

Abstract
Criteria for diagnosis of arrhythmogenic cardiomyopathy (ACM) were first proposed in 1994 and revised in 2010 by a Task Force. Although the Task Force criteria demonstrated a good accuracy for diagnosis of the original right ventricular phenotype (arrhythmogenic right ventricular cardiomyopathy), they lacked sensitivity for identification of the expanding phenotypic spectrum of ACM, which includes left‐sided variants and did not incorporate late‐gadolinium enhancement findings by cardiac magnetic resonance. The 2020 International criteria (“Padua criteria”) have been developed by International experts with the aim to improve the diagnosis of ACM by providing new criteria for the diagnosis of left ventricular phenotypic features. The key upgrade was the incorporation of tissue characterization findings by cardiac magnetic resonance for noninvasive detection of late‐gadolinium enhancement/myocardial fibrosis that are determinants for characterization of arrhythmogenic biventricular and left ventricular cardiomyopathy. The 2020 International criteria are heavily dependent on cardiac magnetic resonance, which has become mandatory to characterize the ACM phenotype and to exclude other diagnoses. New criteria regarding left ventricular depolarization and repolarization ECG abnormalities and ventricular arrhythmias of left ventricular origin were also provided. This article reviews the evolving approach to diagnosis of ACM, going back to the 1994 and 2010 International Task Force criteria and then grapple with the modern 2020 International criteria.
Keywords: cardiac magnetic resonance, cardiomyopathy, diagnosis, sudden death, ventricular arrhythmia
Subject Categories: Electrophysiology, Cardiomyopathy, Magnetic Resonance Imaging (MRI)
Nonstandard Abbreviations and Acronyms
- ACM
arrhythmogenic cardiomyopathy
- ALVC
arrhythmogenic left ventricular cardiomyopathy
- ARVC
arrhythmogenic right ventricular cardiomyopathy
- DCM
dilated cardiomyopathy
- EMB
endomyocardial biopsy
- HRS
Heart Rhythm Society
- LGE
late gadolinium enhancement
- PVBs
premature ventricular beats
- TF
Task Force
In 1982 Marcus et al published their pioneering clinical and electrophysiologic observations on a previously overlooked heart muscle disease specifically affecting the right ventricle (RV) and manifesting with right ventricular tachycardia (VT). 1 The disease was originally designated as “arrhythmogenic right ventricular dysplasia” because it was thought to be a congenital defect in the development of the right ventricular (RV) myocardium. In 1988, Thiene et al reported a series of effort‐related sudden cardiac death in young people, and the clinical and pathologic features of these deaths were suggestive of arrhythmogenic right ventricular dysplasia. 2 The ECG recordings before deaths showed in all of these patients the presence of negative T‐waves in the right precordial leads and ventricular arrhythmias with a left‐bundle branch block (LBBB) morphology. On postmortem examination of the heart there was histopathologic evidence of progressive loss of the RV myocardium, with foci of inflammation, degeneration, and necrosis, a pathologic scenario more consistent with a heart muscle disease developing after birth. This led to the adoption in the article of the more appropriate disease designation of arrhythmogenic right ventricular “cardiomyopathy” (ARVC) instead of “dysplasia.” For the first time, ARVC was recognized as a leading cause of sudden cardiac death in young people and athletes.
Since the original reports by Marcus and Thiene, there have been substantial advances in our understanding of the pathogenesis, clinical manifestations, and long‐term outcome of the disorder. 3 The discovery that the disease is caused by a genetic defect in the cardiac desmosomes has led to its definitive recognition as a cardiomyopathy and its inclusion in the classification of cardiomyopathies by the American Heart Association. The phenotypic spectrum of the disease has become broader than initially thought because of the inclusion of biventricular and left‐dominant disease variants, which has led to the use of the more comprehensive designation of “arrhythmogenic cardiomyopathy (ACM).” 4
The clinical diagnosis of ACM is complex because of the lack of the “Holy Grail” (ie, a sensitive and specific diagnostic test) and the several problems with the specificity of the ECG abnormalities, different potential causes of ventricular arrhythmias, evaluation of cardiac imaging findings, and characterization of the myocardial tissue structure. In addition, the interpretation of molecular testing for gene defects associated with ACM may be problematic because of the limitations of current understanding of the genetic background and the large “genetic noise” that translates into the risk of disease misdiagnosis. 5
Because of these difficulties in the clinical diagnosis, a number of diagnostic criteria sets have been proposed over the last 35 years. This article reviews the evolving approach to diagnosis of ACM, from the Task Force (TF) criteria first introduced in 1994 6 and subsequently revised in the 2010 7 to the most recent 2020 International criteria. 8
The 1994 Original TF Criteria for ARVC
In 1994, a TF of experienced clinicians in the field of cardiomyopathy was convened under the auspices of the European Society of Cardiology (Working Group on Myocardial and Pericardial Diseases) and the International Society and Federation of Cardiology (Scientific Council) to establish formal criteria aimed at facilitating and standardizing the clinical diagnosis of the original phenotype of the disease characterized by typical involvement of the RV. 6 Because single clinical features and instrumental tests had low diagnostic accuracy, the TF strategy consisted of reaching a clinical diagnosis by combining multiple aspects of diagnostic information such as family history, ECG, arrhythmic, structural, functional, and histopathological findings. The TF criteria were set up on the basis of their specificity for the right‐dominant variant of ARVC in order to avoid misdiagnosis because of conditions with an overlapping phenotype such as biventricular dilated cardiomyopathy (DCM) or idiopathic right ventricular outflow tract (RVOT) VT. The TF diagnostic criteria were grouped into 6 different categories encompassing the spectrum of clinical manifestations of ARVC. The criteria were classified in “major” and “minor” according to their specificity for ARVC and the diagnosis fulfilled in the presence of 2 major criteria or 1 major plus 2 minor or 4 minor criteria from different categories.
In general, the major limitations of the 1994 TF diagnostic criteria concerned the qualitative and subjective assessment of the clinical features of the disease. With regard to the category of “morpho‐functional abnormalities,” the TF criteria lacked quantitative values for grading of RV dilation/dysfunction. The measurement of volumes, dimensions, and systolic function of the RV by echocardiography or angiography was difficult because the distinct geometry of the RV made unsuitable the mathematical models used for evaluation of the left ventricle (LV). 9 The global dilatation of the RV by echocardiography or angiography was defined as “mild” or “severe” based on 2 to 3 SD and >3 SD from normal, respectively; “moderate” dilatation was not defined. Moreover, the identification of either global systolic function and regional wall motion abnormalities of the RV relied on subjective judgment. To increase the diagnostic specificity and to prevent misdiagnosis of ARVC in patients affected by DCM with RV involvement, a prerequisite was that diagnostic criteria of morphofunctional RV abnormalities were fulfilled in the presence of no (or only mild) LV impairment. Although ventricular arrhythmogenesis was a recognized cardinal manifestation of the disease, either recording >1000 premature ventricular beats (PVBs) during a 24‐hour Holter monitoring or documentation of sustained or nonsustained VT with a LBBB morphology by standard ECG, Holter monitoring or exercise testing were listed as minor diagnostic criteria. The explanation is that the finding of frequent PVBs or VT with a LBBB morphology can occur in conditions other than ARVC, particularly the RVOT arrhythmia/VT, which is the most common “idiopathic” ventricular arrhythmia, caused by focal enhanced automaticity/triggered activity from the RVOT in the absence of structural heart disease and, thus, characterized by a benign prognosis. 10 The classification of ventricular arrhythmia as a minor criterion mostly aimed to limit misdiagnosis of common and benign RVOT ventricular arrhythmias as ARVC‐related ventricular arrhythmias at risk of sudden cardiac death.
Since the 1994 TF, fibrofatty replacement of myocardium on endomyocardial biopsy (EMB) has been classified as a major criterion for diagnosis of ARVC because it was deemed as highly specific and strongly supporting any other clinical findings from other categories. Of note, in the original TF criteria the histopathologic definition of “fibrofatty replacement” was not provided. The EMB findings lack sensitivity for diagnosis of ARVC because of sampling error: fibrofatty myocardial replacement may be spotty and myocardial samples may be obtained from unaffected RV regions. 5 , 11
Another important limitation of the 1994 TF criteria was the absence of a definition of the “epsilon waves,” which was a major diagnostic criterion in the category of ECG depolarization findings. 12
In summary, the 1994 original TF criteria were designed to guarantee an adequate specificity for ARVC diagnosis among index cases with overt clinical manifestations. However, the criteria were largely qualitative (rather than quantitative) and revealed flaws in practical application. Moreover, the criteria lacked sensitivity for identification of early/minor phenotypes, which is a major challenge, particularly in the setting of familial ARVC. 13
The 2010 Revised TF Criteria for ARVC
In the 2010 Revised Criteria, the TF panel reconvened to revise the original criteria with the goal of improving diagnostic sensitivity but with the important requisite of maintaining diagnostic specificity. 7 The approach of the original 1994 TF criteria to classify structural, histopathological, ECG, arrhythmic, and genetic features of the disease as major and minor criteria was maintained by the revised 2010 TF criteria (Table 1). According to the revised criteria, a definite diagnosis of ARVC was fulfilled by 2 major or 1 major and 2 minor criteria or 4 minor criteria from different categories; a borderline diagnosis by 1 major and 1 minor or 3 minor criteria from different categories; and a possible diagnosis by 1 major or 2 minor criteria from different categories.
Table 1.
