

Summary
Glucosylceramide (GluCer) was accumulated in sphingomyelin synthase 1 (SMS1) but not SMS2 deficient mouse tissues. In current study, we studied GluCer accumulation-mediated metabolic consequences. Livers from liver-specific Sms1/global Sms2 double-knockout (dKO) exhibited severe steatosis under a high-fat diet. Moreover, chow diet-fed ≥6-month-old dKO mice had liver impairment, inflammation, and fibrosis, compared with wild type and Sms2 KO mice. RNA sequencing showed 3- to 12-fold increases in various genes which are involved in lipogenesis, inflammation, and fibrosis. Further, we found that direct GluCer treatment (in vitro and in vivo) promoted hepatocyte to secrete more activated TGFβ1, which stimulated more collagen 1α1 production in hepatic stellate cells. Additionally, GluCer promoted more β-catenin translocation into the nucleus, thus promoting tumorigenesis. Importantly, human NASH patients had higher liver GluCer synthase and higher plasma GluCer. These findings implicated that GluCer accumulation is one of triggers promoting the development of NAFLD into NASH, then, fibrosis, and tumorigenesis.
Subject areas: Genetics, Molecular genetics, Omics
Graphical abstract
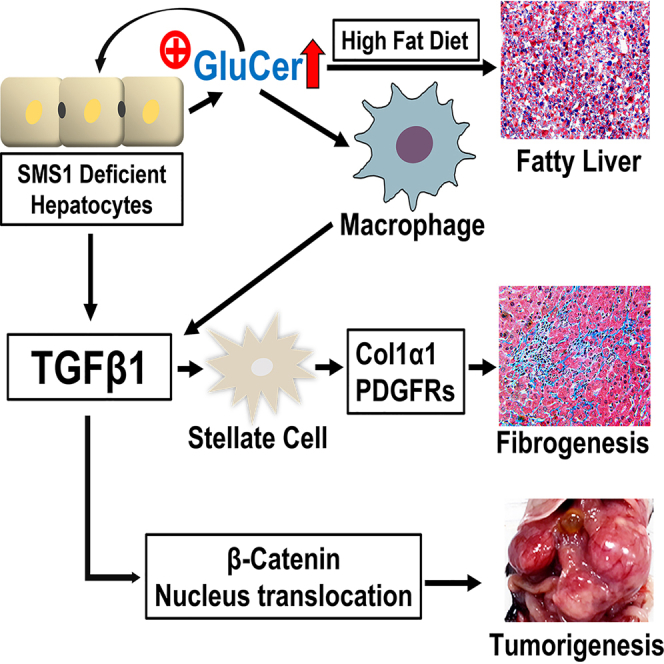
Highlights
-
•
Sphingomyelin synthase 1 deficiency causes glucosylceramide accumulation
-
•
Glucosylceramide accelerates liver steatosis, steatohepatitis, and tumorigenesis
-
•
Glucosylceramide stimulates TGFβ1 activation, which mediates liver fibrosis
-
•
Human NASH patients have higher glucosylceramide synthase in their livers
Genetics; Molecular genetics; Omics
Introduction
Gaucher disease is a rare and autosomal recessive genetic disorder, caused by glucocerebrosidase deficiency and then glucosylceramide (GluCer) accumulation in macrophages (Stirnemann et al., 2017). Recent studies clearly indicated that Gaucher disease is closely related with metabolic dysfunction-associated liver steatosis, fibrosis, and cirrhosis (Nascimbeni et al., 2021). Thus, macrophage GluCer accumulation might not be the only reason for the relationship between Gaucher disease and metabolic disorders.
Nonalcoholic fatty liver disease (NAFLD) is strongly associated with metabolic syndrome and type II diabetes (Loomba et al., 2012). The disease spectrum of NAFLD can range from naive steatosis to steatosis with liver inflammation and fibrosis, which is referred to as nonalcoholic steatohepatitis (NASH) (Tiniakos et al., 2010). Approximately 15% of patients with NAFLD have NASH, and 15% of patients with NASH are at risk for the development of liver tumors (Day, 2011; Sanyal et al., 2011). It remains unclear what factor(s) can trigger the development of NAFLD into NASH. Thus, an understanding of the molecular mechanisms underlying the development of NASH is an urgently needed.
NAFLD is reflected by abnormal fat deposition in the liver. Triglycerides are mostly associated with the pathology. However, other lipids, including sphingolipids, may also play an important role in the development and severity of NAFLD (Regnier et al., 2019). Ceramide could be a factor for inducing (Chaurasia et al., 2019) or reducing hepatic steatosis (Li et al., 2013). Glycosphingolipids, such as GluCer, also contribute to hepatic steatosis (Zhao et al., 2009).
Sphingolipid biosynthesis occurs via the actions of serine palmitoyltransferase (SPT), 3-ketosphinganine reductase, ceramide synthase, and dihydroceramide desaturase to produce ceramide, which is the central substrate for the production of sphingomyelin (SM; through sphingomyelin synthase, SMS), sphingosine-1-phosphate [S1P; through ceramidase/sphingosine kinase, (SPHK)], and glucosylceramide [GluCer; through glucosylceramide synthase (GCS)] (Merrill, 1983).
The de novo sphingolipid synthesis pathway is considered a promising target for pharmacological intervention in insulin resistance and obesity. It has been shown that inhibition of SPT increases insulin sensitivity (Holland et al., 2007; Li et al., 2011). However, this mechanism is incompletely understood, as such an inhibition decreases many bioactive sphingolipids, including ceramide, S1P, and glycosphingolipids (Merrill, 1983). We showed that heterozygous Sptlc2 knockout (KO) mice were protected from high-fat diet-induced obesity and insulin resistance (Li et al., 2011). However, liver tumorigenesis is promoted in homozygous liver-specific Sptlc2 KO mice (Li et al., 2016). To investigate the effect of sphingolipids on metabolic diseases, we chose to study SMS, as it catalyzes the conversion of ceramide to SM and acts downstream of SPT. Thus, SMS activity should influence ceramide, S1P, glycosphingolipid, and SM (Figure S1).
SMS has two isoforms, namely SMS1 and SMS2 (Huitema et al., 2004; Yamaoka et al., 2004), with different cellular localizations. SMS1 resides in the Golgi, whereas SMS2 is located in the Golgi and plasma membrane (Huitema et al., 2004). The major isoform of SMS in macrophages is SMS1 (Li et al., 2012), whereas SMS2 is the major isoform in the liver (Li et al., 2013). Global SMS1 deficiency leads to moderate neonatal lethality in mice (Li et al., 2012; Yano et al., 2011). In contrast, global SMS2 deficiency prevents the development of NAFLD (Li et al., 2013) and atherosclerosis (Fan et al., 2010) in mice.
In this study, we utilized wild type (WT), Sms2 KO, and Sms1/Sms2 double KO mice to specifically evaluate the effect of total SMS deficiency on the liver. We found that total SMS depletion in hepatocytes promoted NAFLD, induced by high-fat diet, NASH, fibrosis, and tumorigenesis. All of these pathological phenotypes are associated with SMS1 deficiency-mediated GluCer accumulation, but not with ceramide and SM levels.
Results
Effect of SMS1/SMS2 deficiency on diet-induced liver steatosis
We previously demonstrated that disruption of liver Sms in 2- to 3-month-old mice results in a significant reduction of triglyceride-rich lipoprotein production (Li et al., 2021). In addition, livers from 2-month-old Sms1/Sms2 KO mice appear to be normal, compared with those from 2-month-old WT and Sms2 KO mice (Li et al., 2021).
We next sought to feed 3-month-old female mice with a high-fat diet for 5 weeks and found that Sms1/Sms2 double KO mice had significantly higher liver triglyceride and cholesterol levels (Figures 1A and 1B) than that of Sms2 KO or WT mice. Staining the liver frozen sections with Oil Red O revealed that there was substantially more lipid accumulation in the liver in Sms1/Sms2 double KO mice, compared with Sms2 KO or WT mice (Figures 1C and 1D). Trichrome staining further confirmed lipid accumulation in the double KO liver (Figure 1E). Additionally, Cd36, FSP27, Pparγ1, and Pparγ2 were upregulated (Figure 1F).
Figure 1.

SMS1 deficiency accelerates diet-induced fatty liver
Three-month-old wild-type (WT), Sms2 knockout (KO), and Sms1/Sms2 double KO female mice were fed with the high-fat diet for 5 weeks.
