

Abstract
Early embryonic arrest and fragmentation (EEAF) is a common phenomenon leading to female infertility, but the genetic determinants remain largely unknown. The Moloney sarcoma oncogene (MOS) encodes a serine/threonine kinase that activates the ERK signaling cascade during oocyte maturation in vertebrates. Here, we identified four rare variants of MOS in three infertile female individuals with EEAF that followed a recessive inheritance pattern. These MOS variants encoded proteins that resulted in decreased phosphorylated ERK1/2 level in cells and oocytes, and displayed attenuated rescuing effects on cortical F‐actin assembly. Using oocyte‐specific Erk1/2 knockout mice, we verified that MOS‐ERK signal pathway inactivation in oocytes caused EEAF as human. The RNA sequencing data revealed that maternal mRNA clearance was disrupted in human mature oocytes either with MOS homozygous variant or with U0126 treatment, especially genes relative to mitochondrial function. Mitochondrial dysfunction was observed in oocytes with ERK1/2 deficiency or inactivation. In conclusion, this study not only uncovers biallelic MOS variants causes EEAF but also demonstrates that MOS‐ERK signaling pathway drives human oocyte cytoplasmic maturation to prevent EEAF.
Keywords: female infertility, human oocyte, maternal mRNA decay, mitochondria, MOS
Subject Categories: Development, Urogenital System
Biallelic variants in MOS gene cause recurrent early embryonic arrest and fragmentation (EEAF) and female infertility. MOS variants impair activation of MOS‐ERK signal cascade, prevent substantial maternal mRNAs decay and hamper embryonic development both in human and mice.
The paper explained.
Problem
Early embryonic arrest and fragmentation (EEAF) is a compound phenotype of cleavage arrest and embryo fragmentation, leading to recurrent failure of assisted reproductive technology treatment and female infertility. In recent years, several pathogenic variants in maternal genes could account for 10–15% of the etiology of embryonic arrest. However, the underlying genetic determinants and mechanism of EEAF are largely unknown.
Results
We identified three patients bearing biallelic mutations in Moloney sarcoma oncogene (MOS) gene displaying EEAF. They carry homozygous missense variant c.285C>A (p. Asn95Lys), homozygous nonsense variant c.960C>A (p. Cys320Ter), and compound heterozygous missense variants c.416T>C (p. Met139Thr) and c.737G>A (p. Arg246His), respectively. Compared with wild‐type MOS, these MOS variants impaired ERK1/2 activation, resulting in oocyte meiotic resumption and cytoskeleton assembly defects. RNA‐sequencing results revealed that substantial maternal mRNAs’ clearance during oocyte maturation was retarded either in mature oocytes carrying MOSAsn95Lys variant or in U0126‐treated oocytes, in which the most affected transcripts were enriched in mitochondrial biological process. Furthermore, inactivation of MOS‐ERK signal cascade caused mitochondrial dysfunction of mature oocytes. These results indicate MOS‐ERK1/2 signal pathway is vital for embryo development through driving oocyte cytoplasmic maturation in human and mouse.
Clinical impact
This study firstly provides direct evidence that biallelic MOS mutation is associated with EEAF phenotype and female infertility, which may serve a target gene for early diagnosis and therapeutic strategy development.
Introduction
Early embryo fragmentation is defined as anucleate cell fragments derived from the blastomere, and it is a common feature of in vitro fertilization (IVF) or intracytoplasmic sperm injection (ICSI) cycles. Embryos with more than 25% fragmentation are closely associated with deleterious outcomes, including decreased blastocyst formation and implantation rate, and increased malformation rate after pregnancy (Ebner et al, 2001). Several hypotheses for pathogenesis of embryo fragmentation concerns on apoptosis, telomere length, cytoskeleton abnormality, increasing maternal age, and DNA fragment in sperm, with no definitive conclusion having been drawn (Fujimoto et al, 2011). Recurrent early embryonic arrest with fragmentation (EEAF) is a severe type of embryo developmental failure that results in no embryo transfer, which performed as a compound phenotype of cleavage arrest and embryo fragmentation. Despite increasing studies support that EEAF may be of maternal origin defects, however, the causative gene and the underlying mechanism remain largely unknown.
In recent years, variants in single maternal‐effect gene, including TUBB8 (MIM: 616768), PADI6 (MIM: 610363), TLE6 (MIM: 612399), KHDC3L (MIM: 611687), NLRP2 (MIM: 609364), and NLRP5 (MIM: 609658) (Alazami et al, 2015; Feng et al, 2016; Xu et al, 2016; Mu et al, 2019; Zhang et al, 2019b; Zheng et al, 2020a), have been found to be responsible for human recurrent embryonic arrest in IVF/ICSI attempts. Additionally, we previously indicated variants in human BTG4 (MIM: 605673), causing large‐scale maternal‐effect gene decay defect, resulted in zygotic cleavage failure (Zheng et al, 2020b). However, none of these genes can explain the EEAF mechanistically.
The Moloney sarcoma oncogene (Mos) gene encodes a serine/threonine protein kinase, which was first identified as a cytostatic factor to maintain oocyte metaphase II (MII) arrest via activation of the ERK pathway by phosphorylating MEK (Sagata et al, 1989; Verlhac et al, 2000). MOS‐ERK signal pathway‐mediated MII arrest relied on EMI2‐mediated APCCDC20 inhibition to prevent cyclin B1 from degradation (Shoji et al, 2006; Suzuki et al, 2010; Sako et al, 2014). The MOS mRNA is highly expressed in oocyte, and MOS protein is actively translated during oocyte maturation and rapidly degraded after fertilization, in several vertebrates, including human (Sagata et al, 1988; Watanabe et al, 1991; Sha et al, 2020b). Despite that MOS protein is nearly untranslated in oocyte at GV stage, precocious activation MOS‐ERK signal cascade by microinjection of Mos mRNAs in mouse immature oocytes could promote cyclin B1 translation and maturation promoting factor (MPF) activation, leading to oocyte meiotic maturation resumption (Choi et al, 1996b; Cao et al, 2020). Deficiency of Mos or Erk1/2 in mice results in oocyte MII arrest failure, spindle abnormality, large polar body, and early embryo developmental arrest (Colledge et al, 1994; Hashimoto et al, 1994; Araki et al, 1996; Choi et al, 1996a; Zhang et al, 2015). Until now, MOS (OMIM ID: 190060; NM_005372.1) has not been found to be associated with any human disease definitely.
In this study, we identified four pathogenic variants in MOS that are responsible for human recurrent EEAF. Both affected individuals had biallelic variants that followed a Mendelian recessive inheritance pattern. We verified the pathogenic effects of MOS variants on protein expression and ERK1/2 activation. Inactivation of MOS‐ERK signal pathway in oocyte caused abnormal F‐actin assembly disorder, maternal‐effect genes decay defect, and mitochondrial dysfunction, resulting in EEAF. Our findings established the causal relationship between MOS and the phenotype of EEAF in human, which may provide targets for therapeutic strategies in future.
Results
Clinical characteristics of the affected individuals
We recruited three independent female probands with infertility of unknown causes. All affected individuals belong to Chinese Han population and are from three families without potential fertility problems in China. They had normal menstrual cycles and sex hormone levels (Appendix Table S1). Proband II‐1 from a consanguineous family (family 1) had undergone one IVF and two ICSI failed attempts. All 15 oocytes retrieved in the three attempts were mature. However, half of the retrieved oocytes displayed multiple polar bodies or enriched granules in the cytoplasm, with a low normal fertilization (2PN) rate (20%, 25%, and 50%, respectively), and nine zygotes were cleaved at the two‐ to seven‐cell stage on day 3 and arrested during subsequent blastocyst culture. Three of nine arrested embryos were accompanied by severe fragmentation at the cleavage stage (Table 1 and Fig 1D). Proband II‐1 in family 2 had undergone four separate ICSI attempts, and three previous failed attempts in other hospitals were diagnosed as embryonic arrest (arrested at the two‐ to six‐cell stage, with embryo fragmentation). In her fourth ICSI attempt, we used time‐lapse to observe embryonic development. Overall, 11 of the 14 oocytes retrieved were mature, and the couple chose to freeze four MII oocytes in this attempt. Then, three 2PN, two 1PN, and two 3PN zygotes were formed; all zygotes consistently arrested at the two‐ to five‐cell stage, and six of seven arrested embryos with severe fragmentation were produced during the first cleavage (Table 1 and Fig 1D). Proband II‐1 from a consanguineous family (family 3) had undergone one IVF with ICSI rescue in other hospital and one ICSI attempt in our hospital. In her two attempts, partial immature oocytes were observed, including 2 metaphase I (MI) oocytes of 12 retrieved oocytes and 2 MI oocytes of 5 retrieved oocytes. In her first attempt, four zygotes were derived yet only one developed to five‐cell stage accompanied by severe fragmentation and other three were arrested at one‐cell stage. In her second attempt, all three 2PN zygotes arrested at two‐ and three‐cell stage (Table 1 and Fig 1D).
Table 1.
Oocyte and embryo characteristics of IVF and ICSI attempts for the affected individuals.
| Family NO. | Age (years) | Duration of infertility (years) | IVF/ICSI Attempt | Retrieved oocytes | Immature oocytes | MII oocytes | Oocyte with abnormal morphology | Fertilized outcomes (2PN + 1PN + MPN + 0PN) | Cleaved/Fragmented embryo | Embryo outcomes |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 40 | 12 | IVF | 5 | 0 | 5 | multi‐polar body | 1 + 0+1 + 3 | 3/0 | 2*3‐cell, 1*2‐cell, arrested |
| ICSI | 4 | 0 | 4 | multi‐polar body, dark cytoplasm | 1 + 0+0 + 3 | 3/2 | 1*6‐cell, 1*4‐cell, 1*2‐cell, arrested | |||
| ICSI | 6 | 0 | 6 | multi‐polar body, dark cytoplasm | 3 + 0+0 + 3 | 3/1 | 1*7‐cell, 1*4‐cell, 1*5‐cell, arrested | |||
| 2 | 31 | 4 | ICSI | 9 | 0 | 9 | N/A | NA | 3/N/A | All arrested at 2‐ to 6‐cell a |
| ICSI | 13 | 0 | 13 | N/A | NA | 2/2 | All arrested at 2‐ to 6‐cell a | |||
| ICSI+AOA | 16 | 0 | 16 | 4 degradations | 4 + 5+1 + 2 | 6/N/A | All arrested at 2‐ to 6‐cell a | |||
| ICSI | 14 | 3 | 11 (4 frozen) | N/A | 3 + 2+2 + 0 | 7/6 | 3*5‐cell, 4*2‐cell, arrested | |||
| 3 | 34 | 7 | IVF+Rescue ICSI | 12 | 2 | 10 | N/A | 4 | 1/1 | 1*5‐cell, arrested a |
| ICSI | 5 | 2 | 3 | N/A | 3 + 0+0 + 0 | 3/0 | 2*3‐cell, 1*2‐cell, arrested |
Arrested, arrested during subsequent blastocyst culture; MII, Metaphase II; MPN, multi‐pronucleus; N/A, not available; PN, pronucleus.
The attempt at other hospitals.
Figure 1. Identification of MOS variants in female patients with early embryonic arrest and fragmentation.
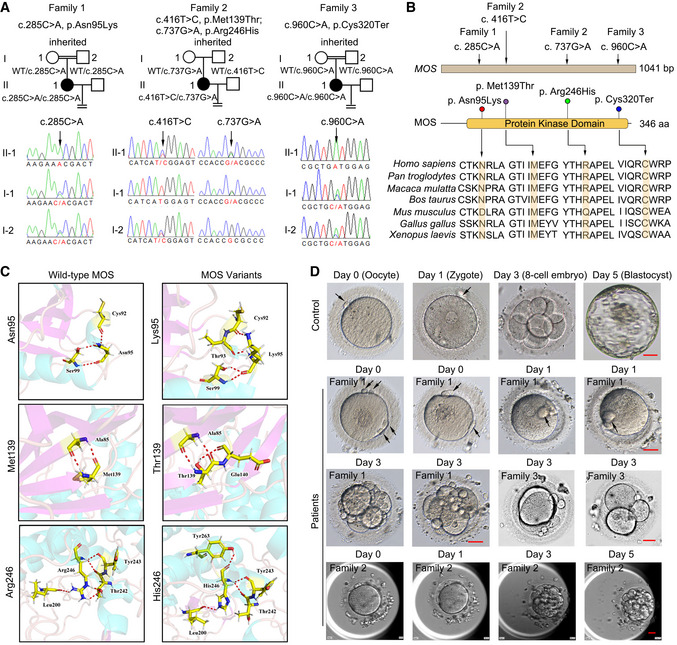
- Three pedigrees and four MOS variants were identified, including homozygous missense variant Asn95Lys in patient 1 (family 1), compound heterozygous missense variants Met139Thr and Arg246His in patient 2 (family 2), and homozygous nonsense variant Cys320Ter in patient 3 (family 3). The four MOS variants were inherited and identified via Sanger sequencing. WT indicates wild‐type.
