

Abstract
Background
Pathophysiologic mechanisms underlying cardiac structural and functional changes in obesity are complex and linked to adipocytokines released from pericardial adipose tissue (PAT) and cardiomyocyte apoptosis. Although leptin is involved in various pathological conditions, its role in paracrine action of pericardial adipose tissue on myocardial apoptosis remains unknown. This study was designed to investigate the role of PAT‐derived leptin on myocardial apoptosis in high‐fat diet–induced obese rats.
Methods and Results
Hearts were isolated from lean or high‐fat diet–induced obese Wistar rats for myocardial remodeling studies. Obese rats had abnormal myocardial structure, diastolic dysfunction, greatly elevated cardiac apoptosis, enhanced cardiac fibrosis, and increased oxidative stress level. ELISA detected significantly higher than circulating leptin level in PAT of obese, but not lean, rats. Western blot and immunohistochemical analyses demonstrated increased leptin receptor density in obese hearts. H9c2 cardiomyoblasts, after being exposed to PAT‐conditioned medium of obese rats, exhibited pronounced reactive oxygen species–mediated apoptosis, which was partially reversed by leptin antagonist. Moreover, leptin derived from PAT of obese rats inhibited Na+/K+‐ATPase activity of H9c2 cells through stimulating reactive oxygen species, thereby activating calcium‐dependent apoptosis. Pretreatment with specific inhibitors revealed that Janus kinase 2/signal transducer and activator of transcription 3 and phosphoinositide 3‐kinase/protein kinase B signaling pathways were involved in leptin‐induced myocardial apoptosis.
Conclusions
PAT‐derived leptin induces myocardial apoptosis in high‐fat diet–induced obese rats via activating Janus kinase 2/signal transducer and activator of transcription 3/reactive oxygen species signaling pathway and inhibiting its downstream Na+/K+‐ATPase activity.
Keywords: leptin, myocardial apoptosis, Na+/K+‐ATPase, obesity, paracrine, pericardial adipose tissue
Subject Categories: Cardiomyopathy, Hypertrophy, Metabolic Syndrome, Remodeling
Nonstandard Abbreviations and Acronyms
- Akt
protein kinase B
- CM
conditioned medium
- HFD
high‐fat diet
- JAK2
Janus kinase 2
- NAC
n‐acetyl‐amino acid
- NKA
Na+/K+‐ATPase
- PAT
pericardial adipose tissue
- PI3K
phosphoinositide 3‐kinase
- ROS
reactive oxygen species
- STAT
signal transducer and activator of transcription
Clinical Perspective
What Is New?
In obesity, pericardial adipose tissue can directly affect the adjacent myocardium via paracrine mechanism.
In vitro, leptin induces cardiomyocyte apoptosis with a time/concentration‐dependent manner.
We demonstrate, for the first time, that pericardial adipose tissue–derived leptin induces cardiomyocyte apoptosis and myocardial remodeling via activation of reactive oxygen species and then inhibition of Na+/K+‐ATPase.
What Are the Clinical Implications?
Our research offered a novel insight into monitor and interference with obesity individuals, which is to focus on the amount and function of visceral adipose tissue around the heart, and try to minimize myocardial damage induced by pericardial adipose tissue–derived adipocytokines.
In the future, pericardial adipose tissue–derived leptin may become a novel biomarker and a new therapeutic target of cardiovascular diseases, based on our results.
Obesity, defined by an excess accumulation of adipose tissue, is the major risk factor for cardiovascular diseases (CVDs). 1 It can defunctionalize adipose tissue by altering adipokines, including leptin, tumor necrosis factor‐α, interleukin‐6, resistin, and adiponectin, thus resulting in inflammation and cardiometabolic dysregulation. 2 Although adipokines have been proposed to be molecular links between obesity and CVD, the exact pathogenic mechanism by which adipokines influence many physiological and pathophysiologic conditions remains unclear.
Studies have implicated that pericardial adipose tissue (PAT), defined as paracardial fat plus all adipose tissue located internal to the parietal pericardium, is a local source of adipocytokines that contribute to the initiation of cardiac remodeling and CVD. 3 , 4 , 5 For example, PAT can secrete some cytokines, such as tumor necrosis factor and interleukin‐6, and free fatty acids, causing electromechanical changes in heart tissues. 6 This contention is supported by data indicating that PAT products are able to induce cardiac mitochondrial dysfunction and cardiomyocyte apoptosis in obese minipigs. Furthermore, high‐fat diet (HFD) causes a different fatty acid profile in PAT, all of which lead to cardiac fibrosis and eventually result in impaired heart functions. 7 Although these findings established a mechanistic link between local PAT and cardiac injury, little evidence has directly proven the molecular mechanism that PAT actually leads to harmful cardiac structural and functional changes in the context of obesity.
A recent study has shown that leptin enhances reactive oxygen species (ROS)–mediated, signal transducer and activator of transcription (STAT) 3–dependent apoptosis in H9c2 cardiomyoblasts. 8 In addition, we have previously demonstrated that leptin released by PAT of obese animals significantly promotes myocardial fibroblast proliferation and cardiac remodeling. Therefore, in this study, we hypothesized that augmented PAT‐derived leptin exacerbates myocardial apoptosis in obesity via oxidative stress action and tested the hypothesis by directly exposing myocardial cells to PAT‐conditioned medium (PAT‐CM) from obese rats. Our findings might offer novel insights into the potential role of PAT‐derived leptin in obesity‐related CVD.
Methods
Animal Modeling and Grouping
A total of 20 specific pathogen‐free Wistar rats (8 weeks old; weighing 200±50 g) were obtained from the Zoological Research Center of Peking Union Hospital, Chinese Academy of Medical Sciences (Animal Certification Number: SCXK, Beijing 2019‐0005). These rats were individually housed at 25±0.5 °C in a constant air‐conditioned environment with a 12‐hour day‐night cycle and relative humidity of 55% to 60%. All animals were allowed free access to food and water. Rats were randomly divided into 2 groups and habituated to research assistants for 1 week before experiments. Rats in the control group (n=10) were fed on a normal diet containing 50% carbohydrate, 20% protein, 8% fat, and 0% cholesterol. Rats in the obese group (n=10) were fed on an HFD containing 30% carbohydrate, 4.8% protein, 12% fat, 20% sucrose, 10% lard, 2.5% cholesterol, and 1.4% sodium cholate. The duration of the diet period was 20 weeks for both control and obese rats. All laboratory chows were purchased from Xi’an Jiaotong University Laboratory Animal Center (Shaanxi, China). Experiments were approved by the Ethical Committee for Animal Experimentation of the First Affiliated Hospital of Xi’an Jiaotong University. The study conforms to the Guide for the Care and Use of Laboratory Animals, published by the US National Institutes of Health (publication No. 85‐23, revised 1996). All supporting data are available within the article and its online supplementary files.
Metabolic Parameters
Blood samples were collected from each rat. After centrifugation, plasma was obtained and then snap frozen at −20 °C. Plasma glucose, total cholesterol, high‐density lipoprotein cholesterol, low‐density lipoprotein cholesterol, triglyceride, and insulin levels were determined using a Hitachi 7600‐010 Automatic Biochemical Analyzer (Hitachi High Technologies Corp, Japan). The Homeostasis Model Assessment index was used to assess insulin resistance.
Echocardiographic Studies and Hemodynamic Assessments
Blinded echocardiography was performed on chloral hydrate–anesthetized rats using a Vivid7 imaging system (Shenzhen MeruiBio‐medical Electronics Co, LTD, China) equipped with a 14‐MHz sectorial probe, described previously. 9 Two‐dimensional images were recorded in parasternal long‐ and short‐axis projections with guided M‐mode recordings at the midventricular level in both views. Parameters measured included end‐diastolic left ventricular posterior wall thickness, end‐diastolic interventricular septal thickness, left ventricular end‐diastolic diameter, left ventricular ejection fraction, and heart rate. A catheter filled with heparinized saline was connected to the BL‐420F biological and functional experimental system and inserted into the left ventricle via the right carotid artery. Data recorded included left ventricular systolic pressure, the maximum rising rate of left ventricular systolic pressure, the maximum descent rate of left ventricular systolic pressure, and left ventricular end‐diastolic pressure.
Histological Analysis
At the end of the experiment, rats were anesthetized by intraperitoneal injection of 20% urethane, and the chest cavity was cut open to remove the heart and its surrounding adipose tissue. Ultrastructural studies of cardiac tissue by electron microscopy were done as reported previously. 10 Briefly, cardiac tissues were fixed in cold 2.5% glutaraldehyde/1% paraformaldehyde, post fixed in 2% osmium tetroxide, embedded in resin, and sectioned. Hematoxylin‐eosin, Masson, and TdT‐mediated DUTP nick end labeling stainings of heart cryosections were done according to the standard procedures. The collagen volume fraction was quantified using the ImageJ software. 11 The expressions of cardiac collagen fiber 1, matrix metalloproteinase‐1, tissue inhibitor of metalloproteinase‐1, and α‐smooth muscle actin protein were detected by Western blot. Immunohistochemistry was performed as described by the manufacturer (Vectors Laboratories) using the primary rat monoclonal anti‐leptin antibody (Abcam) and anti–α 1 Na+/K+ ATPase (NKA) antibody (Abcam).
Preparation of PAT‐CM and Cell Treatment
PAT‐CM was prepared and concentrated, as described previously. 12 Briefly, PAT from control and obese rats was separated, and each 200 mg was cultured at 37 °C for 24 hours in 1 mL of serum‐free DMEM (Thermo Scientific) containing 0.2% BSA (Sigma Chemical). The PAT‐CM was then centrifuged, frozen, and stored at −80°C until use.
