

Highlights
-
•
GNF-7, a multi-targeted kinase inhibitor, is highly potent against FGFR4.
-
•
GNF-7 and SIJ1263 are highly potent on Ba/F3 cells with wtFGFR4 or mtFGFR4.
-
•
GNF-7 and SIJ1263 are highly potent on HCC cells with FGFR4 activation.
-
•
GNF-7 and SIJ1263 strongly block FGFR signaling and induce apoptosis in HCC cells.
-
•
GNF-7 and SIJ1263 strongly suppress migration/invasion/AIG of HCC cells.
Keywords: FGFR4 kinase, HCC, FGFR4 inhibitor, GNF-7, SAR
Abbreviations: HCC, hepatocellular carcinoma; FGFR, fibroblast growth factor receptor; FGF, fibroblast growth factor; KLB, klotho beta; PARP, poly (ADP-ribose) polymerase; EMT, epithelial-mesenchymal transition
Abstract
Hepatocellular carcinoma (HCC) is disease with a high mortality rate and limited treatment options. Alterations of fibroblast growth factor receptor 4 (FGFR4) has been regarded as an oncogenic driver for HCC and a promising target for HCC therapeutics. Herein, we report that GNF-7, a multi-targeted kinase inhibitor, and its derivatives including SIJ1263 (IC50 < 1 nM against FGFR4) are highly potent FGFR4 inhibitors and are capable of strongly suppressing proliferation of HCC cells and Ba/F3 cells transformed with wtFGFR4 or mtFGFR4. Compared with known FGFR4 inhibitors, both GNF-7 and SIJ1263 possess much higher (up to 100-fold) anti-proliferative activities via FGFR signaling blockade and apoptosis on HCC cells. Especially, SIJ1263 is 80-fold more potent (GI50 = 24 nM) on TEL-FGFR4 V550E Ba/F3 cells than BLU9931, which suggests that SIJ1263 would be effective for overriding drug resistance. In addition, both substances strongly suppress migration/invasion and colony formation of HCC cells. It is worth noting that SIJ1263 is superior to GNF-7 with regards to the fact that activities of SIJ1263 are higher than those of GNF-7 in all assays performed in this study. Collectively, this study provides insight into designing highly potent FGFR4 inhibitors capable of potentially overcoming drug-resistance for the treatment of HCC patients.
Introduction
Hepatocellular carcinoma (HCC) is one of the diseases with high mortality1. Even though surgical treatments including liver resection and transplant increase survival for patients with HCC, most HCC patients have poor post-operative prognosis due to multiple relapses and intrahepatic metastasis. Despite advancements in therapeutic strategies against HCC, response rates and overall survival rates remain low2, 3. Since sorafenib, a multi-targeted kinase inhibitor, has been approved in 2007 as a targeted therapy for HCC patients4, other multi-targeted kinase inhibitors (lenvatinib5, regorafenib6, and cabozantinib7) and monoclonal antibodies (nivolumab8 and ramucirumab9) have been demonstrated to improve the overall survival benefit of patients10. However, there are still significant unmet medical needs11 in the treatment of HCC patients due to rapid emergence of drug resistance and off-target toxicities12, 13. Among the various molecular targets and pathways14, 15 involved in the initiation and progression of HCC, FGF19/FGFR4 signaling is significantly associated with HCC and particularly involved in drug-resistance in HCC[16], [17], [18]. Klotho beta (KLB) co-receptor is required for FGF19 binding specifically to FGFR4 and FGF19/FGFR4/KLB complex abundantly expressed in hepatocytes is associated with the progression of HCC19, 20. Therefore, blockage of FGFR4/FGF19 signaling has been regarded as an effective strategy against HCC21. To this end, selective and covalent FGFR4 inhibitors targeting Cys552 located in the hinge region of ATP site have been developed22, 23. Among irreversible-covalent FGFR4 inhibitors including BLU993124, BLU55425, 26, INCB62079, and H3B-652727, both BLU554 and H3B-6527 have entered clinical trials. FGF401, a reversible-covalent inhibitor, is also under clinical trials against on HCC[28], [29], [30]. Despite attempts to treat HCC patients with selective FGFR4 inhibitors, unmet needs for current HCC therapeutics including drug resistance remain31. Considerable efforts have been devoted to overcoming drug resistance in HCC16, 17, 32. In pursuit of overriding drug resistance that contributes to the progression of aggressive HCC, multi-targeted strategy such as multi-targeted pan-FGFR inhibitor would be more effective than single-targeted therapeutics33, 34. It is of note that FGFR3 has been reported to be associated with higher-grade, poorly differentiated HCC21, 35. In addition, the gatekeeper mutation V550E and molecular brake mutation N535K of FGFR4 are implicated with drug resistance36, which suggests that potent FGFR inhibitors against mutants of FGFR4 are necessary to override the drug resistance25, 29, 36.
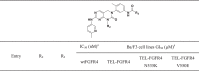
We have previously reported GNF-7, a multi-targeted type II kinase inhibitor, possesses excellent potency against Bcr-Abl T315I gatekeeper mutant37 and suppresses potently and selectively AML cells expressing NRAS mutant38, 39. In addition, it strongly blocks strongly proliferation of cancer cells harboring BRAF class I/II/III mutations40. Type II kinase inhibitors such as imatinib and GNF-7 bind to ATP-binding pocket of the kinase with the “DFG-out” inactive conformation. We found that GNF-7 is highly potent against FGFR1-4 in biochemical kinase assay and possesses potent anti-proliferative activities against various HCC cells. In contrast to GNF-7, most pan-FGFR inhibitors such as PD173074, BGJ398, and AZD4547 bind to the ATP-binding pocket of the “DFG-in” active conformation41. These pan-FGFR inhibitors are potent against FGFR1-3 but have limited activities against FGFR4 in contrast to GNF-7. It worth recalling that FGFR3 and FGFR4 are mainly expressed in liver tissue and involved in the mechanism of tumorigenesis in HCC42. Especially, FGFR3 has been reported to be associated with higher-grade, poorly differentiated HCC21, 35.
Herein, we report FGFR4-inhibitory activities of GNF-7 and its 23 derivatives against TEL-FGFR4, TEL-FGFR4 N535K, TEL-FGFR4 V550E Ba/F3 and HCC cell lines harboring FGFR4. We found that among the 23 synthetic GNF-7 derivatives, which we have previously reported by us for other studies39, both GNF-7 and SIJ1263 possess excellent enzymatic activities against FGFR4 and have remarkable anti-proliferative activities on HCC cells as well as on Ba/F3 cells transformed with wtFGFR4 and mtFGFR4. In addition, both substances are capable of blocking the FGFR signaling and suppressing the migration and invasion capacity of HCC cells.
Materials and Methods
Chemistry
General Information
Unless otherwise described, all commercial reagents and solvents were purchased from commercial suppliers and used without further purification. All reactions were performed under a N2 atmosphere in flame-dried glassware. Reactions were monitored by using TLC with 0.25 mm E. Merck precoated silica gel plates (60 F254). Reaction progress was monitored by using TLC analysis using a UV lamp, ninhydrin, or p-anisaldehyde stain for detection purposes. All solvents were purified by using standard techniques. Purification of reaction products was carried out by using silica gel column chromatography with Kieselgel 60 Art. 9385 (230–400 mesh). Purities of all compounds were > 95%, and mass spectra and purities of all compounds was accessed using Waters LC/MS system (Waters QDA Detector, Waters 2998 Photodiode Array Detector, Waters SFO System Fluidics Organizer, Water 2545 Binary Gradient Module, Waters 2767 Sample Manager) using SunFire™ C18 column (4.6 × 50 mm, 5 μm particle size): solvent gradient = 90% A at 0 min, 0% A at 5 min. Solvent A = 0.10% TFA in H2O; Solvent B = 0.10% TFA in MeOH; flow rate: 0.8 mL/min. 1H and 13C NMR spectra were obtained using Bruker 400 MHz FT-NMR (400 MHz for 1H, and 100 MHz for 13C) spectrometer and Bruker 300 MHz FT-NMR (300 MHz for 1H, and 75.5 MHz for 13C). Standard abbreviations are used for denoting the signal multiplicities. The synthesis of all compounds is described in our previous report39.
