

Abstract

In this work, we study how the cation identity and concentration alter the kinetics of the hydrogen evolution reaction (HER) on platinum and gold electrodes. A previous work suggested an inverted activity trend as a function of alkali metal cation when comparing the performance of platinum and gold catalysts in alkaline media. We show that weakly hydrated cations (K+) favor HER on gold only at low overpotentials (or lower alkalinity), whereas in more alkaline pH (or high overpotentials), a higher activity is observed using electrolytes containing strongly hydrated cations (Li+). We find a similar trend for platinum; however, the inhibition of HER by weakly hydrated cations on platinum is observed already at lower alkalinity and lower cation concentrations, suggesting that platinum interacts more strongly with metal cations than gold. We propose that weakly hydrated cations stabilize the transition state of the water dissociation step more favorably due to their higher near-surface concentration in comparison to a strongly hydrated cation such as Li+. However, at high pH and consequently higher near-surface cation concentrations, the accumulation of these species at the outer Helmholtz plane inhibits HER. This is especially pronounced on platinum, where a change in the rate-determining step is observed at pH 13 when using a Li+- or K+-containing electrolyte.
Keywords: hydrogen evolution, platinum, gold, cation effect, local pH, alkaline media
Introduction
Efficient water electrolysis is an important technology toward a more sustainable society.1 While water electrolysis in acidic media leads to the highest activity, it utilizes scarce and expensive materials (platinum and iridium); in alkaline media, more abundant materials can be used but at the expense of a lower activity. To understand this difference in activity between the two media, the cathode reaction, that is, the hydrogen evolution reaction (HER), has been studied extensively both in acidic and alkaline media. The focus of most studies has been on tailoring the catalyst surface.2,3 However, metal cations in the electrolyte have been shown to have a significant effect on the activity of HER, although usually not taken into account in reaction mechanisms.4−7 In fact, the underlying mechanisms of the noncovalent interactions between cations and HER intermediates remain incompletely understood. Therefore, systematically studying electrolyte-related phenomena on different metal surfaces is desired to improve our understanding of the underlying phenomena and to assist in further optimization of alkaline and proton exchange membrane electrolyzers.
Platinum and gold are two model catalysts for understanding electrocatalytic reactions, including hydrogen evolution. Although these are two noble metals, it has been shown that the interaction of these surfaces with adsorbed species (reaction intermediates) and water is very different.8,9 Platinum is considered an optimal catalyst for HER (in acidic media), a consequence of the optimal hydrogen binding energy (ΔGHads ≈ 0). On the other hand, on gold electrodes, hydrogen binds weakly and the activity for HER at low overpotentials is significantly inferior to the activity found on Pt. Very recently, efforts have been dedicated to the effect that metal cations have on the activity of HER on different metal surfaces. A main contribution has been made by Xue et al.7 who studied the HER activity of Pt, Ir, Au, and Ag in alkaline media as a function of the cation identity (Li+, Na+, K+, Rb+, and Cs+). They reported that weakly hydrated cations such as Cs+ favor HER on Au and Ag, while they are detrimental to the reaction activity on Pt and Ir. The authors explain this “inverted” trend based on the binding energy of hydrogen to the metal, which weakens as a function of the alkali metal cation: Cs+ > Rb+ > K+ > Na+ > Li+. They proposed that weakly hydrated cations such as Cs+ are beneficial to HER on Au and Ag electrodes, which lie on the weak binding side of the activity volcano, while being detrimental to HER on Pt and Ir due to over-stabilization of the adsorbed intermediates.
The hydrogen adsorption energy is generally considered to be an accurate descriptor for HER activity in acidic media, where the adsorbed hydrogen is formed from proton/hydronium (H3O+) reduction. However, in alkaline media, where water dissociation has to occur in order for hydrogen to adsorb, other descriptors appear to be needed to describe activity trends.8 On platinum, the hydrogen binding energy (HBE) is derived from the underpotential hydrogen region (Hupd) in the blank voltammetry.10,11 A positive shift of the Hupd peak, for example as a function of pH, has been ascribed to an increase in the HBE and an associated lower activity for HER. However, it has been shown that this positive shift is actually associated with weakening of OH adsorption on Pt(100) and Pt(110) steps and facets due to the presence of alkali metal cations at the reaction interface.12 The so-called Hupd region is therefore a hydrogen-cation-hydroxyl region, and therefore, it cannot serve as a simple activity descriptor. The nature of the rate-determining step for HER in alkaline media is still under debate. The study of Markovic et al. suggests that on platinum, in alkaline media, water dissociation is the rate-determining step.5 However, Liu et al.4 suggest that for a similar system, the driving force for OH desorption is larger for Li+ than for Na+ and K+, and this trend gives rise to the HER activity trend of LiOH > NaOH > KOH at pH 13. Gold is a much less investigated catalyst for HER but of interest as a catalyst for the electrocatalytic reduction of carbon dioxide, for which HER is a competing reaction.13 As discussed above, an inverted activity trend (K+ > Na+ > Li+), compared to platinum, has been reported for gold electrodes in 0.1 M XOH.7 Our recent study on polycrystalline gold and Au(111) in alkaline media (NaOH) shows that up to pH 11, increasing the Na+ concentration significantly enhances the HER activity.14 At higher pH (>12) and consequently high (near-surface) cation concentrations, the activity for HER decreases likely due to a blockage effect caused by cation accumulation at the interface. However, this phenomenon has not yet been investigated for other metal surfaces as a function of the cation identity.
