

SUMMARY
Adult mammalian tissues such as heart, brain, retina, and the sensory structures of the inner ear do not effectively regenerate, although a latent capacity for regeneration exists at embryonic and perinatal times. We explored the epigenetic basis for this latent regenerative potential in the mouse inner ear and its rapid loss during maturation. In perinatal supporting cells, whose fate is maintained by Notch-mediated lateral inhibition, the hair cell enhancer network is epigenetically primed (H3K4me1) but silenced (active H3K27 de-acetylation and trimethylation). Blocking Notch signaling during the perinatal period of plasticity rapidly eliminates epigenetic silencing and allows supporting cells to transdifferentiate into hair cells. Importantly, H3K4me1 priming of the hair cell enhancers in supporting cells is removed during the first post-natal week, coinciding with the loss of transdifferentiation potential. We hypothesize that enhancer decommissioning during cochlear maturation contributes to the failure of hair cell regeneration in the mature organ of Corti.
Graphical abstract

In brief
Mammals are unable to regenerate cochlear sensory hair cells, while other vertebrates preserve robust inner ear regenerative capacity. We show that epigenetic decommissioning of hair-cell-specific enhancers (removal of H3K4me1) in supporting cells during perinatal maturation contributes to loss of regenerative capacity in the mammalian inner ear.
INTRODUCTION
Failure to regenerate damaged tissues is a major cause of human disability (Kwan et al., 2009; Ming and Song, 2011; Goldman, 2014; Little, 2016; Uygur and Lee, 2016). In mammals, many adult tissues lack stem cell reserves and thus even a limited ability for repair. However, during a brief neonatal period, some tissues in mice, such as the heart, retina, and kidney, show a limited ability to regenerate after damage (Hartman et al., 2007; Porrello et al., 2011; Haubner et al., 2012; Ueki et al., 2015). Similarly, cardiac regeneration, neonephrogenesis, and even digit tip re-growth have been observed in young children (Illingworth, 1974; Faa et al., 2010; Rupp and Schranz, 2015). These observations suggest that regenerative capacity may be present in a latent form but that specific mechanisms associated with mammalian maturation may cause the subsequent failure of regeneration in adults. In particular, changes in the epigenetic status of cell-type-specific enhancer networks during maturation has been proposed to underlie the failure of many mammalian tissues to regenerate (Ong and Corces, 2012; Kang et al., 2016; Yang and Kang, 2019; Nicetto and Zaret, 2019; Goldman and Poss, 2020).
The organ of Corti, the auditory portion of the mammalian inner ear, consists of an ordered mosaic of mechanosensitive hair cells and associated supporting cells within the cochlear duct (Kelley, 2006). Hair cells and supporting cells differentiate from a post-mitotic progenitor population that is established between E12.5 and E14.5 in the mouse (Ruben, 1967; Lee et al., 2006; Driver et al., 2013). This population remains un-committed to a hair or supporting cell fate until the onset of Atoh1 expression (Woods et al., 2004; Abdolazimi et al., 2016; Tateya et al., 2019), a bHLH transcription factor that is both necessary and sufficient for hair cell differentiation (Bermingham et al., 1999; Zheng and Gao, 2000; Chen et al., 2002; Woods et al., 2004; Gubbels et al., 2008; Kelly et al., 2012). ATOH1 regulates expression of the Notch ligands Jag2 and Dll1 in hair cells, leading to lateral inhibition and suppression of a hair cell fate in the surrounding cells, which subsequently differentiate as supporting cells (Woods et al., 2004; Kiernan et al., 2005; Groves, 2010; Neves et al., 2013; Abdolazimi et al., 2016).
In mammals, the failure to replace hair cells after environmental trauma or aging leads to permanent deafness (Furness, 2015; Wong and Ryan, 2015; Wagner and Shin, 2019). In contrast, non-mammalian vertebrates mount a robust regenerative response after damage, in which supporting cells can proliferate and transdifferentiate into new hair cells (Cotanche, 1987; Cruz et al., 1987; Corwin and Cotanche, 1988; Ryals and Rubel, 1988; Balak et al., 1990; Harris et al., 2003). Interestingly, mouse post-natal day 1 (P1) organ of Corti supporting cells harbor a latent capacity to produce hair cells (White et al., 2006) that can be stimulated by blocking Notch signaling, leading to the upregulation of Atoh1 (Yamamoto et al., 2006; Zhao et al., 2011; Korrapati et al., 2013). However, this transdifferentiation potential is transient, and by P6, neither Notch inhibition nor ectopic Atoh1 overexpression is sufficient to stimulate regeneration (Liu et al., 2012; Kelly et al., 2012; Maass et al., 2015; Figure 4A). The mechanisms underlying this rapid loss of regenerative potential are poorly understood.
Figure 4. Decommissioning of hair cell gene enhancers (H3K4me1 removal) accompanies post-natal maturation of supporting cells.
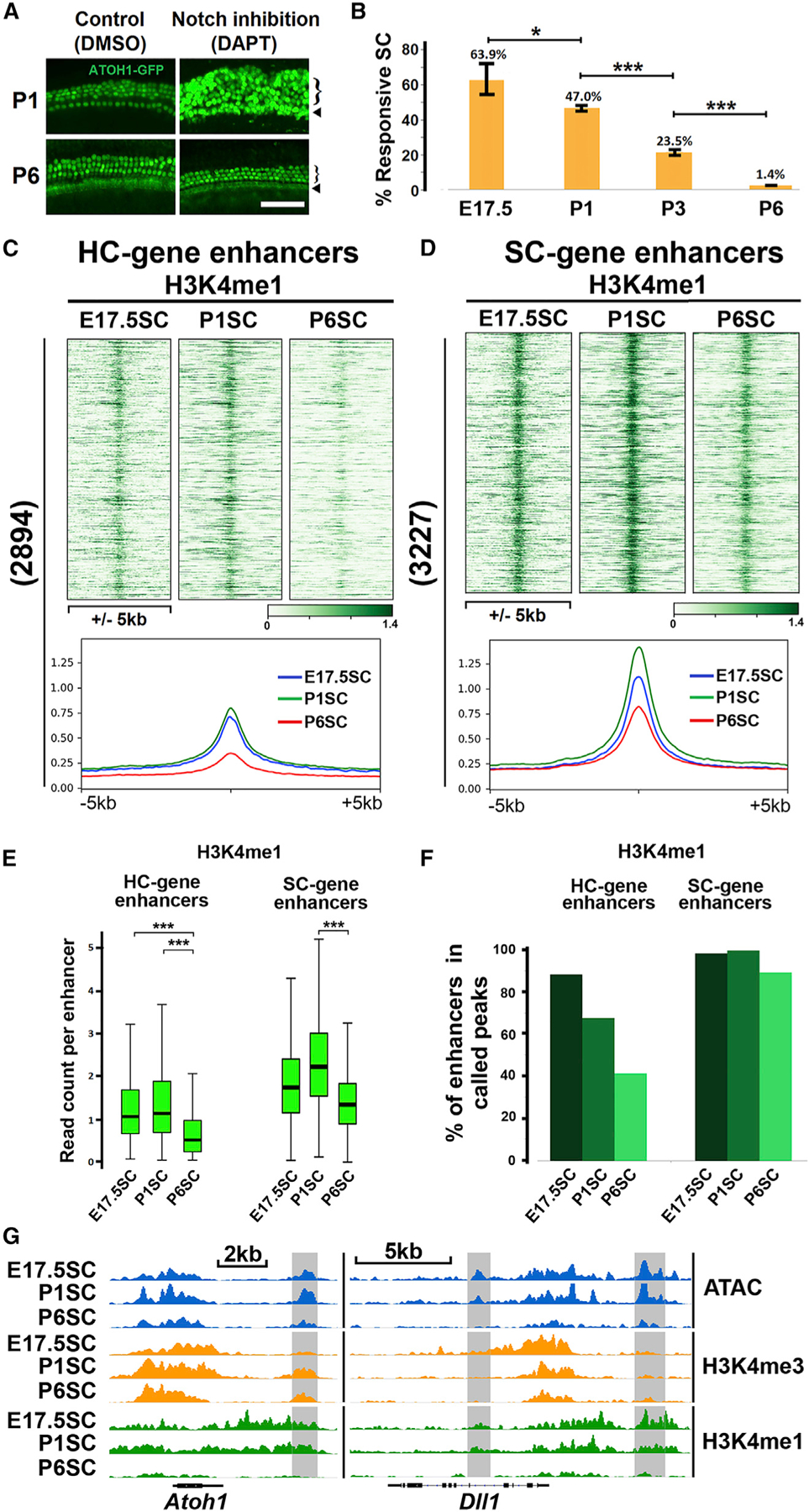
(A) Cochlear supporting cells lose the potential to transdifferentiate into hair cells with age. Organ of Corti explants from P1 and P6 cochleae were treated with the Notch inhibitor (DAPT, 10 μM) or DMSO control for 72 h to induce transdifferentiation of supporting cells to hair cells. Three rows of outer hair cells (bracket) and one row of inner hair cells (arrow) were observed in both P1 and P6 control organs. Notch inhibition induces large numbers of supernumerary hair cells (ATOH−GFP+) in P1 organs, but not in P6 organs. Scale bar, 50 μm.
(B) Progressive loss of transdifferentiation potential within the supporting cell population from E17.5 to P6. Lineage-labeled organ of Corti explants (Figure 3A) from E17.5, P1, P3, and P6 were treated with DAPT for 48 h, and responsive supporting cells (tdTomato and ATOH1-GFP double positive, Figure 3B) were quantified by FACS. The percentage of responsive supporting cells (calculated by dividing the number of tdTomato and GFP-double-positive cells by the number of all tdTomato positive cells) declines as the cochlea matures (63.9% ± 10.2% in E17.5 organs, 47.0% ± 4.8% in P1, 23.5% ± 2.5% in P3, and 1.4% ± 0.2% in P6) (Error bars, standard deviation; *, **, and ***, adjusted p value < 0.1; < 0.01; < 0.001, respectively; n ≥ 3).
(C) H3K4me1 priming at hair cell gene enhancers in supporting cells is strongly diminished between E17.5 and P6. Heatmaps and average signal profiles show decreased H3K4me1 epigenetic profiles surrounding hair cell gene enhancers in supporting cells during perinatal maturation.
(D) Supporting cell gene enhancers remain primed by H3K4me1 in E17.5, P1, and P6 supporting cells, as shown by heatmaps and average signal profiles.
(E) Quartile distribution of normalized read count per enhancer (±500 bp, boxplots) shows significant reduction of the enhancer priming mark H3K4me1 at hair cell gene enhancers in P6 supporting cells. H3K4me1 at supporting cell gene enhancers is modestly reduced in P6 supporting cells but maintained at a level much higher than hair cell gene enhancers (***, adjusted p value < 0.001, paired Wilcoxon test).
(F) Quantification of hair cell and supporting cell gene enhancers that overlap with H3K4me1 peaks in E17.5, P1, and P6 supporting cells. There is a clear age-dependent decline in the percentage of hair cell gene enhancers that overlap with H3K4me1 peaks (86% in E17.5, 68% in P1, and 41% in P6 supporting cells), while >90% of supporting cell gene enhancers remain H3K4me1 positive.
(G) IGV genome browser tracks showing enhancers (gray boxes) associated with Atoh1 and Dll1, depleted of H3K4me1 in P6 supporting cells, while H3K4me3 is maintained at their promoters.
We hypothesized that epigenetic silencing may contribute to the age-dependent failure of hair cell regeneration in mammals. We first analyzed the transcriptomes, chromatin accessibility profiles, and histone modifications of hair cells and supporting cells in neonatal mice, when supporting cells are still capable of transdifferentiation. We discovered that hair cell genes are silenced in supporting cells by active histone deacetylation and the presence of H3K27me3. However, the hair cell regulatory network remains epigenetically ‘‘poised’’ and ‘‘primed’’ by H3K4me3 and H3K4me1 at hair cell gene promoters and enhancers, respectively. We show that when supporting cells are driven to transdifferentiate into hair cells by Notch inhibition, this primed-but-silenced state is rapidly changed by loss of H3K27me3 and an increase in H3K27ac. We further show that hair cell enhancer priming by H3K4me1 is gradually and specifically lost with age, and this enhancer decommissioning process correlates with the loss of transdifferentiation potential. Blocking decommissioning by inhibiting H3K4me1 removal prolongs the transdifferentiation potential of supporting cells. Finally, unlike in the cochlea, H3K4me1 commissioning of hair cell enhancers in supporting cells of the vestibular system persists into adulthood, correlating with the limited capacity of the vestibular system to regenerate (Golub et al., 2012). We hypothesize that the epigenetic changes that we describe may contribute to the loss of regenerative potential of the mammalian cochlea with age.