- Distribution of MOS variants in genome and the corresponding amino acid sequences. Multiple sequence alignment of four segments of MOS and the orthologs, with mutated residue marked in yellow.
- MOS variants encoding amino acid disrupted the ion pairs formed by wild‐type MOS protein.
- The morphology of oocytes and embryos from female patients with MOS variants. The day of oocyte retrieval was defined as day 0. Polar bodies were indicated by black arrows. Scale bar = 20 µm.
Source data are available online for this figure.
Identification of MOS pathogenic variants
Whole‐exome sequencing (WES) was used to identify pathogenic variants. After filtering using the criteria described in the materials and methods section, only biallelic variants of MOS (OMIM ID: 190060; NM_005372.1) were found in all affected individuals yet absent in our 100 controls undergoing natural conception, suggesting the possible genetic contribution of MOS to human EEAF. Patient 1 and patient 3 had a homozygous missense variant c. 285C>A (p. Asn95Lys) and nonsense variant c. 960C>A (p. Cys320Ter), respectively, both variants inherited from their parents as confirmed by Sanger sequencing (Fig 1A). Patient 2 carried two missense variants, c. 416T>C (p. Met139Thr) and c. 737G>A (p. Arg246His), inherited from her father and mother, respectively (Fig 1A). All three families followed a recessive inheritance pattern. The variant p. Asn95Lys had a low frequency of 0.0000163 in the gnomAD exome database and 0.0000329 in the ExAC database (Table 2). The other three variants have not been reported in those public databases (Table 2). All four residues were located in the protein kinase domain, comprising amino acids 60th to 341st, and they are highly conserved in different species, from Xenopus laevis to Homo sapiens (Fig 1B). Based on the three‐dimensional (3D) structures of the MOS protein used to assess the effect of missense variants, the three variants had no obvious effects on the protein structure but resulted in some new hydrogen bonds produced, such as Thr93, Glu140, and Thr263, to the residues Lys95, Thr139, and His246, respectively (Fig 1C), which might have further effects on MOS protein property. In addition, the four variants were predicted pathogenic by SFIT or Polyphen with possibly or probably damaging and all had CADD scores > 20 (Table 2). The nonsense variant c. 960C>A results in premature termination at cysteine at 320th amino residue, disrupting the integrity of MOS kinase domain. Collectively, these results indicated that MOS variants are likely to be pathogenic.
Table 2.
Overview of the MOS variants observed in the families.
| Probands in Families | Genomic Position on chr 8 (bp) | cDNA Change | Protein Change | Mutation Type | Genotype | gnomAD Allele frequency | ExAC Allele frequency | Polyphen Score | Polyphen | SIFT Score | SIFT | CADD_phread Score |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Family 1 | 57026257 | c.285C>A | p. Asn95Lys | missense | homozygous | 1.63x10‐5 | 3.29x10‐5 | 0.843 | P | 0 | D | 28.5 |
| Family 2 | 57026126 | c.416T>C | p. Met139Thr | missense | compound heterozygous | N/A | N/A | 0.995 | D | 0 | D | 27.2 |
| 57025805 | c.737G>A | p. Arg246His | missense | N/A | N/A | 0.874 | D | 0.001 | D | 28.1 | ||
| Family 3 | 57025582 | c.960C>A | p. Cys320Ter | nonsense | homozygous | N/A | N/A | N/A | N/A | N/A | N/A | 41.0 |
D, Deleterious, MOS, MOS proto‐oncogene, serine/threonine kinase; N/A, not available; P, Possibly damaging.
Pathogenic effects of MOS variants in protein expression and ERK1/2 activation
Moloney sarcoma oncogene translation mediates MEK1/2 phosphorylation, leading to ERK1/2 activation during oocyte maturation, which has been demonstrated in various vertebrate oocytes (Sagata et al, 1988, 1989; Verlhac et al, 2000), except humans. Thus, we examined the expression of MOS in human oocytes. Through analysis of the reported RNA‐seq data of human oocytes and early embryos (Data ref: Yan et al, 2013), we found that MOS mRNA was highly expressed in oocytes, remarkably decreased after fertilization, and slightly increased in four‐cell stage embryos, followed by a gradual decrease from eight‐cell to blastocyst embryos (Fig 2A). Next, we examined ERK1/2 activation by staining for phosphorylated ERK1/2 (pERK1/2). The pERK1/2 signal was weak in human GV oocytes and zygotes, but was strong in MII oocytes. The staining signal is highly specific to pERK1/2, as it is undetectable in in vitro matured MII oocytes with U0126 (20 µM) treatment for 24 h (Fig 2B). Moreover, the pERK1/2 levels were significantly decreased in the unfertilized oocytes of patient 1 compared with those in the control oocytes from unidentified patients (Fig 2B). These results indicate that ERK1/2 is highly activated during human oocyte maturation and is suddenly inactivated after fertilization, and the MOS Asn95Lys variant affects ERK1/2 activation.
Figure 2. The effects of MOS variants on protein expression and ERK1/2 activation in cells and oocytes.

- The per million mapped reads (RPKM) values (extracted from GSE36552) showing the MOS expression level in human oocytes and early embryos (n = over 3 biological replicates).
- The immunofluorescence images show the pERK1/2 (red) dynamics in immature (GV, n = 5), mature (MII, n = 10), and fertilized oocytes from unidentified control patients (n = 6), as well as in mature oocytes from patient 1 carrying homozygous MOSAsn95Lys variants. pERK1/2 activation was inhibited after U0126 treatment (n = 6). FITC‐α‐tubulin (Green) and DAPI (blue) were used for co‐staining. Scale bar = 10 µm.
- Western blot analysis of the MEK1/2 and ERK1/2 activation after transfection of FLAG‐tagged wild‐type MOS and identified MOS variants in HEK 293 cells.
- Statistical analysis of the protein expression level of FLAG, pERK1/2, and pMEK1/2 (n = 2 technical replicates). The protein intensities were quantified by Image J software.
- Diagram showing the experimental design, including procedure of MOS mRNAs injection and oocyte culture. GV oocytes were overexpressed via microinjection with wild‐type MOS or MOS variant mRNAs combined with mCherry mRNAs, followed by culture for 4 h in medium containing 2.5 µM milrinone.
- Immunofluorescence of FLAG (green) and pERK1/2 (grey) in oocytes after injection of different MOS variant mRNAs combined with mCherry mRNAs. The signal of mCherry was directly captured after fixation, with DAPI staining for DNA visualization (blue). (n = over 30 oocytes in each group.) Scale bar = 10 µm.
Data information: In A and D, data are presented as mean ± SD. **P < 0.01, ***P < 0.001; ns., no significance (two‐way ANOVA with Tukey’s multiple comparisons test in D). Detailed P value as indicated.
Source data are available online for this figure.
Next, we used HEK293 cells to assess the functional properties of these MOS variants. Transfection of FLAG‐labeled expression plasmids revealed that wild‐type MOS remarkably enhanced pERK1/2 levels, whereas overexpression of the MOS variants could not effectively activate pMEK1/2 and pERK1/2 in HEK293 cells (Fig 2C and D). In addition, MOSAsn95Lys and MOSCys320Ter variants encoded MOS protein with slight decrease (Fig 2C and D). The four MOS variants exhibited similar subcellular localization but could not activate pERK1/2 in HEK293 cells except for MOSArg246His, whose overexpression just caused a decrease in pERK1/2 level (Appendix Fig S1A). Since MOS directly interacts and phosphorylates MEK1/2 to activate ERK1/2, the binding capacity of these four MOS variants with MEK1 was assessed by co‐immunoprecipitation. We found MOSCys320Ter variant hardly interacted with MEK1, and both MOSAsn95Lys and MOSArg246His variants interacted with MEK1 weaker than that of wild‐type MOS (Appendix Fig S1B and C). Therefore, we concluded that MOS variants mainly lead to MEK/ERK inactivation through the decreased MOS protein or weaker MEK1 interaction.
Since MOS is an oocyte‐specific gene, we overexpressed wild‐type MOS or four MOS variants into mouse GV oocytes to determine their expression. The MOS variants plasmids were transcribed into mRNA, and then, the same amounts of mRNAs were microinjected into GV oocytes and maintained in M2 medium with 2.5 µM milrinone for 4 h to assure the mRNA translation. The milrinone is a PDE3 inhibitor, which is used to prevent oocyte from meiotic resumption by maintaining elevated cAMP levels in oocytes. The mCherry mRNAs were co‐injected with MOS variants mRNAs to indicate successful translation. After the mCherry signal was observed by fluorescent microscope at 4 h after microinjection, the GV oocytes were collected for immunofluorescent staining to detect pERK1/2 and FLAG level (Fig 2E). We found that wild‐type MOS mRNA group activated much higher pERK1/2 than all four MOS variants in mCherry‐positive oocytes (Fig 2F).
Despite MOS protein’s translation after GVBD, previous studies have revealed that precocious MOS translation in GV oocytes facilitates meiotic maturation (Cao et al, 2020). To test whether these MOS variants altered this function, we microinjected mCherry and MOS variants mRNAs (500 ng/ul for the homozygous variants, and each 250 ng/µl with mixed total 500 ng/µl for the compound heterozygous variants) in GV oocytes and maintained in milrinone‐treated medium for 24 h, and then counted the numbers of GV, GVBD, and MII oocytes (Fig EV1A). Although oocytes were cultured in the presence of milrinone, meiosis resumption (GVBD or MII stage) occurs in 7.5% oocytes in the vector group. In contrast, approximately 60% of oocytes resumed meiosis maturation in the wild‐type MOS group, which was significantly higher than those in the other three groups with MOS variants (50% reduction with MOSCys320Ter or MOSAsn95Lys and only 20% reduction with the combined MOSMet139Thr and MOSArg246His) (Fig EV1B and C). By western blot analysis using these oocytes, we found pERK1/2 level deceased with varying degrees in three MOS variants group than wild‐type MOS group, as the MOSCys320Ter variant almost completely inactivated pERK1/2, however, combined MOSMet139Thr and MOSArg246His variants just had slight decrease (Fig EV1D).
Figure EV1. The effect of MOS variants on promoting oocyte meiosis maturation.
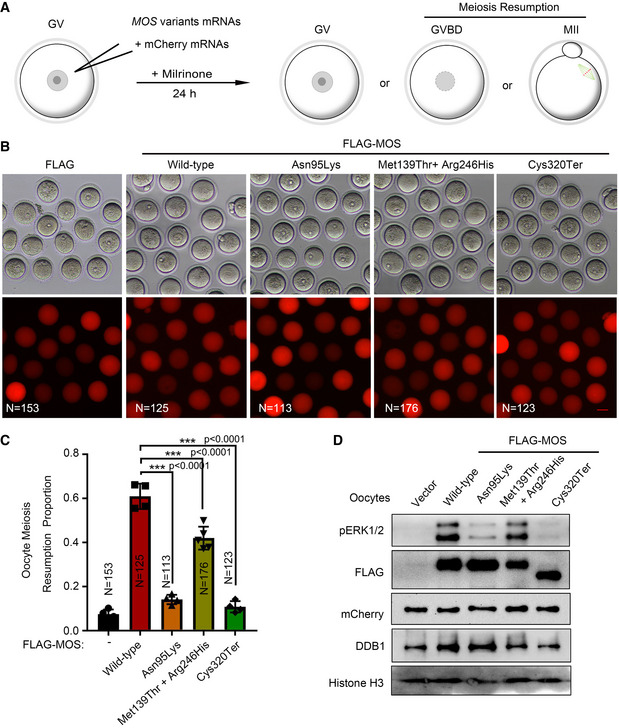
- The diagram showing the experimental procedure. Geminal vesical (GV) oocytes were microinjected with FLAG‐tagged MOS, MOS Asn95Lys, MOS Met139Thr and MOS Arg246His, MOS Cys320Ter, or FLAG mRNAs combined with mCherry mRNAs, and maintained in milrinone‐treated M2 medium for 24 h. The number of GV oocytes, geminal vesical breakdown (GVBD), or metaphase II (MII) oocytes was counted.