Comparison of 2010 TF Criteria and 2020 International Criteria for Diagnosis of ARVC
| Category | 2010 TF criteria | 2020 International criteria |
|---|---|---|
| I. Global or regional dysfunction and structural alteration |
Major By 2D echocardiogram:
By MRI:
and one of the following:
By RV angiography:
Minor By 2D echocardiogram:
By MRI:
|
Major By 2D echocardiogram, CMR, or angiography:
plus 1 of the following:
or
Minor By 2D echocardiogram, CMR, or angiography:
|
| II. Tissue characterization |
Major By EMB
Minor By EMB
|
Major By CE‐CMR:
Major By EMB (limited indications):
|
| III. Repolarization abnormalities |
Major
Minor
|
Major
Minor
|
| IV. Depolarization and conduction abnormalities |
Major
Minor
|
Minor
|
| V. Arrhythmias |
Major
Minor
|
Major
Minor
|
| VI. Family history/genetics |
Major
Minor
|
|
Cut‐off values of EDV and EF of the 2020 International criteria for RV dilatation and systolic dysfunction, respectively, are reported in Table 3. ACM indicates arrhythmogenic cardiomyopathy; BSA, body surface area; CMR, cardiac magnetic resonance; EDV, end diastolic volume; EF, ejection fraction; EMB, endomyocardial biopsy; ITF, International Task Force; LBBB, left bundle‐branch block; LGE, late gadolinium enhancement; LV, left ventricle; MRI, magnetic resonance imaging; PLAX, parasternal long axis; PSAX, parasternal short axis; RBBB, right bundle‐branch block; SAECG, signal‐averaged ECG; RV, right ventricle; and RVOT, right ventricular outflow tract.
*The morphology of “Major” ventricular arrhythmias is LBBB with a QRS axis other than inferior (ie, intermediate or superior).
Adapted from Corrado et al 8 with permission, ©2020, Elsevier.
The 2010 revised TF criteria provided quantitative imaging (echocardiography, ventricular angiography, and cardiac magnetic resonance [CMR]) reference values based on sex‐specific volumetric measurements indexed to body surface area (BSA) to define normal RV and to categorize the various degrees of structural and functional RV abnormalities. To optimize the diagnostic specificity of morpho‐functional criteria, the 2010 TF criteria required the association of global RV dilatation or RV systolic dysfunction with regional wall motion abnormalities (ie, akinesia, dyskinesia, aneurysm, or bulging). These criteria were classified as “major” or “minor” based on the severity of RV dilatation and/or systolic impairment.
In addition, the 2010 TF criteria provided a definition and quantitative histomorphometry for proper grading of fibrofatty replacement of the myocardium on EMB. Noninvasive tissue characterization by CMR for demonstration of fibrofatty myocardial replacement was not included among the 2010 TF criteria because it was not considered a reliable technique because of limited experience, difficult interpretation, and low specificity at the time the criteria were developed.
With regard to the ECG and arrhythmic features, in the 2010 TF criteria, T‐wave inversion in V1–V3 as well as VT with a LBBB morphology with superior/indeterminate QRS axis, either sustained or nonsustained, became major criteria. The following findings were included among the minor criteria: (1) T‐wave inversion in V1 and V2 in the absence of right bundle branch block (RBBB) and from V1 to V4 in the presence of complete RBBB 14 ; (2) prolongation of right precordial QRS duration with delayed S‐wave upstroke (terminal activation delay >55 ms) 15 ; (3) positivity of any 1 of the 3 signal‐averaged ECG parameters for late potentials; and (4) PVBs >500 per 24 hours on Holter monitoring.
Finally, the TF diagnostic criteria were modified to include (newly available) molecular genetic information in the category of “family history” criteria. 16 , 17 , 18 The identification of a pathogenetic gene mutation in a first‐degree relative was listed as a major criterion for diagnosis of ARVC.
The clinical experience with the use of 2010 TF criteria over the last decade has highlighted inherent diagnostic limitations and called for the need of an upgrading. 5
The Paradigm Shift in the Diagnosis of ACM
Since 2010, there have been significant insights into our understanding of the phenotypic expression of the disease, and the original concept of a heart muscle disease exclusively affecting the RV has evolved to the current perspective of a cardiomyopathy of both ventricles, with LV involvement that may parallel (“biventricular”) or exceed (“left‐dominant” or arrhythmogenic left ventricular cardiomyopathy [“ALVC”]) the severity of RV involvement. 19 , 20 , 21 , 22 , 23 , 24 , 25 , 26 , 27 , 28 , 29 Thus, the original term “ARVC” was progressively replaced by the new designation of “ACM” that better describes the entire spectrum of the phenotypic variants. 4
A 2019 International Expert report provided an extensive critical appraisal of the clinical performance of the 2010 TF criteria, identifying critical diagnostic flaw backs and potential areas of improvement. 5 Major evidences of the inadequacy of the 2010 TF diagnostic criteria were in regard to the absence of specific criteria for diagnosis of the broader phenotypic spectrum of the disease, which includes left‐sided variants, and the lack of tissue characterization findings by CMR, which has progressively emerged as the most accurate imaging modality for characterization of structural myocardial abnormalities and plays a key role for appropriate diagnosis of left‐sided variants of ACM.
In the same year, a group of experts in the field, under the auspices of the Heart Rhythm Society (HRS), published a consensus statement that intended to provide the clinician with guidance on evaluation and management of ACM and included clinically relevant information on genetics and disease mechanisms. 30 The HRS definition of ACM included, but was not limited to, arrhythmogenic RV and LV cardiomyopathy. According to the HRS consensus statement, ACM was defined as an “arrhythmogenic heart muscle disorder not explained by ischemic, hypertensive, or valvular disease, which incorporates, besides desmosomal gene related–ACM, a broad spectrum of systemic (ie, sarcoidosis, amyloidosis), inflammatory (ie, myocarditis), infectious (ie, Chagas disease), or genetic (ie, lamin A/C, filamin‐C, phospholamban cardiomyopathies) disorders and ion channel diseases.” 21 Hence, in the perspective of the HRS document, the term ACM covered a wide group of heterogeneous heart muscle diseases whose common denominator is the “clinical presentation with symptoms or documentation of atrial fibrillation, conduction disease, and/or RV and/or LV arrhythmia.” This broad HRS definition of ACM, based on “arrhythmic clinical presentation” shared by many heart diseases, differs from the definition more specific and commonly in use in both experimental and clinical settings, which designates a nosographically distinct condition characterized by typical cardiomyopathic features. 4
Both the International Expert report and the HRS consensus statement agreed that the TF criteria, exclusively targeting the original RV phenotype, overshadowed recognition of the sizeable proportion of patients with ACM with LV involvement. 5 , 30 In recent years, the increasing use of more sensitive imaging modalities, such as gadolinium‐enhanced CMR, has led to the recognition of an increasing number of individuals and families with predominantly biventricular or left‐dominant phenotypes. Both documents denounced the limited availability of genetic, diagnostic, and prognostic data for left‐sided ACM, and highlighted the importance of defining diagnostic criteria for guiding experimental and clinical studies aimed at characterizing the cause/pathogenesis and the clinical outcome of these disease variants. Hence, the documents called for the development of internationally recognized clinical criteria analogous to those established for ARVC, for diagnosis of left‐sided ACM in a proband or family member with ECG abnormalities and ventricular arrhythmia of LV origin associated with an underlying LV cardiomyopathy.
The 2020 International Criteria for ACM
In the 2020 criteria, an International expert consensus document provided upgraded diagnostic criteria for ACM (the “Padua Criteria”). 8 The proposed criteria were derived from the diagnostic approach to ACM, which has been developed over 30 years by the multidisciplinary team of basic researchers and clinical cardiologists of the Medical School of the University of Padua. 31 These criteria were reviewed and shared by several international experts, resulting in an International consensus document.
According to the 2020 International document, ACM is defined as a heart muscle disease that affects the RV, the LV, or both, whose hallmark pathologic feature is the fibrofatty myocardial replacement that underlies the impairment of systolic ventricular function and predisposes to potentially lethal ventricular arrhythmias, regardless of the severity of pump failure.
The 2020 classification of ACM includes the following phenotypic variants (Figures 1, 2, 3, 4): (1) the “dominant‐right” variant (ie, the classic ARVC phenotype characterized by the predominant RV involvement, with no LV abnormalities); (2) the “biventricular disease” variant, characterized by the involvement of both RV and LV; and (3) the “dominant‐left” variant (also referred to as ALVC) characterized by LV involvement, with no RV abnormalities.
Figure 1. Clinical features of arrhythmogenic right ventricular cardiomyopathy.