Molecular Docking Study
Co-crystal structures of the wtFGFR4 (PDB code: 4QRC) were retrieved from the Protein Data Bank. The retrieved protein-ligand structures were loaded into Maestro software (Schrödinger Release 2020-4). Protein Preparation Wizard was used for addition of all hydrogens, assignment of bond orders, deletion of all water molecules, and filling of missing residue and loops. Restrained energy minimization was applied using the OPLS3e force field. Docking study of GNF-7 and SIJ1263 on wtFGFR4 kinase domain was carried out using GLIDE module. GNF-7 and SIJ1263 were prepared using the LigPrep module. A docking grid defining FGFR4 kinase domain was generated mainly considering the binding pocket of the wtFGFR4 inhibitors.
Cell culture and reagents
AN3-CA and J82 cells were obtained from ATCC (Manassas, VA, USA). KMS-11 cells were purchased from JCRB. HEP3B, HUH7, HEPG2, MDA-MB-453 and SK-HEP1 cells were purchased from KCLB (Seoul, Korea). HEP3B, AN3-CA, MDA-MB-453 and SK-HEP1 cells were cultured in DMEM (#LM001-05, Welgene). HUH7, HEPG2, J82 and KMS-11 cells were cultured in RPMI1640 (#LM011-01, Welgene). The culture media were supplemented with 10% fetal bovine serum (#S001-01, Welgene), antibiotic-antimycotic solution (#LS203-01, Welgene) containing 10,000U/mL penicillin. The cells were maintained in a humidified atmosphere containing 5% CO2 at 37 °C.
Biochemical in vitro kinase assay
The biochemical inhibitory kinase activity on FGFR1-4 protein kinase were performed at Reaction Biology Corp. Compounds were tested with ATP (10 µΜ) in a 10-dose IC50 mode with 3-fold serial dilution.
Kinome profiling
Kinome-wide profiling was conducted by Reaction Biology Corp. SIJ1263 (0.1 μM) was tested against 317 recombinant human kinases.
Cell proliferation assay
Cells (5 × 103) were plated in 96-well tissue culture plates. Each compound was added to each well at 10 dose points of 3-fold serial dilution. After 72 h, the cellular viability was determined by CellTiter-Glo reagent (# G7572, Promega). Cell proliferation was assessed by measuring the luminescence using a 96-well plate reader (ENVISION). GI50 values were calculated using GraphPad Prism 8.0 software.
RT-PCR
Total RNAs were isolated from HUH7, HEP3B, HEPG2, SK-HEP1, SNU475 and SNU398 cells using TRIzol reagent (#15596026, Invitrogen) according to the manufacturer's instruction. cDNA was synthesized from total RNAs (2 μg) using M-MLV reverse transcriptase (#M170B, Promega). cDNA was amplified using the following primers: hFGFR4 5’- GGCCTCCAGTCCGGTGACAAGC-3’(Forward) hFGFR4 5’- CCACAGCGTTCTCTACCAGG-3’(Reverse) hGAPDH 5’- GGATTTGGTCGTATTGGG-3’(Forward) hGAPDH 5’- GGAAGATGGTGATGGGATT-3’(Reverse)
Cell lysis and western blotting
Cells were extracted and lysed using RIPA buffer, containing 50 mM HEPES (pH 7.4), 1% Triton X-100, 2 mM EDTA, 150 mM NaCl, 2.5 mM NaF, 5 mM Na3VO4, and protease inhibitor cocktail solution (#11-878-5-001, Roche). Protein concentrations were determined by the bradford assay. Proteins were separated using SDS-PAGE electrophoresis and transferred to the NC membrane. Membranes were blocked with 5% skim milk in TBS-T buffer. Polyclonal rabbit antibodies against FGFR4, pFGFR, pERK, pFRS2a, and pPLCγ, were purchased from Cell Signaling Technologies, and polyclonal anti-β-actin rabbit antibodies were purchased from Santa Cruz Biotechnology. Membranes were incubated in primary antibody solution overnight at 4 °C and then in secondary antibody solution for 1 h. Proteins were detected using ECL reagents. ImageJ software program was used for the quantification of western blot analysis.
Cell cycle arrest
Cells were cultured on 60 mm plates at 1 × 105 cells. Cells were treated with GNF-7 for 18 h and fixed using 70% ethanol. Before analyzing the cells using flow cytometry, cells were suspended in 300 μL phosphate-buffered saline solution containing 50 μg/mL propidium iodide.
Apoptosis assay
Cells (1 × 105) were prepared and incubated for 24 h. According to the manufacturer's instruction (Thermo Fisher Scientific), cells were harvested and stained using Alexa Fluor 488 conjugated annexin V (# A13201) and propidium iodide (# P3560). Cells were analyzed using flow cytometry.
Migration assay
Cells were grown in a 6-well plate until 100% confluency on a single layer and scraped with a SPLScarTM Scratcher. The cell layer was washed with media. Media containing concentrations of compounds were added. Cell migration to the scraped area was monitored, and the images of the cells were captured using a microscope.
Invasion assay
The invasion assay was conducted using a CHEMICON QCM 24-well infiltration analysis kit (ECM 554, Chemicon International). Cancer cells were seeded in 8 μm ECMatrix™-coated transwell chambers at a concentration of 5.0 × 104 cells/chamber after 12 h of serum starvation. Cells were incubated at 5% CO2, 37 °C after treating compounds. Invaded cells were stained with 0.005% crystal violet dissolved in phosphate-buffered saline.
Soft agar growth transformation assay
Anchorage-independent growth was evaluated in soft agar for colony formation. Equal volumes of agar (1%) and Dulbecco's modified Eagle medium were mixed at 40 °C, and 0.5% basic agar was produced on a 6-well plate. Cells were suspended in 2 × Dulbecco's modified Eagle medium and 0.7% agar. Next, the cell suspension was added to each well at a final concentration of 5 × 103 cells/well. The upper agar was then covered with the culture medium. Plates were incubated for 3 weeks at 5% CO2 and 37 °C. Cells were then stained with iodonitrotetrazolium chloride (#I10406, Sigma Aldrich) dissolved in phosphate-buffered saline, and the images were acquired.
Statistical analysis
Statistical analysis was performed using GraphPad Prism 8.0. The data were presented as the mean ± S.D. Student's t-test was used to evaluate the significance between two experimental condition, and p < 0.001 was statistically significant.
Results and Discussion
Enzymatic activities of GNF-7 against FGFR1-4 and their mutants
We performed biochemical kinase assays for GNF-7 against FGFR1-4 and their mutants based on our previous report37 that GNF-7 possesses anti-proliferative activity on Ba/F3 cells transformed with FGFR3. The results show that activities of GNF-7 against wtFGFR1-3, FGFR2 N549H, and FGFR3 G697C are comparable (IC50 = 7 to 20 nM) to those of PD173074. It is worth noting that GNF-7 is much more potent against wtFGFR4 (IC50 = 4 nM) and FGFR3 K650M43 (IC50 = 25 nM) than PD173074. Collectively, GNF-7 turned out to be potent against pan-FGFR including wtFGFR4 (IC50 = 4 nM) while PD173074, a pan-FGFR inhibitor, possesses a mild activity (IC50 = 97 nM) against wtFGFR4 (Table 1).
Table 1.
In vitro biochemical IC50 values of GNF-7 against FGFRs
| Kinases | Status | IC50 (µM) a | |
|---|---|---|---|
| GNF-7 | PD173074 | ||
| FGFR1 | WT | 0.011 | 0.005 |
| V561M | 7.538 | 4.650 | |
| FGFR2 | WT | 0.020 | 0.004 |
| N549H | 0.007 | 0.005 | |
| FGFR3 | WT | 0.010 | 0.007 |
| K650E | 0.140 | N.D.b | |
| G697C | 0.008 | 0.008 | |
| K650M | 0.025 | 0.291 | |
| FGFR4 | WT | 0.004 | 0.097 |
Radiometric biochemical kinase assay results.
Not Determined.