RESULTS
Atoh1 plays a prominent role in the gene regulatory network of differentiating hair cells
To identify the genes and regulatory elements needed for direct transdifferentiation of neonatal supporting cells to a hair cell fate, we compared the transcriptomes of P1 hair cells and supporting cells, independently FACS-purified from Atoh1-Gfp and Lfng-Gfp reporter mice, respectively (Rose et al., 2009; Gong et al., 2003; Figure 1A). We identified 949 hair-cell-enriched genes (adjusted p value <0.01; fold change >2; RPKM in hair cells >1; and baseMean >50, DESeq2; Figure 1A). GO term enrichment analysis of hair cell genes revealed terms such as ‘‘cilium assembly’’ and ‘‘sensory perception of sound’’ (Figure 1A) and included known hair cell genes, such as Atoh1, Pou4f3, Gfi1, Myo6, Myo7a, and Jag2 (Table S1).
Figure 1. The hair cell gene regulatory network reveals the prominent role of Atoh1.

(A) Purifition (FACS) and analysis (RNA-seq) of hair cells and supporting cells from P1 mouse cochlea. From top: inner ear (paint fill) in the P1 mouse head; the spiral cochlea expressing ATOH1-GFP; depiction of organ of Corti cross-section (one inner and three outer hair cells [green], and supporting cells [red]); P1 hair cells or supporting cells FACS-purified and analyzed (RNA-seq) for enriched genes (HC genes: log2(fold change) > 1; −log10 (adjusted p value) > 2(green dots); SC genes: log2(fold change) < −1 and −log10 (adjusted p value) > 2 [red dots]). Hair-cell-related terms (gene ontology−biological process−EnrichR; Chen et al., 2013). Lists of hair cell genes, supporting cell genes, and their TSSs can be found in Table S1. Paint-fill courtesy of Drs. Donna Fekete and John Brigande.
(B) Categorical heatmaps showing putative P1 hair cell enhancers based on chromatin accessibility (ATAC-seq), H3K4me1 and H3K27ac. Potential hair cell gene distal regulatory elements categorized by open chromatin structure (ATAC-seq peaks) lying within ± 200 kb of hair cell gene TSS (regions ± 2 kb from TSS were excluded as proximal elements) and ranked by signal strength from ATOH1 CUT&RUN. Putative ‘‘active’’ enhancers enriched for both H3K4me1 and H3K27ac (2,894); ‘‘inactive’’ enhancers marked by H3K4me1-alone (4,239), and ‘‘unmarked’’ (3,407). There are 1,212 active enhancers (above dashed line) overlapping with ATOH1 peaks (MACS2, FDR < 0.01). Regions around peak summit (±5 kb) are shown in the heatmap.
(C) Representative genome browser tracks (IGV) of hair-cell-specific genes Atoh1 and Rasd2. Putative active enhancers (gray bars) identified by epigenetic characteristics (B). A known Atoh1 3′ enhancer is accessible (ATAC peak), H3K4me1 enriched, H3K27ac-positive, and bound by ATOH1. A predicted Rasd2 5′ enhancer is similarly marked and bound by ATOH1.
(D) De novo ATOH1-binding motif in E17.5 hair cells. Motif enrichment shown (p value of 1−2,056 and found in 62.7% of top 2,000 ATOH1 CUT&RUN peaks; analysis performed with Homer (Heinz et al., 2010).
To identify a putative enhancer regulatory network associated with hair cell gene expression, we analyzed chromatin accessibility in FACS-purified hair cells from P1 cochlea using ATAC-seq (Buenrostro et al., 2013) and used CUT&RUN (Skene and Henikoff, 2017) to analyze H3K4me1 and H3K27ac, two epigenetic marks associated with active enhancers (Figure 1B; Creyghton et al., 2010; Rada-Iglesias et al., 2011; Zentner et al., 2011). We filtered accessible elements (ATAC-seq peaks) based on their distance to the transcription start sites (TSSs) of hair cell genes, and only elements lying between 2–200 kb upstream or downstream from the TSS were retained as potential hair cell gene distal regulatory elements for further analysis. We classified these accessible elements into four groups: ‘‘active,’’ ‘‘inactive,’’ ‘‘unannotated,’’ and ‘‘unmarked’’ based on their histone modifications (Figure 1B). We identified 2,894 accessible distal elements positive for both H3K27ac and H3K4me1, which were considered active putative enhancers (hereafter ‘‘active enhancers’’) (Figure 1B; Table S2). This group was used to define the hair cell gene regulatory network analyzed in the subsequent figures. In addition, we identified 4,239 ‘‘inactive’’ enhancers by virtue of being marked by H3K4me1 but lacking H3K27ac. These ‘‘inactive’’ elements, while lacking H3K27ac, may none-the-less have roles to play in regulating gene expression at other stages of hair cell differentiation, or be important for regulation of other more distant genes. Accessible elements (3,407) were identified as ‘‘unmarked,’’ while an additional 256 were positive for H3K27ac but unmarked by H3K4me1, thus likely ‘‘unannotated’’ promoters. The majority of H3K27ac-positive elements were also marked by H3K4me1, consistent with the idea that this conjunction marks active enhancers (Creyghton et al., 2010; Rada-Iglesias et al., 2011). Two examples of this active enhancer signature in hair cells are shown by the presence of an ATAC-seq peak, enriched for both H3K27ac- and H3K4me1-labeled nucleosomes (Figure 1C: the Atoh1 3′ enhancer [Helms et al., 2000] and a predicted Rasd2 5′ enhancer [Jen et al., 2019]). In cultures of dissociated P1 cochlear cells, both enhancers direct GFP expression specifically in hair cells (98.7% ± 2.3% GFP+ cells were also stained positive for MYO6, when GFP expression is regulated by the Atoh1 3′ enhancer; 98.4% ± 2.8% for Rasd2 5′ enhancer; Figure S1B; Table S3). Additional examples of putative hair cell gene enhancers are shown in Figures S1A and S1B.
We also analyzed the supporting cell gene regulatory network (Figures 1A and S2; Table S1), identifying 1,251 supporting-cell-enriched genes, (including Prox1, Jag1, and Hes5; Figures 1A and S2A; Table S1) and 3,227 distal elements predicted as active supporting cell gene enhancers (Figures S2B and S2C; Table S2).
ATOH1 is the first transcription factor newly expressed as hair cells begin to differentiate, and is necessary and sufficient for their differentiation and survival (Bermingham et al., 1999; Zheng and Gao, 2000; Chen et al., 2002; Woods et al., 2004). To identify the ATOH1 targetome within the hair cell gene regulatory network, we used CUT&RUN to analyze the ATOH1-binding sites in FACS-purified hair cells from E17.5 organ of Corti, when expression of Atoh1 peaks (Stojanova et al., 2015). The ATOH1 targetome showed strong correlation to active enhancers (1,212 out of 2,894 H3K27ac and H3K4me1-double-positive active enhancers overlapped with ATOH1 peaks; Figure 1B), including strong signal at the Atoh1 3′ enhancer, and the Rasd2 5′ enhancer (Figure 1C), suggesting ATOH1 uses such putative hair cell enhancers to drive hair cell gene expression and thus differentiation.
We analyzed the DNA sequences of ATOH1 peaks by HOMER (Heinz et al., 2010) and found one significantly enriched 10-bp motif in 62.7% of analyzed regions (Figure 1D). This motif contains a classic E-Box (CANNTG; Atchley and Fitch, 1997) and is similar to the previously identified ‘‘AtEAM’’ ATOH1-binding motif found in cerebellar granule precursors (Klisch et al., 2011) and that found in intestinal precursors (Lo et al., 2017), yet distinct from both at positions 5 and 6 (Figure S1C).
The identification of a large set of hair-cell-enriched genes and the prediction of cell-type-specific gene regulatory networks provides the groundwork for investigating the mechanism by which hair cell genes are maintained in a silent state in supporting cells, as well as the epigenetic changes needed for supporting cell transdifferentiation.
The hair cell gene regulatory network is primed-but-silenced in supporting cells by active H3K27 deacetylation and repressive trimethylation
Neonatal supporting cells are capable of transdifferentiating into hair cells but are normally repressed from doing so by Notch-mediated lateral inhibition (Woods et al., 2004; Kiernan et al., 2005; Groves, 2010; Neves et al., 2013; Abdolazimi et al., 2016). To identify the mechanisms involved in maintaining hair cell genes in a transcriptionally silent state in P1 supporting cells, we compared chromatin accessibility (ATAC-seq) and epigenetic state in both hair cells and supporting cells, focusing on the group of ‘‘active’’ enhancers (Figure 1), which we have defined as the hair cell gene regulatory network (Figure 2A). Analysis revealed comparable levels of accessibility at both promoters and enhancers of hair cell genes in both cell types (Figure 2A). Similarly, hair cell genes in hair cells and supporting cells are marked by H3K4me3 at their promoters, and by H3K4me1 at their enhancers (Figure 2A). However, the active histone mark H3K27ac (Creyghton et al., 2010; Rada-Iglesias et al., 2011), which is present at hair cell gene regulatory elements in hair cells, is lacking at these elements in supporting cells; and instead these elements have elevated levels of the repressive mark H3K27me3 (Figure 2A; Boyer et al., 2006; Barski et al., 2007). This combination of epigenetic marks in supporting cells indicates a developmentally ‘‘poised’’ (Bernstein et al., 2006) and ‘‘primed’’ state (Creyghton et al., 2010; Rada-Iglesias et al., 2011) at promoters and enhancers, respectively. This primed-but-silenced epigenetic state of hair cell gene enhancers in supporting cells correlates with the low expression of hair cell genes in P1 supporting cells (Figure 1A) and is consistent with their potential to transdifferentiate.
Figure 2. The hair cell gene regulatory network is primed (H3K4me1) but silenced by both H3K27 deacetylation and H3K27 trimethylation in P1 supporting cells.

(A) Chromatin structure (ATAC-seq) and epigenetic analysis (H3K4me3, H3K4me1, H3K27ac, and H3K27me3) of FACS-purified P1 hair cells (P1HC, ATOH1-GFP+) and supporting cells (P1SC, Lfng-GFP+). Heatmaps with signal intensity profiles of promoters (defined by ± 2 kb of TSS of expressed genes) and active enhancers (±5 kb around peak summit) defined in Figure 1B). Hair cell gene promoters are accessible (ATAC-seq) and marked by H3K4me3 in both P1HC and P1SC. Hair cell gene enhancers are accessible and primed by H3K4me1 in both P1HC and P1SC. Normalized read counts for H3K27ac and H3K27me3 at each promoter or enhancer (±500 bp) in P1HCs are significantly different from that in P1SC (quartile distribution, boxplots). HC gene promoters and enhancers show lower H3K27ac and higher H3K27me3 in P1SC relative to P1HC (***, adjusted p value < 0.001 by paired Wilcoxon test).
(B) Quantification of poised hair cell gene promoters (H3K4me3+/H3K27ac−) and primed hair cell gene enhancers (H3K4me1+/H3K27ac−) in P1HC and P1SC analyzed by ChromHMM. 52% of hair cell gene promoters (498 promoters) are poised in P1SC, while less than 7% (72 promoters) are classified as poised in P1HC. 40% of hair cell gene enhancers (1,159 enhancers) are primed in P1SC, while only 3% (86 enhancers) are classified as primed in P1HC.
(C) Genome browser tracks (IGV) of hair cell genes Atoh1 and Rasd2, illustrating the differential epigenetic state in P1 hair cells and supporting cells. The Atoh1 promoter is accessible and marked by H3K4me3 in both HCs and SCs but enriched for H3K27ac only in P1HCs. The Atoh1 3′ enhancer (gray box) is accessible and enriched for H3K4me1 in both cell types but lacks H3K27ac in P1SC. Similarly, the Rasd2 promoter is marked by H3K4me3, and the Rasd2 enhancer (gray box) is marked by H3K4me1 in both P1HC and P1SC, but both elements are marked by H3K27me3 in P1SC instead of H3K27ac.
(D) Inhibition of H3K27 deacetylase leads to upregulation of hair cell gene expression in supporting cells. Organ of Corti explants were treated with either DMSO or a histone deacetylase (HDAC) inhibitor (TSA; 200 nM) for 48 h. Supporting cells (Lfng−GFP+) were FACS-purified for RNA-seq analysis. Compared with DMSO control, HDAC inhibition leads to significant upregulation of four hair cell genes in supporting cells (Atoh1, Pou4f3, Gfi1, and Rasd2); while the hair cell gene Mfng remains unchanged (Table S4) (Error bars, data range; ***, **, and *, adjusted p value < 0.001; < 0.01; < 0.05, respectively).
