramos B, del Río JC, Vitorica J, Ruano D. Cellular environment facilitates protein accumulation in aged rat hippocampus. Neurobiol Aging. 2006;27:973–982. DOI: 10.1016/j.neurobiolaging.2005.05.010. [DOI] [PubMed] [Google Scholar]
- 15. Minamino T, Komuro I, Kitakaze M. Endoplasmic reticulum stress as a therapeutic target in cardiovascular disease. Circ Res. 2010;107:1071–1082. DOI: 10.1161/CIRCRESAHA.110.227819. [DOI] [PubMed] [Google Scholar]
- 16. Nakashima Y, Plump AS, Raines EW, Breslow JL, Ross R. ApoE‐deficient mice develop lesions of all phases of atherosclerosis throughout the arterial tree. Arterioscler Thromb. 1994;14:133–140. DOI: 10.1161/01.ATV.14.1.133. [DOI] [PubMed] [Google Scholar]
- 17. Han J, Weisbrod RM, Shao DI, Watanabe Y, Yin X, Bachschmid MM, Seta F, Janssen‐Heininger YMW, Matsui R, Zang M, et al. The redox mechanism for vascular barrier dysfunction associated with metabolic disorders: glutathionylation of Rac1 in endothelial cells. Redox Biol. 2016;9:306–319. DOI: 10.1016/j.redox.2016.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Seo HS, Lombardi DM, Polinsky P, Powell‐Braxton L, Bunting S, Schwartz SM, Rosenfeld ME. Peripheral vascular stenosis in apolipoprotein E‐deficient mice—potential roles of lipid deposition, medial atrophy, and adventitial inflammation. Arterioscler Thromb Vasc Biol. 1997;17:3593–3601. DOI: 10.1161/01.ATV.17.12.3593. [DOI] [PubMed] [Google Scholar]
- 19. Yoshida H, Matsui T, Yamamoto A, Okada T, Mori K. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell. 2001;107:881–891. DOI: 10.1016/S0092-8674(01)00611-0. [DOI] [PubMed] [Google Scholar]
- 20. Marciniak SJ, Ron D. Endoplasmic reticulum stress signaling in disease. Physiol Rev. 2006;86:1133–1149. DOI: 10.1152/physrev.00015.2006. [DOI] [PubMed] [Google Scholar]
- 21. Rabek JP, Boylston WH III, Papaconstantinou J. Carbonylation of ER chaperone proteins in aged mouse liver. Biochem Biophys Res Commun. 2003;305:566–572. DOI: 10.1016/S0006-291X(03)00826-X. [DOI] [PubMed] [Google Scholar]
- 22. Fribley A, Zhang K, Kaufman RJ. Regulation of apoptosis by the unfolded protein response. Methods Mol Biol. 2009;559:191–204. DOI: 10.1007/978-1-60327-017-5_14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zhou AX, Tabas I. The UPR in atherosclerosis. Semin Immunopathol. 2013;35:321–332. DOI: 10.1007/s00281-013-0372-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Garbin U, Stranieri C, Pasini A, Baggio E, Lipari G, Solani E, Mozzini C, Vallerio P, Cominacini L, Fratta Pasini AM. Do oxidized polyunsaturated fatty acids affect endoplasmic reticulum stress‐induced apoptosis in human carotid plaques? Antioxid Redox Signal. 2014;21:850–858. DOI: 10.1089/ars.2014.5870. [DOI] [PubMed] [Google Scholar]
- 25. Kedi XU, Ming Y, Yongping W, Yi Y, Xiaoxiang Z. Free cholesterol overloading induced smooth muscle cells death and activated both ER‐ and mitochondrial‐dependent death pathway. Atherosclerosis. 2009;207:123–130. DOI: 10.1016/j.atherosclerosis.2009.04.019. [DOI] [PubMed] [Google Scholar]
- 26. Moore KJ, Tabas I. Macrophages in the pathogenesis of atherosclerosis. Cell. 2011;145:341–355. DOI: 10.1016/j.cell.2011.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zulli A, Lau E, Wijaya BPP, Jin X, Sutarga K, Schwartz GD, Learmont J, Wookey PJ, Zinellu A, Carru C, et al. High dietary taurine reduces apoptosis and atherosclerosis in the left main coronary artery association with reduced CCAAT/enhancer binding protein homologous protein and total plasma homocysteine but not lipidemia. Hypertension. 2009;53:1017–1022. DOI: 10.1161/HYPERTENSIONAHA.109.129924. [DOI] [PubMed] [Google Scholar]
- 28. Nuss JE, Choksi KB, DeFord JH, Papaconstantinou J. Decreased enzyme activities of chaperones PD1 and BiP in aged mouse livers. Biochem Biophys Res Commun. 2008;365:355–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. van der Vlies D, Woudenberg J, Post JA. Protein oxidation in aging: endoplasmic reticulum as a target. Amino Acids. 2003;25:397–407. DOI: 10.1007/s00726-003-0025-9. [DOI] [PubMed] [Google Scholar]
