

Abstract
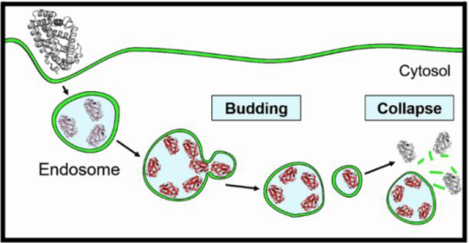
Bacterial protein toxins autonomously enter the cytosol of the target cell where they modify the activities of host components to exert their toxic effects. Many of the toxins enter the host cell by endocytosis followed by endosomal escape. However, their mechanism of endosomal escape remains unresolved. We show herein that diphtheria toxin (DT) and NleC of enteropathogenic Escherichia coli exit the endosome by inducing budding and collapse of small toxin-enriched vesicles from the endosomal membrane.
Keywords: Bacterial toxin, diphtheria toxin, endosomal escape, membrane translocation, NleC
Graphical Abstract
INTRODUCTION
Proteins are generally impermeable to the cell membrane, because their large size and hydrophilicity prevent passive diffusion across the lipid bilayer. Nevertheless, some naturally occurring proteins such as bacterial toxins have evolved the capability of reaching the cytosol of target cells, where they modify the activities of host components, leading to changes in cellular physiology and promoting bacterial pathogenesis.1 Bacterial protein toxins can enter host cells by at least three different pathways.2,3 Some toxins bind to the host cell surface and translocate directly across the plasma membrane to reach the cytosol. Alternatively, other toxins bind to cell-surface receptors, enter the cell by endocytosis, and subsequently escape from the endosomal compartments into the cytosol. Still other toxins are trafficked to the endoplasmic reticulum (ER) where they undergo retrograde translocation. Regardless of the specific cell entry pathway, the toxin must topologically cross a lipid bilayer at some point to reach the cytosolic space. However, how bacterial toxins (or any protein) autonomously translocate across a lipid bilayer remains poorly understood.
Diphtheria toxin (DT) is a prototypical and well-studied member of the “AB class” of bacterial toxins which typically consist of an enzymatic A moiety and a B moiety that mediates the cellular entry of the A moiety.4 Cellular entry of DT starts with its receptor-binding domain (R-domain) recognizing the heparin-binding EGF-like growth factor (HB-EGF) receptor on the host cell surface, leading to the endocytosis of the receptor-DT complex. Acidification of the endosomal compartment induces a conformational change of the translocation domain (T-domain) and a portion of the T-domain inserts into the endosomal membrane to form an ion-conducting pore/channel in the membrane. The membrane-imbedded T domain subsequently causes the translocation of the catalytic domain (C-domain) across the endosomal membrane. Once inside the cytosol, the C-domain inhibits host protein synthesis by ADP-ribosylating and inactivating eukaryotic elongation factor 2 (eEF-2). However, the precise mechanism of the T-domain-mediated translocation remains elusive despite decades of intense investigation. A long-standing hypothesis states that helices 5 through 9 of the T-domain form a pore/channel in the endosomal membrane, through which the unfolded C-domain threads into the cytosol.5,6 This elegant mechanism has been challenged by many experimental observations over the years (Table 1). For example, some mutations in helix 5 of the T-domain eliminate the ion channel activity but have little effect on the translocation of the C-domain.7 On the other hand, mutations in TH1 helix of the T-domain prevent translocation but not the channel activity.8,9 Hyperstable cargo proteins fused to the C-domain,10 as well as nucleic acids noncovalently associated with the T-domain,11,12 have been delivered into the cytosol, suggesting that DT is capable of translocating cargos in their folded states. Lastly, endosomal escape of DT exhibits “quantal” kinetics, namely, each endosomal escape event releases a bolus of ~80 DT molecules into the cytosol simultaneously,13 whereas DT release through a pore would be sequential. It should be noted that the mechanism of endosomal escape has been a mystery for not only DT and other bacterial toxins, but also biomolecules in general.14,15
Table 1.
Experimental Evidence for (√), Consistent with (○), or against (×) the Proposed Endosomal Escape Mechanisms for DT
| Experimental Observation | Extrusion through Pore/Channel | Vesicle Budding and Collapse |
|---|---|---|
| H322Q mutation in TH5 blocks ion channel activity but not translocation7 | x | ○ |
| Mutations in TH1 increase or decrease translocation but have no effect on ion channel activity8,9 | x | √ |
| Delivery of cargo proteins in their folded states10 | x | √ |
| Delivery of noncovalently associated nucleic acids11,12 | x | √ |
| “Quantal” kinetics of endosomal release13 | x | √ |
| Requirement of endosomal acidification for translocation31 | √ | ○ |
| Dose-dependent lag between endosomal acidification and translocation13,28 | x | √ |
| Role of membrane insertion for translocation32 | √ | ○ |
| Endosomal release of dye-labeled dextran29 | x | √ |
We recently discovered that cell-penetrating peptides (CPPs) escape the endosome through a vesicle budding-and-collapse (VBC) mechanism.15–18 Examination of previous observations (Table 1) led us to hypothesize that bacterial toxins might exit the endosome by a similar mechanism (Figure 1A).15 Briefly, bacterial toxins bind to the endosomal membrane to form toxin-enriched lipid domains, which bud off the endosomal membrane as small toxin-loaded vesicles. The budded vesicle spontaneously disintegrates into an amorphous lipid/toxin aggregate, thereby releasing its luminal contents into the cytosol as a “bolus”. Note that the endosome remains intact during and after each VBC event. In this work, we provide direct evidence that DT and NleC, a metalloprotease produced by enterohemorrhagic and enteropathogenic Escherichia coli,19 both escape the endosome by the VBC mechanism and reconcile many previously enigmatic observations in the literature.
