

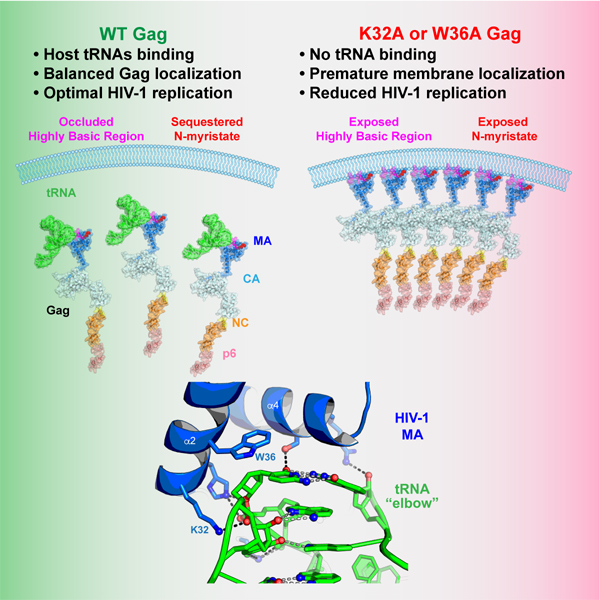
Summary
The HIV-1 virion structural polyprotein, Gag, is directed to particle assembly sites at the plasma membrane by its N-terminal Matrix (MA) domain. MA also binds to host tRNAs. To understand the molecular basis of MA-tRNA interaction and its potential function, we present a co-crystal structure of HIV-1 MA-tRNALys3 complex. The structure reveals a specialized group of MA basic and aromatic residues preconfigured to recognize the distinctive structure of the tRNA elbow. Mutational, crosslinking, fluorescence and NMR analyses show that the crystallographically defined interface drives MA-tRNA binding in solution and living cells. The structure indicates that MA is unlikely to bind tRNA and membrane simultaneously. Accordingly, single amino-acid substitutions that abolish MA-tRNA binding caused striking redistribution of Gag to the plasma membrane and reduced HIV-1 replication. Thus, HIV-1 exploits host tRNAs to occlude a membrane localization signal and control the subcellular distribution of its major structural protein.
Graphical Abstract
eTOC Blurb
Host tRNAs interact with HIV-1 Gag matrix domain which targets Gag to the plasma membrane. Bou-Nader et al., solve a co-crystal structure of HIV-1 matrix bound to human tRNALys3, and reveal that specific recognition of tRNA “elbow” allows HIV-1 to exploit host tRNAs to regulate Gag localization and virion assembly.
Introduction
The HIV-1 Gag protein is the major virion structural protein that determines the location of particle assembly and the morphology that particles adopt. Gag also recruits the viral genome as well as host cell proteins that complete particle envelopment (Sundquist and Kräusslich, 2012). Expression of the Gag protein alone in human cells drives the formation of virus-like particles that are nearly indistinguishable in morphology from immature virions.
Subcellular targeting of HIV-1 Gag protein and the site of particle assembly is governed by the N-terminal matrix (MA) domain. MA is composed of an N-terminal globular head and a C-terminal helical stalk that connects to the remaining Gag domains (Hill et al., 1996; Massiah et al., 1994; Tang et al., 2002). Two key features of the MA globular head specify the location of particle assembly. Specifically, an N-terminal myristoyl moiety is required for stable membrane association (Bryant and Ratner, 1990; Göttlinger et al., 1989) while basic amino acids on the MA globular head surface form a highly basic region (HBR) (Zhou et al., 1994) that is important for specific interactions between MA and phosphatidylinositol-4,5-bisphosphate (PIP2), a plasma membrane resident phospholipid (Mücksch et al., 2017; Ono et al., 2004). Notably, at low concentrations where Gag tends to be monomeric, myristate is sequestered in a hydrophobic pocket. As Gag accumulates in infected cells, multimerization through the capsid (CA) and nucleocapsid (NC) domains drives MA domain trimerization that in turn triggers myristate exposure and stable association with membrane (Perez-Caballero et al., 2004; Tang et al., 2004).
MA and NC both bind to RNA but with very different specificities. NC binds to specific 5′ leader RNA structures and purine-rich RNA sequences to mediate genome packaging (Bieniasz and Telesnitsky, 2018; Kuzembayeva et al., 2014; Lu et al., 2011). Conversely, crosslinking immunoprecipitation sequencing (CLIP-seq) studies reveal that MA binds to tRNA (Kutluay et al., 2014). MA:tRNA interaction is selective; GluCUC, GluUUC, GlyGCC, GlyCCC, LysCUU, LysUUU, ValAAC and ValCAC tRNAs are selectively bound and MA-tRNA binding was the most frequently detected binding event involving Gag. Notably, biochemical assays indicate that tRNA can inhibit Gag binding to membrane (Kutluay et al., 2014) and RNA can block MA binding specifically to liposomes that lack acidic phospholipids (Chukkapalli et al., 2010; Dick et al., 2013). Occlusion of MA basic residues by RNA has been proposed to block nonproductive assembly at intracellular membranes and impart greater selectivity for PIP2-rich plasma membrane. However, NMR studies showing that tRNA inhibits MA association with PIP2-enriched liposomes are inconsistent with that model (Gaines et al., 2018b). Alternatively, MA-tRNA binding may temporally regulate assembly in conjunction with myristate sequestration to inhibit Gag binding to membrane under conditions of low Gag concentration, shortly after infection (Holmes et al., 2015).
MA also binds several in vitro selected RNA aptamers and exhibits significant sequence preference, with Kds ranging from 3 to 500 nM (Lochrie et al., 1997; Purohit et al., 2001). These studies hint at potential sequence or structural specificities beyond charge complementarity underlying MA-RNA interactions. To define the RNA-binding specificity of MA, particularly its strong preference for tRNAs, and to elucidate the biological role of the interaction in cells, we undertook a detailed analysis of MA-tRNA interactions in cells and in vitro and solved a co-crystal structure of a MA-tRNA complex. The structure reveals highly specific contacts between a specialized portion of the MA HBR and the “elbow” (or “outer corner”) structure of the tRNA, as well as fortuitous contacts to the inner corner region of tRNA only observed in crystals. We validate these interfacial contacts and define binding specificities using calorimetry, analytical ultracentrifugation, fluorescence and NMR analyses in vitro and crosslinking immunoprecipitation and live-cell imaging analyses in cells. Remarkably, mutation of a single amino acid (K32A or W36A) that abolished MA-tRNA interaction but maintained MA-PIP2 interaction, changed the subcellular distribution of the HIV-1 Gag protein or the isolated MA domain, and caused cell type-dependent deficits in HIV-1 replication.