Taking HBE as a descriptor cannot explain the different cation trends for HER on Pt and Au as a function of the cation identity. This is especially relevant in alkaline media where our recent study has shown that the local pH and cation concentration are two interrelated variables.13−15 Therefore, in this study, we have systematically investigated the effect of pH and cation concentration on the activity for HER on polycrystalline Pt and Au, comparing electrolytes containing weakly (K+) and strongly (Li+) hydrated cations. In alkaline media, on gold, we see that the surface blockage phenomena (previously observed at pH 13 in a Na+-containing electrolyte) are not present in a Li+-containing electrolyte due to the less-pronounced accumulation of Li+ ions near the surface even at high pH. In contrast, K+ ions are detrimental to the reaction activity above pH 13. A similar behavior is observed on platinum however already at pH 11 and at low cation concentrations, suggesting that cations interact more strongly with Pt than with Au electrodes. These results show that on Pt in alkaline media, cations typically act as inhibitors, whereas on gold, cations can act as either a promotor or an inhibitor depending on their local accumulation in the double layer.
Experimental Section
Materials
The following chemicals were used to prepare the electrolytes used in this study: Li2SO4 (Sigma-Aldrich, 99.99%, metal basis), K2SO4 (Alfa Aesar, Puratronic, 99.997%, metal basis), H2SO4 (Merck, Suprapur, 96%), LiClO4 (Sigma-Aldrich, 99.99%, trace metal basis), KClO4 (Sigma-Aldrich, ≥99.99%, trace metal basis), LiOH (Sigma-Aldrich, 99.995%, monohydrate), NaOH (Merck, 30% solution, Suprapur), and KOH (Merck, 99.995%, Suprapur). Sulfate salts were used for the experiments in acidic media, and hydroxide salts were used for the experiments in alkaline media. For the measurements in alkaline media in which the cation concentration was varied, appropriate amounts of perchlorate salts were added to the electrolyte. Gold and platinum disc electrodes were cut from polycrystalline gold (0.5 mm thick, MaTecK, 99.995%) and platinum (0.5 mm thick, MaTecK, 99.99%) foils and prepared by first grinding with a silicon carbide paper (grit size 600, MaTecK). The electrodes were then polished using a microcloth (Buehler) with diamond suspensions (MetaDi 3, 1, 0.25, and 0.05 μm, Buehler) for 1 min with each size.16 In between the polishing steps, the electrodes were cleaned in an ultrasonic bath (Bandelin SONOREX RK 52H) in ultrapure (>18.2 MΩ cm, Millipore Milli-Q) water for 3 min. After the last step, the electrodes were sonicated in ethanol for 3 min and subsequently in ultrapure water for 10 min. All glassware used was cleaned by immersion in a potassium permanganate solution overnight (1 g/L KMnO4 dissolved in 0.5 M H2SO4), followed by immersion in a dilute Piranha solution. The glassware was further boiled in ultrapure water at least five times before use.
Electrochemical Measurements
All electrochemical measurements were carried out using a BioLogic two-channel potentiostat/galvanostat/EIS (SP-300). The hanging meniscus and rotating disc electrode (RDE) experiments were carried out in a one-compartment glass cell. A gold wire (0.5 mm diameter, MaTecK, 99.9%) or a platinum wire (0.5 mm diameter, MaTecK, 99.9%) was used as the counter electrode (depending on the working electrode), and a reversible hydrogen electrode (RHE, Gaskatel, HydroFlex) was used as a reference. Argon (6.0 purity, Linde) was purged through the solution for 20 min prior to the experiments. The argon flow was also kept above the solution during the experiments in order to avoid oxygen diffusing into the electrolyte. Before the measurements, the gold electrode was prepared by flame annealing using the procedure described in our previous study.16 The platinum electrode was also flame-annealed and subsequently cycled 200 times between 0.06 and 1.65 V versus RHE in 0.1 M H2SO4 at 1 V s–1 in order to yield a reproducible blank voltammogram. Blank cyclic voltammograms (CVs) were recorded in argon-saturated 0.1 M H2SO4 prior to every measurement. The electrochemically active surface area (ECSA) of gold was calculated based on the charge corresponding to gold oxide reduction and a surface charge density of 386 μC cm–2.17 For platinum, the integral of the hydrogen desorption region 0.06 < E < 0.6 V was used (after subtraction of the double-layer charge), and the ECSA was calculated based on a surface charge density of 230 μC cm–2 reported for a polycrystalline Pt surface in sulfuric acid.18 RDE experiments were performed using an MSR electrode rotator (Pine Research Instrumentation) equipped with an AFE6M shaft and gold and platinum discs (diameter = 5 mm, Pine Research Instrumentation). The disc electrodes were polished with a polycrystalline diamond suspension of 0.25 μm (MetaDi, Buehler) and then sonicated (Bandelin SONOREX RK 52H) in ethanol and ultrapure water (>18.2 MΩ cm, Millipore Milli-Q) for 10 min before the measurements. For all hydrogen evolution experiments, the solution resistance was determined by performing potentiometric electrochemical impedance spectroscopy, and the electrode potential was always automatically compensated for 85% of the ohmic drop.