- The representative bright‐field images and fluorescent images were captured at 24 h after microinjection using a Nikon microscope. Over 100 oocytes in each group. Scale bar = 100 µm.
- The percentage of meiosis resumption (GVBD and MII) oocytes at 24 h after microinjection with different MOS mRNAs in (B). N number was labeled in each graph.
- Western blot analysis of FLAG, pERK1/2, and mCherry using oocytes after microinjection with different MOS mRNAs combined with mCherry mRNAs for 24 h. Histone H3 and DDB1 were used as loading controls. N = 100 oocytes per sample.
Data information: For C, data are expressed as mean ± SD. One‐way ANOVA followed by post hoc Tukey’s test for multiple comparisons, ***P < 0.0001.
Source data are available online for this figure.
We further used another method to verify the pathogenic effects of MOS variants in oocytes. We injected negative control or mouse Mos siRNAs for 24 h, and then complemented with different MOS variants mRNAs into mouse GV oocytes in a medium with milrinone, 4 h later, followed by in vitro maturation (Fig EV2A). We found that pERK1/2 was completely undetectable in the siMos group with high knockdown efficiency (Fig EV2B and C), and complement of wild‐type human MOS mRNA reversed the pERK1/2 level (Fig EV2D). However, pERK1/2 levels were much lower in other three groups supplemented with MOS Asn95Lys, MOS Cys320Ter, or MOS Met139Thr combined with MOS Arg246His mRNAs than that in the wild‐type MOS mRNAs after depletion of inner Mos in mouse oocytes (Fig EV2D).
Figure EV2. The RNAi effect of Mos and the rescuing effects of different MOS variants in oocytes.
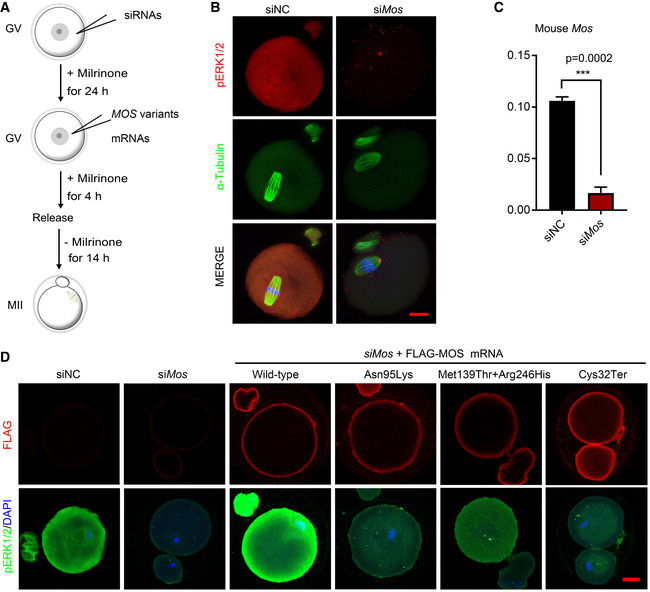
- Schematic diagram of oocyte cultures and experimental procedure. Mouse GV oocytes were microinjected with siRNAs and cultured for 24 h in the presence of 2.5 µM milrinone, followed by microinjection of MOS variants mRNAs, and cultured for 4 h before release to fresh M2 medium for another 14 h culture. Then, the RNAi efficiency of siRNA targeting mouse Mos and rescuing effects of different human MOS variants were determined.
- The representative images showing the decreased pERK1/2 (red) and abnormal spindle (FITC‐α‐tubulin, green) after Mos siRNA injection. DAPI (blue) was stained for visualization of DNA. Scale bar = 10 µm.
- RT‐qPCR results showing the mouse Mos mRNA expression levels in oocytes with negative control siRNAs or Mos siRNAs (n = 3).
- Representative immunofluorescence images showing the FLAG (red) and pERK1/2 (green) levels in mature oocytes after microinjection of siMos or combined with indicated human MOS variants in GV oocytes. At least 30 oocytes each group were used for immunofluorescence. Scale bar = 10 µm.
Data information: In C, data are expressed as mean ± SD of three biological replicates. ***P = 0.0002 (unpaired two‐tailed Student’s t‐test).
Source data are available online for this figure.
Oocyte MOS‐ERK pathway is required for cytoskeleton assembly and prevents embryo from fragmentation
One of the most obvious phenotypes caused by MOS variants herein is embryonic arrest with sever fragmentation. Since the MOS‐ERK pathway is involved in the regulation of spindle assembly and cortical F‐actin thickness in mouse oocytes (Choi et al, 1996a; Chaigne et al, 2013), we investigated whether embryo fragmentation is derived from cytoskeleton assembly defects under the ERK1/2 inactivation. First, we evaluated the effects of MOS variants on the thickness of cortical F‐actin. We observed that F‐actin intensity in MII oocytes was associated with MOS and pERK1/2 levels, with much weakened F‐actin signal in Mos‐deficient oocytes; however, only wild‐type MOS supplement could reverse the siMos‐mediated F‐actin intensity (Fig 3A and B). We used U0126, a MEK1/2 inhibitor, to inactivate ERK1/2 during oocyte maturation and found decreased cortical F‐actin in both human and mouse oocytes (Appendix Fig S2A–C). We generated ERK1 −/− ;ERK2f/f; Gdf9‐Cre (Erk1/2oo −/−) mice to specifically delete ERK1/2 in oocytes. As expected, ERK1/2 deficiency in oocytes caused very low cortical F‐actin and TPX2 signals, in which the latter one is a marker of spindle assembly (Fig 3C and D). In addition, Mos knockdown or ERK1/2 inactivation caused α‐tubulin instability (Fig EV2B and Appendix Fig S2). These results indicated that the MOS‐mediated ERK1/2 signaling pathway is essential for oocyte cytoskeleton homeostasis.
Figure 3. Oocyte MOS‐ERK1/2 pathway inactivation perturbs cortical F‐actin assembly and causes early embryonic arrest and fragmentation.
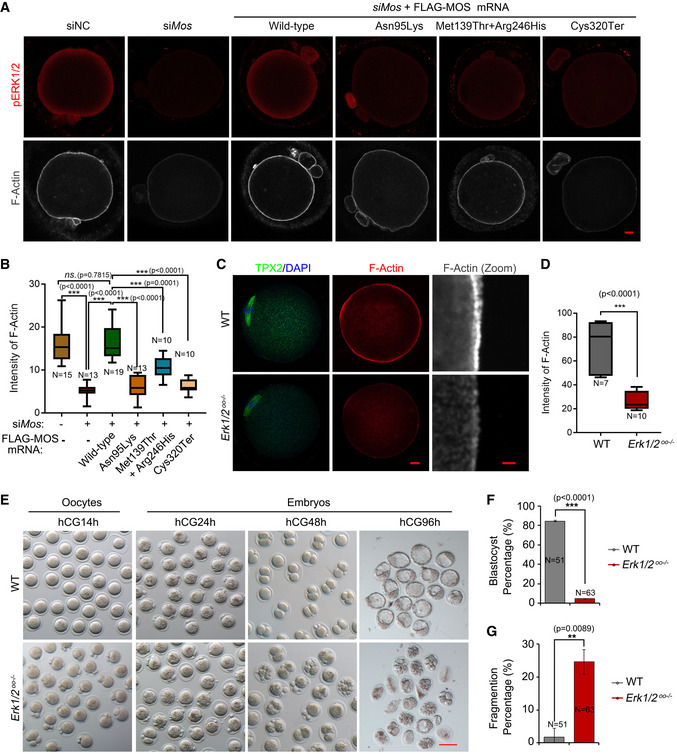
-
AImmunofluorescence of FLAG (red) and F‐actin (gray) in oocytes after microinjection of negative control or mouse Mos siRNAs combined with or without wild‐type human MOS or MOS variants mRNAs and culture in medium with 2.5 µM milrinone for 24 h, followed by release to maturation (n = 15–20 oocytes each group). Scale bar = 10 µm.
-
BThe relative intensities of F‐actin were measured and compared among the indicated groups. The oocyte numbers for analyzed were labeled in each group. The box plot showing the F‐actin intensities distribution in each group quantified using Image J (n = 10–19/group).
-
CThe immunofluorescence of F‐actin (red) and TPX2 (green) in WT and Erk1/2oo −/− oocytes, using DAPI (blue) co‐staining (n = 15–20 oocytes for each group). The zoomed images of F‐actin are displayed in gray. Scale bar = 10 µm.
-
DThe box plot summarizing the relative intensities of F‐actin between control and ERK1/2oo −/− oocytes (n = 7–10 oocytes for each group).
-
ERepresentative bright‐field images showing the morphology of oocytes and embryos from wild‐type and ERK1/2oo −/− mice. The MII oocyte, zygote, two‐cell, and blastocyst embryos were harvested in vivo at 14, 24, 48, and 96 h post‐hCG, respectively (n = 3 mice for each time points in two groups). Scale bar = 100 µm.
-
F, GBar graphs shows the blastocyst percentage (F) and fragment percentage (G) in wild‐type (n = 51) and maternal ERK1/2 deletion embryos (n = 63).
Data information: For B and D, the line in the box plot represents the median value of F‐actin intensity. Main box represents the values from the lower to upper quartile (25th to 75th percentile). The lower and upper whiskers indicate the min to max values. For (F) and (G), data are represented as mean ± SD. One‐way ANOVA with Tukey’s multiple comparisons test (B) and unpaired two‐tailed Student’s t‐test (D, F, and G) were used for statistical analysis. **P < 0.01, ***P < 0.001; ns., no significance.
Source data are available online for this figure.
We then examined the fragmentation percentage derived from maternal wild‐type or Erk1/2oo −/− oocytes. All super‐ovulated Erk1/2oo −/− oocytes were mature, but with large and multiple polar bodies (Fig 3E). After fertilization, approximately 85% of embryos reached blastocysts from the wild‐type group 96 h post‐hCG injection (Fig 3F) with rarely fragmentation (1.7%, Fig 3G). However, among the embryos derived from maternal Erk1/2oo −/− oocyte, only 4.7% of the embryos developed into blastocysts (Fig 3F), 25% embryos were severely fragmented, and the remaining embryos are arrested or degraded (Fig 3E and G).
Human MOS/ERK cascade governs maternal mRNA decay during oocyte maturation
Our previous studies demonstrated that ERK1/2 regulates the maternal mRNA translation and decay in mouse oocytes from the GV to MII stage (Sha et al, 2017; Jiang et al, 2021), and defects in maternal mRNA degradation is associate with early embryonic arrest (Sha et al, 2020a, 2020b). However, whether MOS is required for maternal mRNA decay in human oocytes is unclear. Thus, we performed RNA sequencing (RNA‐seq) using GV and MII oocytes from unidentified control individuals and MII oocytes from the patient 1 carrying homozygous MOS Asn95Lys variant. Using Spearman correlation analysis, the mRNA profiles of replicates in the groups were determined to be comparable (Fig 4A). From GV to MII oocytes, 5,290 gene transcripts were downregulated and 1,111 transcripts were upregulated with a threshold of twofold, indicating that substantial mRNAs were robustly removed by mRNA decay during human oocyte maturation (Fig 4B). These 5,290 genes in this study were referred to as “maturation‐directed decay genes”.However, the mRNA expression profile of MOS Asn95Lys MII oocytes was very different from that of the three control MII oocytes, but it was more similar to that of GV stage oocytes (Fig 4A). By comparing the log‐transformed FPKM+1 of MOS Asn95Lys oocytes with those in the control oocytes, 2,894 and 813 genes were identified as upregulated and downregulated more than twofold, respectively (Fig 4C). Additionally, more transcripts with high abundance were observed in the 2,894 upregulated genes in MOS Asn95Lys oocytes compared with the control MII oocytes (Fig 4D). Among the 2,894 upregulated transcripts in MOS Asn95Lys oocytes, 2,460 genes belong to the maturation‐directed decay genes from human GV to MII oocytes (Fig 4 E and F and Dataset EV1), which were named “MOS‐regulated decay genes”. The Gene Oncology (GO) analysis results revealed that these MOS‐regulated decay genes were mainly enriched in mitochondrial function, mitochondrial translation, and translation (Fig 4G). We selected several MOS‐regulated decay genes, including genes responsible for mitochondrial function (NDUFA8, NDUFB5, MRPL36, and MRPL37) and translation (RPL6, RPL19, and RPL23), as well as maternal genes (MOS, ZP2, TLE6, and SLBP) that were highly expressed in oocytes, as shown in Fig 4H. Based on these results, we conclude that MOS governs maternal mRNA decay during human oocyte maturation.
