

Abstract
SARS‐CoV‐2 spike protein (S) binds to human angiotensin‐converting enzyme 2 (hACE2), allowing virus to dock on cell membrane follow by viral entry. Here, we use high‐speed atomic force microscopy (HS‐AFM) for real‐time visualization of S, and its interaction with hACE2 and small extracellular vesicles (sEVs). Results show conformational heterogeneity of S, flexibility of S stalk and receptor‐binding domain (RBD), and pH/temperature‐induced conformational change of S. S in an S‐ACE2 complex appears as an all‐RBD up conformation. The complex acquires a distinct topology upon acidification. S and S2 subunit demonstrate different membrane docking mechanisms on sEVs. S‐hACE2 interaction facilitates S to dock on sEVs, implying the feasibility of ACE2‐expressing sEVs for viral neutralization. In contrary, S2 subunit docks on lipid layer and enters sEV using its fusion peptide, mimicking the viral entry scenario. Altogether, our study provides a platform that is suitable for real‐time visualization of various entry inhibitors, neutralizing antibodies, and sEV‐based decoy in blocking viral entry.
Teaser: Comprehensive observation of SARS‐CoV‐2 spike and its interaction with receptor ACE2 and sEV‐based decoy in real time using HS‐AFM.
Keywords: EV, exosomes, High‐speed AFM, SARS‐CoV‐2 spike, ACE2
1. INTRODUCTION
Coronavirus Disease 2019 (COVID‐19), a pandemic caused by a newly emerged beta‐coronavirus (β‐CoV), SARS‐CoV‐2, has brought devastating socioeconomic impacts to many countries (Guan et al., 2020; Huang et al., 2020; Sajidah et al., 2021). SARS‐CoV‐2 is an enveloped, positive sense, single‐stranded RNA virus. Like other enveloped viruses, SARS‐CoV‐2 entry requires membrane fusion, and this process is orchestrated by a group of heavily glycosylated homotrimeric class I fusion proteins, known as spike (S) proteins (Cai et al., 2020). An S protein consists of a globular head S1 subunit and a stalk‐like S2 subunit. The S1 subunit mediates host recognition whereas the S2 subunit manages membrane fusion. The S1 subunit possess four domains: N‐terminal domain (NTD), receptor‐binding domain (RBD), and two structurally conserved subdomains (CTD1 or SD1, and CTD2 or SD2) (Henderson et al., 2020). Heptad repeats (HR1 and HR2) and the highly hydrophobic fusion peptide, important components for membrane fusion, are located in the S2 subunit (Epand, 2003; Tang et al., 2020). The S proteins of both SARS‐CoV‐1 and SARS‐CoV‐2 bind to the same receptor, angiotensin‐converting enzyme 2 (ACE2) (Hoffmann, Kleine‐Weber, & Pöhlmann, 2020; Hoffmann, Kleine‐Weber, Schroeder, et al., 2020; Walls et al., 2020; Yan et al., 2020). The RBD of SARS‐CoV‐2 S protein has higher binding affinity to ACE2 compared with the RBD of SARS‐CoV‐1 S protein (Wang et al., 2020). SARS‐CoV‐2 S protein has two cleavage sites, S1/S2 and S2’. The S1/S2 site is cleaved by a host proprotein convertase, furin, converting S protein into a metastable state (Fuentes‐Prior, 2020). Upon receptor binding, either host surface serine proteases (transmembrane serine protease 2, TMPRSS2) or host endosomal cysteine protease cathepsin B and L (CatB/L) cleaves S protein at the S2’ site to remove the S1 subunit (Hoffmann, Kleine‐Weber, & Pöhlmann, 2020; Hoffmann, Kleine‐Weber, Schroeder et al., 2020). S1 subunit dissociation exposes the S2 subunit and triggers its rearrangement to complete membrane fusion (Cai et al., 2020).
Understanding the protein conformation and dynamic behaviour of S protein is crucial to block SARS‐CoV‐2 entry and expedite synthetic vaccine and target drug development. Several groups have reported that the S protein conformation is heterogeneous, as the protein exists in two different forms depending on the position of the RBD: a receptor‐inaccessible form (closed conformation; all RBDs down) and a receptor‐accessible form (open conformation; at least one RBD up) (Cai et al., 2020; Walls et al., 2020; Wrapp et al., 2020; Wrobel et al., 2020; Xiong et al., 2020). Transition from a closed conformation to an open conformation depends on the interdomain interaction in the S1 subunit, in which the interaction can be perturbed by external factors such as furin cleavage (Wrobel et al., 2020), pH (Zhou et al., 2020), temperature (Rath & Kumar, 2020), and receptor (ACE2) binding (Lu et al., 2020; Zhou et al., 2020). Dynamic interdomain interactions confer the conformational plasticity of S protein, and it is essential for SARS‐CoV‐2 to evade host immunity while retaining S protein fusion ability (Rey & Lok, 2018). Besides the RBD, the conformational dynamics of S stalk have also been resolved by cryo‐electron microscopy (cryo‐EM) imaging. Turoňová et al. (2020) and Ke et al. (2020) revealed that S stalk can bend and rotate on the viral surface. This dynamic property improves receptor accessibility and hence enhances S protein attachment on the host surface. To date, the conformational dynamics of S protein have been mainly dissected by cryo‐EM imaging. Although cryo‐EM is a powerful tool for structural analysis, many cryo‐EM images are rather static snapshots. Furthermore, SARS‐CoV‐2 viruses used for cryo‐EM imaging have to be fixed with 4% paraformaldehyde (Ke et al., 2020; Turoňová et al., 2020) and the conformational dynamics of S protein may therefore not be fully recapitulated. Single‐molecule fluorescence (Förster) resonance energy transfer (smFRET) has a high temporal resolution in capturing the conformational dynamics of S protein (Lu et al., 2020); however, this approach only detects fluorescent signals.
High‐speed atomic force microscopy (HS‐AFM) is feasible for real‐time observation of biomolecules with high spatiotemporal resolution at nano‐scale range. We have performed biomolecular imaging using HS‐AFM for structural characterization, observation of conformational dynamics, and to reveal the dynamic interactions of biomolecules and organelles (Ando, 2019; Mohamed et al., 2017; Nishide et al., 2021). Recently, we used HS‐AFM to investigate the conformational dynamics of influenza A fusion protein, the hemagglutinin (Lim, Mohamed et al., 2020; Lim, Kodera et al., 2020). In this study, we used HS‐AFM to observe the native conformation and conformational dynamics of SARS‐CoV‐2 S ectodomain in different physicochemical conditions, ACE2 binding, and interaction with small extracellular vesicles (sEVs). In addition, we also studied the interaction between S2 subunit and sEVs.
2. MATERIALS AND METHODS
2.1. Materials
Recombinant proteins including SARS‐CoV‐2 S protein (S1+S2 ectodomain; 40589‐V08B1), SARS‐CoV‐2 spike S2 subunit ectodomain (40590‐V08B), and human ACE2 ectodomain (10108‐H08B) were purchased from Sino Biological Inc (Beijing, China). These recombinant proteins were reconstituted in Milli‐Q water at 0.25 mg/ml and then further diluted with appropriate buffers for HS‐AFM recording.
2.2. Cell line
PC‐9 cell line was purchased from the Immuno‐Biological Laboratories Co (Gunma, Japan). HEC50B and A549 cell lines were purchased from the Japanese Collection of Research Bioresources (JCRB) Cell Bank (Osaka, Japan). HEC50B was cultured in Minimum Essential Medium (MEM; Nacalai Tesque, Kyoto, Japan) with 10% foetal bovine serum (FBS; Life Technologies, CA, USA) and 1% Penicillin‐Streptomycin (Nacalai Tesque, Kyoto, Japan). A549 and PC‐9 were maintained in Roswell Park Memorial Institute (RPMI) 1640 culture medium (Gibco, MA, USA) supplemented with 10% FBS and 1% Penicillin‐Streptomycin. All cell lines were cultured at 37°C in a humidified CO2 incubator.
2.3. Preparation, purification, and characterization of sEV
sEVs of HEC50B, PC‐9, and A549 were obtained by using the protocol mentioned in our previous study (Lim, Mohamed et al., 2020; Lim, Kodera et al., 2020). Briefly, cells were cultured for 4 days, and then cell‐conditioned medium was subjected to sequential centrifugation to remove cells, debris and large EVs. Supernatant was then filtered through a 0.22 μm Millex‐GV filter (Merck Millipore, MA, USA) to produce a final pre‐cleared cell conditioned medium, designated as the 10K sup. After that, sEVs in the 10K sup were purified by using a MagCapture Exosome Isolation Kit PS (Wako, Osaka, Japan) according to the manufacturer's manual. The purified sEVs were then transferred into a dialysis membrane (3500 MWCO) for overnight dialysis in PBS (‐). Concentration of sEV was measured by nanoparticle tracking analysis (NTA) using the NanoSIGHT LM10 (Malvern Panalytical, Malvern, UK). For detecting exosome markers, first, HEC50B, PC‐9, and A549 cells were lysed with RIPA buffer. Then, cell lysates were collected by centrifugation at 15,000 × g for 30 min. Protein concentration of all samples were measured using BCA protein assay kit (Thermo Fisher Scientific, MA, USA). 400 ng of sEV and cell lysate were used for SDS‐PAGE and western blotting. We follow the Minimal Information for Studies of Extracellular Vesicle 2018 (MISEV2018) guidelines (Théry et al., 2018) and have submitted all relevant data of our experiments (EV‐TRACK ID: EV210256) to the EV‐TRACK knowledgebase (Van Deun et al., 2017).
2.4. Furin cleavage of S protein
Furin cleavage was carried out by incubating furin (P8077, NEB, MA, USA) with recombinant S proteins in a reaction buffer at 25°C for 1 h. The reaction buffer was made of 50 mM HEPES‐KOH (pH7.4), 150 mM NaCl, 5 mM MgCl2, and 2 mM CaCl2. A negative control (without furin treatment) was prepared. Samples were added with 4× SDS sample buffer and heated at 95 °C for 10 min to stop furin activity, then proceeded to SDS‐PAGE and Western blotting.
2.5. SDS‐PAGE and Western blotting
Protein samples were separated in 5%–20% SDS polyacrylamide gels and then transferred onto PVDF membranes. Blocking was performed using 5% skim milk in PBST, followed by overnight primary antibody incubation at 4°C. Primary antibodies used in this study were SARS‐CoV‐2 spike antibody (40591‐MM42; Sino Biological Inc, Beijing, China), antibodies for exosome markers including CD9 (312102; BioLegend, CA, USA), CD63 (353039, BioLegend), CD81 (349502, BioLegend), TSG101 (GTX118736, GeneTex, CA, USA) and VPS37B (HPA038217; ATLAS ANTIBODIES, Stockholm, Sweden), antibody for cellular marker GAPDH (M171‐3; MBL, Nagoya, Japan). After primary antibody incubation, PVDF blots were incubated with secondary horseradish‐conjugated anti‐mouse (Cell Signaling Technology, MA, USA) or anti‐rabbit (406401, BioLegend) at room temperature for an hour. Finally, Western blot images were developed using horseradish peroxidase (Millipore, MA, USA), and captured using an image analyser LAS 4000 (Fujifilm, Tokyo, Japan).
