

Abstract
Background
Aldehyde dehydrogenase‐2 (ALDH2), a mitochondrial enzyme, detoxifies reactive aldehydes such as 4‐hydroxy‐2‐nonenal (4HNE). A highly prevalent E487K mutation in ALDH2 (ALDH2*2) in East Asian people with intrinsic low ALDH2 activity is implicated in diabetic complications. 4HNE‐induced cardiomyocyte dysfunction was studied in diabetic cardiac damage; however, coronary endothelial cell (CEC) injury in myocardial ischemia‐reperfusion injury (IRI) in diabetic mice has not been studied. Therefore, we hypothesize that the lack of ALDH2 activity exacerbates 4HNE‐induced CEC dysfunction which leads to cardiac damage in ALDH2*2 mutant diabetic mice subjected to myocardial IRI.
Methods and Results
Three weeks after diabetes mellitus (DM) induction, hearts were subjected to IRI either in vivo via left anterior descending artery occlusion and release or ex vivo IRI by using the Langendorff system. The cardiac performance was assessed by conscious echocardiography in mice or by inserting a balloon catheter in the left ventricle in the ex vivo model. Just 3 weeks of DM led to an increase in cardiac 4HNE protein adducts and, cardiac dysfunction, and a decrease in the number of CECs along with reduced myocardial ALDH2 activity in ALDH2*2 mutant diabetic mice compared with their wild‐type counterparts. Systemic pretreatment with Alda‐1 (10 mg/kg per day), an activator of both ALDH2 and ALDH2*2, led to a reduction in myocardial infarct size and dysfunction, and coronary perfusion pressure upon cardiac IRI by increasing CEC population and coronary arteriole opening.
Conclusions
Low ALDH2 activity exacerbates 4HNE‐mediated CEC injury and thereby cardiac dysfunction in diabetic mouse hearts subjected to IRI, which can be reversed by ALDH2 activation.
Keywords: aldehyde dehydrogenase‐2, cardiac dysfunction, coronary endothelial cells, diabetic heart, ischemia‐reperfusion injury, Langendorff system, 4‐hydroxy‐2‐nonenal
Subject Categories: Angiogenesis, Animal Models of Human Disease, Ischemia, Oxidant Stress, Pathophysiology
Nonstandard Abbreviations and Acronyms
- 4HNE
4‐hydroxy‐2‐nonenal
- ALDH2
aldehyde dehydrogenase‐2
- CECs
coronary endothelial cells
- DM
diabetes mellitus
- IR
ischemia‐reperfusion
- IRI
ischemia‐reperfusion injury
- WT
wild type
Clinical Perspective
What Is New?
We demonstrated that 4‐hydroxy‐2‐nonenal exacerbated coronary endothelial cell injury in the aldehyde dehydrogenase 2 allele diabetic heart and this damage was further enhanced during ischemia‐reperfusion injury.
Direct 4‐hydroxy‐2‐nonenal infusion to the myocardium decreased the number of open arterioles and increased the number of closed arterioles demonstrating the possible mechanism of how 4‐hydroxy‐2‐nonenal exerts its toxic effects in the diabetic myocardium.
Systemic treatment of aldehyde dehydrogenase 2 activator, Alda‐1, for just 1 day was sufficient to confer cardio protection in diabetic hearts that are subjected to ischemia‐reperfusion injury; this is the first report that shows the acute effects of Alda‐1 treatment on preventing ischemia‐reperfusion‐induced injury in diabetic hearts.
What Are the Clinical Implications?
Treatment with Alda‐1 like aldehyde dehydrogenase 2 activators or strategies that mitigate 4‐hydroxy‐2‐nonenal toxicity can be considered as treatment strategy of patients with diabetes mellitus (DM) and myocardial ischemic events, more so for patients with DM and the aldehyde dehydrogenase 2 allele mutation.
4‐hydroxy‐2‐nonenal (4HNE) is generated upon lipid peroxidation, which is increased during oxidative stress in the pathological state. 4HNE is a highly reactive aliphatic aldehyde that can form adducts with cellular macromolecules 1 , 2 and contributes to cytotoxicity. 3 , 4 , 5 , 6 , 7 , 8 , 9 Increased 4HNE adducts were found in the diabetic myocardium. 10 , 11 Earlier studies have pointed out that both ischemia and reperfusion (IR) contribute to the release of 4HNE. 12 , 13 Accelerated removal of 4HNE during IR injury (IRI) may protect the myocardium. 14 Aldehyde dehydrogenase (ALDH) is one of the primary enzyme systems that detoxify 4HNE. 15 Reduced ALDH2 activity has been implicated in cardiac pathologies including diabetic cardiac damage. 16 , 17 A single‐point mutation (E487K) in ALDH2, termed as ALDH2*2, leads to low intrinsic ALDH2 activity in ≈30% of East Asian people. 18 , 19 , 20 , 21 , 22 Several epidemiological studies in East Asian people indicate that ALDH2 is critical during cardiovascular diseases. Inactive ALDH2 genotype is associated with a higher incidence of myocardial infarction (MI), 23 , 24 angina, 25 and diabetes mellitus (DM) 26 and its complications. 27 , 28
The myocardial cell population consists of cardiomyocytes as well as noncardiomyocytes including coronary endothelial cells (CECs), fibroblasts, smooth muscle cells, nerve cells, and inflammatory cells. Among these cell types, earlier studies have focused on cardiomyocytes, the primary functional cells of the myocardium in the streptozotocin‐induced DM and myocardial IR‐induced heart failure, which implicates low ALDH2 activity mediated 4HNE‐induced cardiac pathogenesis. 17 Among the noncardiomyocytes, fibroblasts have also been studied. 29 However, in a variety of diabetic complications, microvascular angiopathy has been implicated as a culprit of pathogenesis but not in diabetic cardiac damage. Thus, in this study, we wanted to focus on the CECs in diabetic cardiac damage because CECs are the first cardiac cells to come in direct contact with blood as they form the inner lining of the blood vessels. Because of this direct contact, these cells get exposed to hyperglycemia, hyperinsulinemia, oxidative stress, and inflammatory mediators that are generated during DM. These abnormal metabolic milieus generate oxidative stress and inflammation in CECs. This oxidative stress should enhance 4HNE generation in the CECs in diabetic hearts subjected to IRI. Because we use ALDH2*2 mice that intrinsically have low ALDH2 activity, studying 4HNE‐induced cellular damage in CECs in diabetic hearts with IRI will provide new information to understand the pathogenesis for new treatment strategies. Thus, we hypothesized that 4HNE exacerbates CEC injury in ALDH2*2 diabetic hearts and this damage is further enhanced during IRI.
We induced myocardial IRI in the ALDH2*2 mutant diabetic mouse hearts via ex vivo and in vivo approaches to mimic the clinical scenarios where patients with DM experience myocardial ischemic diseases, such as MI and angina. Alda‐1, an agonist of ALDH2 was shown to protect the myocardium against acute IRI. 14 Most important, Alda‐1 can activate ALDH2*2 30 by widening the catalytic tunnel of mutant ALDH2 by acting as a chemical chaperone. We treated the diabetic ALDH2*2 mice systemically with Alda‐1 (10 mg/kg per day) acutely for 1 day before subjecting them to IRI ex vivo, by using the Langendorff perfusion system for the first time, as opposed to intramyocardial acute Alda‐1 treatments in vivo (5 minutes before coronary artery occlusion) to induce IR and ex vivo (20 minutes before global IR), as reported in a previous study. 14 We chose a 1‐day treatment because this is the shortest period that an osmotic minipump is available for sustained systemic delivery. First, we treated ALDH2*2 mice with 3 weeks of short‐term DM. We then determined if this 1‐day Alda‐1 treatment protects the myocardium from IRI if the DM duration is chronic, that is, 3 months. Because the only variable in these 2 protocols is the diabetic duration, we were able to identify whether activating ALDH2 systemically during DM in vivo can protect the heart from IRI. Next, we increased the Alda‐1 treatment period to 2 weeks with a 3‐month diabetic period. At the end of the protocols, we evaluated the beneficial effects of systemic Alda‐1 treatment to ALDH2*2 diabetic mouse hearts that were subjected to IRI. The Alda‐1 dose was chosen from previous studies. 29 , 31 Finally, to understand the direct role of 4HNE in inducing CEC injury, we perfused the control ALDH2*2 hearts with 50 μmol/L 4HNE (with or without Alda‐1 pretreatment) using Langendorff perfusion system. We assert that our data provide critical insights for treatment strategy of patients with DM and myocardial ischemic events, especially for patients with the ALDH2*2 mutation.
Methods
The authors declare that all supporting data are available within the article and its online supplementary files.
ALDH2*2 E487K Knock‐In Mutant Mice
ALDH2*2 E487K knock‐in mutant mice that mimic E487K mutation in East Asian people were developed by Mochly‐Rosen as reported earlier. 32 Briefly, ALDH2*2 knock‐in mutation was introduced as a single nucleotide substitution (G to C) within exon 12 of the ALDH2 genomic fragment corresponding to the position of human E487K mutation in mice with a C57/BL6 background by homologous recombination. This was transferred to embryonic stem cells and then positive embryonic stem cells were microinjected into C57/BL6 blastocytes and implanted into pseudo pregnant females. Thus, E487K knock‐in mutant mice were generated. We obtained the ALDH2*2 mice from Mochly‐Rosen group. The mice were bred and grown in the animal care facility at Henry Ford Health System and they were genotyped by Transnetyx Inc. (Cordova, TN). For this study, we used mice of either sex as DM affects both men and women. We used C57/BL6 wild type (WT) mice as controls. The animal protocols were approved by the Henry Ford Health System Institutional Animal Care and Use Committee. It adheres to the guiding principles of care and use of experimental animals per the National Institutes of Health guidelines. Henry Ford Hospital operates as an American Association for Accreditation of Laboratory Animal Care certified animal facility.
Experimental Protocols
Protocol 1: Determine If Streptozotocin‐Induced Type‐1 DM Leads to Cardiac Damage in ALDH2*2 Mutant Mice
We induced type 1 DM with a single intraperitoneal injection of streptozotocin at a dose of 150 mg/kg in WT and ALDH2*2 mice at the age of ≈3 months. After 3 weeks of hyperglycemia (>250 mg/dL), we measured their cardiac function using echocardiography and euthanized the mice to perform biochemical and histopathological analysis. We explained the methodologies in detail in Data S1.