Results
Identification of RNA-binding site on MA in cells using CLIP
To identify MA determinants important for tRNA binding, we adopted a ‘surface scanning’ mutagenesis approach. Wild-type (WT) and mutated forms of the isolated MA protein, appended with a C-terminal 3xHA tag, were expressed in 293T cells and subjected to an abbreviated CLIP experiment. Cells were fed with 4-thiouridine (4sU) and UV-irradiated to crosslink proteins to RNA. After treatment of cell lysates with RNase A, the tagged MA proteins were immunoprecipitated, and labelled with polynucleotide kinase and γ32P-ATP. We have previously shown that this approach yields 32P-labelled MA-RNA adducts, comprised of MA-3xHA crosslinked to the 5ʹ halves of various tRNA species that migrate as a nearly homogeneous species on SDS-PAGE (Kutluay et al., 2014). We tested a panel of 42 MA variants containing alanines substituted for solvent-exposed amino acids on the upper surface of the MA globular head (Figure 1A). Most of these mutations, including those that targeted HBR residues important for membrane binding, had little effect on tRNA crosslinking (Figure 1B). However, several alanine substitutions, including L21A, R22A, K27A and R76A reduced MA-tRNA crosslinking. Most notably, the K32A or W36A substitutions nearly abolished MA:tRNA crosslinking. Thus, the key tRNA-binding determinants on MA consist of a surprisingly small set of amino acids within the HBR. This suggests a specific MA-tRNA interaction, rather than a diffuse, interchangeable cluster of electrostatic contacts.
Figure 1. CLIP, mutational and biophysical analyses of MA:tRNA interactions.
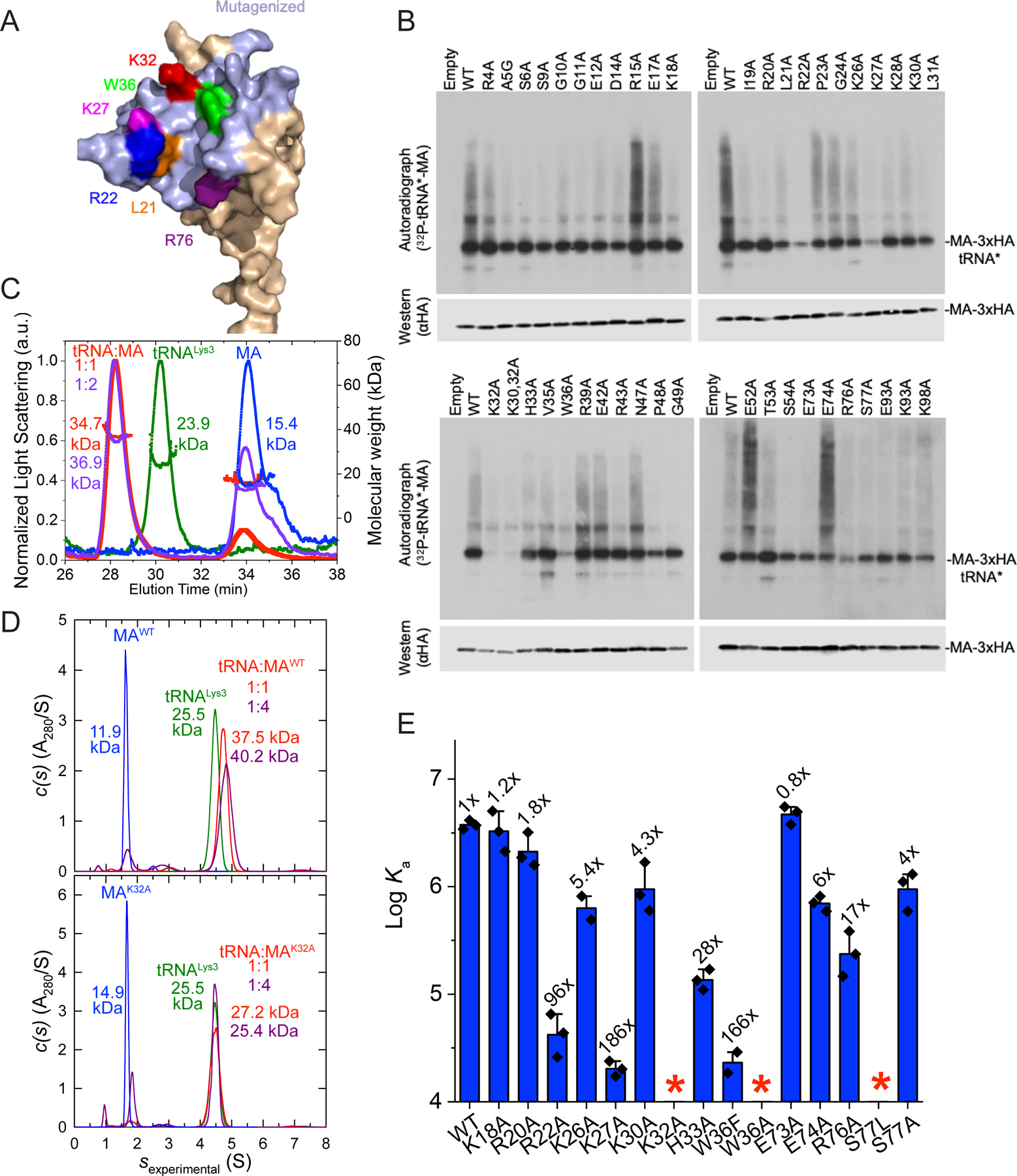
(A) Surface-exposed, mutated MA residues are colored light blue; others in beige. Residues important for tRNA crosslinking are in red, magenta, purple, green and blue.
(B) CLIP analysis of C-terminally 3xHA tagged, WT and mutant MA proteins in 293T cells. Upper panel: autoradiography of γ32P-ATP-labelled tRNA 5′ halves crosslinked to MA. Lower panel: anti-HA western blot.
(C) Analysis of WT MA (blue), tRNALys3 (green), and tRNA Lys3:MA mixtures at 1:1 (red) and 1:2 (purple) molar ratios by SEC-MALS. Molecular weights (MWs) are indicated.
(D) Analysis of WT (upper panel) and K32A (lower panel) MA (blue), tRNALys3 (green), and tRNA Lys3:MA mixtures at 1:1 (red) and 1:4 (dark red) molar ratios by AUC. MWs are indicated.
(E) Mutational and ITC analyses of select MA residues. *: no significant binding detected. Data are represented as mean ± s.d.