2.6. Preparation of 3D protein structure and simulated HS‐AFM images
The PDB files of S protein, spike S2 subunit, ACE2, and S protein‐ACE2 complex were obtained from the RCSB PDB website (https://www.rcsb.org). PyMOL (The PyMOL Molecular Graphics System, Version 2.1.0; Schrödinger Inc, NY, USA) was used to visualize the 3D protein structures and to label important domains of these proteins. BioAFMviewer software (WPI Nano LSI) (Amyot & Flechsig, 2020) was used to generate simulated 3D HS‐AFM images of these proteins.
2.7. Calculation of protein net charge and generation of surface electrostatic map
Total net charges of S protein, spike S2 subunit, and ACE2 at extracellular pH (7.4) were calculated using Prot Pi website (https://www.protpi.ch/). PDB2PQR (Dolinsky et al., 2004) and APBS (Adaptive Poisson‐Boltzmann Solver) (Baker et al., 2001) analysis were performed in the APBS‐PDB2PQR webserver (https://server.poissonboltzmann.org/). After that, 3D surface electrostatic maps of these proteins were generated using the PyMOL software.
2.8. Per‐residue prediction of intrinsic disorder tendency
Prediction of the intrinsic disorder tendency in S protein and ACE2 were conducted using seven predictors including members of the PONDR (Predictor of Natural Disordered Regions) family such as PONDR VLXT (Romero et al., 2001), PONDR VL3 (Peng et al., 2005), PONDR VSL2 (Peng et al., 2006), PONDR FIT (Xue et al., 2010), IUPred for predicting long (≥30 residues) (Erdős & Dosztányi, 2020) and short IDPRs (<30 residues) (Mészáros et al., 2018), and PrDos (Ishida & Kinoshita, 2007). Residues with predicted disordered propensity scores exceeding the threshold value of 0.5 are considered as intrinsically disordered residues. In contrast, residues with score in between 0.2 and 0.5 are considered flexible. Complete predicted percentage of intrinsic disordered (PPID) and complete predicted percent of flexibility (PPF) of these proteins were computed from outputs of seven predictors. In addition, mean PPID and mean PPF were also calculated by averaging the outputs. Intrinsically disordered residues of S protein and ACE2 were labelled in their respective 3D structure using the PyMOL software.
2.9. Generation of sharp cantilever tip by using electron‐beam deposition (EBD)
BL‐AC10DS‐A2 cantilever was purchased from Olympus (Tokyo, Japan), and used as a scanning probe to image recombinant proteins and sEVs. The cantilever has a spring constant (k) of 0.1 N/m and a resonance frequency (f) of 0.6 MHz in water (1.5 MHz in air). The dimension of the cantilever is 9 μm (length), 2 μm (width), and 0.13 μm (thickness). EBD generates a long, sharp, and small apical radius tip on a cantilever to enhance image resolution. First, a cantilever was cleaned by UV/O3 and then in piranha solution, containing sulfuric acid and hydrogen peroxide. Next, EBD was performed on the cantilever at 30 kV accelerating voltage and 2 min of irradiation using a field emission scanning electron microscope, ELS‐7500 (Tokyo, Japan).
2.10. HS‐AFM visualization
HS‐AFM images were obtained with our laboratory‐build HS‐AFM microscope, as previously described (Lim, Mohamed et al., 2020; Lim, Kodera et al., 2020). A laser beam (λ = 670 nm) passed through a 20× objective lens (CFI S Plan Fluor ELWD, Nikon, Tokyo, Japan) and focused on an EBD‐processed cantilever tip. The dynamic cantilever deflection was detected by sensing the position of the laser beam reflected by the cantilever with a position‐sensing two‐segmented photodiode. A small tip‐sample loading force, which is crucial to protect sample structure, was generated by tuning the free oscillation amplitude of the cantilever (A0) to 1.5–2.5 nm and the set point to 80%–90% of the free amplitude. A glass stage glued with a stack of muscovite mica layers (∼0.1 mm thickness) was mounted on the HS‐AFM scanner. The muscovite mica layers served as a substrate for adsorbing target proteins and sEVs.
To scan native conformation of S protein, spike S2 subunit, and ACE2, recombinant proteins diluted with a scanning buffer (50 mM Tris HCl, 150 mM NaCl, pH7.4) were loaded on a suitable mica substrate and then incubated for 10 min. After that, proteins were washed twice with the scanning buffer and proceeded to HS‐AFM scanning. Bare mica was used as a substrate to adsorb S protein and spike S2 subunit whereas nickel (Ni2+)‐coated mica was used to adsorb ACE2. To prepare nickel (Ni2+)‐coated mica, 3 μl of 0.5 M nickel chloride was added on bare mica and incubated for 15 min, then washed twice with the scanning buffer.
To observe conformational dynamic of S protein at different pH, neutral scanning buffer (50 mM sodium acetate, pH 7.4) was replaced with acidic scanning buffer (50 mM sodium acetate, pH 5) during HS‐AFM scanning. For neutralization, the acidic scanning buffer was then replaced with the neutral scanning buffer during the scanning.
To visualize the topology of S protein after exposed to different temperatures, S protein was first either incubated at room temperature, 37°C, 60°C, or 90°C for 15 min. After that, the S protein was loaded on bare mica for HS‐AFM observation under a neutral scanning buffer (50 mM Tris HCl, 150 mM NaCl, pH7.4).
For real‐time observation of S protein‐ACE2 interaction, empty nickel (Ni2+)‐coated mica was scanned to obtain background image. After that, ACE2 was added into chamber containing scanning buffer (50 mM sodium acetate pH7.4). Once the nickel (Ni2+)‐coated mica had been filled with ACE2, ACE2‐containing scanning buffer was replaced with a plain scanning buffer to remove excess ACE2. Lastly, S protein was added into chamber during HS‐AFM scanning to observe the S protein‐ACE2 interaction in a real‐time fashion. To observe the conformational dynamic of S protein‐ACE2 complex at low pH, the neutral scanning buffer was replaced with an acidic scanning buffer (50 mM sodium acetate pH5). In the premix approach, S protein and ACE2 were mixed and incubated in a scanning buffer (50 mM Tris HCl, 150 mM NaCl, pH7.4) for 30 min. After that, sample were loaded on bare mica for HS‐AFM scanning.
To visualize S protein‐ or S2 subunit‐sEV interaction in a real‐time manner, sEVs were first loaded on a poly‐l‐lysine (PLL)‐coated mica and incubated for 10 min. After that, sEVs were washed with a scanning buffer (50 mM Tris HCl, 150 mM NaCl, pH7.4). Once an sEV had been found, either S protein or S2 subunit was added into chamber during HS‐AFM scanning. For premix experiment, S protein or S2 subunit and sEVs were incubated at room temperature for 3 h prior to HS‐AFM imaging. A negative control set (sEV only) was also prepared.
All HS‐AFM scanning experiments were repeated at least three times to ensure the reproducibility of the experiments. Since the same batches of recombinant proteins were used for HS‐AFM scanning, repeated measurements were considered to be technical replicates.
2.11. Force curve measurement
sEVs were incubated with S2 subunit at room temperature for 3 h before AFM imaging. Negative control (sEV only) was also prepared. Then, samples were immobilized on PLL‐coated mica substrate, which was previously affixed on a 3‐cm petri dish. Scanning buffer (50 mM Tris HCl, 150 mM NaCl, pH7.4) was used. A qp‐BioAC CB3 cantilever (Nanosensors, Neuchatel, Switzerland) with a tip (tip radius < 10 nm, spring constant: 0.09 N/m, resonance frequency: 23–37 kHz in air) was equipped on a Nanowizard‐II AFM scanner (JPK Instrument, Berlin, Germany) combined with an AxioObserver D1 inverted optical microscope (Carl Zeiss, Germany) to image sEVs and to measure force–distance curve of sEV membrane. The AFM setting was identical for all force mapping measurement. The imaging force was 0.1–1 nN, time per curve was 0.5 s, Z‐length was 0.5 μm, and speed of curve acquisition was 1.0 μm/s. The optical lever sensitivity and the spring constant of the individual probes in liquid were obtained from the force curve and the thermal noise measurements on the JPK SPM software. All images and force–distance curves (FDCs) were analysed using the JPK Data Processing software. Extend segment of each FDC was fitted to a Hertzian/Sneddon model modified for a paraboloid AFM probe to obtain the Young's modulus value. The Poisson ratio of sEVs were assumed to be constant, with a value of 0.5. Mean Young's moduli between control group and S2 subunit‐sEV group were compared using SPSS software.
2.12. Analysis of HS‐AFM images
All HS‐AFM images were processed by using the ImageJ software (https://imagej.nih.gov/ij/). The HS‐AFM images were first filtered by a fit polynomial filter (order for both x‐and y‐directions: 1) then by a low pass filter (Gaussian blur, Sigma radius: 2 nm) to suppress noise and enhance image features. For some HS‐AFM images, a Fast Fourier Transform (FFT) bandpass filter was used to perform pixel filtration to remove both large and small structures in order to increase image details. A 3D surface plot plugin in the ImageJ was used to generate 3D images of the recorded HS‐AFM images. Besides image processing, parameters such as cross‐sectional height, area, and circularity were also measured by using the same software. A circularity value of 1.0 represents a perfect circle. Values approaching zero indicate an increasingly elongated polygon. The circularity of S protein and sEV is given by 4πSL−2, where L and S are the perimeter and the area surrounded by the object, respectively. The processed images were converted to videos (AVI format). These videos were then arranged, edited, and compiled using Adobe Premiere Pro CS6 software (Adobe).
2.13. Statistical analysis
Graphs for descriptive statistics were plotted using GraphPad Prism version 7 (GraphPad, CA, USA). Statistical analysis was performed using SPSS version 22 (IBM Group, NY, USA). Comparative study of two‐related samples was performed using the Wilcoxon Signed‐Rank test. Mann‐Whitney U test and Kruskal‐Wallis H test were conducted to compare independent samples. The statistically significant level was set at p < 0.05 with a confidence interval of 95%. Sample size was determined based on our earlier similar study (Lim, Mohamed et al., 2020; Lim, Kodera et al., 2020).