Results
We initially studied how cations affect the HER on polycrystalline platinum and gold electrodes using stationary cyclic voltammetry, and the results are shown in Figure 1a–c. We carried out the experiments in alkaline media (pH 13) and found that K+ cations promote water reduction on gold, while they are detrimental to the reaction on platinum, in agreement with the results found by Xue et al. for Au(111) and polycrystalline Pt at the same pH and similar overpotentials.7 We have also recorded the blank voltammetry of the polycrystalline platinum electrode in alkaline media, and a positive shift of the Hupd peak in the K+-containing electrolyte was also found at pH 13 (Figure 1c). Similar measurements were performed in acidic media (see Figure S1a–c and discussion in the Supporting Information). There, water reduction is promoted by weakly hydrated cations on both gold and platinum, and a similar (but less pronounced) positive shift in the Hupd peak of the platinum electrode is found in the K+ electrolyte. Although the effect of the different cations on water reduction on platinum is inverted for acidic and alkaline media, it seems that at both pH, weakly hydrated cations interact with the adsorbed H/OH in a similar fashion, suggesting that the mechanism behind this inverted trend goes beyond just an effect on the HBE, as proposed by Xue et al.7 A more pronounced positive shift in the Hupd peak at pH 13 in comparison to that at pH 3 was also observed by Chen et al.12 In the same study, density functional theory (DFT) calculations suggest that higher coverages of alkali metal cations adsorbed along the steps of Pt(553) become more favorable on increasing the electrolyte pH (at the same potential on an RHE scale). Therefore, we expect that the larger shift we observe on polycrystalline platinum for the experiment carried out at pH 13 (Figure 1b) is due to an increase in the cation–OHads–Hads interactions at higher cation coverages. The blank voltammetry of the polycrystalline gold and platinum electrodes, taken before the measurements shown in Figure 1, is shown in Figure S2 in the Supporting Information.
Figure 1.

Hydrogen evolution on stationary electrodes (a) gold and (b) platinum in alkaline media (0.1 M MOH, pH = 13), together with (c) the blank voltammetry of the platinum electrode. M = Li+ or K+. The voltammetry was recorded at 50 mV s–1.
In Figure 2, we show the Tafel slopes, derived from the CVs from Figures 1 and S1 (Supporting Information), as a function of the applied potential. Eqs 1–5 show the three accepted reaction steps during proton (H3O+) or water reduction with the rate-determining step in the mechanism that can be derived from the Tafel slope analysis in parenthesis.19 Here, * represents a free surface adsorption site. Tafel slopes of around 120 mV dec–1 are obtained for the gold electrode in the Li+ and K+ electrolytes, in both acidic and alkaline media. This indicates that at the potentials studied, in both cases, the activity for HER is controlled by the first electron transfer step (Volmer step, eq 1), which in alkaline media is in fact governed by the barrier of the electrochemical water dissociation (eq 2). The latter is in agreement with our previous studies in a Na+-containing electrolyte.14 For platinum, in acidic media, Tafel slopes of around 40 mV dec–1 imply that the second electron transfer Heyrovsky step (eq 3) is the rate-determining step, in both Li+ and K+ electrolytes.20 In contrast, in alkaline media, where we see higher HER activity in Li+ than K+, the Tafel slopes indicate that in Li+, the Heyrovsky step is still rate-determining, while there seems to be a change in the reaction mechanism in the K+ electrolyte. A Tafel slope of 112 mV dec–1 suggests either the Volmer step or a Heyrovsky step at high coverage of adsorbed hydrogen (at high overpotentials) as the rate-limiting step. Considering that the Tafel analysis was performed using the current response at low overpotentials, the Volmer step is more likely to be the rate-determining step here. This change in the reaction mechanism on platinum suggests that the cation identity plays an important role on how accessible the surface is for hydrogen adsorption.
| 1 |
| 2 |
| 3 |
| 4 |
| 5 |
Figure 2.
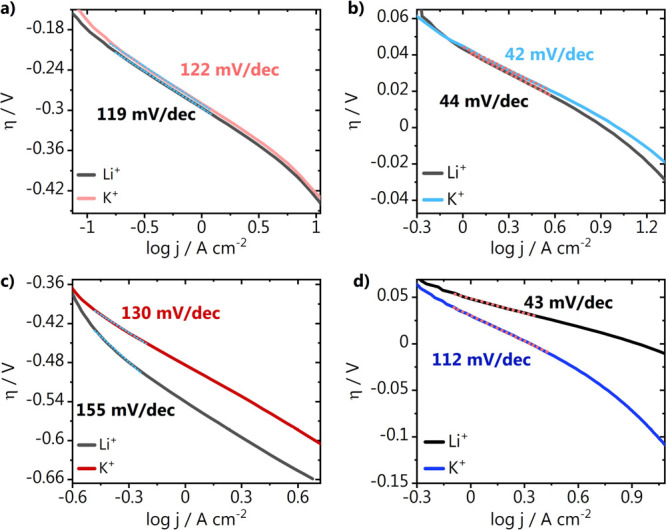
Tafel slopes derived from the cyclic voltammetry on stationary electrodes recorded on (a) gold and (b) platinum in acidic media (0.1 M M2SO4, pH = 3) as well as for (c) gold and (d) platinum in alkaline media (0.1 M MOH, pH = 13). M = Li+ or K+. The range used for the curve fitting is indicated in all plots with a dotted line.