Figure 1.

DT-induced VBC from the endosomal membrane in live HeLa cells. (A) Proposed mechanism of endosomal escape by DT. (B, C) Time-lapse confocal microscopy of HeLa cells undergoing DT-induced vesicle budding and collapse. Cells were incubated with 1.5 μM PSTopFluor (green channel) for 10 min, washed with DPBS, and treated with 5 μM DTpHAb (red channel) for 30 min. Cells were washed with DMEM to remove DTpHAb, treated with 5 μM unlabeled DT, and imaged. B, Merged image of several HeLa cells (DIC, green and red channels) at 23 min after the addition of unlabeled DT. C, Enlarged images of the boxed area in (B) during the period of 4–48 s after the initiation of imaging. Images were acquired every 4 s. A budded vesicle emerged from an endosome (marked by a white arrow) at 28 s, completely separated from the endosome at 32 s, and collapsed at 40 s. Scale bars, 1 and 5 μm.
RESULTS AND DISCUSSION
DT Induces VBC from the Endosomal Membrane.
To test our hypothesis, we treated HeLa (human cervical cancer) cells with a catalytically inactive DT mutant (K51E/E148K) labeled on a surface lysine residue(s) with a pH sensitive dye, 3′,6′-bis(4-(4-sulfobutyl)piperazin-1-yl) rhodamine (5′,6′) carboxylic acid (pHAb).20 With an apparent pKa value of 6.2, pHAb emits strong fluorescence (λEM 560 nm; red channel) when inside the acidic extracellular space (pH 7.4). The cells were also treated with a environments of the endosome (pH 5.5–6.5) and lysosome (pH membrane marker, 1-palmitoyl-2-(dipyrrometheneboron 4.5–5.5) but is only weakly fluorescent in the cytosolic/nuclear or difluoride)undecanoyl-sn-glycero-3-phospho-l-serine (PSTopFluor; λEM = 503 nm; green channel). To increase the abundance of VBC events, we first treated HeLa cells with a high concentration of pHAb-labeled DT (DTpHAb, 5 μM) and imaged the cells by time-lapse, live-cell confocal microscopy at 4-s intervals. According to the proposed mechanism (Figure 1A), endosomes (and lysosomes) of the treated cells should emit both red (DTpHAb) and green fluorescence (PSTopFluor). Immediately after a budding event, the budded vesicle as well as the remaining endosome should also have both red and green fluorescence. However, once the budded vesicle collapses, exposing the lipid/toxin complex to the cytosolic pH (7.4), DTpHAb fluorescence (red) would immediately disappear whereas PSTopFluor fluorescence would remain, until the aggregate is gradually dispersed into the cytosolic milieu. As shown in Figure 1B–C, Figure S1, and Movies S1–S2, this chain of events was captured in real time by confocal microscopy in live HeLa cells, providing direct evidence for the VBC mechanism. Repetition of the experiment at lower concentrations of DTpHAb (0.2 and 1.0 μM) showed similar VBC events (Figures 2 and S2). In addition, at lower DTpHAb concentrations, we observed the formation and collapse of tubular structures at the endosomal membrane (Figure S3 and Movie S3), as previously reported for other bacterial toxins.21 Note that due to the limitations of confocal microscopy, we could only observe the rare events during which both the budded vesicle and the remaining endosome stayed within the same focal plane over the entire VBC process. As such, capturing these rare events became progressively more difficult as the DT concentration decreased.
Figure 2.

A VBC event induced by a lower concentration of DT. HeLa cells were incubated with PSTopFluor (1.5 μM) and DTpHAb (200 nM) for 30 min, washed with DMEM, and imaged by time-lapse confocal microscopy every ~8 s. Images of a single endosome at 15–55 s after initiation of imaging are shown, with the budded/collapsed vesicle indicated by white arrows. Scale bars = 1 μm.
Visualization of VBC Intermediates.
