

Abstract
Circular RNAs (circRNAs) have important potential in modulating vascular smooth muscle cell (VSMC) activity, but their roles in abdominal aortic aneurysm (AAA) are unknown. We performed in situ hybridization and immunohistochemistry and determined that circChordc1 (cysteine and histidine-rich domain containing 1) was markedly downregulated in aneurysm tissue compared with normal arteries. A gene gain and loss strategy was used to confirm that circChordc1 transformed VSMCs into a contracted phenotype and improved their growth, which significantly suppressed aneurysm formation and reduced the risk of rupture in mouse models of angiotensin (Ang) II- and CaCl2-induced AAA. RNA pull-down, immunoprecipitation, and immunoblotting indicated that circChordc1 facilitated the VSMC phenotype and growth determination by binding to vimentin and ANXA2 (annexin A2), which not only increased vimentin phosphorylation to promote its degradation but also promoted the interaction between ANXA2 and glycogen synthase kinase 3 beta (GSK3β) to induce the nuclear entry of β-catenin. Thus, our present study revealed that circChordc1 optimized the VSMC phenotype and improved their growth by inducing vimentin degradation and increasing the activity of the GSK3β/β-catenin pathway, thereby extenuating vascular wall remodeling and reversing pathological aneurysm progression.
Keywords: abdominal aortic aneurysm, VSMCs, apoptosis, phenotypic switching, CircRNA, ANXA2, vimentin
Graphical abstract
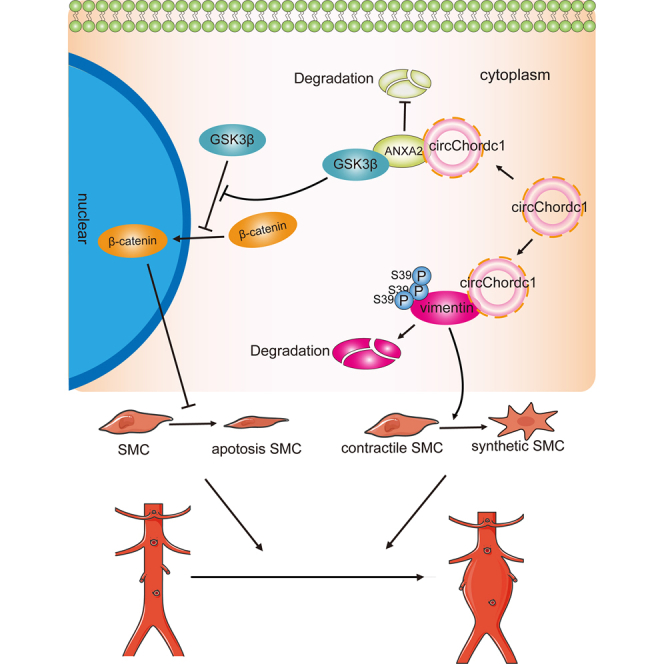
Our study identified a VSMC-specific circChordc1, which optimized the VSMC phenotype and improved their growth by inducing vimentin degradation and activating the GSK3β/β-catenin pathway, thereby extenuating vascular wall remodeling, could be an ideal target for preventing abdominal aortic aneurysm formation and progression.
Introduction
The vascular smooth muscle cell (VSMC) phenotype and growth characteristics determine the course of abdominal aortic aneurysm (AAA), a common harmful vascular disorder associated with a high risk of morbidity and mortality.1,2 In response to the effects of various risk factors, such as hypertension, dyslipidemia, and smoking, VSMCs are initially induced to adopt a degradative phenotype and then gradually progress to terminal death. The orchestrated interaction between these processes causes cellular and extracellular matrix (ECM) abnormalities in the medial layer of the aortic wall, thus inducing AAA formation and aggravating the disease course.3 Although an increasing number of studies have suggested that genetic-based interventions can aid in regulating the VSMC phenotype and growth,4,5 VSMC behavior is not fundamentally changed, which may still lead to aortic dilation and aneurysm rupture. The explanation for this outcome is that the appropriate function of VSMCs and their quantity are not addressed simultaneously by these interventions, which often lead to the excessive proliferation of degraded VSMCs, thus worsening medial degeneration and exacerbating AAA progression.6 An increased understanding of VSMC behavior will enable the identification of strategies to improve vessel wall remodeling, which may reverse the outcome of aneurysms.7 According to previous studies, several coding genes and noncoding genes, including microRNAs and long noncoding RNAs with easy degradation or nonspecific expression patterns, are involved in the VSMC phenotypic switch or growth in vitro,8,9 but their roles in determining the AAA prognosis is unknown.
Circular RNAs (circRNAs), a novel class of noncoding RNAs characterized by covalently closed loop structures with neither 5′ to 3′ polarity nor a polyadenylated tail, are expressed in a tissue specific and stable manner.10 CircRNAs are involved in various vascular diseases, including vascular dysfunction, carotid atherosclerosis disease, diabetes mellitus-related retinal vascular dysfunction, and chronic thromboembolic pulmonary hypertension.11 Recently, research has revealed abundant differentially expressed circRNAs between human AAA groups and control groups; moreover, circRNAs are involved in VSMC proliferation, differentiation, and migration, which are core mechanisms involved in the pathology of aneurysms.12, 13, 14 Of crucial importance, certain circRNAs have been found to interact with glycogen synthase kinase 3 beta (GSK3β) and α-smooth muscle actin, which have been validated to modulate VSMC growth and the contractile apparatus,15,16 the key component in vascular wall remodeling along with the deterioration of AAA, Therefore, circRNAs may be a key factor mediating the pathogenesis of AAA. Furthermore, a previous study profiling circRNA expression in AAA and normal mice found that circRNA-001265, originating from cysteine and histidine-rich domain containing 1 (Chordc1), was significantly reduced in diseased arteries and related to VSMC dysfunction and loss,17 suggesting that it may have the potential to control VSMC phenotype and growth.
In this study, we named this newly discovered VSMC-enriched circRNA circChordc1 and, using angiotensin (Ang) II- and CaCl2-induced AAA mouse models, identified its role in VSMC behavior and AAA outcomes. CircChordc1 prevented AAA pathology by inducing VSMCs to a contractile phenotype and improving their survival. These effects of circChordc1 were achieved through its promotion of the phosphorylation-induced degradation of vimentin and inhibition of GSK3β/β-catenin pathway activity. Based on our results, circChordc1 is a novel molecular target that mitigates AAA formation and exacerbation by contributing to VSMC phenotype and growth and improving vascular wall remodeling.
Results
Aortic tissues from patients with AAA exhibit phenotype switching and increased apoptosis of VSMCs
Human AAA and corresponding adjacent normal aortic samples were collected from 10 patients who underwent AAA resection surgery. The clinical and laboratory features of the patients are shown in Table S3. Computed tomography angiography revealed dilation of the abdominal aorta in the patients with AAA (Figures S1A and S1B). H&E staining of aortic aneurysm and adjacent normal tissues showed that VSMCs exhibited a disordered arrangement in aneurysm samples, while they were neatly arranged in parallel in adjacent normal tissues (Figure S1C). Masson's trichrome staining showed attenuated collagen deposition in the aorta of the patients with AAA (Figure S1D). Immunohistochemical (IHC) staining results revealed that the expression of the VSMC contractile marker smooth muscle 22 (SM22) was substantially decreased in AAA tissues compared with corresponding adjacent normal aortic tissues (Figures S1E and S1F) and was accompanied by significantly increased synthetic marker vimentin (Figure S1G). The patterns of contractile markers (sm22 and calponin) and synthetic markers (vimentin and osteopontin) identified using western blotting were concordant with those of the IHC staining (Figures S1H and S1I). IHC staining confirmed that the levels of α-SMA, a marker of SMCs, were substantially decreased in human aortic aneurysm tissues, compared with those in adjacent nonaneurysmal SMCs. In contrast, the caspase-3 protein, which serves as a crucial executor of cell apoptosis, was expressed at high levels (Figures S2A–S2D). In addition, TUNEL staining revealed that the apoptotic rate of VSMCs was dramatically upregulated in AAA tissues (Figures S2E and S2F). Taken together, these data show that there were phenotypic transforming and increased apoptosis of VSMCs in the human AAA samples.