Figure 4. MOS deficiency retards maternal mRNA decay during human oocyte maturation.
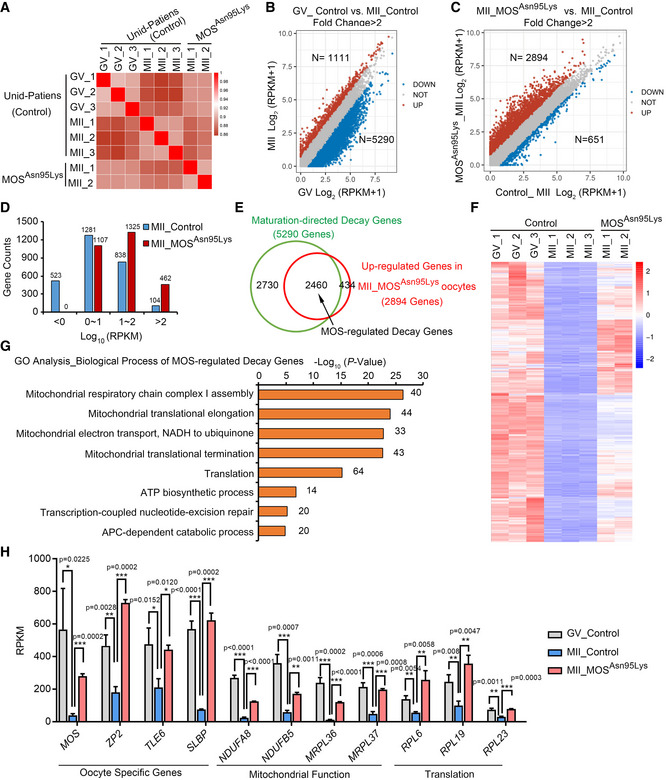
-
AThe Pearson correlation analysis of RNA‐seq results of oocyte samples from unidentified control patients and MOS Asn95Lys patient 1.
-
B, CScatter plots showing transcriptional changes between human GV and MII stage oocytes from unidentified control patients (B) and between control and MOS Asn95Lys MII oocytes (C), separately. Genes that were increased or decreased more than twofolds are indicated in red and blue, respectively. RPKM, reads per kilobase per million mapped reads.
-
DUpregulated gene numbers based on different expression level in control and MOS Asn95Lys MII oocytes.
-
EVenn diagram showing the MOS‐regulated decay genes, overlapped between the maturation‐directed decay genes (downregulated transcripts from GV to MII oocytes from control individuals) and the upregulated genes in MOS Asn95Lys oocytes.
-
FHeatmap showing the expression dynamics of MOS‐regulated decay genes in GV and MII oocytes from control or MOS Asn95Lys patient.
-
GThe enriched biological process of upregulated MOS‐regulated decay genes in MOS Asn95Lys oocytes, obtained via Gene Ontology (GO) analysis. The transcripts numbers of each biological process were labeled on the right.
-
HThe bar graphs show the expression levels of selected MOS‐regulated decay genes (n = 2 or 3 biological replicates).
Data information: In H, the RPKM values from RNA‐seq results are represented as mean ± SD. Unpaired two‐tailed Student’s t‐test was used for analysis. *P < 0.05, **P < 0.01, ***P < 0.001. Detailed P‐value as indicated.
Source data are available online for this figure.
To further determine the role of human MOS on regulating maternal mRNA clearance through ERK1/2 signal cascade, we performed RNA‐seq using in vitro matured human oocytes with U0126 treatment, a MEK1/2 inhibitor, to inhibit ERK1/2 activation. Samples from control and U0126 group were analyzed in duplicate, and the two independent replicates exhibited high correlations (Fig 5A and B). Gene expression levels were assessed as reads per kilobase of transcript per million mapped reads (RPKM) with ERCC as a spike‐in. As expected, substantial transcripts were upregulated compared with the control MII oocytes. Of the approximately 18,000 genes detected in RNA‐seq data of oocytes, 4,251 and 594 genes were upregulated and downregulated, respectively, by a twofold change threshold (Fig 5C and Dataset EV2). The gene ontology (GO) analysis revealed that the upregulated genes were relative to mitochondrial translation and mitochondrial respiratory chain complex assembly in oocytes either with MOSAsn95Lys variant or U0126 inhibition (Fig 5D). Over half of the upregulated transcripts in MII oocytes with MOSAsn95Lys variant (1,597 in 2,894) overlapped with the upregulated genes in oocytes with U0126 treatment (Fig 5E). Consistently, about 60% transcripts in MOS‐directed decay genes (1,505/2,460) belong to the transcripts whose level increased in U0126‐treated oocytes (Fig 5E). Among the 4,845 differential expressed genes (with 4,251 upregulation and 594 downregulation) in U0126‐treated oocytes, 2,364 genes with upregulation and 29 with downregulation belong to maturation‐directed decay genes (5,290, Fig 5F and G). Many genes that should be degraded during oocyte maturation were significantly upregulated both in MOSAsn95Lys oocytes and U0126‐treated oocytes, including maternal genes with high expression in oocyte (MOS, ZAR1L, ZP1, ZP2, ZP4, and SLBP), genes relative to mitochondrial translation (MRPSs and MRPLs) and mitochondrial respiratory chain complex assembly (NDUFs) (Fig 5H and Appendix Fig S3A and B). Therefore, MOS mutation or ERK1/2 inhibition mainly prevented the decrease in transcripts that should be removed from human GV to MII oocytes. Together, these results suggest MOS‐ERK signal cascade governs maternal mRNA clearance during human oocyte maturation.
Figure 5. ERK1/2 inactivation disrupts maternal mRNA clearance during human oocyte maturation.
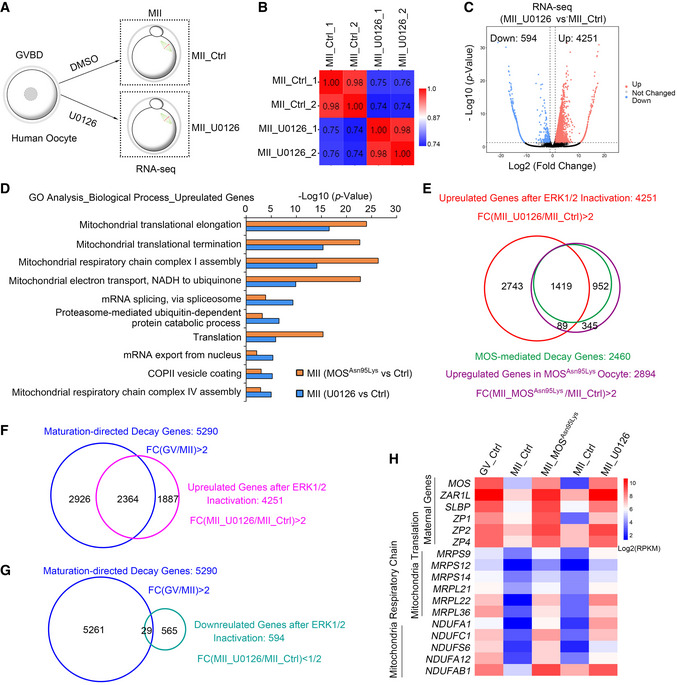
-
ASchematic presentation of human oocyte sample collection for RNA‐seq. Human immature oocytes (GVBD stage) from unidentified control women were in vitro matured with DMSO or U0126 treatment for 24 h, followed by collection of MII oocytes for RNA‐seq. Ctrl means control.
-
BThe Pearson correlation analysis of RNA‐seq results of human oocyte samples from control and U0126 group (n = 2 samples for each group). Scale bar indicates the range of the correlation coefficients (r) displayed.
-
CVolcano plot of RNA‐seq data obtained from human oocytes in control and U0126 group. “Up” and “Down” are the upregulated and downregulated transcripts in oocytes treated with U0126. The genes with upregulation and downregulation (abs[log2FC] ≥ 1; FDR ≤ 0.05) were labeled in red and blue, respectively.
-
DGene Ontology (GO) analysis of upregulated genes from RNA‐seq data of U0126 group and MOSAsn95Lys MII oocytes compared with each control oocytes.
-
EVenn diagram showing the upregulated genes in oocytes after U0126 treatment versus MOS‐regulated decay genes (green) or upregulated genes in MOSAsn95Lys oocytes (purple). FC, fold change.
-
F, GThe overlapped genes between the maturation‐directed decay genes and the upregulated (F) or downregulated (G) genes in U0126‐treated MII oocytes. FC, fold change.
-
HHeatmap showing the expression level of selected maternal genes, mitochondrial translation genes, and mitochondrial respiratory chain genes regulated by MOS mutation or ERK1/2 inactivation.
Source data are available online for this figure.
MOS/ERK cascade maintains oocyte mitochondrial function through facilitating mitochondrial mRNA clearance
As a vital organelle for energy metabolism, mitochondria become gradually activated during oocyte maturation by increasing mtDNA copy number, changing structure, and subcellular distribution to coordinate with oocyte meiosis division (Richani et al, 2021; Trebichalska et al, 2021). Mitochondrial dysfunction causes poor oocyte quality and interferes with early embryonic development (Morimoto et al, 2020; Zhang et al, 2021). Since the common retarded transcripts are enriched in mitochondrial biological process in oocytes with MOS mutation or after U0126 treatment indicated by RNA‐seq results. As expected, compared with the control group, ATP levels were significantly decreased in both human and mouse MII oocytes after U0126 treatment (Fig 6A and B), and the mitochondrial membrane potential was also obviously decreased (Fig 6C and D). In addition, we observed that the mitochondrial distribution in U0126‐treated oocytes was extremely different from the MII oocytes of control group (Fig 6C). We also determined the mitochondrial distribution and membrane potential using in vivo matured oocytes from wild‐type and Erk1/2oo −/− mice through staining JC‐1, which exhibits potential‐dependent accumulation from monomer to aggregates in mitochondria. Mitochondria distribution was comparable in either wild‐type or Erk1/2oo −/− GV oocytes. However, JC‐1 aggregates in mitochondria were enriched in the polar bodies of Erk1/2oo −/− oocytes, which is distinctly different from the uniform distribution of JC‐1 aggregates of mitochondria in wild‐type MII ooplasm (Fig 6E and F). Compared with wild‐type oocytes, the mitochondrial membrane potential and ATP level were significantly decreased in MII oocytes rather than GV oocytes from Erk1/2oo −/− mice (Fig 6G–I). Together, these results indicate human MOS/ERK cascade is required for maintaining mitochondrial function through accelerating mitochondrial mRNA clearance during oocyte maturation.
Figure 6. ERK1/2 inactivation causes oocyte mitochondrial dysfunction during meiotic maturation.

-
AThe scatter plots showing the ATP levels of human MII oocyte matured in medium with DMSO (control, Ctrl, n = 11) or 20 µM U0126 treatment (n = 12).
-
BThe scatter plots showing the ATP levels of mouse MII oocytes after in vitro maturation with DMSO (control, Ctrl, n = 19) or 20 µM U0126 (n = 17).