(A) Liver triglycerides were measured.
(B) Liver cholesterol was measured.
(C) Liver sections were stained with Oil Red O.
(D) Oil red O staining quantification.
(E) Liver sections were stained with trichrome.
(F) Lipogenesis gene mRNA expression was measured using real-time PCR (PCR). Data are represented as mean ± SD, n = 5–6. ∗p < 0.01.
Effect of SMS1/SMS2 deficiency on liver inflammation and fibrosis
Compared with WT and Sms2 KO mice, we observed liver abnormalities in 6-month-old Sms1/Sms2 double KO mice that were on a chow diet. The following studies were performed on mice that were at least 6 months old.
We utilized WT, Sms2 KO, and Sms1/Sms2 double KO female mice and visualized the mouse hepatocyte basal membrane by immunostaining with Na+-taurocholate co-transporting polypeptide (NTCP), which is a hepatocyte basal membrane marker (Watashi et al., 2014). As shown in Figure 2A, NTCP is precisely localized to both WT and Sms2 KO hepatocyte basal membranes, whereas NTCP is diffused into the cytosol of the double KO hepatocytes, and a defined basal membrane was barely seen. We also stained for bile salt export pump (BSEP), which is located on the bile canaliculi membrane (Noe et al., 2002), and found that the double KO hepatocytes showed diffuse staining, compared with WT and Sms2 KO cells (Figure 2B). Thus, SMS deficiency caused hepatocyte impairment.
Figure 2.

SMS1 deficiency-mediated hepatocyte impairment
Liver sections from 6-month-old WT, Sms2 KO, and Sms1/Sms2 KO female mice were utilized.
(A) Na+-taurocholate co-transporting polypeptide (NTCP) immunostaining for hepatocyte basal membrane.
(B) Bile salt export pump (BSEP) immunostaining for hepatocyte apical membrane. The pictures are the representatives of four mice/group.
(C) Plasma was collected from WT, Sms2 KO, and Sms1/Sms2 KO mice, and jaundice was observed.
(D) Fluorogram of plasma bilirubin (conjugated) levels.
(E) Fluorogram of plasma total bile acid levels.
(F and G) Fluorogram of plasma alanine aminotransferase (ALT) and aspartate aminotransferase (AST) levels. Data are represented as mean ± SD, n = 4–6, ∗p < 0.01.
We also found that male and female mice with total liver SMS depletion, but not SMS2 depletion, developed jaundice with different levels of severity (Figure 2C). Measurement of plasma bilirubin levels showed a dramatic increase in conjugated (direct) bilirubin in double KO mice (Figure 2D). Plasma bile acid levels were also dramatically increased in double KO mice (Figure 2E). Moreover, both plasma alanine transaminase (ALT) and aspartate transaminase (AST) activities were significantly increased (Figures 2F and 2G).
We further stained 6-month-old female mouse liver sections and found that the double KO, but not WT and Sms2 KO, mouse livers had more macrophages, stained by MOMA-2 antibody (Abcam) (Figure 3A) and fibrosis, stained by trichrome (Figure 3B). Fibrosis worsened when the mice were one-year old (Figures 3C, S2A, and S2B), suggesting that Sms1 deficiency can cause liver fibrosis in a time-dependent manner. We also noticed that the double KO mice suffered from liver cirrhosis, and it was very difficult for us to obtain primary hepatocytes using the collagenase perfusion procedure. In contrast, we had no difficulties with obtaining primary hepatocytes from WT and Sms2 KO mouse livers, regardless of age. One obvious pathology of one-year-old Sms1/Sms2 double KO mice was that they had liver tumors of various sizes, whereas one-year-old WT and Sms2 KO mice did not have any tumors (Figures 3D and S3). Similar phenotypes were observed in double KO male mice (data not shown).
Figure 3.

SMS1 deficiency-mediated liver inflammation, fibrosis, and tumorigenesis.
(A) Liver sections from 6-month-old WT, Sms2 KO, and Sms1/Sms2 KO female mice were stained with MOMA-2 for macrophages.
(B) Trichrome staining for fibrosis.
(C) Liver sections from 1-year-old WT, Sms2 KO, and Sms1/Sms2 KO mice were stained with trichrome for fibrosis.
(D) Liver tumor observation in 1-year-old mice. The pictures are the representatives of six mice/group.
Next, we measured SM, ceramide, and S1P in the livers of 6-month-old female double KO, Sms2 KO, and control mice. Compared with control mice, SM levels were significantly reduced in both Sms2 KO and Sms1/Sms2 double KO mice. However, the difference between Sms2 KO and the double KO mice was not significant (Table 1). Moreover, both single and double KO mouse livers had comparable ceramide (Table 1) and S1P levels (Figure S4). This suggests that liver SM, ceramide, and S1P might not be involved in the observed pathology. Similar phenotypes were observed in double KO male mice (data not shown).
Table 1.
Sphingomyelin and ceramide measurements in female mouse livers
| Mice (pmol/mg protein) | C16:0 | C18:0 | C22:0 | C24:0 | C24:1 |
|---|---|---|---|---|---|
| Sphingomyelin | |||||
| WT | 483±35a | 99±5a | --- | 71±4a | 438±16a |
| Sms2 KO | 277±44b | 74±2b | --- | 39±5b | 254±12b |
| Sms1/2 KO | 250±5b | 67±6b | --- | 35±3b | 239±19b |
| Ceramide | |||||
| WT | 65±6a | 9±4a | 27±2a | 99±10a | 84±5a |
| Sms2 KO | 78±5b | 12±6a | 23±4a | 98±4a | 87±6a |
| Sms1/2 KO | 81±9b | 13±2a | 18±1b | 82±6b | 81±9a |
WT, wild type; SMS, sphingomyelin synthase; KO, knockout. Columns labeled with different lowercase letters are statistically different, p <0.05. Values are mean ± SD; n = 5.
Effect of SMS1/SMS2 deficiency on the expression of genes that are related to lipogenesis, inflammation, fibrosis, and tumorigenesis
We next isolated primary hepatocytes from 2-month-old, chow-fed, Sms1/Sms2 double KO and WT female mice and performed total RNA-seq. There were 866 upregulated genes and 110 downregulated genes (Figure S5) in Sms1/Sms2 double KO hepatocytes, compared with the controls. Many lipogenesis genes were upregulated; these included Cd36 and Fsp27 (Figure 4A). Approximately 40 inflammatory genes, including Tgfβ1, Tnf, Il6, and Il1, were also upregulated. Moreover, many fibrosis- and tumorigenesis-related genes were upregulated (Figure 4A). These included 5- to 12-fold increases in Tgfβ1, Col1α1, Timp1, Pdgfrα, Pdgfrβ, and Mycn, which are the most relevant factors in liver fibrosis and tumor (Borkham-Kamphorst and Weiskirchen, 2016; Zhu et al., 2018). Results from real-time PCR confirmed what we observed from RNA-seq (Figure 4B). We then performed immunoblotting for TGFβ1, collagen 1α1, PDGFRα, and PDGFRβ. The levels of all four proteins were significantly increased with SMS1/SMS2 double deficiency, but not with SMS2 deficiency, compared with the control (Figures 4C and 4D). Moreover, inflammation array analysis indicated that approximately 20 cytokines were upregulated (Figure 4E).
Figure 4.

SMS1/SMS2 deficiency-mediated lipogenesis, inflammation, fibrosis, and tumorigenesis gene expression
(A) Heatmap for genes related to lipogenesis, inflammation, fibrosis, and tumorigenesis in WT vs. Sms1/Sms2 KO mouse hepatocytes.
(B) Real-time PCR for related genes.
(C and D) Western blot for liver TGFβ1, collagen 1α1, PDGFRα, and PDGFRβ, as well as quantifications.
(E) Inflammation array analysis. Inflammatory cytokines were measured by using the Mouse Inflammation Array C1 kit (RayBiotch). Each measurement was performed in duplicate.
GluCer promotes liver fibrosis through TGFβ1 activation
Previously, we found that the levels of three glycosphingolipids [(GluCer, lactosylceramide, and monosialodihexosylganglioside(GM3)] are significantly increased in Sms1/Sms2 double KO, but not Sms2 KO, mouse hepatocytes, compared with controls (Li et al., 2021). Here, we first confirmed that GluCer levels were indeed significantly increased in double KO mouse livers (Figure 5A).
Figure 5.