CircChordc1 is significantly downregulated in human and mouse AAA tissues
A previous expression profiling study identified that circRNAs are differentially expressed between normal and AAA tissues. From this transcriptomic dataset, the top 10 downregulated circRNAs were chosen as candidates. According to the conservation between humans and mice, and the correlation with apoptosis, three circRNAs, including circChordc1, circFrivd4b, and circSh3rf1, were selected, and subsequent quantitative real-time PCR assays suggested that circChordc1 was the most significantly differentially expressed circRNA between human and mouse normal and AAA groups (Figures 1A–1D). CircChordc1 has a length of 265 bp and is formed by the circularization of exon 2, exon 3, and exon 4 of the Chordc1 gene, which is located on chromosome 9 (qA2), as reported in circBase (mmu_circ_0001747) (Figure 1E). Furthermore, circChordc1 is conserved among humans and mice (Figure S3A). Among cells that comprise the vessel tissue, smooth muscle cells (SMCs) were the main cells expressing circChordc1, followed by endothelial cells and fibroblasts (Figure S3B). Next, we verified that circChordc1 was more stable than the linear transcript (Figures 1F and 1G). We performed immunofluorescence staining (Figure 1H) and used quantitative real-time PCR to isolate the cytoplasmic and nuclear RNAs of VSMCs (Figure 1I) to identify the distribution of circChordc1 in subcellular components. CircChordc1 was predominantly located in the cytoplasm of VSMCs. Quantitative real-time PCR assays (Figure 1J) suggested markedly higher circChordc1 levels in the corresponding adjacent normal aortic tissues than in human AAA tissues. In addition, an angiotensin II (Ang II)-induced mouse AAA model and a CaCl2-induced mouse AAA model were established to investigate the different circChordc1 levels in the aortas of AAA mice and control mice. As macroscopically observed, both types of AAA model mice showed more pronounced aortic bulges than the control mice, confirming that we had successfully established well-accepted AAA models. Quantitative real-time PCR assays revealed dramatically higher levels of circChordc1 in control mice than in Ang II-induced and CaCl2-induced AAA mice (p < 0.05; Figures 2K–2N).
Figure 1.

CircChordc1 is significantly downregulated in human and mouse AAA tissues
(A) The process used to screen the circChordc1 expression profile in AAA. (B–D) Quantitative real-time PCR validation of circChordc1, circFrivd4b, and circSh3rf1 in human normal and abdominal aortic aneurysm tissues and mouse control and abdominal aortic aneurysm samples. (E) The mouse loci of circChordc1 in the chordc1 gene. (F) The abundances of circChordc1 and chordc1 mRNA in SMCs treated with RNase R normalized to those measured in the control. n = 6, ∗p < 0.05 versus RNase treatment. (G) The amount of circChordc1 and chordc1 mRNA in SMCs treated with actinomycin D at the indicated time points. n = 6, ∗p < 0.05 versus chordc1 mRNA at each time point. (H) Fluorescence in situ hybridization to determine the circChordc1 levels in SMCs. Scale bar, = 50 μm. (I) Quantitative real-time PCR for the abundance of circChordc1 in either the cytoplasm or nucleus of SMCs. (J) Quantitative real-time PCR for detecting relative expression of circChordc1 in human tissue. n = 10, ∗p < 0.05 versus control tissues. (K) Images of the macroscopic features of ApoE−/− mouse aortas stimulated with saline or Ang II. (L) Quantitative real-time PCR for detecting the relative expression of circChordc1 in Ang II-induced AAA or control mice. n = 10, ∗p < 0.05 versus control tissues. (M) Images of the macroscopic features of C57BL/6J mouse aortas stimulated with saline or CaCl2. (N) Quantitative real-time PCR for detecting the relative expression of circChordc1 in CaCl2-induced AAA or control mice. n = 10, ∗p < 0.05 versus control tissues.
Figure 2.

CircChordc1 regulates the proliferation, apoptosis, and phenotypic switching of VSMCs
(A and B) The results of the FCM analysis and the relative expression in VSMCs after transfection with scr-RNA or si-chordc1. n = 6, ∗p < 0.05 versus scr-RNA group. (C) TUNEL immunofluorescence staining in VSMCs after treatment with scr-RNA or sh-circChordc1. The white arrows indicate TUNEL-positive VSMCs. Scale bar = 50 μm. (D) Quantification of the percentage of TUNEL-positive VSMCs in the scr-RNA and sh-circChordc1 groups. n = 6, ∗p < 0.05 versus scr-RNA group. (E) G0/G1 phase cell-cycle analysis after VSMCs were treated with scr-RNA or sh-circChordc1. n = 6, ∗p < 0.05 versus scr-RNA group. (F) Quantification of the percentage of VSMCs in the G0/G1 phase. n = 6, ∗p < 0.05 versus scr-RNA group. (G) Immunofluorescence staining for Ki-67 in VSMCs treated with scr-RNA or si-chordc1. The white arrows indicate Ki-67-positive VSMCs. Scale bar, = 50 μm. (H) Quantification of Ki-67-positive VSMCs. n = 6, ∗p < 0.05 versus scr-RNA group. (I) Immunofluorescence staining for PH3 in VSMCs treated with scr-RNA or sh-circChordc1. The white arrows indicate pH3-positive VSMCs. Scale bar, 50 μm. (J) Quantification of pH3-positive VSMCs. n = 6, ∗p < 0.05 versus scr-RNA group. (K and L) Western blotting results and densitometric analysis of PH3 and caspase-3 protein levels in the scr-RNA and si-circChordc1 groups. ∗p < 0.05; n = 6 per group. (M) Immunofluorescence staining for SM22 and vimentin in VSMCs treated with scr-RNA or si-circChordc1. Scale bar, 50 μm. (N and O) Western blotting results and densitometric analysis of SM22, calponin, vimentin, and OPN protein levels in the scr-RNA and si-circChordc1 groups. ∗p < 0.05; n = 6 per group.
CircChordc1 regulates the proliferation, apoptosis, and phenotypic switching of VSMCs
To further explore the role of circChordc1 in vascular VSMC physiology in vitro, VSMCs were rendered deficient in circChordc1 through transfection of three different small interfering RNAs (siRNAs) for 48 h. We chose the siRNA with the highest silencing efficiency for subsequent in vitro experiments (Figure S4A). Moreover, we designed circChordc1 constructs to increase circChordc1 levels in VSMCs (Figure S4B). No significant differences were seen in the mRNA and protein levels of Chordc1 between the circChordc1 intervention groups and control groups (Figures S4C–S4F). Knockdown of circChordc1 enhanced the apoptotic rate of VSMCs (Figures 2A–2D), increased the proportion of cells in the G0/G1 phase of VSMCs (Figures 2E and 2F), and decreased the proportion of Ki-67-positive and PH3-positive VSMCs (Figures 2G–2J), as assessed by flow cytometry, TUNEL staining, and immunofluorescence. Furthermore, as shown by western blotting, the caspase-3 protein was markedly increased in VSMCs with reduced circChordc1, while the expression level of PH3 was significantly decreased (Figures 2K–2L). Accumulating evidence has proven that the phenotypic switching of SMCs can trigger their apoptosis. We subsequently investigated whether circChordc1 affects SMC phenotype switching. Interestingly, we found that circChordc1 knockdown in vitro reduced the levels of some contractile markers, including SM22 and calponin, and increased the expression of several synthetic markers, including vimentin and osteopontin (Figures 2M–2O).
Targeting circChordc1 influences Ang II-induced AAA formation
To investigate a potential causal relationship between circChordc1 and AAA development, we obtained overexpressed circChordc1 (circChordc1-OE) and their respective control viruses (adeno-associated virus [AAV]-green fluorescent protein [GFP]) to perform function gained experiments. Immunofluorescence staining confirmed that an enhanced aortic GFP signal was detected in the virus intervention groups, indicating that the virus was successfully transfected into the aortic wall (Figures 3A and S5A). Beginning on the 30th day, overexpression interventions yielded a significant increase in circChordc1 levels, as confirmed by quantitative real-time PCR (Figure S5B). The mice in the control group exhibited a more obvious bulge in the abdominal aorta than those in the circChordc1-overexpressing group (Figure 3B). In addition, enhanced circChordc1 triggered a substantial decrease in the maximal abdominal aortic diameter (Figure 3C). The incidence of AAA formation (Figure 3D) and the rupture rate of AAA (Figure 3E) were nearly absent in the circChordc1-overexpressing group. Altogether, these data indicated that circChordc1 inhibited Ang II-induced AAA formation. Furthermore, we wondered whether the inhibitory effect of circChordc1 on AAA formation was related to the phenotypic switching and apoptosis of SMCs. Consistent with our hypothesis, overexpressed circChordc1 significantly increased the protein levels of contractile markers (sm22 and calponin) and reduced the protein levels of synthetic markers (vimentin and osteopontin), as assessed by western blotting (Figures 3F and 3G) and immunofluorescence staining (Figure 3H). In addition, the TUNEL assay (Figures 3I and 3J) and the protein levels of caspase-3 and α-SMA (Figures 3K–3N) unanimously indicated that the apoptotic rate of SMCs was significantly decreased in the circChordc1-overexpressing group. Western blots produce similar results to those findings (Figures 3O and 3P).
Figure 3.