Previous study reported higher HER activity for gold in electrolytes containing weakly hydrated cations in the order Cs+ > K+ > Na+ > Li+, and the opposite trend was observed for platinum electrodes in alkaline media.7 However, experiments on gold have usually been carried out in a limited potential range as at large overpotentials, currents become impractical, that is, due to bubble formation and a possible loss of potential control. To further elucidate the nature of this “inverted” trend, we have performed HER on gold at pH 11 and 13 over a wide potential range in electrolytes containing Li+, Na+, and K+ cations (Figure 3). As shown in Figure 3a, at pH 13 and low overpotentials, indeed the activity trend is found to be K+ > Na+ > Li+. Remarkably, at more negative overpotentials (−0.8 V vs RHE), the trend inverts, and a higher current is obtained in the Li+ electrolyte. This strongly suggests that on gold, upon an increase in the local alkalinity at high overpotentials, and a consequent increase in the near-surface cation concentration, weakly hydrated cations are actually detrimental to water reduction. This is likely due to accumulation at the reaction interface and blockage of the surface, as proposed in our previous study.14 This blockage effect is less pronounced in Na+ and even less in the Li+ electrolyte as the stronger interaction of water molecules in the hydration shell of these cations prohibits their accumulation at the outer Helmholtz plane (OHP), as we have also shown previously through DFT-based ab initio molecular dynamics simulations on the interaction of alkali cations with Au(111).21 To further support these observations, we have performed the same experiment at pH 11, where there is in principle a lower driving force for cations to accumulate near the surface than at pH 13 due to the lower interfacial electric field strength. As shown in Figure 3b, at pH 11, the inversion of the HER activity trend as a function of the cation identity happens at ca. 0.25 V more negative potential than at pH 13. This confirms that the local alkalinity defines the concentration of cations near the surface and that weakly hydrated cations hinder HER on gold if their local concentration is high enough. This behavior is similar to platinum although on platinum this effect is already observed at lower overpotentials, whereas on gold more driving force is required. At pH 11, more current is required to reach high local alkalinity, while at pH 13, this happens at lower overpotentials. It is important to point out that the fluctuations in the current shown in Figure 3b at large overpotentials are due to bubble formation at these high currents. However, several repetitions of the experiment (not shown) resulted in the same trend, showing that the bubbles are therefore not detrimental to the final conclusions.
Figure 3.
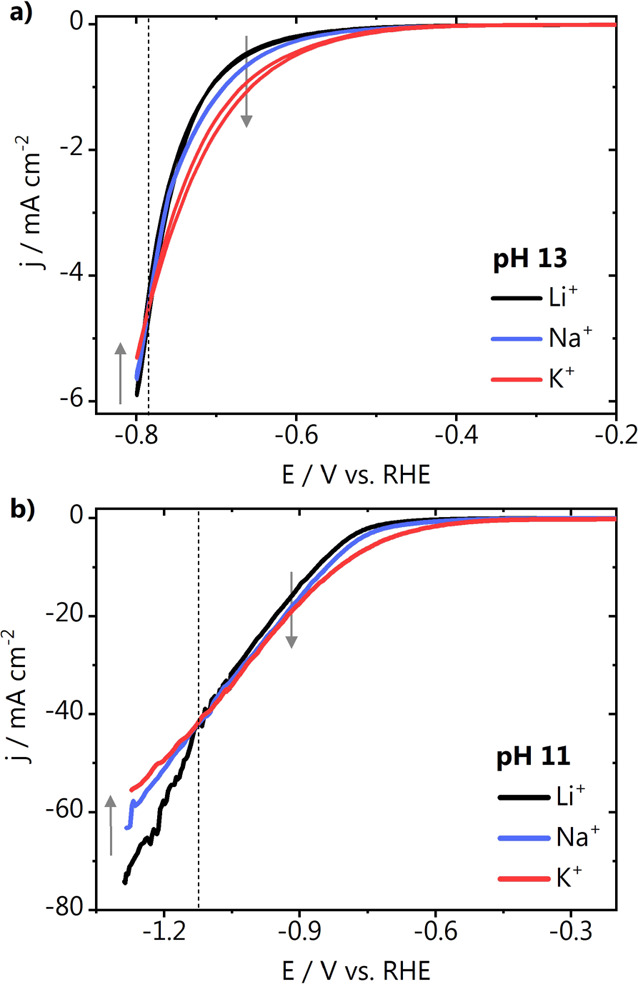
Hydrogen evolution cyclic voltammetry on a stationary gold electrode at pH (a) 13 and (b) 11 recorded in 0.1 M MOH and 0.001 M MOH, with M = Li+, Na+, or K+ at 50 mV s–1. The dotted gray lines indicate the potential at which the CVs cross, and the arrows point going from the strongly to the weakly hydrated metal cation species.