3. RESULTS
3.1. Native conformation of SARS‐CoV‐2 S protein as visualized by HS‐AFM
Before HS‐AFM imaging, we first obtained simulated HS‐AFM images of S protein (Figure 1a), determined a suitable mica substrate (Figure 1b and c), and predicted the potential intrinsically disordered region (IDR) in S protein (Figure 1d). To generate simulated HS‐AFM images of S protein in closed and open conformations, we downloaded PDB files of the cryo‐EM structure of S protein from the RCSB PDB data bank (PDB code, Closed: 6XR8 (Cai et al., 2020), Open 1‐Up: 6VSB (Wrapp et al., 2020), Open 2‐Up: 7A93 (Benton et al., 2020)). Simulation was performed using the BioAFMviewer software (Amyot & Flechsig, 2020). The protein surface charge is a key parameter for S protein adsorption on a mica substrate. We calculated the net charge (Figure 1c) and computed a surface electrostatic potential map (Figure 1b) (Baker et al., 2001; Dolinsky et al., 2004) for S protein, which indicated that bare mica was feasible for S protein adsorption with sufficient mobility at pH 7.4 in the presence of salt (150 mM NaCl). A single protein could comprise ordered and disordered domains. These structural properties could affect the protein topology acquired by HS‐AFM imaging. To determine the predisposition of IDR in S protein, we performed IDR prediction using seven IDR predictors, including members of PONDR (PONDR VLXT, PONDR VL3, PONDR VSL2, PONDR FIT), the IUPRED platform (IUPREDLong and IUPREDShort), and PrDos. The output of the seven predictors together with the average of the outputs were illustrated in Figure 1d and Table S1‐3. S was a highly ordered and flexible protein (mean PPID: 1.59%; mean PPF: 31.89%). The S1 subunit had a higher mean PPID than S2 subunit (mean PPIDS1 vs. PPIDS2: 2.53% vs. 0.39%) and S2 subunit was more flexible than S1 subunit (mean PPFS2 vs. PPFS1: 41.41% vs. 23.85%). Two predicted IDRs were highlighted in red and yellow in the 3D structure of S protein (Figure 1d). The first IDR (yellow) was located at the N‐terminal domain (NTD; residues 16–24; mean disorder score ± SD: 0.566 ± 0.038). The second IDR (red) was located at the furin cleavage site (residues 679–688; mean disorder score ± SD: 0.526 ± 0.016). Previous studies had also reported that a furin cleavage site exists as a disordered flexible loop (Wrapp et al., 2020), and this structure favours furin binding. Direct visualization of the S protein in a physiological buffer revealed a globular head and slender stalk topology (Figure 1e and f, Figure S1, B and C, and Movies S1 and S2), comparable to the simulated HS‐AFM images (Figure 1a). We managed to record both closed and open conformations of S protein using HS‐AFM (Figure 1e, Figure S1C, and Movie S2). Similarly, such conformational heterogeneity was also found in formalin‐fixed S protein (Figure S1D and Movie S3). 3D surface plots of both native and fixed S proteins demonstrate the hallmark crown‐like topology of S protein (Figure S1E). Western blotting results implied that S proteins were partially cleaved by innate furin, which is expressed in insect cells, during recombinant protein synthesis (Figure S1F). Therefore, we managed to visualize S proteins in an open conformation without pre‐treating S protein with furin. Using HS‐AFM, the conformation dynamics of S protein in different conformations could be analysed at the single‐molecule level. This idea is similar to single‐cell analysis in which the sample pool is heterogeneous.
FIGURE 1.
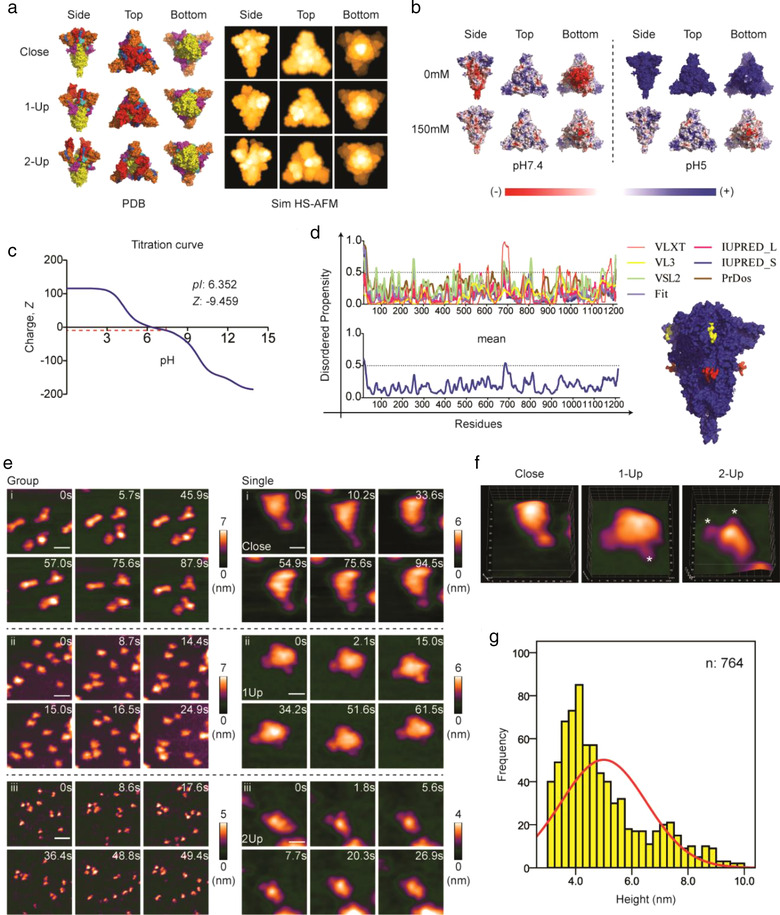
HS‐AFM reveals three distinct conformations of SARS‐CoV‐2 S proteins. (a) Cryo‐EM structures of S protein in close (PDB: 6XR8), 1‐Up (PDB: 6VSB), and 2‐Up (PDB: 7A93) conformations as shown in their 3D structures. An explanation of the colours used is available in Figure S1A. These 3D structures were used to simulate HS‐AFM images (Sim HS‐AFM). (b) PDB2PQR and APBS analysis of S protein were performed to generate surface electrostatic maps at different salt concentrations and pH. (c) Titration curve computed from the Prot pi website using S protein amino acid sequence. The isoelectric point and net charge at pH 7.4 are shown. (d) Prediction of intrinsic disorder region (IDR) predisposition in S protein. The predicted IDR regions are labelled in red and yellow as shown in a 3D structure of S protein. (e) HS‐AFM images showing a group or a single S protein at extracellular pH (scale bar, group, i: 25 nm, ii: 45 nm, iii: 60 nm; single, i: 12 nm, ii: 18 nm, iii: 20 nm). (f) 3D‐surface plot of the S protein in three distinct conformations (* indicates RBD). (g) Height histogram of the S protein
3.2. HS‐AFM imaging reveals the dynamic behaviour of the stalk and RBD of S protein
Previous cryo‐EM findings provided important clues about the dynamics of S stalk in tilting and rotating on the viral surface (Ke et al., 2020; Turoňová et al., 2020; Yao et al., 2020), but the results only represented the dynamics of formalin‐fixed S stalks. In addition, the dynamics of S stalk have been depicted using molecular dynamic simulations (Ke et al., 2020; Turoňová et al., 2020). Direct observation of S proteins on a SARS‐CoV‐2 virion using AFM is feasible (Kiss et al., 2021), yet the image resolution is not ideal for analysis of the conformational dynamics of S stalk. HS‐AFM provides a high spatiotemporal resolution in real‐time imaging that allows us to study the native conformational dynamics of S stalk without fixation. As predicted, the HS‐AFM movie demonstrated the highly dynamic S stalk and the less dynamic S head (Movie S2). The topology of the RBD loop resembled a short tentacle protruding out of the S1 subunit head. We found that the RBD was dynamic but to a lesser extent than the S stalk. This finding is in accordance with our flexibility prediction (mean PPFRBD vs. mean PPFS2: 22.22% vs 41.41%) (Table S2 and S3). We calculated the tilted angle relative to the axis that is perpendicular to the horizontal axis (Figure 2a and Figure S2A). The horizontal axis could be considered as the viral membrane surface. Therefore, dynamic movement of S stalk during HS‐AFM scanning mimicked the dynamic tilting of S protein on the viral surface. We designated a negative tilted angle as left tilt, and a positive tilted angle as right tilt. Our results demonstrate that S stalk tilting was bidirectional. The tilted angle was able to reach close to 90°, indicating that S protein could be tilted to touch the viral surface (Figure 2a). Besides tilting, we found that the S stalk domain was able to extend or retract during HS‐AFM scanning (Figure 2a and Figure S2A). This dynamic behaviour could imply that S protein was able to move upward or downward on the viral surface. We further investigated the conformational dynamics of the RBD loop (Figure 2b, and Figure S2, B and C). Similar to S stalk, the RBD loop tilted and extended or retracted during HS‐AFM scanning (Figure 2b and Figure S2, B and C). Intriguingly, the RBD loop intermittently disappeared during HS‐AFM scanning, when its loop length became zero (Figure S2, B and C). This finding suggested that interdomain interactions in the S1 subunit may play a role in spontaneous conformation masking to hide the RBD. The tilting orientation of an S protein with two RBDs facing upwards was predominantly unidirectional tilting, which may be due to the repulsive force exerted when two RBDs approaching each other.
FIGURE 2.