Overexpressed circChordc1 inhibits Ang II-induced AAA formation
(A) Immunofluorescence staining for virus-borne green fluorescent protein (GFP) in the suprarenal aortas of male mice in different virus-mediated groups and the saline group. (B) Images of the macroscopic features of ApoE−/− mouse aortas with Ang II stimulation or Ang II stimulation and circChordc1 overexpression. (C) Maximal AAA diameter of the abdominal region in ApoE−/− mice with Ang II stimulation alone or Ang II stimulation and circChordc1 overexpression. (D) The incidence of AAA in the Ang II-induced AAA group or circChordc1-overexpression group. ∗p < 0.05; n = 20 per group (Fisher's exact test). (E) The rupture rate of AAA in the Ang II-induced AAA group or circChordc1-overexpression group. ∗p < 0.05; n = 20 per group (Fisher's exact test). (F and G) Western blotting results and densitometric analysis of SM22, calponin, vimentin, and OPN protein levels in ApoE−/− mouse aortas with Ang II stimulation or Ang II stimulation and circChordc1 overexpression. n = 6, ∗p < 0.05 versus Ang II + AAV-GFP group. (H) Immunofluorescence staining for SM22 and vimentin in ApoE−/− mouse aortas with Ang II stimulation alone or Ang II stimulation and circChordc1 overexpression. Scale bar, = 50 μm. (I and J) TUNEL immunofluorescence staining in ApoE−/− mouse aortas with Ang II stimulation or Ang II stimulation and circChordc1 overexpression and quantification of TUNEL-positive cells. n = 6, ∗p < 0.05 versus Ang II + AAV-GFP group. Scale bar, 50 μm. (K–N) Immunohistochemistry staining for caspase-3 and α-SMA in ApoE−/− mouse aortas with Ang II stimulation alone or Ang II stimulation and circChordc1 overexpression and quantification of caspase-3 and α-SMA expression levels in ApoE−/− mouse aortas. n = 6, ∗p < 0.05 versus Ang II + AAV-GFP group. Scale bar, 50 μm. (O) Western blot assay detecting the expression levels of caspase-3 and α-SMA in ApoE−/− mouse aortas with Ang II stimulation or Ang II stimulation and circChordc1 overexpression. (P) Quantification of caspase-3 and α-SMA protein levels in the aortas. n = 6, ∗p < 0.05 versus Ang II + AAV-GFP group.
AAVs containing circChordc1 interference constructs (sh-circChordc1) and scr-RNA were obtained to reduce circChordc1 level. Immunofluorescence staining indicated that the AAV containing sh-circChordc1 was successfully transfected into the aortic wall (Figure 4A). The circChordc1 level was significantly decreased in the sh-circChordc1 group (Figure S5C). The expression of circChordc1 in aorta, hepatic, and skeletal muscle after modulation is shown in Figure S5D. Generally, the expression of circChordc1 in hepatic and skeletal muscle was changed. To investigate whether circChordc1 modulated AAA formation in normolipidemic circumstances, we infused Ang II into wild-type male C57BL/6J mice for 28 days. As a result, the bulge in the abdominal aorta in scr-RNA mice was less obvious than that in circChordc1 knockdown mice (Figure 4B). The maximal abdominal aortic diameter (Figure 4C), AAA incidence (Figure 4D), and the rupture rate of AAA (Figure 4E) were substantially higher in the circChordc1 knockdown group than in the scr-RNA group. Consistent with these findings, western blotting (Figures 4F and 4G) and immunofluorescence staining (Figure 4H) indicated that knockdown of circChordc1 significantly reduced SM22 and calponin levels while promoting the expression of vimentin and osteopontin. In addition, knockdown of circChordc1 promoted SMC apoptosis, as reflected by TUNEL assays (Figures 4I and 4J), and caspase-3 and α-SMA expression levels (Figures 4K–4N). Furthermore, western blotting displayed an identical trendency with those findings (Figures 4O and 4P). Based on these results, the circChordc1 deficiency was sufficiently potent to instigate AAA onset by prompting SMCs to acquire a synthetic phenotype and undergo apoptosis upon Ang II treatment.
Figure 4.

Reduced circChordc1 promotes Ang II-induced AAA formation
(A) Immunofluorescence staining of virus-borne GFP in the suprarenal aortas of male mice in different virus-mediated groups and the saline group. (B) Images of the macroscopic features of C57BL/6J mouse aortas with Ang II stimulation or Ang II stimulation alone and circChordc1 knockdown (C). Maximal AAA diameter of the abdominal region in C57BL/6J mice with Ang II stimulation alone or Ang II stimulation and circChordc1 knockdown. (D) The incidence of AAA in the Ang II-induced AAA group or circChordc1-knockdown group. ∗p < 0.05; n = 20 per group (Fisher's exact test). (E) The rupture rate of AAA in the Ang II-induced AAA group or circChordc1-knockdown group. ∗p < 0.05; n = 20 per group (Fisher's exact test). (F and G) Western blotting results and densitometric analysis of SM22, calponin, vimentin, and OPN protein levels in aortas from C57BL/6J mice with Ang II stimulation or Ang II stimulation and circChordc1 knockdown. n = 6, ∗p < 0.05 versus Ang II + scr-RNA group. (H) Immunofluorescence staining for SM22 and vimentin in C57BL/6J mouse aortas with Ang II stimulation or Ang II stimulation alone and circChordc1 knockdown. Scale bar, 50 μm. (I and J) TUNEL immunofluorescence staining in C57BL/6J mouse aortas with Ang II stimulation or Ang II stimulation and circChordc1 knockdown and quantification of TUNEL-positive cells. n = 6, ∗p < 0.05 versus Ang II + scr-RNA group. Scale bar, 50 μm. (K–N) Immunohistochemistry staining for caspase-3 and α-SMA in C57BL/6J mouse aortas with Ang II stimulation alone or Ang II stimulation and circChordc1 knockdown and quantification of caspase-3 and α-SMA expression levels in C57BL/6J mouse aortas. n = 6, ∗p < 0.05 versus Ang II + scr-RNA group. Scale bar, 50 μm. (O) Western blot assay detecting the expression levels of caspase-3 and α-SMA in C57BL/6J mouse aortas with Ang II stimulation alone or Ang II stimulation and circChordc1 knockdown. (P) Quantification of caspase-3 and α-SMA protein levels in aortas. n = 6, ∗p < 0.05 versus Ang II + scr-RNA group.
Modulation of circChordc1 regulates AAA formation in CaCl2-treated mice
To further examine whether the inhibitory effects of circChordc1 on AAA formation are independent of Ang II stimulation, we established a CaCl2-induced AAA model, another well-accepted type of AAA model, to explore the role of circChordc1. Three weeks after administration of CaCl2 treatment to the infrarenal aorta, the maximal abdominal aortic diameter was substantially increased in circChordc1 knockdown mice (Figures 5A and 5B), consistent with the findings in Ang II-infused mice. Moreover, the protein expression patterns of SM22, calponin, vimentin, and osteopontin identified using western blotting (Figures 5C and 5D) and immunofluorescence staining (Figure 5E) were identical to those in the Ang II-induced AAA model when circChordc1 expression was reduced. Decreased circChordc1 expression also increased the apoptotic rate of SMCs, as rrdemonstrated by the TUNEL assay (Figures 5F and 5G), and caspase-3 and α-SMA expression levels (Figures 5H–5K). In addition, western blotting displayed an identical trendency with those findings (Figures 5L–5M). These results therefore indicated that circChordc1 exerted a protective effect in the CaCl2-induced mouse AAA model.
Figure 5.

Modulation of circChordc1 regulates AAA formation in CaCl2-treated mice
(A) Images of the macroscopic features of C57BL/6J mouse aortas with CaCl2 stimulation alone or CaCl2 stimulation and circChordc1 knockdown. (B) Maximal AAA diameter of the abdominal region in C57BL/6J mice with CaCl2 stimulation or CaCl2 stimulation alone and circChordc1 knockdown. (C and D) Western blotting results and densitometric analysis of SM22, calponin, vimentin, and OPN protein levels in aortas from C57BL/6J mice with CaCl2 stimulation alone or CaCl2 stimulation alone and circChordc1 knockdown. n = 6, ∗p < 0.05 versus CaCl2+scr-RNA group. (E) Immunofluorescence staining for SM22 and vimentin in C57BL/6J mouse aortas with CaCl2 stimulation alone or CaCl2 stimulation and circChordc1 knockdown. Scale bar, 50 μm. (F and G) TUNEL immunofluorescence staining in C57BL/6J mouse aortas with CaCl2 stimulation alone or CaCl2 stimulation and circChordc1 knockdown and quantification of TUNEL-positive cells. n = 6, ∗p < 0.05 versus CaCl2+scr-RNA group. Scale bar, 50 μm. (H–K) Immunohistochemistry staining for caspase-3 and α-SMA in C57BL/6J mouse aortas with CaCl2 stimulation alone or CaCl2 stimulation and circChordc1 knockdown and quantification of caspase-3 and α-SMA expression levels in C57BL/6J mouse aortas. n = 6, ∗p < 0.05 versus CaCl2+scr-RNA group. Scale bar, 50 μm. (L) Western blot assay detecting the expression levels of caspase-3 and α-SMA in C57BL/6J mouse aortas with CaCl2 stimulation alone or CaCl2 stimulation and circChordc1 knockdown. (M) Quantification of caspase-3 and α-SMA protein levels in aortas. n = 6, ∗p < 0.05 versus CaCl2+scr-RNA group.