H9c2 rat cardiomyoblasts were obtained from the Experimental Animal Center of Xi’an Jiaotong University. The cells were cultured in a modified culture medium containing 10% fetal bovine serum at 37 ℃ with 5% CO2 and 95% air. Before each experiment, cells were serum starved for 6 hours in DMEM containing 5.5 mmol/L glucose and antibiotics (streptomycin, 100 mg/mL; and penicillin, 100 U/mL). H9c2 cells were treated with exogenous recombinant rat leptin (Peprotech), leptin plus leptin antagonist (leptin+leptin‐tA), or leptin‐tA in DMEM for 48 hours. H9c2 cells in PAT‐CM group were incubated with PAT‐conditioned medium from nonobese (nonobese CM) or obese rats (obese CM) for 48 hours. To determine whether PAT‐derived leptin promotes cardiac apoptosis, cells were pretreated with 50 ng/mL recombinant triple leptin mutant (leptin‐tA) (Protein Laboratories, Israel) that has been reported to function as a competitive leptin inhibitor 13 for 24 hours before incubation with nonobese CM (nonobese CM+leptin‐tA) or obese CM (obese CM+leptin‐tA). The medium was changed every 8 hours to prevent adipokine depletion. Besides, to elucidate whether leptin promotes H9c2 cell apoptosis via activation of Janus kinase 2 (JAK2)/STAT3 and phosphoinositide 3‐kinase (PI3K)/protein kinase B (Akt) signaling pathways, leptin was added for another 48 hours after 1 hour of pretreatment with JAK2 inhibitor AG490 (20 µmol/L; Sigma‐Aldrich), STAT3 inhibitor S3I‐201 (10 µm/L; Sigma‐Aldrich), PI3K inhibitor LY294002 (20 µmol/L; Cell Signaling Technology, Inc), Akt inhibitor GSK690693 (1 µmol/L; Sigma‐Aldrich), ROS inducer 2‐(di‐2‐pyridinylmethylene)‐N,N‐dimethyl‐hydrazinecarbothioamide (30 nmol/L; Beyotime, China), and scavenger n‐acetyl‐amino acid (NAC) (1 mmol/L; Beyotime, China).
Cell Proliferation and Apoptosis Assays
Cell viability was measured with methyl thiazolyl tetrazolium method. Briefly, H9c2 cells were seeded in 96‐well plates at 1×104 cells/well and cultured overnight. After treatment as designed, the culture medium was aspirated and 200 µL of fresh medium containing 0.5 mg/mL methyl thiazolyl tetrazolium was added into each well. After incubation at 37 ℃ for 4 hours, the culture medium containing methyl thiazolyl tetrazolium was removed. Dimethyl sulfoxide (150 µL) was then added into each well, and the absorbance at 570 nm was measured using a Safire2 spectrophotometric plate reader (Tecan Group Ltd, Switzerland). To visualize nuclear morphology, cells were fixed with 4% paraformaldehyde and stained with Hoechst 33342. Uniformly stained nuclei were scored as healthy viable cells. Condensed or fragmented nuclei were scored as apoptotic. To obtain unbiased counting, Petri dishes were coded, and cells were scored blindly without knowledge of their treatment. Transmission electron microscopy (Hitachi, HT7800) was used to observe the ultrastructure of cells under different treatments.
ROS Measurement
ROS generation was measured using fluorescent probe 2,7‐dichlorodihydrofluorescein diacetate (DCFH‐DA) with ROS Assay Kit (Beyotime, China). After treated as mentioned above, H9c2 cells were washed with DMEM for 3 times, incubated for 20 minutes at 37 ℃ in serum‐free DMEM containing 10 µmol/L DCFH‐DA to label intracellular ROS, and then washed again. ROS generation was detected by flow cytometry.
Determination of Malondialdehyde and Superoxide Dismutase Contents
After treatment, cells were collected, and cellular malondialdehyde and superoxide dismutase contents were determined using malondialdehyde as well as superoxide dismutase assay kits (Cayman Chemical Co), respectively, according to the manufacturer’s instruction. Loss of mitochondrial membrane potential was assessed by 5,5’,6,6’‐tetrachloro‐1,1’,3,3’‐tetraethylbenzimidazolylcarbocyanine iodide (JC‐1) (Beyotime) fluorescence probe. 14 In brief, after treatment, cells were washed with PBS and stained with JC‐1 for 20 minutes at 37 ℃. Cells were then rinsed twice with warm DMEM, incubated in 1 ml of culture medium, and visualized under a fluorescence microscope (Olympus). Fluorescence intensity was quantified using ImageJ software (National Institutes of Health).
Mitochondrial Permeability Assay
To analyze the opening of mitochondrial permeability transition pore, H9c2 cells were loaded with 1.0 mmol/L of calcein AM (Molecular Probes) and 1.0 mmol/L CoCl2 in Hanks’ balanced salt solution (Gibco) for 20 minutes at 37 ℃ and then observed and photographed under a fluorescence microscope. The mean fluorescence intensity of calcein AM from 5 random fields, which was considered as an index of mitochondrial permeability transition pore opening level, was analyzed using the ImageJ 1.47i software.
NKA Activity Assay
NKA activity in H9c2 cells was indirectly determined by measuring the phosphorous liberated after incubation of cell homogenate in a reaction mixture containing substrate ATP with cosubstrate elements at 37 ℃ for 10 minutes using NKA assay kit (Nanjing Jiancheng Bioengineering Institute), according to the manufacturer’s instruction. ATPase activity was expressed as µmol Pi/h per mg protein. Immunofluorescence analysis of α1 subunit of NKA was performed as described previously. 15
Cytosolic Ca2+ Measurement
Cytosolic Ca2+ concentration was determined using Fluo‐3 AM (Sigma) fluorescence probe, as described previously. 16 Briefly, an equal number of cells were seeded in black 96‐well plates or 35‐mm dishes and cultured overnight. At the end of treatment, cells were incubated with 0.5 µmol/L Fluo‐3 AM reagent at 37 ℃ for 30 minutes in the dark, washed with warm Hank’s balanced salt solution, and subjected to fluorescence measurement under a fluorescent microscope at an excitation wavelength of 506 nm and an emission wavelength of 526 nm.
Western Blot Analysis
Expression levels of leptin, caspase 3, Bax, Bcl‐2, NKA, calpain, sodium‐calcium exchanger 1, and β‐actin proteins were determined by Western blot, as described previously. 17 The bands of target proteins and internal reference β‐actin were scanned and analyzed by Quantity One software (Media Cybernetics Inc). The results are shown as the ratio of integrated optical density of the target proteins/the internal control and statistically analyzed. The primary antibodies used were anti–β‐actin antibody (JC‐PA002; 1:1000; Jingcai Biology, China), anti–caspase 3 antibody (ab4051; 1:500; Abcam), anti‐Bax antibody (1:1000; Abcam), anti‐leptin antibody (ab3583; 1:1000; Abcam), anti–Bcl‐2 antibody (ab59348; 1:1000; Abcam), anti–cytochrome C antibody (ab133504; 1:1000; Abcam), anti–calpain 1 antibody (ab108400; 1:5000; Abcam), and anti‐α1 NKA antibody (ab7671; 1:1000; Abcam).
Statistical Analysis
Statistical analysis was performed with SPSS version 22.0 (SPSS Inc). Data are presented as mean±SEM. Data with normal distribution were analyzed using independent t test, using ANOVA followed by the Scheffe multiple comparisons to identify significant differences among multiple groups. Data with nonnormal distribution were analyzed using Mann‐Whitney U test to identify significant differences between 2 groups and Kruskal‐Wallis test followed by Dunn multiple comparison post hoc tests to identify significant differences among multiple groups. P<0.05 was considered statistically significant.
Results
Obese Rat Model
To construct an animal model of obesity, rats were administrated by 20 weeks of HFD. Several metabolic parameters were examined in normal diet and HFD rats. At baseline levels (week 0), there was no statistically significant difference of phenotypic characteristics between the control and HFD rats (Table S1). However, at the end of the experiment (week 20), HFD rats showed a 27% increase in body weight, a 63% increase in total cholesterol, a 111% increase in triglyceride levels, a 116% increase in low‐density lipoprotein cholesterol levels, and a 70% increase in fasting blood glucose (P<0.05 or P<0.01; Table S2). More important, HFD rats also exhibited lower levels of high‐density lipoprotein cholesterol, compared with their normal diet counterparts (P<0.05; Table S2). Besides, as shown in Figure 1A, HFD rats were obese, and the fur lacked luster and appeared dull compared with control rats. Therefore, HFD rats were obese, hyperlipidemic, and hyperglycemic. We hence successfully constructed an animal model of obesity.
Figure 1. Comparison of control and obese rats at the end of the experiment.
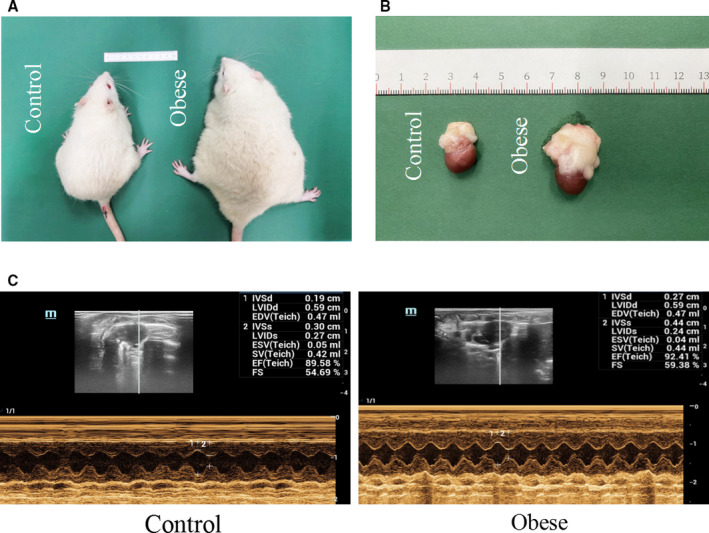
A, Rats were fed with normal diet (left) and high‐fat diet (right) for 20 weeks. B, After 20 weeks of feeding, rats were euthanized and their hearts and pericardial adipose tissue (PAT) were separated. PAT was used to make the conditioned medium. C, Echocardiography examination of control and obese rats. EDV indicates end‐diastolic velocity; EF, ejection fraction; ESV, end‐systolic velocity; FS, shortening fraction; IVSd, ventricular septum diastolic thickness; IVSs, ventricular septum systolic thickness; LVIDd, left ventricular internal diameter at end diastole; LVIDs, left ventricular internal diameter at end systole; and SV, stroke volume.
Myocardial Remodeling and Dysfunction of Obese Rats
Compared with their lean counterparts, obese rats had markedly higher heart/body mass (P<0.01; Table). In addition, they also had elevated left ventricular systolic pressure and left ventricular end‐diastolic pressure as well as greater ventricular septum diastolic thickness, ventricular septum systolic thickness, and left ventricular posterior wall end‐systolic thickness. Moreover, these rats began to exhibit diastolic dysfunction, as evidenced by significant decreases in the maximum rate of pressure development in the left ventricle (–maximum rising rate of left ventricular systolic pressure, 3862.174±62.589 versus 3368.731±216.928; P<0.05) and in the ratio of early/late transmitral peak flow velocities (1.46±0.29 versus 1.08±0.14; P<0.05; Table). More important, ejection fraction values were comparable between the 2 groups (Figure 1C and Table). Electron microscopy and hematoxylin‐eosin staining demonstrated a normal myocardial physiological structure and clear layer boundaries in control rats, but disordered myocardial structure and loss layer arrangement in obese rats (Figure 2A and 2B). These results indicate the occurrence of myocardial remodeling and defunctionalizing in obese rats.