Protocol 2: Evaluating Potentiation of High‐Glucose Induced Injury in Cultured Endothelial Cells Along With ALDH2 Inhibition
We pretreated mouse CEC line with 10 μmol/L disulfiram (DSF+), an inhibitor of ALDH2 or vehicle (DSF−) for 2 hours, and then subjected them to high‐glucose stress 33 mmol/L d‐glucose (HG) or 33 mmol/L D‐mannitol as an osmotic control (Ctrl.) for 24 hours. Earlier we reported that this dose of DSF inhibited ALDH2 activity and increased 4HNE adducts in cultured cells. 33 We described the methods for the following measurements that we performed on the cells in Data S1: ALDH2 activity, 4HNE protein adducts, mitochondrial reactive oxygen species, mitochondrial membrane potential (ΔΨm), and angiogenesis‐tube formation assay. We pretreated the CECs with either Alda‐1 (20 μmol/L), an activator of ALDH2, with DSF alone or HG+DSF.
Protocol 3: Induction of Ex Vivo Myocardial IRI in ALDH2*2 Mutant Diabetic Mice Hearts
We euthanized ALDH2*2 mutant diabetic mice after 3 to 4 weeks of DM induction and, excised and mounted the hearts on the cannula of the Langendorff perfusion apparatus (AD Instruments). Then we subjected them to 30 minutes ischemia and 120 minutes reperfusion protocol. After, we conducted an experimental analysis for injury (details in Data S1).
Protocol 4: Induction of In Vivo Myocardial IRI in ALDH2*2 Mutant Diabetic Mice
Under anesthesia, a midline cervical incision was created by using a surgical microscope to separate the skin, muscle, and tissue covering the trachea using a surgical microscope. An endotracheal tube was inserted and connected to a rodent ventilator. Under artificial ventilation (110 per minute, 17–18 cm H2O), left‐sided thoracotomy between the athird and the fourth rib was performed. Left anterior descending coronary artery (LAD) was identified between the pulmonary artery and the left auricle and the ligation of the LAD was performed. The mice were placed on a 37°C heat pad for 60 minutes for ischemia. Then the ligation was released for 2 hours, that is, reperfusion.
Protocol 5: Effect of Acute Treatment With ALDH2 Activator, Alda‐1 in ALDH2*2 Mutant Mice With Short‐Term and Long‐Term Type‐1 DM
We treated ALDH2*2 mutant diabetic mice after 3 weeks of DM induction (short term) and 3 months of DM induction (long term) with Alda‐1 (10 mg/kg per day) or vehicle (dimethyl sulfoxide) acutely for 1 day using Alzet minipumps. At the end of the treatment period, we euthanized the mice and subjected the hearts to ex vivo and in vivo IRI protocol as mentioned previously. Next, we evaluated the extent of cardioprotection against IRI by Alda‐1 by measuring infarct size, cardiac function, and coronary perfusion pressure (details in Data S1).
Protocol 6: Effect of Chronic Treatment With ALDH2 Activator, Alda‐1 in ALDH2*2 Mutant Mice With Long‐Term Type‐1 DM
We treated ALDH2*2 mutant diabetic mice after 3 months of DM induction (chronic) with Alda‐1 (10 mg/kg per day) or vehicle (dimethyl sulfoxide) chronically for 2 weeks using Alzet mini pumps. At the end of the treatment period, we euthanized the mice and subjected the hearts to IRI protocol and evaluated the extent of cardioprotection against IRI by Alda‐1 as mentioned.
Protocol 7: Effect of Acute Alda‐1 Treatment on Acute 4HNE Challenge Mediated CEC Injury, Ex Vivo
We challenged the isolated hearts of ALDH2*2 control mice (nondiabetic) with 4HNE (50 μmol/L or dimethyl sulfoxide for 30 minutes) in Langendorff apparatus. We chose 50 μmol/L of 4HNE; as per previous studies, this dose damaged microvascular endothelial cells 34 and CECs. 35 In another set of mice, before the 4HNE challenge, we treated the ALDH2*2 control mice with Alda‐1 acutely for 1 day using Alzet mini pumps. Finally, we measured coronary perfusion pressure, cardiac function, and endothelial cell damage, as mentioned earlier.
Statistical Analysis
Data are presented as mean±SEM. The means among groups were compared using 1‐way ANOVA. Student t test was used when we compared the cardiac function between 4HNE infusion with or without Alda‐1. At the least, P<0.05 was considered statistically significant.
Results
Type‐1 DM Exacerbates Cardiac Dysfunction and Fibrosis in ALDH2*2 Mice Compared With Their WT Counterparts
Injection of streptozotocin‐induced type 1 DM in both WT and ALDH2*2 mutant mice as compared with citrate buffer injected WT and ALDH2*2 mutant mice (Ctrl.) (Table). After 3 weeks of hyperglycemia, we found increased diastolic and systolic dysfunction in ALDH2*2 diabetic mice compared with WT diabetic mice by using conscious echocardiography (Figure 1A through 1G) and balloon catheter in Langendorff perfusion system (Figure S1). We also observed an increase in cardiac fibrosis in both ALDH2*2 mutant and WT diabetic mice compared with ALDH2*2 mutant and WT control mice (Figure 1H through 1L). This increase in cardiac fibrosis was higher in ALDH2*2 diabetic mice compared with WT diabetic mice (Figure 1H through 1L).
Table 1.
Change in Body Weight, Heart Weight, and Blood Glucose Levels
| Item | Unit | WT Ctrl. (n=5) | WT DM (n=6) | ALDH2*2 Ctrl. (n=6) | ALDH2*2 DM (n=6) |
|---|---|---|---|---|---|
| BW | g | 31.92±2.10 | 23.52±2.24* | 32.08±2.05 † | 22.20±1.68* , ‡ |
| HW | g | 0.14±0.01 | 0.13±0.01 | 0.14±0.01 | 0.14±0.02 |
| HW/BW | % | 0.45±0.01 | 0.54±0.07* | 0.44±0.02 † | 0.59±0.09* , ‡ |
| HW to tibia length | mg/mm | 6.37±0.37 | 5.58±0.35* | 6.35±0.68 † | 6.07±0.66 |
| Blood glucose | mg/dL | 168.00±22.00 | 411.67±107.81* | 167.00±31.62 † | 401.67±105.55* , ‡ |
ALDH2*2 indicates aldehyde dehydrogenase 2 mutant allele; BW, body weight; Ctrl., control; DM, diabetes mellitus; HW, heart weight; HW/BW, heart weight to body weight ratio; HW/TL, heart weight to tibia length; and WT, wild type.
P<0.05 vs WT Ctrl.
P<0.05 vs WT DM.
P<0.05 vs ALDH2*2 Ctrl.
Figure 1. ALDH2*2 mutant and wild type (WT) mice with type‐1 diabetes mellitus (DM) decreased cardiac function and increased cardiac fibrosis when compared with ALDH2*2 and WT control mice without DM.
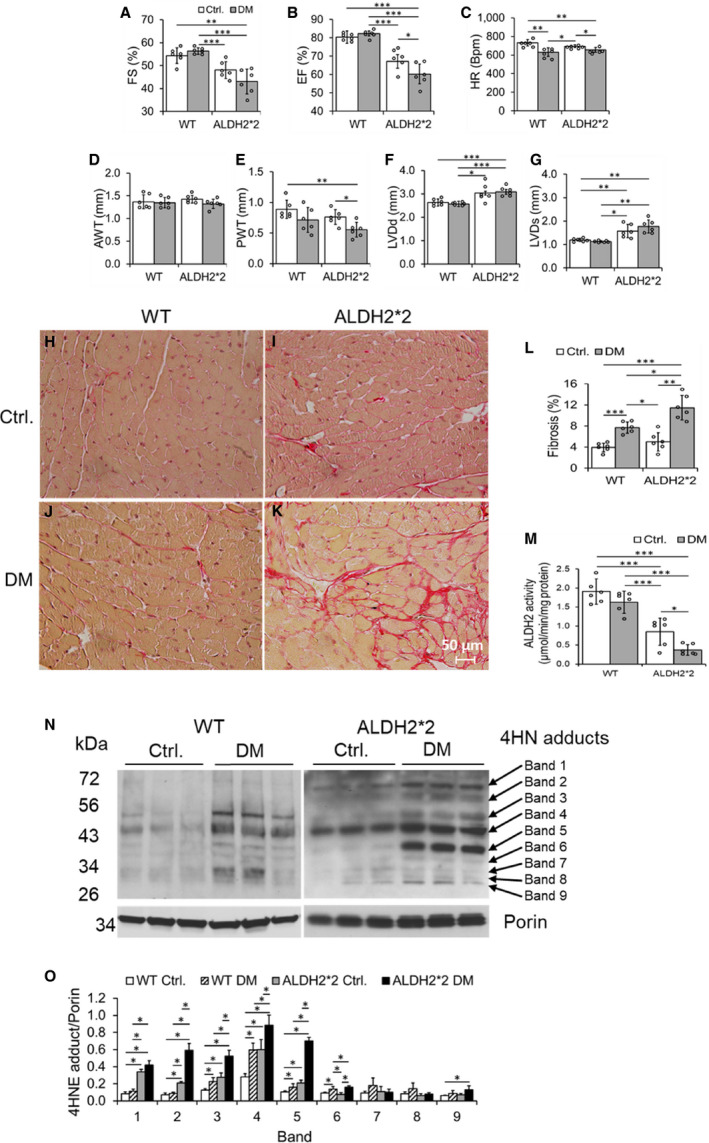
Fractional shortening (FS) % (A), ejection fraction (EF) % (B), heart rate (HR), (C), anterior wall thickness (AWT) (D), posterior wall thickness (PWT) (E), left ventricular end‐diastolic diameter (LVDd) (F), left ventricular end‐systolic diameter (LVDs) (G), microscopic images of cardiac fibrosis (H through K) and its quantification (L), ALDH2 activity (M), Western blots of 4‐hydroxy‐2‐nonenal (4HNE) adducts (N) and its quantification data (O) were shown from ALDH2*2 mutant diabetic mice and nondiabetic control ALDH2*2 mutant mice (Ctrl.) and respective WT mice. Data are presented as mean±SEM. *P<0.05, **P<0.01, and ***P<0.001. N=6. ALDH2*2 indicates aldehyde dehydrogenase 2 mutant allele; Ctrl., control.