CircChordc1 interacts with ANXA2 to activate Wnt/β-catenin signaling
Notably, circRNAs exert their functions through RNA-protein interactions. We performed pull-down assays and mass spectrometry analysis to determine proteins that potentially interact with circChordc1 and to explore the underlying mechanism by which circChordc1 regulates SMC growth (Figure 6A). ANXA2 was detected to specifically bind to circChordc1 (Figure 6B), which was verified by western blotting (Figure 6C). We then confirmed the interactions between circChordc1 and ANXA2 by performing RNA immunoprecipitation (RIP) assays (Figure 6D). RNA-FISH (fluorescence in situ hybridization) and immunofluorescence assay results revealed that circChordc1 was colocalized with ANXA2 in the cytoplasm of human aortic SMCs (Figure 6E). A substantial increase in ANXA2 expression was observed in AAA aortas compared with that in normal aortas of humans and mice (Figures S6A–S6I). Our data showed that ANXA2, which has been established to play a critical role in regulating cell survival, could significantly promote VSMC proliferation (Figures S7A and S7B). We next investigated whether the interactions between circChordc1 and ANXA2 affected ANXA2 levels. The mRNA and protein expression of ANXA2 was detected when circChordc1 was overexpressed or knocked down. Enhanced circChordc1 significantly increased ANXA2 protein levels in VSMCs (Figures 6F and 6G), while reduced circChordc1 decreased the level of ANXA2 (Figures 6H and 6I). However, no significant differences in the mRNA level of ANXA2 were noted between the intervention groups (Figure S7C), which was further confirmed in the Ang II-induced and CaCl2-induced AAA models (p < 0.05; Figures S7D–S7I). To further clarify the molecular regulatory mechanism underlying the interaction between circChordc1 and ANXA2, we treated circChordc1-knockdown VSMCs with the protein synthesis inhibitor cycloheximide (CHX). The half-life of ANXA2 in circChordc1-knockdown VSMCs was notably shorter than that in the control VSMCs, which demonstrated that circChordc1 could delay ANXA2 degradation (Figure 6J). It has been reported that ANXA2 can alter the subcellular localization of β-catenin and bind to GSK3β, disrupting the formation of the GSK3β/β-catenin complex in hepatocellular carcinoma cells.18 Coimmunoprecipitation (coIP) and immunofluorescence assays were performed to confirm that ANXA2 could directly bind with GSK-3β in VSMCs (Figures 6K–6L). We observed that GSK-3β interacted with more endogenous β-catenin in ANXA2-knockdown VSMCs than in control VSMCs using coIP assays (Figure 6M). coIP assays indicated that ANXA2 directly bound GSK3β, and knockdown of circChordc1 attenuated the binding of ANXA2/GSK3β, thus increasing GSK3β/β-catenin interactions (Figure 6N). Furthermore, immunoblotting revealed that ANXA2 knockdown significantly inhibited the nuclear translocation of β-catenin (Figures 6O–6P). Overexpressed circChordc1 accelerated VSMC proliferation, which was blocked by the inhibition of ANXA2 (Figures 6Q–6R). Thus, circChordc1 promotes VSMC proliferation and apoptosis by regulating the ANXA2/GSK-3β/β-catenin pathway.
Figure 6.

CircChordc1 interacts with ANXA2 to activate Wnt/β-catenin signaling
(A) ChIRP assays were performed with a probe against circChordc1 and a control probe. Silver-stained SDS-PAGE gel bands marked by a red frame were excised for mass spectrometry to identify interacting proteins. (B) ANXA2 was identified by mass spectrometry. (C) ANXA2 was detected by western blotting. (D) RIP was performed using an anti-ANXA2 antibody or a negative IgG antibody. n = 5 per group, ∗p < 0.05 versus IgG (Student's t test). RNA was used for quantitative real-time PCR analysis, and the circChordc1 level was normalized against the input. (E) Colocalization of circChordc1 (red) and ANXA2 (green) was visualized by FISH and immunofluorescence assays. Scale bar, 50 μm. (F–I) Western blot assays detecting the expression levels of ANXA2 when circChordc1 was overexpressed or silenced (β-tubulin was the internal reference) and quantification of ANXA2 protein levels in VSMCs. n = 6, ∗p < 0.05 versus vector or si-scr group. (J) circChordc1 was downregulated in VSMCs, and the cells were treated with cycloheximide (CHX) (20 μg/mL) for the indicated times. ANXA2 protein levels were assessed by western blotting (β-tubulin is the internal reference). (K) coIP experiments indicated that ANXA2 could interact with GSK-3β. (L) Colocalization of GSK-3β (red) and ANXA2 (green) was visualized by immunofluorescence assays. Scale bar, 50 μm. (M) The interactions between GSK-3β and β-catenin were explored by coIP assay in ANXA2-silenced cells. (N) coIP experiments were performed with ANXA2 and GSK-3β in circChordc1 knockdown cells. (O and P) Protein expression levels of β-catenin in the nucleus and cytosol in VSMCs treated with si-scr or si-circChordc1 as detected by western blotting. n = 6, ∗p < 0.05 versus si-scr group. (Q and R) Immunofluorescence staining for Ki-67 in VSMCs with circChordc1 overexpression or circChordc1 overexpression and ANXA2 knockdown and quantification of Ki-67-positive VSMCs. n = 6, ∗p < 0.05 versus vector group. Scale bar, 50 μm.
CircChordc1 targeted vimentin to regulate SMC phenotype switching
We studied several phenotype switching-related RNA binding proteins identified by mass spectrometry to investigate the molecular mechanism by which circChordc1 regulates VSMC phenotype switching. Vimentin, the VSMC synthetic phenotype marker, was the most abundant among these proteins (Figure 7A). Furthermore, we performed western blotting and RIP to confirm the interaction between circChordc1 and vimentin (Figures 7B and 7C). We sought to determine whether circChordc1 attenuated VSMC phenotypic transformation through effects on vimentin. The immunofluorescence staining indicated that vimentin was substantially upregulated in mouse AAA samples compared with control samples (Figure 7D). In addition, western blotting revealed that enhanced circChordc1 inhibited vimentin expression levels (Figures 7E, 7F, S8A, and S8B). It has been reported that several serine residues within the N-terminal head domain of vimentin are phosphorylated by various kinases, and phosphorylated vimentin is in turn disassembled and presents in the soluble fraction in the cytoplasm. Therefore, we applied two antibodies to detect phosphorylation at two sites (Ser83 and Ser39) in vimentin to examine whether circChordc1 could regulate vimentin phosphorylation in VSMCs. Interestingly, we found that circChordc1 significantly increased the phosphorylation level of vimentin at Ser39 but not at Ser83 in VSMCs (Figure 7G). Moreover, overexpressed circChordc1 increased the expression of the VSMC contractile phenotype marker sm22, which were offset by enhanced vimentin (Figures 7H–7J). A similar situation was also observed in the effect of circChordc1 on caspase-3 (Figure 7K–7L). The above findings collectively suggest that circChordc1 increases vimentin phosphorylation to promote its degradation and determines the phenotype and growth of VSMCs.
Figure 7.

CircChordc1 targets vimentin to regulate SMC phenotype switching
(A) Vimentin was identified by mass spectrometry. (B) Vimentin was detected in the specific band by western blotting. (C) RIP was performed using an anti-vimentin antibody or a negative IgG antibody. n = 5 per group, ∗p < 0.05 versus IgG (Student's t test). RNA was used for quantitative real-time PCR analysis, and the level of circChordc1 was normalized against the input. (D) Immunofluorescence staining in ApoE−/− mouse aortas from the control group and Ang II-induced AAA group. Scale bar, 50 μm. (E and F) Western blot assay detecting the expression levels of vimentin when circChordc1 was overexpressed and quantification of vimentin protein levels in VSMCs. n = 6, ∗p < 0.05 versus vector group. (G) RNA pull-down and western blotting assays showing the target sites for circChordc1 on vimentin. (H) SM22 immunofluorescence staining in AngII + circChordc1-overexpressing ApoE−/− mice and Ang II + circChordc1-overexpressing + vimentin-overexpression ApoE−/− mice. Scale bar, 50 μm. (I and J) Western blotting and densitometric analysis of SM22 in Ang II + circChordc1-overexpressing ApoE−/− mice and Ang II + circChordc1-overexpressing + vimentin-overexpression ApoE−/− mice. n = 6, ∗p < 0.05 versus Ang II group. (K and L) Immunohistochemistry staining for caspase-3 in Ang II + circChordc1-overexpression ApoE−/− mice and Ang II + circChordc1-overexpression + vimentin-overexpression ApoE−/− mice and quantification of caspase-3 expression levels in ApoE−/− mouse aortas. n = 6, ∗p < 0.05 versus Ang II group. Scale bar, 50 μm.