Structure of a human tRNALys3 - HIV-1 MA complex
We next solved a co-crystal structure of MA bound to tRNALys3 at 3.15 Å resolution (Figure 2, S1, S2, Table S1). We used tRNALys3 because it was among the human tRNAs that was most frequently crosslinked to MA in HIV-infected cells (Kutluay et al., 2014). MA exhibited robust, stoichiometric binding (1:1) with tRNALys3 in solution with a Kd of ~267 nM, as evidenced by size-exclusion chromatography coupled with multi-angle light scattering (SEC-MALS, Figure 1C), isothermal titration calorimetry (ITC, Figure 1E, S3, Table S2), and analytical ultracentrifugation (AUC, Figure 1D, S4, Table S3). An electrophoretic mobility shift assay (EMSA) also only detected 1:1 complexes (Figure S1). Despite forming primarily 1:1 complexes in solution, the co-crystal structure revealed two MA molecules bound to each tRNALys3 (Figure 2A-B); one at the “elbow” (also known as the “outer corner”) and the other at the “inner corner” of the central region of the tRNA, burying 533 Å2 and 828 Å2 of solvent-accessible surface areas, respectively, (Figure 2, S2). The tRNA elbow is a characteristically flat, surface-exposed feature located at the vertex of its “L” shape while the inner corner is located on the opposite, interior side. Both MA molecules are nearly identical to each other and to an isolated MA structure, with overall root-mean-square deviations (RMSDs) of ~0.6 Å and ~0.5 Å over 89 Cα atoms, respectively (Figure S1) (Hill et al., 1996). The tRNALys3 in the MA complex exhibits minor conformational differences compared to an isolated, post-transcriptionally modified tRNALys3 structure (RMSD of 2.1 Å over 64 C1′ residues), with the most pronounced differences in the flexible D-loop (Figure S1) (Benas et al., 2000). We conclude that MA approaches tRNA largely as a rigid body but appears to induce minor, local conformational adaptations near the tRNA elbow especially in the D-loop.
Figure 2. Co-crystal structure of tRNALys3 in complex with HIV-1 MA.
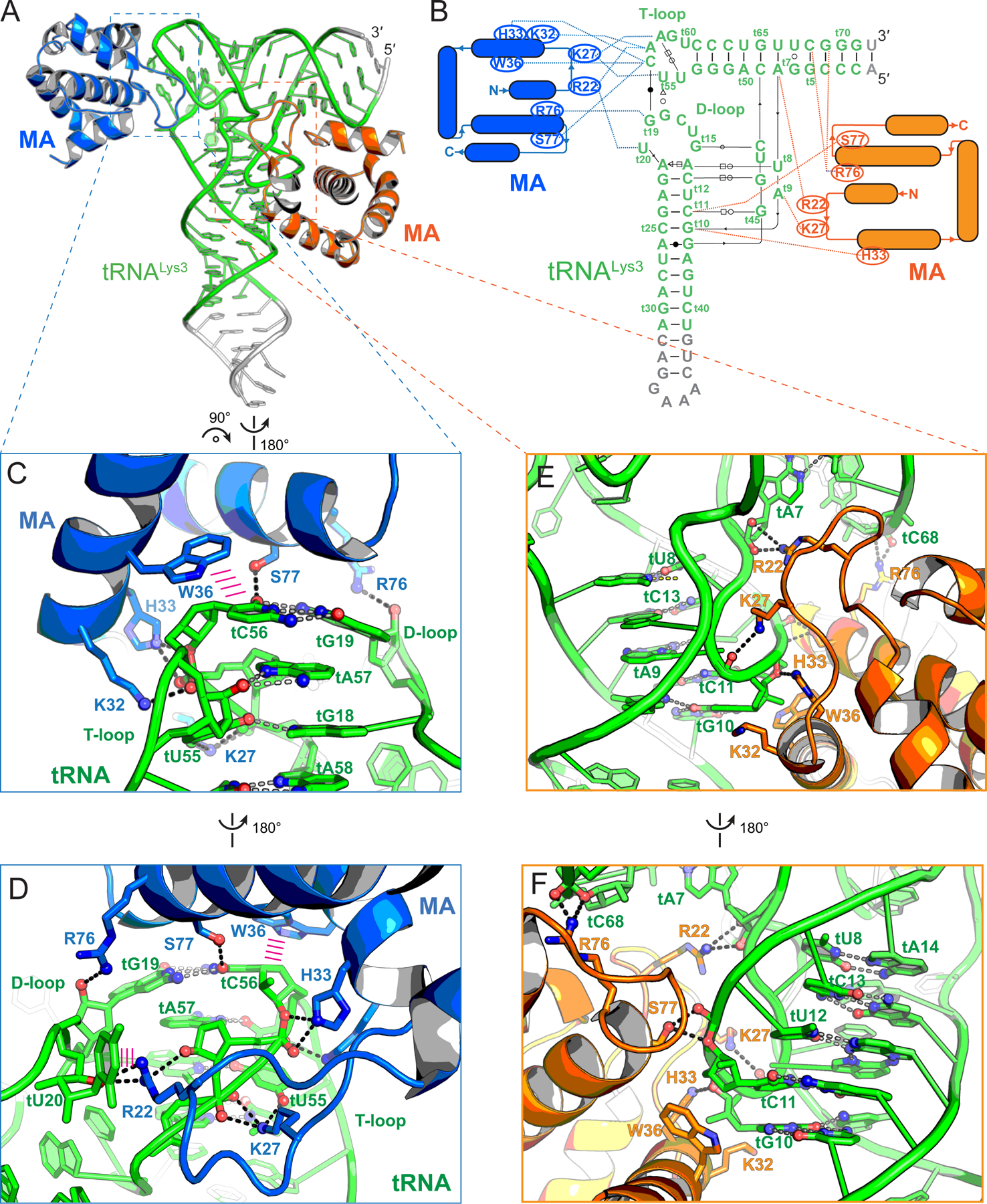
(A) Overall structure of tRNALys3 (green) bound to two MA molecules (blue and orange), which bind the tRNA elbow and inner corner regions, respectively. Engineered tRNA regions to promote crystallization shown in gray.
(B) Secondary structures and interactions of the MA-tRNALys3 complex. Noncanonical base pairs are indicated using Leontis-Westhof symbols (Leontis and Westhof, 2001). The tRNALys3 is numbered according to standard convention with a ‘t’ prefix. MA-tRNALys3 contacts as dashed lines.
(C & D) Specific recognition of tRNALys3 elbow structure by MA. Hydrogen bonds as dashed lines; stacking interactions as parallel magenta lines.
(E & F) Fortuitous interactions of tRNALys3 inner corner region with MA in crystallo.
MA specifically recognizes the characteristic structure of the tRNA elbow
Intriguingly, out of a larger panel of available basic residues in the HBR (K18-H33), only a subset of basic and aromatic residues primarily recognizes the well-defined backbone structure of the tRNA elbow (Figures 1, 2). The elbow structure is conserved among known cytosolic elongator tRNAs and is formed by the intercalation of the D-loop (t15-t20, tRNA numbering preceded by a “t”) into the adjacent T-loop (t54-t58). The T-loop is a recurring pentanucleotide structural motif that bends the RNA backbone by 180° and leaves a stacking gap between tA57 and tA58 (Figure 2, 3A). This gap is filled by the intercalating tG18 from the D-loop, which further forms a bifurcated hydrogen bond with tU55 (Figure 2C, 3A). A nearly invariant tertiary base pair between tC56 at the apex of the T-loop and tG19 of the D-loop caps and completes the elbow structure. Notably, the tRNA elbow region harbors the major site of MA-tRNA crosslinking (tU20) in 4sU-driven CLIP assays (Kutluay et al., 2014).
Figure 3. tRNA determinants for MA binding.