GluCer supplementation in vitro and in vivo
(A) Liver homogenates were prepared from WT, Sms2 KO, and Sms1/Sms2 KO female mice. GluCer was measured by LC/MS/MS.
Studies in vitro: (B) Raw 264.7 cells were treated with the indicated dose of GluCer, and activated TGFβ1 (12.5 kDa) was measured in the culture medium.
(C) Huh7 cells and LX2 cells were used to set up a co-culture system, which was treated with the indicated dose of GluCer. TGFβ1 was measured in the co-culture medium, and the level of collagen 1α1 in LX2 cells was measured by Western blot analysis.
(D) LX2 cells were treated with the indicated dose of GluCer, and collagen 1α1 was measured by Western blot analysis.
(E) Fluorogram of Western blots from the macrophages, co-culture system, and LX2 cells. Columns that are labeled with different lowercase letters are statistically different. Studies in vivo: 8-week-old Sms2 KO female mice were injected with vehicle or GluCer (10 μg/g body weight/day) for 2 weeks.
(F) Liver real-time PCR analysis.
(G) Western blot analysis.
(H) Quantification of the Western blots. Data are represented as mean ± SD, n = 3–4, ∗p < 0.01.
Next, we treated Raw 264.7 (a mouse macrophage cell line) cells with GluCer and found that the active form of TGFβ1 (12.5 kDa) was accumulated in the medium in a dose-saturated manner (Figures 5B and 5E). We then utilized Huh7 cells (human liver hepatoma cell line) and LX2 cells (human hepatic stellate cell line) to set up a hepatocyte/hepatic stellate cell co-culture system to evaluate the effect of exogenous GluCer. We measured active form of TGFβ1 (12.5 kDa) in the co-culture medium and then measured the level of collagen 1α1 protein, a key marker for liver fibrosis, in LX2 homogenates as a consequence of TGFβ1 activation (Dewidar et al., 2019). We found that GluCer treatment promoted TGFβ1 accumulation in the co-culture medium (Figures 5C and 5E). Furthermore, collagen 1α1 protein level in LX2 cells was significantly increased in a dose-saturated manner (Figures 5C and 5E). The effect on LX2 cells was hepatocyte-dependent because GluCer had no direct effect on LX2 (Figures 5D and 5E).
To directly examine the effect of GluCer on liver lipogenesis, inflammation, fibrosis, and tumorigenesis genes, we injected it (i.p. once a day for two weeks) into Sms2 KO mice and then used real-time PCR to measure mRNA levels of Cd36, Tnfα, Tgfβ1, Col1α1, Pdgfrα, Pdgfrβ, Timp1, and Mycn. The mRNA levels of Cd36, Tnfα, Tgfβ1, Col1α1, Pdgfrα, and Mycn were significantly upregulated after GluCer treatment (Figure 5F). Western blotting confirmed the upregulation of collagen 1α1, PDGFRα, and PDGFRβ at the protein level (Figures 5G and 5H).
TGFβ1 can disrupt the cadherin/β-catenin complex and promote β-catenin nuclear translocation, thus promoting tumorigenesis (Taiyab et al., 2019; Vogelmann et al., 2005). Thus, we next sought to determine the effect of SMS1 deficiency-mediated GluCer on β-catenin cellular distribution. The plasma membrane, cytosol, and nucleus were isolated from liver homogenates. SMS1/SMS2 double deficiency significantly reduced plasma membrane cadherin (72%, P<0.01) (Figures 6A and 6B) and significantly induced phosphorylated cadherin, indicating its degradation (Fujita et al., 2002), in the cytosol (Figures 6A and 6B). We also found significant changes in the subcellular distribution of β-catenin, which was barely detectable on the plasma membrane and was increased in the cytosol (3.3-fold) and nucleus (1.9-fold) (Figures 6A and 6B). Moreover, E-cadherin, N-cadherin, and β-catenin mRNA levels were unchanged (Figure 6C), suggesting that SMS1 depletion–mediated cadherin/β-catenin regulation occurs post-transcriptionally.
Figure 6.

SMS1/SMS2 deficiency-mediated β-catenin cellular re-distribution
(A) Western blot analyses of cadherin, phosphorylated-cadherin, β-catenin, and α-catenin on the plasma membrane, cytoplasm, and nucleus from WT, Sms2 KO, and Sms1/Sms2 double female KO mice.
(B) Fluorogram of cadherin, β-catenin, and α-catenin levels.
(C) Real-time PCR for liver E-cadherin, N-cadherin, and β-catenin.
(D) Huh7 cells were treated with the indicated dose of GluCer, and the mRNA levels were measured by real-time PCR. Data are represented as mean ± SD, n = 3, ∗p < 0.01.
To further explore the effect of GluCer on lipogenesis, inflammation, fibrosis, and tumorigenesis, we treated Huh7 cells with exogenous GluCer. Then, mRNA levels of Cd36, Pparγ2, Tnfα, Tgfβ1, and Mycn were measured using real-time PCR. All of these mRNA levels were significantly increased after GluCer treatment (Figure 6D).
Finally, we evaluated the relationship between GCS expression and NASH. Liver samples from patients with NASH and controls were immunostained with GCS antibody. The protein expression of GCS was dramatically induced in the diseased livers (Figures 7A and 7B). Moreover, patients with NASH have significantly higher GluCer in their circulation (Figure 7C).
Figure 7.

Human NASH patient livers have significantly higher GCS levels
(A) Liver samples from NASH patients and controls were immunostained with GluCer synthase (GCS) antibody.
(B) Quantification of GCS.
(C) Human plasma GluCer measured by LC/MS/MS. Data are represented as mean ± SD, n = 5–7 for control, and n = 7–12 for NASH patients, ∗p < 0.01.
Discussion
In this study, we demonstrate that disruption of hepatocyte Sms1 in mice resulted in the 1) induction of hepatocyte GluCer levels; 2) promotion of high-fat diet-induced NAFLD; 3) stimulation of NASH; 4) upregulation of major fibrosis-related genes, including Tgfβ1, Col1α1, Pdgfrα, and Pdgfrβ in the liver, thus promoting liver fibrosis; and 5) acceleration of β-catenin nuclear translocation, thus upregulating tumor-related genes and inducing tumorigenesis. Finally, we found that human NASH patients had significantly higher levels of GCS in the liver and higher GluCer in the blood.
SMS1 deficiency-mediated GluCer deposition in hepatocytes is a mechanism for the observed pathology. SMS2 is the major isoform in the liver (Li et al., 2013). The differences in SM, ceramide, and S1P, between SMS2-deficient and SMS1/SMS2-deficient mouse livers, were negligible (Table 1 and Figure S4). Notably, systemic SMS2 deficiency increases insulin sensitivity and prevents diet-induced obesity, compared with controls (Li et al., 2011). The deficiency, which is associated with a ceramide induction, also attenuates diet-induced hepatic steatosis (Li et al., 2013) and reduces atherosclerosis in a mouse model (Fan et al., 2010). Moreover, SMS2 liver overexpression accelerates diet-induced hepatic steatosis, and this is related to the upregulation of lipogenesis genes (Li et al., 2013). Importantly, normal liver GluCer levels are observed in the setting of SMS2 deficiency (Li et al., 2012). Thus, we and others have suggested that SMS2 could be a therapeutic target for human metabolic diseases (Li et al., 2019; Mo et al., 2018; Sugimoto et al., 2016). In contrast, mice with global SMS1 deficiency exhibit moderate neonatal lethality (Li et al., 2012; Yano et al., 2011), as more than 30% of homozygous Sms1 global KO mice die postnatally (the remainder can grow to adulthood). Yano et al. reported that SMS1 deficiency results in mitochondrial dysfunction and impairs insulin secretion (Yano et al., 2011), and the KO mice have lipodystrophy (Yano et al., 2013). We previously found that SMS1 deficiency dramatically increases GluCer levels in the liver, plasma, and macrophages (Li et al., 2012).