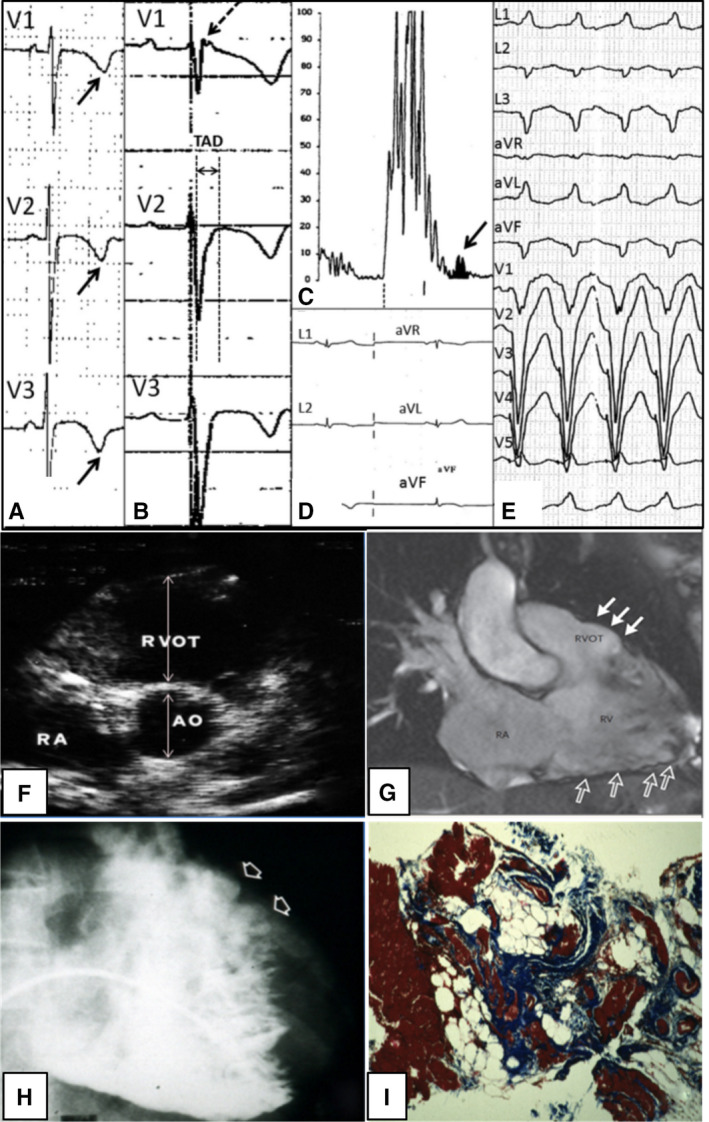
Right precordial negative T waves in leads V1 to V3 and prolongation of QRS complex because of delayed S‐wave upstroke leading to a significant terminal activation delay (A), epsilon waves (arrow) (B), late potentials on signal‐averaged ECG (arrow) (C), low QRS voltages (<0.5 mV) in the limb leads (D), and ventricular tachycardia with a left bundle branch block (E). Two‐dimensional echocardiogram (parasternal short‐axis view), showing dilatation of the RVOT (parasternal long axis‐RVOT=37 mm) (F). Cardiac magnetic resonance imaging scan (systolic frame of right ventricular 2‐chamber long‐axis view on cine sequences) evidencing an aneurysm (with dyskinesia, not shown) of the RVOT (solid arrows) and multiple sacculations of the inferior and apical regions (open arrows) (G). Angiography showing RV dilatation with a bulging of the RVOT (arrows) (H). Endomyocardial biopsy revealing myocyte loss with fibrofatty replacement (I). AO indicates aorta; RA, right atrium; RV, right ventricle; and RVOT, right ventricular outflow tract. Adapted from Corrado et al. 4
Figure 2. Clinical and histopathologic features of arrhythmogenic left ventricular cardiomyopathy.
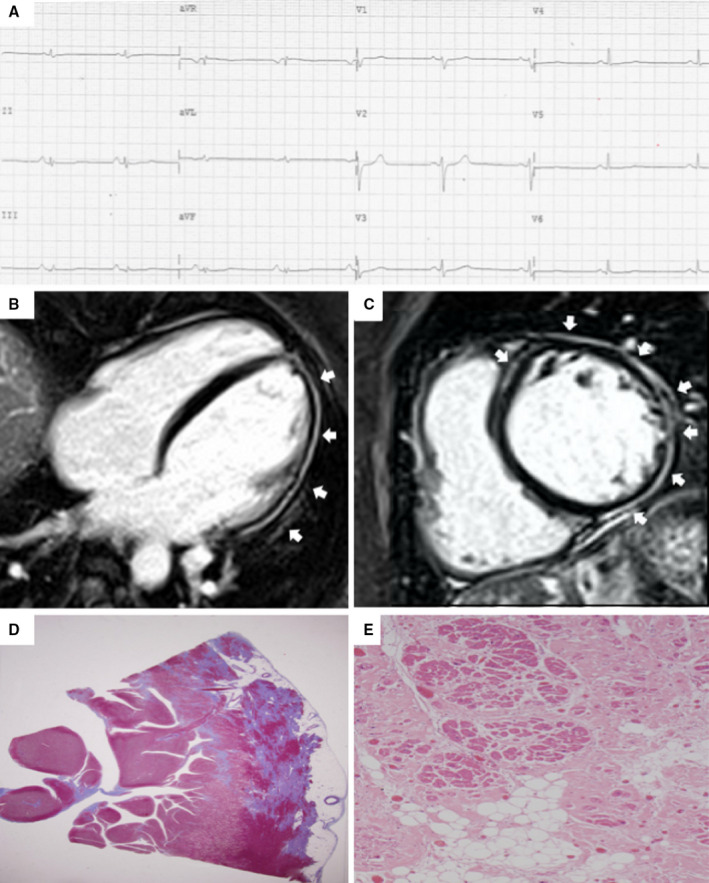
ECG, and CMR findings of a patient with ALVC related to a DSP gene defect. Basal ECG showing low voltages in limb leads and flattened T‐waves in the inferolateral leads (A). Postcontrast CMR images in long‐axis (B) and short‐axis (C) views showing normal LV cavity size and subepicardial LGE (white arrows) involving the whole LV free wall and the anterior septum (“ring‐like” pattern), from basal to apical regions. Histology of the LV inferolateral region showing fibrofatty myocardial replacement affecting the subepicardial layer (Heidenhain trichrome stain) (D); close‐up detailing residual myocytes embedded within fibrous and fatty tissue (hematoxylin and eosin stain) (E). ALVC indicates arrhythmogenic left ventricular cardiomyopathy; CMR, cardiac magnetic resonance; DSP, desmoplakin gene; LGE, late gadolinium enhancement; and LV, left ventricle. Adapted from Cipriani et al. 29
Figure 3. Clinical features of biventricular arrhythmogenic cardiomyopathy.
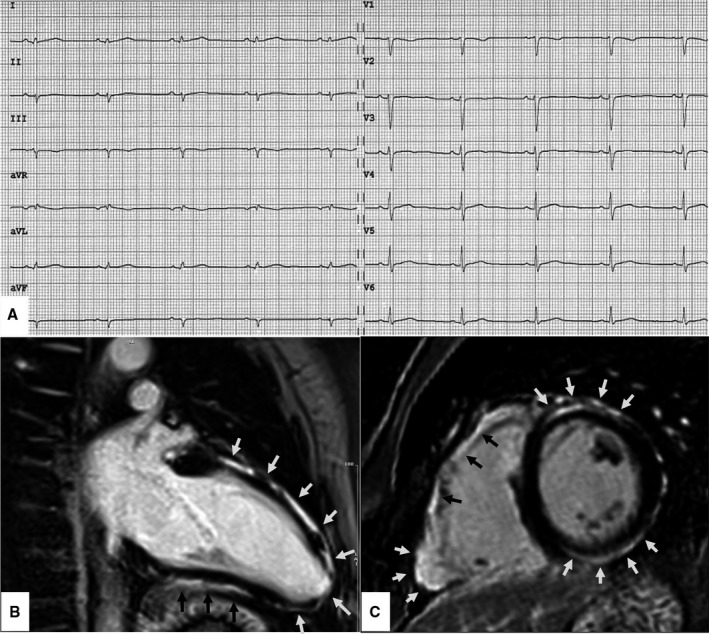
ECG and CMR findings in a 40‐year‐old patient with biventricular ACM caused by a pathogenic DSG‐2 gene mutation. Basal ECG showing low QRS voltages (<0.5 mV, peak to peak) in the limb leads, in the absence of other repolarization and depolarization ECG abnormalities. Premature ventricular beats on Holter monitoring <500/24 hours; no sustained or nonsustained ventricular tachycardia (not shown) (A). Postcontrast cardiovascular magnetic resonance images—end‐diastolic frame on long‐axis view (B) and short‐axis view (C)—showing normal cavity size of both RV and LV and LGE of the myocardium of the basal anterolateral right ventricular wall and anterior and inferior LV wall (arrows). On cine sequences (not shown) the RV shows regional akinesia with a mild reduction of the ejection fraction (ie, 50%) and the LV an inferolateral ipokinesia with a preserved systolic function. While this phenotypic variant of ACM does not fulfill the 2010 TF criteria, it is diagnosed according to the 2020 International criteria as definite biventricular ACM based on the low QRS voltages, the regional akinesia of the RV, the regional hypokinesia of the LV, and the biventricular LGE other than the pathogenic gene mutation. ACM indicates arrhythmogenic cardiomyopathy; CMR, cardiac magnetic resonance; DSG‐2, desmoglein‐2 gene; LGE, late gadolinium enhancement; LV, left ventricle; RV, right ventricle; and TF, task force.
Figure 4. Diagnostic flow‐chart for ACM phenotypic variants.
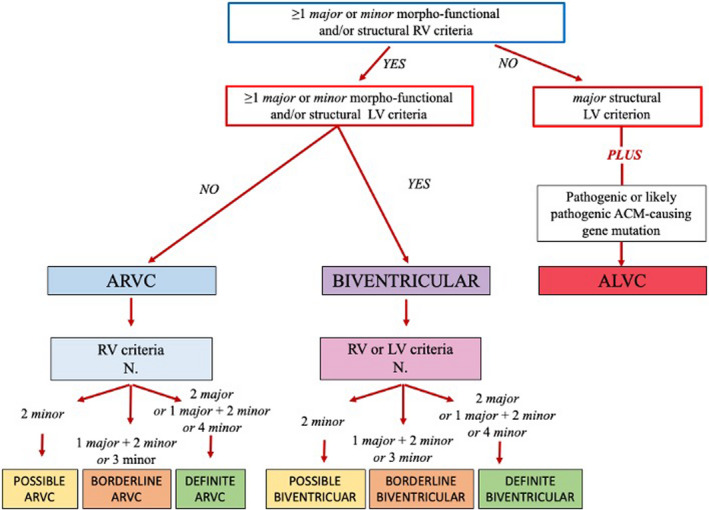
According to the 2020 International criteria, any diagnosis of ACM requires that at least 1 criterion either major or minor from category I (ie, morpho‐functional abnormalities) or II (ie, structural abnormalities) be fulfilled. For diagnosis of possible, borderline, or definite biventricular ACM, besides the need for ≥1 morpho‐functional and/or structural criteria from both the RV and LV, the remaining criteria are from either the RV or the LV (see text for details). ACM indicates arrhythmogenic left ventricular cardiomyopathy; ALVC, arrhythmogenic left ventricular cardiomyopathy; ARVC, arrhythmogenic right ventricular cardiomyopathy; LV, left ventricle; and RV, right ventricle.