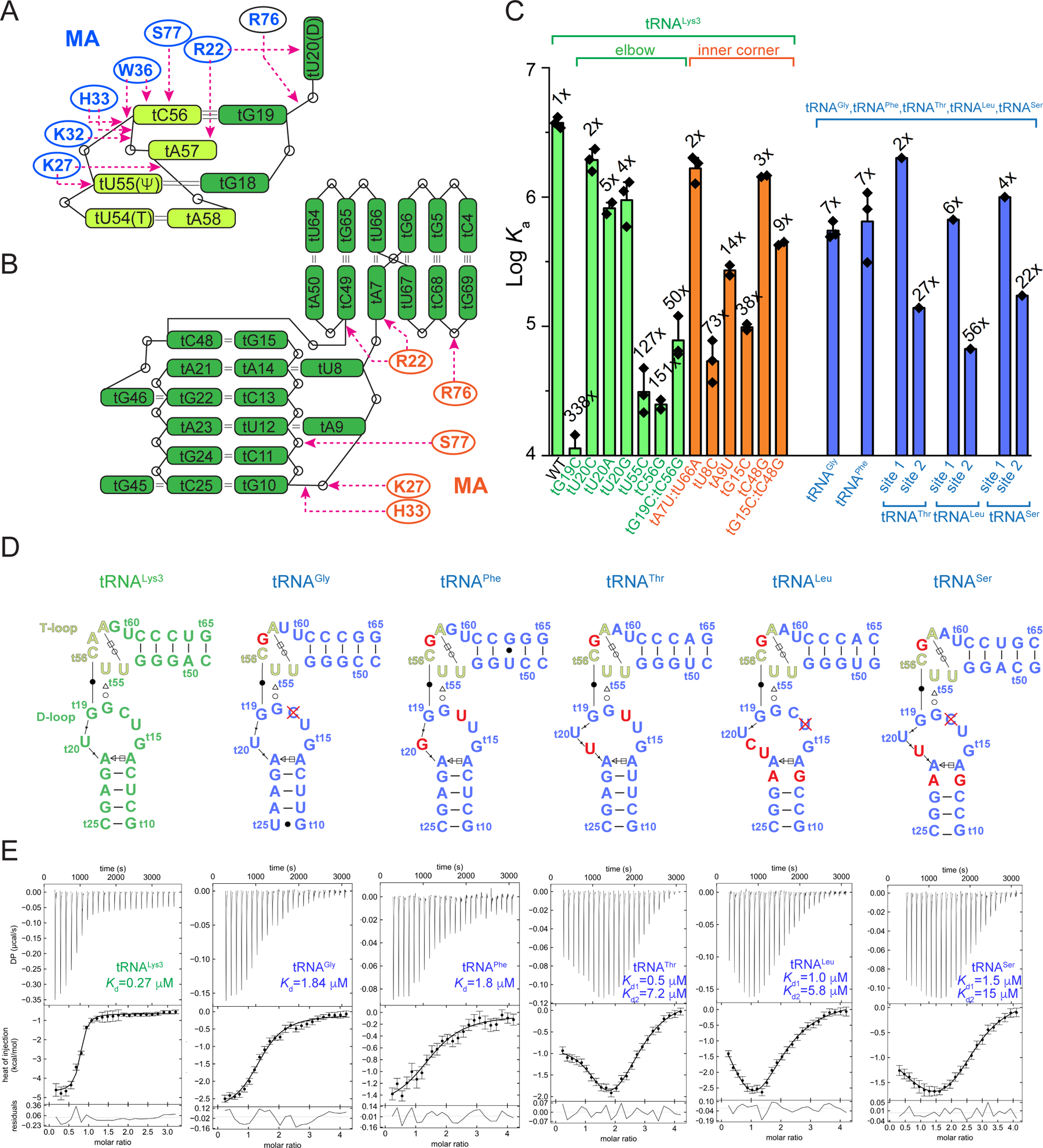
(A & B) Schematic representations of MA-tRNALys3 interactions at the elbow (A) and inner corner (B). The T-loop motif (t54-t58) is shown in light green.
(C) Mutational and ITC analyses of human tRNALys3, tRNAGly, tRNAPhe, tRNAThr, tRNALeu and tRNASer. Data are represented as mean ± s.d.
(D) Secondary structures of the elbow regions of the six tRNAs assayed in (C). Red crosses denote deletion.
(E) Representative ITC isotherms of MA binding to the six tRNAs in (D). The error bars indicate uncertainties in individual injection heats estimated by NITPIC (Methods).
MA employs three highly conserved basic residues K27, K32 and H33, located in the α1-α2 loop (the loop connecting helices 1 and 2) and helix 2, to track the “U”-shaped curvature of the T-loop backbone. In addition, MA clutches the tC56 nucleobase located at the T-loop apex through stacking with W36 — a large, bicyclic side chain (Figure 2C-D, S1). Consistent with these observed direct contacts and CLIP assay results, alanine substitutions of any of these MA side chains led to dramatic drops in tRNA binding in solution, with K32A and W36A completely abolishing binding (Figure 1E). In striking contrast, two immediately adjacent residues of the HBR that do not contact tRNA in the structure had little effect when mutated, with K26A and K30A exhibiting ~5- and ~4-fold deficits, respectively. Interestingly, even a conservative W36F substitution caused a 166-fold reduction in binding, suggesting a requirement for an aromatic ring larger than phenylalanine to achieve optimal stacking with tC56. In addition to two hydrogen bonds to the phosphate oxygens of tA58, K27 makes a third hydrogen bond to the O4 carbonyl of tU55, in one of the few nucleobase-selective contacts to the tRNA (Figure 2C, 2D). Consistent with these extensive K27A contacts, a K27A substitution in MA led to 186-fold drop in binding affinity (Figure 1E). In reciprocity, the tU55C substitution in the tRNA, which K27 contacts, also weakened binding by 127-fold (Figure 3C). This defect is likely caused by a partial loss of the K27 contacts as well as destabilization of the internal T-loop structure. The surface-exposed tG19-tC56 tertiary base pair is evidently required for robust MA binding, as mismatches here reduced MA binding by 150- to 340-fold (Figure 3C). Notably, exchanging the elbow tertiary G-C pair for C-G only partially restored binding (60-fold defect), suggesting a sequence specificity for MA interaction beyond structural selectivity. To assess if the deleterious effects of tG19-tC56 mismatches on MA binding are caused by disruption of the local elbow interface, or by global destruction of the tRNA tertiary structure, we measured tRNA thermal denaturation profiles using Differential Scanning Calorimetry (DSC). As expected, WT tRNALys3 mostly unfolded via a single, cooperative transition centered at ~73.6 °C in the presence of 1 mM Mg2+ (Figure S4), in congruence with the strong coupling of secondary and tertiary structures reported for several bacterial and yeast tRNAs (Leamy et al., 2019; Strulson et al., 2014). Both mismatch mutations (tG19C, tC56G) lowered the Tm by a modest 1.6 °C to 72.0 °C, without drastically impacting the overall tRNA fold. This analysis suggests that local disruption of the elbow surface is principally responsible for the reduction in MA binding.