Table 1.
Comparison of Hemodynamic Properties Among Control and Obese Rats
| Variable | Control rats (n=8) | Obese rats (n=6) |
|---|---|---|
| Heart weight, g | 1.54±0.27 | 1.88±0.37 |
| Heart weight/body weight | 0.0028±0.0003 | 0.0036±0.0003* |
| Heart rate, bpm | 400.00±44.43 | 396.17±53.17 |
| LVSP, mm Hg | 117.40±15.65 | 139.70±10.19* |
| LVEDP, mm Hg | 5.45±3.26 | 12.06±2.24 † |
| +dp/dtmax, mm Hg/s | 4810.263±62.323 | 4932.598±228.904 |
| –dp/dtmin, mm Hg/s | 3862.174±62.589 | 3368.731±216.928 † |
| IVSd, cm | 0.20±0.01 | 0.24±0.02* |
| IVSs, cm | 0.31±0.03 | 0.39±0.04* |
| LVPWd, cm | 0.19±0.03 | 0.21±0.02 |
| LVPWs, cm | 0.30±0.04 | 0.36±0.03 † |
| SV, mL | 0.56±0.09 | 0.62±0.11 |
| EF, % | 82.70±5.08 | 86.52±3.03 |
| E/A | 1.46±0.29 | 1.08±0.14* |
The t‐test was used for all data; data are presented as mean±SD. +dp/dtmax indicates maximum rate of pressure increase in the left chamber; –dp/dtmin, maximum rate of pressure decrease in the left chamber; A, late peak transmitral flow velocity; bpm, beats per minute; E, early peak transmitral flow velocity; EF, ejection fraction; IVSd, ventricular septum diastolic thickness; IVSs, ventricular septum systolic thickness; LVEDP, left ventricular end‐diastolic pressure; LVPWd, left ventricular posterior wall end‐diastolic thickness; LVPWs, left ventricular posterior wall end‐systolic thickness; LVSP, left ventricular systolic pressure; and SV, stroke volume.
P<0.01 vs control group.
P<0.05 vs control group.
Figure 2. The myocardial structure was disordered and cardiac apoptosis was increased in obese rats.
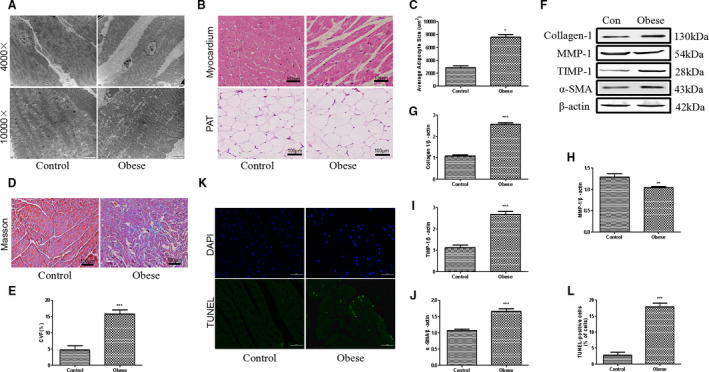
A, Representative images of electron microscope of myocardium of control (Con) and obese rats. Images were enlarged by 4000 and 10,000 times. B, Hematoxylin‐eosin staining of heart cryosections and pericardial adipose tissue (PAT) of rats; bar=100 µm. C, Average adipocyte size of PAT from control and obese rats. D, Masson staining of myocardium from control and obese rats. E, Collagen volume fraction (CVF) of myocardium from 2 experimental groups. F through J, Western blot analysis of cardiac collagen 1, matrix metalloproteinase‐1 (MMP‐1), tissue inhibitor of metalloproteinase‐1 (TIMP‐1), and α‐smooth muscle actin (α‐SMA) protein expressions of control and obese rats. K, Representative images of heart sections following TdT‐mediated dUTP nick end labeling (TUNEL) staining. Blue indicates the nucleus by 4′,6‐diamidino‐2‐phenylindole (DAPI) staining. Bar=50 µm. L, Statistical analysis of TUNEL‐positive cardiomyocytes in 2 groups of rats. Mann‐Whitney U test was used for C, E, and L; t‐test was used for G‐J. Data are presented as mean±SEM. For C, E, and L, n=5/group; for G through J, n=3/group. *P<0.05 vs control, **P<0.01 vs control, and ***P<0.001 vs control.
Increased Cardiac Apoptosis and PAT Accumulation in Obese Rats
Rats in both control and obese groups were euthanized, and their hearts were carefully isolated and subjected to subultrastructural analysis. Obese rats had significantly enlarged hearts surrounded by a large amount of PAT (Figure 1B). Electron microscopy showed abundant infiltrated adipose tissues among the myocardium of obese rats (Figure 2A). Hematoxylin‐eosin staining of PAT found remarkably enlarged, fused, and disorganized adipocytes in obese rats, but not in lean rats (Figure 2B and 2C). Masson staining in the left ventricle exhibited prominent cardiac fibrosis in obese rats, as evaluated by collagen volume fraction (Figure 2D and 2E). Meanwhile, collagen fiber 1, tissue inhibitor of metalloproteinase‐1, and α‐smooth muscle actin protein expressions were greatly increased, whereas the matrix metalloproteinase‐1 expression decreased, in the hearts of obese rats, compared with the control hearts (Figure 2F through 2J). Moreover, TdT‐mediated dUTP nick‐end labeling staining found 18% to 20% positive cells in the heart tissue of obese rats and only 3% to 5% positive cells in the control rats (Figure 2K and 2L). Herein, cardiac apoptosis as well as fibrosis are clearly correlated with abnormal myocardial structure and diastolic dysfunction in obese rats.
Upregulation of PAT‐Derived Leptin in Obese Rats Is Accompanied by Elevated Oxidative Stress Level and Inhibition of NKA Activity in the Hearts
To further investigate the underlying mechanisms, PAT of both control and obese rats was dissected, minced, and incubated with serum‐free DMEM for 24 hours, and the conditioned medium (PAT‐CM) was collected. The contents of leptin in serum and PAT‐CM, measured by ELISA, were remarkably elevated in obese rats compared with the control rats (Figure 3A). Moreover, leptin protein expression in PAT of obese rats was significantly higher than that of control‐PAT (Figure S1). Both immunohistochemistry and Western blot demonstrated similar cardiac leptin expression in the control and obese rats (Figure 3B, 3C, 3H, and 3J). However, the protein expression of leptin receptor (OB‐Rb) was greatly increased in the hearts of obese rats in comparison with the control rats (Figure 3H and 3K). Furthermore, obese rats demonstrated elevated cardiac oxidative stress levels, as evaluated by lipid peroxidation end product malondialdehyde as well as antioxidant superoxide dismutase (Figure 3D and 3E). Immunohistochemistry assay indicated that obese rats had lower cardiac NKA activity than control rats (Figure 3F and 3G). Western blot analysis also showed less protein expression of α1 subunit of NKA (Figure 3H and 3I).
Figure 3. Obese rats exhibited an increase in pericardial adipose tissue (PAT)–derived leptin and enhanced cardiac oxidative stress accompanied by a decrease in Na+/K+‐ATPase (NKA) activity.
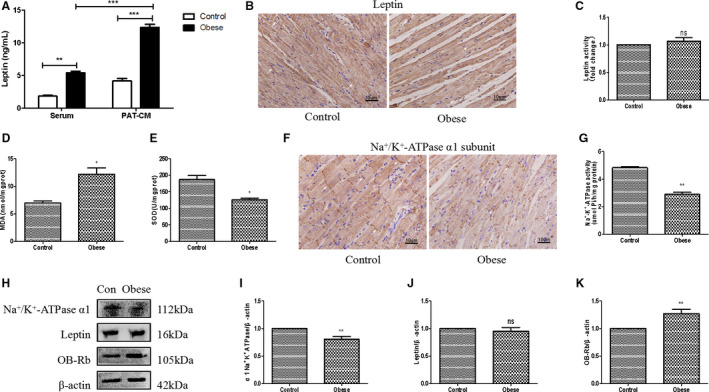
A, ELISA analysis of leptin levels in both serum and PAT conditioned medium (PAT‐CM) of control and obese rats. B and C, Immunohistochemical (IHC) analysis of cardiac leptin expression of control and obese rats. D, Detection of cardiac malonaldehyde (MDA) assay of control and obese rats. E, Detection of cardiac superoxide dismutase (SOD) assay of rats in 2 groups. F and G, IHC analysis of cardiac NKA α1 subunit expression of control and obese rats. H through K, Western blot analysis of cardiac α1 subunit of NKA, leptin, and leptin receptor (OB‐Rb) of control and obese rats. Two‐way ANOVA was used for A; t‐test was used for D, E, and I through K; and Mann‐Whitney U test was used for C and G. Data are presented as mean±SEM. For D, E, and I through K, n=3/group; for A, C, and G, n=5/group. Ns indicates insignificant. *P<0.05 vs control, **P<0.01 vs control, and ***P<0.001 vs control.
Leptin Stimulates Apoptosis of H9c2 Cardiomyoblasts Only After Long‐Term Administration
Given that obese rats had pronounced cardiac apoptosis, H9c2 cells were used in vitro to elucidate the apoptotic effect of leptin. The survival rate of H9c2 cells was determined by methyl thiazolyl tetrazolium after incubation with leptin at different concentrations (0, 20, 40, 80, 100, and 120 ng/mL) for 0, 15, and 30 minutes and 1, 8, 12, 24, 48, and 72 hours. The results showed that short‐term administration of leptin (15 and 30 minutes) promoted proliferation of H9c2 cells, whereas this effect gradually weakened, even reversed with prolonged incubation (Figure S2A through S2K), in particular with 40 ng/mL leptin treatment. In addition, leptin treatment at 24 hours after intervention significantly enhanced expression of caspase 3, a crucial downstream activator of apoptotic progress, as determined by Western blot, and this proapoptotic effect reached its peak at 48 hours (Figure S2L and S2M). Overall, leptin induction of H9c2 cell apoptosis requires a relatively long‐term administration.