One very important question is why is a deficiency in SMS1, but not SMS2, related to GluCer accumulation? SMS and GCS are involved in a sphingolipid metabolic branchpoint (Figure S1). SMS catalyzes the transfer of phosphocholine from phosphatidylcholine to ceramide to form SM, whereas GCS catalyzes the transfer of glucose from UDP-glucose to ceramide to form GluCer (Ichikawa et al., 1996). Similar to SMS1 (Huitema et al., 2004), GCS is located in the Golgi apparatus (Futerman and Pagano, 1991). A recent study revealed that there is an interaction between the SMS1 N-terminal Sterile alpha motif (SAM) domain and GCS C-terminal domain to form a SMS1–GCS complex in the Golgi, controlling the metabolic fate of ceramide in the organelle (Hayashi et al., 2018). Although SMS2 is also present in the Golgi, it has no SAM domain (Huitema et al., 2004). The formation of the SMS1–GCS complex increases SM synthesis and decreases GCS synthesis (Hayashi et al., 2018). In normal hepatocytes, the formation of the SMS1–GCS complex may serve as a switch that controls the steady state levels of GluCer. However, in SMS1 deficiency, the complex is depleted, and this can greatly release GCS activity and promote GluCer biosynthesis, which was observed in our previous study (Li et al., 2012) and current study (Figure 5A).
GluCer plays an important role in mammalian cells (Aerts et al., 2011). Gaucher disease is an autosomal recessive disease that is caused by a mutation of the GluCer hydrolysis enzyme β-glucosidase that results in GluCer accumulation (Sidransky, 2012). The prolonged overabundance of GluCer is detrimental (Cho et al., 2000; Langeveld and Aerts, 2009; Nilsson and Svennerholm, 1982). The storage and deposition of GluCer within macrophages result in the appearance of Gaucher cells (Parkin and Brunning, 1982). Changes in GluCer levels are also observed in cells and tissues in response to cardiovascular disease, diabetes, and tumors (Messner and Cabot, 2010). Although we found an accumulation of GluCer in the livers of the double KO mice, no Gaucher cells were seen (data not shown). Thus, all observed phenotypes in this study were a result of hepatocyte GluCer disposition. A very obvious question is, is there a direct link between hepatocyte GluCer deposition and the observed disorders?
The first key finding of this study was that SMS1 deficiency-mediated GluCer accumulation in hepatocytes promoted liver steatosis. Based on published reports, the effect of GluCer on hepatic steatosis is controversial. On one hand, many studies have indicated that inhibiting GCS ameliorates hepatic steatosis in mouse models (Aerts et al., 2011; Bijl et al., 2009; Zhao et al., 2009). Moreover, GCS inhibition enhances insulin sensitivity (Aerts et al., 2011). On the other hand, one group of researchers indicated that oral administration of GluCer can correct hepatic steatosis in Cohen diabetic rats (Zigmond et al., 2009). Although fatty liver was not observed in chow diet-fed double KO mice in the current study, we found that a high-fat diet could induce more lipid accumulation, causing severe fatty liver in double KO mice, compared with Sms2 KO and control mice (Figure 1). Furthermore, GluCer treatment directly stimulated lipogenesis gene expression in mouse livers and Huh7 cells (Figures 5F and 6D). Simple fatty liver is considered benign, but the initiation of inflammation and subsequent fibrosis and cirrhosis are critical steps in NAFLD with NASH (Day, 2011; Kneeman et al., 2012; Ratziu et al., 2010).
The second key finding was that GluCer accumulation in hepatocytes was associated with NASH. Both RNA-seq and protein dot blot analyses indicated that more than 20 inflammatory cytokines were upregulated in double KO mouse livers compared with controls (Figures 4A and 4E). These included TGFβ1 (Figures 4A and 4D), which was one of the major players in the observed pathology. TGFβ1 is an important cytokine that performs many cellular functions, including inflammation, cell proliferation, cell differentiation, and tumor initiation (Meng et al., 2016). The TGFβ1 precursor has 391 amino acids, with a molecular weight of ∼43 kDa (Poniatowski et al., 2015). The mature form of TGFβ1 is a dimer (∼25 KDa) with a disulfide bond that can be reduced to form a secretory molecule of TGFβ1 with a molecular weight of ∼12.5 kDa (Poniatowski et al., 2015). Direct treatment with GluCer stimulated the secretion of the active form of TGFβ1 from macrophages (Figures 5B and 5E). It is known that GCS inhibitors can counteract TGFβ1-induced abnormalities in insulin signaling (Aerts et al., 2011) and attenuate lipopolysaccharide-induced inflammation (Mobarak et al., 2018). However, an opposite effect of GluCer has also been reported in the Psammomys obesus model (Zigmond et al., 2014), and we could not explain the difference in findings between mouse models and the obesus model.
The third key finding of this study was that GluCer accumulation in hepatocytes caused fibrosis. Liver fibrosis is a pathology that is common to many different chronic liver diseases, and the predominant cell type responsible for fibrogenesis is hepatic stellate cells (HSCs) (Friedman, 2008; Kent et al., 1976). The TGFβ1 signaling pathway is related to liver fibrosis and HSC activation (Dewidar et al., 2019). Collagen 1α1 has been associated with hepatic fibrosis (Koilan et al., 2010) and is controlled by TGFβ1 (Fang et al., 2014; Pan et al., 2013). In the hepatocyte/hepatic stellate cell co-culture system, GluCer treatment stimulated TGFβ1 production from hepatocytes and then promoted the HSCs to produce collagen 1α1 in a dose-saturated manner (Figures 5C and 5E). Importantly, in vivo treatment with GluCer also significantly increased the expression of liver fibrosis-related genes (Figures 5F–5H).
Another key finding of the current study was that GluCer accumulation in hepatocyte caused tumor formation. It is known that only a small portion of liver fibrosis can result in tumors (Day, 2011; Sanyal et al., 2011). However, we found that the double KO mice not only had severe liver fibrosis but also had liver tumors, which progressed with age. There are some mechanisms linking liver fibrosis to liver tumor formation, and TGFβ1 could also be one of the mediators. TGFβ1 activation can stimulate the canonical Wnt/β-catenin pathway to promote tumor formation (Akhmetshina et al., 2012; Vallee et al., 2017). TGFβ1 can also disrupt the cell plasma membrane cadherin/β-catenin complex, which is the major component in adherens junctions, and promote β-catenin nuclear translocation (Taiyab et al., 2019). Previously, we showed that liver SPT deficiency affects the cellular distribution of β-catenin, which is transferred from the plasma membrane into the nucleus, to stimulate tumor gene expression (Li et al., 2016). In this study, we also observed the same pathology in Sms1/Sms2 double KO mice (Figures 6A and 6B), and the mechanism could be related to SMS1-deficiency-mediated GluCer accumulation in the liver, followed by TGFβ1 activation.
It is known that mouse bile duct ligation is a model for liver fibrosis study (Tag et al., 2015). We believe that SMS1/2 deficiency-mediated liver fibrosis is different from bile duct ligation for the following reasons: 1) Sms dKO mice developed jaundice when they were old (6 months or older) (Figure 2C); and 2) Bile duct ligation-caused cholestasis should promote liver fibrosis quickly and severely, compared with SMS1/2 deficiency. It is known that TGFβ1 signaling mediates hepatic bile acid metabolism (Matsubara et al., 2012). Thus, bile accumulation could be a consequence instead of cause of GluCer accumulation-mediated TGFβ1 activation.
We were unable to exclude the possibility that the observed pathology was related to other glycosphingolipids, such as lactosylceramide and GM3, which have also been shown to be increased in the double KO mouse liver (Li et al., 2021). Lactosylceramide is enriched in the cell plasma membrane and may be involved in cell migration and phagocytosis (Iwabuchi et al., 2015). Lactosylceramide is also a lipid second messenger in neuroinflammatory disease (Won et al., 2007). GM3, on the other hand, is able to dissociate the insulin receptor/caveolin-1 complex, thus causing insulin receptor dysfunctionality (Kabayama et al., 2007). GM3 levels are strongly associated with human tumors, such as lung cancer, brain cancer, and melanoma (Zheng et al., 2019), as well as with Parkinson's disease (Chan et al., 2017). Certain human lysosomal storage diseases are also associated with certain glycosphingolipid accumulation. For example, Tay-Sachs's disease is related to GM2 accumulation, and Fabry disease is related to Gb3 accumulation (Winchester et al., 2000). Thus, the effect of GluCer observed in the current study might only partially explain the liver pathology. Further studies investigating the effect of other glycosphingolipids on NAFLD, NASH, fibrosis, and tumorigenesis are warranted.
In summary, findings from the study demonstrate that SMS1 deficiency-mediated GluCer accumulation directly promoted NAFLD, NASH, liver fibrosis, and liver tumor formation.