Anti-proliferative activities of GNF-7 on FGFR driven cancer cells
Given that GNF-7 is potent against FGFRs including FGFR4, we further investigated anti-proliferative activities of GNF-7 against FGFRs-driven cancer and FGFR4-overexpressed HCC cells (Table 2). Seven FGFRs-driven cancer cell lines were treated with GNF-7, PD173074, BLU9931, and BGJ398 for 72 h, viable cells were determined using the CellTiter-Glo (CTG) assay. Known FGFR inhibitors such as PD173074, BLU9931, and BGJ398 were also evaluated as reference compounds. Both PD173074 and BGJ398 bind to the ATP-binding pocket of the “DFG-in” active conformation and BLU9931 is an irreversible FGFR4 inhibitor that covalently binds to a unique Cys552 in FGFR4. Our results show that GNF-7 is capable of suppressing strongly proliferation FGFRs-driven cancer cell lines with GI50 values ranging 15 nM to 443 nM. All seven cancer cell lines tested are more sensitive to GNF-7 in dose-dependent manner than to PD173074 and BGJ398. The cellular activity of GNF-7 on J82 cells harboring FGFR3 K652E is over 20-fold higher than three references (BLU9931, PD173074, and BGJ398). Notably, anti-proliferative activities of GNF-7 on three HCC cell lines (HEP3B, HUH7, and HEPG2) harboring activated FGFR4 are 3 to 50 times higher than three references. It is worthwhile to note that GNF-7 is over 12-fold more potent on three HCC cells overexpressed with FGFR4 than BLU9931, a potent and selective FGFR4 inhibitor.
Table 2.
Anti-proliferative activities of GNF-7 against FGFR driven cancer cell lines
| Cell lines | FGFR Family | Status | Tissue origin | GI50 (µM) a | |||
|---|---|---|---|---|---|---|---|
| GNF-7 | BLU9931 | PD173074 | BGJ398 | ||||
| AN3-CA | FGFR2 | N549K/K310R | endometrium | 0.017 ± 0.00 | 9.200 ± 0.45 | 0.973 ± 2.55 | 0.057 ± 0.01 |
| KMS-11 | FGFR3 | Y373C | myeloma | 0.015 ± 0.00 | 0.541 ± 0.01 | 0.064 ± 0.02 | 0.143 ± 0.10 |
| J82 | FGFR3 | K652E | urinary bladder | 0.050 ± 0.01 | 5.871 ± 0.58 | 0.975 ± 0.45 | 2.843 ± 1.57 |
| HEP3B | FGFR4 | overexpression | HCC | 0.373 ± 0.15 | 4.221 ± 1.03 | 2.503 ± 0.49 | 1.369 ± 0.14 |
| HUH7 | FGFR4 | overexpression | HCC | 0.060 ± 0.01 | 0.715 ± 0.10 | 0.598 ± 0.24 | 0.157 ± 0.12 |
| HEPG2 | FGFR4 | overexpression | HCC | 0.366 ± 0.07 | > 10 | > 10 | 5.367 ± 1.10 |
| MDA-MB-453 | FGFR4 | overexpression | breast cancer | 0.443 ± 0.07 | 2.148 ± 0.38 | > 10 | 2.643 ± 0.28 |
All cancer cells were treated with inhibitors for 72 h in a dose dependent manner. Average GI50 values with S.D. (n = 3, duplicate) are shown.
Structure-Activity Relationships
We performed SAR studies with respect to growth-inhibitory capability. In order to determine the growth-inhibitory activities of GNF-7 and its derivatives against FGFR4 and their mutants, we utilized three Ba/F3 cell lines transformed with the TEL-FGFR4, TEL-FGFR4 N535K (molecular brake mutant) and TEL-FGFR4 V550E (gatekeeper mutant). In order to carry out the SAR studies, we first focused on the exploration of the R1 substituent as depicted in Table 3. The R1 substituent, phenyl group of 1 causes 18-fold decreased activity against TEL-FGFR4 V550E compared to that of GNF-7 while activities of 1 on both TEL-FGFR4 and TEL-FGFR4 N535K are comparable to those of GNF-7. Installation of the ethylpiperazine group (2, 3) adopted in the pan-FGFR inhibitor BGJ39841 brings about slightly increased activities on both TEL-FGFR4 and TEL-FGFR4 N535K compared with 1 and GNF-7. Among ten R1 substituents investigated, the R1 substituent bearing acetylpiperazine group of 4 results in the highest activities on TEL-FGFR4 (GI50 = 0.015 µM), TEL-FGFR4 N535K (GI50 = 0.036 µM), and TEL-FGFR4 V550E (GI50 = 0.247 µM). Introduction of the carbonylated ethylpiperazine group (5) gives rise to 3 to 8-fold less activities (GI50s = 0.135 to 1.563 µM) relative to 2 and 3, respectively. The pyrazole derivatives (6 and 7) result in comparable activities to those of GNF-7. Aliphatic groups (8 and 9) and hydrogen (10) cause substantially decreased activities compared to GNF-7. Collectively, it was found that the R1 group of 4 in the optimization of R1 substituent causes the highest anti-proliferative activities on the three Ba/F3 cells.
Table 3.
Anti-proliferative activities of GNF-7 and its derivatives on TEL-FGFR4, TEL-FGFR4 N535K and TEL-FGFR4 V550E Ba/F3 cell lines
 |
 |
aRadiometric biochemical kinase assay results.
bAll cells were treated with inhibitors for 72 h in a dose-dependent manner. Average GI50 values with S.D. (n = 3, duplicate).
cNot Determined.
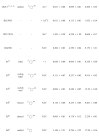
As shown in Table 4, we next investigated the effect of R2 groups on the anti-proliferative activity. Among R2 groups explored, only ethyl group (11) is comparable to the methyl group (GNF-7) in terms of anti-proliferative activities against TEL-FGFR4 (GI50 = 0.020 µM), TEL-FGFR4 N535K (GI50= 0.066 µM), and TEL-FGFR4 V550E (GI50 = 0.328 µM) Ba/F3 cell lines. Much bigger R2 groups such as cyclopropyl (12), cyclohexyl (13), benzyl (14), and phenyl (15) compared with the methyl group (GNF-7) results in 3 to 50-fold decreased anti-proliferative activities (GI50s = 0.084 to 12.39 µM). In particular, cyclohexyl group (13) causes almost no activity on TEL-FGFR4 V550E (GI50 = 12.39 µM) Ba/F3 cells. It was found that methyl group is the optimal R2 substituent in terms of cellular potency.
Table 4.
Anti-proliferative activities of GNF-7 derivatives on TEL-FGFR4, TEL-FGFR4 N535K and TEL-FGFR4 V550E Ba/F3 cell lines
 |
 |
 |
aRadiometric biochemical kinase assay results.
bAll cells were treated with inhibitors for 72 h in a dose-dependent manner. Average GI50 values with S.D. (n = 3, duplicate).
cNot Determined.
Next, we focused on the optimization studies for R3 group as described in Table 4. In order to assess effect of CF3 group in GNF-7, the activities of 16 lacking the CF3 group were evaluated. Derivative 16 exhibited 55 to 133-fold diminished anti-proliferative activities compared to GNF-7, which indicates that the CF3 group is essential for anti-proliferative activities on Ba/F3 cells transformed with wtFGFR4 and mtFGFR4. Additional functional groups were introduced at the meta and para position in the phenyl group bearing the CF3 group in GNF-7. The both substituents (4-piperidinol in 17 and 4-methylimidazole in 18) at meta-position result in largely comparable activities relative to GNF-7 and anti-proliferative activity of 18 on TEL-FGFR4 V550E cells is 2-fold higher than that of GNF-7.
The effects of additional substituents at para position of the tail phenyl ring in GNF-7 tail were also investigated. The results show that both methylpiperazine (19) and methylpiperazinylmethylene (20) substituents cause decreased anti-proliferative activities on Ba/F3 cells transformed with wtFGFR4 and FGFR4 N535K compared with those of GNF-7. However, the 3-(dimethylamino)pyrrolidinnylmethylene substituent (21) results in slightly enhanced anti-proliferative activities against TEL-FGFR4, TEL-FGFR4 N353K and TEL-FGFR4 V550E Ba/F3 cells (GI50 = 0.028 µM, 0.062 µM, and 0.233 µM, respectively) relative to those of GNF-7. On the basis of this results, the tail group of 21 was selected for further optimization.