Type‐1 DM Exacerbates the Decrease in ALDH2 Activity and Increase in 4HNE Adducts in ALDH2*2 Mice
Short‐term hyperglycemia of type 1 DM (ie, 3 weeks) significantly decreased myocardial ALDH2 activity in ALDH2*2 mutant mice compared with their nondiabetic controls (Figure 1M). However, the ALDH2 activity was slightly decreased in WT diabetic mice compared with their nondiabetic controls (Figure 1M). Furthermore, among the diabetic groups, the myocardial ALDH2 activity was significantly reduced in ALDH2*2 mutant diabetic mice compared with their WT counterparts (Figure 1M). As previously established in the literature, we noted that myocardial ALDH2 activity was significantly lower in nondiabetic ALDH2*2 mice (Figure 1M). On the other hand, type 1 DM significantly increased cardiac tissue 4HNE adduct levels in WT and ALDH2*2 mice compared with their nondiabetic controls (Figure 1N and 1O); This increase was higher in ALDH2*2 diabetic mice compared with WT diabetic mice (Figure 1N and 1O). Again, even in normal condition, cardiac 4HNE protein adducts were significantly higher in ALDH2*2 mutant mice compared with WT mice (Figure 1N and 1O). Several proteins showed increased 4HNE adductions as evident from increased intensity in the immunoblotting bands in both ALDH2*2 mutant and WT mice with DM compared with their respective nondiabetic controls (Figure 1N and 1O). The band intensities were higher in ALDH2*2 diabetic samples compared with WT diabetic samples (Figure 1N and 1O). The ALDH2 protein levels in CD31+ CECs were lower in diabetic hearts of both WT and ALDH2*2 mice compared with their nondiabetic counterparts (Figure S2). Also, it is noteworthy that ALDH2 levels were lower in nondiabetic ALDH2*2 mutant mice compared with nondiabetic WT mice (Figure S2). The decrease in ALDH2 levels in the tissues of ALDH2*2 mutant mice is due to low stability of ALDH2*2 protein as demonstrated by Jin et al. 36 These results suggest that DM and ALDH2*2 mutation contribute to the changes in cardiac ALDH2 activity and 4HNE adducts level. Furthermore ALDH2*2 mutations augments DM‐mediated decrease in ALDH2 activity and increase in 4HNE adducts level.
Type 1 DM Exacerbates a Decrease in the Number and Function of CECs in ALDH2*2 Mice
We found that type 1 DM decreased the number of CECs in the myocardium of WT slightly as compared with nondiabetic WT controls (Figure 2A through 2E). However, this decrease was significantly exacerbated in ALDH2*2 diabetic mice (Figure 2A through 2E). We also found a significant increase in the coronary perfusion pressure in ALDH2*2 diabetic mice compared with its controls (Figure 2F); Also, coronary perfusion pressure increase was significantly enhanced in ALDH2*2 diabetic mice compared with WT diabetic mice (Figure 2F). Next, we found a decrease in endothelial nitric‐oxide synthase (eNOS) in CD31+ CECs in diabetic heart tissue sections as compared with nondiabetic controls (Figure 2G through 2R) and this decrease was displayed higher in the hearts of ALDH2*2 diabetic mice compared with WT diabetic mice. We also found a decrease in eNOS protein levels (Figure 2S and 2T), eNOS phosphorylation at serine 1177 (Figure 2S and 2U), and the ratio of phospho eNOS and eNOS (Figure 2V) in the cardiac lysates of ALDH2*2 mutant diabetic mice as compared with WT diabetic mice. We inferred from these data that in diabetic cardiomyopathy, low ALDH2 activity contributes to the CEC loss, decreased coronary function, and reduced eNOS levels and their phosphorylation in CECs.
Figure 2. Decrease in coronary endothelial cell (CEC)s, endothelial nitric oxide (eNOS) and eNOS phosphorylation in type‐1 diabetic hearts when compared between ALDH2*2 mutant and WT control mice.

Representative micrographs of CD31+ immunostaining of CECs of cardiac sections (A through D) and the quantification of the number of CECs in cardiac sections (E) were shown from WT and ALDH2*2 control (Ctrl.) and diabetic mellitus (DM) mice. Quantification data of myocardial coronary perfusion pressure (F) were shown from WT and ALDH2*2 Ctrl. and DM mice. Data are presented as mean±SEM. Representative micrographs of double staining of CD31+ and endothelial nitric‐oxide synthase (eNOS) immunofluorescence staining in CECs of cardiac sections were shown from WT and ALDH2*2 Ctrl. and DM (G through R). The quantification data of CD31+ cells (S) and eNOS + cells (T) were shown. Representative Western blot images of eNOS, phospho eNOS (p‐eNOS), and porin levels (U) and eNOS quantification data (V), phospho eNOS S1177 (W), and the ratio of phospho eNOS S1177/eNOS (X) were shown. Data are presented as mean±SEM. *P<0.05, **P<0.01, and ***P<0.001. N=6. ALDH2*2 indicates aldehyde dehydrogenase 2 allele; and WT, wild type.
ALDH2 Inhibition Potentiates High Glucose‐Induced CEC Injury
ALDH2 inhibition by DSF+ augmented hHG stress induced decrease in ALDH2 activity (Figure S3A) and increase in 4HNE adduction (Figure S3B and S3C) in cultured CECs.
DSF+ augmented HG‐induced decrease in viability of CECs (Figure 3A through 3E) as compared with cells treated with HG with DSF− and control cells. Similarly, DSF+ enhanced HG mediated decrease in blood vessel tube formation (angiogenesis), a functional assay of CECs (Figure 3F through 3J). Pretreatment with DSF+ augmented HG induced an increase in mitochondrial membrane potential (Figure 3K through 3O) and mitochondrial reactive oxygen species (Figure 3P) in cultured CECs. We also found that the 4HNE was significantly colocalized with mitochondria (Figure S4). Once again, the immunostaining data also showed increased 4HNE adducts with HG stress, which was potentiated with ALDH2 inhibition via DSF+ (Figure S4).
Figure 3. High glucose decreases cell viability and angiogenesis (tube formation) and increases mitochondrial membrane potential and oxidative stress (mitosox) in cultured coronary endothelial cell (CEC) lines.
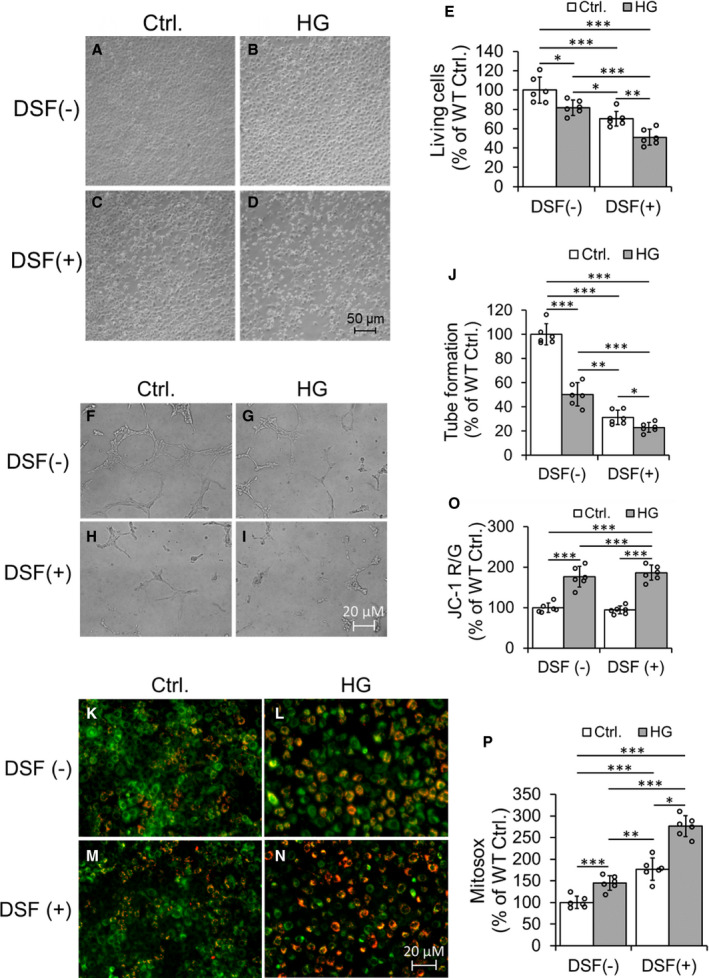
Representative Hoffman modulation‐contrast microscopic images of CECs were shown (A through D). The data quantification of viable cells was shown (E). Representative micrographs of tube formation of cultured CECs (F through I) and quantification of tube formation (J). Representative fluorescent images of JC‐1 staining of CECs (K through N) and quantification of the ratio of JC‐1 Red and JC‐1 Green (O) were shown. Data quantification of mitosox staining of cultured CECs (P). Data are presented as mean± SEM. *P<0.05, **P<0.01, and ***P<0.001. N=6. DSF indicates disulfiram; HG, high glucose; and WT, wild type.
Though HG alone did not decrease eNOS levels (ratio of dimer and monmer), phospho eNOS S1177 or increase phospho eNOS T495 levels (Figure S5A through S5E), it reduced the ratio of phospho eNOS S1177 and phospho eNOS T495 (Figure S5F). These changes were enhanced by ALDH2 inhibition with DSF either alone or combined with DSF (Figure S5A through S5F). Our in vitro studies established that low ALDH2 activity via DSF mediated ALDH2 inhibition potentiated HG stress‐mediated CEC death (decreased cell viability), whereas dysfunction (decrease in tube formation and S1177 phospho eNOS level as well as increase in phospho eNOS T495) and metabolic abnormality (increased mitochondrial membrane potential and mitochondrial reactive oxygen species) occurred perhaps via augmented formation of 4HNE adducts. The results of this hyperglycemic cell culture model were in accordance with our in vivo data. This reaffirmed our notion that low ALDH2 activity mediated increase in 4HNE may form adducts with eNOS and thus decrease its phosphorylation and levels, and thus it ultimately enhances CEC damage in diabetic conditions.