The results shown in Figure 3a,b suggest that the cation trends for HER on platinum and gold are not actually “inverted” as previously stated7 but just depend on the reaction conditions, such as bulk/local pH, near-surface cation concentration, and cation identity. In order to further elucidate this, we have performed HER under well-defined mass transport conditions using gold and platinum disc electrodes. We changed the cation concentration in electrolytes having different bulk pH values and observed the effect of those variables on the HER kinetics. The HER cyclic voltammetry on gold is shown in Figure S4 in the Supporting Information, and the corresponding reaction order plots are shown in Figure 4. In the Li+-containing electrolyte, a positive reaction order is found both at pH 11 and 13, unlike what we observed for instance for Na+ in our previous study.14 In the case of K+, at pH 11, a higher cation concentration promotes HER, as shown by the higher reaction order than that found in Li+. However, at pH 13, we see a negative reaction order, with HER being inhibited as the concentration of K+ increases.
Figure 4.

Reaction order plot of HER on polycrystalline gold in the cation concentration at pH 11 and 13 in (a, b) Li+- and (c, d) K+-containing electrolytes. The current is reported for 50 mV potential steps (vs RHE) plotted as a function of the logarithm of the current density on the y-axis and logarithm of the cation concentration on the x-axis. The slopes extracted from the linear fit of the current response (reaction orders) are shown next to the plots. The slope at the top corresponds to the most negative potential applied and the slope at the bottom to the most positive, as also indicated by the colors. CVs are shown in Figure S4 and were recorded using an RDE at 25 mV s–1 and 2500 rpm.
The HER inhibition in alkaline pH seems to be more pronounced for weakly hydrated cations and needs less driving force to be observed on platinum than on gold, as we have shown in Figure 1. To better define the experimental conditions at which this phenomenon takes place, we performed similar experiments as shown in Figure 4 on platinum, also using an RDE. The cyclic voltammetry is shown in Figure S5, and the corresponding reaction order plots are shown in Figure 5. It is important to point out that although the electrolytes in this study were used as received, no metal contaminants are expected to affect our measurements as evidenced by the overlapping consecutive CVs shown in Figure S6 in the Supporting Information.22 As shown in Figure 5a,b, similar to gold, on platinum, increasing the concentration of Li+ cations promotes HER both at pH 11 and 13. In contrast, for the K+ electrolyte, at pH 9 and 10, we observe a positive reaction order at a low K+ concentration and at the highest concentrations, HER starts to be inhibited, confirming the relationship between local alkalinity and the concentration of weakly hydrated cations at the reaction interface. At pH 11, while on gold we see a promotion of HER upon increasing the K+ concentration, for platinum, we observe a negative reaction order with even more negative values at pH 12. This indicates that weakly hydrated metal cations in the electrolyte interact more strongly with platinum than with gold electrodes under the same experimental conditions.
Figure 5.

Reaction order plot of HER on polycrystalline platinum in the cation concentration at pH 11 and 13 in (a, b) Li+-containing electrolyte and at pH 9–12 in (c, d) K+-containing electrolyte. The current is reported for 50 mV potential steps (vs RHE) plotted as a function of the logarithm of the current density on y-axis and logarithm of the cation concentration on x-axis. The slopes extracted from the linear fit of the current response (reaction orders) are shown next to the plots. The slope at the top corresponds to the most negative potential applied and the slope at the bottom to the most positive, as also indicated by the colors. CVs are shown in Figure S5 and were recorded using an RDE at 25 mV s–1 and 2500 rpm.
Discussion
The opposite cation trends for HER on platinum and gold electrodes in alkaline media have previously been explained using HBE as a descriptor for HER.7 However, our recent study has shown that although this might be a suitable descriptor for HER in acidic media, on gold, in alkaline media, the rate of this reaction depends on the near-surface pH and near-surface cation concentration.14 In our present study, we elucidate that the inverted cation trend between platinum and gold strongly depends on the cation identity, the electrolyte bulk pH, and the local alkalinity. We see that on gold, HER is promoted by weakly hydrated cations only at low current densities and moderately alkaline pH, as shown in Figure 3. This is confirmed by the different cation reaction orders on polycrystalline Pt and Au as a function of pH, potential, and cation identity. For gold, we find a positive reaction order in a Li+ electrolyte at pH 13, while in the K+ electrolyte, there is an inhibition of HER at the same pH. The same is observed for platinum; however, on this electrode, HER starts to be inhibited already at lower pH and lower cation concentrations, indicating that metal cations interact more strongly with platinum than with gold electrodes. Additionally, on platinum, we see a change in the reaction mechanism as a function of the electrolyte cation: in the Li+ electrolyte, the Heyrovsky step is the rate-determining step, and in the K+ electrolyte, the Volmer step seems to be rate-determining. These differences in cation interactions with gold and platinum have also been suggested by other studies. For instance, DFT calculations presented in the study of Hersbach et al.23 indicate that cation adsorption is more energetically favorable on platinum (111) and (100) facets than on gold. Additionally, a study from our group has reported an anomalously large diffuse layer Gouy-Chapman capacitance for the Pt(111) aqueous electrolyte interface in comparison to Au(111) and Hg.24 It shows that the double layer of Pt(111) (and to some extent also of Au(111)) is more compact than predicted by the Gouy-Chapman theory because the electrolyte interacts more strongly with Pt than with Au and Hg. Therefore, we suggest that at the cathodic potentials at which HER takes place, the driving force for cations to accumulate near the platinum surface is higher.