In addition, MA uses the N-terminal region of helix 4 to augment overall tRNA binding. On the T-loop side of the tertiary base pair, E74 and S77 make single hydrogen bonds to tC56 (Figure 2C, 2D, 3A). On the D-loop side, E73 packs against the nucleobase of tG19 (distance: ~3.0 Å) while R76 uses its guanidinium group to recognize the 2′-OH of tG19. Compared to the critical α1-α2 loop and helix 2 region, helix 4 plays an auxiliary role in tRNA recognition, as evidenced by relatively minor effects of alanine substitutions here (up to 17-fold by R76A) (Figure 1E). An E73A substitution actually slightly increased tRNA binding, likely due to improved hydrophobic interactions conferred by the methyl side chain of alanine with the tG19 nucleobase. Notably, while S77A had only 4-fold reduction in tRNA binding, S77L abrogated binding, presumably due to amplified steric clash with the elbow base pair (Figure 1E, 2C).
Finally, R22 of MA occupies the space between the T- and D-loops. Its terminal guanidinium moiety bridges the ribose 2′-OH of tA57 (T-loop) and ribose O4 of tU20 (D-loop) and engages a cation-π interaction with the nucleobase of the extrahelically flipped tU20 (Figure 2D, 3A, S4). tU20 also packs with the hydrophobic alkyl chain of R22 and points its O4 carbonyl group towards K98, less than 5 Å away. This observation readily explains the prominent 4sU-mediated crosslinking between the tRNA D-loop and MA seen in CLIP experiments (Kutluay et al., 2014). Notably, tU20 is frequently post-transcriptionally modified to dihydrouridine (D, the eponym of D-loop), a non-aromatic post-transcriptional modification that increases the flexibility of the D-loop and facilitates its flipping (Bou-Nader et al., 2019; Bou-Nader et al., 2015; Dalluge et al., 1997). Consistent with the nucleobase non-selective nature of the cation-π interaction, substitution of tU20 by C, A or G had modest effects on MA binding, increasing the Kd by 2-, 5- and 4-fold, respectively (Figure 3C). On the MA side, in contrast to the 96-fold defect of the R22A substitution, the neighboring K18A and R20A substitutions, also key residues of the HBR for membrane binding, had less than 2-fold effects on tRNA binding, consistent with their negligible effects in the cell-based tRNA crosslinking assay (Figure 1B, 1E). These findings not only strongly support the contacts observed in the complex structure, but further suggest a surprising tRNA-binding specificity that involves only a handful of HBR residues.
Overall, the structure reveals that instead of non-specific, electrostatic interactions generally believed to drive MA-nucleic acids interactions, MA employs a precise set of strategically placed basic (R22, K27, K32, and to a lesser extent, H33) and aromatic (W36) side chains that are pre-configured to recognize the peculiar structure of the tRNA elbow, one of the most salient surface features of this abundant host RNA. Notably, there was striking concordance among the effects of substitutions on tRNA binding in CLIP assays in cells, in in vitro solution binding assays, and the tRNA:MA contacts revealed by the structure. By contrast, other HBR residues in the immediate vicinity (K18, R20, K26, K30) that contribute to membrane binding play little roles in tRNA binding, in solution or in cells, suggesting that a specific subset of HBR residues have become specialized for tRNA recognition and cannot be substituted by adjacent basic residues. This remarkable specificity allows HIV-1 Gag to selectively hijack host tRNAs amongst a plethora of host and viral RNA molecules with comparable charges in the cytoplasm of infected cells.
Fortuitous MA binding to the tRNA inner corner in crystallo but not in solution
While one MA molecule specifically recognized the tRNA elbow, in co-crystals a second MA molecule unexpectedly bound the inner corner of the tRNA, likely due to the negative charges congregated here (Figure 2E, 2F), and MA-MA contacts in the crystal. The inner corner is characterized by a hairpin turn trajectory which exposes a cluster of backbone phosphates towards the solvent, whilst pointing their nucleobases inward to form three base triples. Thus, this region acts as a “linchpin” to stabilize the D-stem and the tertiary fold of tRNA (Hamann and Hou, 2000; Oliva et al., 2006; Westhof and Auffinger, 2001).
While the MA residues that bind the elbow and the inner corner overlap significantly, there are also prominent differences. Specifically, R22, K27, S77, and R76 bind non-bridging phosphate oxygens of tU8, tG10, tU12, and tG72, respectively (Figure 2E, 2F, 3B). R22 also makes a single nucleobase-selective hydrogen bond between its guanidinium moiety and the N3 of A7. To assess the importance of this contact, we swapped the tA7-tU66 pair with an isosteric U-A pair, removing the N3 contact. However, this had a mere 2-fold effect on MA binding, suggesting that the sole base-specific contact to this tRNA region is not important for binding in solution. Crucially, neither K32 nor W36, both MA residues that are clearly essential for tRNA binding in solution and in cells, are seen in contact with tRNA at the inner corner site (Figure 2B, 2E, 2F, 3B). These observations suggest that the inner corner site is unlikely a biologically relevant MA binding site that functions in vivo. Nevertheless, as this interface may potentially inform our understanding of MA binding to other RNAs such as MA aptamers, we continued to characterize it. Single-nucleotide substitutions here led to 2–73-fold deficits in MA binding, significantly less than the 60–340-fold deficits by the elbow substitutions (excluding tU20) (Figure 3C). The most deleterious inner corner mutation, tU8C, is expected to prevent a base triple formation with tA14 and tA21. Indeed, it significantly perturbed the tRNA fold producing two discrete transitions at 70.4°C and 82.3°C (Figure S4). Other inner corner mutations, tA9U, tG15C, and tC48G, had intermediate impacts, supporting the notion that the tRNA fold is resilient to most single substitutions (Guy et al., 2014; Strulson et al., 2014). Given the short distance (30–40 Å) and close chain connectivity between the elbow and the inner corner, it is likely that mutations in the inner corner could lead to subtle changes in the elbow. As MA binding is exquisitely sensitive to the elbow backbone trajectory and tertiary pair, inner corner mutations likely indirectly impacted MA binding.
To ascertain if the inner corner can bind MA at elevated concentrations, we monitored the formation of MA-tRNA complexes using AUC. AUC readily detected 1:1 (tRNA:MA) complexes but no 1:2 complexes even at high micromolar MA concentrations (Figure 1D, S4, Table S3). Taken together, we conclude that MA binding to the tRNALys3 inner corner occurs exclusively in crystallo and does not occur in solution under reasonable conditions.