Because of the small size of endosomes (~0.5 μM in diameter), their detailed structural features cannot be well resolved by standard confocal microscopy. We therefore enlarged the endosomes of HeLa cells (to ~2 μm in diameter) by treatment with YM201636, an inhibitor of phosphatidylinositol-3-phosphate kinase-FYVE.22 The cells were next treated with tetramethylrhodamine (TMR)-labeled DT (DTTMR, red channel) and a fluid-phase endocytosis marker, AlexaFluor488-labeled dextran (DextranAlexa488, green channel), for 30 min and imaged by confocal microscopy. This allowed us to visualize the intermediates along the endosomal escape pathway. At the very early stage (stage I), DTTMR was localized to and evenly distributed over the endosomal membrane, whereas DextranAlexa488 was uniformly distributed throughout the endosomal lumen (Figure 3A). At stage II, DTTMR molecules clustered into one or more highly fluorescent foci on the endosomal membrane, which likely represent DT-enriched lipid domain(s). During stage III, endosomes that were undergoing or had just undergone vesicle budding were observed. At stage IV, collapsed vesicles were evident as irregularly shaped objects that emitted red (DTTMR) but not green fluorescence (DextranAlexa488), usually in the vicinity of an intact endosome(s). Note that TMR is a pH-insensitive fluorophore, while DextranAlexa488 has no appreciable binding affinity for the lipid/toxin aggregate and rapidly diffuses away from the latter upon vesicle collapse. A representative VBC event encompassing all above stages is shown in Figure S4 and Movie S4. The observed endosomal structures/intermediates are very similar to those generated by CPP12 (Figure 3B), a highly active cyclic CPP which has previously been demonstrated to exit the endosome by the VBC mechanism.17
Figure 3.

DT- (A), CPP12- (B), and NleC-induced (C) VBC intermediates in enlarged endosomes. HeLa cells were pretreated with YM201636 (800 nM) for 2 h. DextranAlexa (50 μg mL−1) and DTTMR (5 μM), CPP12TMR (2 μM), or NleCTMR (2.5 or 5 μM) were added. After incubation for another 30 min, the cells were washed and imaged by live-cell confocal microscopy. Individual endosomes at different stages of endosomal escape (I–IV) are shown, with each vertical panel (green channel, red channel, and merge) representing a different endosome(s). Scale bars, 1 μm for (A) and (C) and 2 μm for (B). Part (B) was reproduced from ref 17.
Induction of VBC by NleC.
To determine whether the VBC mechanism is unique to DT, we next examined the cellular entry of NleC, a metalloprotease which cleaves NF-κB and depresses downstream transcription events that lead to inflammation.19 NleC consists of a single catalytic domain of 330 residues23,24 and yet shares many characteristics of the AB type bacterial toxins in that it autonomously enters the cytosol of a target cell via lipid-raft mediated endocytosis and endosomal escape.25 Unlike DT, NleC appears to enter the cell in its native conformation, as NleC mutants lacking the native structure are defective in cellular entry.25 HeLa cells were treated with 5 or 0.5 μM pHAb-labeled NleC (NleCpHAb) and imaged by confocal microscopy. Robust VBC events were observed at both concentrations (Figure 4, Movies S5–S6, and Figures S5 and S6). Furthermore, NleC generated similar VBC intermediates to those observed for DT and CPP12 (Figure 3C). Some of the endosomes also formed the same tubular structures as those observed for DT (Figure S7). These results demonstrate that NleC also escapes the endosome by the VBC mechanism.
Figure 4.

Time-lapse confocal microscopy of a HeLa cell undergoing NleC-induced VBC. Cells were incubated with PSTopFluor (1.5 μM) and NleCpHAb (5 μM) for 30 min, washed with DMEM, and imaged after ~7 min. (A) Merged image of a HeLa cell (DIC, green and red channels). (B) Enlarged images of the boxed area in (A) during 15–118 s after the initiation of imaging. Images were acquired every ~8 s. A budded vesicle emerged from an endosome (marked by a white arrow) at 79 s, separated from the endosome at 110 s, and collapsed at 118 s. Scale bars, 1 and 10 μm.
Discussion.