(E) Inhibition of H3K27 methyltransferase leads to upregulation of hair cell gene expression in supporting cells. Organ of Corti explants were treated with either DMSO or H3K27 histone methyltransferase inhibitor (EZH2 inhibitor GSK-343, 5 μM) for 48 h. Methyltransferase inhibition leads to upregulation of hair cell genes (Atoh1, Pou4f3, Gfi1, and Rasd2) in supporting cells; while the hair cell gene Mfng remains unchanged (Table S4) (Error bars, data range; ***, **, and *, adjusted p value < 0.001; < 0.01; < 0.05, respectively).
We next quantified the percentage of hair cell gene elements classified as ‘‘poised’’ or ‘‘primed’’ (H3K4me1/3+ H3K27ac−) by ChromHMM (Ernst and Kellis, 2012) in supporting cells. Almost 60% of hair cell gene promoters (498 promoters) were poised, and 47% of enhancers (1,159) were primed in P1 supporting cells (Figure 2B). Epigenetic signal tracks at Atoh1 and Rasd2 are representative of this general observation (Figure 2C). As a control, we analyzed the epigenetic state of supporting cell gene elements in P1 hair cells and supporting cells. Supporting cell gene elements were epigenetically active in supporting cells, and primed-but-silenced in hair cells (Figures S3A and S3B). Housekeeping gene promoter regions are marked by similar levels of H3K27ac and H3K27me3 in P1 hair cells and supporting cells (Figure S3C).
Our data indicated that hair cell gene silencing in neonatal supporting cells is accomplished by maintaining both promoters and enhancers in a primed-but-silenced state by a combination of relatively high levels of H3K27me3 and low levels of H3K27ac. To test this further, we disrupted ongoing histone modification by treating P1 organ of Corti cultures with inhibitors of either HDACs (TSA; Figure 2D), or PRC2 methyltransferases (GSK-343; Figure 2E). Following 48 h treatment with TSA or GSK-343, we FACS-purified supporting cells from these cultures and used RNA-seq to show that a number of hair cell genes (Atoh1, Pou4f3, Gfi1, and Rasd2) were upregulated in supporting cells (Figures 2D and 2E; Table S4). These results suggest that both active histone deacetylation and ongoing histone H3K27 methylation are necessary for maintaining the transcriptionally repressed state of hair cell genes in neonatal supporting cells at the time when hair cell gene enhancers are still epigenetically primed.
Transdifferentiation of supporting cells into hair cells is accompanied by H3K27 acetylation and increased chromatin accessibility of hair cell gene regulatory elements
Blocking Notch signaling between hair cells and supporting cells with gamma-secretase inhibitor promotes transdifferentiation of supporting cells to a hair cell fate (Takebayashi et al., 2007; Doetzlhofer et al., 2009; Korrapati et al., 2013; Maass et al., 2015). However, several reports indicate that supporting cell transdifferentiation at P1 is predominantly found in the apical half of the cochlea (Figure 3A; Doetzlhofer et al., 2009; Korrapati et al., 2013; Maass et al., 2015), consistent with the maturation wave from the base to the apex of the cochlear duct (Chen et al., 2002; Cai et al., 2013). To better analyze the changes in epigenetic status of neonatal supporting cells that are able to transdifferentiate, we lineage-labeled supporting cells by administering tamoxifen to a transgenic mouse line harboring a Lfng-CreERT2 transgene (Semerci et al., 2017), tdTomato reporter (Madisen et al., 2010), as well as an Atoh1-Gfp transgene (Rose et al., 2009) to track hair cell gene expression (Figure 3B). Supporting cells that transdifferentiate to hair cells in response to the gamma-secretase inhibitor DAPT, are double labeled by both ATOH1-GFP and tdTomato (yellow in accompanying FACS plot), while supporting cells that do not respond to DAPT in the same organs, or in control organs treated with DMSO, are only labeled with the tdTomato reporter (Figure 3B). RNA-seq analysis of DAPT-responsive and non-responsive supporting cells indicated that hair cell genes such as Atoh1, Pou4f3, Lhx3, and Gfi1 are strongly upregulated in DAPT-responsive supporting cells, but not in the non-responsive population (Figure 3B; Table S5). In contrast, specific Notch targets, such as Hes5, are significantly downregulated in both responsive and non-responsive supporting cells after DAPT treatment (Figure 3B; Table S5).
Figure 3. Transdifferentiation of supporting cells is accompanied by hair cell enhancer activation (H3K27ac) and increased chromatin accessibility (ATAC-seq).

(A) Hair cells differentiate in the organ of Corti in a wave from base to apex. Inhibition of Notch signaling by DAPT causes supporting cells to transdifferentiate into hair cells, but this ability is lost as supporting cells mature. Whole mounts of DAPT-treated cochlea show that supernumerary hair cells (ATOH1−GFP+, tdTomato+ and POU4F3+) are mainly found in the apical, less mature, half of the organ of Corti. Scale bar, 50 µm.
(B) Purification of supporting cell subpopulations for transcriptome analysis. P1 organ of Corti explants with Lfng-CreERT2/tdTomato reporter, and the Atoh1-Gfp transgene, were treated with tamoxifen for 24 h to lineage-label supporting cells (red), while hair cells are labeled by ATOH1-GFP (green). Organs were then treated with DMSO or the Notch inhibitor DAPT (10 µM) for 48 h. FACS-purified supporting cells undergoing transdifferentiation to hair cells express both reporters (yellow box in FACS plot, ‘‘responsive’’), while supporting cells refractory to Notch inhibition remain tdTomato single-positive (red box, ‘‘non-responsive’’) were analyzed by RNA-seq. Control supporting cells (pink box) were not treated with DAPT. Scatter plots show gene expression changes in non-responsive and responsive subpopulations of supporting cells relative to control. Hair cell genes with significant expression changes (fold change > 2 or < −2, and adjusted p value <0.01) are green, supporting cell genes with significant expression change are red, and genes unchanged are gray. Hair cell genes strongly induced in DAPT-responsive supporting cells (Atoh1, Pou4f3, Lhx3, and Gfi1) are indicated.
(C) Chromatin structure (ATAC) and epigenetic changes (H3K27ac) accompanying transdifferentiation were compared at HC gene promoters and enhancers in control, responsive (transdifferentiating), and non-responsive supporting cells. Comparison of heatmaps, average signal profiles, and quartile distributions of normalized read count per element (±500 bp, boxplots) for each group indicate that HC gene enhancers gain both accessibility and H3K27ac during transdifferentiation (responsive versus non-responsive or control), while promoters are accessible in all subpopulations but gain modest H3K27ac during transdifferentiation (***, adjusted p value < 0.001 by paired Wilcoxon test).
(D) Examples of changing epigenetic status upon transdifferentiation. Promoters and enhancers (gray boxes) of Atoh1 and Pou4f3 gain accessibility (higher ATAC peaks) and accumulate H3K27ac levels in responsive supporting cells.
To investigate changes in epigenetic modification of hair cell gene regulatory elements during transdifferentiation, we profiled chromatin accessibility and the active histone mark H3K27ac in FACS-purified supporting cells 48 h after blocking Notch signaling with DAPT (Figure 3C). We analyzed heatmaps comprising the previously defined hair cell gene promoters and enhancers (Figure 2A; Table S2). Chromatin accessibility remained relatively unchanged at hair cell gene promoters in supporting cells; however, accessibility increased dramatically at hair cell gene enhancers in DAPT-responsive supporting cells (Figure 3C). In contrast, the accessibility of supporting cell gene enhancers remained largely unchanged (Figures S4A and S4B). Analysis of H3K27ac levels showed a dramatic increase at hair cell gene enhancers and a modest increase at promoters in DAPT-responsive supporting cells (Figure 3C). Interestingly, non-responsive supporting cells have a slightly higher level of H3K27ac at supporting cell gene promoters and enhancers, suggesting their commitment to a supporting cell fate (Figures S4A and S4B). This increased chromatin accessibility and the accumulation of H3K27ac at hair cell gene elements is observed in individual signal tracks of the hair cell genes Atoh1 and Pou4f3 in DAPT-responsive supporting cells (Figure 3D).
Together, our data indicate that the potential of perinatal supporting cells to transdifferentiate into hair cells, stems from the primed-but-silenced epigenetic state of hair cell gene regulatory elements, and that this repressed state is rapidly reversed during transdifferentiation induced by Notch inhibition.
Decommissioning of hair cell gene enhancers by H3K4me1 removal correlates with the loss of regenerative potential of supporting cells
As noted above, many supporting cells in the neonatal organ of Corti respond to Notch inhibition by transdifferentiating into hair cells, but this transdifferentiation potential is rapidly lost during the first week of post-natal development. By 6 days after birth, almost no supporting cell transdifferentiation is seen in cochlear explants when Notch is inhibited (Maass et al., 2015; Figure 4A). To quantify the progressive loss of transdifferentiation potential, we used the same lineage tracing scheme (Figure 3B) to mark supporting cells as they transdifferentiate into hair cells. We observed a steady loss of transdifferentiation between E17.5 and P6, with 63.9% ± 10.2% supporting cells transdifferentiating in response to DAPT at E17.5, diminishing to 47.0% ± 4.8% in P1, 23.5% ± 2.5% in P3 and 1.4% ± 0.2% by P6 (Figure 4B).
We hypothesized that epigenetic silencing of hair cell gene regulatory elements (Figure 1) may underlie the loss of transdifferentiation potential in supporting cells during neonatal maturation. To test this, we purified supporting cells (Lfng-GFP) from E17.5, P1, and P6 animals and profiled two histone marks that prime either promoters (H3K4me3; Figure S5A) or enhancers (H3K4me1; Figure 4C). Heatmaps and normalized read counts show that the majority of hair cell gene promoters in supporting cells remain primed by the promoter mark, H3K4me3 (Figures S5A, S5C, and S5D). In contrast, hair cell gene enhancers lose the H3K4me1 modification between E17.5 to P6 (Figures 4C and 4E). We also quantified the extent of hair cell enhancer decommissioning with age in supporting cells. The number of H3K4me1-called peaks at hair cell enhancers in supporting cells decreases between E17.5 to P6 (H3K4me1 peaks overlapping with 86% hair cell enhancers in E17.5 supporting cells, 68% in P1 supporting cells and 41% in P6 supporting cells; FDR < 0.01 for peak calling; Figure 4F). These global changes are illustrated at the individual hair cell gene loci (Atoh1 and Dll1; Figure 4G). As controls, we analyzed supporting cell gene elements in E17.5, P1, and P6 supporting cells and found that these elements remained H3K4me3 or H3K4me1 primed during postnatal maturation (Figures S5B–S5E and 4D–4F). Though H3K4me1 levels at supporting cell gene enhancers are lower in P6 supporting cells (Figures 4D and 4E), supporting cell gene enhancers remain primed based on the observation that >90% of these enhancers overlap with H3K4me1-called peaks at all stages (Figure 4F). In contrast, only 41% of hair cell gene enhancers overlap with H3K4me1 peaks in P6 supporting cells (FDR < 0.01 for peak calling; Figure 4F). As further control, we assayed housekeeping genes (Table S7) for changes in H3K4me3 and H3K4me1 and observed no difference in E17.5, P1, and P6 supporting cells (Figures S5E and S5F).
Many aspects of differentiation and maturation of the organ of Corti are known to occur in a basal-to-apical wave (Chen et al., 2002; Cai et al., 2013). As previously reported, in DAPT-treated P1 cochlear cultures, more supporting cells in the apical half than in the basal half transdifferentiate into hair cells after DAPT, indicating the existence of a base-to-apex loss of transdifferentiation potential (Figure 3A; Doetzlhofer et al., 2009; Korrapati et al., 2013; Maass et al., 2015). To test if loss of hair cell gene enhancer priming by H3K4me1 correlated with this loss of transdifferentiation potential, we profiled H3K4me1 in FACS-purified supporting cells (Lfng−GFP+) from the basal half versus the apical half of the cochlea. H3K4me1 levels at hair cell enhancers in supporting cells are sharply reduced in the basal half compared with the apical half (Figure S6A). The reduction of H3K4me1 levels at hair cell gene enhancers is illustrated at the Atoh1 and Rasd2 loci (Figure S6B). As a control, housekeeping gene elements did not show a base-to-apex gradient of H3K4me1 in supporting cells (Figure S6C).
Together, the changes we observe in both temporal and spatial epigenetic modifications strongly suggest a link between hair cell gene enhancer decommissioning (H3K4me1 removal) in supporting cells and the loss of supporting cell transdifferentiation potential with age.