Derivatives containing a combination of the optimal R1 (4 and 6) and R3 (21) substituent were evaluated on Ba/F3 cells (TEL-FGFR4, TEL-FGFR4 N535K and TEL-FGFR4 V550E) (Table 5). Compared with GNF-7, 22 has comparable anti-proliferative activities on Ba/F3 cells transformed with TEL-FGFR4 (GI50 = 0.019 µM) and TEL-FGFR4 N535K (GI 50 = 0.096 µM) while the activity of 22 on TEL-FGFR4 V550E Ba/F3 cells is higher than that of GNF-7. To our delight, SIJ1263 exhibits significantly enhanced anti-proliferative activities (TEL-FGFR4 GI50 = 0.006 µM, TEL-FGFR4 N535K GI 50 = 0.012 µM and TEL-FGFR4 V550E GI50 = 0.024 µM) compared to GNF-7. In particular, the anti-proliferative activity of SIJ1263 on TEL-FGFR4 V550E Ba/F3 cell was increased by 21-fold compared with that of GNF-7. From our investigation on SAR study, we have identified promising GNF-7 derivatives possessing excellent anti-proliferative activities on Ba/F3 cells transformed with FGFR4 and its mutant. Furthermore, 8 derivatives with excellent cellular activities (GI 50 < 50 nM) on TEL-FGFR4 Ba/F3 cells were selected to assess their enzymatic activities against FGFR4 in biochemical kinase assay. As a result, 2 has comparable enzymatic activity (IC50 = 5.82 nM) to that of GNF-7 (IC50 = 4.17 nM). In addition, enzymatic activities of other 7 derivatives (3, 4, 6, 7, 11, 22 and SIJ1263 are higher (IC50 < 1 nM) than that of GNF-7. It is worth noting that the enzymatic activities of these 7 derivatives are comparable to that of BLU9931, highly potent FGFR4 inhibitor. In conclusion, the combined SAR results reveal that SIJ1263 possesses the highest anti-proliferative activity against TEL-FGFR4, TEL-FGFR4 N535K and TEL-FGFR4 V550E Ba/F3 cells as well as highest enzymatic activity against wtFGFR4 Ba/F3 cells among the 23 GNF-7 derivatives.
Table 5.
Anti-proliferative activities of GNF-7 derivatives on TEL-FGFR4, TEL-FGFR4 N535K and TEL-FGFR4 V550E Ba/F3 cell lines
 |
aRadiometric biochemical kinase assay results.
bAll cells were treated with inhibitors for 72 h in a dose-dependent manner. Average GI50 values with S.D. (n = 3, duplicate).
Molecular docking studies of GNF-7 and SIJ1263 on wtFGFR4 and FGFR4 V550E
Molecular docking studies were carried out to predict the binding modes of GNF-7 and SIJ1263 on wtFGFR4 and FGFR4 V550E. The analysis of docking studies reveals that GNF-7 and SIJ1263 make a hinge contact through hydrogen bond with Ala553 of wtFGFR4 and FGFR4 V550E and also forms additional H-bond with Glu520, which is a key feature in the binding mode manifested by type II kinase inhibitors (Figure 1). Also, mutation of FGFR4 Val550 to Glu550 has little influence on the binding of GNF-7 and SIJ1263 (Figure 1. B, D). In particular, SIJ1263 forms additional hydrogen bond with backbone carbonyl group of Ile609, which contributes to activity against wtFGFR4 and FGFR4 V550E (Figure 1. B, D). Based on the docking studies, we concluded that both GNF-7 and SIJ1263 would be active on wtFGFR4 and FGFR4 V550E, which is consistent with results obtained from the biochemical kinase assays and cellular activity against TEL-FGFR4 and TEL-FGFR4 V550E Ba/F3 cells.
Figure 1.

Predicted docking models of GNF-7 and SIJ1263 on wtFGFR4 and FGFR4 V550E (PDB: 4QRC) and kinome-wide inhibition profiling of SIJ1263. Binding model of GNF-7 on (A) wtFGFR4 and (B) FGFR4 V550E. Binding model of SIJ1263 on (C) wtFGFR4 and (D) FGFR4 V550E. The dash lines indicate hydrogen bond interactions. (E) Kinome-wide inhibition profiling of SIJ1263 at 0.1μM. Kinases showing > 90% inhibition were indicated by red circles. Illustration is reproduced courtesy of Cell Signaling Technology, Inc. (www.cellsignal.com). (F) List of 37 kinases inhibited > 90% by SIJ1263 out of 317 kinases.
Kinome-wide inhibition profiling of SIJ1263
Among 23 derivatives, SIJ1263 has most potent enzymatic activity and highest cellular activity on TEL-FGFR4 and TEL-FGFR4 V550E Ba/F3 cells. We performed kinome-wide inhibition profiling of SIJ1263 at 0.1 µM concentration against 317 kinases. Among the 317 kinases, 37 kinases are inhibited more than 90% (S-Score (10) = 0.111) by SIJ1263, which include several tyrosine kinases (SRC, BTK, FER, LYNB, YES, RET, EPHB3, LYN, EPHB1, EPHB2, CSK, ABL2, ABL1, FGR, EPHA2, JAK2, PDGFRa, FES, ERBB4, LCK, ERBB2, FYN, EPHA1, c-Kit, FLT3, PDGFRb, TYK2) and other kinases (LIMK1, RSK1, MLK1, NEK4, p38a, NEK11) as well as FGFR1-4 (Figure 1. E, F). The kinome-wide selectivity profiling reveals that SIJ1263 is capable of inhibiting several kinases besides FGFR1-4, indicating that off-target effects are associated with the anti-proliferation activities of SIJ1263.
Anti-proliferative activities of GNF-7 and SIJ1263 on HCC cells expressing high level of FGFR4
Increased FGFR4 mRNA and protein expression has been reported in various cancers including HCC18, 44. Based on the results obtained from our SAR study using Ba/F3 cells transformed with wtFGFR4 and mtFGFR4, we decided to examine the effects of GNF-7 analogues on HCC cells. To this end, we first measured FGFR4 protein and mRNA levels in six HCC cells by RT-PCR and western blot analysis and found that FGFR4 is overexpressed in HUH7, HEP3B and HEPG2 cells among the six HCC cells (Figure 2. A-D). Also, we investigated whether the levels of FGFR4 in HCC cells are correlated with the anti-proliferative activities of GNF-7 on the HCC cells and observed that the three HCC cells (HUH7, HEP3B, and HEPG2) expressing high level of FGFR4 are more sensitive to cell death induced by GNF-7 than other HCC cells (SNU449, SK-HEP1, and SNU398) with lower level of FGFR4 (Figure 2. E).
Figure 2.

FGFR4 expression levels in 6 HCC cell lines. (A, C) FGFR4 mRNA expression levels and (B, D) protein expression levels. (E) Percent inhibition of GNF-7 (0.5 μM) on six HCC cells. The correlations between the expression level of FGFR4 and anti-proliferative activity of GNF-7 on HCC cells. The values are represented as the percentage of viable cells compared with DMSO-treated control.
Next, the anti-proliferative activities of GNF-7 analogues on human HCC cell lines, HUH7, HEP3B and HEPG2 were assessed (Table 6). The four GNF-7 derivatives, 2, 3, 4, and 6 have excellent potencies on HUH7 cells (GI50s range from 0.020 to 0.025 µM) and high activities on HEP3B and HEPG2 cells (GI50s range from 0.115 to 0.252 µM and 0.342 to 0.440 µM). In particular, SIJ1263 possesses excellent anti-proliferative activities on HUH7, HEP3B and HEPG2 cells (GI50 = 0.006 µM, 0.008 µM and 0.076 µM respectively). These results show that SIJ1263 is superior to GNF-7 in terms of anti-proliferative activities on HCC cells, which indicates that we successfully identified promising GNF-7 analogue that has very strong anti-proliferative activities against FGFR4 overexpressed HCC cells. It is worth noting that SIJ1263 is capable of strongly suppressing proliferation of HCC cells as well as TEL-FGFR4, TEL-FGFR4 mutant Ba/F3 cells.