In this work, we provide direct evidence that DT and NleC escape the endosome by inducing VBC from the endosomal membrane. This mechanism reconciles many of the previously enigmatic observations documented in the lietrature, which in turn provide further support for our mechanism (Table 1). Since the toxin molecules do not physically traverse any cell membrane, the VBC mechanism readily explains how bacterial toxins enter the cytosol in their folded states. Similarly, macromolecular cargoes (e.g., proteins10 and nucleic acids11,12) can be delivered into the cytosol in their folded states irrespective of their size or the nature of conjugation to DT (covalent attachment10 or noncovalent association11,12), so long as they do not interfere with the binding of DT to the cell-surface receptor or the endosomal membrane. The fact that NleC enters host cells in its native conformation25 demonstrates that acid-mediated conformational change and membrane insertion, although observed for many bacterial toxins,1–3 are not a prerequisite for membrane translocation. We hypothesize that DT, NleC and other protein toxins bind selectively to the budding neck and stabilize the negative Gaussian curvature (i.e., simultaneous positive and negative membrane curvatures in orthogonal directions) at the budding neck, thereby promoting VBC from the endosomal membrane.18 In the case of DT, amphipathic helices 1 and 2 of the T-domain, which contain both positively charged and hydrophobic residues, are likely resposnible for membrane binding8,9 and inducing negative Gaussian curvature at the budding neck. Membrane insertion of the T-domain likely increases the binding affinity of the toxin for the endosomal membrane, enabling VBC at low toxin concentrations (vide infra). The acidic pH inside the endosome likely facilitates VBC by promoting conformational change, partial unfolding, and membrane insertion of the T-domain.26 The acidic pH may also increase the binding affinity of the toxins for the endosomal membrane by protonating a membrane component(s), as previously observed for CPPs.16
Due to technical limitations, we have not yet been able to observe VBC events at low nM or pM DT concentrations, raising the question whether the VBC mechanism is operative under physiologically relevant DT concentrations. For a VBC event to take place, an endosome must contain a minimum number of DT molecules. Hudson and Neville showed that each endosomal escape event releases ~80 DT molecules into the cytosol simultaneously, irrespective of the extracellular DT concentration.13 This suggests that for DT, the minimum number of molecules required to induce a VBC event is ≥80, corresponding to an intraluminal concentration of ~2 μM (by assuming that an average endosome has a diameter of 0.5 μm). The latter concentration is orders of magnitude higher than the physiological (extracellular) DT concentration (pM to low nM), indicating that DT molecules must accumulate into one or more endosomes in order to escape from the endosome(s). Note that the concentration event is necessary for any endosomal escape mechanism, as one molecule/endosome equals to ~25 nM, which is still higher than the physiological DT concentration.
How do ≥80 DT molecules accumulate into an endosome when the extracellular concentration is in the pM range? Receptor-mediated endocytic uptake likely causes significant concentration of DT from the extracellular environment into the endosomes. In addition, the endosomes of a mammalian cell are interconnected through constant vesicle fusion and fission events. Lateral fusion of endosomes coupled with clustering of DT molecules into lipid domains (Figure 3A) likely also plays a role in gradually concentrating the endocytosed DT molecules into fewer endosomes, which culminate to reach the “quantum” for VBC.27 Membrane insertion of the T-domain should increase the binding affinity of DT for the endosomal membrane, ensuring that most of the ≥80 DT molecules inside an endosome stay bound to the endosomal membrane to promote VBC. This scenario is supported by the previous observation that there exists a DT concentration-dependent lag between the time of endocytic uptake and endosomal acidification and the time of DT release into the cytosol (i.e., longer lag at lower DT concentration).13,28 It also suggests that the VBC mechanism should operate under any toxin concentraion; it merely requires a longer period of time for toxin molecules to accumulate into one or more endosomes and reach the “quantum” for VBC when the toxin concentration is low. Finally, each VBC event is accompanied by the release of a small endosomal volume into the cytosol. This would result in the release of unassociated cargoes (e.g., dye-labeled dextran) from the endosome into the cytosol.29
CONCLUSION
In conclusion, we have demonstrated that two structurally different bacterial protein toxins, DT and NleC, both escape the endosome by the VBC mechanism. To our knowledge, this is the first experimentally validated mechanism by which a folded, biologically active protein autonomously translocates across a lipid bilayer. Since this mechanism is also shared by CPPs and non-peptidic cell-penetrating molecules,17 we propose that it may be a common mechanism for other bacterial toxins, non-enveloped viruses, and synthetic drug delivery systems.15
EXPERIMENTAL SECTION
Materials.
Cell culture media, fetal bovine serum (FBS), penicillin-streptomycin, 0.25% trypsin-EDTA, and DPBS (2.7 mM potassium chloride, 1.5 mM monopotassium phosphate, 8.9 mM disodium hydrogen phosphate, and 137 mM sodium chloride) were purchased from Sigma-Aldrich (St. Louis, MO). 5(6)-Carboxytetramethylrhodamine succinimidyl ester (TMR-NHS) and HisPur™ Ni-NTA resin were purchased from Thermo Fisher Scientific (Waltham, MA). Fluorescent pHAb dye (amine reactive) was purchased from Promega (Madison, WI). TopFluor PS (1-palmitoyl-2-(dipyrrometheneboron difluoride)undecanoyl-snglycero-3-phospho-L-serine, ammonium salt) was purchased from Avanti Polar Lipids (Alabaster, AL). Endosome maturation inhibitor YM201636 was purchased from Cayman Chemical Company (Ann Arbor, MI). Dextran Alexa Fluor 488 (10,000 MW, anionic) was purchased from Invitrogen (Carlsbad, CA). All other chemical reagents were obtained from Sigma-Aldrich, Chem-Impex (Wood Dale, IL), Thermo Fisher Scientific, Acros Organics (Fair Lawn, NJ) or VWR (West Chester, PA) and were used without further purification. The plasmid used to express Diphtheria toxin (K51E/E148K mutant), pET-22b DT 51E/148K, was a generous gift from Professor Dmitri Kudryashov (The Ohio State University, Columbus, OH) (original source - John Collier, Addgene plasmid #11081; http://n2t.net/addgene:11081; RRID: Addgene_11081). The plasmid used to express wild-type NleC, pET-28a NleC, was a generous gift from Professor Fengyi Wan (Johns Hopkins University, Baltimore, MD). HeLa cells were obtained from ATCC (Manassas, VA).