Inhibiting decommissioning of hair cell enhancers marked by H3K4me1 prolongs the regenerative potential of supporting cells
Our data suggest that the loss of transdifferentiation potential in supporting cells between E17.5 and P6 correlates with the loss of H3K4me1 commissioning at hair cell gene enhancers (Figures 4 and S5). To test this further, we clustered the ATOH1-bound enhancers of hair cell genes (1,212 enhancers, Figure 1B) into groups that were H3K4me1-positive or H3K4me1-negative in P1 supporting cells and correlated these differences in H3K4me1 status with differences in RNA expression of the associated genes in response to Notch inhibition with DAPT (Figure 5A). The genes associated with H3K4me1-positive enhancers in P1 supporting cells were upregulated after 24 h of DAPT treatment, compared with those associated with enhancers that were H3K4me1-negative (p < 0.001; Figure 5A; Table S6). This correlation between H3K4me1 priming and DAPT-induced transcriptional induction strongly supports the essential role of hair cell gene enhancer priming by H3K4me1.
Figure 5. The primed state (H3K4me1 positive) of hair cell gene enhancers in supporting cells determines the transcriptional response during Notch inhibition-induced transdifferentiation.

(A) Primed enhancers preferentially drive hair cell gene expression during P1 supporting cell transdifferentiation. RNA expression of genes associated with primed ATOH1-bound enhancers (H3K4me1+ in P1SC) and compared with genes associated with ‘‘decommissioned’’ ATOH1 targets (H3K4me1− in P1SC). Comparison of quartile distribution of RNA-seq (log2[fold change]) following Notch inhibition (DAPT 10 nM for 24 h). (Error bar data range; ***, p value < 0.001, Mann-Whitney U test).
(B and C) Inhibition of H3K4-demethylation (GSK-LSD1) prolongs the transdifferentiation potential of supporting cells.
(B) Organ of Corti explants from P1 Atoh1-Gfp and Lfng-CreERT2/tdTomato transgenic mice (as in Figure 3) were divided into basal and apical halves and cultured with and without GSK-LSD1 (20 µM) for 24 h. After this pretreatment, the organs were treated for another 48 h with either Notch inhibitor (DAPT alone; 10 µM), or DAPT plus continued GSK-LSD1 (20 µM). DAPT-responsive supporting cells that transdifferentiated into hair cells were quantified by FACS analysis as in Figure 3. A significant increase in the number of responsive supporting cells is seen in the basal half of the P1 cochlea in the presence of GSK-LSD1 inhibitor (7.4% ± 1.0%), compared with DAPT alone (1.1% ± 0.7%). No significant change is observed in the apical half of P1 cochlea (41.3% ± 9.1% in DAPT + GSK-LSD1 versus 42.9% ± 2.0% in DAPT alone).
(C) An increase in the percentage of DAPT-responsive supporting cells is seen at both P1 and P3 following inhibition of LSD1. The same experimental paradigm was used as in (B), except whole organs of Corti were compared (P1 organs: 31.8% ± 6.1% responsive to DAPT alone versus 42.3% ± 6.4% by DAPT + GSK-LSD1; P3 organs: 19.8% ± 2.6% responsive to DAPT alone versus 29.6% ± 6.8% to DAPT + GSK-LSD1) (Error bar, standard deviation; **, p value < 0.01; and n ≥ 4 for B, n ≥ 8 for C).
H3K4me1 can be demethylated by the lysine-specific demethylase KDM1A/LSD1 (Shi et al., 2004). We reasoned that blocking demethylation of H3K4me1 in organ of Corti cultures using a KDM1A inhibitor (GSK-LSD1; Mohammad et al., 2015) would delay or reverse the loss of supporting cell transdifferentiation potential with age. Since the basal half of the P1 cochlea has lost a portion of its transdifferentiation potential by P1 (Figure 3A), we hypothesized that KDM1A inhibition might allow the more mature basal half of the cochlea to respond when Notch signaling is blocked, while the less mature apical half, and thus H3K4me1-marked, would remain responsive to DAPT with or without inhibitor.
To test this, we pre-incubated P1 organ of Corti explants from both the apex and base of the cochlea with either control (DMSO) or GSK-LSD1 inhibitor for 24 h, and then both halves were treated with DAPT for another 48 h to block Notch signaling and induce transdifferentiation of supporting cells. FACS analysis of lineage-labeled supporting cells was used to quantify the response (Figure 5B). Blocking the removal of H3K4me1 significantly increased the number of supporting cells that transdifferentiated in the basal half of the organ of Corti in response to DAPT (Figure 5B; 1.2% ± 0.7% responsive supporting cells with DAPT alone versus 7.4% ± 1.0% with DAPT and GSK-LSD1, p < 0.01). As expected, no change in transdifferentiation potential was observed in the apical half (Figure 5B; 42.9% ± 2.0% DAPT-responsive supporting cells with DAPT alone versus 41.3% ± 9.1% with DAPT and GSK-LSD1, p = 0.7). When whole organ of Corti explants from P1 and P3 cochlea were tested, an increase in DAPT-responsive supporting cells was observed following KDM1A inhibition (Figure 5C; 31.8% ± 6.1% responsive supporting cells with DAPT alone versus 42.3% ± 6.4% with DAPT and GSK-LSD1 in P1 organs, p < 0.01; 19.8% ± 2.6% with DAPT alone versus 29.6% ± 6.8% with DAPT and GSK-LSD1 in P3 organs, p < 0.01). Consistent with the idea that H3K4 demethylation has already occurred by P6, blocking of H3K4me1 demethylation by GSK-LSD1 had no effect on transdifferentiation of P6 explant cultures (data not shown). Our results suggest that blocking ongoing H3K4me1 demethylation in the organ of Corti prolongs the potential of supporting cells to transdifferentiate into hair cells.
Enhancer decommissioning through loss of H3K4me1 does not occur in utricular supporting cells of the mature vestibular system
The mature vestibular system in mammals retains a modest degree of regenerative potential (Kawamoto et al., 2009; Golub et al., 2012; Bucks et al., 2017), and we have previously shown that mature utricular supporting cells retain a relatively open chromatin configuration of the hair cell enhancer network at P21 (Jen et al., 2019). These observations suggest that enhancer licensing by H3K4me1 is retained in mature utricular supporting cells. To test this, we FACS-purified supporting cells (Lfng-GFP) from P21 cochlea and utricle and analyzed their chromatin structure (ATAC-seq) and enhancer priming (H3K4me1) status. As previously reported, hair cell gene enhancers are quantitatively more accessible in P21 utricular supporting cells than cochlear supporting cells (Jen et al., 2019; Figures 6A and S7D). Interestingly, hair cell gene enhancers in P21 utricular supporting cells are also quantitatively more heavily marked by H3K4me1 in the utricle than in the cochlea (Figures 6A and S7C), while supporting cell gene enhancers, and housekeeping gene elements, are marked by similar levels in both utricle and cochlea (Figures S7A–S7D). This is also observed at the gene level, for instance at the Atoh1 and Rasd2 enhancer loci (Figure 6B, gray bars). The persistence of hair cell gene enhancer commissioning by H3K4me1 in mature utricular supporting cells may help explain the persistence of some regenerative capacity in mature utricles and supports our hypothesis that decommissioning of the hair cell gene enhancer network leads to the loss of transdifferentiation potential.
Figure 6. Hair cell gene enhancer decommissioning does not occur in mature utricular supporting cells.

(A) Hair cell gene enhancers in mature (P21) utricular supporting cells are significantly more accessible (heatmaps and average signal profiles) and have significantly higher H3K4me1 labeling than in P21 cochlear supporting cells, as shown by quartile distributions of normalized read count per element (±500 bp, boxplots) (***p value < 0.001 by paired Wilcoxon test).
(B) Representative genome browser track views (IGV) of two hair cell genes (Atoh1 and Rasd2). Enhancer priming (H3K4me1) is preserved in P21 utricular supporting cells at the Atoh1 3′ enhancer (gray box), and the Rasd2 5′ enhancer (gray box) but is depleted at these enhancers in P21 cochlear supporting cells.
DISCUSSION
Regeneration requires changes in expression of hundreds of genes in a pattern that often recapitulates embryonic development (Goldman and Poss, 2020; Rodriguez and Kang, 2020). Many tissues are capable of mounting a regenerative response in young animals that can restore function, but they lose this ability as tissues mature. In our study, we have identified epigenetic mechanisms that contribute to the inability of the mature mammalian cochlea to regenerate its auditory hair cells. Understanding and manipulating these changes in the inner ear may provide a means to promote hair cell regeneration therapeutically.
Hair cells and supporting cells derive from a common progenitor population in the embryonic cochlea. The first sign of hair cell differentiation in this population is the expression of Atoh1 (Bermingham et al., 1999; Chen et al., 2002), which activates expression of Notch ligands in hair cells and through lateral inhibition blocks the surrounding progenitors from acquiring a hair cell fate (Lanford et al., 1999; Eddison et al., 2000; Woods et al., 2004). This segregation of Atoh1-expressing and non-expressing cells is plastic−as lineage tracing with Atoh1-Cre mice revealed that at least 50% of supporting cells transiently express Atoh1 early in their normal development (Yang et al., 2010; Driver et al., 2013). Moreover, disruption of Notch signaling genetically or pharmacologically can drive immature supporting cells to transdifferentiate into hair cells (Lanford et al., 1999; Zine et al., 2001; Yamamoto et al., 2006; Doetzlhofer et al., 2009; Maass et al., 2015; Figure 3A). Here, we identified an epigenetic signature of this plasticity (Figure 7) in which differentiating supporting cells harbor hair cell gene regulatory elements marked by H3K4me1, a mark of either primed or active enhancers (Creyghton et al., 2010; Rada-Iglesias et al., 2011). Although primed by H3K4me1, these enhancers lack H3K27ac, a mark of active enhancers (Creyghton et al., 2010; Figure 2A), maintained by active histone deacetylation and consequent H3K27 trimethylation by PRC2/EZH2 (Figure 2D). When Notch signaling between hair cells and supporting cells is blocked, this primed-but-silenced signature is converted to an active state by the loss of H3K27me3 and the acquisition of H3K27ac at hair cell gene enhancers (Figures 3C and 7A). This conversion allows the activation of hundreds of hair cell genes including Atoh1, Pou4f3, Gfi1, and Lhx3, and the transformation of supporting cells into hair-cell-like cells (Maass et al., 2015; Figures 3B and 3C).
Figure 7. Model of loss of the transdifferentiation potential of supporting cells caused by decommissioning the enhancers of hair cell genes.

(A) Hair cell gene enhancers and promoters are maintained in a primed/poised epigenetic state in young supporting cells. The organ of Corti matures in a base-to-apex wave and supporting cells that can respond to Notch inhibition by transdifferentiating into hair cells are found mainly in the apical half of the cochlea at P1. In these responsive supporting cells, hair cell gene promoters are poised by the co-existence of H3K4me3 and H3K27me3, and hair cell gene enhancers are primed by a combination of H3K4me1 and H3K27me3 (top panel). Upon Notch inhibition, hair cell gene promoters and enhancers are epigenetically activated by H3K27ac, facilitating the binding of ATOH1 and other hair cell transcription factors that induce hair cell gene transcription and drive transdifferentiation of supporting cells to the hair cell fate (bottom panel).
(B) Hair cell gene enhancers are decommissioned in supporting cells with increasing age. The enhancer priming mark H3K4me1 is removed from hair cell gene enhancers in P6 supporting cells, leading to enhancer inactivation, and preventing induction of hair cell genes. Without properly configured enhancers, ATOH1 and other hair cell transcription factors cannot bind to their targets and hair cell genes cannot be actively transcribed in service to transdifferentiation.
The plasticity of supporting cells to transdifferentiate into hair cells begins to be lost from the cochlea around the time of birth, and this loss is essentially complete by P6 (Figure 4A), about 8 days before the onset of hearing in mice (Ehret, 1976). We show that this loss of regenerative potential in supporting cells is accompanied by removal of H3K4me1 priming at hair cell gene enhancers (Figures 4D and 7B). We demonstrate that this demethylation occurs along the basal-to-apical axis of the cochlea (Figure S6B), with the less mature, apical half of the cochlea having a greater proportion of H3K4me1-marked hair cell gene enhancers than the more mature basal half. Inhibition of H3K4me1 decommissioning by lysine demethylase LSD1 inhibitors extends the time of transdifferentiation potential of supporting cells (Figure 5B), demonstrating that the epigenetic switch from the primed-but-silenced state to a decommissioned state underlies the loss of transdifferentiation potential in the cochlea.