Table 6.
Anti-proliferative activities of GNF-7 and its derivatives on HUH7, HEP3B and HEPG2 cells
| Entry | HCC cells GI50 (µM)a | ||
|---|---|---|---|
| HUH7 | HEP3B | HEPG2 | |
| GNF-737,39,40 | 0.060 ± 0.01 | 0.373 ± 0.15 | 0.366 ± 0.07 |
| sorafenib | 9.589 ± 2.06 | 5.419 ± 1.12 | 8.564 ± 1.45 |
| BLU9931 | 0.715 ± 0.10 | 4.221 ± 1.03 | 20.33 ± 6.73 |
| PD173074 | 0.598 ± 0.24 | 2.503 ± 0.49 | 13.36 ± 3.60 |
| BGJ398 | 0.157 ± 0.12 | 1.369 ± 0.14 | 5.367 ± 1.10 |
| 139 | 0.066 ± 0.02 | 0.847 ± 0.18 | 1.887 ± 0.93 |
| 239 | 0.025 ± 0.02 | 0.115 ± 0.02 | 0.342 ± 0.20 |
| 339 | 0.021 ± 0.01 | 0.139 ± 0.02 | 0.440 ± 0.12 |
| 439 | 0.021 ± 0.01 | 0.159 ± 0.11 | 0.369 ± 0.16 |
| 539 | 0.073 ± 0.06 | 0.886 ± 0.34 | 0.748 ± 0.33 |
| 639 | 0.020 ± 0.00 | 0.252 ± 0.16 | 0.397 ± 0.22 |
| 739 | 0.056 ± 0.03 | 0.586 ± 0.17 | 0.597 ± 0.43 |
| 839 | 1.471 ± 0.75 | 5.732 ± 4.00 | 3.778 ± 0.58 |
| 939 | 0.607 ± 0.22 | 2.737 ± 0.54 | 3.574 ± 1.85 |
| 1039 | 0.531 ± 0.19 | 2.703 ± 1.12 | 2.692 ± 0.66 |
| 1139 | 0.057 ± 0.01 | 0.652 ± 0.07 | 1.313 ± 1.02 |
| 1239 | 0.158 ± 0.08 | 1.066 ± 0.46 | 0.878 ± 0.26 |
| 1339 | 0.093 ± 0.02 | 1.537 ± 0.92 | 1.579 ± 0.52 |
| 1439 | 0.118 ± 0.02 | 0.633 ± 0.14 | 1.272 ± 0.56 |
| 1539 | 0.162 ± 0.03 | 1.508 ± 0.74 | 15.91 ± 4.07 |
| 1639 | 1.390 ± 0.90 | 8.661 ± 3.51 | 6.688 ± 7.65 |
| 1739 | 0.037 ± 0.01 | 0.201 ± 0.07 | 0.743 ± 0.49 |
| 1839 | 0.037 ± 0.02 | 0.153 ± 0.02 | 0.879 ± 0.42 |
| 1939 | 0.070 ± 0.05 | 0.138 ± 0.02 | 0.876 ± 0.88 |
| 2039 | 0.173 ± 0.20 | 0.350 ± 0.21 | 1.324 ± 0.25 |
| 2139 | 0.034 ± 0.02 | 0.120 ± 0.03 | 0.458 ± 0.42 |
| 2239 | 0.021 ± 0.02 | 0.094 ± 0.02 | 0.405 ± 0.15 |
| SIJ1263 (23)39 | 0.006 ± 0.00 | 0.008 ± 0.00 | 0.076 ± 0.04 |
All cells were treated with inhibitors for 72 h in a dose dependent manner. Average GI50 values with S.D. (n = 3, duplicate) are shown.
Effects of GNF-7 and SIJ1263 on FGFR signaling in HUH7 and HEP3B cells
In order to examine the effects of GNF-7 and SIJ1263 on activation of FGFR and its downstream molecules in a cellular context, we carried out western blot analysis using HUH7 and HEP3B cells. As shown in Figure 3, both GNF-7 and SIJ1263 are capable of inhibiting significantly phosphorylation of FGFR and its downstream signaling molecules (PLCγ, FRS2α, AKT, and ERK1/2) in HUH7 and HEP3B cells in a dose-dependent manner, which indicates that both GNF-7 and SIJ1263 substantially suppress the activation of signaling molecules closely related with HCC cell survival18, 33. Moreover, both GNF-7 and SIJ1263 block FGFR signaling more significantly than PD173074, which is in agreement with their anti-proliferative activities (Figure 3. A-D). On the other hand, these compounds inhibit less strongly phosphorylation of FGFR and its downstream molecules in SK-HEP1 cells expressing low level of FGFR4 than in HUH7 and HEP3B cells expressing high level of FGFR4 (Figure 3. E, F). It is of note that SIJ1263 inhibits more strongly phosphorylation of FGFR downstream signaling molecules in HUH7 and HEP3B cell lines compared with GNF-7. Collectively, both GNF-7 and SIJ1263 are capable of blocking very strongly FGFR signaling, which is in accordance with their potent anti-proliferative activities on HUH7 and HEP3B cells.
Figure 3.

The effects of GNF-7 and SIJ1263 on FGFR signaling. Autophosphorylation of FGFR and its downstream signaling molecules is inhibited by GNF-7 and SIJ1263 in (A, B) HUH7 (C, D) HEP3B (E, F) SK-HEP1 cells. Bar graph shows the average and SD. *** p < 0.0001, ** p < 0.001, * p < 0.01).
GNF-7 and SIJ1263 induce apoptosis and cell cycle arrest in HCC cells
We next investigated whether apoptosis and cell cycle arrest are involved in anti-proliferative effects of GNF-7 and SIJ1263 on HCC cells. It was observed that both GNF-7 and SIJ1263 induce G0/G1 phase cell cycle arrest with decrease in the S phase on HUH7 and HEP3B cells (Figure 4. A, B). In addition, analysis of the FACS data reveals that both GNF-7 and SIJ1263 are capable of inducing apoptosis of HUH7 and HEP3B cells (Figure 4. D, F). On the other hand, both GNF-7 and SIJ1263 have little effects on induction of G0/G1 phase cell cycle arrest and apoptosis in SK-HEP1 cells (Figure 4. C, H, I). It is worth noting that SIJ1263 (0.1 or 0.5 µM) induces apoptosis of the two HCC cells more strongly than GNF-7 (1 or 5 µM) (Figure 4. D, F). In addition, both GNF-7 and SIJ1263 result in upregulation of cleaved PARP and cleaved caspase-3 in HEP3B and HUH7 cells (Figure 4. E, G), indicating that both GNF-7 and SIJ1263 effectively induce cell cycle arrest and apoptosis in FGFR4 overexpressed HCC cells.
Figure 4.

Cell cycle distribution and apoptosis induction. GNF-7 and SIJ1263 induced apoptosis and cell cycle arrest. Cell cycle distribution in (A) HUH7 cell line (B) HEP3B (C) SK-HEP1 cells after treatment of GNF-7 or SIJ1263 for 18 h. Treatment of GNF-7 and SIJ1263 induced apoptosis for 24 h in (D) HUH7, (F) HEP3B and (H) SK-HEP1 cells. Treatment of GNF-7 and SIJ1263 increased cleaved PARP and cleaved caspase-3 levels in (E) HUH7 (G) HEP3B cells, but not in (I) SK-HEP1 cells. Bar graph shows the average and SD. *** p < 0.0001, ** p < 0.001, * p < 0.01.