-
CThe representative images showing the JC‐1 monomers (green) and aggregates (red) of mouse oocytes after treatment with DMSO (control, Ctrl, n = 34) or U0126 (n = 40) in in vitro culture medium for 16 h. Scale bar = 100 µm.
-
DThe bar graph of ratio of intensities of JC‐1 aggregates to those of monomers in (C). The intensities of JC‐1 were quantified by Image J software. The oocytes number used was labeled.
-
ERepresentative images displaying JC‐1 monomers (green), aggregates (red), and bright filed images of GV and MII oocytes from wild‐type (WT) and Erk1/2oo −/− oocytes (n = 3 mice in each group). Erk1/2oo −/− oocytes exhibited big and multiple polar bodies. Scale bar = 100 µm.
-
FConfocal images of JC‐1 monomers (green) and aggregates (red) of GV and MII oocytes from wild‐type (WT) and Erk1/2oo −/− oocytes (n = 3 mice in each group). Erk1/2oo −/− oocytes exhibited big and multiple polar bodies, indicated by red arrows. Hoechst 33342 (blue) was co‐stained to represent nucleus or chromosome. Scale bar = 10 µm.
-
GThe graph showing the ratio of intensities of JC‐1 aggregates to those of monomers in (E).
-
H, IBox plot showing the ATP contents of mouse GV oocytes (H) or MII oocytes (I) from wild‐type (WT) and Erk1/2oo −/− oocytes (N number was labeled in each graph).
Data information: For A, B, D, and G, data represent mean ± SD. For H and I, the central line in the box indicates the median value of ATP level. The lower and upper limits of main boxes represent the first and third quartiles, respectively. The lower and upper whiskers represent the limits of extreme measurements. ns., no significance, **P < 0.01, ***P < 0.0001 (unpaired two‐tailed Student’s t‐test).
Source data are available online for this figure.
Discussion
Early embryonic arrest is a cause of female infertility and occurs frequently in IVF or ICSI cycles. Fragmentation is commonly accompanied with early embryonic arrest; however, the genetic determinants of fragmentation formation are still unclear. In recent years, several maternal‐effect genes have been identified to cause human early embryonic arrest: the products of TUBB8 is vital for oocyte spindle assembly and meiosis progression (Feng et al, 2016); the products of TLE6, PADI6, NLRP5, NLRP2, and KHDC3L can form subcortical maternal complex in oocytes (Alazami et al, 2015; Xu et al, 2016; Mu et al, 2019; Zhang et al, 2019b); and the product of BTG4 is responsible for maternal mRNA decay during oocyte maturation and fertilization (Alazami et al, 2015; Feng et al, 2016; Mu et al, 2019; Zhang et al, 2019b; Sha et al, 2020a; Zheng et al, 2020b). These present studies have not elucidated the origination of embryo fragmentation. In this study, we described three infertile patients characterized by EEAF, carrying MOS biallelic variants followed by a recessive inheritance pattern.
Moloney sarcoma oncogene was initially discovered as a cytostatic factor (CSF) in oocytes to maintain MII arrest, in 1989 (Sagata et al, 1989). In the last four decades, the physiological function of the MOS‐mediated ERK signaling pathway has been well characterized in studies on Xenopus and rodents. Oocytes from Mos or Erk1/2 knockout mice show parthenogenetic activation or fragmentation with longer duration of culture (Colledge et al, 1994; Hashimoto et al, 1994), while displaying a high frequency of abnormal spindle assembly, large and multiple polar bodies, and an even asymmetric division, resulting in a low fertilization rate and few offspring numbers (Araki et al, 1996; Choi et al, 1996a, 1996b). Similar to other species, human MOS mRNA is abundantly transcribed and stored in GV oocytes, and then, it is translationally activated after GVBD (Prasad et al, 2008). Only one study demonstrates that microinjection of MOS siRNA prevents meiosis maturation in human oocytes (Hashiba et al, 2001). To date, little is known about the function of human MOS in oocyte maturation and early embryos. In this study, we observed that the affected female individuals have similar reproductive phenotype previously identified in Mos −/− mice. The retrieved oocytes extruded multiple polar bodies and large polar body after fertilization in proband from family 1 with homozygous MOS variant (c.285C>A, p. Asn95Lys) (Fig 1D). However, the oocytes are morphologically normal from other two individuals carrying MOS nonsense (c.960C>A, p. Cys320Ter, family 3) or compound missense variants (c.416T>C, p. Met139Thr and c.737G>A, p. Arg246His, family 2), we speculate the phenotype difference among these three individuals is probably due to the variant locations. Consistent with the two‐cell stage arrest from Mos −/− mice (Colledge et al, 1994), most of the embryos from these three affected individuals displayed EEAF with development not past seven‐cell stage (Table 1), all arrested before embryonic genome activation (EGA). Moreover, the differences in the EEAF phenotype among the three patients are correlated with these differences of MOS variants in ERK1/2 inhibition. The patient 3 carries a homozygous nonsense variant (c.960C>A, p. Cys320Ter) in MOS gene, which displayed the weakest interaction with MEK1 (Appendix Fig S1B and C) and resulted in lowest ERK1/2 activation (Figs 2C and EV1D). Consistently, the patient 3 exhibited the earliest embryonic arrest (two‐ to three‐cell stage) than other two patients (two‐ to seven‐cell stage). This study is the first to identify pathogenic MOS variants and provides direct evidence of the role of MOS in oocyte maturation and early embryo development in human.
In addition, we observed a minor inhibition of MEK1/2 and ERK1/2 activation when we microinjected the same amount of mRNAs of MOSMet139Thr and MOSArg246His variants (each with 250 ng/µl) into mouse immature oocytes (Fig EV1C). Based on the results in Appendix Fig S1, the MOSMet139Thr variant loss function on ERK1/2 activation, similar to the other two homozygous variants (MOSAsn95Lys and MOSCys320Ter). However, the MOSArg246His variant displayed reduced binding ability with MEK1, leading to decreased pERK1/2 level to some extent (Fig Appendix S1A–C). On the one hand, we neglected there existed allelic gene expression imbalance in human. We guess that MOSMet139Thr variant is expressed more than that of MOSArg246His variant in oocytes of patient 2. This needs further investigation and validation due to lack of oocyte samples so far. On the other hand, 500 ng/µl mRNAs for microinjection is a commonly used concentration, which is sufficient and even overdosed. We performed a range of concentration of MOS mRNAs for microinjection and found MOS mRNAs from 125 ng/µl to 500 ng/µl had similar effects (50–60%) on promoting oocyte maturation, whereas the 50 ng/µl MOS mRNAs only promotes 35% oocyte to resume meiosis maturation (Appendix Fig S4A). This suggests that there exists a threshold of MOS mRNAs to activate ERK1/2 in oocyte. Then, a total of 125 ng/µl mRNAs with combined MOSMet139Thr and MOSArg246His variants were microinjected into GV oocytes with milrinone, and only 15% oocytes resumed meiotic maturation (Appendix Fig S4B and C), significantly lower than that (40% maturation) after 500 ng/µl mRNAs microinjection (Fig EV1C). Thus, we supposed that the minor inhibition of ERK1/2 activation of combined MOSMet139Thr and MOSArg246His variants might result from these two factors: the imbalanced allelic gene expression and overdose of microinjected mRNAs.
Fragmentation is often used as a morphological parameter to assess human embryo quality. However, the cause of embryo fragmentation remains unclear. Firstly, cytoskeleton disorders along with membrane instability have been proposed to involve in fragmentation as the importance of actin filaments–microtubules interplay in coordinating human oocytes spindle assembly (Kawahara et al, 2002; Fujimoto et al, 2011; Stigliani et al, 2013; Holubcova et al, 2015; Roeles & Tsiavaliaris, 2019). Mos −/− mouse oocytes displayed cytoskeleton assembly defects during meiotic maturation and severe fragmentation with long‐time culture (Choi et al, 1996a; Chaigne et al, 2013). In our study, human MOS functions as a driver for normal cytoskeleton assembly, and all the four MOS variants displayed pathogenic roles in cortical F‐actin assembly (Fig 3A and B). The oocytes from ERK1/2‐deficient mice exhibited cortical F‐actin and spindle assembly defects, leading to EEAF as patients (Fig 3C–G). We suggest that MOS/ERK pathway activation is required for cytoskeleton assembly and oocyte membrane stability maintenance. Mitochondria, the primary source of ATP generation, is another factor to be associated with embryo fragmentation, as more mitochondrial DNAs have been detected in the culture medium of embryos having higher fragmentation rates (Stigliani et al, 2013). Decreased mitochondrial membrane potential and increased reactive oxygen species are observed in fragmented embryos compared with normal embryos (Fujimoto et al, 2011). A recent study demonstrated mitochondrial transfer from induced pluripotent stem cells rescues developmental potential of in vitro fertilized embryos from aging female mouse, further validating the vital role of oocyte mitochondria in promoting early embryo development. Through our RNA‐seq analysis, we also found dysregulated mitochondrial function in MOS Asn95Lys oocytes or U0126‐treated oocytes, such as mitochondrial respiratory chain complex I assembly and mitochondrial translation elongation and termination (Fig 4G). We further found that MOS‐ERK signal cascade inactivation caused decreases in mitochondrial membrane potential and ATP content in MII oocytes (Fig 6). MOS‐ERK signal pathway inactivation in oocytes caused embryo fragmentation probably due to the dual roles of cytoskeleton disorder and mitochondrial dysregulation. Since each independent role of mitochondria or cytoskeleton on fragmentation is unclear, the molecular basis underlying oocyte‐caused embryo fragmentation requires further investigation.
Abundant maternal mRNAs are synthesized and stored during oocyte growth, and were translationally activated followed by degradation during maternal to zygotic transition (MZT) (Sha et al, 2019). We previously found that ERK1/2 facilitates CPEB1 phosphorylation and protein degradation, accelerating maternal mRNA translation and subsequent decay process during mouse oocyte maturation. When maternal mRNA decay is blocked by the deletion of relative genes, such as Btg4, MZT failure results in early embryonic arrest (Yu et al, 2016; Zhao et al, 2017, 2020). As MOS is a maternal‐effect gene that is transiently translated during oocyte maturation, and has been reported to regulate the translation of some mRNAs (de Moor & Richter, 1997), we suspected that MOS‐ERK signal cascade may participate in mRNA decay from the GV to MII stage in human oocyte. Using in vivo matured oocyte with MOS mutation and in vitro matured oocytes by blocking ERK1/2 activation, this study demonstrated MOS/ERK signal pathway facilitated maternal mRNA decay during human oocyte maturation, as substantial transcripts that should be degraded from GV to MII oocytes were abolished both in MOS Asn95Lys oocytes (Fig 4C–F) and U0126‐treated oocytes (Fig 5C–E). EGA in early embryos is dependent on maternal mRNA translation and followed by degradation (Chan et al, 2019; Sha et al, 2019). The abnormality of maternal mRNA removal is associated with human early embryonic arrest and EGA failure (Sha et al, 2020a). We infer that MOS variants‐caused early embryonic arrest is probably due to maternal mRNA decay disorder. Collectively, this study provides the first and direct evidence that the human MOS/ERK pathway governs maternal mRNA decay during human oocytes maturation.
ERK1/2 signal pathway is the best known for its role in controlling cell proliferation, differentiation, and cell survival and cell fate decision through connecting activated growth factor receptors to change gene expression (Cook et al, 2017). ERK1/2 pathway also promotes genes expression of pro‐survival BCL2 family proteins (such as BCL2 and MCL1) and represses or inhibits pro‐death proteins (such as BAD and BMF) by a variety mechanism, and thereby inhibits mitochondrial pathway of apoptosis (Boucher et al, 2000; She et al, 2002; VanBrocklin et al, 2009; Booy et al, 2011). Activated ERK1/2 also translocates to mitochondria and interacts with mitochondrial proteins. For example, ERK1/2 directly phosphorylates DRP1 and MFN1 to promote mitochondrial fission and fragmentation (Kashatus et al, 2015; Pyakurel et al, 2015). Moreover, a study reported that ERK1/2 activation‐mediated mitochondrial fission and fragmentation is required for reprogramming somatic cells to induced pluripotent stem cells (Prieto et al, 2016). Our study demonstrated that MOS‐ERK pathway facilitates genes degradation relative to mitochondria in oocyte, unveiling a new way of ERK1/2 signal pathway for regulating mitochondrial function. However, it needs further investigation that the underlying mechanism of MOS/ERK signal pathway regulates mitochondria‐relative transcripts’ stability.