Fluorescence and NMR probing of MA-tRNA interfaces corroborate elbow binding in solution
To obtain direct physical evidence in solution that MA binds the tRNA elbow, we employed 2-aminopurine (2AP) fluorescence probing. tRNALys3 was site-specifically labelled either at tG10 of the inner corner or tG19 of the elbow with 2AP, a fluorescent analogue of adenine and guanine. The quantum yield of 2AP is highly sensitive to its local chemical environment, principally from stacking interactions with nearest neighboring nucleobases (Jean and Hall, 2001; Zhang and Ferré-D’Amaré, 2014). When increasing amounts of MA were titrated into the labelled tRNAs, a dose-dependent increase of 2AP fluorescence intensity was observed for tRNA-2AP19 (elbow) but not tRNA-2AP10 (inner corner) (Figure 4A). The considerable fluorescence enhancement of 2AP19 (up to 50%) upon MA binding is highly congruent with the observed π - π stacking interactions between W36 and tC56, which presumably destabilized the latter’s pairing with 2AP19, thus liberating the 2AP and increasing its quantum yield. As expected, the tRNA binding-incompetent K32A MA variant did not alter 2AP19 fluorescence, nor did the WT MA when titrated into the 2AP-labelled tRNA 5′ half (t1-t33) (Figure 4A). These data indicate that the fluorescence enhancement requires both binding competency by MA and full tRNA structure. On the inner corner side, the lack of MA effect on 2AP10 fluorescence is consistent with the absence of MA binding to that site in solution.
Figure 4. Fluorescence and NMR analyses of MA-tRNA interactions.
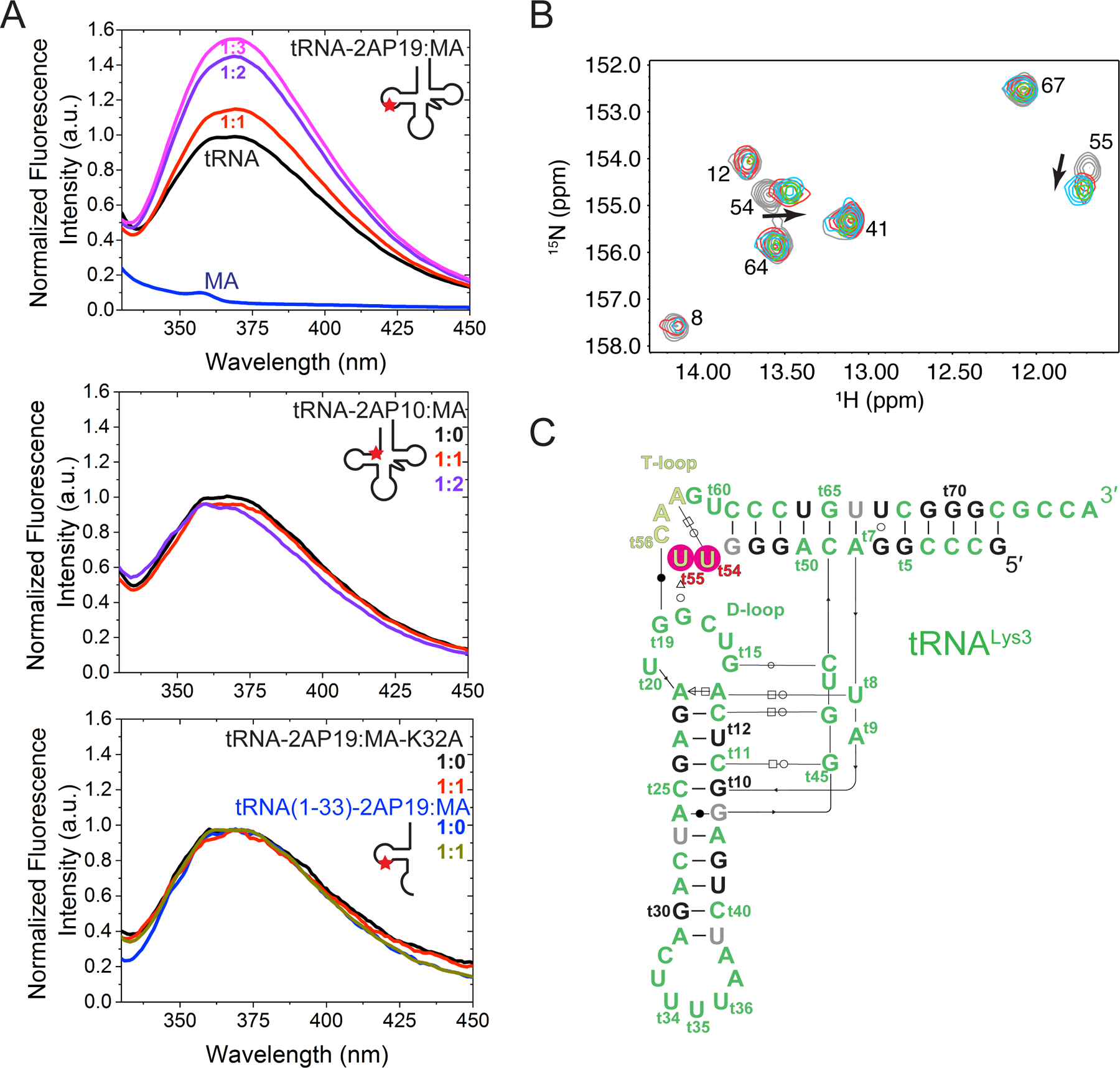
(A) Fluorescence emission spectra of tRNALys3-2AP19 (elbow) in the absence (black) or presence (red, purple and magenta) of WT MA (top panel), tRNALys3-2AP10 (inner corner) in the absence (black) or presence of WT MA (red and purple) (middle panel), tRNALys3-2AP19 in the absence (black) or presence (red) of binding-incompetent K32A MA (bottom panel), and of the 5′ half of tRNALys3-2AP19 (nt 1–33) in the absence (blue) or presence (gray) of WT MA (bottom panel).
(B) Portion of 2D 1H-15N HSQC spectra obtained for 15N-labeled tRNALys3 upon titration with MA [MA]/[tRNA] = 0.0 (gray), 0.5 (red), 1.0 (green), 1.5 (cyan).
(C) Secondary structure of tRNALys3 with resolved residues shown in black, unassigned residues in gray, and residues that shift upon MA titration in red.
To further characterize tRNA-MA binding in solution, 2D-[1H-15N] heteronuclear single quantum coherence data were collected on uniformly 15N-G, -U enriched tRNALys3 to monitor base paired G and U nucleotides upon titration with MA (Figure 4B, 4C) (Puglisi and Puglisi, 2007). These data show chemical shift perturbations of the imino signals of U54, which participates in a Hoogsteen base pair with A58 (Puglisi and Puglisi, 2007), and U55 within the T-loop (Figure 4B, 4C). No chemical shift perturbations were detected for G5, G6, U12, G22, G24, G69, G70, and G71 located near the inner corner site (Figure 4C, black residues). However, significant signal broadening was observed at MA:tRNA molar ratios greater than 1.5:1, and we cannot rule out the possibility that a second MA molecule binds more weakly and with greater exchange dynamics. Taken together, both fluorescence and NMR analyses provide direct physical evidence that the tRNA elbow is the primary, high-affinity site engaged by MA in solution, consistent with crosslinking analysis in living cells.
Comparison of the tRNA-MA complex structure with the MA structure in complex with PIP2 analog di-C4-phosphatidylinositol-(4,5)-bisphosphate reveals extensive overlap between the tRNA and PIP2 binding sites (Figure 5A-C, S2). The phosphate moieties of PIP2 are recognized by R22, K27 and R76 while the 2′-acyl chain docks inside the hydrophobic cleft formed between W36 and L21 (Figure 5C). tRNA nearly completely occludes the PIP2 binding site on MA, thereby providing a molecular basis for the direct competition between tRNA and membrane for binding MA observed in vitro (Gaines et al., 2018b; Kroupa et al., 2020; Todd et al., 2017).