GNF-7 and SIJ1263 suppress migration and invasion of HCC cells
It has been reported that the FGF19-FGFR4-GSK3β axis plays a pivotal role in epithelial-mesenchymal transition (EMT)45, 46. Thus, the effect of GNF-7 and SIJ1263 on the migration capability of HCC cells was investigated by using scratch wound healing assay. The wound was almost completely healed after 36 h with DMSO-treatment and PD173074 (0.1 μM) moderately reduced the wound healing, whereas the wound healing area was substantially reduced with the treatment of low concentrations of GNF-7 (0.01 or 0.05 μM) and SIJ1263 (0.002 or 0.005 μM) in HUH7 and HEP3B cells (Figure 5. A, B), indicating that both substances at even low concentrations are capable of suppressing remarkably migration of HUH7 and HEP3B cells. In addition, both GNF-7 and SIJ1263 at even low concentrations remarkably attenuate invasion of HUH7 and HEP3B cells (Figure 5. D, E), while PD173074 has little effect on invasion capability of the HCC cells. Both GNF-7 and SIJ1263 at 0.5 or 5 μM concentration suppress less significantly migration and invasion of SK-HEP1 cells than those of HUH7 and HEP3B cells (Figure 5. C, F). It was observed that GNF-7 inhibits GSK3β phosphorylation in HUH7 and HEP3B cells (Figure 5. G), which suggests that both GNF-7 and SIJ1263 are capable of suppressing migration and invasion of HCC cells by suppressing phosphorylation of GSK3β. It is worthwhile to note that SIJ1263 is superior to GNF-7 in terms of capability to suppress migration and invasion of HCC cells.
Figure 5.

Suppression of cellular migration and invasion by GNF-7 and SIJ1263. Inhibitory effect of GNF-7 and SIJ1263 on the migration of (A) HUH7 and (B) HEP3B (C) SK-HEP1 cell lines. After a monolayer of cells was grown 100% confluence and scratched to create wound, the cells were treated with GNF-7 and SIJ1263. The migration of the cells was observed for 36 h or 24 h. Representative image of cells from three independent experiments in the original magnification was 4 × . Migration ratio was calculated using migration area using Image J. Invasion capabilities of (D) HUH7 and (E) HEP3B (F) SK-HEP1 cells were measured by GNF-7 and SIJ1263 using cell invasion assay (QCM™ Collagen Cell Invasion Assay) (G) Inhibition of migration/invasion by GNF-7 and SIJ1263 mediated pGSK3β inhibition. Bar graph shows the average and SD. *** p < 0.0001, ** p < 0.001, * p < 0.01.
Suppression of anchorage-independent growth by GNF-7 and SIJ1263
In order to investigate whether GNF-7 and SIJ1263 inhibit tumorigenesis formation in FGFR4 overexpressed HCC cells, we performed a soft agar assay using HUH7 (Figure 6. A, B), HEP3B (Figure 6. C, D) and SK-HEP1 (Figure 6. E, F) cells. It was observed that incubation with either GNF-7 (0.01 or 0.05 µM) or SIJ1263 (0.002 or 0.005 µM) for 3 weeks causes HUH7 and HEP3B cells to form in substantially lower colony numbers. Both GNF-7 and SIJ1263 suppress anchorage independent growth of HCC cells more significantly than PD173074. In particular, both GNF-7 (0.1 or 0.5 µM) and SIJ1263 (0.01 or 0.05 µM) are capable of suppressing almost completely colony-formation of HUH7 and HEP3B cells expressing high level of FGFR4, while GNF-7 and SIJ1263 at 5 µM concentration block colony formation of SK-HEP1 cells by 11% and 40%, respectively. It is worth noting that SIJ1263 is superior to GNF-7 in terms of capability to inhibit anchorage independent growth of HCC cells.
Figure 6.

Anchorage-independent growth inhibition of GNF-7 and SIJ1263 in HUH7, HEP3B and SK-HEP1 cell lines. (A) HUH7, (C) HEP3B and (E) SK-HEP1 cells seeded in 0.35% agar were incubated for 3 weeks at indicated concentration of GNF-7 and SIJ1263. And colonies were observed (B, D, F) average number of colonies per well was counted using Image J software. Bar graph shows the average (n = 3) and SD. ** p < 0.001, * p < 0.01.
Conclusion
HCC is disease with a high mortality rate with limited therapeutic options and a poor prognosis. Sorafenib has been approved as the standard treatment option for patients diagnosed with advanced HCC for a decade, but its use is limited due to development of drug resistance10, 18, 32. Recent studies have reported FGFR4 as an attractive molecular target for overcoming drug resistance for HCC and a few FGFR4 inhibitors are undergoing clinical trials16, 33. As part of our endeavor to expand applicability of GNF-7, we evaluated inhibitory activities of GNF-7 against FGFRs in vitro. These results showed that GNF-7, a type II multi-targeted kinase inhibitor, is capable of strongly inhibiting not only FGFR1-3, but also FGFR4. Based on the findings, we performed SAR study by assessing anti-proliferative activities of 23 derivatives of GNF-7 on HCC cells harboring FGFR4 activation as well as on Ba/F3 cells transformed with TEL-FGFR4, TEL-FGFR4 N535K, TEL-FGFR4 V550E. Among 23 derivatives, especially, it is noteworthy that SIJ1263 has extremely potent enzymatic activity (IC50 < 1 nM) against FGFR4 and possesses excellent anti-proliferative activities on TEL-FGFR4 (GI50 of 0.006 μM), TEL-FGFR4 N535K (GI50 of 0.012 μM), TEL-FGFR4 V550E (GI50 of 0.024 μM) Ba/F3 cells, HUH7 (GI50 of 0.006 μM), HEP3B (GI50 of 0.008 μM), and HEPG2 (GI50 of 0.076 μM) cells. Analysis of molecular docking studies reveals that SIJ1263, compared with GNF-7, forms additional hydrogen bond with backbone carbonyl group of Ile609 contributing to its enhanced activities against wtFGFR4 and FGFR4 V550E. Both GNF-7 and SIJ1263 possess much higher (up to 100-fold) anti-proliferative activities compared with known FGFR4 inhibitors and are capable of blocking strongly FGFR signaling and remarkably inducing G0/G1 arrest and apoptosis in HCC cells. Moreover, both substances remarkably suppress migration, invasion, and anchorage-independent growth of HCC cells. It is worth noting that SIJ1263 is superior to GNF-7 with regards to the fact that activities of SIJ1263 are higher than those of GNF-7 in all assays performed in this study. Also, it is worthwhile to recall that SIJ1263 is 80-fold more potent (GI50 = 24 nM) on TEL-FGFR4 V550E Ba/F3 cells than BLU9931, a highly selective and potent FGFR4 inhibitor, which suggests that SIJ1263 would be effective for overriding drug resistance caused by gatekeeper mutation of FGFR4.
We believe that the results would provide insights into designing highly potent FGFR4 inhibitors capable of potentially overcoming drug-resistance for the treatment of HCC patients.
Acknowledgments
Author Contributions
Y.N, I.S, Y.K, and S.R drafted the work. I.S and E.J synthesized SIJ1263 and its derivatives. Y.N conducted biological experiments. N.K conducted docking study. T.S conceived and supervised the manuscript. All authors have read and agreed to the published version of the manuscript.
Acknowledgments
This research was supported by Basic Science Research Program (NRF-2021R1A2C3011992) from the National Research Foundation in Korea, Korea Institute of Science and Technology (KIST), Brain Korea 21 Project, and the KU-KIST Graduate School of Converging Science and Technology Program.