Site blocking by metal cations has been previously proposed in various studies. An early report by Herasymenko and Šlendyk assumed a simple competitive Langmuir adsorption model to rationalize cation trends for HER, which was later further rationalized in the study of Frumkin considering the effect of cation adsorption on reaction rates.25,26 The specific adsorption of cations on catalytic sites for HER has been extensively discussed in the recent review of Waegele et al.,27 showing that despite various theoretical and experimental evidence, it is still unclear under which specific conditions (potential, pH, concentration, and electrode surface) cations specifically adsorb, if they do at all. Another site-blocking theory has been put forward by Markovic and co-workers in which they propose that noncovalent interactions between hydrated alkali metal cations M+(H2O)x and adsorbed OH (OHad) give rise to OHad–M+(H2O)x clusters at the interface. They proposed that the concentration of these clusters increases in the same order as the hydration energies of the corresponding cations (Li+ ≫ Na+ > K+ > Cs+). However, we can rule out this being the reason for the inhibition of HER that we observe in our experiments as we find positive reaction orders in the Li+ electrolyte both on gold and platinum (see Figures 4 and 5), and the inhibition effect is only seen in the K+ electrolyte (and in our previous study in Na+).14
Based on our results and the study discussed above, we propose that the model that we recently formulated for cation effects in HER on gold in alkaline media is more broadly applicable. This model has two regimes depending on the local cation concentration: a promotion regime and an inhibition regime (see Figure 6). In the promotion regime (low cation concentration and low pH), both strongly and weakly hydrated cations stabilize the transition state of the first electron transfer step, the water dissociation (eq 6).14 This interaction is somewhat similar to the effect suggested by Singh et al.,28 but we emphasize that in our model, the local cation concentration is the main parameter determining the reaction rate, rather than changes in the cation’s pKa of hydrolysis near the surface.
| 6 |
Figure 6.
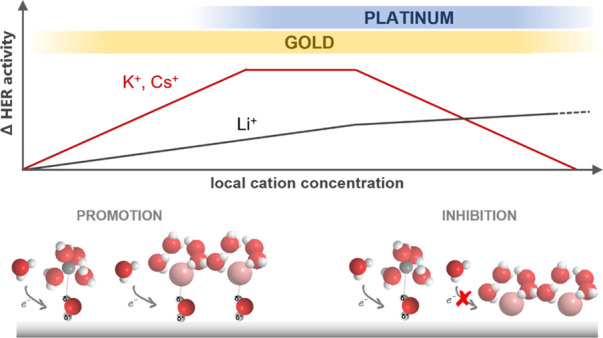
Schematic representation of the effect of cation concentration on the activity of HER comparing platinum and gold together with a pictorial description of the mechanism through which weakly (K+ and Cs+) and strongly (Li+) hydrated cations promote and/or inhibit HER.
In our previous study, we showed both through experiments and AIMD and DFT simulations that alkali cations with a large ionic radius have a larger driving force to accumulate at the electrochemical interface.21 This was done by comparing the energy of a cation in the bulk with that in the OHP at U = 0 V versus SHE. Therefore, our model shows that weakly hydrated cations lead to higher activity due to their stronger driving force to accumulate at the OHP and their consequently higher concentration at the interface. We see that this promotion regime is more limited for platinum than for gold due to stronger interaction of metal cations with platinum, already pronounced in weakly alkaline conditions and moderate cation concentrations. In case of such a very strong interaction, the promotion regime develops into the inhibition regime, and weakly hydrated cations accumulate near the surface to such an extent that they hinder the access of water to the reaction interface or lower the availability of free surface sites. In this regime, the HER has a negative reaction order on cation concentration. Experiments with Pt electrodes are typically in this inhibition regime, whereas experiments with Au are typically in the promotion regime. This model explains the inverted cation dependence between Pt and Au, observed here as well as in a previous study.7
Our findings may have important implications for electrolyzers in which HER is the cathode reaction. Due to the high current densities of operation, a high local alkalinity can develop near the cathode surface. As our study shows, depending on the electrolyte cation identity, the activity for HER can be compromised upon local alkalization of the reaction interface. Tuning the cathode geometry and consequently enhancing the transport of species, or selecting a specific cation, can help to mitigate these negative effects.
Conclusions
In this study, we have elucidated the cation effects on HER on gold and platinum electrodes in alkaline media. Through cyclic voltammetry and RDE experiments, we investigated HER in alkaline media in electrolytes containing strongly and weakly hydrated cations, namely, Li+, Na+, and K+. In agreement with the model that we recently formulated for HER on gold, we show here that for platinum, there are also two distinct regimes for how cations affect HER according to the cation concentration. At a low cation concentration and mildly alkaline media, both weakly and strongly hydrated cations promote HER on gold and platinum. At more alkaline pH and consequently higher near-surface cation concentrations, HER is inhibited by weakly hydrated cations. This inhibition regime is observed for platinum at lower alkalinity and cation concentration than for gold electrodes as platinum interacts more strongly with cations in the electrolyte. On platinum, based on Tafel slopes, we find a change in the reaction mechanism at pH 13 from the Heyrovsky to the Volmer step when the reaction is carried out in a Li+ or K+ electrolyte, respectively. This can be understood as inhibition of the Volmer step in K+ electrolyte, whereas Li+ actually promotes the Volmer step. The observation that HER on gold occurs mostly in the promotion regime whereas HER on platinum occurs mostly in the inhibition regime explains the previously observed inverted cation dependence of HER on gold and platinum. Our study shows the complexity of the electrode–electrolyte interface during hydrogen evolution on platinum and gold and that to achieve high activity, cation identity and near-surface cation concentration are crucial activity descriptors, with the latter depending on the electrolyte pH and the metal surface employed.