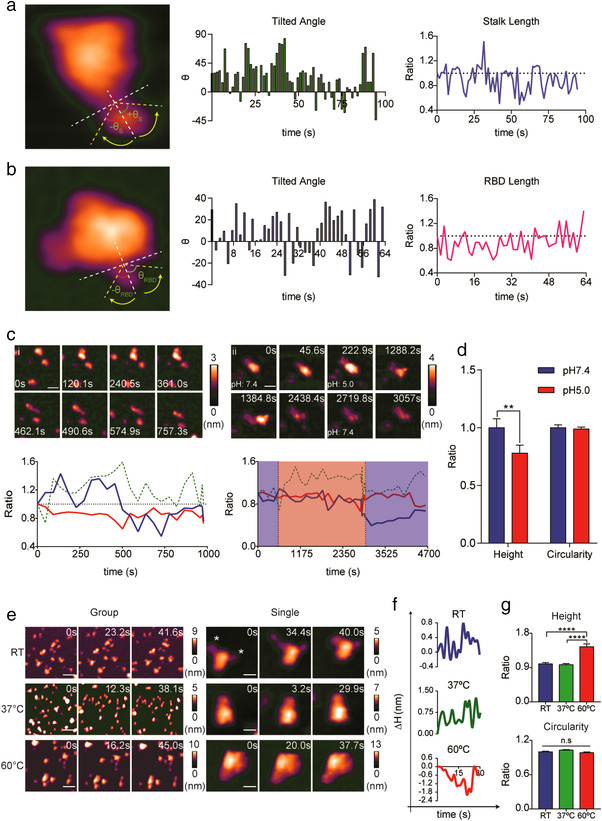
Biophysical and morphological properties of the S protein. (a, b) HS‐AFM scanning revealed a flexible S stalk (a) and RBD (b). Real‐time tilted angle and length change are shown in bar graphs and line graphs respectively (length ratio: length t = n/length t = 0). (c) Dissociation of the S head (i and ii) and elongation of the S stalk (i) upon acidification (scale bar, i: 30 nm, ii: 25 nm). Real‐time ratio changes in height (h t = n/h t = 0), area (A t = n/A t = 0), and circularity (C t = n / C t = 0) were calculated and are illustrated in line graphs (blue: height, green dotted: area, red: circularity; blue area: pH 7.4, red area: pH5). (d) Low pH induced significant reduction in S protein height. Data are presented as the mean ratio ± SEM of height before and after acidification, analysed by Wilcoxon Signed‐Rank test (n: 21; p = 0.0057, **p < 0.01). (e) Topology of S protein after exposed to different temperatures (scale bar, group, RT: 60 nm, 37°C: 75 nm, 60°C: 60 nm; single: RT and 37°C: 15 nm, 60°C: 20 nm). (f) Real‐time height changes (Δ h = h t = n ˗ h t = 0) in the S protein exposed to different temperatures. (g) High temperatures significantly increased the height of the S protein. Both height and circularity values are expressed in ratios relative to the samples treated at RT. Data are presented as the mean ratio ± SEM (n, RT: 94, 37°C: 80, 60°C: 104; ****p < 0.0001, ns: no statistically significant)
3.3. Effect of pH and temperature on S protein conformation
Next, we investigated the effects of two physicochemical factors (pH and temperature) on S protein conformation. Low pH is not the primary triggering factor for S protein to undergo fusogenic transition. Nevertheless, it could promote the evasion of SARS‐CoV‐2 from neutralizing antibodies via antibody shedding (Zhou et al., 2020). Using HS‐AFM, we aimed to study the conformational dynamics of S protein induced by acidification followed by neutralization. For S protein in a closed conformation, we observed dissociation of the S head after acidification (Figure 2c, Figure S3A, and Movie S4, A‐C). Following S head dissociation, the remaining S stalk underwent elongation at low pH (Figure 2ci and Movie S4A). In Figure 2 cii (see also Movie S4B), we only visualized dissociation of the S head but no elongation of S stalk because we performed neutralization shortly after dissociation of the S head. HS‐AFM images in Figure S3B (see also Movie S4D) were captured in the middle of the acidification process, the early phase of acidification was observed in another scanning area, so we were only able to observe the elongation process. We found that elongation of S stalk was a reversible process upon neutralization. Multiple S stalks changed their conformation from globular to slender rods at low pH, then reverted back to their original globular conformation after neutralization. For S protein in open conformation, dissociation of the S head did not occur. Instead, our data demonstrated that the frequency of RBD appearance was gradually reduced after exposure to low pH conditions for a prolonged period of time (Figure S3C and Movie S5). Surprisingly, the RBD was always visible after neutralization. The real‐time height and circularity profiles of S protein in both the closed and open conformations appeared to follow a similar trend, with both height and circularity decreasing during acidification, which was then reversed upon neutralization. We further analysed the effects of acidification on a group of S proteins and found that the height but not the circularity of S protein was significantly reduced (Figure 2d). These findings implied that decreasing height was a general effect, whereas the loss of circularity only occurs in S proteins with conformational changes. The decrease in height may be explained in two ways. First, as mentioned above, dissociation of the S1 subunit reduced S protein height. Second, protonation of S protein rendered its surface charge more positive (Figure 1b), strengthening its adsorption onto a bare mica surface via electrostatic interactions. Consequently, the height of S protein becomes reduced. This further explains why S protein regains its height upon neutralization because its surface charge becomes less positive, thereby weakening its adsorption onto a bare mica surface.
To investigate the temperature effect on S protein conformation, we heated S proteins at 37°C (S37°C), 60°C (S60°C), and 90°C (S90°C) for 15 min before HS‐AFM scanning. S protein incubated at room temperature (SRT) was used as a control. S proteins were found to be denatured at 90°C and failed to adsorb onto the mica surface (Figure S4A), whereas both S37°C and S60°C proteins abundantly attached onto the mica surface (Figure 2e, Figure S4A, and Movie S6, C‐F). Surprisingly, we found that S37°C proteins forming a thread‐like pattern (Figure S4, A and B, and Movie S6D), which was not observed with the SRT (Figure 2e, Figure S4A, and Movie S, 6A and B) and S60°C (Figure 2e, Figure S4A, and Movie S6, E and F) proteins. Open conformations were found for the S37°C proteins (Figure S4C, 37°C1 and 37°C2, and Movie S7, E and F). Two RBDs protruded out from the S37°C protein during scanning (Figure S4C, 37°C2 and Movie S7F). Although S60°C protein retained its spike conformation, most of the proteins appeared in a closed conformation (Figure 2e, Figure S4C, and Movie S7, G, H, and I). We plotted the real‐time height change profile (ΔH; ΔH = Ht = n ˗ Ht = 0) of the three groups of S proteins (SRT, S37°C, and S60°C) and found that S protein could have changed its elasticity as the temperature increased (Figure 2f and Figure S4D). In addition, a high temperature could promote S protein expansion as the height of S60°C was significantly higher than that of SRT and S37°C (Figure 2g).
3.4. Conformation dynamics of S protein in the S‐ACE2 interaction observed by HS‐AFM in real‐time
The S protein–ACE2 interaction primes S protein for cleavage by host proteases, releasing S1 subunit and exposing the buried fusion peptide, followed by the rearrangement of S2 subunit to orchestrate membrane fusion. Simulated HS‐AFM images of the ACE2 ectodomain were computed based on its 3D‐model, which was generated by homology modelling using a cryo‐EM structure template (PDB: 6M18 (Yan et al., 2020)). ACE2 consists of a globular N‐terminal peptidase domain (PD) and a tail‐like structure C‐terminal collectrin‐like domain (CLD) (Figure S5A). The net charge of ACE2 at pH 7.4 was highly negative (Z = ˗24.13), and negative charges were mainly distributed at the PD (Figure S5, B and C). IDR prediction analysis showed that ACE2 is a highly ordered and highly flexible protein (PPID: 2.4%; PPF: 42.6%) (Table S4). The IDR region of ACE2 is located at the CLD (Figure S5D). For ACE2 adsorption, a Ni2+‐coated mica substrate was used. Specific interaction between polyhistidine tag in C‐terminus of ACE2 and Ni2+ enabled ACE2 adsorbed on Ni2+‐coated mica substrate in an orientation similar to ACE2 on cell membrane. Therefore, majority of ACE2 appeared in their top view (globular) in HS‐AFM imaging (Figure 3c, Figure S5, E and F, and Movie S8). A small portion of ACE2 laterally adsorbed on the nickel (Ni2+)‐coated mica substrate due to its high negatively charges at neutral pH. In this orientation, both PD and CLD are clearly visible on HS‐AFM images (Figure S5Fi and Movie S8D). To visualize the interaction between S proteins and ACE2, we first computed simulated HS‐AFM images of single‐, double‐, and triple‐ACE2‐bound S proteins (Figure 3a) according to their respective cryo‐EM models (Benton et al., 2020) (PDB: single: 7A94; double: 7A97; triple: 7A98). The S protein–ACE2 interaction takes place at the PD region (blue) of ACE2 (Figure 3c). We deleted the S2 subunit in the triple‐ACE2‐bound S protein structure (PDB: 7A98) to simulate a triple‐ACE2‐bound S1 subunit complex, considering that S2 subunits might dissociate after S protein bound to the ACE2‐coated mica surface during HS‐AFM scanning (Figure 3b). Visualization of the S–ACE2 interaction was carried out via two approaches: real‐time observation and premix incubation followed by HS‐AFM scanning. The results of real‐time observation of the S–ACE2 interaction are illustrated in Figure 3d–g (see also Movie S9). We first added ACE2 protein into the chamber during HS‐AFM scanning. ACE2 proteins approached and adhered to the nickel (Ni2+)‐coated mica substrate surface over time (Figure 3di and Movie S9, ACE2 Filling). After ACE2 proteins had covered the substrate, excessive ACE2 was removed. Finally, we added S proteins into the chamber and observed the interaction in real‐time for about 13 min (800 s). S proteins were able to bind to the ACE2‐coated mica surface, but we did not detect any noticeable change in S protein conformation throughout the scan (Figure 3dii and Movie S9, S protein added). ACE2‐bound S proteins appeared to be either in a 2‐Up or a 3‐Up open conformation (Figure S6A). After observation of the S–ACE2 interaction at pH 7.4, we then performed acidification to observe the conformational dynamics of ACE2‐bound S proteins (Figure 3diii and Movie S9, Acidification). We further focused on two areas, designated as “a” and “b”, to closely observe the conformation dynamics of S protein starting from the addition of S protein to acidification of S protein (Figure 3e and Movie S10). A noticeable rapid drop in both the height and area of ACE2‐bound S proteins occurred (Figure 3f and Movie S10). We did not observe dissociation of the S head throughout the acidification event (Movie S10). At higher magnification, we observed several objects with similar topologies resembling five small balls connected to a central ball (Figure 3, Div and g, and Movie S11). A schematic diagram is illustrated in Figure S6B to depict low pH‐induced conformation transition in S‐ACE2 complex. Since the endosomal entry route of SARS‐CoV‐2 is mainly mediated by cysteine proteases (cathepsin B/L) (Hoffmann, Kleine‐Weber, & Pöhlmann, 2020; Hoffmann, Kleine‐Weber, Schroeder et al., 2020), we postulated that this structure could be the intermediate conformation of a triple‐ACE2‐bound S protein after acidification, priming the protease cleavage to reveal S2 subunit to complete membrane fusion. In the premix experiment, we only observed the topology of the S–ACE2 complex at pH 7.4. We found that S proteins predominantly appeared in a 3‐Up open conformation (Figure 3h and Movie S12). In real‐time observations, ACE2‐bound S protein could appear in a 3‐Up open conformation, yet the contact point was beneath the S protein, making it appear like a 2‐Up open conformation. The S stalk remained mobile after the S protein bound to ACE2 (Figure 3i and Movie S13). This result may imply that the mobility of S protein on the SARS‐CoV‐2 surface enables the virus to readily interact with the adjacent ACE2, maximizing its attachment on the target cell surface.
FIGURE 3.