Protein Expression and Purification.
Enzymatically inactive DT mutant (K51E/E148K) containing an N-terminal pelB leader sequence and a C-terminal hexa-His affinity tag was expressed in E. coli BL21(DE3) cells and purified by modifying a previously published protocol.30 Briefly, 10 mL of Luria-Bertani (LB) medium containing 75 mg L−1 ampicillin was inoculated with a single colony and incubated overnight at 37 °C with shaking (180 rpm). The next day, the seed culture was diluted to 1 L of LB media supplemented with 75 mg L− ampicillin and incubated at 37 °C until OD600 reached 0.6−0.8, at which point the culture was induced with 1 mM isopropyl β-D-thiogalactopyranoside (IPTG) and incubated for 16 h at 18 °C. The cells were pelleted by centrifugation at 5000 rpm (Sorvall SLA-3000 rotor) for 30 min at 4 °C, and the pellet was stored at −80 °C. For extraction of the periplasmic DT, the pelleted cells (from ½ L of culture) were first resuspended in 100 mL of Tris buffer (30 mM, pH 8.0) containing 20% sucrose and 1 mM EDTA. After 20 min at 4 °C, the cells were centrifuged at 15000 rpm (Sorvall SS-34 rotor) for 30 min at 4 °C, and the supernatant was discarded. The cell pellet was resuspended in 100 mL of ice-cold 5 mM MgSO4 and incubated on ice for 15 min. The cells were again pelleted by centrifugation at 15000 rpm and the supernatant containing DT was loaded onto a Ni-NTA affinity column equilibrated with Tris buffer (20 mM, pH 8.0) containing 300 mM NaCl and 25 mM imidazole. It was allowed to incubate with the resin at 4 °C for 1 h to ensure complete binding. Bound protein was eluted with Tris buffer (20 mM, pH 8.0) containing 300 mM NaCl and 300 mM imidazole. Fractions containing the desired protein were pooled, concentrated, and exchanged into PBS (pH 7.4) using an Amicon Ultra-15 centrifugal concentrator (30K MWCO, EMD Millipore).
Recombinant wild-type NleC, bearing an N-terminal hexa-histidine affinity tag, was similarly expressed in E. coli BL21(DE3) cells in LB media containing 50 mg L−1 kanamycin. Cells were induced with 0.3 mM IPTG and incubated for 16 h at 18 °C. The cell pellet obtained after centrifugation (from 1 L of culture) was lysed in 50 mL of lysis buffer [20 mM Tris buffer, pH 8.0, 150 mM NaCl, 20 mM imidazole, 0.5 mg mL−1 lysozyme, 1 mM PMSF, 2 mM DTT, and two EDTA-free protease inhibitor cocktail tablets (Roche)] by sonication. After centrifugation for 30 min at 15000 rpm (Sorvall SS-34 rotor) at 4 °C, the clear cell lysate was loaded onto a Ni-NTA affinity column equilibrated with Tris buffer (20 mM, pH 8.0) containing 150 mM NaCl, 2 mM DTT and 20 mM imidazole. It was allowed to incubate with the resin at 4 °C for 1 h to ensure complete binding. The bound protein was eluted with Tris buffer (20 mM, pH 8.0) containing 150 mM NaCl, 2 mM DTT and 300 mM imidazole. Fractions containing the desired protein were pooled, concentrated, and exchanged into PBS (pH 7.4, containing 2 mM DTT) using an Amicon Ultra-15 centrifugal concentrator (30K MWCO, EMD Millipore).
The purity of both proteins was judged to be ≥95% by SDS-PAGE. Protein concentration was determined by the Bradford assay using bovine serum albumin as standard. The yields of DT and NleC were ~9 and 11 mg per liter of culture, respectively. Protein solutions were mixed with glycerol (20% final), aliquoted, quickly frozen in a dry ice/isopropanol bath, and stored at –80 °C.
Fluorescent Labeling of Proteins.
Protein labeling was carried out by treating freshly thawed protein (3–7 mg mL−1 in PBS, pH 7.4) with 4–6 equivalents of a proper dye molecule (pHAb-NHS or TMR-NHS, dissolved in minimal volume of DMF) at 4 °C overnight in the dark. For labeling of NleC, 2 mM tris(carboxyethyl)phosphine (TCEP) was added to the reaction solution to prevent disulfide formation (Note: Reducing agent should not be used for DT labeling, to avoid reduction of the internal disulfide bonds). Unreacted dye was removed by passing the reaction mixture through a desalting spin column (Bio-Rad, 6K MWCO). Protein concentration was measured by the Bradford assay. The stoichiometry of dye labeling (dye/protein ratio) was determined by comparing the absorbances of the labeled protein at 280 nm and λmax of the dye:
where ε is the molar extinction coefficient and C.F. is the correction factor of the dye. The λmax, ε, and C.F. values for the dyes and proteins used are listed in Table 2.