Acute Myocardial IRI Was Exacerbated in ALDH2*2 Diabetic Mice
Infarct size was increased in the hearts of both ALDH2*2 diabetic and WT diabetic mice that were subjected to IRI compared with ALDH2*2 diabetic and WT diabetic mouse hearts that were just subjected to sham operation. This increase in IRI‐induced infarction was higher in hearts of ALDH2*2 diabetic mice compared with the hearts of WT diabetic mice (Figure 4A and 4B). Cardiac functional deterioration (HR [Figure 4C], LVP [Figure 4D], +dP/dt [Figure 4E] and −dP/dt [Figure 4F]) was also observed to be significantly higher in ALDH2*2 diabetic mice compared with WT diabetic mice when measured using the balloon catheter. It is noteworthy to mention that LVP (Figure 4D) and +dP/dt (Figure 4E) were significantly decreased in ALDH2*2 diabetic mice compared with the hearts of WT diabetic mice. Myocardial perfusion pressure was significantly increased in ALDH2*2 diabetic mice compared with their non‐diabetic controls, and it was also significantly increased in ALDH2*2 diabetic mice compared with WT diabetic mice (Figure 4G). These IRI studies confirmed that ALDH2*2 diabetic hearts are prone to IRI compared with WT mice, which indorse the salutary role of ALDH2 activity in IRI in the diabetic myocardium. This notion was again reaffirmed as sham‐operated ALDH2*2 mice show significantly jeopardized cardiac function compared with their WT counterparts.
Figure 4. Type 1 DM increases myocardial ischemia‐reperfusion injury in ALDH2*2 mutant diabetic mice as compared with WT control mice.
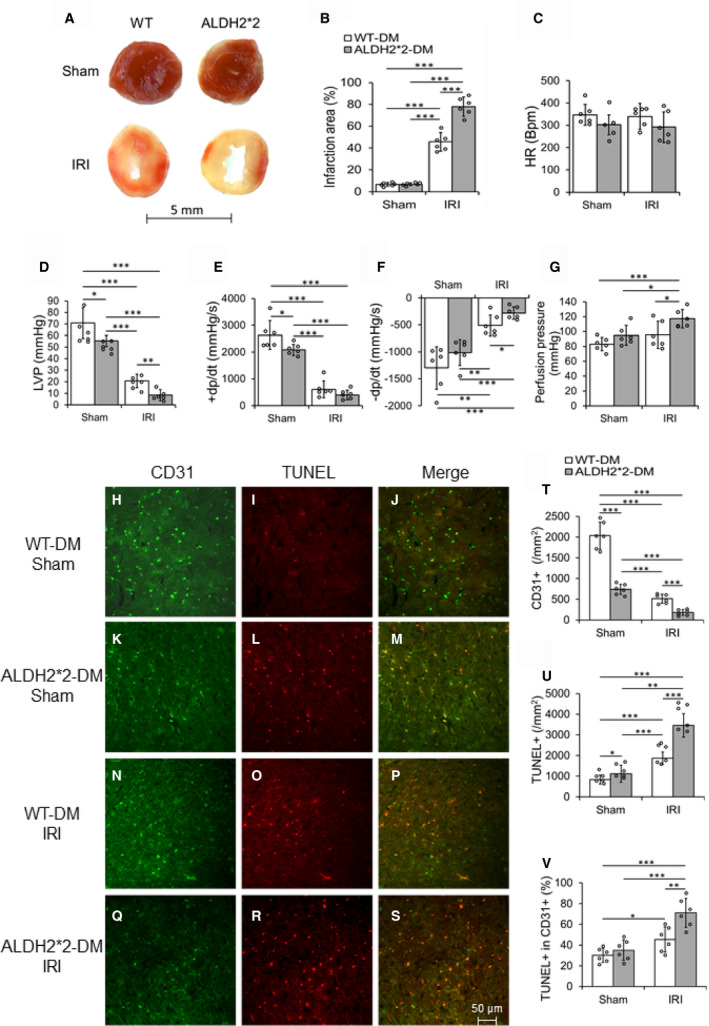
Representative macroscopic cardiac sections with 2,3,5‐triphenyltetrazolium chloride (TTC) staining (A) and its quantification data (B), cardiac functional parameters heart rate (C), Left ventricular pressure (LVP) (D), rate of rise in left ventricular pressure (+dP/dt) (E) and rate of decline in left ventricular pressure (–dP/dt) (F), and quantification data of perfusion pressure (G) were shown in WT and ALDH2*2 diabetic mice with sham operation (Sham) or ischemia‐reperfusion injury (IRI). Representative micrographs of double staining of CD31 and terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) immunofluorescence staining in CECs of cardiac sections (H through S) and the quantification data of staining of CD31 (T), TUNEL (U) and double positive of CD31 and TUNEL (V) were shown in WT and ALDH2*2 diabetic mice with sham operation (Sham) or ischemia reperfusion injury (IRI). Data are presented as mean±SEM. *P<0.05, **P<0.01, and ***P<0.001. N=6. ALDH2*2 indicates aldehyde dehydrogenase 2 mutant allele; DM, diabetes mellitus; and WT, wild type.
Acute Myocardial IRI Worsens CEC Apoptosis in the Myocardium of ALDH2*2 Diabetic Mice
CD31+ CECs were decreased in the myocardium of sham‐operated ALDH2*2 diabetic mice as compared with sham operated WT diabetic mice (Figure 4H, 4K, and 4T). This decrease in CD31+ CECs were further enhanced in the myocardium of ALDH2*2 diabetic mice after IRI as compared with WT diabetic mice with IRI (Figure 4Q, 4N, and 4T). There were significant decreases in CD31+ cells in both ALDH2*2 diabetic and WT diabetic mice with IRI compared with their respective sham‐operated controls (Figure 4T). Apoptotic death of CECs (as evident from increased TUNEL staining on CD31+ CECs [Figure 4I, 4J, 4L, 4M, 4U, and 4V] as well as increased levels of cleaved caspase 3, an apoptotic index in VEGFR2+ CECs of the myocardium [Figure S6]) was found in the myocardium of sham operated ALDH2*2 diabetic mice compared with sham operated WT diabetic mice. Additionally, IRI increased the CEC apoptosis in WT diabetic and ALDH2*2 diabetic mice compared with their sham‐operated counter parts (Figure 4H through 4V and Figure S6). The salient observation here is that this apoptotic death in CECs of the myocardium was further enhanced in ALDH2*2 diabetic mice after IRI as compared with WT diabetic mice with IRI (Figure 4R, 4O, 4S, and 4P as well as Figure 4U and 4V and Figure S6). Our microscopic studies suggest that DM‐induced low ALDH2 activity and ALDH2*2 mutations, which have low ALDH2 activity makes the diabetic heart more susceptible to IRI‐induced CEC apoptosis.
One‐Day Alda‐1 Treatment Protects Diabetic Heart With Short‐Term Hyperglycemia From IR‐Mediated Injury in ALDH2*2 Diabetic Mice in Both the Ex Vivo Langendorff Heart Model and In Vivo Model
One‐day Alda‐1 treatment decreased the IRI‐mediated myocardial infarct size in hearts subjected to IRI via Langendorff apparatus ex vivo (Figure 5A and 5B) and LAD occlusion in vivo (Figure S7A and S7B) of ALDH2*2 mice with short‐term DM (3 weeks of DM). One‐day Alda‐1 treatment improved cardiac function (Figure 5C through 5F) and decreased the myocardial perfusion pressure (Figure 5G) in ALDH2*2 mice with short‐term DM. Alda‐1 decreased apoptotic death in CECs (Figure 5H through 5P and 5R) and increased the CEC density (Figure 5J and 5Q) in ALDH2*2 diabetic mice.
Figure 5. Alda‐1, an ALDH2 activator protects against ischemia‐reperfusion injury in ALDH2*2 diabetic mice.
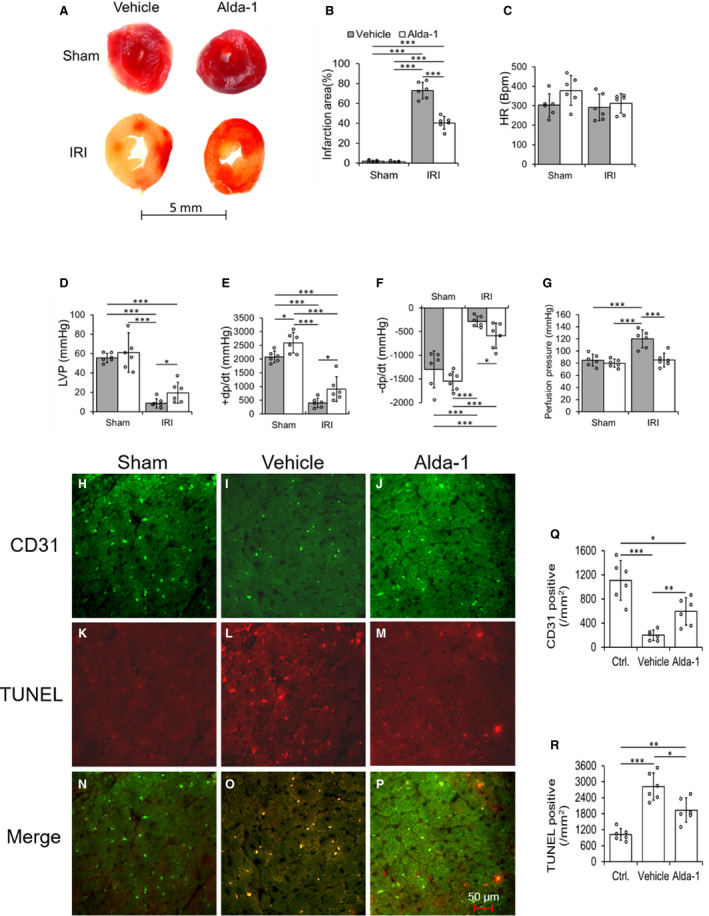
Representative macroscopic cardiac sections with 2,3,5‐triphenyltetrazolium chloride (TTC) staining (A) and its quantification data (B), cardiac functional parameters heart rate (C), left ventricular pressure (LVP) (D), rate of rise in left ventricular pressure (+dP/dt) (E) and rate of decline in left ventricular pressure (–dP/dt) (F), and quantification data of perfusion pressure (G) were shown in ALDH2*2 diabetic mice with 1‐day Vehicle (Vehicle) or Alda‐1 (Alda‐1) pretreatment, and hereafter sham operation (Sham) or ischemia reperfusion injury (IRI). Representative micrographs of double staining of terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) and CD31 immunofluorescence staining and the merging of both staining in CECs of cardiac sections (H through P) and the quantification data of staining of CD31 (Q) and TUNEL (R) in ALDH2*2 diabetic mice with Sham, or pretreated with Vehicle or Alda‐1 before IRI. Data are presented as mean±SEM. *P<0.05, **P<0.01, and ***P<0.001. N=6. ALDH2 indicates aldehyde dehydrogenase 2; ALDH2*2, ALDH2 mutant allele; CEC, coronary endothelial cell; and WT, wild type.