References
- 1.Bray F., Ferlay J., Soerjomataram I., Siegel R.L., Torre L.A., Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA-Cancer J. Clin. 2018;68:394–424. doi: 10.3322/caac.21492. [DOI] [PubMed] [Google Scholar]
- 2.Huang S., He X. The role of microRNAs in liver cancer progression. Br. J. Cancer. 2011;104:235–240. doi: 10.1038/sj.bjc.6606010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kulik L., El-Serag H.B. Epidemiology and management of hepatocellular carcinoma. Gastroenterology. 2019;156:477–491. doi: 10.1053/j.gastro.2018.08.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Llovet J.M., Ricci S., Mazzaferro V., Hilgard P., Gane E., Blanc J.-F., De Oliveira A.C., Santoro A., Raoul J.-L., Forner A. Sorafenib in advanced hepatocellular carcinoma. N. Engl. J. Med. 2008;359:378–390. doi: 10.1056/NEJMoa0708857. [DOI] [PubMed] [Google Scholar]
- 5.Kudo M., Finn R.S., Qin S., Han K.-H., Ikeda K., Piscaglia F., Baron A., Park J.-W., Han G., Jassem J. Lenvatinib versus sorafenib in first-line treatment of patients with unresectable hepatocellular carcinoma: a randomised phase 3 non-inferiority trial. Lancet. 2018;391:1163–1173. doi: 10.1016/S0140-6736(18)30207-1. [DOI] [PubMed] [Google Scholar]
- 6.Bruix J., Qin S., Merle P., Granito A., Huang Y.-H., Bodoky G., Pracht M., Yokosuka O., Rosmorduc O., Breder V. Regorafenib for patients with hepatocellular carcinoma who progressed on sorafenib treatment (RESORCE): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. 2017;389:56–66. doi: 10.1016/S0140-6736(16)32453-9. [DOI] [PubMed] [Google Scholar]
- 7.Abou-Alfa G.K., Meyer T., Cheng A.-L., El-Khoueiry A.B., Rimassa L., Ryoo B.-Y., Cicin I., Merle P., Chen Y.H., Park J.-W., Blanc J.-F., Bolondi L., Klümpen H.-J., Chan S.L., Zagonel V., Pressiani T., Ryu M.-H., Venook A.P., Hessel C., Borgman-Hagey A.E., Schwab G., Kelley R.K. Cabozantinib in patients with advanced and progressing hepatocellular carcinoma. N. Engl. J. Med. 2018;379:54–63. doi: 10.1056/NEJMoa1717002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.El-Khoueiry A.B., Sangro B., Yau T., Crocenzi T.S., Kudo M., Hsu C., Kim T.-Y., Choo S.-P., Trojan J., Welling T.H., 3rd Nivolumab in patients with advanced hepatocellular carcinoma (CheckMate 040): an open-label, non-comparative, phase 1/2 dose escalation and expansion trial. Lancet. 2017;389:2492–2502. doi: 10.1016/S0140-6736(17)31046-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhu A.X., Kang Y.-K., Yen C.-J., Finn R.S., Galle P.R., Llovet J.M., Assenat E., Brandi G., Pracht M., Lim H.Y. Ramucirumab after sorafenib in patients with advanced hepatocellular carcinoma and increased α-fetoprotein concentrations (REACH-2): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2019;20:282–296. doi: 10.1016/S1470-2045(18)30937-9. [DOI] [PubMed] [Google Scholar]
- 10.Villanueva A., Llovet J.M. Second-line therapies in hepatocellular carcinoma: emergence of resistance to sorafenib. Clin. Cancer Res. 2012;18:1824–1826. doi: 10.1158/1078-0432.CCR-12-0151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Balogh J., Victor D., III, Asham E.H., Burroughs S.G., Boktour M., Saharia A., Li X., Ghobrial R.M., Monsour H.P., Jr Hepatocellular carcinoma: a review. J. Hepatocell. Carcinoma. 2016;3:41–53. doi: 10.2147/JHC.S61146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Llovet J.M., Villanueva A., Lachenmayer A., Finn R.S. Advances in targeted therapies for hepatocellular carcinoma in the genomic era. Nat. Rev. Clin. Oncol. 2015;12:408–424. doi: 10.1038/nrclinonc.2015.103. [DOI] [PubMed] [Google Scholar]
- 13.Jindal A., Thadi A., Shailubhai K. Hepatocellular carcinoma: etiology and current and future drugs. J. Clin. Exp. Hepatol. 2019;9:221–232. doi: 10.1016/j.jceh.2019.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Moeini A., Cornellà H., Villanueva A. Emerging signaling pathways in hepatocellular carcinoma. Liver cancer. 2012;1:83–93. doi: 10.1159/000342405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pinyol R., Nault J.C., Quetglas I.M., Zucman-Rossi J., Llovet J.M. Molecular profiling of liver tumors: classification and clinical translation for decision making. Semin. Liver Dis. 2014;34:363–375. doi: 10.1055/s-0034-1394137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gao L., Wang X., Tang Y., Huang S., Hu C.-A.A., Teng Y. FGF19/FGFR4 signaling contributes to the resistance of hepatocellular carcinoma to sorafenib. J. Exp. Clin. Cancer Res. 2017;36:1–10. doi: 10.1186/s13046-016-0478-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gao L., Shay C., Lv F., Wang X., Teng Y. Implications of FGF19 on sorafenib-mediated nitric oxide production in hepatocellular carcinoma cells-a short report. Cell. Oncol. 2018;41:85–91. doi: 10.1007/s13402-017-0354-4. [DOI] [PubMed] [Google Scholar]
- 18.Lang L., Teng Y. Fibroblast growth factor receptor 4 targeting in cancer: new insights into mechanisms and therapeutic strategies. Cells. 2019;8:31–43. doi: 10.3390/cells8010031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li Y., Zhang W., Doughtie A., Cui G., Li X., Pandit H., Yang Y., Li S., Martin R. Up-regulation of fibroblast growth factor 19 and its receptor associates with progression from fatty liver to hepatocellular carcinoma. Oncotarget. 2016;7:52329–52339. doi: 10.18632/oncotarget.10750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Heinzle C. Is fibroblast growth factor receptor 4 a suitable target of cancer therapy? Curr. Pharm. Des. 2014;20:2881–2898. doi: 10.2174/13816128113199990594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cheng A.L., Shen Y.C., Zhu A.X. Targeting fibroblast growth factor receptor signaling in hepatocellular carcinoma. Oncology. 2011;81:372–380. doi: 10.1159/000335472. [DOI] [PubMed] [Google Scholar]
- 22.Rezende Miranda R., Fu Y., Chen X., Perino J., Cao P., Carpten J., Chen Y., Zhang C. Development of a potent and specific FGFR4 inhibitor for the treatment of hepatocellular carcinoma. J. Med. Chem. 2020;63:11484–11497. doi: 10.1021/acs.jmedchem.0c00044. [DOI] [PubMed] [Google Scholar]
- 23.Liu H., Niu D., Tham Sjin R.T., Dubrovskiy A., Zhu Z., McDonald J.J., Fahnoe K., Wang Z., Munson M., Scholte A. Discovery of selective, covalent FGFR4 inhibitors with antitumor activity in models of hepatocellular carcinoma. ACS Med. Chem. Lett. 2020;11:1899–1904. doi: 10.1021/acsmedchemlett.9b00601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hagel M., Miduturu C., Sheets M., Rubin N., Weng W., Stransky N., Bifulco N., Kim J.L., Hodous B., Brooijmans N., Shutes A., Winter C., Lengauer C., Kohl N.E., Guzi T. First selective small molecule inhibitor of FGFR4 for the treatment of hepatocellular carcinomas with an activated FGFR4 signaling pathway. Cancer Discov. 2015;5:424–437. doi: 10.1158/2159-8290.CD-14-1029. [DOI] [PubMed] [Google Scholar]
- 25.Hatlen M.A., Schmidt-Kittler O., Sherwin C.A., Rozsahegyi E., Rubin N., Sheets M.P., Kim J.L., Miduturu C., Bifulco N., Brooijmans N. Acquired on-target clinical resistance validates FGFR4 as a driver of hepatocellular carcinoma. Cancer Discov. 2019;9:1686–1695. doi: 10.1158/2159-8290.CD-19-0367. [DOI] [PubMed] [Google Scholar]