Table 2.
Absorption Properties of Compounds Used in This Study
| λ (nm) | ε (cm−1 M−1) | C.F. | Source | |
|---|---|---|---|---|
| NleC | 280 | 36,330 | N/A | Theoretically determined |
| DT | 280 | 56,060 | N/A | Theoretically determined |
| pHAb | 532 | 75,000 | 0.256 | Manufacturer’s protocol |
| TMR | 555 | 80,000 | 0.34 | Manufacturer’s protocol |
Cell Culture.
HeLa cells were maintained in DMEM supplemented with 10% FBS and 1% penicillin-streptomycin (Abs). Cells were cultured in a humidified incubator at 37 °C in the presence of 5% CO2.
Confocal Microscopy.
All microscopy experiments were performed on live cells. Three hundred mL of HeLa cell suspension (5 × 104 cells/mL) was seeded per compartment in a 35/10 mm glass-bottomed microwell dish with four compartments (Greiner Bio-One) and cultured overnight in DMEM containing 10% FBS and 1% Abs. Unless specified otherwise, phenol red-free DMEM (1% FBS, 1% Abs) was used during incubation and imaging. Cells were imaged on a Nikon A1R live-cell confocal laser scanning microscope (ECLIPSE Ti-E automated, inverted) equipped with a 100× oil objective (1.45 N.A.) and a heated chamber (37 °C) supplied with 5% CO2. Nikon’s Perfect Focus System (PFS) was used for all time-lapse image acquisitions. Differential interference contrast (DIC) images were also obtained to ensure that cells were healthy throughout the imaging process. Data were processed and analyzed on NIS-Elements AR. Typically, images were denoised using Nikon’s “Denoise.ai” feature, and scale bars and time stamps were added where needed. Images were exported in the 8-bit TIFF format. Detailed protocols for cell treatment and imaging parameters are described below.
DTpHAb and PSTopFluor.
Before the experiment, cells were incubated at RT for 10 min and washed twice with DPBS. Media (500 μL per well) containing TopFluor-labeled PS (1.5 μM) was added to the cells, and the cells were incubated at RT for 10 min. The cells were gently washed with DPBS (4 × 300 μL) and incubated with warm phenol red-free DMEM media (300 μL per well) containing 1% FBS, 1% Abs, 5 μM DTpHAb. After 30 min at 37 °C and 5% CO2, the cells were washed with media (2 × 300 μL) and supplemented with 5 μM unlabeled DT in 300 μL of phenol red-free DMEM containing 1% FBS and 1% Abs. Time-lapse imaging was commenced, with images acquired every 2–4 s. For imaging at lower DT concentration, cells were washed as described above, and 300 μL of phenol red-free DMEM containing 1% FBS, 1% Abs, PSTopFluor (1.5 μM), and 200 nM DTpHAb was added to the cells, and the cells were incubated at 37 °C, 5 % CO2 for 30 min. The cells were gently washed with media (2 × 300 μL) and supplemented with fresh media (300 μL of phenol red-free DMEM media containing 1% FBS and 1% Abs). Time-lapse imaging was commenced, with images acquired every ~8 s. For the red channel (DTpHAb), the laser line with λEx 561 nm was used, while for the green channel (PSTopFluor), the laser line with λEx 487 nm was used.
NleCpHAb and PSTopFluor.
Before the experiment, cells were washed twice with DPBS. Three hundred μL of phenol red-free DMEM containing 1% FBS, 1% Abs, PSTopFluor (1.5 μM), and 5 μM NleCpHAb was added to the cells, and the cells were incubated at 37 °C, 5 % CO2 for 30 min. The cells were gently washed with media (2 × 300 μL) and supplemented with fresh media (300 μL of phenol red-free DMEM media containing 1% FBS and 1% Abs). Time-lapse imaging was commenced, with images acquired every 2–8 s. For imaging at lower NleC concentration (500 nM), cells were washed as described above, and 300 μL of phenol red-free DMEM containing 1% FBS, 1% Abs, PSTopFluor (1.5 μM), and 500 nM NleCpHAb was added to the cells, and the cells were incubated at 37 °C, 5 % CO2 for 30 min or 1 h. The cells were gently washed with media (2 × 300 μL) and supplemented with fresh media (300 μL of phenol red-free DMEM media containing 1% FBS and 1% Abs). Time-lapse imaging was commenced, with images acquired every 2–8 s. For the red channel (NleCpHAb), the laser line with λEx 561 nm was used, while for the green channel (PSTopFluor), the laser line with λEx 487 nm was used.
Enlarged Endosomes with YM201636.