Limitations of the study
All the KO mice used in this study were with SMS2 deficient background. Although the comparison between Sms2 KO and Sms1/Sms2 dKO was able to show the effect of SMS1 deficiency, however, we could not answer whether the observed pathology in the dKO mice is dependent or independent on SMS2 deficiency.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit monoclonal anti-PDGF receptor α | Cell Signaling Technology | Cat#3174; RRID:AB_2162345 |
| Rabbit monoclonal anti-PDGF receptor β | Cell Signaling Technology | Cat#3169; RRID:AB_2162497 |
| Rabbit monoclonal anti-β-catenin | Cell Signaling Technology | Cat# 8480; RRID:AB_11127855 |
| Rabbit polyclonal anti- alpha-catenin | Cell Signaling Technology | Cat# 3236; RRID:AB_10827873 |
| Rabbit polyclonal anti-collagen I alpha 1 | Novus Biologicals | Cat#NBP1-30054; RRID:AB_1968486 |
| Rabbit polyclonal anti-GAPDH | Novus Biologicals | Cat#NB300-324; RRID:AB_10002458 |
| Rabbit monoclonal anti-TGF beta 1 | Abcam | Cat#ab179695 |
| Rat monoclonal anti- monocyte + macrophage (MOMA-2) | Abcam | Cat#ab33451 |
| Rabbit polyclonal anti-BSEP | Thermo Fisher Scientific | Cat#PA5-78690; RRID:AB_2745806 |
| Rabbit polyclonal anti-NTCP | Thermo Fisher Scientific | Cat#PA5-80001; RRID:AB_2747116 |
| Rabbit polyclonal anti-pan-cadherin | Thermo Fisher Scientific | Cat#71-7100; RRID:AB_2533992 |
| Rabbit polyclonal anti-phospho-cadherin | ECM Biosciences | Cat#CP1951; RRID:AB_2077405 |
| Rabbit polyclonal anti-UGCG | Proteintech | Cat#12869-1-AP; RRID:AB_10915469 |
| Biological samples | ||
| Human liver microarrays with normal and NASH sections | SEKISUI XenoTech, LLC | Lot#2110289 |
| Mouse inflammation array C1 kit | RayBiotch | CODE: AAM-INF-1-2 |
| Chemicals, peptides, and recombinant proteins | ||
| TRIzol reagent | Life Technologies | Cat#15596018 |
| Glucosylceramide | Avanti Polar Lipids | Cat#131304P |
| Critical commercial assays | ||
| High-capacity cDNA reverse transcription kits | Applied Biosystems | Cat#4368814 |
| PowerUp SYBR green master mix | Applied Biosystems | Cat#A25742 |
| Bilirubin assay kits | BioAssay systems | Cat#DIBR-180 |
| Total bile acid assay kits | Diazyme laboratories | Cat#DZ042A-K |
| Plasma membrane protein extraction kit | BioVision | Cat#K268-50 |
| RNeasy Mini Kit | Qiagen | Cat#74104 |
| Nuclear extraction reagents | Thermo Fisher Scientific | Cat#78833 |
| Experimental models: Cell lines | ||
| LX-2 human hepatic stellate cell cine | Sigma | Cat#SCC064 |
| Raw 264.7 mouse macrophage cell line | ATCC | Cat#TIB-71 |
| Experimental models: Organisms/strains | ||
| Liver-specific Sms1 KO/global Sms2 KO mice | This paper | N/A |
| Sms2 KO mice | This paper | N/A |
| Oligonucleotides | ||
| Primers for Real-time PCR see Table S1 | This paper | N/A |
| Software and algorithms | ||
| ImageJ | NIH | https://imagej.nih.gov/ij/ |
| Other | ||
| RNA sequencing | BGI Genomics Company | https://www.bgi.com/ |
| High-fat diet | Envigo | Cat#TD88137 |
Resource availability
Lead contact
Requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Xian-Cheng Jiang (xjiang@downstate.edu).
Materials availability
All unique/stable reagents generated in this study are available from the Lead Contact with a completed Materials Transfer Agreement and reasonable compensation by requestor for shipping and handling charges.
Experimental model and subject details
We crossed SMS1-Flox mice with global SMS2 KO mice, yielding homozygous SMS1-Flox/SMS2 KO mice. We then crossed SMS1-Flox/SMS2 KO mice with albumin-Cre transgenic mice to get liver-specific Sms1 KO/global Sms2 KO Mice (Li et al., 2021). We used both male and female mice which had a C57BL/6 genetic background. Experiments involving animals were conducted with the approval of the Institutional Animal Care and Use Committee at SUNY Downstate Medical Center.
High-fat diet induced fatty liver study
Three-month-old wild-type (WT), SMS2 knockout (KO), and SMS1/SMS2 double KO mice were fed with a high-fat diet (Envigo, Cat# TD88137) for 5 weeks (5-6 mice each group). Liver triglycerides and cholesterol were measured. Liver sections were stained with Oil Red O or trichrome. Lipogenesis gene mRNA expression was measured using real-time PCR.
Real-time polymerase chain reaction (PCR)
Total RNA was isolated from liver tissue using TRIzol, according to the manufacturer’s instructions. Complementary DNA (cDNA) was synthesized using high-capacity cDNA reverse transcription kits (Applied Biosystems). Real-time PCR was performed on the StepOnePlus Real-time PCR system using the SYBR Green Master Mix System (Applied Biosystems). Primers used for real-time PCR are listed in Table S1.
Western blot analysis
Tissue or cell homogenates were subjected to Western blotting as previously described (Li et al., 2015). The following primary antibodies were used: Platelet-derived growth factor receptor (PDGFR)α, Cell Signaling Technology Catalog No.3174; PDGFRβ, Cell Signaling Technology Catalog No. 3169; Collagen 1 α 1, Novus Biologicals, Catalog No. NBP1-30054; Transforming growth factor (TGF)β1, Abcam, Catalog No. 179695; and Glyceraldehyde-3-phosphate dehydrogenase (GAPDH), Novus Biologicals, Catalog No. NB 300-324. GAPDH was used as a loading control.
Hematoxylin & eosin (H&E) staining
Mouse livers were dissected and fixed overnight in 4% formalin. The tissues were embedded in paraffin and then sliced into sections that were 5 μm in thickness. Each slice was deparaffinized and stained with H&E.
Immunofluorescence staining and immunohistochemistry
Mouse livers were fixed in 4% formalin overnight at 4°C before preparation of 5-μm paraffin sections. Prior to antibody staining, the sections were deparaffinized in xylene and rehydrated in a gradient series of ethanol, after which they were subjected to high-temperature antigen retrieval in 50 mM Tris-HCl (pH 9.0), 1 mM EDTA. The sections were permeabilized and blocked in Tris-HCl with 0.5% Triton X-100 and 5% horse serum. Then, the sections were incubated overnight at 4°C with the primary antibodies for Na+-taurocholate co-transporting polypeptide (NTCP; Invitrogen Cat# PA5-80001), bile salt export pump (BSEP; Invitrogen Cat# PA5-78690), GCS (UGCG) (Proteintech Cat# 12869-1-AP), and macrophages (MOMA-2, Abcam, ab33451). Slides were then incubated with fluorescent-labeled secondary antibodies. Formalin-fixed liver tissues were sent to Histowiz Inc. (SUNY Biotechnology Incubator) for H&E and trichrome staining.
Bilirubin and bile acid measurements
Plasma bilirubin and bile acid were measured using bilirubin assay kits (BioAssay systems, DIBR-180) and total bile acid assay kits (Diazyme laboratories, DZ042A-K), respectively, according to the manufacturers’ protocols.
RNA sequencing (RNA-seq)
Hepatocytes were isolated by a two-step collagenase method. Perfused hepatocytes were cultured in DMEM with 10% FBS at 37°C in 5% CO2. Cells were harvested after attachment. RNA was extracted from mouse hepatocytes using the RNeasy Mini Kit (Qiagen), following the manufacturer’s protocol. DNase digestion was performed. The quality of extracted RNAs was controlled by the 2200 TapeStation system (Agilent). RNA samples were then sent to a global genomics organization (BGI) for RNA sequencing, on a fee-for-service basis.
Sphingolipid analyses by liquid chromatography with tandem mass spectrometry (LC/MS/MS)
Sphingolipid levels were measured in wild-type (WT) and KO mouse livers by LC/MS/MS, as we did before (Hailemariam et al., 2008; Liu et al., 2009).
GluCer supplemental in vivo study
Eight-week-old Sms2 KO mice were injected with vehicle or GluCer (Avanti Polar Lipids, Cat# 131304P), 10 μg/g body weight/day, once a day for 2 weeks. Afterward, mouse livers were perfused with PBS and collected. Real-time PCR and Western blot analyses were performed.