At variance with the 1994 and 2010 TF criteria, the 2020 International document addressed the entire spectrum of ACM variants, also providing diagnostic criteria for the LV phenotype. The multiparametric diagnostic approach was maintained with criteria grouped in 6 categories encompassing functional and structural ventricular abnormalities, tissue characterization findings, depolarization and repolarization electrocardiographic alterations, ventricular arrhythmias, and familial/genetic factors.
The main innovation of the 2020 International criteria was the introduction of tissue characterization findings by late gadolinium enhancement (LGE) for detection of fibro(‐fatty) myocardial replacement of both ventricles. The available data indicate that tissue characterization by CMR shows a high concordance with EMB for identification of myocardial fibrosis and provides added value for identification of the different phenotypic variants of ACM by virtue of the distribution of LGE in the RV, LV, or both. 21 , 32
2020 International Criteria Versus 2010 TF Criteria for RV Phenotype
A comparison of the 2020 International criteria with the 2010 TF criteria by different categories for the diagnosis of the RV phenotype is reported in Tables 1, 2, 3.
Table 2.
Summary of Changes in the 2020 International Criteria for Diagnosis of ARVC*
| Categories | Criteria | Changes |
|---|---|---|
| I. Global or regional dysfunction and structural alteration |
|
|
| II. Tissue characterization |
|
|
| III. Repolarization abnormalities |
|
|
| IV. Depolarization and conduction abnormalities |
|
|
| V. Arrhythmias |
|
|
| VI. Family history/genetics |
|
|
ARVC indicates arrhythmogenic right ventricular cardiomyopathy; BSA, body surface area; CMR, cardiac magnetic resonance; EDV, end diastolic volume; EF, ejection fraction; EMB, endomyocardial biopsy; LBBB, left bundle‐branch block; LGE, late gadolinium enhancement; LV, left ventricle; RV, right ventricle; SAECG, signal averaged ECG; SCD, sudden cardiac death; TF, Task Force; VT, ventricular tachycardia; and WMA, wall motion abnormalities (ie, regional RV akinesia, dyskinesia, or bulging).
*The 2020 International criteria for diagnosis of arrhythmogenic left ventricular cardiomyopathy were not included in the 2010 TF criteria (see text for details).
Table 3.
Ventricular Dilatation and Systolic Dysfunction by CMR: 2010 TFC Versus 2020 IC
| Women | Men | Athletes | ||||
|---|---|---|---|---|---|---|
| 2010 TFC | 2020 IC | 2010 TFC | 2020 IC | 2010 TFC | 2020 IC | |
| Right ventricular dilatation and systolic dysfunction | ||||||
| EDV/BSA, mL/m2 | ≥90 | >112 | ≥100 | >121 | … | >130 |
| EF (%) | ≤45 | <51 | ≤45 | <52 | … | <52 |
| Left ventricular dilatation and systolic dysfunction | ||||||
| EDV/BSA, mL/m2 | … | >96 | … | >105 | … | >122 |
| EF (%) | … | <57 | … | <57 | … | <58 |
Cardiac magnetic resonance (CMR) cutoff values of EDV and EF for nonathletes (±2 SD from the mean, respectively) derived from Petersen et al 33 and for athletes (99% CI) from D’Ascenzi et al. 34 BSA indicates body surface area; EDV, end‐diastolic volume; EF, ejection fraction; IC, International Criteria; and TFC, Task Force Criteria
I. Morpho‐Functional Ventricular Abnormalities (ie, Global and Regional Ventricular Dilatation and Systolic Dysfunction)
In recent years, CMR has become the criterion standard tool for assessing volumes, systolic function, and regional wall motion, as well as characterizing myocardial tissue composition. 29 , 35 , 36 , 37 , 38 Because of the high spatial resolution and unlimited imaging planes, CMR offers the potential to optimally evaluate dilatation/dysfunction, regional dyssynergies, and structural changes of the RV. To optimize the diagnostic accuracy of morpho‐functional criteria, the 2010 TF guidelines required that global RV dilatation (based on sex‐specific volumetric measurements and indexed to BSA or RV systolic dysfunction) had to be associated with major regional wall motion abnormalities (ie, akinesia, dyskinesia, aneurysm, or bulging). Morpho‐functional abnormalities were listed as major or minor criteria based on the severity of RV dilatation or systolic dysfunction associated with the RV wall motion abnormalities. However, the distinction appears more useful for prognostic than for diagnostic purposes; accordingly, in the 2020 International document, the morpho‐functional criterion, if fulfilled, is considered major regardless of the severity of RV dilatation/dysfunction.
On the other hand, a sizeable proportion of patients do not show increase of RV volume and/or decrease of systolic function. 29 , 36 , 37 , 38 This finding reflects the segmental nature of fibrofatty myocardial replacement areas that may not compromise the global hemodynamics of the RV. For this reason, a “new” minor morpho‐functional criterion has been introduced in the 2020 International document, i.e. isolated regional RV wall motion abnormalities in the absence of global RV dilation/dysfunction.
The available data demonstrate that RV wall motion abnormalities (regional akinesia or dyskinesia) have a high degree of diagnostic specificity. 32 , 33 , 34 , 35 , 36 , 37 , 38 , 39 , 40 , 41 , 42 , 43 , 44 However, it should be recognized that the diagnostic accuracy may be limited by the potential misinterpretation (either over‐ or underdiagnosis) because of inherently imperfect subjective evaluation of cine‐CMR images and the potential pitfall of nonpathologic RV wall motion alterations such as (1) the “tethered” appearance of RV wall because of a band of pericardial connective tissue that joins the anterior free wall of the RV to the posterior aspect of the sternum; (2), the “apicolateral bulge” at the insertion site of the moderator band in the apico‐lateral wall; and (3) a “butterfly apex” with unusual prominence (misinterpreted as an apical aneurysm) of the RV apex. 38 For these reasons, the new criterion of regional RV wall motion abnormalities in isolation was classified as minor.
In the 2010 TF criteria, reference values of normal RV end‐diastolic volumes (up to 110 mL/m2 in men and 100 mL/m2 in women) were derived from 462 healthy controls of the MESA (multiethnic study of atherosclerosis), in which quantification of ventricular volumes was obtained using the older fast gradient echo CMR cine technique that underestimates volumes because of an incomplete and lower endocardial border definition. 39 Cardiac chamber volumes are more accurately measured by the modern CMR cine technique with steady‐state free procession images, which provide superior contrast between blood and endocardium at the endocardial border with less blood flow dependence. In order to increase the diagnostic accuracy of CMR imaging findings, the revised criteria recommend using reference values for RV cavity size and systolic function normalized for age, sex, and BSA, according to current nomograms provided by international societies of cardiovascular imaging (Table 3). 33
The normal reference value of 100 mL/m2 (men) and 90 mL/m2 (women) of RV end‐diastolic volume/BSA proposed by the 2010 International TF criteria may lack specificity for ACM, especially because of the overlap with physiologic adaptive changes of the athlete’s heart, which can produce an increase of both LV and RV volumes that is well beyond the upper limit of normality reported in the general population. 40 In this regard, proper reference values for RV volume in the athlete’s heart are currently available and recommended by the 2020 International criteria for differential diagnosis of physiologic versus pathologic RV dilatation, especially if engaged in sports such as rowing or canoeing associated with the greatest RV dimensional remodeling (Table 3). 34
II. Structural Myocardial Abnormalities (ie, Fibrous or Fibro‐Fatty Myocardial Replacement)
Transvenous EMB has been part of the diagnostic work‐up of ACM since 1994. 6 The technique offers the potential for an “in vivo” histologic tissue characterization with demonstration of the hallmark lesion of ACM, ie, the loss of RV myocytes with fibro‐fatty replacement, which has been longer considered the criterion standard for clinical diagnosis of ACM (major diagnostic criterion). Because of the invasive nature with the inherent risk of complications, in the 2020 International document the indication for EMB is reserved for selected cases, namely, probands with a sporadic form of the disease and negative genotyping, in whom the diagnosis of ACM depends on histologic study of the myocardium to exclude phenocopies (mostly cardiac sarcoidosis). 5 , 32 Histologic demonstration of replacement‐type fibrosis, with or without fatty tissue, on EMB samples remains a major structural criterion for diagnosis, while the distinction of EMB findings in major and minor is no longer proposed.
Noninvasive tissue characterization findings by CMR were excluded from the 2010 TF criteria. According to the 2020 International criteria, demonstration by CMR myocardial tissue characterization of transmural LGE/fibrosis affecting ≥1 RV region(s) in 2 orthogonal views, with or without fatty tissue replacement on dedicated sequences, is classified as a major structural myocardial criterion. The available data indicate a high degree of diagnostic specificity of RV LGE, but a limited sensitivity. 32 , 41 , 42 , 43 , 44 Low sensitivity has been ascribed to the poor quality spectral resolution and the suboptimal contrast/noise ratio of current CMR technology to accurately quantify the thin RV wall. 5 , 35 The best diagnostic accuracy by CMR is achieved by combining myocardial tissue characterization with regional RV wall motion assessment. 44 , 45 , 46 Detection of an underlying fibro‐fatty myocardial scar on CMR increases the diagnostic specificity of RV wall motion abnormalities.