Figure 5. Overlapping MA interfaces for binding tRNA, PIP2 and membranes.
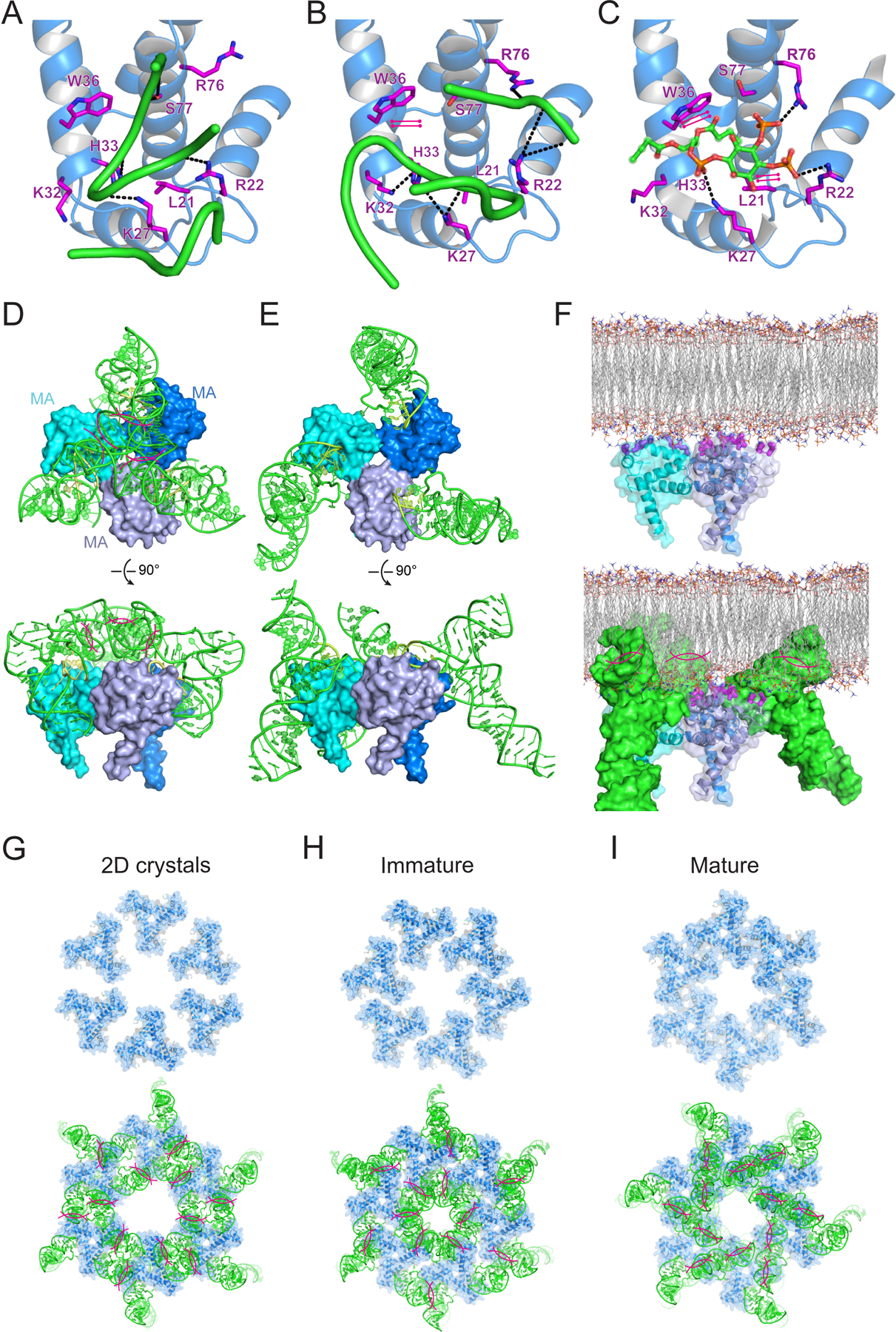
(A-C) MA interfaces with tRNALys3 inner corner (A), elbow (B), and PIP2 (C). Hydrogen bonds as dashed lines; stacking interactions as parallel magenta lines.
(D) Superposition of MA bound to tRNALys3 inner corner onto the trimeric MA structure (Hill et al., 1996). Clashes are highlighted by magenta lines.
(E) Superposition of MA bound to tRNALys3 elbow region onto trimeric MA shows no clashes.
(F) Structural model of trimeric MA binding to the lipid membrane (upper) and effect of tRNA binding via the elbow (lower). HBR as thick magenta sticks.
(G-I) Top: structural models of MA hexamers of trimers for (G) 2D crystals assembled on membranes, (H) in immature HIV-1 virions and (I) in mature HIV-1 virions. Bottom: Superposition of elbow-bound MA-tRNA complexes onto the lattices of MA hexamers of trimers above.
See also Figure S2.
We then asked if natural variations of elbow sequence and structure among distinct tRNA species may explain differential MA crosslinking in cells. To this end, we measured MA binding to human tRNAPhe, tRNAThr, tRNALeu, and tRNASer, all of which exhibited reduced or no crosslinking to MA in vivo, and tRNAGly which had comparable crosslinking to tRNALys3 (Kutluay et al., 2014). Lower binding affinities were observed by ITC for all these tRNA species (Figure 3C-E, Table S3), consistent with their generally diminished crosslinking. Unexpectedly, several tRNAs (tRNAThr, tRNALeu, and tRNASer) exhibited biphasic binding profiles showing two binding events with distinct thermodynamic signatures, albeit both weaker than the single site on tRNALys3. Similar endothermic phases observed upon binding of the HIV-1 nucleocapsid (NC) to RNAs have been attributed to concomitant, localized RNA unwinding and/or remodeling at the NC binding site (Ding et al., 2020; Heng et al., 2012). In agreement with ITC, 1:2 (tRNA:MA) complexes were detected by AUC (Figure S4). Neither binding event is likely driven by the crystallographically observed inner corner interface because K32A abolished all tRNA binding at both sites (Figure S4). Comparison of the six tRNAs assayed reveals major divergences in the length and sequence of the D-loop, while the T-loops are nearly identical (Figure 3D). Mutating or swapping the D-loop sequences had significant effects on binding affinities, with one chimeric tRNA losing all MA binding (Figure S3, Table S2). These data suggest that natural divergences of the flexible D-loop are primarily responsible for the variable crosslinking in cells (Kutluay et al., 2014) and accentuate the remarkable sensitivity of MA binding to the fine elbow structure.