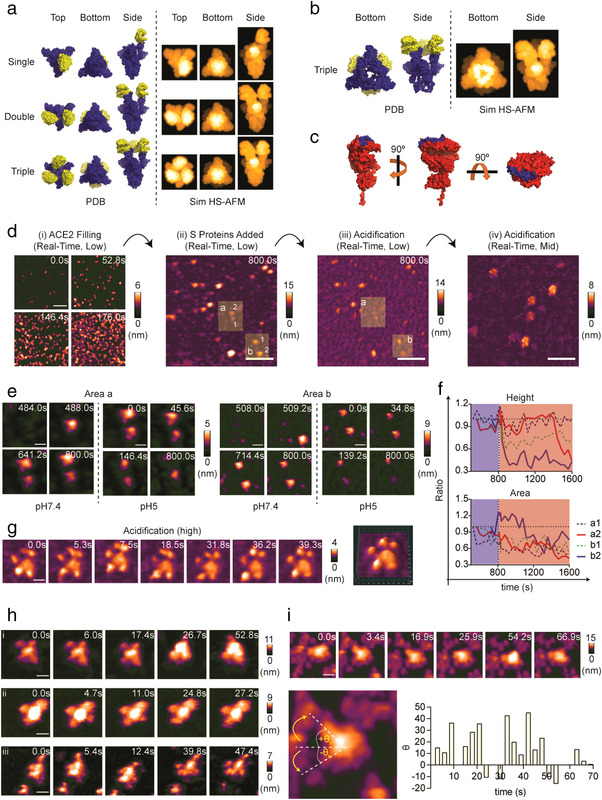
Conformational dynamics of the S protein when interacting with monomeric ACE2. (a) Cryo‐EM structures of single‐ (PDB: 7A94), double‐ (PDB: 7A97), and triple‐ACE2 (PDB: 7A98)‐bound S proteins are illustrated as molecular surface diagrams (blue: S protein; yellow: monomeric ACE2). These structures were used to simulate their respective HS‐AFM images. (b) Cryo‐EM structure of the triple‐ACE2‐bound S protein (PDB: 7A98) was edited by removing the S2 subunit to simulate the HS‐AFM image of the triple‐ACE2‐bound S1 subunit. (c) Cryo‐EM structure of the monomeric ACE2 ectodomain (PDB: 6M18) is depicted in the molecular surface diagram. The blue region indicates the binding site of RBD of S protein. (d) Real‐time observation of the S protein–ACE2 interaction. HS‐AFM observations were started at low magnification with ACE2 filling on the nickel (Ni2+)‐coated mica. Extra ACE2 was removed, followed by the addition of S protein. Real‐time interactions between S protein and ACE2 at pH 7.4 were observed for 800 s, followed by acidification for another 800 s. Then, multiple sites were screened at mid magnification for the S protein–ACE2 complex under acidic conditions (scale bar, i: 150 nm, ii and iii: 250 nm, and iv: 125 nm). (e) Two areas (a and b) cropped from the figures in d(ii) and d(iii) delineated the real‐time conformational dynamics of S protein during interaction with ACE2 from neutral to acidic conditions (scale bar, Area a: 30 nm; Area b: 60 nm). (f) Real‐time height and area changes in the S protein in E were measured and are presented as ratios relative to their respective initial height (ht = 0) and area (At = 0). The results showed rapid declination in height and area upon acidification (blue area: pH 7.4, red area: pH 5). (g) S protein–ACE2 conformation after acidification for 800 s as shown by high magnification HS‐AFM scanning (scale bar: 13 nm). (h) HS‐AFM images of the S protein–ACE2 complex after incubation of S protein and ACE2 for 30 min at room temperature (scale bar, i: 25 nm; ii: 38 nm; iii: 25 nm). (i) HS‐AFM observation revealed the ACE2‐bound S protein with a dynamic S stalk (scale bar: 30 nm). Tilted angles were measured and are plotted in a bar graph
3.5. S2 subunit disrupted the lipid bilayer stability of the sEV
Previously, we successfully recorded the real‐time interaction between HA of human influenza A virus and sEVs under low pH conditions using HS‐AFM (Lim, Mohamed et al., 2020; Lim, Kodera et al., 2020). Our results clearly showed the insertion of HA into the sEV lipid layer after fusogenic transition. Using a similar approach, we observed the interaction between the soluble S2 subunit and sEVs because the S2 subunit possesses the lipophilic fusion peptide. To observe the native conformation of the soluble S2 subunit, we first prepared simulated HS‐AFM images of the S2 subunit (Figure S7A) using two cryo‐EM structures (PDB: 6ZWV for S2 homology modeling (Ke et al., 2020) and 6XRA (Cai et al., 2020)) and determined its net charge and surface electrostatic potential at pH7.4 (Figure S7, B and C). Then, we scanned the S2 subunit on bare mica in a physiological scanning buffer (50 mM Tris HCl, 150 mM NaCl, pH7.4). The S2 conformation was heterogeneous, either presenting as a slender rod or a bulky rod (Figure S7, D–F, and Movie S14). Such heterogeneity could be due to rearrangement of the S2 subunit (from the bulky rod to the slender rod). The S2 subunit actively interacted with sEVs (Figure S8 and S9, and Movie S15), and we grouped these interactions into three categories: less than 1 s, less than 10 s, and more than 10 s (Figure 4a). A stable S2 subunit–sEV interaction could promote insertion of the S2 subunit into the sEV (Figure 4b and c, Figure S10A, and Movie S16). The height and length of the S2 subunit abruptly dropped to zero upon insertion (Figure 4d). We hypothesized that the S2 subunit insertion could disrupt the stability of the exosomal layer, as we observed in our previous study (Lim, Mohamed et al., 2020; Lim, Kodera et al., 2020). When we continued HS‐AFM scanning over a longer period after S2 insertion, a declining trend in the height of sEVs was observed (Figure 4e and Figure S10B). We also observed shrinkage of the sEVs followed by deformation of the sEVs after two consecutive S2 subunit insertions (Figure S10, A and B, and Movie S16B). We further investigated this phenomenon by incubating the S2 subunit with sEVs at two time points: 20 min and 2 h. We found that the S2 subunit was bound to sEVs at 20 min, and the sEV topology appeared rugged (Figure 4f and Movie S17B). After 2 h, sEVs appeared shrunken and completely lost their spherical fluid‐filled topology (Figure 4f and Movie S17C). AFM is a useful tool to measure various mechanical properties of biological samples (Ikai et al., 2018), and we used it to determine the effect of S2 subunit‐sEV interaction on sEV membrane stiffness. Result showed that sEV membrane stiffness was significantly reduced after sEVs interacted with S2 subunit (Figure S10C), indicating that S2 subunit disrupted sEV membrane stability.
FIGURE 4.
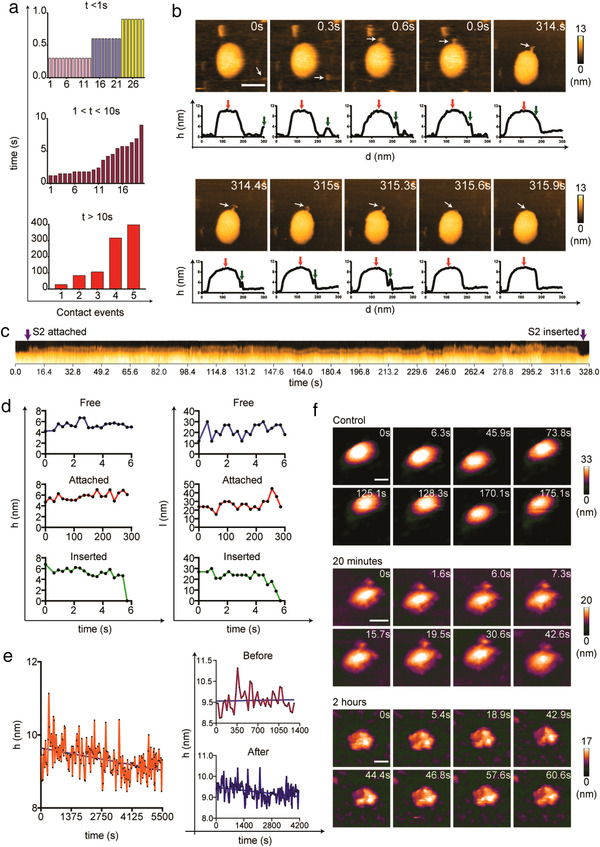
Real‐time interaction between the SARS‐CoV‐2 Spike S2 subunit and sEVs. (a‐e) A representative HS‐AFM visualization of S2 subunit‐sEV interaction (n: 9). (a) Multiple S2 subunits interacted with sEVs for different lengths of time. The durations of the interactions were recorded and categorized into three groups. Data are presented in bar graphs. (b) HS‐AFM images show a S2 subunit (indicated by a white arrow) in contact with a sEV and subsequent insertion into the sEV (scale bar: 100 nm). Graphs beneath the HS‐AFM images indicate the distance between the S2 subunit and the sEV during the interaction (red arrow: sEV, blue arrow: S2 subunit). (c) A kymograph shows a S2 subunit stably bound onto a sEV surface and eventually inserting into the sEV lipid layer. (d) Real‐time height (h) and length (l) changes in the S2 subunit throughout the interaction with sEVs. The changes 6 s before contact (Free) and 6 s before insertion (Inserted) are illustrated in different line graphs. (e) Longer term observations of the S2 subunit–sEV interaction revealed that insertion of the S2 subunit disrupted the integrity of the exosomal layer, as indicated by a declining trend in sEV height. (f) Disruption of the exosomal shape in S2 subunit–sEV premixed samples. The S2 subunit was found to be attached to the sEV surface after incubation for 20 min and the sEV lost its spherical shape after 2 h of incubation with S2 subunit. A control sEV maintained a perfect spherical shape (scale bar: 50 nm)
3.6. S protein potentially interacts with ACE2 receptor to stably dock on an sEV
Several studies have revealed that extracellular vesicles (EV) secreted by host cells, express viral receptors that can neutralize viruses to prevent viral entry (Conzelmann et al., 2020; de Carvalho et al., 2014; F. Cocozza et al., 2020). For example, T cells secreted EV containing the HIV virus receptor, CD4+, to neutralize HIV virus (de Carvalho et al., 2014). A recent study reported that ACE2‐expressing human epithelial cells, Calu‐3 (lung) and Caco‐2 (intestine), secreted ACE2‐expressing EVs (F. Cocozza et al., 2020). The cell line we used for sEV production, PC‐9, is an ACE2‐expressing cell line (Puray‐Chavez et al., 2021). We found that several S proteins could either transiently or stably attach onto the PC‐9‐derived sEV surface (Figure 5a, Figure S11, A‐C, and Movie S19). The transient S protein–sEV interaction showed a reversible interaction pattern (detaching and reattaching to the same sEV) during HS‐AFM scanning (Figure 5b and Movie S18). Multiple S proteins were able to bind to the same sEV (Figure 5, b, d, and e, Movie S18). To further emphasize the role of ACE2 in S protein‐sEV interaction, by referring to data obtained from the Cancer Cell Line Encyclopedia (CCLE, c‐Bioportal), we isolated sEVs of high ACE2‐expressing HEC50B and negligible ACE2‐expressing A549 (Figure S12A). Results show that HEC50B sEVs bound with several S proteins, but A549 sEVs did not (Figure S12, B and C). The S protein–sEV interaction did not impose any effects on the S protein conformation because no sudden change in height was observed throughout the interaction (Figure 5c). Although a recent study reported that the S1 subunit permeated and destabilized a neutral phospholipid bilayer (Asandei et al., 2020), our results revealed no prominent declination in sEV height after a prolonged exposure to S proteins, suggesting that the S protein–sEV interaction did not disrupt sEV membrane integrity (Figure 5f, Figure S12, B and C).
FIGURE 5.