Leptin Derived From Obese Rat PAT Induces Apoptosis of H9c2 Cells
We incubated H9c2 cells in DMEM with exogenous leptin (40 ng/mL), leptin‐tA, and leptin combined with leptin‐tA for 48 hours to illustrate the effect of leptin‐tA. Meanwhile, cells were grown in nonobese or obese PAT‐CM for 48 hours, as mentioned previously, to mimic in vivo conditions. As shown in Figure 4D and 4E, leptin promoted OB‐Rb expression in H9c2 cells and leptin‐tA greatly blunted it, suggesting that leptin‐tA antagonizes the activation effect of leptin to OB‐Rb. As such, leptin‐tA per se had no statistically significant effect on caspase 3 protein expression as well as apoptosis of H9c2 cells compared with the control group, but it significantly inhibited leptin‐induced apoptosis (Figure 4A, 4B, 4D, and 4F). These results demonstrated that leptin antagonist is endowed with the antiapoptotic ability only in the presence of leptin. Moreover, in contrast to the control, leptin promoted cell shrinkage and nuclear condensation (Figure 4A and 4C), whereas incubation with obese PAT‐CM for 48 hours further aggravated that, as evidenced by the formation of apoptotic bodies, one of the typical characteristics of apoptosis (Figure 4C). After pretreatment with leptin antagonist, the proapoptotic effect of obese CM was attenuated. Nevertheless, nonobese CM barely promoted cell apoptosis relative to the control (Figure 4A through 4C). The above results were further illustrated by Western blot analysis of caspase 3, Bax, cytochrome C, and antiapoptotic protein Bcl‐2 (Figure 4D and 4F through 4I). These results indicate that leptin derived from PAT of obese but not nonobese rats exerts proapoptotic effects on H9c2 cells.
Figure 4. Obese rat pericardial adipose tissue–derived leptin induces H9c2 cell apoptosis.
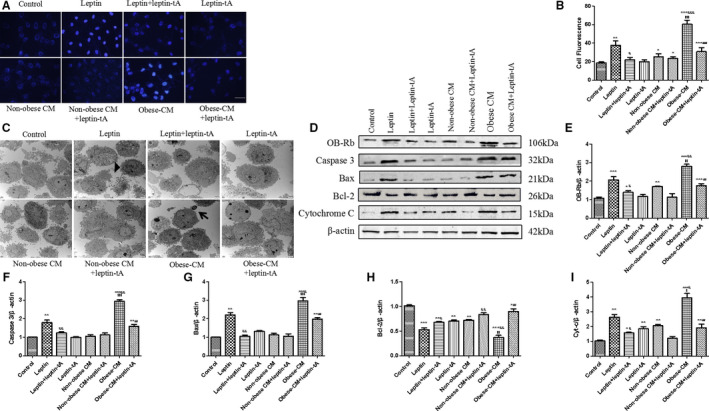
A, Representative images of cell apoptosis by Hoechst 33342 staining; bar=50 µm. B, Statistical analysis of relative fluorescence intensity of apoptosis. C, Transmission electron microscope was used to observe ultrastructure of cells under different treatments at 4000 times of magnification. Apoptotic cells shrink in size, cytoplasm condenses, chromatin aggregates (black triangle), and some form apoptotic bodies (black arrow). D through I, Western blot analysis of leptin receptor (OB‐Rb), caspase 3, Bax, cytochrome c (Cyt‐c), and Bcl‐2 protein expressions in experimental groups. Kruskal‐Wallis test was used for B; and 1‐way ANOVA with Bonferroni correction was used for E through I. Data are presented as mean±SEM. For B, n=5/group; for E through I, n=3/group. CM indicates conditioned medium. * P<0.05 vs control, ** P<0.01 vs control, *** P<0.001 vs control, & P<0.05 vs leptin, && P<0.01 vs leptin, &&& P<0.001 vs leptin, $$$ P<0.001 vs nonobese CM, and ### P<0.001 vs obese CM.
Leptin Derived From Obese Rat PAT Induces Oxidative Stress and Mitochondrial Dysfunction of H9c2 Cells
Incubation with exogenous leptin (40 ng/mL) for 48 hours enhanced oxidative stress of H9c2 cells, as demonstrated by increased cellular ROS and malondialdehyde levels and decreased superoxide dismutase content. Obese PAT‐CM treatment significantly strengthened oxidative stress of H9c2 cells, and this strengthened stress was partly prevented by leptin antagonist (Figure 5A through 5D). Mitochondrial membrane potential is a critical factor for maintaining mitochondria integrity and subsequent apoptosis regulation. Compound JC‐1 has been used as an indicator for apoptotic status of mitochondria, because it exists as aggregates in normal hyperpolarized mitochondria, emitting red fluorescence, whereas it exists as monomers in apoptotic cells, emitting green fluorescence. In the control group, cells exhibit numerous brightly stained mitochondria that emit red fluorescence. However, cells exposed to exogenous leptin had fewer red JC‐1 aggregates and more green JC‐1 monomers, which were aggravated by obese PAT‐CM, indicating the severe dissipation of mitochondrial membrane potential (Figure 5E and 5F) in these cells. Likewise, these cells exhibited higher degree of mitochondrial permeability transition pore opening, another indicator of mitochondrial function (Figure 5G and 5H). More important, administration of a recombinant leptin antagonist attenuated all changes initiated by obese PAT‐CM (Figure 5E through 5H). These results suggest that oxidative stress and mitochondria play a role in the process of apoptosis of H9c2 cells induced by leptin derived from obese rat PAT.
Figure 5. Pericardial adipose tissue–derived leptin of obese rats promotes oxidative stress and mitochondrial dysfunction of H9c2 cells.
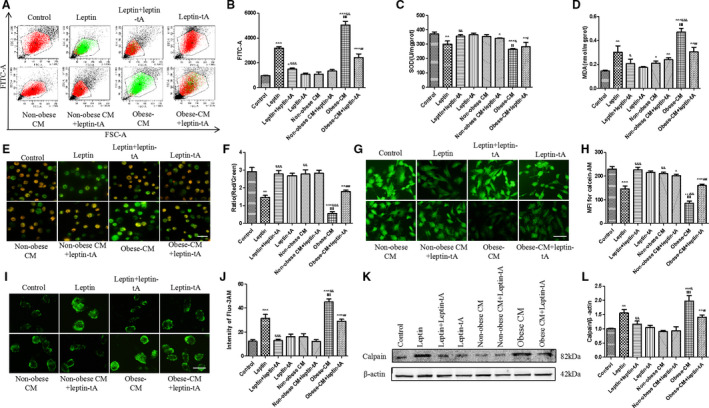
A and B, Representative scatter diagram and analysis of reactive oxygen species (ROS) production by flow cytometry. Cells were incubated for 20 minutes with free fetal bovine serum containing 10 µmol/L DCFH‐DA to label intracellular ROS, as stated before. C, Effects of PAT‐derived leptin on superoxide dismutase (SOD) generation of H9c2 cells. D, Effects of PAT‐derived leptin on malondialdehyde (MDA) generation of H9c2 cells. E and F, Representative images of JC‐1 staining on mitochondrial membrane potential. The red dye represents normal potential, whereas the green is considered as decreased mitochondrial membrane potential; bar=50 µm. G and H, Degrees of mitochondrial permeability transition pore opening by calcein‐AM staining; bar=50 µm. I and J, Fluo3‐AM staining of Ca2+ concentration of experimental groups; bar=50 µm. K and L, Western blot analysis on calpain protein expression in 8 experimental groups. Kruskal‐Wallis test was used for B, F, H, and J; and 1‐way ANOVA with Bonferroni correction was used for C, D, and L. Data are presented as mean±SEM. For B, F, H, and J, n=5/group; for C, D, and L, n=3/group. CM indicates conditioned medium; FSC, forward scatter; and MFI, mean fluorescence intensity. *P<0.05 vs control, **P<0.01 vs control, ***P<0.001 vs control, & P<0.05 vs leptin, && P<0.01 vs leptin, &&& P<0.001 vs leptin, $$ P<0.01 vs nonobese CM, $$$ P<0.001 vs nonobese CM, # P<0.05 vs obese CM, ## P<0.01 vs obese CM, and ### P<0.001 vs obese CM.
Obese Rat PAT‐Derived Leptin Promoted Ca2+/Calpain‐Dependent Apoptosis via Inhibiting NKA Activity in H9c2 Cells
As stated above, cardiac NKA activity was decreased in obese rats accompanied by an increase in oxidative stress level, which can also be induced by obese PAT‐derived leptin. Because inhibition of NKA could lead to apoptosis in cardiomyocytes, 18 whether the role of leptin in apoptosis was related to NKA impairment was investigated in the present study. Leptin (40 ng/mL) intervention for 48 hours inhibited NKA activity, and this inhibition was greatly abrogated by NAC, a selective inhibitor of ROS (Figure 6A and 6B), indicating that leptin inhibits NKA activity mostly through elevated oxidative stress in H9c2 cells. NKA affects intracellular Ca2+ concentration through Na+ and K+ exchange inside and outside the cells. Obese PAT‐CM administration markedly increased intracellular Ca2+ gathering, and this increase was attenuated by leptin antagonist (Figure 5I, and 5J). Moreover, calpain, which acts as an apoptosis regulator downstream of Ca2+, exhibited same trend as Ca2+ (Figure 5K and 5L). Thus, ROS/NKA/calpain signaling pathway is also involved in obese rat PAT‐derived leptin‐induced myocardial apoptosis.
Figure 6. Janus kinase 2 (JAK2)/signal transducer and activator of transcription 3 (STAT3) and phosphoinositide 3‐kinase (PI3K)/protein kinase B (Akt) signaling pathways are involved in the leptin‐mediated H9c2 cell apoptosis.
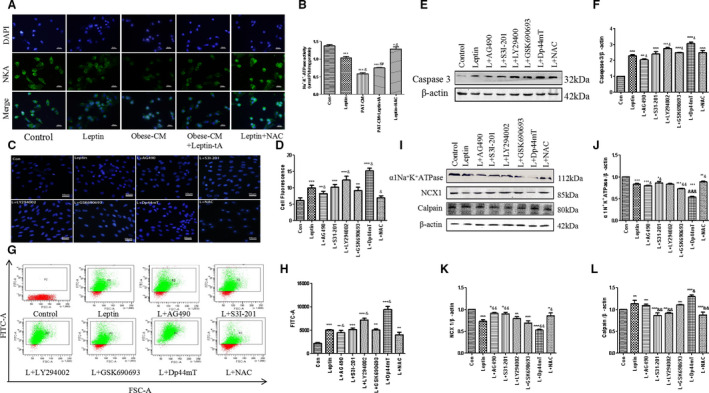
Leptin was added for another 48 hours after 60 minutes of pretreatment of cells with reactive oxygen species (ROS) scavenger n‐acetyl‐amino acid (NAC); moreover, JAK2 inhibitor AG490, STAT3 inhibitor S3I‐201, PI3K inhibitor LY294002, Akt inhibitor GSK690693, and ROS inducer 2‐(di‐2‐pyridinylmethylene)‐N,N‐dimethyl‐hydrazinecarbothioamide (Dp44mT) were preincubated as well. A and B, Na+/K+‐ATPase (NKA) expression assay in cells by immunofluorescence staining. The nuclei were stained by 4′,6‐diamidino‐2‐phenylindole (DAPI) (blue); bar=50 µm. C and D, Hoechst 33342 fluorescence staining in each group. Intensity was calculated using the Image J program; bar=50 µm. E and F, Western blot analysis of caspase 3 protein expression in 8 groups. G and H, Detection of ROS production in 8 groups. I through L, Western blot analysis of NKA, sodium‐calcium exchanger 1 (NCX1), and calpain protein expressions in 8 groups. Kruskal‐Wallis test was used for B, D, and H; and 1‐way ANOVA with Bonferroni correction was used for F and J through L. Data are presented as mean±SEM. For B, D, and H, n=5/group; for F and J through L, n=3/group. CM indicates conditioned medium; Con, control; and PAT, pericardial adipose tissue. *P<0.05 vs control, **P<0.01 vs control, ***P<0.001 vs control, & P<0.05 vs leptin, && P<0.01 vs leptin, and &&& P<0.001 vs leptin.