Cells were washed twice with warm DPBS and then incubated with 800 nM YM201636 in phenol red-free DMEM containing 1% FBS and 1% Abs (300 μL per well) for 2 h. DTTMR or NleCTMR (2.5 or 5 μM) and DextranAlexa488 (50 μg mL−1) were added to the cells, and the cells were incubated for another 30 min. The cells were gently washed with warm DPBS (twice) to remove the compounds and incubated in phenol red-free DMEM (300 μL). Cells with swollen intracellular vesicles were isolated and subjected to time-lapse imaging at 10 to 30-s intervals. For the green channel (DextranAlexa488) the laser line with λEx 487 nm was used, and for the red channel (DTTMR or NleCTMR) the laser line with λEx 561 nm was used.
Supplementary Material
ACKNOWLEDGMENT
We thank D. Kudryashov (The Ohio State University) for the plasmid encoding DT and F. Wan (Johns Hopkins University) for the plasmid encoding NleC.
Funding Sources
This work was supported by the National Institutes of Health (GM122459). Images presented in this report were generated using the instruments and services at the Campus Microscopy and Imaging Facility, The Ohio State University. This facility is supported in part by grant P30 CA016058, National Cancer Institute, Bethesda, MD.
ABBREVIATIONS
- CPP
cell-penetrating peptide
- DT
diphtheria toxin
- PBS
phosphate buffered saline
- VBC
vesicle budding and collapse
Footnotes
Supporting Information. Additional confocal images (Figures S1–S7) and movies (S1–S6) showing vesicle budding-and-collapse events from the endosomes in live cells. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interests.
REFERENCES
- (1).Henkel JS, Baldwin MR, and Barbieri JT (2010) Toxins from bacteria. EXS 100, 1–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Williams JM and Tsai B (2016) Intracellular trafficking of bacterial toxins. Curr. Opin. Cell Biol 41, 51–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Beilhartz GL, Sugiman-Marangos SN, and Melnyk RA (2017) Repurposing bacterial toxins for intracellular delivery of therapeutic proteins. Biochem. Pharmacol 142, 13–20. [DOI] [PubMed] [Google Scholar]
- (4).Parveen S, Bishai WR, and Murphy JR (2019) Corynebacterium diphtheriae: diphtheria toxin, the tox operon, and its regulation by Fe2+ activation of apo-DtxR. Microbiol. Spectrum 7, GPP3–0063-2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Boquet P, Silverman MS, Pappenheimer AM Jr., and Vernon WB (1976) Binding of triton X-100 to diphtheria toxin, crossreacting material 45, and their fragments. Proc. Natl. Acad. Sci. U. S. A 73, 4449–4453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Donovan JJ, Simon MI, Draper RK, and Montal M (1981) Diphtheria toxin forms transmembrane channels in planar lipid bilayers. Proc. Natl. Acad. Sci. U. S. A 78, 172–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Ladokhin AS, Vargas-Uribe M, Rodnin MV, Ghatak C, and Sharma O (2017) Cellular entry of the diphtheria toxin does not require the formation of the open-channel state by its translocation domain. Toxins 9, 299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Madshus IH (1994) The N-terminal alpha-helix of fragment B of diphtheria toxin promotes translocation of fragment A into the cytoplasm of eukaryotic cells. J. Biol. Chem 269, 17723–17729. [PubMed] [Google Scholar]
- (9).Ratts R, Trujillo C, Bharti A, vanderSpek J, Harrison R, and Murphy JR (2005) A conserved motif in transmembrane helix 1 of diphtheria toxin mediates catalytic domain delivery to the cytosol. Proc. Natl. Acad. Sci. U. S. A 102, 15635–15640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Auger A, Park M, Nitschke F, Minassian LM, Beilhartz GL, Minassian BA, and Melnyk RA (2015) Efficient delivery of structurally diverse protein cargo into mammalian cells by a bacterial toxin. Mol. Pharm 12, 2962–2971. [DOI] [PubMed] [Google Scholar]
- (11).Kakimoto S, Hamada T, Komatsu Y, Takagi M, Tanabe T, Azuma H, Shinkai S, and Nagasaki T (2009) The conjugation of diphtheria toxin T domain to poly(ethylenimine) based vectors for enhanced endosomal escape during gene transfection. Biomaterials 30, 402–408. [DOI] [PubMed] [Google Scholar]
- (12).Arnold AE, Smith LJ, Beilhartz GL, Bahlmann LC, Jameson E, Melnyk RA, and Shoichet MS (2020) Attenuated diphtheria toxin mediates siRNA delivery. Sci. Adv 6, eaaz4848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Hudson TH, and Neville DM Jr. (1985) Quantal entry of diphtheria toxin to the cytosol. J. Biol. Chem 260, 2675–2680. [PubMed] [Google Scholar]