- 26.Kim R.D., Sarker D., Meyer T., Yau T., Macarulla T., Park J.-W., Choo S.P., Hollebecque A., Sung M.W., Lim H.-Y. First-in-human phase I study of fisogatinib (BLU-554) validates aberrant FGF19 signaling as a driver event in hepatocellular carcinoma. Cancer Discov. 2019;9:1696–1707. doi: 10.1158/2159-8290.CD-19-0555. [DOI] [PubMed] [Google Scholar]
- 27.Joshi J.J., Coffey H., Corcoran E., Tsai J., Huang C.-L., Ichikawa K., Prajapati S., Hao M.-H., Bailey S., Wu J. H3B-6527 is a potent and selective inhibitor of FGFR4 in FGF19-driven hepatocellular carcinoma. Cancer Res. 2017;77:6999–7013. doi: 10.1158/0008-5472.CAN-17-1865. [DOI] [PubMed] [Google Scholar]
- 28.Deng W., Chen X., Jiang K., Song X., Huang M., Tu Z.-C., Zhang Z., Lin X., Ortega R., Patterson A.V. Investigation of covalent warheads in the design of 2-aminopyrimidine-based FGFR4 inhibitors. ACS Med. Chem. Lett. 2021;12:647–652. doi: 10.1021/acsmedchemlett.1c00052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Weiss A., Adler F., Buhles A., Stamm C., Fairhurst R.A., Kiffe M., Sterker D., Centeleghe M., Wartmann M., Kinyamu-Akunda J. FGF401, a first-in-class highly selective and potent FGFR4 inhibitor for the treatment of FGF19-driven hepatocellular cancer. Mol. Cancer Ther. 2019;18:2194–2206. doi: 10.1158/1535-7163.MCT-18-1291. [DOI] [PubMed] [Google Scholar]
- 30.Fairhurst R.A., Knoepfel T., Buschmann N., Leblanc C., Mah R., Todorov M., Nimsgern P., Ripoche S., Niklaus M., Warin N. Discovery of roblitinib (FGF401) as a reversible-covalent inhibitor of the kinase activity of fibroblast growth factor receptor 4. J. Med. Chem. 2020;63:12542–12573. doi: 10.1021/acs.jmedchem.0c01019. [DOI] [PubMed] [Google Scholar]
- 31.Lu X., Chen H., Patterson A.V., Smaill J.B., Ding K. Fibroblast Growth Factor Receptor 4 (FGFR4) Selective inhibitors as hepatocellular carcinoma therapy: advances and prospects: miniperspective. J. Med. Chem. 2019;62:2905–2915. doi: 10.1021/acs.jmedchem.8b01531. [DOI] [PubMed] [Google Scholar]
- 32.van Malenstein H., Dekervel J., Verslype C., Van Cutsem E., Windmolders P., Nevens F., van Pelt J. Long-term exposure to sorafenib of liver cancer cells induces resistance with epithelial-to-mesenchymal transition, increased invasion and risk of rebound growth. Cancer Lett. 2013;329:74–83. doi: 10.1016/j.canlet.2012.10.021. [DOI] [PubMed] [Google Scholar]
- 33.Kanzaki H., Chiba T., Ao J., Koroki K., Kanayama K., Maruta S., Maeda T., Kusakabe Y., Kobayashi K., Kanogawa N. The impact of FGF19/FGFR4 signaling inhibition in antitumor activity of multi-kinase inhibitors in hepatocellular carcinoma. Sci. Rep. 2021;11:5303. doi: 10.1038/s41598-021-84117-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tovar V., Cornella H., Moeini A., Vidal S., Hoshida Y., Sia D., Peix J., Cabellos L., Alsinet C., Torrecilla S. Tumour initiating cells and IGF/FGF signalling contribute to sorafenib resistance in hepatocellular carcinoma. Gut. 2017;66:530–540. doi: 10.1136/gutjnl-2015-309501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Qiu W.-H., Zhou B.-S., Chu P.G., Chen W.-G., Chung C., Shih J., Hwu P., Yeh C., Lopez R., Yen Y. Over-expression of fibroblast growth factor receptor 3 in human hepatocellular carcinoma. World J. Gastroenterol. 2005;11:5266–5272. doi: 10.3748/wjg.v11.i34.5266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vi J.G.T., Cheuk A.T., Tsang P.S., Chung J.-Y., Song Y.K., Desai K., Yu Y., Chen Q.-R., Shah K., Youngblood V. Identification of FGFR4-activating mutations in human rhabdomyosarcomas that promote metastasis in xenotransplanted models. J. Clin. Invest. 2009;119:3395–3407. doi: 10.1172/JCI39703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Choi H.G., Ren P.D., Adrian F., Sun F.X., Lee H.S., Wang X., Ding Q.A., Zhang G.B., Xie Y.P., Zhang J.M., Liu Y., Tuntland T., Warmuth M., Manley P.W., Mestan J., Gray N.S., Sim T. A Type-II kinase inhibitor capable of inhibiting the T315I "Gatekeeper" mutant of Bcr-Abl. J. Med. Chem. 2010;53:5439–5448. doi: 10.1021/jm901808w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nonami A., Sattler M., Weisberg E., Liu Q.S., Zhang J.M., Patricelli M.P., Christie A.L., Saur A.M., Kohl N.E., Kung A.L., Yoon H., Sim T., Gray N.S., Griffin J.D. Identification of novel therapeutic targets in acute leukemias with NRAS mutations using a pharmacologic approach. Blood. 2015;125:3133–3143. doi: 10.1182/blood-2014-12-615906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cho H., Shin I., Ju E., Choi S., Hur W., Kim H., Hong E., Kim N.D., Choi H.G., Gray N.S., Sim T. First SAR study for overriding NRAS mutant driven acute myeloid leukemia. J. Med. Chem. 2018;61:8353–8373. doi: 10.1021/acs.jmedchem.8b00882. [DOI] [PubMed] [Google Scholar]
- 40.Choi S.-H., Shin I., Kim N., Nam Y., Sim T. The first small molecules capable of strongly suppressing proliferation of cancer cells harboring BRAF class I/II/III mutations. Biochem. Biophys. Res. Commun. 2020;532:315–320. doi: 10.1016/j.bbrc.2020.07.110. [DOI] [PubMed] [Google Scholar]
- 41.Guagnano V., Furet P., Spanka C., Bordas V., Le Douget M., Stamm C., Brueggen J., Jensen M.R., Schnell C., Schmid H. Discovery of 3-(2, 6-dichloro-3, 5-dimethoxy-phenyl)-1-{6-[4-(4-ethyl-piperazin-1-yl)-phenylamino]-pyrimidin-4-yl}-1-methyl-urea (NVP-BGJ398), a potent and selective inhibitor of the fibroblast growth factor receptor family of receptor tyrosine kinase. J. Med. Chem. 2011;54:7066–7083. doi: 10.1021/jm2006222. [DOI] [PubMed] [Google Scholar]
- 42.Hughes S.E. Differential expression of the fibroblast growth factor receptor (FGFR) multigene family in normal human adult tissues. J. Histochem. Cytochem. 1997;45:1005–1019. doi: 10.1177/002215549704500710. [DOI] [PubMed] [Google Scholar]
- 43.Greulich H., Pollock P.M. Targeting mutant fibroblast growth factor receptors in cancer. Trends Mol. Med. 2011;17:283–292. doi: 10.1016/j.molmed.2011.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ho H.K., Pok S., Streit S., Ruhe J.E., Hart S., Lim K.S., Loo H.L., Aung M.O., Lim S.G., Ullrich A. Fibroblast growth factor receptor 4 regulates proliferation, anti-apoptosis and alpha-fetoprotein secretion during hepatocellular carcinoma progression and represents a potential target for therapeutic intervention. J. Hepatol. 2009;50:118–127. doi: 10.1016/j.jhep.2008.08.015. [DOI] [PubMed] [Google Scholar]
- 45.Zhao H., Lv F., Liang G., Huang X., Wu G., Zhang W., Yu L., Shi L., Teng Y. FGF19 promotes epithelial-mesenchymal transition in hepatocellular carcinoma cells by modulating the GSK3β/β-catenin signaling cascade via FGFR4 activation. Oncotarget. 2016;7:13575–13586. doi: 10.18632/oncotarget.6185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Huang J., Qiu M., Wan L., Wang G., Huang T., Chen Z., Jiang S., Li X., Xie L., Cai L. TGF-β1 promotes hepatocellular carcinoma invasion and metastasis via ERK pathway-mediated FGFR4 expression. Cell. Physiol. Biochem. 2018;45:1690–1699. doi: 10.1159/000487737. [DOI] [PubMed] [Google Scholar]