Leptin Induces H9c2 Cell Apoptosis via Activating JAK2/STAT3 and Blocking PI3K/Akt Signaling Pathways
We next examined signaling mechanisms involved in the obese rat PAT‐derived leptin‐induced H9c2 cell apoptosis. To avoid interference from other adipocytokines, exogenous leptin was used in this part of the experiment. Pretreatment with JAK2 inhibitor AG490 (20 µmol/L), STAT3 inhibitor S3I‐201 (10 µmol/L), and ROS scavenger NAC (1 mmol/L) partly blocked leptin‐induced H9c2 cell apoptosis. In contrast, preadministration of PI3K inhibitor LY294002 (20 µmol/L), Akt inhibitor GSK690693 (1 µmol/L), as well as ROS inducer 2‐(di‐2‐pyridinylmethylene)‐N,N‐dimethyl‐hydrazinecarbothioamide (30 nmol/L) enhanced the proapoptotic effect of leptin (Figure 6C through 6F). ROS level was also increased by AG490, S3I‐201, and NAC and decreased by LY294002, GSK690693, and 2‐(di‐2‐pyridinylmethylene)‐N,N‐dimethyl‐hydrazinecarbothioamide (Figure 6G and 6H). Besides, leptin administration significantly inhibited expression of NKA and sodium‐calcium exchanger 1 proteins, whereas AG490 and S3I‐201 reversed these effects (Figure 6I through 6K). More important, inhibition of PI3K/Akt signaling pathway by LY294002 and GSK690693 had little effect on the expression of both NKA and sodium‐calcium exchanger 1 (Figure 6I through 6K). Moreover, calpain protein expression, measured by Western blot, was opposite to that of NKA (Figure 6I and 6L). These results suggest that both JAK2/STAT3 and PI3K/Akt signaling pathways partly participate in the leptin‐induced apoptosis of H9c2 cells.
In summary, our findings demonstrated that PAT‐derived leptin enhances cardiac oxidative stress mostly via JAK2/STAT3 signaling pathway and results in mitochondria‐ as well as NKA‐dependent apoptosis, eventually leading to myocardial remodeling of rats with obesity. Figure 7 summarizes the model diagram of this study.
Figure 7. Schematic drawing representing the proposed role of pericardial adipose tissue–derived leptin in myocardial apoptosis of obese rats.
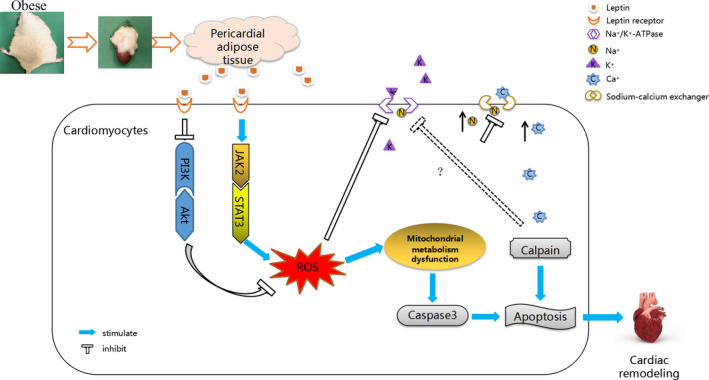
Akt indicates protein kinase B; JAK2, Janus kinase 2; PI3K, phosphoinositide 3‐kinase; ROS, reactive oxygen species; and STAT3, signal transducer and activator of transcription 3.
Discussion
PAT has been found in clinical studies to be significantly associated with key characteristics of obesity, and it has been proposed as a new indicator of CVDs. 19 , 20 , 21 Indeed, because of the close proximity to the coronary artery and the underlying myocardium, PAT may exert a local toxic effect on the nearby organs rather than a systemic effect as a cardiometabolic risk factor. However, the above conclusion is barely supported by direct evidence. Accordingly, this investigation was designed to examine the effects of obese PAT on cardiac remodeling, elucidate the role of PAT‐derived adipokine leptin and the signaling pathway involved, and revealed the following findings. First, HFD‐induced obese rats exhibit decreased diastolic functions and increased heart and PAT mass. Second, leptin secretion from PAT of obese rats is enhanced, and expression of leptin receptor OB‐Rb is upregulated while leptin expression remains unchanged in obese hearts. Third, obese rats exhibit higher cardiac oxidative stress level, myocardial apoptosis, and fibrosis but lower NKA activity in the hearts. Fourth, leptin from obese rat PAT induces mitochondrial dysfunction and caspase 3–dependent apoptosis via JAK2/STAT3 signaling pathway in cardiomyocytes. Fifth, PAT‐derived leptin promotes Ca2+/calpain‐dependent apoptosis through inhibiting NKA activity via activating ROS in cardiomyocytes.
The effects of obesity on heart structure and function have been extensively studied. 22 , 23 , 24 , 25 The present study found that obese rats exhibit cardiac diastolic dysfunction, increased heart/body weight, abnormal changes of myocardial structure, prominent cardiac apoptosis, and fibrosis. These data support that these obese rats have undergone myocardial remodeling, in concordance with previous findings. 22 , 26 Obese rats induced by an HFD in our study also display significantly increased PAT volume. Under normal conditions, PAT protects the heart by paracrine cytokines, such as adiponectin and adrenomedullin. 27 However, if PAT is abnormally deposited, production and secretion of protective adipocytokines are downregulated, whereas destructive adipocytokines, such as leptin, tumor necrosis factor, and interleukin, are upregulated. 6 , 27 , 28 As one of the most important destructive adipocytokines, the effect of leptin on cardiac remodeling has been well documented. For example, leptin has been shown to induce neonatal cardiomyocyte hypertrophy via PI3K/mechanistic target of rapamycin signaling pathways. 29 Huby et al 30 demonstrated that leptin is a direct regulator of aldosterone release, by which it promotes cardiac fibrosis in obese animals. Martinez‐Abundis et al 8 reported that exposure of neonatal rat ventricular myocytes to 3.1 nmol/L (50 ng/mL) leptin for 24 hours promotes high glucose–induced mitochondrial damages, cardiomyocyte injuries, apoptosis, cytotoxicity, and ROS generation. However, until now, supporting evidence is rare as to whether leptin secreted by PAT directly affects cardiac apoptosis and participates in myocardial remodeling in obese individuals.
The present study found significant apoptosis of H9c2 cardiomyocytes after incubation with PAT‐CM from obese rats. Although leptin antagonist only partially inhibits the proapoptotic effect of obese CM, it explains the paracrine role of leptin as a signaling molecule derived from obese rat PAT. This investigation is of great significance compared with those using exogenous leptin alone. 8 , 31 , 32 , 33 In addition, our results showed that the concentration of exogenous leptin needed for promoting apoptosis is much higher than that detected in vivo, in either PAT locally or the circulation of obese rats, suggesting that exogenous leptin plays a role at superphysiological concentrations. It is worth noting that H9c2 cells incubated with nonobese rat PAT‐CM were not comparable to those incubated with obese CM, even leptin, in terms of the degree of cell apoptosis and proapoptotic protein expressions after 48 hours of intervention. Although previous literature suggested a protective role of PAT under normal conditions, 27 the exact proof remains to be explored.
In agreement with a previous finding that there is a large amount of local leptin accumulation at PAT in the context of obesity, 6 the present study also demonstrated that obese PAT secretes vast quantities of leptin. In contrast to PAT, leptin expression in myocardium is not significantly different between control and obese rats. Although the increased production of leptin by failing human hearts has previously been reported, 34 mechanical unloading of the failing hearts downregulates leptin expression, suggesting a potential compensatory role for leptin in cardiomyocytes in response to cardiac overloading, which is limited in the setting of obesity. Also, it has been pointed out that there is no positive correlation between leptin level and body mass index in patients with heart failure (HF), and leptin expression in patients with HF with normal body mass index is also greatly upregulated. 35 Thus, leptin regulation may be perturbed secondary to the hormonal and inflammatory aspects of patients with HF rather than obesity. 36 Research that used dogs with HF, by Fonfara et al, 37 has found that myocardial leptin expression varies with different HF stages, and its production in the late HF stage is higher than that in the early stage. Hence, as an initial form of HF, elevation of cardiac leptin in obesity may not be significant. It is known that leptin exerts its biological effects primarily through its long‐form receptor OB‐Rb. OB‐Rb mainly presents in the hypothalamus, heart, and pancreas, where it mediates signaling pathways. Hence, it is also referred to as the “signaling isoform.” 38 This study demonstrated that OB‐Rb expression in obese hearts is distinctly elevated. Nascimento et al 39 showed that long‐term HFD (15 or 45 weeks) decreases leptin receptors in the hearts of obese rats. However, they measured total leptin receptors rather than OB‐Rb. Thus, this discrepancy could be explained by the attenuated expression of other leptin receptor isoforms in obese hearts. 38 In summary, our data suggest that leptin derived from PAT of obese rats activates OB‐Rb in the heart through paracrine action and mediates myocardial apoptosis.
In this study, we did not conduct electrocardiographic examinations, so we could not determine whether there was atrial arrhythmia in obese rats. Nonetheless, previous data have shown that abnormal PAT accumulation can increase the incidence of atrial arrhythmia, especially atrial fibrillation (AF). 5 , 40 , 41 A recent clinical study demonstrated that plasma leptin is a risk factor for AF in patients with CVDs. For every 1‐unit increase in leptin, the risk of AF increased by 2%. 42 Wang et al 43 have found that leptin level of PAT in patients with AF is upregulated relative to people with sinus rhythm, although they did not prove a direct relationship between leptin secreted by PAT and the occurrence of AF. Another study using ob/ob and Zucker mice confirmed that leptin signaling is an essential link in the development of atrial fibrosis and AF. 44 In the present study, leptin derived from obese rat PAT promotes myocardial fibrosis as well as cardiac remodeling, which is the main pathophysiological mechanism of AF. Therefore, although the incidence of AF was not recorded, this study, to some extent, emphasizes the potential association between PAT paracrine leptin and AF in clinically obese individuals.