Acknowledgments
This work was supported by the European Commission under contract 722614 (Innovative training network Elcorel) and by the Advanced Research Center for Chemical Building Blocks Consortium (ARC CBBC), cofinanced by the Netherlands Organization for Scientific Research (NWO) and Shell Global Solutions B.V.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acscatal.1c04268.
HER in acidic media, blank voltammetry of the electrodes used, and HER cyclic voltammetry (PDF)
Author Contributions
M.C.O.M. wrote the manuscript with input from M.T.M.K. and A.G. All authors contributed equally to the design of the experiments and interpretation of the data.
The authors declare no competing financial interest.
Supplementary Material
References
- Shiva Kumar S.; Himabindu V. Hydrogen Production by PEM Water Electrolysis – A Review. Mater. Sci. Energy Technol. 2019, 2, 442–454. 10.1016/j.mset.2019.03.002. [DOI] [Google Scholar]
- Strmcnik D.; Lopes P. P.; Genorio B.; Stamenkovic V. R.; Markovic N. M. Design Principles for Hydrogen Evolution Reaction Catalyst Materials. Nano Energy 2016, 29, 29–36. 10.1016/j.nanoen.2016.04.017. [DOI] [Google Scholar]
- Zhou Z.; Pei Z.; Wei L.; Zhao S.; Jian X.; Chen Y. Electrocatalytic Hydrogen Evolution under Neutral PH Conditions: Current Understandings, Recent Advances, and Future Prospects. Energy Environ. Sci. 2020, 13, 3185–3206. 10.1039/D0EE01856B. [DOI] [Google Scholar]
- Liu E.; Li J.; Jiao L.; Doan H. T. T.; Liu Z.; Zhao Z.; Huang Y.; Abraham K. M.; Mukerjee S.; Jia Q. Unifying the Hydrogen Evolution and Oxidation Reactions Kinetics in Base by Identifying the Catalytic Roles of Hydroxyl-Water-Cation Adducts. J. Am. Chem. Soc. 2019, 141, 3232–3239. 10.1021/jacs.8b13228. [DOI] [PubMed] [Google Scholar]
- Subbaraman R.; Tripkovic D.; Chang K.-C.; Strmcnik D.; Paulikas A. P.; Hirunsit P.; Chan M.; Greeley J.; Stamenkovic V.; Markovic N. M. Trends in Activity for the Water Electrolyser Reactions on 3d M(Ni,Co,Fe,Mn) Hydr(Oxy)Oxide Catalysts. Nat. Mater. 2012, 11, 550–557. 10.1038/nmat3313. [DOI] [PubMed] [Google Scholar]
- Huang B.; Muy S.; Feng S.; Katayama Y.; Lu Y. C.; Chen G.; Shao-Horn Y. Non-Covalent Interactions in Electrochemical Reactions and Implications in Clean Energy Applications. Phys. Chem. Chem. Phys. 2018, 20, 15680–15686. 10.1039/C8CP02512F. [DOI] [PubMed] [Google Scholar]
- Xue S.; Garlyyev B.; Watzele S.; Liang Y.; Fichtner J.; Pohl M. D.; Bandarenka A. S. Influence of Alkali Metal Cations on the Hydrogen Evolution Reaction Activity of Pt, Ir, Au, and Ag Electrodes in Alkaline Electrolytes. ChemElectroChem 2018, 5, 2326–2329. 10.1002/celc.201800690. [DOI] [Google Scholar]
- Dubouis N.; Grimaud A. The Hydrogen Evolution Reaction: From Material to Interfacial Descriptors. Chem. Sci. 2019, 10, 9165–9181. 10.1039/C9SC03831K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang B.; Myint K. H.; Wang Y.; Zhang Y.; Rao R. R.; Sun J.; Muy S.; Katayama Y.; Corchado Garcia J.; Fraggedakis D.; Grossman J. C.; Bazant M. Z.; Xu K.; Willard A. P.; Shao-Horn Y. Cation-Dependent Interfacial Structures and Kinetics for Outer-Sphere Electron-Transfer Reactions. J. Phys. Chem. C 2021, 125, 4397–4411. 10.1021/acs.jpcc.0c10492. [DOI] [Google Scholar]
- Jiao L.; Liu E.; Mukerjee S.; Jia Q. In Situ Identification of Non-Specific Adsorption of Alkali Metal Cations on Pt Surfaces and Their Catalytic Roles in Alkaline Solutions. ACS Catal. 2020, 10, 11099–11109. 10.1021/acscatal.0c02762. [DOI] [Google Scholar]
- Weber D. J.; Janssen M.; Oezaslan M. Effect of Monovalent Cations on the HOR/HER Activity for Pt in Alkaline Environment. J. Electrochem. Soc. 2019, 166, F66–F73. 10.1149/2.0301902jes. [DOI] [Google Scholar]