In conclusion, we identified and verified the pathogenic MOS variants in infertile females with EEAF. Further experiments elucidated the critical role of the MOS‐ERK pathway in the regulation of human oocyte cytoplasmic maturation, including cytoskeleton assembly, maternal mRNA decay, and mitochondrial function. Inactivation of the MOS‐ERK signaling pathway in human and mouse oocytes results in embryo fragmentation. We believe that this study will provide a better understanding of human oocyte maturation, embryo development, and embryo fragment formation.
Materials and Methods
Clinical samples
We recruited 120 infertile individuals diagnosed with EEAF and controls undergoing natural conception from the Sir Run Run Shaw Hospital and Reproductive and Genetic Hospital of CITIC‐Xiangya. The EEAF patients were screened according to the following criteria: primary infertility; normal karyotype in both couples; male factors excluded; normal endocrine level in female; experienced recurrent failure of IVF/ICSI attempts suffering embryonic arrest; and more than 50% of the cleaved embryo represented severer embryo fragmentation that occurred in at least one attempt. All blood samples and immature oocytes were donated for investigation after informed consent was obtained. Studies of human subjects were approved by the Ethics Committee of Sir Run Run Shaw Hospital and the Ethics Committee of the Reproductive and Genetic Hospital of CITIC‐Xiangya. Informed consents were obtained from all human subjects and the experiments were performed according to the principles set out in the World Medical Association (WMA) Declaration of Helsinki and the Department of Health and Human Services Belmont Report.
Whole‐exome sequencing (WES) and variant screening
Blood samples from patients were subjected to WES. The variant score and functional prediction were assessed using the SIFT, Polyphen, and CADD programs. Variants were filtered using the following criteria (Zheng et al, 2020b): (i) variations with minor allele frequencies < 1% in the genome aggregation database (gnomAD) and Exome Aggregation Consortium (ExAC) databases; (ii) exonic non‐synonymous or splice site variants or coding indels; (iii) biallelic variants in the proband; and (iv) mRNAs/proteins that were highly expressed or specifically expressed in oocytes.
Sanger sequencing
Specific primers flanking the variants in the MOS gene were used for amplification by PCR, followed by analysis using an ABI 3100 DNA analyzer (Applied Biosystems, Foster City, CA, USA).
Molecular modeling and evolutionary conservation analysis
The three‐dimensional structure of wild‐type MOS (NP_005363.1) was predicted using the Swiss Model web server. Molecular graphics and analysis were performed using the PyMol software. Evolutionary conservation analysis was performed using the UniProt software.
Animals
Wild‐type C57BL/6 mice were purchased from the Shanghai SLAC Laboratory Animal Co., Ltd (Shanghai, China). Erk1 −/− ; Erk2f/f; Gdf9‐Cre mice were generated as previously described (Zhang et al, 2015). Female mice (7–8 weeks old) were used in all experiments. These mice were maintained under specific pathogen‐free (SPF) conditions on a 12 h light/12 h dark cycle at 21–23°C with free access to food and water. All animal experiments were performed in accordance with the guidelines of the Animal Committee of Zhejiang University.
Plasmid construction and cell transfection
Full‐length human MOS cDNA (NM_005372.1) was obtained from the human Ultimate ORF clone library. After sequencing, the QuikChange II site‐directed mutagenesis kit (200524, Agilent Technologies, Santa Clara, CA, USA) was used to introduce four variants (c.285C>A, c.416T>C, c.737G>A, and c.960C>A). The primers for mutagenesis are listed in Appendix Table S2. FLAG‐labeled expression plasmids were constructed using LR clonase (11791019, Thermo Fisher Scientific, Waltham, MA, USA), according to the manufacturer’s instructions. The pcDNA3.1+‐mCherry plasmid was kindly provided by Prof. Heng‐Yu Fan. All plasmids were verified by sequencing prior to transfection.
Human HEK293 cells, obtained from American Tissue Culture and Collection (ATCC), were cultured in DMEM medium (Gibco, Waltham, MA, USA) supplemented with 10% fetal bovine serum (FBS, Gibco), penicillin (100 U/ml), and streptomycin (100 μg/ml) at 37°C in 5% CO2. When cells reached 60–70% confluence, the same amount of plasmids was transfected using Lipofectime2000 (11668500, Thermo Fisher Scientific) according to the manufacturer’s instructions. Approximately 24–48 h after transfection, cells were fixed for immunofluorescence, western blotting, or immunoprecipitation.
Oocyte culture and embryo collection
The 7‐ to 8‐week‐old female mice were injected with 7.5 IU pregnant mare serum gonadotropin (PMSG, Ningbo San Sheng Biotech, Ningbo, China) and humanly killed 44–48 h later. GV oocytes were harvested by puncturing the large ovarian follicles in L15 medium containing 0.2% BSA, 100 U/ml penicillin, 100 μg/ml streptomycin, and 2.5 µM milrinone (HY14252, MCE, Neodesha, KS, USA) to prevent oocyte from undergoing germinal vesicle breakdown (GVBD). The GV oocytes were maintained in milrinone‐treated M2 medium (M7167, Sigma‐Aldrich, St. Louis, MO, USA) for subsequent microinjection. For collection of MII oocytes in vivo, each mouse was injected with 7.5 IU human chorionic gonadotropin (hCG, Ningbo San Sheng Biotech) at 44–48 h post‐PMSG, and the mice were then euthanized after 14 h. The oocyte and cumulus cell complexes were digested with 3 mg/ml hyaluronidase (H4272, Sigma‐Aldrich) dissolved in M2 medium. Wild‐type and Erk1 −/− ; Erk2f/f; Gdf9‐Cre female mice were mated with wild‐type adult male mice and sacrificed at 24, 48, and 96 h post‐hCG for collection of zygotes, two‐cell embryos, and blastocyst embryos, respectively.
For in vitro maturation of mouse oocyte, GV oocytes were cultured in M2 medium with indicated drugs for 14 h in an incubator at 37°C in 5% CO2. For in vitro maturation of human oocyte, immature oocyte (GVBD stage) was cultured in a commercial maturation medium (M2215, Easy Check, China) containing DMSO (C6164, Sigma‐Aldrich) or U0126 (HY‐12031A, MCE) for 24 h in an incubator at 37°C with 6% CO2.
In vitro mRNA transcription
MOS mRNA variants were synthesized in vitro as previously described (Rong et al, 2019). Briefly, FLAG‐tagged MOS plasmids were liberalized using KpnI restriction enzymes. 5'‐capped mRNAs were transcribed using Sp6 or T7 mMESSAGE mMACHINE Kits (AM1340, AM1344, Invitrogen, Carlsbad, CA, USA), followed by poly(A) tail addition using a Poly (A) Tailing Kit (AM1350, Invitrogen). Synthesized poly(A) mRNAs were recovered with lithium chloride precipitation at −20°C, cleared with 70% ethanol, and finally, dissolved in nuclease‐free water.
Microinjection of siRNAs or mRNAs
All microinjections were performed using a Narishige micromanipulator. Approximately 10 pl of mRNAs (˜500 ng/µl), siRNAs (10 µM), or their mixture was microinjected into the ooplasm. To mimic the compound heterozygous variants in patient 2, we microinjected 250 ng/µl mRNAs for each variant, reaching a total dose of 500 ng/µl MOS mRNAs as another two patients with homozygous variants. GV oocytes were cultured in M2 medium with 2.5 µM milrinone (HY14252, MCE) for 24 h for mRNA translation or siRNA knockdown, prior to being released into fresh M2 medium. The sequences of siRNAs are listed in Appendix Table S2.
Real‐time quantitative PCR (qRT‐PCR)
The mRNA levels were determined using qRT‐PCR and calculated using the comparative Ct method, as previously described (Zhang et al, 2015). Total RNA from 20 oocytes was extracted using the RNA‐easy Micro Kit (74004, Qiagen, Hilden, Germany), according to the manufacturer’s protocol. First‐strand cDNA was generated using SuperScript II Reverse Transcriptase (18064014, Thermo Fisher Scientific). Real‐time PCR was conducted with SYBR Green mix using the CFX96 Real‐Time System (Bio‐Rad, Hercules, CA, USA). Mouse Gapdh or human ACTB expression was used as an internal control. The primers used are listed in Appendix Table S2.
Immunofluorescence
Human or mouse oocytes were fixed in 3.7% paraformaldehyde diluted in PBS for 30 min at room temperature and then permeabilized with 0.2% TritonX‐100 for 15 min. After incubation for 1 h in blocking buffer (1% BSA diluted in PBS with 0.1% TritonX‐100), oocytes were stained with the indicated primary antibodies diluted in blocking buffer overnight at 4°C. After three washes, samples were incubated with Alexa Fluor 568‐conjugated goat anti‐rabbit (A11036, Invitrogen), Alexa Fluor 488‐conjugated goat anti‐rabbit (A11001, Invitrogen), Alexa Fluor 568‐conjugated donkey anti‐mouse (A10037, Invitrogen) secondary antibodies with dilution of 1:300, in combination with 1 µg/ml DAPI (236276, Roche, Basel, Switzerland), with or without FITC‐α‐tubulin (1:400, F2168, Sigma‐Aldrich), or Alexa Fluor 647‐conjugated phalloidin (1:200, A22287, Invitrogen) for 1 h at room temperature. After washing four times, the oocytes were mounted on slides with anti‐fade medium and imaged using a laser‐scanning confocal microscope (LSM800, Carl Zeiss, Jena, Germany). The primary antibodies used were as follows: mouse monoclonal anti‐FLAG (1:500, F1804, Sigma‐Aldrich), rabbit monoclonal anti‐pERK1/2 (1:400, #4370, Cell Signaling Technology, Danvers, MA, USA), and rabbit polyclonal anti‐TPX2 (1:200, NB500‐179, Novus Biologicals, Littleton, CO, USA).
Western blotting
Cells transiently expressing the indicated plasmids were lysed using 1× Laemmli sample buffer (1610747, Bio‐Rad). Proteins were separated via SDS‐PAGE and transferred onto a polyvinylidene difluoride (PVDF) membrane. After blocking in TBS containing 5% defat milk for 1 h at room temperature, membranes were incubated with primary antibodies (1:1,000 dilution) overnight at 4°C. After washing three times in TBS‐Tween‐20 (0.05%), membranes were incubated with goat anti‐rabbit horseradish peroxidase‐conjugated (1:5,000, 111‐035‐003, Jackson ImmunoResearch, West Grove, PA, USA) or goat anti‐mouse (1:5,000, 115‐035‐003, Jackson ImmunoResearch) secondary antibodies for 1 h at room temperature. Signals were detected using enhanced chemiluminescence (ECL; Millipore, Burlington, MA, USA). The primary antibodies used were as follows: mouse monoclonal anti‐FLAG (1:1,000, F1804, Sigma‐Aldrich), rabbit monoclonal anti‐pERK1/2 (1:1,000, #4370, Cell Signaling Technology), rabbit monoclonal anti‐ERK1/2 (1:1,000, #4695, Cell Signaling Technology), rabbit anti‐pMEK1/2 (1:500, #9154, Cell Signaling Technology), mouse anti‐MEK1/2 (1:500, #4694, Cell Signaling Technology), rabbit anti‐Histone H3 (1:1,000, #4499, Cell Signaling Technology), rabbit anti‐mCherry (1:1,000, ab183628, Abcam), rabbit anti‐Vinculin (1:1,000, ab207440, Abcam), and mouse polyclonal anti‐α‐tubulin (1:1,000, 32‐2500, Thermo Fisher Scientific).