Discussion
In this study, we identified that VSMC-specific circChordc1 could prevent the switch of VSMCs to a degradative phenotype and promote their growth, thus significantly reducing AAA formation and deterioration. CircChordc1 directly binds to vimentin and ANXA2, which not only induces vimentin degradation but also induces GSK3β signaling and promotes β-catenin entry into the nucleus to optimize the VSMC phenotype and increase growth (Figure 8). Based on these findings, circChordc1, which determines VSMC behavior, potentially represents an effective therapeutic target for AAA.
Figure 8.

A schematic representing the role of circChordc1 in preventing abdominal aortic aneurysm
CircChordc1 directly binds to vimentin and ANXA2, which achieves the phosphorylation of vimentin at Ser39 to induce its degradation and promotes the activation of the GSK3β/β-catenin pathway to optimize the VSMCs phenotype and inhibit their apoptosis, thereby preventing AAA formation and progression.
VSMCs featuring an impaired contractile apparatus and disturbed growth are proposed to be the key factors leading to the initiation and deterioration of AAA. In this study, we confirmed the effects of circChordc1 on the VSMC phenotype and growth and the role of circChordc1 in reversing the course of aneurysm. As shown in this study, circChordc1 could inhibit the degradation of VSMCs and promote their growth, thus reducing the probability of aneurysm formation and rupture by approximately 70% in mice. Accumulating studies have reported alterations in the phenotype and growth of VSMCs following interference with some gene targets,19, 20, 21 while in vivo evidence regarding the role of such modifications in AAA is lacking. This study provided in vivo data indicating the critical roles of circChordc1 in regulating the VSMC phenotype and growth and aneurysm course. More importantly, our results showed that, when the degradation of vimentin was blocked, enhanced circChordc1 did not significantly improve the prognosis of aneurysms. It has been suggested that methods that reduce the apoptosis of VSMCs while failing to alter their phenotype are ineffective in reversing thoracic aortic aneurysms, as the phenotype of the proliferating cells is central to disease outcome.6,22 Our study further indicated the critical effects of VSMC phenotype and growth on AAA progression. On the other hand, this study showed that circChordc1 interacts with other molecules to control the function and quantity of VSMCs. Although combination strategies targeting multiple factors to induce complementary effects may be used to improve VSMC phenotype and growth, unfortunately these strategies are limited by the ineffective cooperation and low efficiency reported in previous studies.23,24 Our current study proved that circChordc1 efficiently regulates the VSMC phenotype and growth and significantly improves the prognosis of aneurysms, suggesting that it may be an ideal target for AAA intervention. Finally, it is worth mentioning that we verified the effect of circChordc1 on aneurysm outcomes using Ang II- and CaCl2-induced AAA models, which further suggested that VSMC phenotype and growth modulation by circChordc1 meaningfully affected aneurysm development. Therefore, our study first identified that circChordc1 can decrease vascular wall vulnerability to dilatation and slow AAA development by controlling the VSMC phenotype and growth.
In this study, we verified that a novel circRNA affected the development of AAA by modulating the VSMC phenotype and growth. With enhanced circChordc1 expression, VSMCs transitioned to the contractile phenotype marked by SM22, and VSMC proliferation, as assessed by PH3 and Ki-67, increased following a significant decrease in VSMC apoptosis, as assessed by TUNEL and caspase-3 staining. These effects significantly reduced the rate of aortic aneurysm formation and the risk of aortic aneurysm rupture. Given the potential influence of circRNAs on cell behavior and vascular diseases,25,26 this study further confirmed the critical role of circChordc1 in regulating the phenotype and growth of VSMCs and aneurysms. Recently, noncoding RNAs (mainly microRNAs and long noncoding RNAs) have been shown to exert an important effect on VSMC behavior and to serve as key regulators of the development and progression of aortic aneurysms,27 but their unstable structure, poor conversation, and uncertain potency in vertebrates have been challenges plaguing their utilization.28,29 This study indicated that circChordc1, which has a covalent bond ring structure, could be an ideal target to control the VSMC phenotype and growth. First, we found that circChordc1 tolerated RNase treatment without being degraded and participated in VSMC phenotypic transition and growth, two key processes during aneurysm development. Moreover, circChordc1 was enriched in the human aorta, and the circChordc1 levels detected in human aortic aneurysm specimens were lower than those in normal aorta specimens, which suggested the value of circChordc1 in clinical interventions. Finally, circChordc1 was enriched in VSMCs, and targeting circChordc1 would avoid the effect on other cells, including endothelial cells and macrophages, which may lead to intimal hyperplasia and inflammation. Collectively, these findings suggest that circChordc1 could be an ideal target involved in VSMC phenotype and growth that could be utilized to improve the prognosis of AAA.
CircRNAs generally perform biological functions by acting on RNA binding proteins that subsequently influence the activity of associated proteins.30 This study revealed that the contribution of circChordc1 to VSMC phenotype and growth was achieved by its interaction with vimentin and ANXA2. A previous study indicated that the level of vimentin, serving as a marker of the dedifferentiated phenotype for VSMCs, was positively correlated with the degeneration of contractile VSMCs.31 With RNA pull-down and RIP assays, we determined that circChordc1 could bind to vimentin directly and reduce its level. Furthermore, we performed immunoprecipitation assays and found that circChordc1 could bind to vimentin, and this interaction further recruited phosphorylating groups to induce phosphorylation of vimentin at Ser39. It has been reported that phosphorylation of serine residues on vimentin inhibits subunit polymerization, thus promoting the decomposition of vimentin filaments and increasing the solubility of the protein,32 and these effects were shown to be involved in VSMC behavior during aneurysm. Moreover, we showed that ANXA2 was also a protein interacting with circChordc1. ANXA2 exhibited a feedback increase in aneurysm as an attempt to protect the aorta from further expansion and ultimate rupture. We found the presence of ANXA2 in pull-down products by mass spectrometry analysis, which was further confirmed by immunoblot. CircRNAs have been found to perform a variety of biological functions through regulation of proteins at the posttranscriptional level.33 The level of circChordc1 was positively related to the ANXA2 protein level but did not affect its gene expression, which indicates that the regulation of ANXA2 by circChordc1 mainly occurs at the translation level. This was further verified by the addition of CHX, which suggested that circChordc1 could increase ANXA2 expression by reducing its degradation. ANXA2 has been found to activate the GSK3β/β-catenin signaling pathway and facilitate VSMC growth. This study indicated that silencing ANXA2 could offset the proliferative effect of overexpressed circChordc1 on VSMCs, further suggesting the critical role of ANXA2 in VSMC growth. β-Catenin entering the nucleus has been shown to promote VSMC proliferation and reduce VSMC apoptosis.34 In line with this, the present immunoprecipitation and western blotting results showed that silenced circChordc1 reduced the level of ANXA2, which bind to GSK3β and promoted the β-catenin nucleation. Therefore, circChordc1 could reduce vimentin levels and increase ANXA2 expression to recruit GSK3β and promote the nucleation of β-catenin, thus contributing to shifting the phenotype of VSMCs and improving their growth.
Nevertheless, there are still some limitations of our study. The present results showed that circChordc1 increased the ANXA2 level by blocking its degradation, but the detailed mechanism that mediates the regulation of ANXA2 degradation needs to be further explored. While circChordc1 derived from linear RNA splicing was decreased in the AAA samples, whether Chordc1 participates in the formation of aneurysms and the regulation of circChordc1 still needs to be investigated. CircRNAs are known to carry out their functions by serving as molecular sponges, and numerous miRNAs have been confirmed to be key contributors to aneurysm pathology. Although we have suggested that circChordc1 plays a regulatory role in VSMC phenotype and growth by interacting with downstream proteins, examining whether circChordc1 acts on miRNAs is also warranted.
In conclusion, VSMC-specific circChordc1 extenuates the formation and outcome of AAA by facilitating VSMC phenotype and growth. CircChordc1 interacts with vimentin and ANXA2, which promotes the degradation of vimentin, and binds to GSK3β to increase β-catenin nucleation, thus transitioning VSMCs to the contracted phenotype and reducing their apoptosis. Thus, circChordc1 is involved in VSMC phenotype and growth and could be an ideal target for preventing AAA formation and progression.