One‐day Alda‐1 treatment did not protect the hearts of ALDH2*2 mice with chronic DM from IRI (3 months of DM) (Figure S8A through S8D), however, chronic Alda‐1 treatment ie for 2 weeks, protected hearts of ALDH2*2 mice with chronic DM from IRI (Figure S8E through S8H). Finally, we found that Alda‐1 increased ALDH2 activity (Figure S9A) and decreased 4HNE adduct levels (Figure S9B and S9C) compared with vehicle, which could be the underlying mechanism for the observed beneficial effects of Alda‐1. These treatment strategies provided the critical information that we can increase ALDH2 activity in ALDH2*2 diabetic mice, which intrinsically have low ALDH2 activity that is further reduced by DM. However, the treatment duration is critical. If the duration of DM is longer, then we need longer ALDH2 activation. This information is very critical to translate such preclinical studies into clinic.
Alda‐1 Protected 4HNE Induced CEC Injury in the Heart of ALDH2*2 Mice
From above protocols, we drew a notion that ALDH2 activity is very important in diabetic hearts to attenuate damage after IR insult for decreasing 4HNE‐mediated tissue damage. Finally, we wanted to determine the direct role for 4HNE in inducing CEC injury in the myocardium. Therefore, we infused a pathological dose of 4HNE (50 µmol/L) which increased cardiac dysfunction acutely (Figure 6A through 6C) and perfusion pressure (Figure 6D) in the isolated hearts of control ALDH2*2 mice (having low ALDH2 activity will not allow for metabolism of 4HNE efficiently) compared with the basal value. Alda‐1 pretreatment attenuated 4HNE‐induced cardiac dysfunction (Figure 6A through 6C) and perfusion pressure (Figure 6D) in isolated hearts from non‐diabetic ALDH2*2 mice. 4HNE infusion deceased number of open arterioles and increased the number of closed arterioles in the myocardium of ALDH2*2 non‐diabetic mice compared with controls (Figure 6E through 6L). Alda‐1 pretreatment increased the number of open arterioles and decreased the number of closed coronary arterioles after 4HNE infusion in the hearts of ALDH2*2 non‐diabetic mice (Figure 6E through 6L).
Figure 6. Pretreatment of mice with systemically delivered Alda‐1 protects coronary endothelial cell (CEC)s from 4‐hydroxy‐2‐nonenal (4HNE) induced damage in control ALDH2*2 mice.
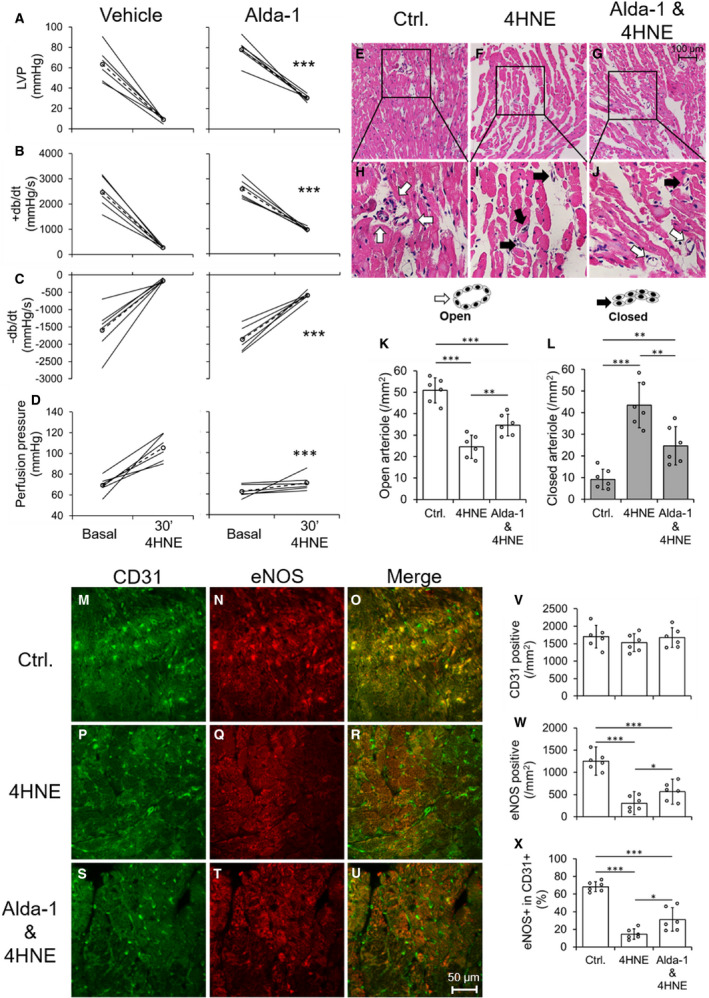
The quantification data of left ventricular pressure (LVP) (A), rate of rise in left ventricular pressure (+dP/dt) (B) rate of decline in left ventricular pressure (–dP/dt) (C) and perfusion pressure (D) were shown from 4HNE‐perfused hearts of nondiabetic ALDH2*2 mice with vehicle (Vehicle) or Alda‐1 pretreatment (Alda‐1). Representative micrographs of hematoxylin‐eosin (H‐E) stained cardiac sections were shown from 4HNE‐perfused hearts of nondiabetic ALDH2*2 mice with control (Ctrl or no 4HNE perfusion) (E), 4HNE perfusion (4HNE) (F) and Alda‐1 pretreatment before 4HNE perfusion (Alda‐1 & 4HNE) (G). The magnifications of the inset boxes were shown in (H through J) micrographs were in the interstitial region of the midventricle section. The cartoons of open and closed arterioles were shown. The quantification of data of open (K) and closed (L) coronary arterioles were shown from the interstitial region of the midventricle section. Representative micrographs of double staining of CD31+ and endothelial nitric‐oxide synthase (eNOS) immunofluorescence staining in CECs of cardiac sections were shown from Ctrl. (M through O), 4HNE (P through R) and Alda‐1 & 4HNE groups (S through U). The quantification of data of CD31 positive (V), eNOS positive (W) and the percentage eNOS positive in CD31 positive CECs (X). Data are presented as mean±SEM. *P<0.05, **P<0.01, and ***P<0.001. N=6. ALDH2*2 indicates aldehyde dehydrogenase 2 mutant allele.
CD31+ CEC density was not changed in the hearts of ALDH2*2 mice with 4HNE infusion and Alda‐1 treatment (Figure 6M, 6P, 6S, and 6V) as they are normal non‐diabetic mice. However, 4HNE infusion decreased eNOS levels in the hearts of ALDH2*2 control mice compared with untreated ALDH2*2 control mice while Alda‐1 pretreatment increased eNOS levels in the hearts of ALDH2*2 control mice infused with 4HNE (Figure 6M through 6X). Next, we found 4HNE infusion decreased eNOS+CD31+ CECs in the hearts of ALDH2*2 control mice compared with untreated controls which was increased by Alda‐1 in the hearts of ALDH2*2 control mice infused with 4HNE (Figure 6M through 6X). Alda‐1 attenuated DSF and DSF+HG mediated increases in 4HNE adducts in CECs by preventing ALDH2 impairment as well as it also improved eNOS signaling as it was evident from increases in eNOS, phospho eNOS S1177 and decrease in phospho eNOS T495.
This direct and acute infusion of 4HNE implicated that 4HNE can affect coronary arterioles and CECs directly and can lead to cardiac dysfunction when they have low ALDH2 activity. This 4HNE‐induced CEC damage and associated myocardial dysfunction can be reverted by activating ALDH2*2 with Alda‐1.
Discussion
In this study, we found cardiac dysfunction and fibrosis were enhanced significantly in ALDH2*2 mice compared with age‐matched WT mice after 3 weeks of type 1 DM. We found reduction in the population of CD31+ CECs in the hearts of ALDH2*2 diabetic mice compared with WT diabetic mice. Additionally, a significant decrease in phospho eNOS (ser 1177) and eNOS levels was observed in the myocardium, specifically in CD31+ CECs in ALDH2*2 diabetic mice as compared with WT diabetic mice. As a result of decrease in CECs and eNOS phosphorylation, there was an increase in coronary perfusion pressure, a measure of poor coronary perfusion, in ALDH2*2 diabetic mice. When we subjected the ALDH2*2 diabetic hearts to ex vivo and in vivo IRI, the myocardial infarct size was increased in ALDH2*2 diabetic mice as compared with WT diabetic mice. Cardiac dysfunction and coronary perfusion pressure were enhanced in ALDH2*2 diabetic mice as compared with WT diabetic mice when subjected to IRI. We also observed increased apoptosis in CD31+ CECs in the cardiac sections from ALDH2*2 diabetic mice subjected to IRI. This may be the cause of the loss of CD31+ CECs in the myocardium of ALDH2*2 diabetic mice subjected to IRI.
Since the myocardium of ALDH2*2 diabetic mice was damaged when subjected to IRI, we planned to treat the ALDH2*2 diabetic mice with Alda‐1 to increase the activity of ALDH2*2. Next, systemic Alda‐1 treatment for just 1‐day conferred cardio protection. This was done by increasing myocardial ALDH2 activity against global IRI (Langendorff ex vivo model) and local IRI (in vivo model via LAD occlusion) in the ALDH2*2 diabetic mouse heart with 3 weeks of DM.
Alda‐1 reduced % infarct size and improved cardiac function by decreasing DM‐mediated 4HNE induced CEC injury, which is evident from both reduced coronary perfusion pressure and cell death of CD31+ CECs in ALDH2*2 diabetic mice subjected to IRI. To further directly elucidate the mechanism, we directly infused 4HNE to the isolated hearts of normal control ALDH2*2 mice. As these mice have low ALDH2 activity this procedure satisfies the purpose perfectly. Exogenous 4HNE increased the number of arteriole closure and decreased the number of open arterioles compared with vehicle. Most important, this direct 4HNE infusion increased coronary perfusion pressure, implicating coronary endothelial cell dysfunction and ultimately resulted in cardiac dysfunction. To determine the ameliorative effect of Alda‐1 on direct 4HNE infusion‐mediated deleterious effects, we pretreated the ALDH2*2 mutant mice systemically with Alda‐1 for just 1 day. This Alda‐1 pretreatment protected coronary arterioles from closure and increased both the number of open arterioles and eNOS+ CD31+ CECs, and thereby improved cardiac function. To encapsulate, either with DM alone or subject the diabetic hearts to IRI, we found 4HNE mediated CEC injury was reduced by ALDH2 activation.