Hepatocyte/hepatic stellate cell co-culture in vitro study
The GluCer stock solution (35 mM) was prepared first. Briefly, 0.5 mg GluCer was dissolved in ethanol, dried under nitrogen gas, suspended in 10 ml DMEM, sonicated for 15 minutes in a water bath at room temperature, and then centrifuged at 10,000 rpm for 5 minutes. The GluCer in the supernatant (solubilized) was assessed by thin layer chromatography, which showed that 50% of GluCer was dissolved after sonication. The supernatant was then filtered with a 0.22-μm filter and used for cell culture. Human liver hepatoma 7 (Huh7) cells and LX-2 cells were co-cultured in DMEM containing 0.5% BSA with exogenous GluCer (0, 0.05, 0.5, and 5 μM) for 24 hrs.
Inflammatory cytokine measurement (dot blot assay)
Inflammatory cytokines were measured using the Mouse Inflammation Array C1 kit (RayBiotch). Briefly, mouse plasma samples with 2-fold dilution were incubated with antibody array membranes (Dots) overnight at 4°C after blocking. The membranes were then incubated with a biotinylated antibody cocktail, followed by with horseradish peroxidase-streptavidin for detection. The intensity of each signal was measured using ImageJ software.
Isolation of the liver plasma membrane, cytosol, and nucleus
The liver plasma membrane and nucleus were isolated using the plasma membrane protein extraction kit (BioVision, Catalog No. K268-50) and nuclear extraction reagents (ThermoFisher, Catalog No.78833), respectively, according to manufacturers’ protocols.
Information on human samples
Human liver microarrays with normal and NASH sections were purchased from SEKISUI XenoTech, LLC (Lot. 2010171), and GCS immunostaining was performed. The descriptions of these samples were listed (Table S2).
Quantification and statistical analysis
Data are expressed as the mean ± standard deviation (SD). Differences between two groups were analyzed by the unpaired two-tailed Student’s t-test, and differences among multiple groups were assessed by analysis of variance, followed by the Student-Newman-Keuls test. A P-value < 0.05 was considered significant.
Acknowledgments
We thank Dr. Guanghou Shui (LipidALL Technologies Co., Beijing, China) for his effort to measure glycosphingolipids, phosphatidylcholine, and diacylglycerol. This work was supported by grants VA Merit 000900-01, NIH HL139582, and NIH RO1HL149730 to X.C.J.
Author contributions
Z.L. and Y.P.C performed 80% of the experiments, analyzed the data, and modified the manuscript. M.H. performed some cell culture, real-time PCR, Western blot analysis, and lipid analysis. H.Z. assisted with measuring glycosphingolipids. T.S.W. measured sphingolipids using LC/MS/MS and modified the manuscript. X.C.J. conceived the ideas, designed, and discussed the experiments, supervised progress, and wrote the manuscript.
Declaration of interests
The authors declare no competing financial interests.
Published: December 17, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2021.103449.
Supplemental information
Data and code availability
RNA-seq data are available at the NCBI Gene Expression Omnibus with the series record GSE146900.
References
- Aerts J.M., Boot R.G., van Eijk M., Groener J., Bijl N., Lombardo E., Bietrix F.M., Dekker N., Groen A.K., Ottenhoff R., et al. Glycosphingolipids and insulin resistance. Adv. Exp. Med. Biol. 2011;721:99–119. doi: 10.1007/978-1-4614-0650-1_7. [DOI] [PubMed] [Google Scholar]
- Akhmetshina A., Palumbo K., Dees C., Bergmann C., Venalis P., Zerr P., Horn A., Kireva T., Beyer C., Zwerina J., et al. Activation of canonical Wnt signalling is required for TGF-beta-mediated fibrosis. Nat. Commun. 2012;3:735. doi: 10.1038/ncomms1734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bijl N., Sokolovic M., Vrins C., Langeveld M., Moerland P.D., Ottenhoff R., van Roomen C.P., Claessen N., Boot R.G., Aten J., et al. Modulation of glycosphingolipid metabolism significantly improves hepatic insulin sensitivity and reverses hepatic steatosis in mice. Hepatology. 2009;50:1431–1441. doi: 10.1002/hep.23175. [DOI] [PubMed] [Google Scholar]
- Borkham-Kamphorst E., Weiskirchen R. The PDGF system and its antagonists in liver fibrosis. Cytokine Growth Factor Rev. 2016;28:53–61. doi: 10.1016/j.cytogfr.2015.10.002. [DOI] [PubMed] [Google Scholar]
- Chan R.B., Perotte A.J., Zhou B., Liong C., Shorr E.J., Marder K.S., Kang U.J., Waters C.H., Levy O.A., Xu Y., et al. Elevated GM3 plasma concentration in idiopathic Parkinson's disease: A lipidomic analysis. PLoS One. 2017;12:e0172348. doi: 10.1371/journal.pone.0172348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaurasia B., Tippetts T.S., Mayoral Monibas R., Liu J., Li Y., Wang L., Wilkerson J.L., Sweeney C.R., Pereira R.F., Sumida D.H., et al. Targeting a ceramide double bond improves insulin resistance and hepatic steatosis. Science. 2019;365:386–392. doi: 10.1126/science.aav3722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho L., Lytle B.W., Moodie D.S. Type IIIC Gaucher's disease. Circulation. 2000;102:E69–E70. doi: 10.1161/01.cir.102.10.e69. [DOI] [PubMed] [Google Scholar]
- Day C.P. Non-alcoholic fatty liver disease: A massive problem. Clin. Med. (Lond.) 2011;11:176–178. doi: 10.7861/clinmedicine.11-2-176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dewidar B., Meyer C., Dooley S., Meindl-Beinker A.N. TGF-beta in hepatic stellate cell activation and liver fibrogenesis-updated 2019. Cells. 2019;8:1419. doi: 10.3390/cells8111419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan Y., Shi F., Liu J., Dong J., Bui H.H., Peake D.A., Kuo M.S., Cao G., Jiang X.C. Selective reduction in the sphingomyelin content of atherogenic lipoproteins inhibits their retention in murine aortas and the subsequent development of atherosclerosis. Arterioscler.Thromb.Vasc.Biol. 2010;30:2114–2120. doi: 10.1161/ATVBAHA.110.213363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang L., Huang C., Meng X., Wu B., Ma T., Liu X., Zhu Q., Zhan S., Li J. TGF-beta1-elevated TRPM7 channel regulates collagen expression in hepatic stellate cells via TGF-beta1/Smad pathway. Toxicol. Appl. Pharmacol. 2014;280:335–344. doi: 10.1016/j.taap.2014.08.006. [DOI] [PubMed] [Google Scholar]
- Friedman S.L. Hepatic stellate cells: Protean, multifunctional, and enigmatic cells of the liver. Physiol. Rev. 2008;88:125–172. doi: 10.1152/physrev.00013.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita Y., Krause G., Scheffner M., Zechner D., Leddy H.E., Behrens J., Sommer T., Birchmeier W. Hakai, a c-Cbl-like protein, ubiquitinates and induces endocytosis of the E-cadherin complex. Nat. Cell. Biol. 2002;4:222–231. doi: 10.1038/ncb758. [DOI] [PubMed] [Google Scholar]
- Futerman A.H., Pagano R.E. Determination of the intracellular sites and topology of glucosylceramide synthesis in rat liver. Biochem. J. 1991;280(Pt 2):295–302. doi: 10.1042/bj2800295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hailemariam T.K., Huan C., Liu J., Li Z., Roman C., Kalbfeisch M., Bui H.H., Peake D.A., Kuo M.S., Cao G., et al. Sphingomyelin synthase 2 deficiency attenuates NFkappaB activation. Arterioscler.Thromb.Vasc. Biol. 2008;28:1519–1526. doi: 10.1161/ATVBAHA.108.168682. [DOI] [PubMed] [Google Scholar]