Materials and methods
The human tissue protocols conformed to the principles in the Declaration of Helsinki and were approved by Zhongshan People's Hospital and NanFang Hospital (ethical approval number: NFEC-2019-086; Tables S1 and S2). Informed consent was provided before inclusion of participants in the study. The animal protocols were approved by the Animal Research Committee at Southern Medical University, and all procedures were performed in accordance with the Guide for the Care and Use of Laboratory Animals published by the National Institutes of Health (Eighth Edition).
Patient specimens
Aneurysm samples and adjacent normal aortic samples were collected from patients who underwent AAA resection surgery at Zhongshan People's Hospital and NanFang Hospital. The detailed characteristics, including age, sex, smoking status, and hypertension status, of subjects with AAA are presented in Table S3. Aneurysm tissues were fixed with formalin for IHC staining or frozen fresh in liquid nitrogen for western blotting and quantitative real-time PCR.
Experimental animals and AAA models
We used 10- to 12-week-old male C57BL/6 mice (CaCl2 model and Ang II infusion model) and 12- to 16-week-old male ApoE−/− mice on a C57BL/6 background (Ang II infusion model) for further animal experiments. For the Ang II-induced AAA model, all age-matched male mice were infused with Ang II (1 μg/kg per min, Sigma-Aldrich) or normal saline via an osmotic minipump (Alzet, Model, 2004; DURECT, Cupertino, CA) for 28 days. The detailed manipulation was performed as described previously.35, 36, 37 For the CaCl2-induced AAA model, 10- to 12-week-old male C57BL/6 mice were anesthetized and underwent laparotomy. The abdominal aorta below the renal arteries and bifurcation of the iliac arteries was dissociated from the surrounding retroperitoneal structures. Then, the external surface of the aorta was spread on a cotton gauze moistened with 0.5 mol/L CaCl2 for 15 min. Sham control mice were processed with NaCl (0.9%) instead of CaCl2. The aorta was rinsed with 0.9% sterile saline, and then the incision was sutured. After 3 weeks, CaCl2-induced AAA mice were sacrificed, and the abdominal aortas were harvested for further IHC and immunofluorescence staining.
Real-time PCR
Total RNA was extracted using TRIzol reagent (Invitrogen) according to the manufacturer's protocol. Then, total RNA was synthesized into cDNAs using reverse transcriptase (Takara Biotechnology, Dalian, China) and primer sequences specific for the examined molecules. Quantitative real-time PCR was performed using a SYBR Green RT-PCR Kit (Takara Biotechnology) with the Light Cycler 480 II system (Roche Diagnostics, Basel, Switzerland). Quantitative real-time PCR was performed as described previously.38 Glyceraldehyde-3-phosphate-dehydrogenase (GAPDH) mRNA was used as an internal control. The primer sequences are presented in Table S4.
Injection of AAVs
For in vivo experiments, a circChordc1 AAV was constructed and packaged by Vigene Biosciences (Jinan, China). AAV serotype 9 harboring shRNA sequences was generated by Vigene Biosciences. Mice were injected with one of the viruses listed above (1 × 1011 vg). The shRNA and mouse circChrodc1 sequences are listed in Table S4. Thirty days later, the mice were randomly grouped and treated with Ang II or CaCl2 as described above.
Cell culture and transfection
Human aortic VSMCs were purchased from Guangzhou Geneseed Biotech and cultured in Dulbecco's modified Eagle's medium (DMEM) containing 10% fetal bovine serum (FBS). Human aortic VSMCs were then cultured in serum-free medium and treated with the siRNA targeting circChordc1 or control siRNA (detailed information regarding the siRNAs is provided in Table S4). All the cells were incubated at 37°C in a humidified atmosphere of 5% CO2 for 48 h. VSMCs were used in subsequent in vitro experiments.
Western blotting
Protein was extracted from cells or aortic tissues using a protein extraction kit containing the protease inhibitor phenylmethanesulfonyl fluoride and protein phosphatase inhibitor. The concentration was determined using a BCA Protein Assay Kit (Beyotime, P0010). Proteins were denatured at 100°C for 10 min, electrophoretically separated on a 10% SDS-PAGE gel, and transferred onto a nitrocellulose membrane. Membranes were blocked with 10% bovine serum at room temperature for 1 h. Membranes were then incubated with the primary antibodies at 4°C overnight. Membranes were washed three times for 15 min and probed with a secondary antibody for 1 h at room temperature. Then, the membranes were washed three times for 15 min again. The bands were detected using enhanced chemiluminescence (ECL Advance; no. RPN2235, GE Healthcare Life Sciences). Finally, the signals were recorded using a ChemiDoc imaging system (Bio-Rad Laboratories). Tubulin or β-actin was used as a negative control. Details about the primary antibodies are available in Table S5.
TUNEL assay
Human aortic VSMCs were transfected with three different siRNAs and their respective control siRNAs. Then, the cells were cultured in 10% FBS-DMEM for 48 h. They were fixed with 10% formalin for 1 h, and apoptotic cells were directly stained with a TUNEL kit. DAPI (Beyotime, China) staining was used to stain nuclei. The percentage of apoptotic cells (TUNEL-positive cells/DAPI-positive cells) in the circChordc1-knockdown group was compared with that in the control group. Images were collected using a Leica TCS SP8 fluorescence confocal microscope.
Cell-cycle analysis
Human aortic VSMCs cultured in 6-well plates were transfected with siRNAs or the control siRNA for 48 h. The VSMCs were then trypsinized, harvested, and washed with phosphate-buffered saline (PBS). Next, the cells were fixed with 75% ethanol at 4°C for 2 h, followed by BD Pharmingen PI/RNase staining for 30 min at room temperature in the dark. Finally, the cell-cycle phases were analyzed using a flow cytometer (BD, FACSCalibur; San Jose, CA, USA).
Histological analyses
After sacrificing the mice, aortic tissue from the ascending aorta to the bifurcation of the iliac artery was isolated, and the whole aorta was perfused with saline. Then, the aorta was fixed with 4% paraformaldehyde in PBS for 5 min. Aortic tissues were harvested, fixed for 24 h, and embedded in a paraffin solution. Cross-sections (5 mm each) were prepared at intervals of approximately 500 mm. At least 10 sections were analyzed per mouse. IHC staining and immunofluorescence staining were performed.
Aneurysm quantification
All surviving mice were euthanized and dissected ventrally to verify the occurrence of AAA. Ten milliliters of PBS was injected into the left cardiac ventricle and exited through the severed right atrium. Then, the aorta was dissected under an anatomical microscope, and the aorta was photographed. The suprarenal aorta was identified as the passage below the last pair of intercostal arteries and above the right renal branch.
The maximum width of the abdominal aorta was analyzed using Image-Pro Plus software (Media Cybernetics) to quantify the AAA size. We adopted the established human aneurysm definition, which requires a >50% increase in the external width of the suprarenal aorta of treated mice compared with control mice, to confirm the formation of aneurysms. Necropsies were performed if mice died during the experimental treatment before being euthanized. If mice died before sacrifice, we performed necropsies to verify whether aortic ruptures occurred. Mice that died of aortic rupture were used only for the calculation of mortality and rupture rates and were excluded from the tissue degradation analysis. AAAs were judged by an independent investigator who was blinded to the experimental treatments.
IHC staining
Sections (5 μm) of the specimens were deparaffinized, and 3% (v/v) hydrogen peroxide in 10% PBS was applied for 10 min to quench endogenous peroxidase activity. Subsequently, sections were incubated with 10% bovine serum in PBS to block nonspecific binding sites at room temperature for 1 h. Sections were incubated overnight at 4°C with primary antibodies against caspase-3, α-SMA, and SM22, followed by an incubation with a biotinylated secondary antibody at 37°C for 1 h and subsequent incubation with horseradish peroxidase-labeled streptavidin solution at 37°C. Sections were then stained with diaminobenzidine and counterstained with hematoxylin. Details of the primary antibodies are available in Table S6.
Immunofluorescence staining
Immunofluorescence staining was performed as described previously.39 For VSMCs, the cells were fixed with 4% polyoxymethylene, permeabilized with 0.5% Triton X-100, and blocked with 5% BSA in PBS for 1 h. The cells were incubated with primary antibodies against β-tubulin, Ki67, and PH3 at 4°C overnight. Then, the cells were washed 3 times with PBS and incubated with Alexa Fluor-labeled secondary antibodies (at maximum excitation wavelengths of 488 and 594 nm). For the aorta samples, frozen sections were fixed with acetone and blocked with 5% bovine serum in PBS for 1 h at room temperature. Slides were incubated with antibodies against SM22 and vimentin at 4°C overnight. Then, the sections were washed 3 times with PBS and incubated with Alexa Fluor-labeled secondary antibodies. DAPI was applied to label the cell nuclei. Finally, images were captured using a Leica TCS SP8 fluorescence confocal microscope. Details of the primary antibodies are provided in Table S7.