Co‐immunoprecipitation
Co‐immunoprecipitation assays were performed as previously described (Zhang et al, 2018). Briefly, after transfection of the indicated plasmids for 36 h, cells were washed with cold PBS and then lysed using NP‐40 lysis buffer (P0013F, Beyotime Biotech, China) containing freshly added proteinase inhibitor cocktail (Roche) and 1 mM PMSF (Beyotime Biotech) at 4°C for 30 min. Subsequently, FLAG‐M2 magnetic beads (M8823, Sigma‐Aldrich) were prepared and added to the cell extracts and incubated overnight at 4°C. Beads were washed four to five times with lysis buffer, and the immune complexes were subjected to western blotting.
Mitochondrial function measurement
ATP level was assayed in denuded human or mouse oocytes at the MII stage, after in vitro maturation. ATP content measurement was performed using Enhanced ATP Assay Kit (S0027, Beyotime Biotech) based on the luciferin–luciferase reaction as described previously (Ma et al, 2021). Briefly, a standard curve was generated for each series of analyses. MII oocytes after treatment with or without U0126 were harvested, followed by zona pellucida removal in acidic Tyrode's solution (T1788, Sigma). After three washes in 0.1% BSA in PBS, each oocyte was lysed directly with 20 µl lysis buffer on ice for 5 min. Then, 100 μl of ice‐cold ATP assay working solution was added to the oocyte lysis solution and incubated for 10 min at room temperature in the dark. Next, the luminescence of each oocyte was measured using a luminometer (Model TD‐20/20, Turner Biosystems, Madison, WI, USA). The ATP level was calculated using the formula derived from the linear regression of the standard curve.
The mitochondrial membrane potential was determined using a commercial kit with JC‐1 (C2006, Beyotime Biotech) according to the manufacturer's protocol. Briefly, MII oocytes after in vitro maturation were transferred to M2 medium with JC‐1 and cultured for 1 h in a 5% CO2 incubator at 37°C. Then, oocytes were rinsed several times in fresh M2 medium. Next, the fluorescence images of JC‐1 aggregates and monomer were captured using a fluorescence microscope (Nikon, Japan, Eclipse ts2R), respectively. Notably, we strictly controlled the washing intensities and the exposure time of each group. Fluorescence intensities of aggregates and monomer were quantified using Image J Software (National Institutes of Health, Bethesda, MD, USA). The ratio of aggregates and monomer was calculated to indicate the mitochondrial membrane potential.
Single oocyte RNA sequencing (RNA‐seq)
Human GV and MII oocytes were donated from unidentified control patients and patients with the MOS Asn95Lys homozygous variant. A single oocyte was lysed using 4 µl cell lysis buffer, and cDNA was obtained via reverse transcription using the SMART‐seq2 method, as previously described (Zhang et al, 2019a). In brief, first‐strand cDNA synthesis and amplification were performed using SuperScript II (18064014, Thermo Fisher Scientific) and KAPA Hotstart Hifi ready mix (KK2601, Roche). Sequencing libraries were constructed using TruePrep DNA Library Prep Kit V2 for Illumina (TD503, Vazyme Biotech, Nanjing, China), followed by sequencing on an Illumina NovaSeq 6000 with 150‐bp long paired‐end reads.
RNA‐seq data analysis
RNA‐seq data were processed using standard procedures as previously described (Zhang et al, 2019a). In brief, raw reads were first preprocessed using Trimmomatic (v0.35) to remove adapters, trim low‐quality bases from both read ends, and remove reads < 36 bp in length. Clean reads were then mapped to the human reference genome of GRCh38 using STAR aligner (v2.5.2b). The gene counts were calculated using HTSeq (v0.6.1p1). The expression levels of each gene were quantified using normalized reads per kilobase per million mapped reads (RPKM). Spearman correlation coefficients were analyzed in R. Scatter plots and volcano plot were created using the ggplot2 packages in R, and heatmaps were plotted using pheaetmap packages in R. Gene Ontology (GO) term analysis was performed using DAVID online tool (https://david.ncifcrf.gov). The Venn diagram plots were drawn based on the results of Bioinformatics and Evolutionary Genomics online tool (http://bioinformatics.psb.ugent.be/webtools/Venn/).
Image acquisition and quantification
For bright‐field image acquisition of oocytes and embryos, a Nikon Ts2R microscope with a Hoffman system was used. Fluorescent images were captured using an LSM800 laser scanning confocal microscope (Carl Zeiss). For oocyte F‐actin acquisition, a layer with maximal intensity was chosen. ImageJ software was used for signal quantification. The signal intensity was measured in an area with an equivalent size selected close to the F‐actin signal, and the mean intensity of the F‐actin was calculated.
Statistical analysis
All collected oocytes from wild‐type mice were randomized into experimental groups for subsequent microinjection. Then, the survived oocytes in each group were used for experiments and analysis. For the oocyte or embryo phenotype analysis from wild‐type and Erk1 −/− ; Erk2f/f; Gdf9‐Cre mice, age‐matched animals were grouped based on their genotype. For the studies using oocytes, N indicates oocyte number or experimental replicates. Two to three independent experiments were conducted.
Statistical analysis was conducted using the GraphPad Prism 7.0 software (GraphPad Software Inc., La Jolla, CA, USA) or Microsoft Office Excel 2016. For the bar graphs, data are expressed as mean ± standard error (SD) for biological replicates or technical replicates. Box plots show the medians, quartiles, and range of continuous data to demonstrate the variability of data and the degree of normality. Differences between two groups were compared using the unpaired two‐tailed Student’s t‐test. In the case of more than two groups, one‐way ANOVA was used, followed by post hoc Tukey’s test for multiple comparisons. P < 0.05 was considered statistically significant.
Author contributions
SZ, H‐YF, and GL conceived the project. Y‐LZ, WZ, and H‐YF designed the experiments. Y‐LZ and WZ performed most of the experiments and analyzed the data. PR microinjected mRNAs or siRNAs in mouse oocytes and performed western blot analysis. XL analyzed the RNA‐seq data. HH, XT, and S‐PZ recruited the patients. HW and JJ collected the human samples. WY, YZ, and YH constructed libraries of single cell RNA‐seq. J‐CJ and YM assisted for animal housing and genotyping for Erk1/2 deficient mice. LC constructed one of the plasmids. YG, LH, KL, FG, and G‐XL assisted for human embryos phenotype monitoring. Y‐LZ and WZ wrote the draft of manuscript, with contributions and approval from all authors. H‐YF, SZ, and GL critically revised the manuscript. All authors read and approved the final manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
For more information
- (i)
-
(ii)
OMIM, https://www.omim.org/
-
(iii)
Uniprot, https://www.uniprot.org/
- (iv)
-
(v)
PyMol software, https://pymol.org/2/
-
(vi)
Swiss Model web server, https://swissmodel.expasy.org/
-
(vii)
ExAC Browser, http://exac.broadinstitute.org/
- (viii)
-
(ix)
PolyPhen‐2, http://genetics.bwh.harvard.edu/pph2/
- (x)
-
(xi)
DAVID, https://david.ncifcrf.gov/
-
(xii)
UCSC Genome Browser, http://genome.ucsc.edu/
- (xiii)
-
(xiv)
PrimerBank, https://pga.mgh.harvard.edu/primerbank/
Supporting information
Appendix
Expanded View Figures PDF
Dataset EV1
Dataset EV2
Source Data for Expanded View and Appendix
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Acknowledgments
We thank all the staff at the IVF center in Sir Run Run Shaw Hospital and at the Reproductive and Genetic Hospital of CITIC‐Xiangya. We thank Chao Yu in Zhejiang University for experimental design of this study. This work was supported by the National Key Research and Development Program of China (2018YFC1004800 and 2017YFC1001500), the National Natural Science Foundation of China (82071640, 31701260, 82001633, and 31930013), the Key Projects Jointly Constructed by the Ministry and the Province of Zhejiang Medical and Health Science and Technology Project (WKJ‐ZJ‐2005), Natural Science Foundation of Zhejiang Province (LY21H040003, D22C068649), China Postdoctoral Science Foundation (2020M682575 and 2021T140198), the Changsha Municipal Natural Science Foundation (kq2007022), and Hunan Provincial Grant for Innovative Province Construction (2019SK4012).
EMBO Mol Med (2021) 13: e14887.
See also: L Wang & Q Sang (December 2021)
Contributor Information
Ge Lin, Email: linggf@hotmail.com.
Heng‐Yu Fan, Email: hyfan@zju.edu.cn.
Songying Zhang, Email: zhangsongying@zju.edu.cn.
Data availability
The processing single‐cell RNA‐seq dataset supporting conclusion is included within the supplemental data. The single‐cell RNA‐seq raw data generated in the manuscript is available in GSA‐Human under the accession code HRA001269 (https://bigd.big.ac.cn/gsa‐human/browse/HRA001269) or from the corresponding author upon reasonable request.