- Hayashi Y., Nemoto-Sasaki Y., Matsumoto N., Hama K., Tanikawa T., Oka S., Saeki T., Kumasaka T., Koizumi T., Arai S., et al. Complex formation of sphingomyelin synthase 1 with glucosylceramide synthase increases sphingomyelin and decreases glucosylceramide levels. J. Biol. Chem. 2018;293:17505–17522. doi: 10.1074/jbc.RA118.002048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holland W.L., Brozinick J.T., Wang L.P., Hawkins E.D., Sargent K.M., Liu Y., Narra K., Hoehn K.L., Knotts T.A., Siesky A., et al. Inhibition of ceramide synthesis ameliorates glucocorticoid-, saturated-fat-, and obesity-induced insulin resistance. Cell Metab. 2007;5:167–179. doi: 10.1016/j.cmet.2007.01.002. [DOI] [PubMed] [Google Scholar]
- Huitema K., van den Dikkenberg J., Brouwers J.F., Holthuis J.C. Identification of a family of animal sphingomyelin synthases. EMBO J. 2004;23:33–44. doi: 10.1038/sj.emboj.7600034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichikawa S., Sakiyama H., Suzuki G., Hidari K.I., Hirabayashi Y. Expression cloning of a cDNA for human ceramide glucosyltransferase that catalyzes the first glycosylation step of glycosphingolipid synthesis. Proc. Natl. Acad. Sci. U S A. 1996;93:4638–4643. doi: 10.1073/pnas.93.10.4638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwabuchi K., Masuda H., Kaga N., Nakayama H., Matsumoto R., Iwahara C., Yoshizaki F., Tamaki Y., Kobayashi T., Hayakawa T., et al. Properties and functions of lactosylceramide from mouse neutrophils. Glycobiology. 2015;25:655–668. doi: 10.1093/glycob/cwv008. [DOI] [PubMed] [Google Scholar]
- Kabayama K., Sato T., Saito K., Loberto N., Prinetti A., Sonnino S., Kinjo M., Igarashi Y., Inokuchi J. Dissociation of the insulin receptor and caveolin-1 complex by ganglioside GM3 in the state of insulin resistance. Proc. Natl. Acad. Sci. U S A. 2007;104:13678–13683. doi: 10.1073/pnas.0703650104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent G., Gay S., Inouye T., Bahu R., Minick O.T., Popper H. Vitamin A-containing lipocytes and formation of type III collagen in liver injury. Proc. Natl. Acad. Sci. U S A. 1976;73:3719–3722. doi: 10.1073/pnas.73.10.3719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kneeman J.M., Misdraji J., Corey K.E. Secondary causes of nonalcoholic fatty liver disease. Therap. Adv. Gastroenterol. 2012;5:199–207. doi: 10.1177/1756283X11430859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koilan S., Hamilton D., Baburyan N., Padala M.K., Weber K.T., Guntaka R.V. Prevention of liver fibrosis by triple helix-forming oligodeoxyribonucleotides targeted to the promoter region of type I collagen gene. Oligonucleotides. 2010;20:231–237. doi: 10.1089/oli.2010.0244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langeveld M., Aerts J.M. Glycosphingolipids and insulin resistance. Prog. Lipid Res. 2009;48:196–205. doi: 10.1016/j.plipres.2009.03.002. [DOI] [PubMed] [Google Scholar]
- Li Z., Chiang Y.P., He M., Zhang K., Zheng J., Wu W., Cai J., Chen Y., Chen G., Chen Y., et al. Effect of liver total sphingomyelin synthase deficiency on plasma lipid metabolism. Biochim.Biophys.Acta Mol. Cell Biol. Lipids. 2021;1866:158898–158917. doi: 10.1016/j.bbalip.2021.158898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y., Dong J., Ding T., Kuo M.S., Cao G., Jiang X.C., Li Z. Sphingomyelin synthase 2 activity and liver steatosis: An effect of ceramide-mediated peroxisome proliferator-activated receptor gamma2 suppression. Arterioscler.Thromb.Vasc. Biol. 2013;33:1513–1520. doi: 10.1161/ATVBAHA.113.301498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y., Huang T., Lou B., Ye D., Qi X., Li X., Hu S., Ding T., Chen Y., Cao Y., et al. Discovery, synthesis and anti-atherosclerotic activities of a novel selective sphingomyelin synthase 2 inhibitor. Eur. J. Med. Chem. 2019;163:864–882. doi: 10.1016/j.ejmech.2018.12.028. [DOI] [PubMed] [Google Scholar]
- Li Z., Fan Y., Liu J., Li Y., Huan C., Bui H.H., Kuo M.S., Park T.S., Cao G., Jiang X.C. Impact of sphingomyelin synthase 1 deficiency on sphingolipid metabolism and atherosclerosis in mice. Arterioscler.Thromb.Vasc. Biol. 2012;32:1577–1584. doi: 10.1161/ATVBAHA.112.251538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z., Jiang H., Ding T., Lou C., Bui H.H., Kuo M.S., Jiang X.C. Deficiency in lysophosphatidylcholine acyltransferase 3 reduces plasma levels of lipids by reducing lipid absorption in mice. Gastroenterology. 2015;149:1519–1529. doi: 10.1053/j.gastro.2015.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z., Kabir I., Jiang H., Zhou H., Libien J., Zeng J., Stanek A., Ou P., Li K.R., Zhang S., et al. Liver serine palmitoyltransferase activity deficiency in early life impairs adherens junctions and promotes tumorigenesis. Hepatology. 2016;64:2089–2102. doi: 10.1002/hep.28845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z., Zhang H., Liu J., Liang C.P., Li Y., Li Y., Teitelman G., Beyer T., Bui H.H., Peake D.A., et al. Reducing plasma membrane sphingomyelin increases insulin sensitivity. Mol. Cell Biol. 2011;31:4205–4218. doi: 10.1128/MCB.05893-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J.Y., Lo P.C., Jiang X.J., Fong W.P., Ng D.K. Synthesis and in vitro photodynamic activities of di-alpha-substituted zinc(ii) phthalocyanine derivatives. Dalton Trans. 2009:4129–4135. doi: 10.1039/b817940a. [DOI] [PubMed] [Google Scholar]
- Loomba R., Abraham M., Unalp A., Wilson L., Lavine J., Doo E., Bass N.M., Nonalcoholic Steatohepatitis Clinical Research Network Association between diabetes, family history of diabetes, and risk of nonalcoholic steatohepatitis and fibrosis. Hepatology. 2012;56:943–951. doi: 10.1002/hep.25772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsubara T., Tanaka N., Sato M., Kang D.W., Krausz K.W., Flanders K.C., Ikeda K., Luecke H., Wakefield L.M., Gonzalez F.J. TGF-beta-SMAD3 signaling mediates hepatic bile acid and phospholipid metabolism following lithocholic acid-induced liver injury. J. Lipid Res. 2012;53:2698–2707. doi: 10.1194/jlr.M031773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng X.M., Nikolic-Paterson D.J., Lan H.Y. TGF-beta: The master regulator of fibrosis. Nat. Rev. Nephrol. 2016;12:325–338. doi: 10.1038/nrneph.2016.48. [DOI] [PubMed] [Google Scholar]
- Merrill A.H., Jr. Characterization of serine palmitoyltransferase activity in Chinese hamster ovary cells. Biochim.Biophys. Acta. 1983;754:284–291. doi: 10.1016/0005-2760(83)90144-3. [DOI] [PubMed] [Google Scholar]
- Messner M.C., Cabot M.C. Glucosylceramide in humans. Adv. Exp. Med. Biol. 2010;688:156–164. doi: 10.1007/978-1-4419-6741-1_11. [DOI] [PubMed] [Google Scholar]
- Mo M., Yang J., Jiang X.C., Cao Y., Fei J., Chen Y., Qi X., Chu Y., Zhou L., Ye D. Discovery of 4-benzyloxybenzo[ d]isoxazole-3-amine derivatives as highly selective and orally efficacious human sphingomyelin synthase 2 inhibitors that reduce chronic inflammation in db/db mice. J. Med. Chem. 2018;61:8241–8254. doi: 10.1021/acs.jmedchem.8b00727. [DOI] [PubMed] [Google Scholar]
- Mobarak E., Haversen L., Manna M., Rutberg M., Levin M., Perkins R., Rog T., Vattulainen I., Boren J. Glucosylceramide modifies the LPS-induced inflammatory response in macrophages and the orientation of the LPS/TLR4 complex in silico. Sci. Rep. 2018;8:13600. doi: 10.1038/s41598-018-31926-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nascimbeni F., Lugari S., Andreone P., Carubbi F. The time has come to look for metabolic dysfunction-associated fatty liver disease in adult patients with type 1 Gaucher disease. Liver Int. 2021;41:224–225. doi: 10.1111/liv.14707. [DOI] [PubMed] [Google Scholar]