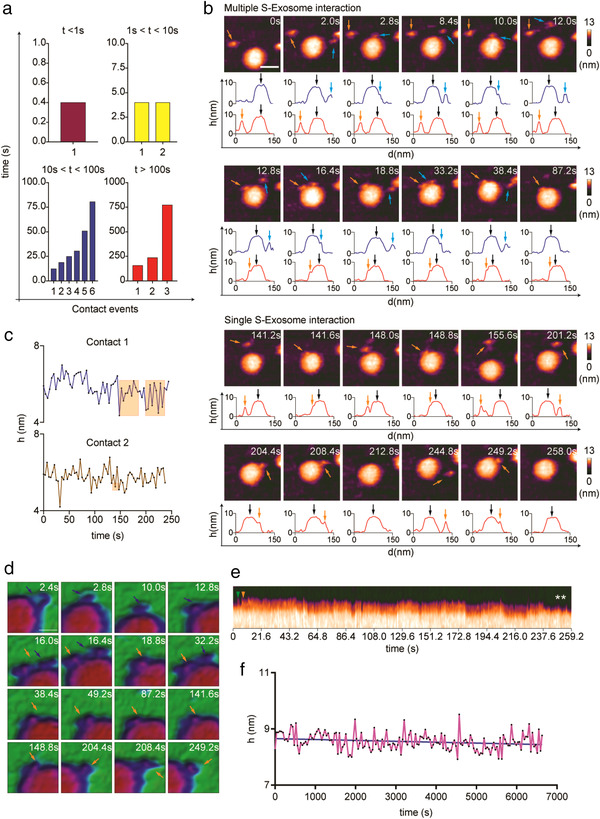
Real‐time interaction between S protein and an sEV. A representative HS‐AFM visualization of S protein‐sEV interaction (n: 6). (a) Multiple S proteins interacted with a sEV for different lengths of time. The durations of the interactions were recorded and categorized into four groups. Data are presented in bar graphs. (b) The interaction between multiple S proteins or a single S protein and an sEV, as captured by HS‐AFM. Graphs beneath the HS‐AFM images indicate the distance between the S protein and the sEV (scale bar: 50 nm, blue and orange arrows: S protein, black arrow: sEV). (c) Real‐time height changes for the two S proteins during their interaction with the sEV. Orange areas indicate the height of S proteins when they were free from the sEV. (d) Close observation of the S protein–sEV interaction illustrates that the S protein slid along the sEV lipid layer (scale bar: 15 nm). S proteins are labelled with arrows. (e) A kymograph shows two S proteins touching (arrows) the sEV surface to form stable interactions, with both S proteins subsequently leaving (asterisk) the sEV. (f) Real‐time height change demonstrate that the sEV height did not reduce after a long period of interaction with S protein. This result shows that the S protein–sEV interaction did not disrupt the integrity of the sEV layer
4. DISCUSSION
HS‐AFM has been proven to be a feasible methodology to study the native conformation and conformational dynamics of viral fusion proteins, and their interactions with sEVs (Lim, Mohamed, et al., 2020; Lim, Kodera et al., 2020). In this study, the SARS‐CoV‐2 S protein topology captured using HS‐AFM was comparable to previously reported cryo‐EM structures. Besides the head and stalk, we managed to observe the native closed and open conformations of S protein at the single‐molecule level. The advantage of HS‐AFM is that its high spatiotemporal resolution enabled us to visualize the highly dynamic behaviour of the S stalk and RBD loop. The data acquired reflected the real‐time active tilting, extension, or retraction of the S stalk or RBD during HS‐AFM scanning. These observations would be challenging to achieve by cryo‐EM. Bioinformatic tools for IDR prediction can complement HS‐AFM observations. For example, the outputs indicate that S protein is highly ordered or that S2 subunit is more flexible than S1 subunit, in line with our observations from HS‐AFM imaging. The same advantage of HS‐AFM also enabled us to track a continuous change in S protein structure during acidification follow by neutralization.
Our results demonstrate a complexity in the conformational dynamics of S protein in response to pH changes. Although S1 dissociation is mainly mediated by host proteases, spontaneous S1 dissociation in S protein with a closed conformation after acidification may be possible. By contrast, low pH‐induced conformation masking in S protein with an open conformation supports a finding reported by Zhou et al. (2020). Technical limitations impeded our observations regarding the conformational dynamics of S protein in response to a gradual change in temperature. Nonetheless, our current findings provide important insights into the effects of temperature on S protein conformation. For example, at 37°C, S protein could become adhesive and this property, together with the flexible S stalk, would allow SARS‐CoV‐2 viruses to form stable viral aggregates and spread to adjacent tissue, or form respiratory aerosols or droplets. A high temperature significantly dampened SARS‐CoV‐2 infectivity (Riddell et al., 2020); however, S protein retained its conformation after heating to 60°C and the virus appeared to possess high global thermal stability (Kiss et al., 2021). Our findings regarding the S protein–ACE2 interaction indicate that this interaction does not induce significant conformational change in S protein, but it could increase the propensity of the 2 Up‐ and 3 Up‐ open conformations. These findings are consistent with results reported by other groups using cryo‐EM (Henderson et al., 2020; Zhou et al., 2020) and smFRET (Lu et al., 2020), suggesting that binding of single monomeric ACE2 disturbs interdomain interactions, and subsequently promotes the remaining RBDs to shift up. Future work is required to study the ACE2‐bound S intermediate that occurred after acidification.
ACE2 orientation on mica surface has to be as close as its orientation on cell surface in order to visualize the real scenario of S protein‐ACE2 interaction during infection. Previously, Yang and coworkers used a bifunctional crosslinker, N‐hydroxysuccinimide (NHS) and dimethylaminopropyl carbodiimide (EDC), to adsorb Fc‐tagged recombinant ACE2 protein on a gold‐coated surface to measure S protein‐ACE2 binding force using AFM (Yang et al., 2020). The NHS‐EDC approach is not suitable for HS‐AFM imaging of single ACE2 topology because it promotes protein aggregation (Valueva et al., 2020). Ni2+‐histidine interaction has been shown to be applicable for orientating protein position on Ni2+‐coated substrates (Sumino et al., 2017; Wasserberg et al., 2017; Yamamoto et al., 2010). Therefore, we used Ni2+‐coated mica substrate for ACE2 adsorption because our recombinant ACE2 has a poly‐histidine tag at its C‐terminus. ACE2 mostly appeared in a globular topology (PD) even though some displayed both globular (PD) and tail‐like (CLD) topologies when ACE2 at low concentration. Likewise, Yamamoto et al. demonstrated that his‐tagged p97, a hexameric ATPase, bound to Ni2+‐NTA coated substrate via Ni2+‐histidine interaction at neutral condition. The isoelectric point of p97 is close to the isoelectric point of ACE2 (Mori‐Konya et al., 2009), and hence it is negatively charged at neutral condition. Collectively, these findings indicate that Ni2+‐histidine interaction could supersede non‐specific electrostatic attraction.
EVs secreted by ACE2‐expressing cell lines contain ACE2 receptors (F. Cocozza et al., 2020), and these EVs could work as nano‐decoys to bind with SARS‐CoV‐2 to block viral entry. sEVs also can be used to suppress hyperinflammation and to promote lung recovery in COVID‐19 patients (Inal, 2020). Recently, three clinical trials of mesenchymal stem cells (MSCs)‐derived sEV based therapy (NCT04602442, NCT04276987, and NCT04798716) for COVID‐19 treatment are currently ongoing. Nano‐imaging of S protein–EV interaction could reflect the dynamic of S protein during its interaction with ACE2 on cell surface. According to the International Society for Extracellular Vesicle (ISEV), EV is defined as a non‐replicating lipid bilayer particle released by cell (Théry et al., 2018). We used Tim4 isolation technique to acquire phosphatidylserine (PS)‐enriched EVs from HEC50B, PC‐9, and A549 cell lines (Nakai et al., 2016). With this technique, we managed to harvest sEVs with exosome markers including CD9, CD63, CD81, TSG101, and VPS37B (Figure S13), abiding the MISEV2018 guideline. S1 subunit in S protein mainly mediates ACE2 receptor interaction and it does not disrupt sEV membrane integrity. However, after dissociation, free S1 subunit becomes positively charge, allowing it to adsorb and permeabilize lipid bilayer (Asandei et al., 2020). We also found that the S–sEV interaction was reversible. Further studies are now required to address whether SARS‐CoV‐2 could temporarily bind to ACE2‐expressing EV to evade antibody or to achieve systemic dissemination.
Besides S protein–ACE2 interaction, sEV has been used to visualize the S2 subunit‐lipid bilayer interaction using HS‐AFM. Insertion of the S2 subunit into sEVs at pH 7.4 illustrates the second entry mechanism of SARS‐CoV‐2, in which the S2 subunit orchestrates the fusion between SARS‐CoV‐2 viral membrane and the host plasma membrane to accomplish viral entry (Hoffmann, Kleine‐Weber, & Pöhlmann, 2020; Hoffmann, Kleine‐Weber, Schroeder et al., 2020). According to Ke et al., post‐fusion S proteins (without S1 subunit) were expressed on a SARS‐CoV‐2 virion (Ke et al., 2020). Also, two serine proteases secreted by neutrophils, neutrophil elastase (NE) and protease 3 (PR3), were found able to cleave S protein at S1/S2 site (Mustafa et al., 2021). Additional works are needed to determine whether these soluble proteases could prematurely remove S1 subunit independent of ACE2 receptor and TMPRSS2. S protein without S1 subunit could favour fusion peptide in S2 subunit to interact with lipid bilayer, potentially promoting immune evasion and expediting viral entry.
In near future, HS‐AFM can be considered for investigating the inhibitory effects of various approaches that targeting the SARS‐CoV‐2 entry, either blocking S protein‐ACE2 interaction or blocking S2 subunit‐mediated membrane fusion. These approaches including synthetic anti‐S neutralizing antibodies (Copin et al., 2021), small‐molecule inhibitors (Bojadzic et al., 2021), anti‐S nanoparticles (Rao et al., 2020), or fusion inhibitor (Xia et al., 2020). Furthermore, HS‐AFM nano‐imaging of antigen–antibody complex could provide important insights about antibody‐dependent enhancement effect (ADE), as reported previously (Asarnow et al., 2021; Liu et al., 2021). Altogether, we report that HS‐AFM is an ideal tool for nanoscopic imaging to enhance our knowledge in SARS‐CoV‐2 S protein behaviour, allowing scientists to design effective countermeasures (sEV‐based nano‐decoys and vaccines) to manage COVID‐19.
AUTHOR CONTRIBUTIONS
Keesiang Lim and Richard Wong designed the study and wrote the manuscript. Keesiang Lim and Goro Nishide performed the experiments and prepared figures. Takeshi Yoshida and Rikinari Hanayama prepared the small extracellular vesicles (exosomes). Takahiro Watanabe‐Nakayama performed AFM force–distance curve measurement. Akiko Kobayashi, Masaharu Hazawa and Toshio Ando analysed the data. All authors read and approved the final manuscript. Richard Wong supervised the whole study.
DATA AND MATERIALS AVAILABILITY
Not available.
Supporting information
Supporting information.
Supporting information.
ACKNOWLEDGEMENTS
We thank all members of the Richard Wong laboratory, especially to Mr. Hanbo Wang for helpful discussion. We also thank Prof. Noriyuki Kodera for providing insightful HS‐AFM advices in this study. This project was funded by grants from the MEXT/JSPS KAKENHI (19K23841 (to K.L.), 20K16262 (to K.L.), 21H05744 (to R.W.), 21K19043 (to R.W.)) from MEXT Japan, the TDRP grant (to K.L.) from WPI NanoLSI, the JST CREST (No. JPMJCR18H4 to R.H), from the Kobayashi International Scholarship Foundation (to R.W.), the Shimadzu Science Foundation (to R.W.), the Takeda Science Foundation (to R.W).
Lim, K. , Nishide, G. , Yoshida, T. , Watanabe‐Nakayama, T. , Kobayashi, A. , Hazawa, M. , Hanayama, R. , Ando, T. , & Wong, R. W. (2021). Millisecond dynamic of SARS‐CoV‐2 spike and its interaction with ACE2 receptor and small extracellular vesicles. Journal of Extracellular Vesicles, 10, e12170. 10.1002/jev2.12170
Contributor Information
Keesiang Lim, Email: limkeesiang@staff.kanazawa-u.ac.jp.