Chronically elevated oxidative stress has been demonstrated in humans and animals to play a central role not only in the development of cellular necrosis but also in programmed cellular death or apoptosis. 45 , 46 We found that the level of oxidative stress in the hearts of obese rats is elevated, which was further verified by in vitro experiments to be related to leptin secretion by PAT. The apoptosis of H9c2 cells increases significantly after incubation with obese CM, a variation that is partially attenuated by leptin antagonist. These results suggest that leptin derived from obese rat PAT promotes cardiac apoptosis, partly by enhancing myocardial oxidative stress. Of note, obese CM treatment also leads to mitochondrial dysfunction, mitochondrial permeability transition pore opening, and decreased mitochondrial membrane potential in H9c2 cells, and these effects are reversed only partially by leptin antagonist. We suspect that there may be positive interactions among multiple detrimental adipocytokines that can induce oxidative stress as well as mitochondrial dysfunction in addition to leptin, which, obviously, warrants further investigations.
Since discovered by Skou in 1957, 47 energy‐transducing NKA has been extensively studied for its ion‐pumping function. More important, documents have demonstrated that NKA also acts as a signaling molecule participating in various signaling pathways. For example, energy‐sensitive regulation of NKA by JAK2 has been found in Jurkat cells. 48 In hepatoma HepG2 cell line, targeting NKA α1 subunit induces apoptosis and cell cycle arrest. 49 In recent years, many studies have pointed out an amplification loop between ROS and NKA. 50 , 51 , 52 , 53 As such, researchers have demonstrated oxidative regulation of NKA in the cardiovascular system. 54 A previous study showed that DRm217, a proven specific NKA antibody, attenuates myocardial ischemia‐reperfusion injury via stabilizing plasma membrane NKA, inhibiting NKA/ROS pathway and activating PI3K/Akt and extracellular signal‐regulated kinase 1/2. 15 Research by White et al 55 suggested that a slight increase of ROS inhibits NKA activity in cardiomyocytes. In our study, leptin induces ROS generation in cultured H9c2 cells; it is, therefore, possible that leptin may inhibit NKA. We observed downregulation in obese rats of both NKA activity and protein expression of NKA α1 subunit, which contains ligand binding sites, such as ATP and digitalis, and is considered to be a catalytic subunit. 56 In addition, leptin inhibition of NKA activity in H9c2 cells is reversed after pretreatment with ROS scavenger NAC. These results revealed that ROS is an upstream signal in NKA inhibition in response to elevated PAT‐derived leptin in obese hearts.
In the classic situation, partial inhibition of NKA ion‐exchange activity raises intracellular sodium concentration ([Na+]i), which, in turn, increases intracellular calcium concentration ([Ca2+]i) by coupling with sodium‐calcium exchanger to execute the inotropic effect. 57 However, evidence has been proposed that Ca2+ overload induced by increased intracellular Na+, the so‐called reverse mode, will further activate calpains and induce apoptosis in the end. 58 Calpains belong to a family of calcium‐dependent thiol proteases. Activation of calpain is implicated in ischemia/reperfusion‐induced apoptosis in the heart and plays a role in tumor necrosis factor‐α–mediated apoptosis in cardiomyocytes. 59 Thus, we assessed Ca2+ concentration and calpain expression in PAT‐CM–cultured cells. Leptin derived from obese PAT increases Ca2+ gathering and calpain protein expression concomitant with the impairment of NKA activity in H9c2 cells. On the basis of these data, it is reasonable to assume that NKA/Ca2+/calpain, as a downstream signaling pathway of ROS, is involved in PAT‐derived leptin‐mediated apoptosis of cardiomyocytes. However, recent data have shown that calpain inhibits NKA activity in the heart, which may induce apoptosis via Ca2+ overload. 60 Li et al 61 also found that calpain‐1 activation induces apoptosis through downregulating NKA activity in high glucose–stimulated cardiomyocytes. Thus, the pathophysiological cross talk between NKA and calpain remains to be further explored.
JAK2/STAT3 signal pathway has proven to be a classic downstream channel of leptin. 62 Our previous research showed that leptin promotes myocardial fibrosis and eventually leads to cardiac remodeling by activating JAK2/STAT3 signaling pathway. 63 We found in this study that leptin activates ROS via JAK2/STAT3 and triggers 2 independent apoptosis pathways. PI3K/Akt is one of the downstream signaling pathways of leptin. 64 Evidence shows that one of the primary functions of PI3K/Akt is to promote growth factor–mediated cell survival and prevent programmed cell death or apoptosis. 65 , 66 However, whether the strong antiapoptotic activity of PI3K/Akt still exists in the presence of local accumulation of leptin is unclear. In this experiment, PI3K/Akt signaling pathway is suppressed in the process of leptin‐induced apoptosis. However, it is worth noting that PI3K/Akt plays a minor role in the calpain‐dependent apoptosis because pretreatment with its inhibitors barely changed the effects of leptin on protein expression of NKA or calpain.
Several limitations of the present study should be acknowledged. First, adipose tissue secretes many types of adipocytokines, among which leptin production increases significantly in obese individuals. The cross talk between leptin and other adipokines in the process of apoptosis is not illustrated in our study. Second, NKA‐related ion activity was not detected in this experiment, and the specific mechanism of ion channels was not studied deeply enough. Third, the obese rat model was established via high calorie uptake to explain paracrine effect. Whether the results also standstill in other obese models, such as ob/ob or db/db animal models, is unclear. Interestingly, Barouch et al 67 acknowledged that the signaling pathways of leptin were unchanged, even eliminated, in the genetically modified obese mice, which is apparently distinct from our results. We hypothesized that these genetically engineered mice might have different cardiac remodeling mechanisms from diet‐induced obese ones. In short, further research is needed to confirm this hypothesis.
The present study highlighted the promoting effect of leptin secreted locally from pericardial adipose tissue on myocardial apoptosis and cardiac remodeling under the condition of obesity and demonstrated the link between NKA and ROS in leptin‐induced cardiac apoptosis. Future studies should focus on exploring the pathophysiological relationship between leptin and other adipokines secreted by PAT in cardiac injury. Besides, we plan to inject small interfering RNA into PAT in the future to silence the expression of leptin in local adipose tissue to further verify our results. Finally, the mechanism of NKA‐associated ion channels in leptin‐mediated cardiac injury needs to be further clarified.
In summary, this study indicates that leptin derived from obese rat PAT promotes myocardial apoptosis via both JAK2/ROS/NKA and JAK2/ROS/mitochondria pathways. These data offer novel insights into potential mechanisms of PAT in heart diseases in diet‐related obese individuals.
Sources of Funding
This work was supported by grants from the Nature Science Foundation of China (grant Nos. 81873513, 81600574, 82070549, and 30871042), Key Projects of Shaanxi Science and Technology Research and Development Plan (No. 2018ZDXM‐SF‐049), Shaanxi Science and Technology Research and Development Plan of International Science and Technology (Nos. 2012 kw‐40‐01 and 2014 JM2‐8145), the Dr Li Jieshou (Academician of Chinese Academy of Sciences) Research Fund for Intestinal Barrier (LJS‐201801), and the clinical research award of the First Affiliated Hospital of Xi'an Jiaotong University (No. XJTU1AF‐CRF‐2019‐016).
Disclosures
Drs Wang, Gao, and Zhu report grants from Nature Science Foundation of China, grants from Key Projects of Shaanxi Science and Technology Research and Development Plan, grants from Key Project of Clinical Research in the First Affiliated Hospital of Xi'an Jiaotong University, and grants from Shaanxi Science and Technology Research and Development Plan of International Science and Technology, outside the submitted work. Dr Li reports grants from Nature Science Foundation of China, grants from Key Projects of Shaanxi Science and Technology Research and Development Plan, and grants from Project of Clinical Research in the First Affiliated Hospital of Xi'an Jiaotong University, outside the submitted work. Dr Tian reports grants from Nature Science Foundation of China, grants from Key Project of Clinical Research in the First Affiliated Hospital of Xi'an Jiaotong University, and grants from Shaanxi Science and Technology Research and Development Plan of International Science and Technology, outside the submitted work. The remaining authors have no disclosures to report.
Supporting information
Tables S1–S2
Figures S1–S2
Acknowledgments
We thank Scientific Writing Solutions (https://www.sciwriting.com) for language editing. Dr Wang conducted experiments, interpreted the data, and drafted the initial manuscript. Dr Luo assisted with supplementary experiments and writing of the revised manuscript. Dr Zhu monitored the progress of the research. Dr Song performed cardiac ultrasound of rats. Drs Cao, Luan, and Zheng assisted with data analysis. Dr Li designed and revised the supplementary experiments. Dr Gao took and illustrated the electron microscope pictures. Dr Tian developed the conception and design of the study and obtained the funding. All authors contributed to discussions and critically appraised the manuscript. All authors approved the final manuscript as submitted and agree to be accountable for all aspects of the work.
P. Wang and C. Luo are co–first authors.
For Sources of Funding and Disclosures, see page 14.
Contributor Information
Hao Li, Email: hao.li215@xjtu.edu.cn.
Gang Tian, Email: tiangang@xjtu.edu.cn.