- Nilsson O., Svennerholm L. Accumulation of glucosylceramide and glucosylsphingosine (psychosine) in cerebrum and cerebellum in infantile and juvenile Gaucher disease. J. Neurochem. 1982;39:709–718. doi: 10.1111/j.1471-4159.1982.tb07950.x. [DOI] [PubMed] [Google Scholar]
- Noe J., Stieger B., Meier P.J. Functional expression of the canalicular bile salt export pump of human liver. Gastroenterology. 2002;123:1659–1666. doi: 10.1053/gast.2002.36587. [DOI] [PubMed] [Google Scholar]
- Pan X., Chen Z., Huang R., Yao Y., Ma G. Transforming growth factor beta1 induces the expression of collagen type I by DNA methylation in cardiac fibroblasts. PLoS One. 2013;8:e60335. doi: 10.1371/journal.pone.0060335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkin J.L., Brunning R.D. Pathology of the gaucher cell. Prog.Clin. Biol. Res. 1982;95:151–175. [PubMed] [Google Scholar]
- Poniatowski L.A., Wojdasiewicz P., Gasik R., Szukiewicz D. Transforming growth factor beta family: Insight into the role of growth factors in regulation of fracture healing biology and potential clinical applications. Mediators Inflamm. 2015;2015:137823. doi: 10.1155/2015/137823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratziu V., Bellentani S., Cortez-Pinto H., Day C., Marchesini G. A position statement on NAFLD/NASH based on the EASL 2009 special conference. J. Hepatol. 2010;53:372–384. doi: 10.1016/j.jhep.2010.04.008. [DOI] [PubMed] [Google Scholar]
- Regnier M., Polizzi A., Guillou H., Loiseau N. Sphingolipid metabolism in non-alcoholic fatty liver diseases. Biochimie. 2019;159:9–22. doi: 10.1016/j.biochi.2018.07.021. [DOI] [PubMed] [Google Scholar]
- Sanyal A.J., Brunt E.M., Kleiner D.E., Kowdley K.V., Chalasani N., Lavine J.E., Ratziu V., McCullough A. Endpoints and clinical trial design for nonalcoholic steatohepatitis. Hepatology. 2011;54:344–353. doi: 10.1002/hep.24376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidransky E. Gaucher disease: Insights from a rare Mendelian disorder. Discov. Med. 2012;14:273–281. [PMC free article] [PubMed] [Google Scholar]
- Stirnemann J., Belmatoug N., Camou F., Serratrice C., Froissart R., Caillaud C., Levade T., Astudillo L., Serratrice J., Brassier A., et al. Areview of gaucher disease pathophysiology, clinical presentation and treatments. Int. J. Mol. Sci. 2017;18:441. doi: 10.3390/ijms18020441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugimoto M., Shimizu Y., Zhao S., Ukon N., Nishijima K., Wakabayashi M., Yoshioka T., Higashino K., Numata Y., Okuda T., et al. Characterization of the role of sphingomyelin synthase 2 in glucose metabolism in whole-body and peripheral tissues in mice. Biochim.Biophys. Acta. 2016;1861:688–702. doi: 10.1016/j.bbalip.2016.04.019. [DOI] [PubMed] [Google Scholar]
- Tag C.G., Sauer-Lehnen S., Weiskirchen S., Borkham-Kamphorst E., Tolba R.H., Tacke F., Weiskirchen R. Bile duct ligation in mice: Induction of inflammatory liver injury and fibrosis by obstructive cholestasis. J. Vis. Exp. 2015;10:52438. doi: 10.3791/52438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taiyab A., Holms J., West-Mays J.A. beta-Catenin/Smad3interaction regulates transforming growth factor-beta-induced epithelial to mesenchymal transition in the lens. Int. J. Mol. Sci. 2019;20:2078. doi: 10.3390/ijms20092078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiniakos D.G., Vos M.B., Brunt E.M. Nonalcoholic fatty liver disease: Pathology and pathogenesis. Annu. Rev. Pathol. 2010;5:145–171. doi: 10.1146/annurev-pathol-121808-102132. [DOI] [PubMed] [Google Scholar]
- Vallee A., Lecarpentier Y., Guillevin R., Vallee J.N. Interactions between TGF-beta1, canonical WNT/beta-catenin pathway and PPAR gamma in radiation-induced fibrosis. Oncotarget. 2017;8:90579–90604. doi: 10.18632/oncotarget.21234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogelmann R., Nguyen-Tat M.D., Giehl K., Adler G., Wedlich D., Menke A. TGFbeta-induced downregulation of E-cadherin-based cell-cell adhesion depends on PI3-kinase and PTEN. J. Cell Sci. 2005;118:4901–4912. doi: 10.1242/jcs.02594. [DOI] [PubMed] [Google Scholar]
- Watashi K., Urban S., Li W., Wakita T. NTCP and beyond: Opening the door to unveil hepatitis B virus entry. Int. J. Mol. Sci. 2014;15:2892–2905. doi: 10.3390/ijms15022892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winchester B., Vellodi A., Young E. The molecular basis of lysosomal storage diseases and their treatment. Biochem. Soc. Trans. 2000;28:150–154. doi: 10.1042/bst0280150. [DOI] [PubMed] [Google Scholar]
- Won J.S., Singh A.K., Singh I. Lactosylceramide: A lipid second messenger in neuroinflammatory disease. J. Neurochem. 2007;103(Suppl 1):180–191. doi: 10.1111/j.1471-4159.2007.04822.x. [DOI] [PubMed] [Google Scholar]
- Yamaoka S., Miyaji M., Kitano T., Umehara H., Okazaki T. Expression cloning of a human cDNA restoring sphingomyelin synthesis and cell growth in sphingomyelin synthase-defective lymphoid cells. J. Biol. Chem. 2004;279:18688–18693. doi: 10.1074/jbc.M401205200. [DOI] [PubMed] [Google Scholar]
- Yano M., Watanabe K., Yamamoto T., Ikeda K., Senokuchi T., Lu M., Kadomatsu T., Tsukano H., Ikawa M., Okabe M., et al. Mitochondrial dysfunction and increased reactive oxygen species impair insulin secretion in sphingomyelin synthase 1-null mice. J. Biol. Chem. 2011;286:3992–4002. doi: 10.1074/jbc.M110.179176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yano M., Yamamoto T., Nishimura N., Gotoh T., Watanabe K., Ikeda K., Garan Y., Taguchi R., Node K., Okazaki T., et al. Increased oxidative stress impairs adipose tissue function in sphingomyelin synthase 1 null mice. PLoS One. 2013;8:e61380. doi: 10.1371/journal.pone.0061380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao H., Przybylska M., Wu I.H., Zhang J., Maniatis P., Pacheco J., Piepenhagen P., Copeland D., Arbeeny C., Shayman J.A., et al. Inhibiting glycosphingolipid synthesis ameliorates hepatic steatosis in obese mice. Hepatology. 2009;50:85–93. doi: 10.1002/hep.22970. [DOI] [PubMed] [Google Scholar]
- Zheng C., Terreni M., Sollogoub M., Zhang Y. Ganglioside GM3 and its role in cancer. Curr.Med. Chem. 2019;26:2933–2947. doi: 10.2174/0929867325666180129100619. [DOI] [PubMed] [Google Scholar]
- Zhu C., Kim K., Wang X., Bartolome A., Salomao M., Dongiovanni P., Meroni M., Graham M.J., Yates K.P., Diehl A.M., et al. Hepatocyte notch activation induces liver fibrosis in nonalcoholic steatohepatitis. Sci. Transl. Med. 2018;10:eaat0344. doi: 10.1126/scitranslmed.aat0344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zigmond E., Tayer-Shifman O., Lalazar G., Ben Ya'acov A., Weksler-Zangen S., Shasha D., Sklair-Levy M., Zolotarov L., Shalev Z., Kalman R., et al. beta-glycosphingolipids ameliorated non-alcoholic steatohepatitis in the Psammomys obesus model. J. Inflamm. Res. 2014;7:151–158. doi: 10.2147/JIR.S50508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zigmond E., Zangen S.W., Pappo O., Sklair-Levy M., Lalazar G., Zolotaryova L., Raz I., Ilan Y. Beta-glycosphingolipids improve glucose intolerance and hepatic steatosis of the Cohen diabetic rat. Am. J. Physiol. Endocrinol.Metab. 2009;296:E72–E78. doi: 10.1152/ajpendo.90634.2008. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNA-seq data are available at the NCBI Gene Expression Omnibus with the series record GSE146900.