- 30. Gavilan MP, Vela J, Castano A, Ramos B, del Río JC, Vitorica J, Ruano D. Cellular environment facilitates protein accumulation in aged rat hippocampus. Neurobiol Aging. 2006;27:973–982. DOI: 10.1016/j.neurobiolaging.2005.05.010. [DOI] [PubMed] [Google Scholar]
- 31. Naidoo N. The endoplasmic reticulum stress response and aging. Rev Neurosci. 2009;20:23–37. DOI: 10.1515/REVNEURO.2009.20.1.23. [DOI] [PubMed] [Google Scholar]
- 32. Wu J, Rutkowski DT, Dubois M, Swathirajan J, Saunders T, Wang J, Song B, Yau GDY, Kaufman RJ. ATF6 alpha optimizes long‐term endoplasmic reticulum function to protect cells from chronic stress. Dev Cell. 2007;13:351–364. DOI: 10.1016/j.devcel.2007.07.005. [DOI] [PubMed] [Google Scholar]
- 33. Yamamoto K, Sato T, Matsui T, Sato M, Okada T, Yoshida H, Harada A, Mori K. Transcriptional induction of mammalian ER quality control proteins is mediated by single or combined action of ATF6 alpha and XBP1. Dev Cell. 2007;13:365–376. DOI: 10.1016/j.devcel.2007.07.018. [DOI] [PubMed] [Google Scholar]
- 34. Harding HP, Novoa I, Bertolotti A, Zeng H, Zhang Y, Urano F, Jousse C, Ron D. Translational regulation in the cellular response to biosynthetic load on the endoplasmic reticulum. Cold Spring Harb Symp Quant Biol. 2001;66:499–508. DOI: 10.1101/sqb.2001.66.499. [DOI] [PubMed] [Google Scholar]
- 35. Jousse C, Oyadomari S, Novoa I, Lu P, Zhang Y, Harding HP, Ron D. Inhibition of a constitutive translation initiation factor 2 alpha phosphatase CReP, promotes survival of stressed cells. J Cell Biol. 2003;163:767–775. DOI: 10.1083/jcb.200308075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lopez I, Tournillon A, Martins RP, Karakostis K, Malbert‐Colas L, Nylander K, Fåhraeus R. p53‐mediated suppression of BiP triggers BIK‐induced apoptosis during prolonged endoplasmic reticulum stress. Cell Death Differ. 2017;24:1717–1729. DOI: 10.1038/cdd.2017.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lyoo HR, Park SY, Kim JY, Jeong YS. Constant up‐regulation of BiP/GRP78 expression prevents virus‐induced apoptosis in BHK‐21 cells with Japanese encephalitis virus persistent infection. Virol J. 2015;12:32. DOI: 10.1186/s12985-015-0269-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Vitale M, Bakunts A, Orsi A, Lari F, Tadè L, Danieli A, Rato C, Valetti C, Sitia R, Raimondi A, et al. Inadequate BiP availability defines endoplasmic reticulum stress. Elife. 2019;8:e41168. DOI: 10.7554/eLife.41168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Meyrelles SS, Peotta VA, Pereira TMC, Vasquez EC. Endothelial dysfunction in the apolipoprotein E‐deficient mouse: insights into the influence of diet, gender and aging. Lipids Health Dis. 2011;10:211. DOI: 10.1186/1476-511X-10-211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Plump AS, Smith JD, Hayek T, Aalto‐Setälä K, Walsh A, Verstuyft JG, Rubin EM, Breslow JL. Severe hypercholesterolemia and atherosclerosis in apolipoprotein‐E‐deficient mice created by homologous recombination in ES cells. Cell. 1992;71:343–353. DOI: 10.1016/0092-8674(92)90362-G. [DOI] [PubMed] [Google Scholar]
- 41. Hayek T, Oiknine J, Brook JG, Aviram M. Increased plasma and lipoprotein lipid‐peroxidation in apo E‐deficient mice. Biochem Biophys Res Commun. 1994;201:1567–1574. DOI: 10.1006/bbrc.1994.1883. [DOI] [PubMed] [Google Scholar]
- 42. Wang JC, Bennett M. Aging and atherosclerosis mechanisms, functional consequences, and potential therapeutics for cellular senescence. Circ Res. 2012;111:245–259. DOI: 10.1161/CIRCRESAHA.111.261388. [DOI] [PubMed] [Google Scholar]
- 43. Pereira TMC, Nogueira BV, Lima LCF, Porto ML, Arruda JA, Vasquez EC, Meyrelles SS. Cardiac and vascular changes in elderly atherosclerotic mice: the influence of gender. Lipids Health Dis. 2010;9:87. DOI: 10.1186/1476-511X-9-87. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1
Table S1
Figure S1