- (14).Brock DJ, Kondow-McConaghy HM, Hager EC, and Pellois JP (2019) Endosomal escape and cytosolic penetration of macromolecules mediated by synthetic delivery agents. Bioconjugate Chem. 30, 293–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Pei D, and Buyanova M (2019) Overcoming endosomal entrapment in drug delivery. Bioconjugate Chem. 30, 273–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Qian Z, Martyna A, Hard RL, Wang J, Appiah-Kubi G, Coss C, Phelps MA, Rossman JS, and Pei D (2016) Discovery and mechanism of highly efficient cyclic cell-penetrating peptides. Biochemistry 55, 2601–2612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Sahni A, Qian Z, and Pei D (2020) Cell-penetrating peptides escape the endosome by inducing vesicle budding and collapse. ACS Chem. Biol 15, 2485–2492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Dougherty PG, Sahni A, and Pei D. (2019) Understanding cell penetration of cyclic peptides. Chem Rev. 119, 10241‐10287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Hodgson A, Wier EM, Fu K, Sun X, Yu H; Zheng W, Sham HP, Johnson K, Bailey S, Vallance BA, and Wan F (2015) Metalloprotease NleC suppresses host NF-κB/inflammatory responses by cleaving p65 and interfering with the p65/RPS3 interaction. PLoS Pathog. 11, e1004705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Robers MB, Binkowski BF, Cong M, Zimprich C, Corona C, McDougall M, Otto G, Eggers CT, Hartnett J, Machleidt T, Fan F, and Wood KV (2015) A luminescent assay for real-time measurements of receptor endocytosis in living cells. Anal. Biochem 489, 1–8. [DOI] [PubMed] [Google Scholar]
- (21).Römer W, Berland L, Chambon V, Gaus K, Windschiegl B, Tenza D, Aly MR, Fraisier V, Florent JC, Perrais D, Lamaze C, Raposo G, Steinem C, Sens P, Bassereau P, and Johannes L (2007) Shiga toxin induces tubular membrane invaginations for its uptake into cells. Nature 450, 670–675. [DOI] [PubMed] [Google Scholar]
- (22).Jefferies HBJ, Cooke FT, Jat P, Boucheron C, Koizumi T, Hayakawa M, Kaizawa H, Ohishi T, Workman P, Waterfield MD, and Parker PJ (2008) A selective PIKfyve inhibitor blocks PtdIns(3,5)P(2) production and disrupts endomembrane transport and retroviral budding. EMBO Rep. 9, 164–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Turco MM, and Sousa MC (2014) The structure and specificity of the type III secretion system effector NleC suggest a DNA mimicry mechanism of substrate recognition. Biochemistry 53, 5131–5139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Li W, Liu Y, Sheng X, Yin P, Hu F, Liu Y, Chen C, Li Q, Yan C, and Wang J (2014) Structure and mechanism of a type III secretion protease, NleC. Acta Crystallogr D Biol Crystallogr. 70, 40–47. [DOI] [PubMed] [Google Scholar]
- (25).Stolle AS, Norkowski S, Körner B, Schmitz J, Lüken L, Frankenberg M, Rüter C, and Schmidt MA (2017) T3SS-independent uptake of the short-trip toxin-related recombinant NleC effector of enteropathogenic Escherichia coli leads to NF-κB p65 cleavage. Front. Cell. Infect. Microbiol 13, 119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Zhao JM, and London E (1988) Conformation and model membrane interactions of diphtheria toxin fragment A. J. Biol. Chem 263, 15369–15377. [PubMed] [Google Scholar]
- (27).Antignani A, and Youle RJ (2008) Endosome fusion induced by diphtheria toxin translocation domain. Proc. Natl. Acad. Sci. U. S. A 105, 8020–8025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Marnell MH, Shia SP, Stookey M, and Draper RK (1984) Evidence for penetration of diphtheria toxin to the cytosol through a prelysosomal membrane. Infect. Immun 44, 145–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Sharpe J and London E (1999) Diphtheria toxin forms pores of different sizes depending on its concentration in membranes: Probable relationship to oligomerization. J. Membr. Biol 171, 209–221. [DOI] [PubMed] [Google Scholar]
- (30).Paoletti LC, Peterson DL, Legmann R, and Collier RJ (2001) Preclinical evaluation of group B streptococcal polysaccharide conjugate vaccines prepared with a modified diphtheria toxin and a recombinant duck hepatitis B core antigen. Vaccine 20, 370–376. [DOI] [PubMed] [Google Scholar]
- (31).Umata T, Moriyama Y, Futai M, and Mekada E (1990) The cytotoxic action of diphtheria toxin and its degradation in intact Vero cells are inhibited by bafilomycin A1, a specific inhibitor of vacuolar-type H(+)-ATPase. J. Biol. Chem 265, 21940–21945. [PubMed] [Google Scholar]
- (32).Finkelstein A, Oh KJ, Senzel L, Gordon M, Blaustein RO, and Collier RJ (2000) The diphtheria toxin channel-forming T-domain translocates its own NH2-terminal region and the catalytic domain across planar phospholipid bilayers. Int. J. Med. Microbiol 290, 435–440. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.