III/IV. ECG Abnormalities
The presence of epsilon waves in right precordial leads was classified as a major ECG criterion in both the 1994 and the 2010 TF criteria. However, the diagnostic value of the epsilon wave has been questioned in the last decade because its identification and interpretation are largely influenced by ECG filtering and sampling rate, giving rise to large interobserver variability. 12 Hence, in the 2020 International diagnostic criteria this ECG marker has been downgraded to a minor ECG depolarization criterion.
The presence of late potentials on signal‐averaged ECG, which was a minor criterion in the 2010 TF criteria, is no longer included among the 2020 International criteria, since the use of the signal‐averaged ECG technique for diagnosis of ACM has been abandoned by the majority of Cardiological Centers worldwide because of its limited diagnostic accuracy. 5 According to a 2020 anonymous survey among a panel of International experts from Europe and the United States, the degree of consensus to eliminate signal‐averaged ECG findings from the diagnostic criteria for ACM was >80% (Domenico Corrado, MD, PhD, unpublished data, 2020).
V. Ventricular Arrhythmias
According to the 2020 International diagnostic criteria, PVBs need to be evaluated not only in terms of absolute number (>500/24 hours as recommended by the 2010 TF criteria), but also of morphology of ectopic QRS. By analogy with nonsustained and sustained VT, demonstration of PVBs with a LBBB and superior axis pattern (indicating their origin from the infero‐apical RV wall) has a greater disease specificity (major ventricular arrhythmia criterion) than PVBs showing a LBBB/inferior axis morphology more consistent with idiopathic RVOT arrhythmia (minor ventricular arrhythmia criterion). 47 , 48 , 49 As a corollary, it is clinically relevant to record the ventricular arrhythmia morphology on 12‐ECG leads by exercise testing or 12‐lead 24‐hour Holter monitoring.
The 2020 International Criteria for LV Phenotype
The 2020 International criteria by different categories for diagnosis of the LV phenotype are reported in Tables 3 and 4. In the 2010 TF guidelines, criteria for diagnosis of biventricular ACM or ALVC were lacking.
Table 4.
The 2020 International Criteria for Diagnosis of ALVC
|
Category |
Diagnostic criteria |
|---|---|
| I. Morpho‐functional ventricular abnormalities |
Minor
Minor
|
|
II. Structural myocardial abnormalities |
Major
|
| III. Repolarization abnormalities |
Minor
|
| IV. Depolarization abnormalities |
Minor
|
|
V. Ventricular arrhythmias |
Minor
|
|
VI. Family history/genetics |
Major
Minor
|
ACM indicates arrhythmogenic cardiomyopathy; ALVC, arrhythmogenic left ventricular cardiomyopathy; BSA, body surface area; CMR, cardiac magnetic resonance; EDV, end diastolic volume; EF, ejection fraction; LBBB, left bundle‐branch block; LGE, late gadolinium enhancement; LV, left ventricle; and RBBB, right bundle‐branch block.
*Global LV systolic dysfunction defined as EF <55% on echocardiography and <57% (nonathletes) or <58% (athletes) on cine CMR (see Table 3).
Adapted from Corrado et al 8 with permission, ©2020, Elsevier.
I. Morpho‐Functional Ventricular Abnormalities (ie, Global and Regional Ventricular Dilatation and Systolic Dysfunction)
Global LV systolic dysfunction and regional LV wall motion abnormalities, with or without LV dilatation, are classified as minor morpho‐functional criteria because of the low disease specificity for diagnosing left‐sided ACM variants, given that these morpho‐functional LV abnormalities can be seen in other common conditions such as ischemic or nonischemic DCM. It is noteworthy that even the sensitivity of global LV dilatation/dysfunction for ACM is low because most patients show regional LV involvement without an increase of the cavity size and/or reduction of the global systolic function (Figures 2 and 3). 29 , 37 , 44 This is explained by the limited extent and the nontransmural (subepi‐midmyocardial) arrangement of fibro‐fatty myocardial scars that may not alter the global hemodynamics of the LV. The ensuing pattern of LV remodeling on CMR is usually characterized by a hypokinetic (or normokinetic) LV, with no or mild cavity dilatation. 23 , 24 , 25 , 26 , 27 , 28 , 29 LV systolic dysfunction may become more severe in the advanced stages because of the increasing extent of regional and transmural myocardial fibrosis.
To increase the diagnostic accuracy of morpho‐functional criteria, it is recommended to use reference values for LV cavity size and systolic function normalized for age, sex, and BSA, according to current nomograms provided by international societies of cardiovascular imaging (Table 3).
II. Structural Myocardial Abnormalities (ie, Fibrous or Fibro‐Fatty Myocardial Replacement)
The rationale for the introduction of tissue characterization by CMR in the 2020 International criteria is the unique ability of this technique to identify myocardial fibrosis of the LV myocardium. Structural LV abnormalities are characterized by nonischemic LGE/fibrosis that affects the subepicardial (less often the midmyocardial) layers of the LV free wall, mostly the inferolateral region, with or without septal involvement (Figures 2 and 3). 23 , 24 , 25 , 26 , 27 , 28 , 29 , 37 The circumferential involvement by subepicardial LGE of the LV free wall and septum in short axis view (“ring pattern”) has been consistently reported as highly specific for ALVC (Figure 2). 5 , 24 The coexistence of fatty myocardial infiltration is often observed on dedicated sequences in the same regions of LGE or in remote LV areas.
Although all patients with LV involvement have LGE, wall motion abnormality or global LV systolic dysfunction may be absent. Accordingly, demonstration of LV myocardial LGE/fibrosis in the form of a stria (or band) pattern affecting ≥1 segment (on the traditional “Bull’s Eye” system) is classified as a major and needed structural criterion for diagnosis of left‐sided ACM. 24 , 29 , 37 , 50 , 51 As a corollary, LV involvement in ACM cannot be ruled out based solely on the lack of overt LV functional abnormalities on echocardiography, cine‐CMR, or angiography.
The relationship between the amount of LV LGE and LVEF is of key importance for differential diagnosis of ALVC with DCM. While in patients with ALVC there is a linear correlation between reduction of LVEF and extent of LV LGE expressed as percentage of LV mass, in patients with DCM the LV systolic dysfunction and LGE are unrelated. 29 Although speculative, the severity of LV systolic dysfunction in ALVC appears to be directly related to the amount of LV LGE/myocardial fibrosis because it is the cause of the reduction of the contractile myocardial mass; by contrast, in DCM the systolic dysfunction results from a primary impairment of the myocyte force generation and LGE occurs as an epiphenomenon of the LV remodeling.
Focal or patchy LV LGE is considered nondiagnostic in the absence of other abnormal findings. Of note, the pattern of “junctional” LGE, which is characterized by focal/patchy involvement of the posterior (or less frequently anterior) ventricular septum at the site of RV attachment, is excluded from the diagnosis of ACM because of its nonpathologic nature. 51
III/IV. Depolarization and Repolarization ECG Abnormalities
LV involvement can be predicted on standard ECG by (1) low QRS voltages in the limb leads (“peak‐to‐peak” QRS amplitude <0.5 mV limb in all leads); (2) T‐wave inversion in the leads exploring the lateral or inferolateral leads. Both ECG abnormalities have been classified as minor criteria, because of the low estimated disease specificity compared with other diseases and normal controls. 24 , 29 , 51
The ECG pattern of low QRS voltages in limb leads has been reported as highly specific for LV involvement in the context of ACM. The mechanism involved in the reduction of QRS voltages reasonably consists of the decrease of LV myocardial mass, which mostly accounts for the generation of the electrical activity causing the depolarization current responsible for the QRS complex. Why it mainly affects the limb leads remains to be elucidated. The low sensitivity of low QRS voltages (<30%) may be explained by a dose–effect relationship between myocardial replacement by fibro‐fatty scar and reduction in QRS amplitudes in limb leads. This is in keeping with the significantly higher number of LV segments affected by LGE in patients with low QRS voltages in limb leads than in those without this ECG abnormality. 29
While T‐wave inversion limited to the lateral precordial leads (V5–V6) is an ECG marker of LV involvement, anterolateral T‐wave inversion extending from leads V2 to V6 may be the result of severe RV dilation. In fact, a severely dilated RV is displaced toward the axilla and a greater proportion of the ventricle is positioned under the ECG leads placed more laterally. Hence, the traditional LV leads (V4–V6) explore the electrical activity of the dilated and displaced RV rather than that of the LV. 29
V. Ventricular Arrhythmias
The RBBB morphology of ventricular arrhythmias (net positive QRS complex in V1) may suggest their origin from the LV and has been proposed as a criterion for LV involvement. However, this is classified as a minor diagnostic criterion because of its low specificity not only for the underlying disease but also for the chamber of origin of the arrhythmia.
An available mapping and catheter ablation study demonstrated that a RBBB VT morphology on standard ECG may be inadequate for identifying the LV origin and, thus, diagnosing a LV disease involvement. 52 Two thirds of RBBB VT in this series of patients with ACM originated from the apical/inferior septal regions of a dilated RV and showed an early transition to a negative QRS by V3. Only 17% of RBBB VT actually arose from the LV and characteristically exhibited a broad positive QRS in V1, lack of precordial QRS transition, and rightward QRS axis.