- Chen X.; McCrum I. T.; Schwarz K. A.; Janik M. J.; Koper M. T. M. Co-Adsorption of Cations as the Cause of the Apparent PH Dependence of Hydrogen Adsorption on a Stepped Platinum Single-Crystal Electrode. Angew. Chem. Int. Ed. 2017, 56, 15025–15029. 10.1002/anie.201709455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goyal A.; Marcandalli G.; Mints V. A.; Koper M. T. M. Competition between CO2 Reduction and Hydrogen Evolution on a Gold Electrode under Well-Defined Mass Transport Conditions. J. Am. Chem. Soc. 2020, 142, 4154–4161. 10.1021/jacs.9b10061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goyal A.; Koper M. T. M. The Interrelated Effect of Cations and Electrolyte pH on the Hydrogen Evolution Reaction on Gold Electrodes in Alkaline Media. Angew. Chem. Int. Ed. 2021, 60, 13452–13462. 10.1002/anie.202102803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goyal A.; Koper M. T. M. Understanding the Role of Mass Transport in Tuning the Hydrogen Evolution Kinetics on Gold in Alkaline Media. J. Chem. Phys. 2021, 155, 134705. 10.1063/5.0064330. [DOI] [PubMed] [Google Scholar]
- Monteiro M. C. O.; Koper M. T. M. Alumina Contamination through Polishing and Its Effect on Hydrogen Evolution on Gold Electrodes. Electrochim. Acta 2019, 325, 134915 10.1016/j.electacta.2019.134915. [DOI] [Google Scholar]
- Do U. P.; Seland F.; Johannessen E. A. The Real Area of Nanoporous Catalytic Surfaces of Gold and Palladium in Aqueous Solutions. J. Electrochem. Soc. 2018, 165, H219. 10.1149/2.0341805jes. [DOI] [Google Scholar]
- Chen Q.-S.; Solla-Gullón J.; Sun S.-G.; Feliu J. M. The Potential of Zero Total Charge of Pt Nanoparticles and Polycrystalline Electrodes with Different Surface Structure: The Role of Anion Adsorption in Fundamental Electrocatalysis. Electrochim. Acta 2010, 55, 7982–7994. 10.1016/j.electacta.2010.03.050. [DOI] [Google Scholar]
- Shinagawa T.; Garcia-Esparza A. T.; Takanabe K. Insight on Tafel Slopes from a Microkinetic Analysis of Aqueous Electrocatalysis for Energy Conversion. Sci. Rep. 2015, 5, 13801. 10.1038/srep13801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ledezma-Yanez I.; Wallace W. D. Z.; Sebastián-Pascual P.; Climent V.; Feliu J. M.; Koper M. T. M. Interfacial Water Reorganization as a pH-Dependent Descriptor of the Hydrogen Evolution Rate on Platinum Electrodes. Nat. Energy 2017, 2, 17031. 10.1038/nenergy.2017.31. [DOI] [Google Scholar]
- Monteiro M. C. O.; Dattila F.; Hagedoorn B.; García-Muelas R.; López N.; Koper M. T. M. Absence of CO2 Electroreduction on Copper, Gold and Silver Electrodes without Metal Cations in Solution. Nat. Catal. 2021, 4, 654–662. 10.1038/s41929-021-00655-5. [DOI] [Google Scholar]
- Li X.; Gunathunge C. M.; Agrawal N.; Montalvo-Castro H.; Jin J.; Janik M. J.; Waegele M. M. Impact of Alkali Metal Cations and Iron Impurities on the Evolution of Hydrogen on Cu Electrodes in Alkaline Electrolytes. J. Electrochem. Soc. 2020, 167, 106505. 10.1149/1945-7111/ab987b. [DOI] [Google Scholar]
- Hersbach T. J. P.; McCrum I. T.; Anastasiadou D.; Wever R.; Calle-Vallejo F.; Koper M. T. M. Alkali Metal Cation Effects in Structuring Pt, Rh, and Au Surfaces through Cathodic Corrosion. ACS Appl. Mater. Interfaces 2018, 10, 39363–39379. 10.1021/acsami.8b13883. [DOI] [PubMed] [Google Scholar]
- Ojha K.; Arulmozhi N.; Aranzales D.; Koper M. T. M. Double Layer at the Pt(111)–Aqueous Electrolyte Interface: Potential of Zero Charge and Anomalous Gouy–Chapman Screening. Angew. Chem. Int. Ed. 2020, 59, 711–715. 10.1002/anie.201911929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herasymenko P.; Šlendyk I. Wasserstoffüberspannung Und Adsorption Der Ionen [Hydrogen Evolution Overpotential and Adsorption of Ions]. Z. Phys. Chem. A 1930, 149, 123–139. [Google Scholar]
- Frumkin A. N. Influence of Cation Adsorption on the Kinetics of Electrode Processes. Trans. Faraday Soc. 1959, 55, 156–167. 10.1039/tf9595500156. [DOI] [Google Scholar]
- Waegele M. M.; Gunathunge C. M.; Li J.; Li X. How Cations Affect the Electric Double Layer and the Rates and Selectivity of Electrocatalytic Processes. J. Chem. Phys. 2019, 151, 160902. 10.1063/1.5124878. [DOI] [PubMed] [Google Scholar]
- Singh M. R.; Kwon Y.; Lum Y.; Ager J. W.; Bell A. T. Hydrolysis of Electrolyte Cations Enhances the Electrochemical Reduction of CO2 over Ag and Cu. J. Am. Chem. Soc. 2016, 138, 13006–13012. 10.1021/jacs.6b07612. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.