References
- 1. Francisco B, Ortega CJL, Steven N. Obesity and cardiovascular disease. Circ Res. 2016;118:1752–1770. doi: 10.1161/CIRCRESAHA.115.306883 [DOI] [PubMed] [Google Scholar]
- 2. Juge‐Aubry CE, Henrichot E, Meier CA. Adipose tissue: a regulator of inflammation. Best Pract Res Clin Endocrinol Metab. 2005;19:547–566. doi: 10.1016/j.beem.2005.07.009 [DOI] [PubMed] [Google Scholar]
- 3. Aslanabadi N, Salehi R, Javadrashid A, Tarzamani M, Khodadad B, Enamzadeh E, Montazerghaem H. Epicardial and pericardial fat volume correlate with the severity of coronary artery stenosis. J Cardiovasc Thorac Res. 2014;6:235–239. doi: 10.15171/jcvtr.2014.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Fox CS, Gona P, Hoffmann U, Porter SA, Salton CJ, Massaro JM, Levy D, Larson MG, D'Agostino RB, O'Donnell CJ, et al. Pericardial fat, intra‐thoracic fat, and measures of left ventricular structure and function: the Framingham Heart Study. Circulation. 2009;119:1586–1591. doi: 10.1161/CIRCULATIONAHA.108.828970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Gabriel B, Bernasochi WCB, Curl CL, Upasna VSP, Tare M, Parry LJ, Evdokia DSBH, Nalliah CJ, Jonathan M. Pericardial adipose and aromatase: a new translational target for aging, obesity and arrhythmogenesis? J Mol Cell Cardiol. 2017;10:111 [DOI] [PubMed] [Google Scholar]
- 6. Wang CY, Li SJ, Wu TW, Lin HJ, Chen JW, Mersmann HJ, Ding ST, Chen CY. The role of pericardial adipose tissue in the heart of obese minipigs. Eur J Clin Invest. 2018;48:e12942. doi: 10.1111/eci.12942 [DOI] [PubMed] [Google Scholar]
- 7. Li SJ, LW SJ, Chien MJ, Mersmann HJ, Chen CY. Involvement of pericardial adipose tissue in cardiacfibrosis of dietary‐induced obese minipigs—role of mitochondrial function. Biochim Biophys Acta Mol Cell Biol Lipids. 2019;7:957–965 [DOI] [PubMed] [Google Scholar]
- 8. Martinez‐Abundis E, Rajapurohitam V, Haist JV, Gan XT, Karmazyn M. The obesity‐related peptide leptin sensitizes cardiac mitochondria to calcium‐induced permeability transition pore opening and apoptosis. PLoS One. 2012;7:e41612. doi: 10.1371/journal.pone.0041612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Pchejetski D, Foussal C, Alfarano C, Lairez O, Calise D, Guilbeau‐Frugier C, Schaak S, Seguelas MH, Wanecq E, Valet P, et al. Apelin prevents cardiac fibroblast activation and collagen production through inhibition of sphingosine kinase 1. Eur Heart J. 2012;33:2360–2369. doi: 10.1093/eurheartj/ehr389 [DOI] [PubMed] [Google Scholar]
- 10. Boal F, Roumegoux J, Alfarano C, Timotin A, Calise D, Anesia R, Drougard A, Knauf C, Lagente C, Roncalli J, et al. Apelin regulates FoxO3 translocation to mediate cardioprotective responses to myocardial injury and obesity. Sci Rep. 2015;5:16104. doi: 10.1038/srep16104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Foussal C, Lairez O, Calise D, Pathak A, Guilbeau‐Frugier C, Valet P, Parini A, Kunduzova O. Activation of catalase by apelin prevents oxidative stress‐linked cardiac hypertrophy. FEBS Lett. 2010;584:2363–2370. doi: 10.1016/j.febslet.2010.04.025 [DOI] [PubMed] [Google Scholar]
- 12. Li H, Wang YP, Zhang LN, Tian G. Perivascular adipose tissue‐derived leptin promotes vascular smooth muscle cell phenotypic switching via p38 mitogen‐activated protein kinase in metabolic syndrome rats. Exp Biol Med. 2014;239:1–12. doi: 10.1177/1535370214527903 [DOI] [PubMed] [Google Scholar]
- 13. Payne GA, Borbouse L, Kumar S. Epicardial perivascular adipose‐derived leptin exacerbates coronary endothelial dysfunction in metabolic syndrome via a protein kinase C‐beta pathway. Arterioscler Thromb Vasc Biol. 2010;30:1711–1717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Li Y, Li Q, Wang Z, Liang DI, Liang S, Tang X, Guo L, Zhang R, Zhu D. 15‐HETE suppresses K(+) channel activity and inhibits apoptosis in pulmonary artery smooth muscle cells. Apoptosis. 2009;14:42–51. doi: 10.1007/s10495-008-0286-6 [DOI] [PubMed] [Google Scholar]
- 15. Yan X, Xun M, Wu L, Du X, Zhang F, Zheng J. DRm217 attenuates myocardial ischemia‐reperfusion injury via stabilizing plasma membrane Na+‐K+‐ATPase, inhibiting Na+‐K+‐ATPase/ROS pathway and activating PI3K/Akt and ERK1/2. Toxicol Appl Pharmacol, 2018;349:62–71. doi: 10.1016/j.taap.2018.04.030 [DOI] [PubMed] [Google Scholar]
- 16. Bilgin M, Neuhof C, Doerr O, Benscheid U, Andrade SS, Most A, Abdallah Y, Parahuleva M, Guenduez D, Oliva ML, et al. Bauhinia bauhinioides cruzipain inhibitor reduces endothelial proliferation and induces an increase of the intracellular Ca2+ concentration. J Physiol Biochem. 2010;66:283–290. doi: 10.1007/s13105-010-0032-8 [DOI] [PubMed] [Google Scholar]
- 17. Payne GA, Bohlen HG, Dincer UD. Periadventitial adipose tissue impairs coronary endothelial function via PKC‐beta‐dependent phosphorylation of nitric oxide synthase. Am J Physiol Heart Circ Physiol. 2009;297:H460–H465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ramirez‐Ortega M, Zarco G, Maldonado V, Carrillo JF, Ramos P, Ceballos G, Melendez‐Zajgla J, Garcia N, Zazueta C, Chanona J, et al. Is digitalis compound‐induced cardiotoxicity, mediated through guinea‐pig cardiomyocytes apoptosis? Eur J Pharmacol. 2007;566:34–42 [DOI] [PubMed] [Google Scholar]
- 19. Si Y, Cui Z, Liu J, Ding Z, Han C, Wang R, Liu T, Sun L. Pericardial adipose tissue is an independent risk factor of coronary artery disease and is associated with risk factors of coronary artery disease. J Int Med Res. 2020;48:1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Yang TT, Fish AF, Kong WM, Gao X, Huang J, Feng JT, Zhu JY, Chen T, Lou QQ. Correlates of pericardial adipose tissue volume using multidetector CT scanning in cardiac patients in China. Int J Cardiol. 2017;6:114 [DOI] [PubMed] [Google Scholar]
- 21. Liu J, Fox CS, Hickson D. Pericardial adipose tissue, atherosclerosis, and cardiovascular disease risk factors. Cardiovasc Metab Risk. 2010;33:1635–1639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Knight JA. Diseases and disorders associated with excess body weight. Ann Clin Lab Sci. 2011;41:107–121 [PubMed] [Google Scholar]
- 23. Chen HH, Tseng YJ, Wang SY, Tsai YS, Chang CS, Kuo TC, Yao WJ, Shieh CC, Wu CH, Kuo PH. The metabolome profiling and pathway analysis in metabolic healthy and abnormal obesity. Int J Obes (Lond). 2015;39:1241–1248. doi: 10.1038/ijo.2015.65 [DOI] [PubMed] [Google Scholar]
- 24. Goossens GH. The metabolic phenotype in obesity: fat mass, body fat distribution, and adipose tissue function. Obes Facts. 2017;10:207–215. doi: 10.1159/000471488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. González‐Muniesa P, Mártinez‐González MA, Hu FB, Després J‐P, Matsuzawa Y, Loos RJF, Moreno LA, Bray GA, Martinez JA. Obesity. Nat Rev Dis Primers. 2017;3:17034. doi: 10.1038/nrdp.2017.34 [DOI] [PubMed] [Google Scholar]
- 26. Zeng H, Vaka VR, He X. High‐fat diet induces cardiac remodelling and dysfunction: assessment of the role played by SIRT3 loss. J Cell Mol Med. 2015;19:1847–1856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Fitzgibbons TP, Czech MP. Epicardial and perivascular adipose tissues and their influence on cardiovascular disease: basic mechanisms and clinical associations. J Am Heart Assoc. 2014;3:e000582. doi: 10.1161/JAHA.113.000582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Al‐Dibouni A, Gaspar R, Ige S, Boateng S, Cagampang FR, Gibbins J, Cox RD, Sellayah D. Unique genetic and histological signatures of mouse pericardial adipose tissue. Nutrients. 2020;12:1855. doi: 10.3390/nu12061855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zeidan A, Hunter JC, Javadov S, Karmazyn M. mTOR mediates RhoA‐dependent leptin‐induced cardiomyocyte hypertrophy. Mol Cell Biochem. 2011;352:99–108. doi: 10.1007/s11010-011-0744-2 [DOI] [PubMed] [Google Scholar]
- 30. Huby AC, Antonova G, Groenendyk J, Gomez‐Sanchez CE, Bollag WB, Filosa JA, de Chantemèle EJB. The adipocyte‐derived hormone leptin is a direct regulator of aldosterone secretion, which promotes endothelial dysfunction and cardiac fibrosis. Circulation. 2015;132:2134–2145. doi: 10.1161/CIRCULATIONAHA.115.018226 [DOI] [PubMed] [Google Scholar]
- 31. Huang F, Xiong X, Wang H. Leptin‐induced vascular smooth muscle cell proliferation via regulating cell cycle, activating ERK1/2 and NF‐kappa B. Acta Biochim Biophys Sin (Shanghai). 2010;42:325–331 [DOI] [PubMed] [Google Scholar]
- 32. Shin EJ, Schram K, Zheng XL, Sweeney G. Leptin attenuates hypoxia/reoxygenation‐induced activation of the intrinsic pathway of apoptosis in rat H9c2 cells. J Cell Physiol. 2009;221:490–497. doi: 10.1002/jcp.21883 [DOI] [PubMed] [Google Scholar]
- 33. Eguchi M, Liu Y, Shin E‐J, Sweeney G. Leptin protects H9c2 rat cardiomyocytes from H2O2‐induced apoptosis. FEBS J. 2008;275:3136–3144. doi: 10.1111/j.1742-4658.2008.06465.x [DOI] [PubMed] [Google Scholar]
- 34. Kenneth R, McGaffin CSM, McTiernan CF. Leptin signaling in the failing and mechanically unloaded human heart. Circ Heart Fail. 2009;2:676–683. doi: 10.1161/CIRCHEARTFAILURE.109.869909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Doehner W, Pflaum CD, Rauchhaus M, Godsland IF, Egerer K, Cicoira M, Florea VG, Sharma R, Bolger AP, Coats AJ, et al. Leptin, insulin sensitivity and growth hormone binding protein in chronic heart failure with and without cardiac cachexia. Eur J Endocrinol. 2001;145:727–735. doi: 10.1530/eje.0.1450727 [DOI] [PubMed] [Google Scholar]