VI. Family History/Molecular Genetics
As reported in Tables 1, 2, and 4, the 2020 International criteria for the family history and molecular genetic category remained unchanged when compared with the 2010 TF criteria. However, more restricted indications for genotyping are proposed in the light of the increased awareness of the risk of misdiagnosis. Misinterpretation of molecular genetic results is the consequence of our limited current understanding of the genetic basis of ACM and the high genetic noise, because of frequent disease‐associated genetic variants both in the normal population and other cardiomyopathies. 53 , 54
Accordingly, genotyping: (1) is recommended to identify a pathogenic or likely pathogenic mutation in a proband who already fulfills the diagnosis of right‐dominant or biventricular ACM, with the aim to apply mutation‐specific cascade genetic testing for detection of gene carriers at a preclinical phase among family members 55 ; (2) may be considered to achieve a diagnosis in selected patients with borderline phenotypic manifestations, provided that the results are interpreted by experts on the molecular genetics of ACM 5 ; and (3) is mandatory for diagnosis in patients with an ALVC phenotype and no clinically detectable RV involvement, because demonstration of a pathogenic mutation in ACM‐related genes is the most specific finding linking the LV phenotypic features to ACM. 5
Diagnosis of Phenotypic Variants of ACM
Figure 4 illustrates the flow chart of diagnosis of major phenotypic variants of ACM according to the 2020 International criteria.
Unlike genetically determined cardiac ion channel disorders, pathogenic mutations, ECG abnormalities, or arrhythmias are not sufficient for diagnosis of ACM, which is a structural heart muscle disease. According to the 2020 International criteria, any diagnosis of ACM requires that at least 1 criterion, either major or minor, from category I (ie, morpho‐functional RV abnormalities) or II (ie, structural RV abnormalities) be fulfilled.
“Definite” ARVC (ie, the original “RV dominant” variant) is diagnosed in patients fulfilling 2 major, 1 major and 2 minor, or 4 minor RV diagnostic criteria from different categories. Patients with 1 major and 1 minor, or 3 minor RV diagnostic criteria from different categories are diagnosed with “borderline” ARVC and patients with 1 major or 2 minor RV diagnostic criteria with “possible” ARVC. Although “possible” and “borderline” diagnoses may be justified for everyday clinical activity, particularly in case of limited access to CMR, their use should be discouraged for research studies.
In patients with biventricular ACM, the disease specificity of the LV phenotypic criteria is ensured by the concomitant fulfilment of the criteria for the RV phenotype. In these patients, the diagnosis of “biventricular” ACM can reasonably rely on morpho‐functional and/or structural abnormalities of both ventricles and classified as definite, borderline, or possible depending on the total number of fulfilled criteria for either the RV or LV phenotypes.
In patients with no RV abnormalities, the diagnosis of ALVC cannot be achieved on the basis of the LV phenotypic criteria only. In fact, morpho‐functional and structural LV abnormalities of ACM do not provide sufficient disease specificity because of the overlap with the phenotypic features of other heart muscle diseases such as DCM, myocarditis, or cardiac sarcoidosis. Hence, the diagnosis of ALVC requires, in addition to consistent LV phenotypic features, the demonstration of a positive genotyping for pathogenic or likely pathogenic ACM‐causing gene mutations.
Clinical Impact
To estimate the clinical impact of the 2020 International criteria, we applied the new scoring system “post hoc” to 112 patients diagnosed with ACM over the period 2015 to 2019 at the University of Padua; results for these patients were previously published by our group in the Journal of the American Heart Association. 29 All patients fulfilled the 2010 International Task Force criteria for definite (n=87), borderline (n=15), and possible (n=9) ARVC.
Of the 87 patients previously diagnosed with definite ARVC, 51 also fulfilled the new LV criteria, either morpho‐functional or structural, and were reclassified as biventricular ACM. Of 15 patients previously diagnosed with borderline ARVC, 5 were reclassified as definite ARVC because they met the RV LGE criterion and 6 as biventricular ACM because of evidence of LV LGE (Figure 3). Of 9 patients previously diagnosed with possible ARVC based on the detection of a pathogenic desmosomal‐gene mutation (ie, 4 Desmoplakin, 3 Filamin C, and 2 Desmoglein gene mutations) in the absence of RV morpho‐functional and/or structural abnormalities, 7 were reclassified as ALVC because they met the LV major structural (LV LGE) criterion.
Hence, the clinical impact of the use of the 2020 International criteria was the increase of diagnostic accuracy for ACM and a better characterization of the disease phenotype.
Conclusions
The lack of specific diagnostic criteria for left‐sided variants of ACM has resulted in clinical under‐recognition of patients with phenotypes other than the original ARVC over the 4 decades since the disease discovery. The development of the 2020 International criteria based on the evolving clinical experience with the expanding spectrum of ACM phenotypes was needed to fill the diagnostic gap of the previous 1994 and 2010 TF guidelines and to provide a codification for future translational and clinical research. The 2020 International criteria aimed to improve the diagnosis of ACM by providing new criteria for diagnosis of biventricular and ALVC phenotypes, particularly by means of the incorporation of CMR tissue characterization findings as well as LV depolarization and repolarization ECG abnormalities and ventricular arrhythmias.
The 2020 International criteria are heavily dependent on CMR, which has become mandatory to characterize the ACM phenotype and to exclude other diagnoses. Preliminary data confirm that the clinical use of the 2020 International criteria substantially impacts the diagnostic accuracy and permits a comprehensive identification of the phenotypic variety of ACM, mostly by virtue of demonstration of RV and LV LGE/myocardial fibrosis by CMR.
The validity of this new diagnostic approach needs to be assessed by future studies on large patient populations. The routine application of the 2020 International criteria in real‐world clinical practice will be crucial for future refinement and correlation with therapeutic outcomes.
Disclosures
None.
For Disclosures, see page 15.
REFERENCES
- 1. Marcus FI, Fontaine GH, Guiraudon G, Frank R, Laurenceau JL, Malergue C, Grosgogeat Y. Right ventricular dysplasia: a report of 24 adult cases. Circulation. 1982;65:384–398. DOI: 10.1161/01.CIR.65.2.384. [DOI] [PubMed] [Google Scholar]
- 2. Thiene G, Nava A, Corrado D, Rossi L, Pennelli N. Right ventricular cardiomyopathy and sudden death in young people. N Engl J Med. 1988;318:129–133. DOI: 10.1056/NEJM198801213180301. [DOI] [PubMed] [Google Scholar]
- 3. Corrado D, Link MS, Calkins H. Arrhythmogenic right ventricular cardiomyopathy. N Engl J Med. 2017;376:61–72. DOI: 10.1056/NEJMra1509267. [DOI] [PubMed] [Google Scholar]
- 4. Corrado D, Basso C, Judge DP. Arrhythmogenic cardiomyopathy. Circ Res. 2017;121:784–802. DOI: 10.1161/CIRCRESAHA.117.309345. [DOI] [PubMed] [Google Scholar]
- 5. Corrado D, van Tintelen PJ, McKenna WJ, Hauer RNW, Anastastakis A, Asimaki A, Basso C, Bauce B, Brunckhorst C, Bucciarelli‐Ducci C, International Experts , et al. Arrhythmogenic right ventricular cardiomyopathy: evaluation of the current diagnostic criteria and differential diagnosis. Eur Heart J. 2020;41:1414–1429. DOI: 10.1093/eurheartj/ehz669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. McKenna WJ, Thiene G, Nava A, Fontaliran F, Blomstrom‐Lundqvist C, Fontaine G, Camerini F. Diagnosis of arrhythmogenic right ventricular dysplasia/cardiomyopathy. Task Force of the Working Group Myocardial and Pericardial Disease of the European Society of Cardiology and of the Scientific Council on Cardiomyopathies of the International Society and Federation of Cardiology. Br Heart J. 1994;71:215–218. DOI: 10.1136/hrt.71.3.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Marcus FI, McKenna WJ, Sherrill D, Basso C, Bauce B, Bluemke DA, Calkins H, Corrado D, Cox MGPJ, Daubert JP, et al. Diagnosis of arrhythmogenic right ventricular cardiomyopathy/dysplasia: proposed modification of the task force criteria. Circulation. 2010;121:1533–1541. DOI: 10.1161/CIRCULATIONAHA.108.840827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Corrado D, Perazzolo Marra M, Zorzi A, Beffagna G, Cipriani A, Lazzari MD, Migliore F, Pilichou K, Rampazzo A, Rigato I, et al. Diagnosis of arrhythmogenic cardiomyopathy: the Padua criteria. Int J Cardiol. 2020;319:106–114. DOI: 10.1016/j.ijcard.2020.06.005. [DOI] [PubMed] [Google Scholar]