Several mechanisms have been proposed for how H3K4me1 positively regulates enhancer activity. First, the chromatin re-modeling BAF-complex directly interacts with H3K4me1 (Local et al., 2018), and so its recruitment would thus maintain the accessibility of primed enhancers. Indeed, we observe that hair cell gene enhancers lose accessibility as H3K4me1 levels drop during maturation (Figure 4E). Second, H3K4me1 marks can recruit the Cohesin complex (Yan et al., 2018) and allow long-range interactions between enhancers and target gene promoters that are essential for regulated gene activation (Furlong and Levine, 2018). Finally, H3K4 methylation reduces the interaction between histone H3 and DNMT3L (a subunit for the de novo DNA methylation complex; Ooi et al., 2007), preventing more permanent silencing of enhancers by DNA methylation. Taken together, loss of H3K4me1 at enhancers likely leads to enhancer decommissioning through nucleosome repositioning, disengagement of long-range interactions, and ultimately DNA methylation. We suggest that removal of H3K4me1 during supporting cell post-natal maturation leads to the inactivation of a hair cell gene enhancer network, and with it the subsequent loss of regenerative potential in the cochlea.
The importance of enhancer priming has been observed in pluripotent stem cell induction, stem cell differentiation, transcription-factor-induced reprogramming, and transdifferentiation in vivo. Deletion of Mll3 and Mll4, two genes responsible for mono-methylation of H3K4, prevents induction of pluripotency genes when fibroblasts are reprogrammed to iPS cells with OSKM factors and prevents differentiation of embryonic stem cells (Wang et al., 2016; Yan et al., 2018). H3K4 monome-thylation is necessary to allow the neurogenic transcription factor ASCL1 to access its targets and reprogram fibroblasts or keratinocytes into neurons, despite the fact that ASCL1 has pioneer factor activity and can bind nucleosomal chromatin (Wapinski et al., 2013). In addition, ASCL1 fails to bind to its authentic binding site in keratinocytes in the absence of H3K4me1 (Wapinski et al., 2013), suggesting that an H3K4me1-permissive epigenetic state is necessary for fate conversion during reprogramming. Similar to our current findings, the plasticity of midgut cells to activate foregut genes in vivo after deletion of Cdx2 is dependent on H3K4me1 marking of foregut enhancers (Banerjee et al., 2018), consistent with our hypothesis that loss of enhancer priming compromises transdifferentiation and regenerative potential. The correlation between loss of plasticity and demethylation of H3K4 is also seen in the retina, where the ability to convert Müller glia to amacrine cells, bipolar cells, and photoreceptors is lost by 2 weeks of age and correlates with a loss of accessibility of retinal progenitor genes (Ueki et al., 2015). Moreover, a substantial number of cis-regulatory elements of genes required for retinal neuron and photoreceptor differentiation are kept in a silenced state (H3K4me3/me1 depleted, H3K27me3 enriched; Dvoriantchikova et al., 2019), suggesting that decommissioning of cis-regulatory elements by H3K4 demethylation imposes an epigenetic obstacle for Müller glia transdifferentiation, as we observe in the cochlea.
No significant hair cell regeneration occurs in the mature mammalian cochlea, and hearing loss is therefore progressive and permanent. Our current work suggests that re-commissioning of hair cell gene loci in supporting cells, or other non-sensory cells of the cochlea, might be one component of future regenerative strategies. Indeed, the vestibular organs of the mouse inner ear are capable of modest regeneration even in adult animals (Kawamoto et al., 2009; Golub et al., 2012; Bucks et al., 2017). Our recent work has shown that cis-regulatory elements associated with hair cell genes are kept in a relatively more accessible state in mature vestibular supporting cells compared with their cochlear counterparts (Jen et al., 2019). We now show that the vast majority of hair cell gene enhancers retain their H3K4me1-primed state in supporting cells of the utricle, but not the cochlea (P21, Figure 6). Nevertheless, we believe that decommissioning of enhancers is just the initial step of epigenetic silencing of the hair cell program in supporting cells. It is likely that DNA methylation and deposition of inhibitory histone marks such as H3K27me3 and H3K9me3 at hair cell gene loci increase as supporting cells continue to mature. Direct reprogramming by hair cell transcription factors may be required to re-commission and reactivate hair cell gene loci in surviving supporting cells of long deafened mammals. Overexpression of three hair cell transcription factors, Atoh1, Pou4f3, and Gfi1, can generate hair-cell-like cells by either direct programming of differentiating embryonic stem cells (Costa et al., 2015), or, along with Six1, direct reprogramming of somatic cells (Menendez et al., 2020). In common with other reprogramming strategies (reviewed in Morris, 2016), these direct programming/reprogramming approaches do not give rise to fully mature hair cells, as some hair cell gene enhancers are not engaged and some hair cell genes are not expressed (Menendez et al., 2020). However, these results, together with the work presented here, offer a glimpse of future hearing restoration strategies that make use of epigenetic manipulation by pharmaceutical inhibitors (Huangfu et al., 2008; Liang et al., 2010), epigenetic engineering strategies such as the CRISPR-dCAS9 system (reviewed in Pulecio et al., 2017), and direct reprogramming.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Neil Segil (nsegil@med.usc.edu).
Material availability
This study did not generate new unique reagents.
Data and code availability
The datasets generated during this study are available at NCBI Gene Expression Omnibus(NCBI GEO: GSE150010).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animals
Experiments were conducted in accordance with the policies of the Institutional Animal Care and Use Committee of the Keck School of Medicine of USC. The first postnatal day of mice in this study was designated as postnatal day 0 (P0). For hair cell collection, Atoh1-Gfp knockin animals (MGI: Atoh1tm4.1Hzo ; C57bl6 background) were used (Rose et al., 2009); for supporting cell collection, Lfng-Gfp (MGI: Tg(Lfng-EGFP)HM340Gsat) transgenic animals were used (Gong et al., 2003). To identify supporting cells that underwent transdifferentiation into a hair cell fate after Notch inhibition in cochlear cultures, Lfng-CreERT2 (MGI: Tg(Lfng-cre/ERT2)1Mmsa) / ROSA-tdTomato (ROSA-Ai9, MGI: Gt(ROSA)26Sortm9(CAG-tdTomato)Hze) transgenic animals (CD1 background; Semerci et al., 2017) were mated with Atoh1-Gfp animals, which allowed for supporting cell lineage tracing after tamoxifen administration.
Organotypic cultures and drug treatment
Inner ears were isolated at E17.5, P1, P3 or P6 animals as described previously (Tao and Segil, 2015). Cochleae were then micro-dissected under a stereo microscope in a laminar flow hood in ice-cold Ca2+- and Mg2+-free DPBS. After removal of lateral wall and spiral ganglion tissues, cochleae were cultured on SPI black filter membranes (SPI Supplies) in L15 Leibovitz media (Hyclone) supplemented with N2 (Invitrogen, 1:100), B27 (Gibco, 1:50), Penicillin G (Sigma, 100U/ml), EGF(Shenandoah Biotechnology, 5ng/ml), FGF (Shenandoah Biotechnology, 2.5ng/ml) and HEPES (Invitrogen, 10µM). To lineage-trace supporting cells in Lfng-CreERT2/tdTomato animals, (Z)-4-Hydroxytamoxifen (Sigma) was included in the culture medium at a concentration of 0.02mg/ml for the first 24 hours. The following inhibitors were used at the indicated concentrations: 10µM DAPT (Calbiochem), 10µM GSK-343 (courtesy gift from GlaxoSmithKline PLC), 200nM TSA (Sigma), 20μM GSK-LSD1 (Sigma) or an equivalent amount of DMSO (VWR).
METHOD DETAILS
Experimental design general
Due to the scarcity of hair cells and supporting cells in the inner ear, organs from multiple animals from the same litter were pooled together for FACS-purification/analysis as one biological replicate. Both male and female animals were used without further randomization or stratification. Alternate left and right ears were used as experimental or control samples. At least two biological replicates were collected for transcriptomic and epigenetic analysis as per ENCODE standard. For epigenetic datasets, replicates that failed the ‘‘reproducibility test’’ of the Encode QC pipeline were excluded. Samples were not blinded.
Fluorescence activated cell sorting
To purify hair cells or supporting cells from perinatal organs, dissected or cultured cochlea from multiple animals (>5 mice) were treated with trypsin (Invitrogen, 0.125%) at 37°C for 8 minutes. To inactivate trypsin, FBS (Invitrogen) was added to a final concentration of 10%; and the tissue was dissociated into a single cell suspension by trituration with a P200 pipette. To purify supporting cells from P21 cochleae, inner ears were dissected, vestibular organs were removed, and a few openings were made in the bony wall of the cochlear ducts to expose the organ of Corti. The tissue was incubated in 200µL trypsin (Invitrogen, 0.125%) for 25 min at 37°C with shaking at 600rpm; the trypsin treatment was stopped by addition of 300µL 10% FBS in DPBS, and the tissues were triturated with P1000 pipette for 3 min. The supernatant was passed through a 40μm cell strainer (BD Biosciences) to remove residual undis-sociated tissues. To purify mature vestibular supporting cells, utricles dissected from P21 animals were dissociated into a single cell suspension by incubation in trypsin (Invitrogen, 0.125%) and thermolysin (Promega, 100µg/mL) at 37°C for 25 min, followed by inactivation with 10% FBS and trituration with a P200 pipette for 3 min.
Hair cells or supporting cells were FACS-purified on Aria I or Aria II machines (BD Biosciences, 100um Nozzle). Purity of the sorted cells was confirmed to be > 95% by re-sorting and/or by cell counting. Cells were sorted directly into 300µL RNA lysis buffer (Zymo Research) for RNAseq, or into 500µL PBS supplemented with 10% FBS for ATACseq, ChIPseq and CUT&RUN.
Expression quantification and differential expression analysis by RNAseq
Total RNA was extracted with a Quick-RNA Microprep kit (Zymo Research) from FACS-purified cells, and libraries were constructed with QIAseq FX Single Cell RNA Library Kit (Qiagen) after RNA QC by a Bioanalyzer (Aligent). At least 2 biological replicates were collected, and a minimum of 20 million reads were collected for each replicate. Reads were mapped to the GRCm38/mm10 mouse genome assembly with STAR aligner (Dobin et al., 2013), and counted against transcript by RSEM (Li and Dewey, 2011). For genes with multiple transcript isoforms, the transcription start site of the most abundant isoform was defined as the gene transcription start site; and then the transcript counts of isoforms were collapsed to give a gene count. Differentially expressed protein-coding genes were identified using DESeq2 (Love et al., 2014). Genes with a base mean count over 50, RPKM greater than 1, fold change bigger than 2 (or smaller than −2) and adjusted p-value less than 0.01, were considered as differentially expressed. Gene ontology enrichment analysis was done using EnrichR (Chen et al., 2013).
ATACseq, ChIPseq and CUT&RUN
The ATACseq assay (Buenrostro et al., 2013) was performed with the following modifications: at least 3,000 FACS-purified cells were used for each replicate, and 2 or 3 biological replicates were prepared for each stage or condition; nuclei were transposed without a purification step using home-made Tn5 transposase at 37°C for 30 min. DNA fragments were extracted, repaired, and amplified by PCR to construct libraries. Over 20 million paired-end reads were collected for each replicate.
A histone ChIPseq protocol was developed based on our previously published μChIP-PCR protocol (Stojanova et al., 2015), with the inclusion of an extra Tn5 tagmentation step. Briefly, at least 10,000 cells were used for each replicate, and cells were cross-linked with 1% formaldehyde (Thermo Fisher) for 8 min, quenched with 125mM Glycine (Sigma) for 5 min at room temperature, sonicated using the microtip of a High Intensity Ultrasonic Processor (Sonics & Materials, Newtown, CT; amplitude 50, power 50) for 8X30s with 30s pauses. Chromatin fragments generated by sonication were tagmented with Tn5 transposase for 30 min at 37°C, incubated with antibodies complexed with Dynabeads Protein A (Thermo Fisher) overnight at 37°C, precipitated, washed three times on a magnetic rack, and finally PCR-amplified with primers matching Tn5 adapters. More than 20 million paired-end reads were sequenced for each library, and 5% of tagmented chromatin fragments were also sequenced as an input control.
The CUT&RUN method for protein-DNA interaction analysis has been described previously (Skene and Henikoff, 2017), and was used to profile genome-wide histone modifications and ATOH1 occupancy. More than 5,000 cells were used for each histone modification replicate and more than 10,000 cells were used for each replicate of ATOH1 CUT&RUN. Briefly, FACS-purified cells were permeabilized with 0.06% digitonin (Millipore), incubated with primary antibody at 4°C overnight, washed, incubated with protein-A MNase (courtesy gift from Dr. Henikoff), washed, and digested on ice by adding 0.002M CaCl2. The MNase digestion process was stopped by addition of 2X stop buffer, and DNA fragments were released by incubation at 37°C for 10 min. Finally, DNA fragments were extracted by the phenol-chloroform method. DNA fragments generated by the identical protocol using rabbit-anti-mouse IgG as primary antibody (Abcam) were also collected as negative controls. Accel-NGS 2S Plus DNA prep kits with single index and MIDs (Swift Bioscience) were used to construct CUT&RUN libraries, and more than 20 million paired-end reads were sequenced for each library. The following antibodies were used for ChIPseq and CUT&RUN: rabbit-anti-H3K27ac, rabbit-anti-H3K27m3, rabbit-anti-H3K4me1, rabbit-anti-H3K4me3 (all from Active Motif) and rabbit-anti-GFP (targeting the ATOH1-GFP fusion protein; Torrey Pines Biolabs).