RIP assay
RIP experiments were performed as described previously.35
RNA-FISH
Human aortic VSMCs on coverslips were fixed with 4% paraformaldehyde, washed 3 times with PBS, and then permeabilized with 0.2% Triton X-100 in PBS for 30 min. Human aortic VSMCs were hybridized with hybridization buffer (RiboBio, Guangzhou, China) and incubated with a labeled human circChrodc1 probe (5′-TATGTGCAAGCATCTTGGCCT-3′) overnight at 37°C. The circChrodc1 probe was purchased from BersinBio (Guangzhou, China). The cells were then sequentially washed with 2× SSC, 1× SSC, and 0.5× SSC and incubated with a mouse anti-digoxin antibody conjugated with AP (Boster Biotechnology, Wuhan, China). Subsequently, the cells were incubated with DAPI (Beyotime Biotechnology, Shanghai). Images were obtained using a Leica (TCS Sp8) confocal microscope.
Chromatin isolation by RNA purification
The chromatin isolation by RNA purification (ChIRP) assay was performed using the Magna ChIRP RNA Interactome Kit (Millipore, USA) following the manufacturer's guidelines. In brief, a total of 1 × 107 cells was lysed in complete lysis buffer for each reaction, and the DNA was then sheared into small fragments through sonication. Then the lysate was incubated with biotin-labeled probes that could hybridize with circChrodc1. We divided probes into the “odd” group and the “even” group according to their orders, and probes targeting LacZ were selected as nonspecific controls. Finally, the probes were extracted by streptavidin magnetic beads, and the combined DNA was isolated for quantitative real-time PCR. The material purchasing information is provided in Table S8.
Statistical analyses
Quantitative data are presented as the mean ± standard deviation, which were analyzed using SPSS version 20.0 (SPSS, Chicago, IL, USA). Student's t tests were used for normally distributed datasets when statistically comparing the data from two groups. The differences between multiple groups were analyzed with one-way analysis of variance, followed by Bonferroni tests for equal variances and Dunnett's C test for unequal variances. Fisher's exact test was performed to analyze aneurysm incidence. A p value <0.05 indicated statistical significance.
Acknowledgments
This work was supported by grants awarded to J.B. from the National Natural Science Foundation of China (no. 82070315) and Guangzhou Regenerative Medicine and Health Laboratory of Guangdong (no. 2018GZR110105009), and grants provided to X.L. from the President Foundation of Nanfang Hospital, Southern Medical University (no. 2019Z001), the Natural Science Foundation of Guangdong Province (no. 2020A151501302), the National Natural Science Foundation of China (no. 82000248) and the Outstanding Youths Development Scheme of Nanfang Hospital, Southern Medical University (no. 2019J012). This study was also supported by grants provided to Z.T. from the Science and Technology Foundation of Guangdong Province (no. 2020S00051) and grants provided to S.H. from the National Natural Science Foundation of China (no. 82000282), Guangzhou Basic and Applied Basic Research Fund (no. 202002030198), and the Outstanding Youths Development Scheme of Nanfang Hospital, Southern Medical University (no. 2019J003).
Author contributions
Manuscript writing, X.L. and J.B.; study conception and design, X.H., J.B., and J.X.; experimental design and data acquisition, Y.H., G.C., T.X., D.C., Y.S., S.W., and Y.L.; supervision, Z.T., S.H., W.L., and Y.L.; interpretation of data and critical revision of the manuscript, X.H., X.L., Y.H., J.B., and J.X. All authors take full responsibility for the work.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtn.2021.11.005.
Contributor Information
Jianping Bin, Email: jianpingbin@126.com, jianpingbin@hotmail.com.
Jiancheng Xiu, Email: xiujc@126.com.
Supplemental information
and Tables S1–S8
References
- 1.Sakalihasan N., Limet R., Defawe O.D. Abdominal aortic aneurysm. Lancet. 2005;365:1577–1589. doi: 10.1016/S0140-6736(05)66459-8. [DOI] [PubMed] [Google Scholar]
- 2.Kent K.C. Clinical practice. Abdominal aortic aneurysms. N. Engl. J. Med. 2014;371:2101–2108. doi: 10.1056/NEJMcp1401430. [DOI] [PubMed] [Google Scholar]
- 3.Golledge J., Norman P.E. Current status of medical management for abdominal aortic aneurysm. Atherosclerosis. 2011;217:57–63. doi: 10.1016/j.atherosclerosis.2011.03.006. [DOI] [PubMed] [Google Scholar]
- 4.Elia L., Kunderfranco P., Carullo P., Vacchiano M., Farina F.M., Hall I.F., Mantero S., Panico C., Papait R., Condorelli G., et al. UHRF1 epigenetically orchestrates smooth muscle cell plasticity in arterial disease. J. Clin. Invest. 2018;128:2473–2486. doi: 10.1172/JCI96121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nogi M., Satoh K., Sunamura S., Kikuchi N., Satoh T., Kurosawa R., Omura J., Elias-Al-Mamun M., Abdul Hai Siddique M., Numano K., et al. Small GTP-binding protein GDP dissociation stimulator prevents thoracic aortic aneurysm formation and rupture by phenotypic preservation of aortic smooth muscle cells. Circulation. 2018;138:2413–2433. doi: 10.1161/CIRCULATIONAHA.118.035648. [DOI] [PubMed] [Google Scholar]
- 6.Li G., Wang M., Caulk A.W., Cilfone N.A., Gujja S., Qin L., Chen P.Y., Chen Z., Yousef S., Jiao Y., et al. Chronic mTOR activation induces a degradative smooth muscle cell phenotype. J. Clin. Invest. 2020;130:1233–1251. doi: 10.1172/JCI131048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen P.Y., Qin L., Li G., Malagon-Lopez J., Wang Z., Bergaya S., Gujja S., Caulk A.W., Murtada S.I., Zhang X., et al. Smooth muscle cell reprogramming in aortic aneurysms. Cell Stem Cell. 2020;26:542–557.e11. doi: 10.1016/j.stem.2020.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Leeper N.J., Maegdefessel L. Non-coding RNAs: key regulators of smooth muscle cell fate in vascular disease. Cardiovasc. Res. 2018;114:611–621. doi: 10.1093/cvr/cvx249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Briot A., Jaroszewicz A., Warren C.M., Lu J., Touma M., Rudat C., Hofmann J.J., Airik R., Weinmaster G., Lyons K., et al. Repression of Sox9 by Jag1 is continuously required to suppress the default chondrogenic fate of vascular smooth muscle cells. Dev. Cell. 2014;31:707–721. doi: 10.1016/j.devcel.2014.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Qu S., Yang X., Li X., Wang J., Gao Y., Shang R., Sun W., Dou K., Li H. Circular RNA: a new star of noncoding RNAs. Cancer Lett. 2015;365:141–148. doi: 10.1016/j.canlet.2015.06.003. [DOI] [PubMed] [Google Scholar]
- 11.Ding S., Zhu Y., Liang Y., Huang H., Xu Y., Zhong C. Circular RNAs in vascular functions and diseases. Adv. Exp. Med. Biol. 2018;1087:287–297. doi: 10.1007/978-981-13-1426-1_23. [DOI] [PubMed] [Google Scholar]
- 12.Zhou M., Shi Z., Cai L., Li X., Ding Y., Xie T., Fu W. Circular RNA expression profile and its potential regulative role in human abdominal aortic aneurysm. BMC Cardiovasc. Disord. 2020;20:70. doi: 10.1186/s12872-020-01374-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang Y., Wang Y., Li Y., Wang B., Miao Z., Liu X., Ma Y. Decreased expression of circ_0020397 in intracranial aneurysms may be contributing to decreased vascular smooth muscle cell proliferation via increased expression of miR-138 and subsequent decreased KDR expression. Cell Adh. Migr. 2019;13:220–228. doi: 10.1080/19336918.2019.1619432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hall I.F., Climent M., Quintavalle M., Farina F.M., Schorn T., Zani S., Carullo P., Kunderfranco P., Civilini E., Condorelli G., et al. Circ_Lrp6, a circular RNA enriched in vascular smooth muscle cells, acts as a sponge regulating miRNA-145 function. Circ. Res. 2019;124:498–510. doi: 10.1161/CIRCRESAHA.118.314240. [DOI] [PubMed] [Google Scholar]