- 9. Yoerger DM, Marcus F, Sherrill D, Calkins H, Towbin JA, Zareba W, Picard MH; Multidisciplinary Study of Right Ventricular Dysplasia Investigators . Echocardiographic findings in patients meeting task force criteria for arrhythmogenic right ventricular dysplasia: new insights from the multidisciplinary study of right ventricular dysplasia. J Am Coll Cardiol. 2005;45:860–865. DOI: 10.1016/j.jacc.2004.10.070. [DOI] [PubMed] [Google Scholar]
- 10. Ventura R, Steven D, Klemm HU, Lutomsky B, Mullerleile K, Rostock T, Servatius H, Risius T, Meinertz T, Kuck K‐H, et al. Decennial follow‐up in patients with recurrent tachycardia originating from the right ventricular outflow tract: electrophysiologic characteristics and response to treatment. Eur Heart J. 2007;28:2338–2345. DOI: 10.1093/eurheartj/ehm293. [DOI] [PubMed] [Google Scholar]
- 11. Basso C, Ronco F, Marcus F, Abudureheman A, Rizzo S, Frigo AC, Bauce B, Maddalena F, Nava A, Corrado D, et al. Quantitative assessment of endomyocardial biopsy in arrhythmogenic right ventricular cardiomyopathy/dysplasia: an in vitro validation of diagnostic criteria. Eur Heart J. 2008;29:2760–2771. DOI: 10.1093/eurheartj/ehn415. [DOI] [PubMed] [Google Scholar]
- 12. Platonov PG, Calkins H, Hauer RN, Corrado D, Svendsen JH, Wichter T, Biernacka EK, Saguner AM, Te Riele AS, Zareba W. High interobserver variability in the assessment of epsilon waves: implications for diagnosis of arrhythmogenic right ventricular cardiomyopathy/dysplasia. Heart Rhythm. 2016;13:208–216. DOI: 10.1016/j.hrthm.2015.08.031. [DOI] [PubMed] [Google Scholar]
- 13. Hamid MS, Norman M, Quraishi A, Firoozi S, Thaman R, Gimeno JR, Sachdev B, Rowland E, Elliott PM, McKenna WJ. Prospective evaluation of relatives for familial arrhythmogenic right ventricular cardiomyopathy/dysplasia reveals a need to broaden diagnostic criteria. J Am Coll Cardiol. 2002;40:1445–1450. DOI: 10.1016/S0735-1097(02)02307-0. [DOI] [PubMed] [Google Scholar]
- 14. Migliore F, Zorzi A, Michieli P, Perazzolo Marra M, Siciliano M, Rigato I, Bauce B, Basso C, Toazza D, Schiavon M, et al. Prevalence of cardiomyopathy in Italian asymptomatic children with electrocardiographic T‐wave inversion at preparticipation screening. Circulation. 2012;125:529–538. DOI: 10.1161/CIRCULATIONAHA.111.055673. [DOI] [PubMed] [Google Scholar]
- 15. Cox MGPJ, van der Smagt JJ, Wilde AAM, Wiesfeld ACP, Atsma DE, Nelen MR, Rodriguez L‐M, Loh P, Cramer MJ, Doevendans PA, et al. New ECG criteria in arrhythmogenic right ventricular dysplasia/cardiomyopathy. Circ Arrhythm Electrophysiol. 2009;2:524–530. DOI: 10.1161/CIRCEP.108.832519. [DOI] [PubMed] [Google Scholar]
- 16. Sen‐Chowdhry S, Syrris P, McKenna WJ. Genetics of right ventricular cardiomyopathy. J Cardiovasc Electrophysiol. 2005;16:927–935. DOI: 10.1111/j.1540-8167.2005.40842.x. [DOI] [PubMed] [Google Scholar]
- 17. Corrado D, Thiene G. Arrhythmogenic right ventricular cardiomyopathy/dysplasia: clinical impact of molecular genetic studies. Circulation. 2006;113:1634–1637. DOI: 10.1161/CIRCULATIONAHA.105.616490. [DOI] [PubMed] [Google Scholar]
- 18. Awad MM, Calkins H, Judge DP. Mechanisms of disease: molecular genetics of arrhythmogenic right ventricular dysplasia/cardiomyopathy. Nat Clin Pract Cardiovasc Med. 2008;5:258–267. DOI: 10.1038/ncpcardio1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sen‐Chowdhry S, Syrris P, Prasad SK, Hughes SE, Merrifield R, Ward D, Pennell DJ, McKenna WJ. Left‐dominant arrhythmogenic cardiomyopathy: an under‐recognized clinical entity. J Am Coll Cardiol. 2008;52:2175–2187. DOI: 10.1016/j.jacc.2008.09.019. [DOI] [PubMed] [Google Scholar]
- 20. Sen‐Chowdhry S, Syrris P, Ward D, Asimaki A, Sevdalis E, McKenna WJ. Clinical and genetic characterization of families with arrhythmogenic right ventricular dysplasia/cardiomyopathy provides novel insights into patterns of disease expression. Circulation. 2007;115:1710–1720. DOI: 10.1161/CIRCULATIONAHA.106.660241. [DOI] [PubMed] [Google Scholar]
- 21. Marra MP, Leoni L, Bauce B, Corbetti F, Zorzi A, Migliore F, Silvano M, Rigato I, Tona F, Tarantini G, et al. Imaging study of ventricular scar in arrhythmogenic right ventricular cardiomyopathy: comparison of 3D standard electroanatomical voltage mapping and contrast‐enhanced cardiac magnetic resonance. Circ Arrhythm Electrophysiol. 2012;5:91–100. DOI: 10.1161/CIRCEP.111.964635. [DOI] [PubMed] [Google Scholar]
- 22. Miles C, Finocchiaro G, Papadakis M, Gray B, Westaby J, Ensam B, Basu J, Parry‐Williams G, Papatheodorou E, Paterson C, et al. Sudden death and left ventricular involvement in arrhythmogenic cardiomyopathy. Circulation. 2019;139:1786–1797. DOI: 10.1161/CIRCULATIONAHA.118.037230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Segura‐Rodríguez D, Bermúdez‐Jiménez FJ, Carriel V, López‐Fernández S, González‐Molina M, Oyonarte Ramírez JM, Fernández‐Navarro L, García‐Roa MD, Cabrerizo EM, Durand‐Herrera D, et al. Myocardial fibrosis in arrhythmogenic cardiomyopathy: a genotype‐phenotype correlation study. Eur Heart J Cardiovasc Imaging. 2020;21:378–386. DOI: 10.1093/ehjci/jez277. [DOI] [PubMed] [Google Scholar]
- 24. Augusto JB, Eiros R, Nakou E, Moura‐Ferreira S, Treibel TA, Captur G, Akhtar MM, Protonotarios A, Gossios TD, Savvatis K, et al. Dilated cardiomyopathy and arrhythmogenic left ventricular cardiomyopathy: a comprehensive genotype‐imaging phenotype study. Eur Heart J Cardiovasc Imaging. 2020;21:326–336. DOI: 10.1093/ehjci/jez188. [DOI] [PubMed] [Google Scholar]
- 25. Hall CL, Akhtar MM, Sabater‐Molina M, Futema M, Asimaki A, Protonotarios A, Dalageorgou C, Pittman AM, Suarez MP, Aguilera B, et al. Filamin C variants are associatedwith a distinctive clinical and immunohistochemical arrhythmogenic cardiomyopathy phenotype. Int J Cardiol. 2020;307:101–108. DOI: 10.1016/j.ijcard.2019.09.048. [DOI] [PubMed] [Google Scholar]
- 26. Ortiz‐Genga MF, Cuenca S, Dal Ferro M, Zorio E, Salgado‐Aranda R, Climent V, Padrón‐Barthe L, Duro‐Aguado I, Jiménez‐Jáimez J, Hidalgo‐Olivares VM, et al. Truncating FLNC mutations are associated with high‐risk dilated and arrhythmogenic cardiomyopathies. J Am Coll Cardiol. 2016;68:2440–2451. DOI: 10.1016/j.jacc.2016.09.927. [DOI] [PubMed] [Google Scholar]
- 27. Groeneweg JA, van der Zwaag PA, Olde Nordkamp LRA, Bikker H, Jongbloed JDH, Jongbloed R, Wiesfeld ACP, Cox MGPJ, van der Heijden JF, Atsma DE, et al. Arrhythmogenic right ventricular dysplasia/cardiomyopathyaccording to revised 2010 task force criteria with inclusion of non‐desmosomalphospholamban mutation carriers. Am J Cardiol. 2013;112:1197–1206. DOI: 10.1016/j.amjcard.2013.06.017. [DOI] [PubMed] [Google Scholar]
- 28. Te Rijdt WP, Ten Sande JN, Gorter TM, van der Zwaag PA, van Rijsingen IA, Boekholdt SM, van Tintelen JP, van Haelst PL, Planken RN, de Boer RA, et al. Myocardial fibrosis as an early feature in phospholamban p.Arg14del mutation carriers: phenotypic insights from cardiovascular magnetic resonance imaging. Eur Heart J Cardiovasc Imaging. 2019;20:92–100. DOI: 10.1093/ehjci/jey047. [DOI] [PubMed] [Google Scholar]
- 29. Cipriani A, Bauce B, De Lazzari M, Rigato I, Bariani R, Meneghin S, Pilichou K, Motta R, Aliberti C, Thiene G, et al. Arrhythmogenic right ventricular cardiomyopathy: characterization of left ventricular phenotype and differential diagnosis with dilated cardiomyopathy. J Am Heart Assoc. 2020;9:e014628. DOI: 10.1161/JAHA.119.014628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Towbin JA, McKenna WJ, Abrams DJ, Ackerman MJ, Calkins H, Darrieux FCC, Daubert JP, de Chillou C, DePasquale EC, Desai MY, et al. 2019 HRS expert consensus statement on evaluation, risk stratification, and management of arrhythmogenic cardiomyopathy. Heart Rhythm. 2019;16:e373–e407. DOI: 10.1016/j.hrthm.2019.09.019. [DOI] [PubMed] [Google Scholar]
- 31. Beffagna G, Zorzi A, Pilichou K, Perazzolo Marra M, Rigato I, Corrado D, Migliore F, Rampazzo A, Bauce B, Basso C, et al. Arrhythmogenic cardiomyopathy. Eur Heart J. 2020;41:4457–4462. DOI: 10.1093/eurheartj/ehaa719. [DOI] [PubMed] [Google Scholar]