Chromatin accessibility and histone modification data analysis
ENCODE pipelines were adapted for analysis of ATACseq, ChIPseq, and CUT&RUN data. Briefly, the reads were trimmed to 37bp and aligned to GRCm38/mm10 genome assembly with STAR aligner (Dobin et al., 2013). For ATACseq and ChIPseq, PCR-duplicated reads were marked and removed based on fragment genomic coordinates; For CUT&RUN, read duplicates were removed by MIDs using UMI-tools (Smith et al., 2017). Peaks were called by Model-based analysis of ChIP-Seq (MACS2; Feng et al., 2012) with FDR< 0.01 and disabled dynamic lambda (–nolambda) option for individual replicates, and the peaks with at least 10% reciprocal overlapping between replicates were selected using bedtools (Quinlan and Hall, 2010), and were used for downstream analysis. Elements within ±2kb of TSS were considered as proximal elements, and elements within 2~200kb upstream or downstream of hair cell gene TSS or supporting cell gene TSS were considered as potential distal regulatory elements. BigWig files were generated with deepTools (Ramírez et al., 2014) using the bamCoverage command (bin size of 50 and smoothing length of 150 with CPM normalization). Heatmaps and average signal profiles were generated with deepTools based on the normalized bigWig signal files, and the color intensity range is shown for each heatmap. Quartile distributions of normalized read count per element (±500bp) were plotted as boxplots using R, and the statistical significance was calculated using paired Wilcoxon tests. As a control for signal normalization, promoter regions of housekeeping genes were also analyzed (Table S7). The ChromHMM (Ernst and Kellis, 2012) package was used to classify epigenetic states (bin size of 200bp and p=0.0001 for histone mark binarization). Individual genomic loci were visualized with IGV (Robinson et al., 2011). To identify consensus ATOH1-binding motifs in hair cells, ATOH1 peaks were used for de novo motif enrichment analysis by HOMER (Heinz et al., 2010). To quantify the H3K4me1/3 peaks overlapping with hair cell gene elements or supporting cell elements, the intersectBed command from bedtools (Quinlan and Hall, 2010) was used to extract H3K4me1/3 peak regions with at least 1 bp overlap with query element regions.
Cryosectioning, immunostaining and microscopy
Cultured cochlear organs were fixed with 4% paraformaldehyde (Electron Microscopy Sciences) at room temperature for 5 min. Fixed organs were submerged in 30% sucrose (Sigma) for at least 10 min, transferred into OCT (Sakura) and flash frozen in liquid nitrogen. Organs were sectioned on a Leica cryostation CM3050S at 10µm thickness. Sections or whole mount preparations were permeabilized with 1% Triton X-100 in PBS for 30 min, blocked with 5% goat serum in PBS, incubated with 1:500 diluted primary antibody at room temperature for 2 hours, washed three times with PBS, incubated with 1:1000 diluted secondary antibody at room temperature for 1 hour, and washed three times with PBS before mounting. Mounted slides were imaged with a Zeiss LSM780 confocal microscope. The following antibodies were used for immunostaining: mouse-anti-POU4F3 (Santa Cruz), rabbit-anti-MYO6 (Proteus Biosciences), goat-anti-mouse Alexa 647 and goat-anti-rabbit Alexa 647 (Thermo Fisher Scientific).
Enhancer activity assay in vitro and in vivo
Enhancers identified as specific for hair-cell genes were cloned into the pLKO scramble H2B-Mrfp1 CBFRE-EGFP construct between KpnI and SalI sites (courtesy gift from Dr. Fuchs; Williams et al., 2011). Constructs were packaged into lentivirus by transfection into 293T cells (ATCC) together with helper plasmid psPAX2 and pCMV-VSV-G (Addgene) using Lipofectamine (Invitrogen). Virus was collected from culture medium at 48 hour and 72 hour post-transfection, and concentrated with Lenti-X concentrator (TaKaRa) to reach 1x107 TU/ml. To infect dissociated cells from P1 cochleae, 1µL virus was mixed with 50µL culture medium containing 10 µg/mL Polybrene (Sigma) for each well of a 96 well plate, mixed and incubated for 10 min. Cochlear organs from P1 animals were dissociated as described above. Cells were diluted with culture medium to ~1,000cells/µL; 50µL of cell suspension was added into each well and incubated for 24 hours at 37°C in a 5% CO2 incubator. Medium was changed after 24 hours and cells were cultured for another 96 hours before fixation and immunostaining.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analysis was conducted as indicated in individual Figure Legends. Student t-test (in R) was used to compare data from DAPT-responsive and non-responsive supporting cells (different developmental stages; basal vs apical half of cochlea; DAPT alone and DAPT/GSK-LSD1). For comparison of DAPT-induced expression, fold change of genes associated with H3K4me1-positive ATOH1-bound enhancers and genes associated with H3K4m1-negative ATOH1 bound enhancers, a Mann Whitney U test was used in R. The Bonferroni correction method was used to control for the statistical significance of multiple tests across experimental conditions. Statistical significance test for differential gene expression by RNAseq was performed using DESeq2, which uses the Wald test for p-value after fitting normalized gene counts to the Negative Binomial distribution model and the Benjamini and Hochberg method for multiple testing correction (adjusted p-value). Statistical significance for called peaks was calculated using the MACS2 package, in which a Poisson distribution is used to calculate p-value for individual peaks and the Benjamini and Hochberg method is used for multiple testing correction (FDR value). For bar charts with error bars, the number of biological replicates (n) and the meaning of error bars can be found in figure legends. Box-whisker plots were used to show the quartile distribution, with the median as the center, upper and lower quartiles as the box, and data ranges as the whiskers.
ADDITIONAL RESOURCES
RNA expression and epigenetic datasets generated in this study were deposited at the NCBI (GEO: GSE150010) and gEAR portal: https://umgear.org/p?l=1c98096b&g=atoh1.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
|
| ||
| Rabbit-anti-H3K4me3 | Active motif | Cat#39159; Lot#01609004; RRID: AB_2615077 |
| Rabbit-anti-H3K4me1 | Active motif | Cat#61633; Lot#31814001; RRID: AB_2793712 |
| Rabbit-anti-H3K27ac | Active motif | Cat#39133; Lot#:01518010; RRID: AB_2561016 |
| Rabbit-anti-H3K27me3 | Active motif | Cat#39155; Lot#31618020; RRID: AB_2561020 |
| Rabbit-anti-mouse | Abcam | Cat#ab46540; RRID: AB_2614925 |
| Rabbit-anti-GFP | Torrey Pines Biolabs | Cat#TP401; Lot#071519; RRID: AB_10013661 |
| Rabbit-anti-MYO6 | Proteus Biosciences | Cat#25-6791; RRID: AB_2314836 |
| Mouse-anti-POU4F3 | Santa Cruz | Cat#sc-81980; RRID: AB_2167543 |
| Goat-anti-mouse-Alex-647 | Thermo Fisher | Cat#A-21235; RRID: AB_2535804 |
| Goat-anti-rabbit-Alex-647 | Thermo Fisher | Cat#A-21245; RRID: AB_2535813 |
|
| ||
| Chemicals, Peptides, and Recombinant Proteins | ||
|
| ||
| DAPT | Calbiochem | Cat#565770; CAS# 208255-80-5 |
| GSK-343 | GSK | CAS#1346704-33-3 |
| TSA | Sigma | Cat#T1952; CAS# 58880-19-6 |
| GSK-LSD1 | Sigma | Cat#SML1072; CAS# 1431368-48-7 |
| (Z)-4-Hydroxytamoxifen | Sigma | Cat#H7904; CAS# 68047-06-3 |
| pA-Mnase | Skene and Henikoff, 2017 | N/A |
|
| ||
| Critical Commercial Assays | ||
|
| ||
| Quick RNA-microprep kit | Zymo | Cat#R1051 |
| QIAseq FX Single Cell RNA Library Kit | Qiagen | Cat# 180735 |
| Accel-NGS 2S plus DNA prep kits | Swift Biosciences | Cat#21096 |
| 2S Set A+B MID Indexing Kit | Swift Biosciences | Cat#27396 |
|
| ||
| Deposited Data | ||
|
| ||
| P1HC_RNAseq | this paper | GSE150000 |
| P1SC_RNAseq | this paper | GSE150000 |
| P1SC_TSA_48h_RNAseq | this paper | GSE150001 |
| P1SC_GSK343_48h_RNAseq | this paper | GSE150001 |
| P1SC_DMSO_48h_RNAseq | this paper | GSE150001 |
| P1SC_DAPT_24h_RNAseq | this paper | GSE150002 |
| P1SC_DMSO_24h_RNAseq | this paper | GSE150002 |
| P1SC_control_48h_RNAseq | this paper | GSE150002 |
| P1SC_nonresponsive_48h_RNAseq | this paper | GSE150002 |
| P1SC_responsive_48h_RNAseq | this paper | GSE150002 |
| P1HC_ATACseq | this paper | GSE150000 |
| P1HC_H3K4me1_CUT&RUN | this paper | GSE150000 |
| P1HC_H3K4me3_ChIPseq | this paper | GSE150000 |
| P1HC_H3K27ac_ChIPseq | this paper | GSE150000 |
| P1HC_H3K27me3_ChIPseq | this paper | GSE150000 |
| P1HC_ATOH1_CUT&RUN | this paper | GSE150000 |
| P1SC_ATACseq | this paper | GSE150000 |
| P1SC_H3K4me3_CUT&RUN | this paper | GSE150000 |
| P1SC_H3K4me1_CUT&RUN | this paper | GSE150000 |
| P1SC_H3K27ac_CUT&RUN | this paper | GSE150000 |
| P1SC_H3K27me3_ChIPseq | this paper | GSE150000 |
| P1SC_control_48h_ATACseq | this paper | GSE150002 |
| P1SC_control_48h_H3K27ac_CUT&RUN | this paper | GSE150002 |
| P1SC_nonresponsive_48h_ATACseq | this paper | GSE150002 |
| P1SC_nonresponsive_48h_H3K27ac_CUT&RUN | this paper | GSE150002 |
| P1SC_responsive_48h_ATACseq | this paper | GSE150002 |
| P1SC_responsive_48h_H3K27ac_CUT&RUN | this paper | GSE150002 |
| E17SC_H3K4me3_ChIPseq | this paper | GSE150000 |
| E17SC_H3K4me1_CUT&RUN | this paper | GSE150000 |
| P6SC_H3K4me3_CUT&RUN | this paper | GSE150000 |
| P6SC_H3K4me1_CUT&RUN | this paper | GSE150000 |
| P1SC_Base_H3K4me1_CUT&RUN | this paper | GSE150000 |
| P1SC_Apex_H3K4me1_CUT&RUN | this paper | GSE150000 |
| P21cSC_ATACseq | this paper | GSE150000 |
| P21cSC_H3K4me1_CUT&RUN | this paper | GSE150000 |
| P21uSC_ATACseq | this paper | GSE150000 |
| P21uSC_H3K4me1_CUT&RUN | this paper | GSE150000 |
|
| ||
| Experimental Models: Cell Lines | ||
|
| ||
| 293T cells | ATCC | ATCC®CRL-3216™ |
|
| ||
| Experimental Models: Organisms/Strains | ||
|
| ||
| Mouse: Atoh1-Gfp: B6.129S-Atoh1tm4.1Hzo/J | Jackson Laboratory | JAX: 013593 |
| Mouse: Lfng-Gfp: Tg(Lfng-EGFP)HM340Gsat | Gong et al., 2003 | MGI:4847192 |
| Mouse: Lfng-CreERT2: B6;D-Tg(Lfng-TagRFP/cre/ERT2)8Amc/J | Jackson Laboratory | JAX: 017496 |
| Mouse: tdTomato: B6;129S6-Gt(ROSA)26Sortm14(CAG-tdTomato)Hze/J | Jackson Laboratory | JAX: 007908 |
|
| ||
| Recombinant DNA | ||
|
| ||
| pTXB1-Tn5 | Addgene | Plasmid #60240 |
| pK19pA-MN | Addgene | Plasmid #86973 |
| pLKO-scramble-H2B-mRFP1;CBFRE-EGFP | Elaine Fuchs (Williams et al., 2011) | NA |
| psPAX2 | Addgene | Plasmid #12260 |
| pCMV-VSV-G | Addgene | Plasmid #8454 |
|
| ||
| Software and Algorithms | ||
|
| ||
| STAR aligner | Dobin et al., 2013 | https://github.com/alexdobin/STAR |
| RSEM | Li and Dewey, 2011 | https://github.com/deweylab/RSEM |
| DESeq2 | Love et al., 2014 | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| bedtools | Quinlan and Hall, 2010 | https://bedtools.readthedocs.io/en/latest/ |
| UMI_tools | Smith et al., 2017 | https://github.com/CGATOxford/UMI-tools |
| MACS2 | Feng et al., 2012 | https://github.com/taoliu/MACS |
| ChromHMM | Ernst and Kellis, 2012 | http://compbio.mit.edu/ChromHMM/ |
| IGV | Robinson et al., 2011 | http://software.broadinstitute.org/software/igv/ |
| deepTools | Ramírez et al., 2014 | https://deeptools.readthedocs.io/en/develop/index.html |
| HOMER | Heinz et al., 2010 | http://homer.ucsd.edu/homer/ |
| Tomtom Motif comparison tool | Gupta et al., 2007 | http://meme-suite.org/tools/tomtom |
Highlights.