- 15.Chen J., Zhou X., Yang J., Sun Q., Liu Y., Li N., Zhang Z., Xu H. Circ-GLI1 promotes metastasis in melanoma through interacting with p70S6K2 to activate Hedgehog/GLI1 and Wnt/β-catenin pathways and upregulate Cyr61. Cell Death Dis. 2020;11(7):596. doi: 10.1038/s41419-020-02799-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Weiser-Evans M.C.M. Smooth muscle differentiation control comes full circle: the circular noncoding RNA, circActa2, functions as a miRNA sponge to fine-tune α-SMA expression. Circ. Res. 2017;121:591–593. doi: 10.1161/CIRCRESAHA.117.311722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang J., Sun H., Zhou Y., Huang K., Que J., Peng Y., Wang J., Lin C., Xue Y., Ji K. Circular RNA microarray expression profile in 3,4-benzopyrene/angiotensin II-induced abdominal aortic aneurysm in mice. J. Cell Biochem. 2019;120:10484–10494. doi: 10.1002/jcb.28333. [DOI] [PubMed] [Google Scholar]
- 18.Yan X., Zhang D., Wu W., Wu S., Qian J., Hao Y., Yan F., Zhu P., Wu J., Huang G., et al. Mesenchymal stem cells promote hepatocarcinogenesis via lncRNA-MUF interaction with ANXA2 and miR-34a. Cancer Res. 2017;77:6704–6716. doi: 10.1158/0008-5472.CAN-17-1915. [DOI] [PubMed] [Google Scholar]
- 19.Peterson T.E., Guicciardi M.E., Gulati R., Kleppe L.S., Mueske C.S., Mookadam M., Sowa G., Gores G.J., Sessa W.C., Simari R.D. Caveolin-1 can regulate vascular smooth muscle cell fate by switching platelet-derived growth factor signaling from a proliferative to an apoptotic pathway. Arterioscler Thromb. Vasc. Biol. 2003;23:1521–1527. doi: 10.1161/01.ATV.0000081743.35125.05. [DOI] [PubMed] [Google Scholar]
- 20.Song S.H., Kim K., Jo E.K., Kim Y.W., Kwon J.S., Bae S.S., Sung J.H., Park S.G., Kim J.T., Suh W. Fibroblast growth factor 12 is a novel regulator of vascular smooth muscle cell plasticity and fate. Arterioscler Thromb. Vasc. Biol. 2016;36:1928–1936. doi: 10.1161/ATVBAHA.116.308017. [DOI] [PubMed] [Google Scholar]
- 21.Yuan X., Zhang T., Yao F., Liao Y., Liu F., Ren Z., Han L., Diao L., Li Y., Zhou B., et al. THO complex-dependent posttranscriptional control contributes to vascular smooth muscle cell fate decision. Circ. Res. 2018;123:538–549. doi: 10.1161/CIRCRESAHA.118.313527. [DOI] [PubMed] [Google Scholar]
- 22.El-Hamamsy I., Yacoub M.H. Cellular and molecular mechanisms of thoracic aortic aneurysms. Nat. Rev. Cardiol. 2009;6:771–786. doi: 10.1038/nrcardio.2009.191. [DOI] [PubMed] [Google Scholar]
- 23.Alonso-Carbajo L., Alpizar Y.A., Startek J.B., López-López J.R., Pérez-García M.T., Talavera K. Activation of the cation channel TRPM3 in perivascular nerves induces vasodilation of resistance arteries. J. Mol. Cell Cardiol. 2019;129:219–230. doi: 10.1016/j.yjmcc.2019.03.003. [DOI] [PubMed] [Google Scholar]
- 24.Mohamed T.M.A., Ang Y.S., Radzinsky E., Zhou P., Huang Y., Elfenbein A., Foley A., Magnitsky S., Srivastava D. Regulation of cell cycle to stimulate adult cardiomyocyte proliferation and cardiac regeneration. Cell. 2018;173:104–116.e12. doi: 10.1016/j.cell.2018.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhuang Z., Jia L., Li W., Zheng Y. The emerging roles of circular RNAs in regulating the fate of stem cells. Mol. Cell Biochem. 2020 doi: 10.1007/s11010-020-03900-w. [DOI] [PubMed] [Google Scholar]
- 26.Yang L., Yang F., Zhao H., Wang M., Zhang Y. Circular RNA circCHFR facilitates the proliferation and migration of vascular smooth muscle via miR-370/FOXO1/cyclin D1 pathway. Mol. Ther. Nucleic Acids. 2019;16:434–441. doi: 10.1016/j.omtn.2019.02.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kumar S., Boon R.A., Maegdefessel L., Dimmeler S., Jo H. Role of noncoding RNAs in the pathogenesis of abdominal aortic aneurysm. Circ. Res. 2019;124:619–630. doi: 10.1161/CIRCRESAHA.118.312438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Clark M.B., Johnston R.L., Inostroza-Ponta M., Fox A.H., Fortini E., Moscato P., Dinger M.E., Mattick J.S. Genome-wide analysis of long noncoding RNA stability. Genome Res. 2012;22(5):885–898. doi: 10.1101/gr.131037.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Romaine S.P., Tomaszewski M., Condorelli G., Samani N.J. MicroRNAs in cardiovascular disease: an introduction for clinicians. Heart. 2015;101:921–928. doi: 10.1136/heartjnl-2013-305402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Han B., Chao J., Yao H. Circular RNA and its mechanisms in disease: from the bench to the clinic. Pharmacol. Ther. 2018;187:31–44. doi: 10.1016/j.pharmthera.2018.01.010. [DOI] [PubMed] [Google Scholar]
- 31.Nishida N., Aoki H., Ohno-Urabe S., Nishihara M., Furusho A., Hirakata S., Hayashi M., Ito S., Yamada H., Hirata Y., et al. High salt intake worsens aortic dissection in mice: involvement of IL (interleukin)-17A-dependent ECM (extracellular matrix) metabolism. Arterioscler Thromb. Vasc. Biol. 2020;40:189–205. doi: 10.1161/ATVBAHA.119.313336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yang H., Li X., Meng Q., Sun H., Wu S., Hu W., Liu G., Li X., Yang Y., Chen R. CircPTK2 (hsa_circ_0005273) as a novel therapeutic target for metastatic colorectal cancer. Mol. Cancer. 2020;19(1):13. doi: 10.1186/s12943-020-1139-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Greene J., Baird A.M., Brady L., Lim M., Gray S.G., McDermott R., Finn S.P. Circular RNAs: biogenesis, function and role in human diseases. Front Mol. Biosci. 2017;4:38. doi: 10.3389/fmolb.2017.00038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tsaousi A., Williams H., Lyon C.A., Taylor V., Swain A., Johnson J.L., George S.J. Wnt4/β-catenin signaling induces VSMC proliferation and is associated with intimal thickening. Circ. Res. 2011;108:427–436. doi: 10.1161/CIRCRESAHA.110.233999. [DOI] [PubMed] [Google Scholar]
- 35.He X., Wang S., Li M., Zhong L., Zheng H., Sun Y., Lai Y., Chen X., Wei G., Si X., et al. Long noncoding RNA GAS5 induces abdominal aortic aneurysm formation by promoting smooth muscle apoptosis. Theranostics. 2019;9:5558–5576. doi: 10.7150/thno.34463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhong L., He X., Song H., Sun Y., Chen G., Si X., Sun J., Chen X., Liao W., Liao Y., et al. METTL3 induces AAA development and progression by modulating N6-methyladenosine-dependent primary miR34a processing. Mol. Ther. Nucleic Acids. 2020;21:394–411. doi: 10.1016/j.omtn.2020.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Song H., Xu T., Feng X., Lai Y., Yang Y., Zheng H., He X., Wei G., Liao W., Liao Y., et al. Itaconate prevents abdominal aortic aneurysm formation through inhibiting inflammation via activation of Nrf2. EBioMedicine. 2020;57:102832. doi: 10.1016/j.ebiom.2020.102832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhong L., He X., Si X., Wang H., Li B., Hu Y., Li M., Chen X., Liao W., Liao Y., et al. SM22α (smooth muscle 22α) prevents aortic aneurysm formation by inhibiting smooth muscle cell phenotypic switching through suppressing reactive oxygen species/NF-κB (nuclear factor-κb) Arterioscler Thromb. Vasc. Biol. 2019;39:e10–e25. doi: 10.1161/ATVBAHA.118.311917. [DOI] [PubMed] [Google Scholar]
- 39.Sun Y., Zhong L., He X., Wang S., Lai Y., Wu W., Song H., Chen Y., Yang Y., Liao W., et al. LncRNA H19 promotes vascular inflammation and abdominal aortic aneurysm formation by functioning as a competing endogenous RNA. J. Mol. Cell Cardiol. 2019;131:66–81. doi: 10.1016/j.yjmcc.2019.04.004. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
and Tables S1–S8