Acute MI‐induced deaths in patients with DM are 2 times higher than patients without DM. 37 The post‐MI prognosis is worse in patients with DM than in patients without. 38 , 39 There are multiple mechanisms proposed for acute myocardial IRI. 40 4HNE‐induced toxicity is one such mechanism. 4HNE protein adducts started to form in the heart at around 10 minutes into ischemia and subsequently peaked around 30 minutes. 12 Reperfusion of ischemic cardiac tissue increases 4HNE, which inactivates proteins by forming adducts on them and ultimately inhibiting mitochondrial respiration. 13 Along with this, mitochondrial metabolism of 4HNE and its extrusion from the heart are inhibited in myocardial ischemia, 41 suggesting enhanced accumulation of 4HNE in ischemic conditions. Thus, our study confirms the present concept of 4HNE‐mediated pathophysiology in IRI. Additionally, it provides a new mechanism ie 4HNE mediated CEC dysfunction in clinically relevant IRI models.
Langendorff ex vivo model of IRI mimics the situation of global IRI which occurs in patients having cardiac arrest and cardiopulmonary resuscitation. There are more than 350 000 cardiac arrests annually in the United States. 42 Employing Langendorff ex vivo model of IRI has such clinical relevance besides providing an experimental system devoid of systemic influence and studying the cardiac‐centric effects. Our results from this model are equally important as that of the in vivo IRI model of LAD occlusion. In vivo IRI model via LAD occlusion creates acute MI in a focal area which mimics common clinal MI. Roughly 600, 000 acute MI cases are reported each year in the United States. 42 Our study found that Alda‐1 had the protective effects against IRI in both Langendorff heart and acute MI model, which suggests the potential value of Alda‐1 in clinic.
Diabetes mellitus leads to microvascular complications including retinopathy, nephropathy, neuropathy, stroke, atherosclerosis, and cardiomyopathy. Increased oxidative stress by DM impairs endothelial cell function and is thought to mediate microvascular angiopathy. Endothelial function is usually conducted via eNOS mediated nitric oxide (NO) production and/or its bioavailability. 43 Hyperglycemia and DM lead to an impairment of eNOS activity and impaired NO production and its activity, thus resulting in endothelial cell damage, dysfunction and death. 44 ALDH2*2 contributes to the decrease in the efficacy of sublingual nitroglycerin which implied the role of ALDH2 in NO biotransformation. 45 We also found reduced phosphorylation of Serine 1177 in eNOS in low ALDH2 activity with hyperglycemic stress in vivo and in vitro where 4HNE adduct levels were higher. Thus, we presume 4HNE adduction on eNOS may have prevented its phosphorylation.
In this study, we found that ALDH2*2 mutation exacerbated DM‐induced endothelial cell damage before IRI. Since healthy functional CECs plays a critical role in minimizing myocardial IRI, the increase in the impairment of CECs in ALDH2*2 diabetic mice might have aggravated the outcome of IRI in them. Our in vitro protocol using mouse CECs revealed that high‐glucose stress mediated mitochondrial stress, decreased eNOS function, worsened angiogenesis, and increased death, which were further augmented by ALDH2 inhibition via DSF treatment. These results implicate that ALDH2 mediated 4HNE detoxification is important for maintaining healthy and functional CECs. We also found mitochondrial 4HNE presence was increased with DSF during high‐glucose stress. Thus, we supposed that the 4HNE‐mediated CEC injury is critical in diabetic myocardium and loss of ALDH2 activity exacerbates it. This is evident from our ALDH2*2 mutant mice carrying low ALDH2 activity having higher CEC apoptosis in normal and diabetic conditions. In our recent cell culture studies using mouse CECs, we found 4HNE can directly contribute to decreased CEC cell migration 46 and angiogenesis 35 which were again exacerbated by DSF pretreatment.
In earlier studies, the deleterious effects of reduced ALDH2 activity and increased 4HNE adduct formation in myocardial ischemia or IRI has been demonstrated. 14 , 47 In a landmark study, Chen et al showed the correlation between decrease in infarct size and pharmacological agents that possibly activate ALDH2. 14 For the first time, they demonstrated that ALDH activation by Alda‐1 protected the heart from IRI in the ex vivo and in vivo models of MI. 14 In both models, it was shown that Alda‐1 was infused directly to the myocardium. Levi and colleagues have demonstrated that degranulation of cardiac mast cells by 4HNE induced ventricular arrhythmias during acute IR. 48 Further they described that perfusing Alda‐1 directly to the heart attenuated reperfusion‐induced arrhythmias. 48 In our study, strikingly systemic treatment of Alda‐1 just for 1‐day is sufficient to confer cardio protection in diabetic hearts that are subjected to IRI, which implies potential clinical applicability of Alda‐1. This is the first report which shows the effects of Alda‐1 treatment on preventing IR‐induced injury in diabetic hearts. The IRI in diabetic heart tissue was controversial. 49 We observed increased infarct size and poor cardiac function after IRI in diabetic hearts regardless of their genotypes, but compared with WT mice, ALDH2*2 mice exhibited more severe myocardial IRI.
We induced type‐1 DM in ALDH2*2 mutant mice which mimic East Asians with ALDH2*2 mutation anticipating that DM should further decrease ALDH2 activity in ALDH2*2 mice which were already having intrinsically low ALDH2 activity. In humans, patients with DM and the ALDH2*2 mutation had increased diastolic dysfunction compared with patients without DM and the mutation. 28 Our results showing protective effects with Alda‐1 treatment in ALDH2*2 diabetic hearts emphasize the potential of Alda‐1 as a therapeutic agent in diabetic ALDH2*2 carriers.
We found that 4HNE protein adducts levels in the cardiac homogenates were attenuated by Alda‐1 treatment in ALDH2*2 diabetic mice which implies that increased ALDH2 activity by Alda‐1 led to increased detoxification of 4HNE. This in turn decreased deleterious effects of 4HNE in CECs ie forming adducts with critical proteins like eNOS, ultimately conferred cardio protection. It is well known that increased 4HNE adduct formation in the diabetic heart was reported in multiple models of DM such as db/db mice, 50 streptozotocin‐induced type‐1 DM in rats 16 and mice, 17 , 51 high‐fat diet and low‐dose streptozotocin induced type‐2 DM, 10 and insulin resistance models. 52 It was reported that increased 4HNE adduct formation with the mitochondrial respiratory complex 2 protein, succinyl dehydrogenase was reported in the streptozotocin‐induced type‐1 diabetic heart tissue. 53 Using the same model, we also found that mitochondrial respiratory complex protein V subunit levels were decreased in diabetic heart. 11 However, in the current study, we investigated the mechanism of 4HNE induced CEC injury. We found for the first time that the direct infusion of 4HNE to the isolated hearts causes closure of coronary arterioles along with a decrease in eNOS levels in CECs, which might have led to the increased coronary perfusion pressure and myocardial dysfunction. When we pretreated ALDH2*2 mice with Alda‐1 before the 4HNE challenge, we found that it reduced closure of coronary arterioles and increased the number of open arterioles. Thus, Alda‐1 improved the coronary perfusion and cardiac function. We postulate that acute 4HNE infusion reduced eNOS levels by forming adducts with eNOS and then removing 4HNE adducted eNOS. The duration of acute 4HNE infusion is short time to reduce the CD31+ cells from the heart. However, this ex vivo 4HNE study suggested that perhaps decreasing eNOS due to 4HNE adduction eventually decreased the CD31+ endothelial cell density in hyperglycemic hearts. Our acute 4HNE infusion studies pointed out that all these acute changes by 4HNE led to microvascular endothelial cell damage in hyperglycemic mouse studies. Furthermore, the pretreatment with Alda‐1 reversed those deleterious effects of acute 4HNE infusion suggesting that perhaps Alda‐1 prevented IR‐induced cardiac injury and cardiac dysfunction in type‐1 DM by protecting the CECs and coronary arterioles from DM/IRI induced 4HNE mediated toxicity.
Limitation of the Study
Even though we provided a significant amount of evidence for ALDH2 activation by Alda‐1 mediating beneficial effects through decreasing 4HNE‐mediated deleterious effects, there are multiple other reactive aldehydes such as malondialdehyde (MDA) 54 and acrolein 55 that also can contribute to myocardial injuries. In fact, we found MDA and acrolein protein adducts were increased in the hearts of ALDH2*2 mice with DM compared with controls which were slightly attenuated by Alda‐1 treatment (Figures S10 and S11). Nonetheless, these data also underscore that ALDH2 activity should be important in alleviating tissue damage by decreasing the deleterious effects of reactive aldehydes. Next, it is imperative that 4HNE toxicity as well as ALDH2 activity are critical in all cardiac cell types, not only CECs. However, we focused on CEC dysfunction in this study because there is limited information on ALDH2/4HNE roles in CECs in diabetic cardiac damage. Moreover, our intention was not to implicate CEC injury alone as a cellular mechanism by which IRI injury occurs in the diabetic myocardium, however the importance of CEC damage in this pathogenesis. Because, CECs are the first cardiac cells to encounter diabetic milieu from the systemic circulation, and that pathogenesis starts with CECs and the sequence of events follows to the entire myocardial tissue ie oxidative stress‐inflammation‐cardiac fibrosis‐stiffness and finally cardiac dysfunction. This is further exacerbated by IRI‐mediated pathological stimuli. We presume CEC injury is the gatekeeper of the myocardial damage in DM as well as IRI. Further studies are required to dissect how CEC injury contributes to the following pathogenic events in diabetic heart damage with IRI through 4HNE. Finally, identification of 4HNE adducted proteins from both in vitro and in vivo studies should provide potential cell signaling pathways that contribute to the pathophysiology of diabetic cardiac damage in IRI. We propose this could be our future direction.
Conclusions
Finally, we want to conclude that acute systemic treatment with Alda‐1 even just for 1 day is sufficient to protect the myocardium from IRI in ALDH2*2 diabetic mice perhaps via protecting the CECs from 4HNE‐mediated damage. Our data provides initial evidence to develop pharmacological activators of ALDH2 as a therapeutic option for IR‐induced myocardial injury in DM.
Sources of Funding
Palaniyandi was supported by grants from the National Heart, Lung, and Blood Institute: 1R56HL131891‐01A1 and 1R01HL139877‐01A1, the National Institute of Diabetes and Digestive and Kidney Diseases: 30DK020572 (MDRC); American Heart Association (AHA)‐Scientist Development Grant 14SDG20050030; Henry Ford Health System‐Internal grant from (Palaniyandi) A10249, and Pan was supported by AHA Postdoctoral Fellowship 17POST33680126.