References
- Alazami AM, Awad SM, Coskun S, Al‐Hassan S, Hijazi H, Abdulwahab FM, Poizat C, Alkuraya FS (2015) TLE6 mutation causes the earliest known human embryonic lethality. Genome Biol 16: 240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araki K, Naito K, Haraguchi S, Suzuki R, Yokoyama M, Inoue M, Aizawa S, Toyoda Y, Sato E (1996) Meiotic abnormalities of c‐mos knockout mouse oocytes: activation after first meiosis or entrance into third meiotic metaphase. Biol Reprod 55: 1315–1324 [DOI] [PubMed] [Google Scholar]
- Booy EP, Henson ES, Gibson SB (2011) Epidermal growth factor regulates Mcl‐1 expression through the MAPK‐Elk‐1 signalling pathway contributing to cell survival in breast cancer. Oncogene 30: 2367–2378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boucher MJ, Morisset J, Vachon PH, Reed JC, Laine J, Rivard N (2000) MEK/ERK signaling pathway regulates the expression of Bcl‐2, Bcl‐X(L), and Mcl‐1 and promotes survival of human pancreatic cancer cells. J Cell Biochem 79: 355–369 [PubMed] [Google Scholar]
- Cao LR, Jiang JC, Fan HY (2020) Positive feedback stimulation of Ccnb1 and Mos mRNA translation by MAPK cascade during mouse oocyte maturation. Front Cell Dev Biol 8: 609430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaigne A, Campillo C, Gov NS, Voituriez R, Azoury J, Umaña‐Diaz C, Almonacid M, Queguiner I, Nassoy P, Sykes C et al (2013) A soft cortex is essential for asymmetric spindle positioning in mouse oocytes. Nat Cell Biol 15: 958–966 [DOI] [PubMed] [Google Scholar]
- Chan SH, Tang Y, Miao L, Darwich‐Codore H, Vejnar CE, Beaudoin J‐D, Musaev D, Fernandez JP, Benitez MDJ, Bazzini AA et al (2019) Brd4 and P300 confer transcriptional competency during zygotic genome activation. Dev Cell 49: 867–881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi T, Fukasawa K, Zhou R, Tessarollo L, Borror K, Resau J, Vande Woude GF (1996a) The Mos/mitogen‐activated protein kinase (MAPK) pathway regulates the size and degradation of the first polar body in maturing mouse oocytes. Proc Natl Acad Sci USA 93: 7032–7035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi T, Rulong S, Resau J, Fukasawa K, Matten W, Kuriyama R, Mansour S, Ahn N, Vande Woude GF (1996b) Mos/mitogen‐activated protein kinase can induce early meiotic phenotypes in the absence of maturation‐promoting factor: a novel system for analyzing spindle formation during meiosis I. Proc Natl Acad Sci USA 93: 4730–4735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colledge WH, Carlton MB, Udy GB, Evans MJ (1994) Disruption of c‐mos causes parthenogenetic development of unfertilized mouse eggs. Nature 370: 65–68 [DOI] [PubMed] [Google Scholar]
- Cook SJ, Stuart K, Gilley R, Sale MJ (2017) Control of cell death and mitochondrial fission by ERK1/2 MAP kinase signalling. FEBS J 284: 4177–4195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Moor CH, Richter JD (1997) The Mos pathway regulates cytoplasmic polyadenylation in Xenopus oocytes. Mol Cell Biol 17: 6419–6426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebner T, Yaman C, Moser M, Sommergruber M, Polz W, Tews G (2001) Embryo fragmentation in vitro and its impact on treatment and pregnancy outcome. Fertil Steril 76: 281–285 [DOI] [PubMed] [Google Scholar]
- Feng R, Sang Q, Kuang Y, Sun X, Yan Z, Zhang S, Shi J, Tian G, Luchniak A, Fukuda Y et al (2016) Mutations in TUBB8 and human oocyte meiotic arrest. N Engl J Med 374: 223–232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujimoto VY, Browne RW, Bloom MS, Sakkas D, Alikani M (2011) Pathogenesis, developmental consequences, and clinical correlations of human embryo fragmentation. Fertil Steril 95: 1197–1204 [DOI] [PubMed] [Google Scholar]
- Hashiba Y, Asada Y, Heikinheimo O, Lanzendorf SE, Mizutani S (2001) Microinjection of antisense c‐mos oligonucleotides prevents the progression of meiosis in human and hamster oocytes. Fertil Steril 76: 143–147 [DOI] [PubMed] [Google Scholar]
- Hashimoto N, Watanabe N, Furuta Y, Tamemoto H, Sagata N, Yokoyama M, Okazaki K, Nagayoshi M, Takedat N, Ikawatll Y et al (1994) Parthenogenetic activation of oocytes in c‐mos‐deficient mice. Nature 370: 68–71 [DOI] [PubMed] [Google Scholar]
- Holubcova Z, Blayney M, Elder K, Schuh M (2015) Human oocytes. Error‐prone chromosome‐mediated spindle assembly favors chromosome segregation defects in human oocytes. Science 348: 1143–1147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang JC, Zhang H, Cao LR, Dai XX, Zhao LW, Liu HB, Fan HY (2021) Oocyte meiosis‐coupled poly(A) polymerase alpha phosphorylation and activation trigger maternal mRNA translation in mice. Nucleic Acids Res 49: 5867–5880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kashatus JA, Nascimento A, Myers LJ, Sher A, Byrne FL, Hoehn KL, Counter CM, Kashatus DF (2015) Erk2 phosphorylation of Drp1 promotes mitochondrial fission and MAPK‐driven tumor growth. Mol Cell 57: 537–551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawahara M, Mori T, Tanaka H, Shimizu H (2002) The suppression of fragmentation by stabilization of actin filament in porcine enucleated oocytes. Theriogenology 58: 1081–1095 [DOI] [PubMed] [Google Scholar]
- Ma Y, Yang W, Ren P, Li X, Jin J, Dai Y, Pan Y, Jiang L, Fan H, Zhang Y, & et al (2021) Lysophosphatidic acid improves oocyte quality during IVM by activating the ERK1/2 pathway in cumulus cells and oocytes. Mol Hum Reprod 27: gaab032 [DOI] [PubMed] [Google Scholar]
- Morimoto N, Hashimoto S, Yamanaka M, Nakano T, Satoh M, Nakaoka Y, Iwata H, Fukui A, Morimoto Y, Shibahara H (2020) Mitochondrial oxygen consumption rate of human embryos declines with maternal age. J Assist Reprod Genet 37: 1815–1821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mu J, Wang W, Chen B, Wu L, Li B, Mao X, Zhang Z, Fu J, Kuang Y, Sun X et al (2019) Mutations in NLRP2 and NLRP5 cause female infertility characterised by early embryonic arrest. J Med Genet 56: 471–480 [DOI] [PubMed] [Google Scholar]
- Prasad CK, Mahadevan M, MacNicol MC, MacNicol AM (2008) Mos 3' UTR regulatory differences underlie species‐specific temporal patterns of Mos mRNA cytoplasmic polyadenylation and translational recruitment during oocyte maturation. Mol Reprod Dev 75: 1258–1268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prieto J, Leon M, Ponsoda X, Sendra R, Bort R, Ferrer‐Lorente R, Raya A, Lopez‐Garcia C, Torres J (2016) Early ERK1/2 activation promotes DRP1‐dependent mitochondrial fission necessary for cell reprogramming. Nat Commun 7: 11124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pyakurel A, Savoia C, Hess D, Scorrano L (2015) Extracellular regulated kinase phosphorylates mitofusin 1 to control mitochondrial morphology and apoptosis. Mol Cell 58: 244–254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richani D, Dunning KR, Thompson JG, Gilchrist RB (2021) Metabolic co‐dependence of the oocyte and cumulus cells: essential role in determining oocyte developmental competence. Hum Reprod Update 27: 27–47 [DOI] [PubMed] [Google Scholar]
- Roeles J, Tsiavaliaris G (2019) Actin‐microtubule interplay coordinates spindle assembly in human oocytes. Nat Commun 10: 4651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rong Y, Ji SY, Zhu YZ, Wu YW, Shen L, Fan HY (2019) ZAR1 and ZAR2 are required for oocyte meiotic maturation by regulating the maternal transcriptome and mRNA translational activation. Nucleic Acids Res 47: 11387–11402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sagata N, Oskarsson M, Copeland T, Brumbaugh J, Vande Woude GF (1988) Function of c‐mos proto‐oncogene product in meiotic maturation in Xenopus oocytes . Nature 335: 519–525 [DOI] [PubMed] [Google Scholar]
- Sagata N, Watanabe N, Vande Woude GF, Ikawa Y (1989) The c‐mos proto‐oncogene product is a cytostatic factor responsible for meiotic arrest in vertebrate eggs. Nature 342: 512–518 [DOI] [PubMed] [Google Scholar]
- Sako K, Suzuki K, Isoda M, Yoshikai S, Senoo C, Nakajo N, Ohe M, Sagata N (2014) Emi2 mediates meiotic MII arrest by competitively inhibiting the binding of Ube2S to the APC/C. Nat Commun 5: 3667 [DOI] [PubMed] [Google Scholar]
- Sha QQ, Dai XX, Dang Y, Tang F, Liu J, Zhang YL, Fan HY (2017) A MAPK cascade couples maternal mRNA translation and degradation to meiotic cell cycle progression in mouse oocytes. Development 144: 452–463 [DOI] [PubMed] [Google Scholar]
- Sha QQ, Zhang J, Fan HY (2019) A story of birth and death: mRNA translation and clearance at the onset of maternal‐to‐zygotic transition in mammalsdagger. Biol Reprod 101: 579–590 [DOI] [PubMed] [Google Scholar]
- Sha QQ, Zheng W, Wu YW, Li S, Guo L, Zhang S, Lin G, Ou XH, Fan HY (2020a) Dynamics and clinical relevance of maternal mRNA clearance during the oocyte‐to‐embryo transition in humans. Nat Commun 11: 4917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sha QQ, Zhu YZ, Li S, Jiang Y, Chen L, Sun XH, Shen L, Ou XH, Fan HY (2020b) Characterization of zygotic genome activation‐dependent maternal mRNA clearance in mouse. Nucleic Acids Res 48: 879–894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- She QB, Ma WY, Zhong S, Dong Z (2002) Activation of JNK1, RSK2, and MSK1 is involved in serine 112 phosphorylation of Bad by ultraviolet B radiation. J Biol Chem 277: 24039–24048 [DOI] [PubMed] [Google Scholar]
- Shoji S, Yoshida N, Amanai M, Ohgishi M, Fukui T, Fujimoto S, Nakano Y, Kajikawa E, Perry AC (2006) Mammalian Emi2 mediates cytostatic arrest and transduces the signal for meiotic exit via Cdc20. EMBO J 25: 834–845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stigliani S, Anserini P, Venturini PL, Scaruffi P (2013) Mitochondrial DNA content in embryo culture medium is significantly associated with human embryo fragmentation. Hum Reprod 28: 2652–2660 [DOI] [PubMed] [Google Scholar]
- Suzuki T, Suzuki E, Yoshida N, Kubo A, Li H, Okuda E, Amanai M, Perry AC (2010) Mouse Emi2 as a distinctive regulatory hub in second meiotic metaphase. Development 137: 3281–3291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trebichalska Z, Kyjovska D, Kloudova S, Otevrel P, Hampl A, Holubcova Z (2021) Cytoplasmic maturation in human oocytes: an ultrastructural study dagger. Biol Reprod 104: 106–116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- VanBrocklin MW, Verhaegen M, Soengas MS, Holmen SL (2009) Mitogen‐activated protein kinase inhibition induces translocation of Bmf to promote apoptosis in melanoma. Cancer Res 69: 1985–1994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verlhac MH, Lefebvre C, Kubiak JZ, Umbhauer M, Rassinier P, Colledge W, Maro B (2000) Mos activates MAP kinase in mouse oocytes through two opposite pathways. EMBO J 19: 6065–6074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe N, Hunt T, Ikawa Y, Sagata N (1991) Independent inactivation of MPF and cytostatic factor (Mos) upon fertilization of Xenopus eggs . Nature 352: 247–248 [DOI] [PubMed] [Google Scholar]
- Xu Y, Shi Y, Fu J, Yu M, Feng R, Sang Q, Liang BO, Chen B, Qu R, Li B et al (2016) Mutations in PADI6 cause female infertility characterized by early embryonic arrest. Am J Hum Genet 99: 744–752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan L, Yang M, Guo H, Yang L, Wu J, Li R, Liu P, Lian Y, Zheng X, Yan J et al (2013) Gene Expression Omnibus GSE36552 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE36552). [DATASET]
- Yu C, Ji S‐Y, Sha Q‐Q, Dang Y, Zhou J‐J, Zhang Y‐L, Liu Y, Wang Z‐W, Hu B, Sun Q‐Y et al (2016) BTG4 is a meiotic cell cycle‐coupled maternal‐zygotic‐transition licensing factor in oocytes. Nat Struct Mol Biol 23: 387–394 [DOI] [PubMed] [Google Scholar]
- Zhang C, Tao L, Yue Y, Ren L, Zhang Z, Wang X, Tian J, An L (2021) Mitochondrial transfer from induced pluripotent stem cells rescues developmental potential of in vitro fertilized embryos from aging femalesdagger. Biol Reprod 104: 1114–1125 [DOI] [PubMed] [Google Scholar]
- Zhang J, Zhang Y‐L, Zhao L‐W, Guo J‐X, Yu J‐L, Ji S‐Y, Cao L‐R, Zhang S‐Y, Shen LI, Ou X‐H et al (2019a) Mammalian nucleolar protein DCAF13 is essential for ovarian follicle maintenance and oocyte growth by mediating rRNA processing. Cell Death Differ 26: 1251–1266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Chen Z, Zhang D, Zhao B, Liu L, Xie Z, Yao Y, Zheng P (2019b) KHDC3L mutation causes recurrent pregnancy loss by inducing genomic instability of human early embryonic cells. PLoS Biol 17: e3000468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang YL, Liu XM, Ji SY, Sha QQ, Zhang J, Fan HY (2015) ERK1/2 activities are dispensable for oocyte growth but are required for meiotic maturation and pronuclear formation in mouse. J Genet Genomics 42: 477–485 [DOI] [PubMed] [Google Scholar]
- Zhang YL, Zhao LW, Zhang J, Le R, Ji SY, Chen C, Gao Y, Li D, Gao S, Fan HY (2018) DCAF13 promotes pluripotency by negatively regulating SUV39H1 stability during early embryonic development. EMBO J 37: e98981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao BS, Wang X, Beadell AV, Lu Z, Shi H, Kuuspalu A, Ho RK, He C (2017) m(6)A‐dependent maternal mRNA clearance facilitates zebrafish maternal‐to‐zygotic transition. Nature 542: 475–478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao LW, Zhu YZ, Chen H, Wu YW, Pi SB, Chen L, Shen L, Fan HY (2020) PABPN1L mediates cytoplasmic mRNA decay as a placeholder during the maternal‐to‐zygotic transition. EMBO Rep 21: e49956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng W, Chen L, Dai J, Dai C, Guo J, Lu C, Gong F, Lu G, Lin G (2020a) New biallelic mutations in PADI6 cause recurrent preimplantation embryonic arrest characterized by direct cleavage. J Assist Reprod Genet 37: 205–212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng W, Zhou Z, Sha Q, Niu X, Sun X, Shi J, Zhao L, Zhang S, Dai J, Cai S et al (2020b) Homozygous mutations in BTG4 cause zygotic cleavage failure and female infertility. Am J Hum Genet 107: 24–33 [DOI] [PMC free article] [PubMed] [Google Scholar]