Hair cell enhancers in P1 support cells are epigenetically ‘‘primed but silenced’’
Hair cell enhancers are decommissioned by H3K4me1-loss in maturing support cells
Enhancer decommissioning blocks supporting cell transdifferentiation
Inhibiting H3K4me1 demethylation extends perinatal transdifferentiation potential
ACKNOWLEDGMENTS
We thank Welly Makmura for expert technical assistance, Francis James for adapting the ENCODE pipelines, USC Stem Cell Flow Cytometry Facility for FACS assistance, CHLA Molecular Pathology Genomics Core for Next-generation sequencing, Dr. Richard Sandberge for the Tn5 plasmid, Dr. Steven Henikoff for CUT&RUN reagents, Dr. Elaine Fuchs for the pLKO scramble H2B-Mrfp1 CBFRE-EGFP plasmid, and GlaxoSmithKline PLC for GSK-343. We thank Drs. Donna Fekete and John Brigande and the Company of Biologists for the inner ear paint-fill in Figure 1A. We thank Dr. Ksenia Gnedeva and Dr. Unmesh Jadhav for discussion and careful reading of the manuscript. This work was supported bythe following funding sources: NIH-R01 DC015829 to N.S.; NIH-RO1 DC014832 to A.K.G.; Hearing Restoration Project grants from the Hearing Health Foundation to N.S. and A.K.G.; T.T. was supported by USC HCN training grant T32-DC009975 and F31-DC017376.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.devcel.2021.07.003.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Abdolazimi Y, Stojanova Z, and Segil N (2016). Selection of cell fate in the organ of Corti involves the integration of Hes/Hey signaling at the Atoh1 promoter. Development 143, 841–850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atchley WR, and Fitch WM (1997). A natural classification of the basic helix–loop–helix class of transcription factors. Proc. Natl. Acad. Sci. USA 94, 5172–5176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balak KJ, Corwin JT, and Jones JE (1990). Regenerated hair cells can originate from supporting cell progeny: evidence from phototoxicity and laser ablation experiments in the lateral line system. J. Neurosci 10, 2502–2512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee KK, Saxena M, Kumar N, Chen L, Cavazza A, Toke NH, O’Neill NK, Madha S, Jadhav U, Verzi MP, and Shivdasani RA (2018). Enhancer, transcriptional, and cell fate plasticity precedes intestinal determination during endoderm development. Genes Dev 32, 1430–1442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barski A, Cuddapah S, Cui K, Roh TY, Schones DE, Wang Z, Wei G, Chepelev I, and Zhao K (2007). High-resolution profiling of histone methylations in the human genome. Cell 129, 823–837. [DOI] [PubMed] [Google Scholar]
- Bermingham NA, Hassan BA, Price SD, Vollrath MA, Ben-Arie N, Eatock RA, Bellen HJ, Lysakowski A, and Zoghbi HY (1999). Math1: an essential gene for the generation of inner ear hair cells. Science 284, 1837–1841. [DOI] [PubMed] [Google Scholar]
- Bernstein BE, Mikkelsen TS, Xie X, Kamal M, Huebert DJ, Cuff J, Fry B, Meissner A, Wernig M, Plath K, et al. (2006). A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell 125, 315–326. [DOI] [PubMed] [Google Scholar]
- Boyer LA, Plath K, Zeitlinger J, Brambrink T, Medeiros LA, Lee TI, Levine SS, Wernig M, Tajonar A, Ray MK, et al. (2006). Polycomb complexes repress developmental regulators in murine embryonic stem cells. Nature 441, 349–353. [DOI] [PubMed] [Google Scholar]
- Bucks SA, Cox BC, Vlosich BA, Manning JP, Nguyen TB, and Stone JS (2017). Supporting cells remove and replace sensory receptor hair cells in a balance organ of adult mice. eLife 6, e18128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buenrostro JD, Giresi PG, Zaba LC, Chang HY, and Greenleaf WJ (2013). Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat. Methods 10, 1213–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai T, Seymour ML, Zhang H, Pereira FA, and Groves AK (2013). Conditional deletion of Atoh1 reveals distinct critical periods for survival and function of hair cells in the organ of Corti. J. Neurosci 33, 10110–10122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen EY, Tan CM, Kou Y, Duan Q, Wang Z, Meirelles GV, Clark NR, and Ma’ayan A (2013). Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinformatics 14, 128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen P, Johnson JE, Zoghbi HY, and Segil N (2002). The role of Math1 in inner ear development: uncoupling the establishment of the sensory primordium from hair cell fate determination. Development 129, 2495–2505. [DOI] [PubMed] [Google Scholar]
- Corwin JT, and Cotanche DA (1988). Regeneration of sensory hair cells after acoustic trauma. Science 240, 1772–1774. [DOI] [PubMed] [Google Scholar]
- Costa A, Sanchez-Guardado L, Juniat S, Gale JE, Daudet N, and Henrique D (2015). Generation of sensory hair cells by genetic programming with a combination of transcription factors. Development 142, 1948–1959. [DOI] [PubMed] [Google Scholar]
- Cotanche DA (1987). Regeneration of hair cell stereociliary bundles in the chick cochlea following severe acoustic trauma. Hear. Res 30, 181–195. [DOI] [PubMed] [Google Scholar]
- Creyghton MP, Cheng AW, Welstead GG, Kooistra T, Carey BW, Steine EJ, Hanna J, Lodato MA, Frampton GM, Sharp PA, et al. (2010). Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc. Natl. Acad. Sci. USA 107, 21931–21936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruz RM, Lambert PR, and Rubel EW (1987). Light microscopic evidence of hair cell regeneration After gentamicin toxicity in chick cochlea. Arch. Otolaryngol. Head Neck Surg 113, 1058–1062. [DOI] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doetzlhofer A, Basch ML, Ohyama T, Gessler M, Groves AK, and Segil N (2009). Hey2 regulation by FGF provides a Notch-independent mechanism for maintaining pillar cell fate in the organ of Corti. Dev. Cell 16, 58–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Driver EC, Sillers L, Coate TM, Rose MF, and Kelley MW (2013). The Atoh1-lineage gives rise to hair cells and supporting cells within the mammalian cochlea. Dev. Biol 376, 86–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dvoriantchikova G, Seemungal RJ, and Ivanov D (2019). Development and epigenetic plasticity of murine Müller glia. Biochim. Biophys. Acta Mol. Cell Res 1866, 1584–1594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eddison M, Le Roux I, and Lewis J (2000). Notch signaling in the development of the inner ear: lessons from Drosophila. Proc. Natl. Acad. Sci. USA 97, 11692–11699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehret G (1976). Development of absolute auditory thresholds in the house mouse (Mus musculus). J. Am. Audiol. Soc 1, 179–184. [PubMed] [Google Scholar]
- Ernst J, and Kellis M (2012). ChromHMM: automating chromatin-state discovery and characterization. Nat. Methods 9, 215–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faa G, Gerosa C, Fanni D, Nemolato S, Locci A, Cabras T, Marinelli V, Puddu M, Zaffanello M, Monga G, and Fanos V (2010). Marked interindividual variability in renal maturation of preterm infants: lessons from autopsy. J. Matern. Fetal Neonatal. Med 23 (Supplement 3 ), 129–133. [DOI] [PubMed] [Google Scholar]
- Feng J, Liu T, Qin B, Zhang Y, and Liu XS (2012). Identifying ChIP-seq enrichment using MACS. Nat. Protoc 7, 1728–1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furlong EEM, and Levine M (2018). Developmental enhancers and chromosome topology. Science 361, 1341–1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furness DN (2015). Molecular basis of hair cell loss. Cell Tissue Res 361, 387–399. [DOI] [PubMed] [Google Scholar]
- Goldman D (2014). Müller glial cell reprogramming and retina regeneration. Nat. Rev. Neurosci 15, 431–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldman JA, and Poss KD (2020). Gene regulatory programmes of tissue regeneration. Nat. Rev. Genet 21, 511–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golub JS, Tong L, Ngyuen TB, Hume CR, Palmiter RD, Rubel EW, and Stone JS (2012). Hair cell replacement in adult mouse utricles after targeted ablation of hair cells with diphtheria toxin. J. Neurosci 32, 15093–15105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong S, Zheng C, Doughty ML, Losos K, Didkovsky N, Schambra UB, Nowak NJ, Joyner A, Leblanc G, Hatten ME, and Heintz N (2003). A gene expression atlas of the central nervous system based on bacterial artificial chromosomes. Nature 425, 917–925. [DOI] [PubMed] [Google Scholar]
- Groves AK (2010). The challenge of hair cell regeneration. Exp. Biol. Med. (Maywood) 235, 434–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gubbels SP, Woessner DW, Mitchell JC, Ricci AJ, and Brigande JV (2008). Functional auditory hair cells produced in the mammalian cochlea by in utero gene transfer. Nature 455, 537–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta S, Stamatoyannopoulos JA, Bailey TL, and Noble WS (2007). Quantifying similarity between motifs. Genome Biol 8, R24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris JA, Cheng AG, Cunningham LL, MacDonald G, Raible DW, and Rubel EW (2003). Neomycin-induced hair cell death and rapid regeneration in the lateral line of zebrafish (Danio rerio). J. Assoc. Res. Otolaryngol 4, 219–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartman HA, Lai HL, and Patterson LT (2007). Cessation of renal morphogenesis in mice. Dev. Biol 310, 379–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haubner BJ, Adamowicz-Brice M, Khadayate S, Tiefenthaler V, Metzler B, Aitman T, and Penninger JM (2012). Complete cardiac regeneration in a mouse model of myocardial infarction. Aging (Albany, NY) 4, 966–977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, and Glass CK (2010). Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 38, 576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helms AW, Abney AL, Ben-Arie N, Zoghbi HY, and Johnson JE (2000). Autoregulation and multiple enhancers control Math1 expression in the developing nervous system. Development 127, 1185–1196. [DOI] [PubMed] [Google Scholar]
- Huangfu D, Maehr R, Guo W, Eijkelenboom A, Snitow M, Chen AE, and Melton DA (2008). Induction of pluripotent stem cells by defined factors is greatly improved by small-molecule compounds. Nat. Biotechnol 26, 795–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Illingworth CM (1974). Trapped fingers and amputated finger tips in children. J. Pediatr. Surg 9, 853–858. [DOI] [PubMed] [Google Scholar]
- Jen HI, Hill MC, Tao L, Sheng K, Cao W, Zhang H, Yu HV, Llamas J, Zong C, Martin JF, et al. (2019). Transcriptomic and epigenetic regulation of hair cell regeneration in the mouse utricle and its potentiation by Atoh1. eLife 8, e44328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang J, Hu J, Karra R, Dickson AL, Tornini VA, Nachtrab G, Gemberling M, Goldman JA, Black BL, and Poss KD (2016). Modulation of tissue repair by regeneration enhancer elements. Nature 532, 201–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawamoto K, Izumikawa M, Beyer LA, Atkin GM, and Raphael Y (2009). Spontaneous hair cell regeneration in the mouse utricle following gentamicin ototoxicity. Hear. Res 247, 17–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley MW (2006). Regulation of cell fate in the sensory epithelia of the inner ear. Nat. Rev. Neurosci 7, 837–849. [DOI] [PubMed] [Google Scholar]
- Kelly MC, Chang Q, Pan A, Lin X, and Chen P (2012). Atoh1 directs the formation of sensory mosaics and induces cell proliferation in the postnatal mammalian cochlea in vivo. J. Neurosci 32, 6699–6710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiernan AE, Cordes R, Kopan R, Gossler A, and Gridley T (2005). The Notch ligands DLL1 and JAG2 act synergistically to regulate hair cell development in the mammalian inner ear. Development 132, 4353–4362. [DOI] [PubMed] [Google Scholar]