Disclosures
None.
Supporting information
Acknowledgments
Pan, conducted experimental protocols and assays, analyzed the data, made the figures and contributed to the writing; Roy, contributed to the experimentation, and Palaniyandi, Conceived the study and wrote and edited the article. Palaniyandi is the guarantor of this work and, as such, had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
For Sources of Funding and Disclosures, see page 16.
References
- 1. Roede JR, Jones DP. Reactive species and mitochondrial dysfunction: mechanistic significance of 4‐hydroxynonenal. Environ Mol Mutagen. 2010;51:380–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Esterbauer H, Schaur RJ, Zollner H. Chemistry and biochemistry of 4‐hydroxynonenal, malonaldehyde and related aldehydes. Free Radic Biol Med. 1991;11:81–128. doi: 10.1016/0891-5849(91)90192-6 [DOI] [PubMed] [Google Scholar]
- 3. Bulteau AL, Lundberg KC, Humphries KM, Sadek HA, Szweda PA, Friguet B, Szweda LI. Oxidative modification and inactivation of the proteasome during coronary occlusion/reperfusion. J Biol Chem. 2001;276:30057–30063. doi: 10.1074/jbc.M100142200 [DOI] [PubMed] [Google Scholar]
- 4. Ferrington DA, Kapphahn RJ. Catalytic site‐specific inhibition of the 20s proteasome by 4‐hydroxynonenal. FEBS Lett. 2004;578:217–223. doi: 10.1016/j.febslet.2004.11.003 [DOI] [PubMed] [Google Scholar]
- 5. Farout L, Mary J, Vinh J, Szweda LI, Friguet B. Inactivation of the proteasome by 4‐hydroxy‐2‐nonenal is site specific and dependant on 20s proteasome subtypes. Arch Biochem Biophys. 2006;453:135–142. doi: 10.1016/j.abb.2006.02.003 [DOI] [PubMed] [Google Scholar]
- 6. Akude E, Zherebitskaya E, Roy Chowdhury SK, Girling K, Fernyhough P. 4‐hydroxy‐2‐nonenal induces mitochondrial dysfunction and aberrant axonal outgrowth in adult sensory neurons that mimics features of diabetic neuropathy. Neurotox Res. 2010;17:28–38. doi: 10.1007/s12640-009-9074-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kaplan P, Tatarkova Z, Racay P, Lehotsky J, Pavlikova M, Dobrota D. Oxidative modifications of cardiac mitochondria and inhibition of cytochrome c oxidase activity by 4‐hydroxynonenal. Redox Rep. 2007;12:211–218. [DOI] [PubMed] [Google Scholar]
- 8. Keller JN, Mark RJ, Bruce AJ, Blanc E, Rothstein JD, Uchida K, Waeg G, Mattson MP. 4‐hydroxynonenal, an aldehydic product of membrane lipid peroxidation, impairs glutamate transport and mitochondrial function in synaptosomes. Neuroscience. 1997;80:685–696. doi: 10.1016/S0306-4522(97)00065-1 [DOI] [PubMed] [Google Scholar]
- 9. Keith RJ, Haberzettl P, Vladykovskaya E, Hill BG, Kaiserova K, Srivastava S, Barski O, Bhatnagar A. Aldose reductase decreases endoplasmic reticulum stress in ischemic hearts. Chem Biol Interact. 2009;178:242–249. doi: 10.1016/j.cbi.2008.10.055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Mali VR, Ning R, Chen J, Yang XP, Xu J, Palaniyandi SS. Impairment of aldehyde dehydrogenase‐2 by 4‐hydroxy‐2‐nonenal adduct formation and cardiomyocyte hypertrophy in mice fed a high‐fat diet and injected with low‐dose streptozotocin. Exp Biol Med (Maywood). 2014;239:610–618. doi: 10.1177/1535370213520109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Mali VR, Pan G, Deshpande M, Thandavarayan RA, Xu J, Yang XP, Palaniyandi SS. Cardiac mitochondrial respiratory dysfunction and tissue damage in chronic hyperglycemia correlate with reduced aldehyde dehydrogenase‐2 activity. PLoS One. 2016;11:e0163158. doi: 10.1371/journal.pone.0163158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Eaton P, Li JM, Hearse DJ, Shattock MJ. Formation of 4‐hydroxy‐2‐nonenal‐modified proteins in ischemic rat heart. Am J Physiol. 1999;276:H935–H943. doi: 10.1152/ajpheart.1999.276.3.H935 [DOI] [PubMed] [Google Scholar]
- 13. Lucas DT, Szweda LI. Declines in mitochondrial respiration during cardiac reperfusion: age‐dependent inactivation of alpha‐ketoglutarate dehydrogenase. Proc Natl Acad Sci USA. 1999;96:6689–6693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chen CH, Budas GR, Churchill EN, Disatnik MH, Hurley TD, Mochly‐Rosen D. Activation of aldehyde dehydrogenase‐2 reduces ischemic damage to the heart. Science. 2008;321:1493–1495. doi: 10.1126/science.1158554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Vasiliou V, Pappa A, Petersen DR. Role of aldehyde dehydrogenases in endogenous and xenobiotic metabolism. Chem Biol Interact. 2000;129:1–19. doi: 10.1016/S0009-2797(00)00211-8 [DOI] [PubMed] [Google Scholar]
- 16. Wang J, Wang H, Hao P, Xue L, Wei S, Zhang Y, Chen Y. Inhibition of aldehyde dehydrogenase 2 by oxidative stress is associated with cardiac dysfunction in diabetic rats. Mol Med. 2011;17:172–179. doi: 10.2119/molmed.2010.00114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zhang Y, Babcock SA, Hu N, Maris JR, Wang H, Ren J. Mitochondrial aldehyde dehydrogenase (ALDH2) protects against streptozotocin‐induced diabetic cardiomyopathy: role of GSK3beta and mitochondrial function. BMC Med. 2012;10:40. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 18. Goedde HW, Agarwal DP, Fritze G, Meier‐Tackmann D, Singh S, Beckmann G, Bhatia K, Chen LZ, Fang B, Lisker R, et al. Distribution of ADH2 and ALDH2 genotypes in different populations. Hum Genet. 1992;88:344–346. doi: 10.1007/BF00197271 [DOI] [PubMed] [Google Scholar]
- 19. Luo HR, Wu GS, Pakstis AJ, Tong L, Oota H, Kidd KK, Zhang YP. Origin and dispersal of atypical aldehyde dehydrogenase ALDH2487Lys. Gene. 2009;435:96–103. doi: 10.1016/j.gene.2008.12.021 [DOI] [PubMed] [Google Scholar]
- 20. Harada S, Agarwal DP, Goedde HW, Ishikawa B. Aldehyde dehydrogenase isozyme variation and alcoholism in Japan. Pharmacol Biochem Behav. 1983;18(suppl 1):151–153. doi: 10.1016/0091-3057(83)90163-6 [DOI] [PubMed] [Google Scholar]
- 21. Crabb DW, Edenberg HJ, Bosron WF, Li TK. Genotypes for aldehyde dehydrogenase deficiency and alcohol sensitivity. The inactive ALDH2(2) allele is dominant. J Clin Invest. 1989;83:314–316. doi: 10.1172/JCI113875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zhong Z, Hou J, Li B, Zhang Q, Li C, Liu Z, Yang M, Zhong W, Zhao P. Genetic polymorphisms of the mitochondrial aldehyde dehydrogenase ALDH2 gene in a large ethnic Hakka population in southern China. Med Sci Monit. 2018;24:2038–2044. doi: 10.12659/MSM.906606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Jo SA, Kim EK, Park MH, Han C, Park HY, Jang Y, Song BJ, Jo I. A Glu487Lys polymorphism in the gene for mitochondrial aldehyde dehydrogenase 2 is associated with myocardial infarction in elderly Korean men. Clin Chim Acta. 2007;382:43–47. doi: 10.1016/j.cca.2007.03.016 [DOI] [PubMed] [Google Scholar]
- 24. Takagi S, Iwai N, Yamauchi R, Kojima S, Yasuno S, Baba T, Terashima M, Tsutsumi Y, Suzuki S, Morii I, et al. Aldehyde dehydrogenase 2 gene is a risk factor for myocardial infarction in Japanese men. Hypertens Res. 2002;25:677–681. doi: 10.1291/hypres.25.677 [DOI] [PubMed] [Google Scholar]
- 25. Furfaro AL, Menini S, Patriarca S, Pesce C, Odetti P, Cottalasso D, Marinari UM, Pronzato MA, Traverso N. HNE‐dependent molecular damage in diabetic nephropathy and its possible prevention by N‐acetyl‐cysteine and oxerutin. Biofactors. 2005;24:291–298. doi: 10.1002/biof.5520240134 [DOI] [PubMed] [Google Scholar]
- 26. Murata C, Taniyama M, Kuriyama S, Muramatsu T, Atsumi Y, Matsuoka K, Suzuki Y. Meta‐analysis of three diabetes population studies: association of inactive ALDH2 genotype with maternal inheritance of diabetes. Diabetes Res Clin Pract. 2004;66(suppl 1):S145–S147. doi: 10.1016/j.diabres.2003.11.022 [DOI] [PubMed] [Google Scholar]
- 27. Suzuki Y, Taniyama M, Muramatsu T, Higuchi S, Ohta S, Atsumi Y, Matsuoka K. ALDH2/ADH2 polymorphism associated with vasculopathy and neuropathy in type 2 diabetes. Alcohol Clin Exp Res. 2004;28:111S–116S. doi: 10.1111/j.1530-0277.2004.tb03227.x [DOI] [PubMed] [Google Scholar]
- 28. Wang C, Fan F, Cao Q, Shen C, Zhu H, Wang P, Zhao X, Sun X, Dong Z, Ma X, et al. Mitochondrial aldehyde dehydrogenase 2 deficiency aggravates energy metabolism disturbance and diastolic dysfunction in diabetic mice. J Mol Med (Berl). 2016;94:1229–1240. doi: 10.1007/s00109-016-1449-5 [DOI] [PubMed] [Google Scholar]
- 29. Gomes KMS, Campos JC, Bechara LRG, Queliconi B, Lima VM, Disatnik M‐H, Magno P, Chen C‐H, Brum PC, Kowaltowski AJ, et al. Aldehyde dehydrogenase 2 activation in heart failure restores mitochondrial function and improves ventricular function and remodelling. Cardiovasc Res. 2014;103:498–508. doi: 10.1093/cvr/cvu125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Perez‐Miller S, Younus H, Vanam R, Chen CH, Mochly‐Rosen D, Hurley TD. Alda‐1 is an agonist and chemical chaperone for the common human aldehyde dehydrogenase 2 variant. Nat Struct Mol Biol. 2010;17:159–164. doi: 10.1038/nsmb.1737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sun L, Ferreira JC, Mochly‐Rosen D. ALDH2 activator inhibits increased myocardial infarction injury by nitroglycerin tolerance. Sci Transl Med. 2011;3:107ra111. doi: 10.1126/scitranslmed.3002067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zambelli VO, Gross ER, Chen CH, Gutierrez VP, Cury Y, Mochly‐Rosen D. Aldehyde dehydrogenase‐2 regulates nociception in rodent models of acute inflammatory pain. Sci Transl Med. 2014;6:251ra118. doi: 10.1126/scitranslmed.3009539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Pan G, Deshpande M, Thandavarayan RA, Palaniyandi SS. ALDH2 inhibition potentiates high glucose stress‐induced injury in cultured cardiomyocytes. J Diabetes Res. 2016;2016:1390861. doi: 10.1155/2016/1390861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Usatyuk PV, Parinandi NL, Natarajan V. Redox regulation of 4‐hydroxy‐2‐nonenal‐mediated endothelial barrier dysfunction by focal adhesion, adherens, and tight junction proteins. J Biol Chem. 2006;281:35554–35566. doi: 10.1074/jbc.M607305200 [DOI] [PubMed] [Google Scholar]
- 35. Roy B, Palaniyandi SS. Aldehyde dehydrogenase 2 inhibition potentiates 4‐hydroxy‐2‐nonenal induced decrease in angiogenesis of coronary endothelial cells. Cell Biochem Funct. 2020;38:290–299. doi: 10.1002/cbf.3468 [DOI] [PubMed] [Google Scholar]
- 36. Jin S, Chen J, Chen L, Histen G, Lin Z, Gross S, Hixon J, Chen Y, Kung C, Chen Y, et al. ALDH2(E487K) mutation increases protein turnover and promotes murine hepatocarcinogenesis. Proc Natl Acad Sci USA. 2015;112:9088–9093. doi: 10.1073/pnas.1510757112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Diabetes: facts and figures. 2014. Available at: http://www.idf.org/worlddiabetesday/toolkit/gp/facts‐figures. Accessed September 17, 2019.