Richard W. Wong, Email: rwong@staff.kanazawa-u.ac.jp.
REFERENCES
- Amyot, R. , & Flechsig, H. (2020). BioAFMviewer: An interactive interface for simulated AFM scanning of biomolecular structures and dynamics. PLOS Computational Biology. 16, e1008444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ando, T. (2019). High‐speed atomic force microscopy. Current Opinion in Chemical Biology. 51, 105–112. [DOI] [PubMed] [Google Scholar]
- Asandei, A. , Mereuta, L. , Schiopu, I. , Park, J. , Seo, C. H. , Park, Y. , & Luchian, T. (2020). Non‐receptor‐mediated lipid membrane permeabilization by the SARS‐CoV‐2 spike protein S1 subunit. ACS Applied Materials & Interfaces. 12, 55649–55658. [DOI] [PubMed] [Google Scholar]
- Asarnow, D. , Wang, B. , Lee, W. H. , Hu, Y. , Huang, C. W. , Faust, B. , Ng, P. M. L. , Ngoh, E. Z. X. , Bohn, M. , Bulkley, D. , Pizzorno, A. , Ary, B. , Tan, H. C. , Lee, C. Y. , Minhat, R. A. , Terrier, O. , Soh, M. K. , Teo, F. J. , Yeap, Y. Y. C. , … Wang, C. I. (2021). Structural insight into SARS‐CoV‐2 neutralizing antibodies and modulation of syncytia. Cell. 184, 3192–3204.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker, N. A. , Sept, D. , Joseph, S. , Holst, M. J. , & McCammon, J. A. (2001). Electrostatics of nanosystems: application to microtubules and the ribosome. Proceedings of the National Academy of Sciences of the United States of America. 98, 10037–10041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benton, D. J. , Wrobel, A. G. , Xu, P. , Roustan, C. , Martin, S. R. , Rosenthal, P. B. , Skehel, J. J. , & Gamblin, S. J. (2020). Receptor binding and priming of the spike protein of SARS‐CoV‐2 for membrane fusion. Nature. 588, 327–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bojadzic, D. , Alcazar, O. , Chen, J. , Chuang, S. T. , Condor Capcha, J. M. , Shehadeh, L. A. , & Buchwald, P. (2021). Small‐molecule inhibitors of the coronavirus spike: ACE2 protein‐protein interaction as blockers of viral attachment and entry for SARS‐CoV‐2. ACS Infectious Diseases. 7, 1519–1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai, Y. , Zhang, J. , Xiao, T. , Peng, H. , Sterling, S. M. , Walsh, R. M. , Rawson, S. , Rits‐Volloch, S. , & Chen, B. (2020). Distinct conformational states of SARS‐CoV‐2 spike protein. Science. 369, 1586–1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conzelmann, C. , Groß, R. , Zou, M. , Krüger, F. , Görgens, A. , Gustafsson, M. O. , El Andaloussi, S. , Münch, J. , Müller, J. A. (2020). Salivary extracellular vesicles inhibit Zika virus but not SARS‐CoV‐2 infection. Journal of Extracellular Vesicles. 9, 1808281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Copin, R. , Baum, A. , Wloga, E. , Pascal, K. E. , Giordano, S. , Fulton, B. O. , Zhou, A. , Negron, N. , Lanza, K. , Chan, N. , Coppola, A. , Chiu, J. , Ni, M. , Wei, Y. , Atwal, G. S. , Hernandez, A. R. , Saotome, K. , Zhou, Y. , Franklin, M. C. , … Kyratsous, C. A. (2021). The monoclonal antibody combination REGEN‐COV protects against SARS‐CoV‐2 mutational escape in preclinical and human studies. Cell. 184(15), 3949–3961.e11. 10.1016/j.cell.2021.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Carvalho, J. V. , de Castro, R. O. , da Silva, E. Z. , Silveira, P. P. , da Silva‐Januário, M. E. , Arruda, E. , Jamur, M. C. , Oliver, C. , Aguiar, R. S. , & daSilva, L. L. (2014). Nef neutralizes the ability of exosomes from CD4+ T cells to act as decoys during HIV‐1 infection. Plos One. 9, e113691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolinsky, T. J. , Nielsen, J. E. , McCammon, J. A. , & Baker, N. A. (2004). PDB2PQR: An automated pipeline for the setup of Poisson‐Boltzmann electrostatics calculations. Nucleic Acids Research. 32, W665–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epand, R. M. (2003). Fusion peptides and the mechanism of viral fusion. Biochimica Et Biophysica Acta. 1614, 116–121. [DOI] [PubMed] [Google Scholar]
- Erdős, G. , & Dosztányi, Z. (2020). Analyzing protein disorder with IUPred2A. Curr Protoc Bioinformatics. 70, e99. [DOI] [PubMed] [Google Scholar]
- FCocozza, N. N. , Piovesana, E. , Lahaye, X. , Buchrieser, J. , Schwartz, O. , Manel, N. , Tkach, M. , Théry, C. , & Martin‐Jaular, L. (2020). Extracellular vesicles containing ACE2 efficiently prevent infection by SARS‐CoV‐2 Spike protein‐containing virus. Journal of Extracellular Vesicles. 10, e12050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuentes‐Prior, P. (2020). Priming of SARS‐CoV‐2 S protein by several membrane‐bound serine proteinases could explain enhanced viral infectivity and systemic COVID‐19 infection. Journal of Biological Chemistry. 296, 100135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan, W.‐j. , Ni, Z.‐y. , Hu, Y. , Liang, W.‐h. , Ou, C.‐q. , He, J.‐x. , Liu, L. , Shan, H. , Lei, C.‐l. , Hui, D. S. C. , Du, B. , Li, L.‐j. , Zeng, G. , Yuen, K. ‐ Y. , Chen, R.‐c. , Tang, C.‐l. , Wang, T. , Chen, P.‐y. , Xiang, J. , … Zhong, N.‐s. (2020). Clinical characteristics of Coronavirus Disease 2019 in China. New England Journal of Medicine. 382, 1708–1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson, R. , Edwards, R. J. , Mansouri, K. , Janowska, K. , Stalls, V. , Gobeil, S. , Kopp, M. , Hsu, A. , Borgnia, M. , Parks, R. , Haynes, B. F. , & Acharya, P. (2020). Controlling the SARS‐CoV‐2 spike glycoprotein conformation. Nature Structural & Molecular Biology. 27, 925–933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann, M. , Kleine‐Weber, H. , & Pöhlmann, S. (2020). A multibasic cleavage site in the spike protein of SARS‐CoV‐2 is essential for infection of human lung cells. Molecular Cell. 78, 779–784 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann, M. , Kleine‐Weber, H. , Schroeder, S. , Krüger, N. , Herrler, T. , Erichsen, S. , Schiergens, T. S. , Herrler, G. , Wu, N. H. , Nitsche, A. , Müller, M. A. , Drosten, C. , & Pöhlmann, S. (2020). SARS‐CoV‐2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell. 181, 271–280 e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang, C. , Wang, Y. , Li, X. , Ren, L. , Zhao, J. , Hu, Y. , Zhang, L. , Fan, G. , Xu, J. , Gu, X. , Cheng, Z. , Yu, T. , Xia, J. , Wei, Y. , Wu, W. , Xie, X. , Yin, W. , Li, H. , Liu, M. , … Cao, B. (2020). Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet. 395, 497–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikai, A. , Afrin, R. , Saito, M. , & Watanabe‐Nakayama, T. (2018). Atomic force microscopy as a nano‐ and micrometer scale biological manipulator: A short review. Seminars in Cell & Developmental Biology. 73, 132–144. [DOI] [PubMed] [Google Scholar]
- Inal, J. M. (2020). Decoy ACE2‐expressing extracellular vesicles that competitively bind SARS‐CoV‐2 as a possible COVID‐19 therapy. Clinical Science. 134, 1301–1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishida, T. , & Kinoshita, K. (2007). PrDOS: Prediction of disordered protein regions from amino acid sequence. Nucleic Acids Research. 35, W460–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ke, Z. , Oton, J. , Qu, K. , Cortese, M. , Zila, V. , McKeane, L. , Nakane, T. , Zivanov, J. , Neufeldt, C. J. , Cerikan, B. , Lu, J. M. , Peukes, J. , Xiong, X. , Kräusslich, H. G. , Scheres, S. H. W. , Bartenschlager, R. , & Briggs, J. A. G. (2020). Structures and distributions of SARS‐CoV‐2 spike proteins on intact virions. Nature. 588, 498–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiss, B. , Kis, Z. , Pályi, B. , & Kellermayer, M. S. Z. (2021). Topography, spike dynamics, and nanomechanics of individual native SARS‐CoV‐2 virions. Nano Letters. 21, 2675–2680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim, K. S. , Mohamed, M. S. , Wang, H. , Hartono, M. H. , Kobayashi, A. , Voon, D. C. , Kodera, N. , Ando, T. , & Wong, R. W. (2020). Direct visualization of avian influenza H5N1 hemagglutinin precursor and its conformational change by high‐speed atomic force microscopy. Biochim Biophys Acta Gen Subj. 1864(2), 129313, 10.1016/j.bbagen.2019.02.015. [DOI] [PubMed] [Google Scholar]
- Lim, K. , Kodera, N. , Wang, H. , Mohamed, M. S. , Hazawa, M. , Kobayashi, A. , Yoshida, T. , Hanayama, R. , Yano, S. , Ando, T. , & Wong, R. W. (2020). High‐Speed AFM reveals molecular dynamics of human influenza A hemagglutinin and its interaction with exosomes. Nano Letters. 20, 6320–6328. [DOI] [PubMed] [Google Scholar]
- Liu, Y. , Soh, W. T. , Kishikawa, J. I. , Hirose, M. , Nakayama, E. E. , Li, S. , Sasai, M. , Suzuki, T. , Tada, A. , Arakawa, A. , Matsuoka, S. , Akamatsu, K. , Matsuda, M. , Ono, C. , Torii, S. , Kishida, K. , Jin, H. , Nakai, W. , Arase, N. , … Arase, H. (2021). An infectivity‐enhancing site on the SARS‐CoV‐2 spike protein targeted by antibodies. Cell. 184, 3452–3466.e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu, M. , Uchil, P. D. , Li, W. , Zheng, D. , Terry, D. S. , Gorman, J. , Shi, W. , Zhang, B. , Zhou, T. , Ding, S. , Gasser, R. , Prevost, J. , Beaudoin‐Bussieres, G. , Anand, S. P. , Laumaea, A. , Grover, J. R. , Lihong, L. , Ho, D. D. , Mascola, J. , … Mothes, W. , Real‐time conformational dynamics of SARS‐CoV‐2 spikes on virus particles. Cell Host Microbe. 28, 880–891 e8 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mészáros, B. , Erdos, G. , & Dosztányi, Z. (2018). IUPred2A: Context‐dependent prediction of protein disorder as a function of redox state and protein binding. Nucleic Acids Research. 46, W329–W337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohamed, M. S. , Kobayashi, A. , Taoka, A. , Watanabe‐Nakayama, T. , Kikuchi, Y. , Hazawa, M. , Minamoto, T. , Fukumori, Y. , Kodera, N. , Uchihashi, T. , Ando, T. , & Wong, R. W. (2017). High‐Speed Atomic Force Microscopy reveals loss of nuclear pore resilience as a dying code in colorectal cancer cells. ACS Nano. 11, 5567–5578. [DOI] [PubMed] [Google Scholar]