- Klisch TJ, Xi Y, Flora A, Wang L, Li W, and Zoghbi HY (2011). In vivo Atoh1 targetome reveals how a proneural transcription factor regulates cerebellar development. Proc. Natl. Acad. Sci. USA 108, 3288–3293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korrapati S, Roux I, Glowatzki E, and Doetzlhofer A (2013). Notch signaling limits supporting cell plasticity in the hair cell-damaged early postnatal murine cochlea. PLoS One 8, e73276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwan T, White PM, and Segil N (2009). Development and regeneration of the inner ear. Ann. N. Y. Acad. Sci 1170, 28–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanford PJ, Lan Y, Jiang R, Lindsell C, Weinmaster G, Gridley T, and Kelley MW (1999). Notch signalling pathway mediates hair cell development in mammalian cochlea. Nat. Genet 21, 289–292. [DOI] [PubMed] [Google Scholar]
- Lee YS, Liu F, and Segil N (2006). A morphogenetic wave of p27Kip1 transcription directs cell cycle exit during organ of Corti development. Development 133, 2817–2826. [DOI] [PubMed] [Google Scholar]
- Li B, and Dewey CN (2011). RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12, 323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang G, Taranova O, Xia K, and Zhang Y (2010). Butyrate promotes induced pluripotent stem cell generation. J. Biol. Chem 285, 25516–25521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Little M (2016). Kidney Development, Disease, Repair and Regeneration (Elsevier; ). [Google Scholar]
- Liu Z, Dearman JA, Cox BC, Walters BJ, Zhang L, Ayrault O, Zindy F, Gan L, Roussel MF, and Zuo J (2012). Age-dependent in vivo conversion of mouse cochlear pillar and Deiters’ cells to immature hair cells by Atoh1 ectopic expression. J. Neurosci 32, 6600–6610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo YH, Chung E, Li Z, Wan YW, Mahe MM, Chen MS, Noah TK, Bell KN, Yalamanchili HK, Klisch TJ, et al. (2017). Transcriptional regulation by ATOH1 and its target SPDEF in the intestine. Cell. Mol. Gastroenterol. Hepatol 3, 51–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Local A, Huang H, Albuquerque CP, Singh N, Lee AY, Wang W, Wang C, Hsia JE, Shiau AK, Ge K, et al. (2018). Identification of H3K4me1-associated proteins at mammalian enhancers. Nat. Genet 50, 73–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maass JC, Gu R, Basch ML, Waldhaus J, Lopez EM, Xia A, Oghalai JS, Heller S, and Groves AK (2015). Changes in the regulation of the Notch signaling pathway are temporally correlated with regenerative failure in the mouse cochlea. Front. Cell. Neurosci 9, 110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madisen L, Zwingman TA, Sunkin SM, Oh SW, Zariwala HA, Gu H, Ng LL, Palmiter RD, Hawrylycz MJ, Jones AR, et al. (2010). A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat. Neurosci 13, 133–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menendez L, Trecek T, Gopalakrishnan S, Tao L, Markowitz AL, Yu HV, Wang X, Llamas J, Huang C, Lee J, et al. (2020). Generation of inner ear hair cells by direct lineage conversion of primary somatic cells. eLife 9, e55249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ming GL, and Song H (2011). Adult neurogenesis in the mammalian brain: significant answers and significant questions. Neuron 70, 687–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohammad HP, Smitheman KN, Kamat CD, Soong D, Federowicz KE, Van Aller GS, Schneck JL, Carson JD, Liu Y, Butticello M, et al. (2015). A DNA hypomethylation signature predicts antitumor activity of LSD1 inhibitors in SCLC. Cancer Cell 28, 57–69. [DOI] [PubMed] [Google Scholar]
- Morris SA (2016). Direct lineage reprogramming via pioneer factors; a detour through developmental gene regulatory networks. Development 143, 2696–2705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neves J, Abelló G, Petrovic J, and Giraldez F (2013). Patterning and cell fate in the inner ear: a case for Notch in the chicken embryo. Dev. Growth Differ 55, 96–112. [DOI] [PubMed] [Google Scholar]
- Nicetto D, and Zaret KS (2019). Role of H3K9me3 heterochromatin in cell identity establishment and maintenance. Curr. Opin. Genet. Dev 55, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ong CT, and Corces VG (2012). Enhancers: emerging roles in cell fate specification. EMBO Rep 13, 423–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ooi SKT, Qiu C, Bernstein E, Li K, Jia D, Yang Z, Erdjument-Bromage H, Tempst P, Lin SP, Allis CD, et al. (2007). DNMT3L connects unmethylated lysine 4 of histone H3 to de novo methylation of DNA. Nature 448, 714–717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porrello ER, Mahmoud AI, Simpson E, Hill JA, Richardson JA, Olson EN, and Sadek HA (2011). Transient regenerative potential of the neonatal mouse heart. Science 331, 1078–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pulecio J, Verma N, Mejía-Ramírez E, Huangfu D, and Raya A (2017). CRISPR/Cas9-based engineering of the epigenome. Cell Stem Cell 21, 431–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR, and Hall IM (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rada-Iglesias A, Bajpai R, Swigut T, Brugmann SA, Flynn RA, and Wysocka J (2011). A unique chromatin signature uncovers early developmental enhancers in humans. Nature 470, 279–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramírez F, Dündar F, Diehl S, Grüning BA, and Manke T (2014). deepTools: a flexible platform for exploring deep-sequencing data. Nucleic Acids Res 42, W187–W191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson JT, Thorvaldsdóttir H, Winckler W, Guttman M, Lander ES, Getz G, and Mesirov JP (2011). Integrative genomics viewer. Nat. Biotechnol 29, 24–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez AM, and Kang J (2020). Regeneration enhancers: starting a journey to unravel regulatory events in tissue regeneration. Semin. Cell Dev. Biol 97, 47–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose MF, Ren J, Ahmad KA, Chao HT, Klisch TJ, Flora A, Greer JJ, and Zoghbi HY (2009). Math1 is essential for the development of hindbrain neurons critical for perinatal breathing. Neuron 64, 341–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruben RJ (1967). Development of the inner ear of the mouse: a radioautographic study of terminal mitoses. Acta Otolaryngol 220, 1–44. [PubMed] [Google Scholar]
- Rupp S, and Schranz D (2015). Cardiac regeneration in children. Pediatr. Cardiol 36, 713–718. [DOI] [PubMed] [Google Scholar]
- Ryals BM, and Rubel EW (1988). Hair cell regeneration after acoustic trauma in adult Coturnix quail. Science 240, 1774–1776. [DOI] [PubMed] [Google Scholar]
- Semerci F, Choi WT-S, Bajic A, Thakkar A, Encinas JM, Depreux F, Segil N, Groves AK, and Maletic-Savatic M (2017). Lunatic fringe-mediated Notch signaling regulates adult hippocampal neural stem cell maintenance. eLife 6, e24660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Lan F, Matson C, Mulligan P, Whetstine JR, Cole PA, Casero RA, and Shi Y (2004). Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell 119, 941–953. [DOI] [PubMed] [Google Scholar]
- Skene PJ, and Henikoff S (2017). An efficient targeted nuclease strategy for high-resolution mapping of DNA binding sites. eLife 6, e21856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith T, Heger A, and Sudbery I (2017). UMI-tools: modeling sequencing errors in Unique Molecular Identifiers to improve quantification accuracy. Genome Res 27, 491–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stojanova ZP, Kwan T, and Segil N (2015). Epigenetic regulation of Atoh1 guides hair cell development in the mammalian cochlea. Development 142, 3529–3536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takebayashi S, Yamamoto N, Yabe D, Fukuda H, Kojima K, Ito J, and Honjo T (2007). Multiple roles of Notch signaling in cochlear development. Dev. Biol 307, 165–178. [DOI] [PubMed] [Google Scholar]
- Tao L, and Segil N (2015). Early transcriptional response to aminoglycoside antibiotic suggests alternate pathways leading to apoptosis in sensory hair cells in the mouse inner ear. Front. Cell. Neurosci 9, 190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tateya T, Sakamoto S, Ishidate F, Hirashima T, Imayoshi I, and Kageyama R (2019). Three-dimensional live imaging of Atoh1 reveals the dynamics of hair cell induction and organization in the developing cochlea. Development 146, dev177881. [DOI] [PubMed] [Google Scholar]
- Ueki Y, Wilken MS, Cox KE, Chipman L, Jorstad N, Sternhagen K, Simic M, Ullom K, Nakafuku M, and Reh TA (2015). Transgenic expression of the proneural transcription factor Ascl1 in Müller glia stimulates retinal regeneration in young mice. Proc. Natl. Acad. Sci. USA 112, 13717–13722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uygur A, and Lee RT (2016). Mechanisms of cardiac regeneration. Dev. Cell 36, 362–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner EL, and Shin JB (2019). Mechanisms of hair cell damage and repair. Trends Neurosci 42, 414–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Lee JE, Lai B, Macfarlan TS, Xu S, Zhuang L, Liu C, Peng W, and Ge K (2016). Enhancer priming by H3K4 methyltransferase MLL4 controls cell fate transition. Proc. Natl. Acad. Sci. USA 113, 11871–11876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wapinski OL, Vierbuchen T, Qu K, Lee QY, Chanda S, Fuentes DR, Giresi PG, Ng YH, Marro S, Neff NF, et al. (2013). Hierarchical mechanisms for direct reprogramming of fibroblasts to neurons. Cell 155, 621–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White PM, Doetzlhofer A, Lee YS, Groves AK, and Segil N (2006). Mammalian cochlear supporting cells can divide and trans-differentiate into hair cells. Nature 441, 984–987. [DOI] [PubMed] [Google Scholar]
- Williams SE, Beronja S, Pasolli HA, and Fuchs E (2011). Asymmetric cell divisions promote Notch-dependent epidermal differentiation. Nature 470, 353–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong ACY, and Ryan AF (2015). Mechanisms of sensorineural cell damage, death and survival in the cochlea. Front. Aging Neurosci 7, 58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woods C, Montcouquiol M, and Kelley MW (2004). Math1 regulates development of the sensory epithelium in the mammalian cochlea. Nat. Neurosci 7, 1310–1318. [DOI] [PubMed] [Google Scholar]
- Yamamoto N, Tanigaki K, Tsuji M, Yabe D, Ito J, and Honjo T (2006). Inhibition of Notch/RBP-J signaling induces hair cell formation in neonate mouse cochleas. J. Mol. Med. (Berl.) 84, 37–45. [DOI] [PubMed] [Google Scholar]
- Yan J, Chen S-AA, Local A, Liu T, Qiu Y, Dorighi KM, Preissl S, Rivera CM, Wang C, Ye Z, et al. (2018). Histone H3 lysine 4 monomethylation modulates long-range chromatin interactions at enhancers. Cell Res 28, 204–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, Xie X, Deng M, Chen X, and Gan L (2010). Generation and characterization of Atoh1-Cre knock-in mouse line. Genesis 48, 407–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang K, and Kang J (2019). Tissue regeneration enhancer elements: a way to unlock endogenous healing power. Dev. Dyn 248, 34–42. [DOI] [PubMed] [Google Scholar]
- Zentner GE, Tesar PJ, and Scacheri PC (2011). Epigenetic signatures distinguish multiple classes of enhancers with distinct cellular functions. Genome Res 21, 1273–1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao L-D, Guo W-W, Lin C, Li L-X, Sun J-H, Wu N, Ren L-L, Li X-X, Liu H-Z, Young W-Y, et al. (2011). Effects of DAPT and Atoh1 overexpression on hair cell production and hair bundle orientation in cultured organ of Corti from neonatal rats. PLoS One 6, e23729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng JL, and Gao WQ (2000). Overexpression of Math1 induces robust production of extra hair cells in postnatal rat inner ears. Nat. Neurosci 3, 580–586. [DOI] [PubMed] [Google Scholar]
- Zine A, Aubert A, Qiu J, Therianos S, Guillemot F, Kageyama R, and Ribaupierre F de. (2001). Hes1 and Hes5 activities are required for the normal development of the hair cells in the mammalian inner ear. J. Neurosci 21, 4712–4720. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets generated during this study are available at NCBI Gene Expression Omnibus(NCBI GEO: GSE150010).