- 32. Perazzolo Marra M, Cipriani A, Rizzo S, De Lazzari M, De Gaspari M, Akrami N, Bariani R, Zorzi A, Migliore F, Rigato I, et al. Myocardial tissue characterization in arrhythmogenic cardiomyopathy: comparison between endomyocardial biopsy and cardiac magnetic resonance. JACC Cardiovasc Imaging. 2021;14: 1675–1678. DOI: 10.1016/j.jcmg.2021.02.015. [DOI] [PubMed] [Google Scholar]
- 33. Petersen SE, Khanji MY, Plein S, Lancellotti P, Bucciarelli‐Ducci C. European Association of Cardiovascular Imaging expert consensus paper: a comprehensive review of cardiovascular magnetic resonance normal values of cardiac chamber size and aortic root in adults and recommendations for grading severity. Eur Heart J Cardiovasc Imaging. 2019;20:1321–1331. DOI: 10.1093/ehjci/jez232. [DOI] [PubMed] [Google Scholar]
- 34. D’Ascenzi F, Anselmi F, Piu P, Fiorentini C, Carbone SF, Volterrani L, Focardi M, Bonifazi M, Mondillo S. Cardiac magnetic resonance normal reference values of biventricular size and function in male athlete’s heart. JACC Cardiovasc Imaging. 2019;12:1756–1765. DOI: 10.1016/j.jcmg.2018.09.021. [DOI] [PubMed] [Google Scholar]
- 35. Perazzolo Marra M, Rizzo S, Bauce B, De Lazzari M, Pilichou K, Corrado D, Thiene G, Iliceto S, Basso C. Arrhythmogenic right ventricular cardiomyopathy. Contribution of cardiac magnetic resonance imaging to the diagnosis. Herz. 2015;40:600–606. DOI: 10.1007/s00059-015-4228-0. [DOI] [PubMed] [Google Scholar]
- 36. Sen‐Chowdhry S, Prasad SK, Syrris P, Wage R, Ward D, Merrifield R, Smith GC, Firmin DN, Pennell DJ, McKenna WJ. Cardiovascular magnetic resonance in arrhythmogenic right ventricular cardiomyopathy revisited: comparison with task force criteria and genotype. J Am Coll Cardiol. 2006;48:2132–2140. DOI: 10.1016/j.jacc.2006.07.045. [DOI] [PubMed] [Google Scholar]
- 37. De Lazzari M, Zorzi A, Cipriani A, Susana A, Mastella G, Rizzo A, Rigato I, Bauce B, Giorgi B, Lacognata C, et al. Relationship between electrocardiographic findings and cardiac magnetic resonance phenotypes in arrhythmogenic cardiomyopathy. J Am Heart Assoc. 2018;7:e009855. DOI: 10.1161/JAHA.118.009855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Rastegar N, Burt JR, Corona‐Villalobos CP, Te Riele AS, James CA, Murray B, Calkins H, Tandri H, Bluemke DA, Zimmerman SL, et al. Cardiac MR findings and potential diagnostic pitfalls in patients evaluated for arrhythmogenic right ventricular cardiomyopathy. Radiographics. 2014;34:1553–1570. DOI: 10.1148/rg.346140194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Corrado D, Cipriani A, De Lazzari M, Perazzolo MM. Right ventricular dilatation in arrhythmogenic right ventricular cardiomyopathy: need for a revision of the 2010 International Task Force criteria. Eur Heart J. 2020;41:1452–1453. DOI: 10.1093/eurheartj/ehaa003. [DOI] [PubMed] [Google Scholar]
- 40. Pelliccia A, Caselli S, Sharma S, Basso C, Bax JJ, Corrado D, D’Andrea A, D’Ascenzi F, Di Paolo FM, Edvardsen T, et al. European Association of Preventive Cardiology (EAPC) and European Association of Cardiovascular Imaging (EACVI) joint position statement: recommendations for the indication and interpretation of cardiovascular imaging in the evaluation of the athlete's heart. Eur Heart J. 2018;39:1949–1969. DOI: 10.1093/eurheartj/ehx532. [DOI] [PubMed] [Google Scholar]
- 41. Tandri H, Saranathan M, Rodriguez ER, Martinez C, Bomma C, Nasir K, Rosen B, Lima JA, Calkins H, Bluemke DA. Noninvasive detection of myocardial fibrosis in arrhythmogenic right ventricular cardiomyopathy using delayed‐enhancement magnetic resonance imaging. J Am Coll Cardiol. 2005;45:98–103. DOI: 10.1016/j.jacc.2004.09.053. [DOI] [PubMed] [Google Scholar]
- 42. Hunold P, Wieneke H, Bruder O, Krueger U, Schlosser T, Erbel R, Barkhausen J. Late enhancement: a new feature in MRI of arrhythmogenic right ventricular cardiomyopathy? J Cardiovasc Magn Reson. 2005;7:649–655. DOI: 10.1081/JCMR-200065608. [DOI] [PubMed] [Google Scholar]
- 43. Pfluger HB, Phrommintikul A, Mariani JA, Cherayath JG, Taylor AJ. Utility of myocardial fibrosis and fatty infiltration detected by cardiac magnetic resonance imaging in the diagnosis of arrhythmogenic right ventricular dysplasia—a single centre experience. Heart Lung Circ. 2008;17:478–483. DOI: 10.1016/j.hlc.2008.03.085. [DOI] [PubMed] [Google Scholar]
- 44. Aquaro GD, Barison A, Todiere G, Grigoratos C, Ait Ali L, Di Bella G, Emdin M, Festa P. Usefulness of combined functional assessment by cardiac magnetic resonance and tissue characterization versus task force criteria for diagnosis of arrhythmogenic right ventricular cardiomyopathy. Am J Cardiol. 2016;118:1730–1736. DOI: 10.1016/j.amjcard.2016.08.056. [DOI] [PubMed] [Google Scholar]
- 45. Borgquist R, Haugaa KH, Gilljam T, Bundgaard H, Hansen J, Eschen O, Jensen HK, Holst AG, Edvardsen T, Svendsen JH, et al. The diagnostic performance of imaging methods in ARVC using the 2010 task force criteria. Eur Heart J Cardiovasc Imaging. 2014;15:1219–1225. DOI: 10.1093/ehjci/jeu109. [DOI] [PubMed] [Google Scholar]
- 46. Haugaa KH, Basso C, Badano LP, Bucciarelli‐Ducci C, Cardim N, Gaemperli O, Galderisi M, Habib G, Knuuti J, Lancellotti P, et al. Comprehensive multi‐modality imaging approach in arrhythmogenic cardiomyopathy‐an expert consensus document of the European Association of Cardiovascular Imaging. Eur Heart J Cardiovasc Imaging. 2017;18:237–253. DOI: 10.1093/ehjci/jew229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Novak J, Zorzi A, Castelletti S, Pantasis A, Rigato I, Corrado D, Mckenna W, Lambiase PD. Electrocardiographic differentiation of idiopathic right ventricular outflow tract ectopy from early arrhythmogenic right ventricular cardiomyopathy. Europace. 2017;19:622–628. DOI: 10.1093/europace/euw018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Corrado D, Drezner JA, D'Ascenzi F, Zorzi A. How to evaluate premature ventricular beats in the athlete: critical review and proposal of a diagnostic algorithm. Br J Sports Med. 2020;54:1142–1148. DOI: 10.1136/bjsports-2018-100529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Muser D, Santangeli P, Castro SA, Casado Arroyo R, Maeda S, Benhayon DA, Liuba I, Liang JJ, Sadek MM, Chahal A, et al. Risk stratification of patients with apparently idiopathic premature ventricular contractions. A multicenter international CMR registry. JACC Clin Electrophysiol. 2020;6:722–735. DOI: 10.1016/j.jacep.2019.10.015. [DOI] [PubMed] [Google Scholar]
- 50. Zorzi A, Rigato I, Pilichou K, Perazzolo Marra M, Migliore F, Mazzotti E, Gregori D, Thiene G, Daliento L, Iliceto S, et al. Phenotypic expression is a prerequisite for malignant arrhythmic events and sudden cardiac death in arrhythmogenic right ventricular cardiomyopathy. Europace. 2016;18:1086–1094. DOI: 10.1093/europace/euv205. [DOI] [PubMed] [Google Scholar]
- 51. Zorzi A, Perazzolo Marra M, Rigato I, De Lazzari M, Susana A, Niero A, Pilichou K, Migliore F, Rizzo S, Giorgi B, et al. Nonischemic left ventricular scar as a substrate of life‐threatening ventricular arrhythmias and sudden cardiac death in competitive athletes. Circ Arrhythm Electrophysiol. 2016;9:e004229. DOI: 10.1161/CIRCEP.116.004229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Marchlinski DF, Tschabrunn CM, Zado ES, Santangeli P, Marchlinski FE. Right bundle branch block ventricular tachycardia in arrhythmogenic right ventricular cardiomyopathy more commonly originates from the right ventricle: criteria for identifying chamber of origin. Heart Rhythm. 2021;18:163–171. DOI: 10.1016/j.hrthm.2020.08.016. [DOI] [PubMed] [Google Scholar]
- 53. Kapplinger JD, Landstrom AP, Salisbury BA, Callis TE, Pollevick GD, Tester DJ, Cox MGPJ, Bhuiyan Z, Bikker H, Wiesfeld ACP, et al. Distinguishing arrhythmogenic right ventricular cardiomyopathy/dysplasia‐associated mutations from background genetic noise. J Am Coll Cardiol. 2011;57:2317–2327. DOI: 10.1016/j.jacc.2010.12.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Andreasen C, Nielsen JB, Refsgaard L, Holst AG, Christensen AH, Andreasen L, Sajadieh A, Haunso S, Svendsen JH, Olesen MS. New population‐based exome data are questioning the pathogenicity of previously cardiomyopathy‐associated genetic variants. Eur J Hum Genet. 2013;21:918–928. DOI: 10.1038/ejhg.2012.283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Hoorntje ET, Te Rijdt WP, James CA, Pilichou K, Basso C, Judge DP, Bezzina CR, van Tintelen JP. Arrhythmogenic cardiomyopathy: pathology, genetics, and concepts in pathogenesis. Cardiovasc Res. 2017;113:1521–1531. DOI: 10.1093/cvr/cvx150. [DOI] [PubMed] [Google Scholar]