- Mori‐Konya, C. , Kato, N. , Maeda, R. , Yasuda, K. , Higashimae, N. , Noguchi, M. , Koike, M. , Kimura, Y. , Ohizumi, H. , Hori, S. , & Kakizuka, A. (2009). p97/valosin‐containing protein (VCP) is highly modulated by phosphorylation and acetylation. Genes to Cells. 14, 483–497. [DOI] [PubMed] [Google Scholar]
- Mustafa, Z. , Zhanapiya, A. , Kalbacher, H. , & Burster, T. (2021). Neutrophil Elastase and Proteinase 3 cleavage sites are adjacent to the polybasic sequence within the proteolytic sensitive activation loop of the SARS‐CoV‐2 spike protein. ACS Omega. 6, 7181–7185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakai, W. , Yoshida, T. , Diez, D. , Miyatake, Y. , Nishibu, T. , Imawaka, N. , Naruse, K. , Sadamura, Y. , & Hanayama, R. (2016). A novel affinity‐based method for the isolation of highly purified extracellular vesicles. Scientific Reports. 6, 33935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishide, G. , Lim, K. , Mohamed, M. S. , Kobayashi, A. , Hazawa, M. , Watanabe‐Nakayama, T. , Kodera, N. , Ando, T. , & Wong, R. W. (2021). High‐speed atomic force microscopy reveals spatiotemporal dynamics of histone protein H2A involution by DNA inchworming. Journal of Physical Chemistry Letters. 12, 3837–3846. [DOI] [PubMed] [Google Scholar]
- Peng, K. , Radivojac, P. , Vucetic, S. , Dunker, A. K. , & Obradovic, Z. (2006). Length‐dependent prediction of protein intrinsic disorder. Bmc Bioinformatics. 7, 208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng, K. , Vucetic, S. , Radivojac, P. , Brown, C. J. , Dunker, A. K. , & Obradovic, Z. (2005). Optimizing long intrinsic disorder predictors with protein evolutionary information. Journal of Bioinformatics and Computational Biology. 3, 35–60. [DOI] [PubMed] [Google Scholar]
- Puray‐Chavez, M. , LaPak, K. M. , Schrank, T. P. , Elliott, J. L. , & Bhatt, D. P. , et al. (2021). Systemic analysis of SARS‐CoV‐2 infection of an ACE2‐negative human airway cell. Cell Reports. 36, 109364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao, L. , Xia, S. , Xu, W. , Tian, R. , Yu, G. , Gu, C. , Pan, P. , Meng, Q. F. , Cai, X. , Qu, D. , Lu, L. , Xie, Y. , Jiang, S. , & Chen, X. (2020). Decoy nanoparticles protect against COVID‐19 by concurrently adsorbing viruses and inflammatory cytokines. Proceedings of the National Academy of Sciences of the United States of America. 117, 27141–27147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rath, S. L. , & Kumar, K. (2020). Investigation of the effect of temperature on the structure of SARS‐CoV‐2 spike protein by molecular dynamics simulations. Frontiers in Molecular Bioscience. 7, 583523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rey, F. A. , & Lok, S. M. (2018). Common features of enveloped viruses and implications for immunogen design for next‐generation vaccines. Cell. 172, 1319–1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riddell, S. , Goldie, S. , Hill, A. , Eagles, D. , & Drew, T. W. (2020). The effect of temperature on persistence of SARS‐CoV‐2 on common surfaces. Virology Journal. 17, 145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero, P. , Obradovic, Z. , Li, X. , Garner, E. C. , Brown, C. J. , & Dunker, A. K. (2001). Sequence complexity of disordered protein. Proteins. 42, 38–48. [DOI] [PubMed] [Google Scholar]
- Sajidah, E. S. , Lim, K. , & Wong, R. W. (2021). How SARS‐CoV‐2 and other viruses build an invasion route to hijack the host nucleocytoplasmic trafficking system. Cells. 10, 1424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sumino, A. , Uchihashi, T. , & Oiki, S. (2017). Oriented reconstitution of the full‐length KcsA potassium channel in a lipid bilayer for AFM Imaging. Journal of Physical Chemistry Letters. 8, 785–793. [DOI] [PubMed] [Google Scholar]
- Tang, T. , Bidon, M. , Jaimes, J. A. , Whittaker, G. R. , & Daniel, S. (2020). Coronavirus membrane fusion mechanism offers a potential target for antiviral development. Antiviral Research. 178, 104792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Théry, C. , Witwer, K. W. , Aikawa, E. , Alcaraz, M. J. , & Anderson, J. D. et al. (2018). Minimal information for studies of extracellular vesicles 2018 (MISEV2018): A position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. Journal of Extracellular Vesicles. 7, 1535750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turoňová, B. , Sikora, M. , Schürmann, C. , Hagen, W. J. H. , Welsch, S. , Blanc, F. E. C. , von Bülow, S. , Gecht, M. , Bagola, K. , Hörner, C. , van Zandbergen, G. , Landry, J. , de Azevedo, N. T. D. , Mosalaganti, S. , Schwarz, A. , Covino, R. , Mühlebach, M. D. , Hummer, G. , Krijnse Locker, J. , & Beck, M. (2020). In situ structural analysis of SARS‐CoV‐2 spike reveals flexibility mediated by three hinges. Science. 370, 203–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valueva, A. A. , Shumov, I. D. , Kaysheva, A. L. , Ivanova, I. A. , Ziborov, V. S. , YD, I. , & Pleshakova, T. O. (2020). Covalent protein immobilization onto muscovite mica surface with a photocrosslinker. Minerals. 10, 464. [Google Scholar]
- Van Deun, J. et al. (2017). EV‐TRACK: Transparent reporting and centralizing knowledge in extracellular vesicle research. Nature Methods, 14(3), 228–223. [DOI] [PubMed] [Google Scholar]
- Walls, A. C. , Park, Y. J. , Tortorici, M. A. , Wall, A. , McGuire, A. T. , & Veesler, D. (2020). Structure, function, and antigenicity of the SARS‐CoV‐2 spike glycoprotein. Cell. 181, 281–292 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, Q. , Zhang, Y. , Wu, L. , Niu, S. , Song, C. , Zhang, Z. , Lu, G. , Qiao, C. , Hu, Y. , Yuen, K. Y. , Wang, Q. , Zhou, H. , Yan, J. , & Qi, J. (2020). Structural and functional basis of SARS‐CoV‐2 entry by using human ACE2. Cell. 181, 894–904 e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wasserberg, D. , Cabanas‐Danés, J. , Prangsma, J. , O'Mahony, S. , Cazade, P. A. , Tromp, E. , Blum, C. , Thompson, D. , Huskens, J. , Subramaniam, V. , & Jonkheijm, P. (2017). Controlling protein surface orientation by strategic placement of oligo‐histidine tags. ACS Nano. 11, 9068–9083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wrapp, D. , Wang, N. , Corbett, K. S. , Goldsmith, J. A. , Hsieh, C. L. , Abiona, O. , Graham, B. S. , & McLellan, J. S. (2020). Cryo‐EM structure of the 2019‐nCoV spike in the prefusion conformation. Science. 367, 1260–1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wrobel, A. G. , Benton, D. J. , Xu, P. , Roustan, C. , Martin, S. R. , Rosenthal, P. B. , Skehel, J. J. , & Gamblin, S. J. (2020). SARS‐CoV‐2 and bat RaTG13 spike glycoprotein structures inform on virus evolution and furin‐cleavage effects. Nature Structural & Molecular Biology. 27, 763–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia, S. , Liu, M. , Wang, C. , Xu, W. , Lan, Q. , Feng, S. , Qi, F. , Bao, L. , Du, L. , Liu, S. , Qin, C. , Sun, F. , Shi, Z. , Zhu, Y. , Jiang, S. , & Lu, L. (2020). Inhibition of SARS‐CoV‐2 (previously 2019‐nCoV) infection by a highly potent pan‐coronavirus fusion inhibitor targeting its spike protein that harbors a high capacity to mediate membrane fusion. Cell Research. 30, 343–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong, X. , Qu, K. , Ciazynska, K. A. , Hosmillo, M. , Carter, A. P. , Ebrahimi, S. , Ke, Z. , Scheres, S. H. W. , Bergamaschi, L. , Grice, G. L. , Zhang, Y. , Nathan, J. A. , Baker, S. , James, L. C. , Baxendale, H. E. , Goodfellow, I. , Doffinger, R. , & Briggs, J. A. G. (2020). A thermostable, closed SARS‐CoV‐2 spike protein trimer. Nature Structural & Molecular Biology. 27, 934–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue, B. , Dunbrack, R. L. , Williams, R. W. , Dunker, A. K. , & Uversky, V. N. (2010). PONDR‐FIT: A meta‐predictor of intrinsically disordered amino acids. Biochimica et Biophysica Acta. 1804, 996–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto, D. , Uchihashi, T. , Kodera, N. , Yamashita, H. , Nishikori, S. , Ogura, T. , Shibata, M. , & Ando, T. (2010). High‐speed atomic force microscopy techniques for observing dynamic biomolecular processes. Methods in Enzymology. 475, 541–564. [DOI] [PubMed] [Google Scholar]
- Yan, R. , Zhang, Y. , Li, Y. , Xia, L. , Guo, Y. , & Zhou, Q. (2020). Structural basis for the recognition of SARS‐CoV‐2 by full‐length human ACE2. Science. 367, 1444–1448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, J. , Petitjean, S. J. L. , Koehler, M. , Zhang, Q. , Dumitru, A. C. , Chen, W. , Derclaye, S. , Vincent, S. P. , Soumillion, P. , & Alsteens, D. (2020). Molecular interaction and inhibition of SARS‐CoV‐2 binding to the ACE2 receptor. Nature Communications. 11, 4541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao, H. , Song, Y. , Chen, Y. , Wu, N. , Xu, J. , Sun, C. , Zhang, J. , Weng, T. , Zhang, Z. , Wu, Z. , Cheng, L. , Shi, D. , Lu, X. , Lei, J. , Crispin, M. , Shi, Y. , Li, L. , & Li, S. (2020). Molecular architecture of the SARS‐CoV‐2 virus. Cell. 183, 730–738 e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, T. , Tsybovsky, Y. , Gorman, J. , Rapp, M. , Cerutti, G. , Chuang, G. Y. , Katsamba, P. S. , Sampson, J. M. , Schön, A. , Bimela, J. , Boyington, J. C. , Nazzari, A. , Olia, A. S. , Shi, W. , Sastry, M. , Stephens, T. , Stuckey, J. , Teng, I. T. , Wang, P. , … Kwong, P. D. (2020). Cryo‐EM structures of SARS‐CoV‐2 spike without and with ACE2 Reveal a pH‐dependent switch to mediate endosomal positioning of receptor‐binding domains. Cell Host Microbe. 28, 867–879 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information.
Supporting information.