- 36. Lund LH, Freda P, Williams JJ, LaManca JJ, LeJemtel TH, Mancini DM. Leptin resistance after heart transplantation. Eur J Heart Fail. 2010;12:516–520. doi: 10.1093/eurjhf/hfq026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Fonfara S, Hetzel U, Tew SR, Dukes‐Mcewan J, Cripps P, Clegg PD. Leptin expression in dogs with cardiac disease and congestive heart failure. J Vet Intern Med. 2011;25:1017–1024. doi: 10.1111/j.1939-1676.2011.00782.x [DOI] [PubMed] [Google Scholar]
- 38. Fei H, Okano HJ, Li C, Lee GH, Zhao C, Darnell R, Friedman JM. Anatomic localization of alternatively spliced leptin receptors (Ob‐R) in mouse brain and other tissues. Proc Natl Acad Sci. 1997;94:7001–7005. doi: 10.1073/pnas.94.13.7001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Nascimento AF, Luvizotto RAM, Leopoldo AS, Lima‐Leopoldo AP, Seiva FR, Justulin LA, Silva MDP, Okoshi K, Wang XD, Cicogna AC. Long‐term high‐fat diet‐induced obesity decreases the cardiac leptin receptor without apparent lipotoxicity. Life Sci. 2011;88:1031–1038. doi: 10.1016/j.lfs.2011.03.015 [DOI] [PubMed] [Google Scholar]
- 40. Al Chekakie MO, Welles CC, Metoyer R, Ibrahim A, Shapira AR, Cytron J, Santucci P, Wilber DJ, Akar JG. Pericardial fat is independently associated with human atrial fibrillation. J Am Coll Cardiol. 2010;56:784–788. doi: 10.1016/j.jacc.2010.03.071 [DOI] [PubMed] [Google Scholar]
- 41. Thanassoulis G, Massaro JM, O'Donnell CJ, Hoffmann U, Levy D, Ellinor PT, Wang TJ, Schnabel RB, Vasan RS, Fox CS, et al. Pericardial fat is associated with prevalent atrial fibrillation: the Framingham Heart Study. Circ Arrhythm Electrophysiol. 2010;3:345–350. doi: 10.1161/CIRCEP.109.912055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Anaszewicz M, Wawrzeńczyk A, Czerniak B, Banaś W, Socha E, Lis K, Żbikowska‐Gotz M, Bartuzi Z, Budzyński J. Leptin, adiponectin, tumor necrosis factor α, and irisin concentrations as factors linking obesity with the risk of atrial fibrillation among inpatients with cardiovascular diseases. Kardiol Pol. 2019;77:1055–1061 [DOI] [PubMed] [Google Scholar]
- 43. Wang Q, Xi W, Yin L, Wang J, Shen H, Gao Y, Min J, Zhang Y, Wang Z. Human epicardial adipose tissue cTGF expression is an independent risk factor for atrial fibrillation and highly associated with atrial fibrosis. Sci Rep. 2018;8:3585. doi: 10.1038/s41598-018-21911-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Fukui A, Takahashi N, Nakada C, Masaki T, Kume O, Shinohara T, Teshima Y, Hara M, Saikawa T. Role of leptin signaling in the pathogenesis of angiotensin II–mediated atrial fibrosis and fibrillation. Circ Arrhythm Electrophysiol. 2013;6:402–409. doi: 10.1161/CIRCEP.111.000104 [DOI] [PubMed] [Google Scholar]
- 45. Pallepati P, Averill‐Bates DA. Mild thermotolerance induced at 40°C protects HeLa cells against activation of death receptor‐mediated apoptosis by hydrogen peroxide. Free Radic Biol Med. 2011;50:667–679. doi: 10.1016/j.freeradbiomed.2010.11.022 [DOI] [PubMed] [Google Scholar]
- 46. Chandra J, Samali A, Orrenius S. Triggering and modulation of apoptosis by oxidative stress. Free Radic Biol Med. 2000;29:323–333. doi: 10.1016/S0891-5849(00)00302-6 [DOI] [PubMed] [Google Scholar]
- 47. Skou JC. The influence of some cations on an adenosine triphosphatase from peripheral nerves. J Am Soc Nephrol. 1998;9:2170–2177. doi: 10.1681/ASN.V9112170 [DOI] [PubMed] [Google Scholar]
- 48. Bhavsar SK, Hosseinzadeh Z, Brenner D, Honisch S, Jilani K, Liu G, Szteyn K, Sopjani M, Mak TW, Shumilina E, et al. Energy‐sensitive regulation of Na+/K+‐ATPase by Janus kinase 2. Am J Physiol Cell Physiol. 2013;306:C374–C384 [DOI] [PubMed] [Google Scholar]
- 49. Xu ZW, Wang FM, Gao MJ, Chen XY, Hu WL, Xu RC. Targeting the Na/K‐ATPase 1 subunit of hepatoma hepG2 cell line to induce apoptosis and cell cycle arresting. Biol Pharm Bull. 2010;33:743–751 [DOI] [PubMed] [Google Scholar]
- 50. Pratt R, Brickman C, Cottrill C, Shapiro J, Liu J. The Na/K‐ATPase signaling: from specific ligands to general reactive oxygen species. Int J Mol Sci. 2018;19:2600. doi: 10.3390/ijms19092600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Liu J, Lilly MN, Shapiro JI. Targeting Na/K‐ATPase signaling: a new approach to control oxidative stress. Curr Pharm Des. 2018;24:359–364. doi: 10.2174/1381612824666180110101052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Yan Y, Shapiro AP, Haller S, Katragadda V, Liu L, Tian J, Basrur V, Malhotra D, Xie ZJ, Abraham NG, et al. Involvement of reactive oxygen species in a feed‐forward mechanism of Na/K‐ATPase‐mediated signaling transduction. J Biol Chem. 2013;288:34249–34258. doi: 10.1074/jbc.M113.461020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Dada LA, Chandel NS, Ridge KM, Pedemonte C, Bertorello AM, Sznajder JI. Hypoxia‐induced endocytosis of Na, K‐ATPase in alveolar epithelial cells is mediated by mitochondrial reactive oxygen species and PKC‐ζ. J Clin Invest. 2003;111:1057–1064. doi: 10.1172/JCI16826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Figtree GA, Karimi GK, Liu CC, Rasmussen HH. Oxidative regulation of the Naþ–Kþ pump in the cardiovascular system. Free Radical Biol Med. 2012;53:2263–2268. doi: 10.1016/j.freeradbiomed.2012.10.539 [DOI] [PubMed] [Google Scholar]
- 55. White CN, Liu CC, Garcia A, Hamilton EJ, Chia KKM, Figtree GA, Rasmussen HH. Activation of cAMP‐dependent signaling induces oxidative modification of the cardiac Na‐K pump and inhibits its activity. J Biol Chem. 2010;285:13712–13720. doi: 10.1074/jbc.M109.090225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Felippe Gonçalves‐de‐Albuquerque C, Ribeiro Silva A, Ignácio da Silva C, Caire Castro‐Faria‐Neto H, Burth P. Na/K pump and beyond: Na/K‐ATPase as a modulator of apoptosis and autophagy. Molecules. 2017;22:578. doi: 10.3390/molecules22040578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Tang G, Shen YI, Gao P, Song S‐S, Si L‐Y. Klotho attenuates isoproterenol‐induced hypertrophic response in H9C2 cells by activating Na+/K+‐ATPase and inhibiting the reverse mode of Na+/Ca2+‐exchanger. In Vitro Cell Dev Biol Anim. 2018;54:250–256. doi: 10.1007/s11626-017-0215-5 [DOI] [PubMed] [Google Scholar]
- 58. Yampolsky P, Koenen M, Mosqueira M, Geschwill P, Nauck S, Witzenberger M, Seyler C, Fink T, Kruska M, Bruehl C, et al. Augmentation of myocardial If dysregulates calcium homeostasis and causes adverse cardiac remodeling. Nat Commun. 2019;10:3295. doi: 10.1038/s41467-019-11261-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Gagan Bajaj RKS. TNF‐a‐mediated cardiomyocyte apoptosis involves caspase‐12 and calpain. BBRC. 2006;345:1558–1564 [DOI] [PubMed] [Google Scholar]
- 60. Inserte J, Garciadorado D, Hernando V, Barba I, Solersoler J. Ischemic preconditioning prevents calpain‐mediated impairment of Na+/K+‐ATPase activity during early reperfusion. Cardiovasc Res. 2006;70:364–373. doi: 10.1016/j.cardiores.2006.02.017 [DOI] [PubMed] [Google Scholar]
- 61. Li Y, Li Y, Feng Q, Arnold M, Peng T. Calpain activation contributes to hyperglycaemia‐induced apoptosis in cardiomyocytes. Cardiovasc Res. 2009;84:100–110. doi: 10.1093/cvr/cvp189 [DOI] [PubMed] [Google Scholar]
- 62. Shintani T, Higashi S, Suzuki R, Takeuchi Y, Ikaga R, Yamazaki T, Kobayashi K, Noda M. PTPRJ inhibits leptin signaling, and induction of PTPRJ in the hypothalamus is a cause of the development of leptin resistance. Sci Rep. 2017;7:11627. doi: 10.1038/s41598-017-12070-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Chen H, Li M, Liu L, Danjun Z, Gang T. Highlight article: telmisartan improves myocardial remodeling by inhibiting leptin autocrine activity and activating PPARgamma. Exp Biol Med (Maywood). 2020;245:654–666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Smith CCT, Mocanu MM, Davidson SM, Wynne AM, Simpkin JC, Yellon DM. Leptin, the obesity‐associated hormone, exhibits direct cardioprotective effects. Br J Pharmacol. 2006;149:5–13. doi: 10.1038/sj.bjp.0706834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Dudek H, Datta SR, Franke TF, Birnbaum MJ, Yao R, Cooper GM, Segal RA, Kaplan DR, Greenberg ME. Regulation of neuronal survival by the serine‐threonine protein kinase Akt. Science. 1997;275:661–665. doi: 10.1126/science.275.5300.661 [DOI] [PubMed] [Google Scholar]
- 66. Kennedy SG, Wagner AJ, Conzen SD, Jordan J, Bellacosa A, Tsichlis PN, Hay N. The PI3‐kinase/Akt signaling pathway delivers an anti‐apoptotic signal. Genes Dev. 1997;11:701–713. doi: 10.1101/gad.11.6.701 [DOI] [PubMed] [Google Scholar]
- 67. Barouch LA, Gao D, Chen L, Miller KL, Xu W, Phan AC, Kittleson MM, Minhas KM, Berkowitz DE, Wei C, et al. Cardiac myocyte apoptosis is associated with increased DNA damage and decreased survival in murine models of obesity. Circ Res. 2006;98:119–124. doi: 10.1161/01.RES.0000199348.10580.1d [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Tables S1–S2
Figures S1–S2