- 38. Gustafsson I, Hildebrandt P, Seibaek M, Melchior T, Torp‐Pedersen C, Kober L, Kaiser‐Nielsen P. Long‐term prognosis of diabetic patients with myocardial infarction: relation to antidiabetic treatment regimen. The TRACE Study Group. Eur Heart J. 2000;21:1937–1943. doi: 10.1053/euhj.2000.2244 [DOI] [PubMed] [Google Scholar]
- 39. Herlitz J, Malmberg K, Karlson BW, Ryden L, Hjalmarson A. Mortality and morbidity during a five‐year follow‐up of diabetics with myocardial infarction. Acta Med Scand. 1988;224:31–38. doi: 10.1111/j.0954-6820.1988.tb16735.x [DOI] [PubMed] [Google Scholar]
- 40. Perrelli MG, Pagliaro P, Penna C. Ischemia/reperfusion injury and cardioprotective mechanisms: role of mitochondria and reactive oxygen species. World J Cardiol. 2011;3:186–200. doi: 10.4330/wjc.v3.i6.186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hill BG, Awe SO, Vladykovskaya E, Ahmed Y, Liu SQ, Bhatnagar A, Srivastava S. Myocardial ischaemia inhibits mitochondrial metabolism of 4‐hydroxy‐trans‐2‐nonenal. Biochem J. 2009;417:513–524. doi: 10.1042/BJ20081615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Benjamin EJ, Virani SS, Callaway CW, Chamberlain AM, Chang AR, Cheng S, Chiuve SE, Cushman M, Delling FN, Deo R, et al.; American Heart Association Council on E, Prevention Statistics C, Stroke Statistics S . Heart disease and stroke statistics‐2018 update: a report from the American Heart Association. Circulation. 2018;137:e67–e492. [DOI] [PubMed] [Google Scholar]
- 43. Deanfield JE, Halcox JP, Rabelink TJ. Endothelial function and dysfunction: testing and clinical relevance. Circulation. 2007;115:1285–1295. doi: 10.1161/CIRCULATIONAHA.106.652859 [DOI] [PubMed] [Google Scholar]
- 44. Kolluru GK, Bir SC, Kevil CG. Endothelial dysfunction and diabetes: effects on angiogenesis, vascular remodeling, and wound healing. Int J Vasc Med. 2012;2012:918267. doi: 10.1155/2012/918267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Li Y, Zhang D, Jin W, Shao C, Yan P, Xu C, Sheng H, Liu Y, Yu J, Xie Y, et al. Mitochondrial aldehyde dehydrogenase‐2 (ALDH2) Glu504Lys polymorphism contributes to the variation in efficacy of sublingual nitroglycerin. J Clin Invest. 2006;116:506–511. doi: 10.1172/JCI26564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Roy B, Sundar K, Palaniyandi SS. 4‐hydroxy‐2‐nonenal decreases coronary endothelial cell migration: potentiation by aldehyde dehydrogenase 2 inhibition. Vascul Pharmacol. 2020;131:106762. doi: 10.1016/j.vph.2020.106762 [DOI] [PubMed] [Google Scholar]
- 47. Ma H, Guo R, Yu L, Zhang Y, Ren J. Aldehyde dehydrogenase 2 (ALDH2) rescues myocardial ischaemia/reperfusion injury: role of autophagy paradox and toxic aldehyde. Eur Heart J. 2011;32:1025–1038. doi: 10.1093/eurheartj/ehq253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Koda K, Salazar‐Rodriguez M, Corti F, Chan NY, Estephan R, Silver RB, Mochly‐Rosen D, Levi R. Aldehyde dehydrogenase activation prevents reperfusion arrhythmias by inhibiting local renin release from cardiac mast cells. Circulation. 2010;122:771–781. doi: 10.1161/CIRCULATIONAHA.110.952481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Wider J, Przyklenk K. Ischemic conditioning: the challenge of protecting the diabetic heart. Cardiovasc Diagn Ther. 2014;4:383–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Boudina S, Sena S, Theobald H, Sheng X, Wright JJ, Hu XX, Aziz S, Johnson JI, Bugger H, Zaha VG, et al. Mitochondrial energetics in the heart in obesity‐related diabetes: direct evidence for increased uncoupled respiration and activation of uncoupling proteins. Diabetes. 2007;56:2457–2466. doi: 10.2337/db07-0481 [DOI] [PubMed] [Google Scholar]
- 51. Palaniyandi SS, Disatnik M‐H, Sun L, Vishnumangalam JJ, Xia X, Pavlovic A, Bhalla V, Ashley E, Mochly‐Rosen D. Aldehyde dehydrogenase activator attenuates diabetic cardiomyopathy; a role in improving the quality of resident cardiac stem cells? Experimental Biology Meeting. 2010;24(MEETING ABSTRACT SUPPLEMENTS). doi: 10.1096/fasebj.24.1_supplement.572.3 [DOI] [Google Scholar]
- 52. Mattson MP. Roles of the lipid peroxidation product 4‐hydroxynonenal in obesity, the metabolic syndrome, and associated vascular and neurodegenerative disorders. Exp Gerontol. 2009;44:625–633. doi: 10.1016/j.exger.2009.07.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Lashin OM, Szweda PA, Szweda LI, Romani AM. Decreased complex ii respiration and hne‐modified sdh subunit in diabetic heart. Free Radic Biol Med. 2006;40:886–896. doi: 10.1016/j.freeradbiomed.2005.10.040 [DOI] [PubMed] [Google Scholar]
- 54. Yu P, Zhang J, Yu S, Luo Z, Hua F, Yuan L, Zhou Z, Liu Q, Du X, Chen S, et al. Protective effect of sevoflurane postconditioning against cardiac ischemia/reperfusion injury via ameliorating mitochondrial impairment, oxidative stress and rescuing autophagic clearance. PLoS One. 2015;10:e0134666. doi: 10.1371/journal.pone.0134666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Zhao J, Conklin DJ, Guo Y, Zhang X, Obal D, Guo L, Jagatheesan G, Katragadda K, He L, Yin X, et al. Cardiospecific overexpression of atpgd1 (carnosine synthase) increases histidine dipeptide levels and prevents myocardial ischemia reperfusion injury. J Am Heart Assoc. 2020;9:e015222. doi: 10.1161/JAHA.119.015222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Palaniyandi SS, Ferreira JC, Brum PC, Mochly‐Rosen D. PKCbetaII inhibition attenuates myocardial infarction induced heart failure and is associated with a reduction of fibrosis and pro‐inflammatory responses. J Cell Mol Med. 2011;15:1769–1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Palaniyandi SS, Inagaki K, Mochly‐Rosen D. Mast cells and epsilonPKC: a role in cardiac remodeling in hypertension‐induced heart failure. J Mol Cell Cardiol. 2008;45:779–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Pan G, Munukutla S, Kar A, Gardinier J, Thandavarayan RA, Palaniyandi SS. Type‐2 diabetic aldehyde dehydrogenase 2 mutant mice (ALDH 2*2) exhibiting heart failure with preserved ejection fraction phenotype can be determined by exercise stress echocardiography. PLoS One. 2018;13:e0195796. doi: 10.1371/journal.pone.0195796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Selvakumar D, Drescher MJ, Dowdall JR, Khan KM, Hatfield JS, Ramakrishnan NA, Drescher DG. CNGA3 is expressed in inner ear hair cells and binds to an intracellular C‐terminus domain of EMILIN1. Biochem J. 2012;443:463–476. doi: 10.1042/BJ20111255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Palaniyandi SS, Nagai Y, Watanabe K, Ma M, Veeraveedu PT, Prakash P, Kamal FA, Abe Y, Yamaguchi K, Tachikawa H, et al. Chymase inhibition reduces the progression to heart failure after autoimmune myocarditis in rats. Exp Biol Med (Maywood). 2007;232:1213–1221. doi: 10.3181/0703-RM-85 [DOI] [PubMed] [Google Scholar]
- 61. Gage GJ, Kipke DR, Shain W. Whole animal perfusion fixation for rodents. J Vis Exp. 2012;65:3564. doi: 10.3791/3564 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.