The tRNA binding site on MA regulates HIV-1 Gag subcellular localization
Our previous work showed that RNase digestion of crude cell lysates increased HIV-1 Gag protein association with membranes (Kutluay et al., 2014). To determine whether tRNA binding affects HIV-1 Gag subcellular localization, we determined the localization of HIV-1 Gag-GFP using the K32A and W36A variants of HIV-1 Gag that could no longer bind to tRNA (Figure 6, S5). Specifically, HeLa cells were transfected with plasmids expressing Gag(MAWT)-GFP, Gag(MAK32A)-GFP or Gag(MAW36A)-GFP constructs. Gag-GFP readily assembles into virus-like particles (VLPs), visible as GFP-punctae at the plasma membrane, but a Gag(MAWT)-GFP diffuse cytosolic signal was also observed in most cells (Figure 6A, left panels; Figure S5A), that is assumed to represent Gag molecules that have yet to bind plasma membrane and participate in VLP assembly. Conversely, Gag(MAK32A)-GFP and Gag(MAW36A)-GFP formed Gag punctae at the plasma membrane (Figure 6A, right panels; Figure S5B,C), but the diffuse cytosolic signal was almost completely absent. The absence of cytosolic Gag was reflected by a reduced overall GFP fluorescence in cells expressing Gag(MAK32A)-GFP and Gag(MAW36A)-GFP, as compared to Gag(MAWT)-GFP (Figure 6B, left panel), while the number of Gag(MAWT)-GFP, Gag(MAK32A)-GFP and Gag(MAW36A)-GFP punctae at the ventral plasma membrane was similar (Figure 6B, right panel). Thus, the MA-tRNA interaction is not required for HIV-1 assembly but appears to reduce the proportion of Gag that is localized to the plasma membrane and increase the proportion in the cytosol.
Figure 6. Effect of tRNA binding site mutation on Gag localization.
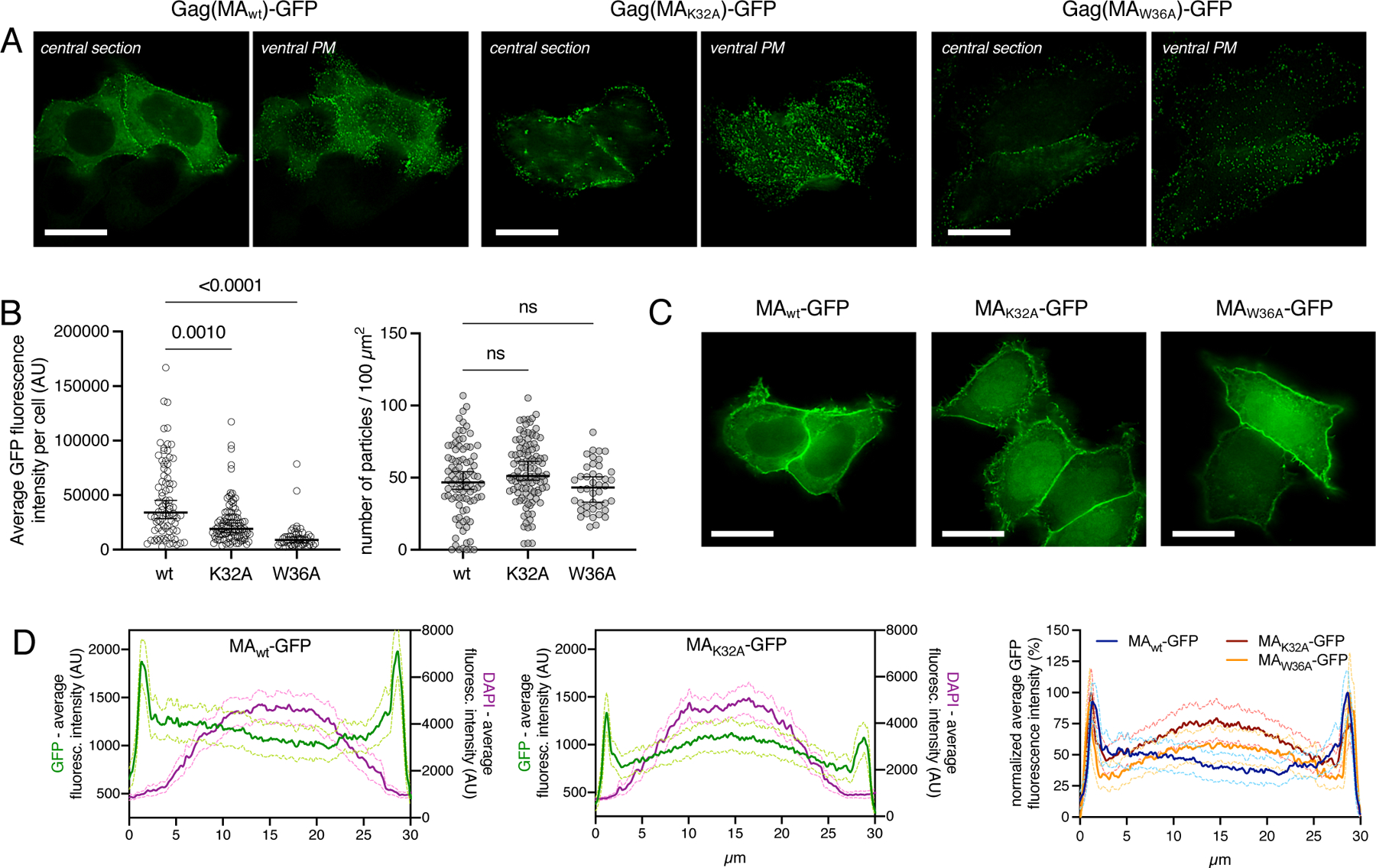
(A) Representative widefield images of a central optical section or ventral plasma membrane (PM), as indicated, of HeLa cells transfected with pCR3.1Gag(MAwt)-GFP, pCR3.1Gag(MAK32A)-GFP or pCR3.1Gag(MAW36A)-GFP. Cells were fixed 12 hours after transfection. Scale bars indicate 20 μm.
(B) Average total GFP-fluorescence per cell (left panel) and number of punctae detected at the ventral membrane per 100 μm2 membrane area (right panel). Horizontal bars and error bars indicate mean and 95 % confidence interval, respectively. Statistical significance was determined using Kruskall-Wallis test and subsequent Dunn’s multiple comparisons test. (C) Representative widefield images of a central optical section of HeLa cells transfected with pCR3.1MAwt-GFP, pCR3.1MAK32A-GFP or pCR3.1MAW36A-GFP. Cells were fixed 12 hours after transfection. Scale bars indicate 20 μm.
(D) Averaged line profiles of selected cells expressing MAwt-GFP (left panel) and MAK32A-GFP (middle panel). Shown is the average fluorescence intensity (arbitrary units, AU) of GFP (green, left y-axis) or DAPI (magenta, right y-axis). Overlay of line profiles showing normalized average GFP fluorescence intensity in cells expressing MAWT-GFP (blue), MAK32A-GFP (red) and MAW36A-GFP (orange) (right panel). Standard Error of the Mean (SEM) in D is shown as light-colored lines that bracket the mean. Additional examples of images in A and C are in Figure S5 and S6, respectively.
We also examined the effect of the K32A and W36A mutations on MA-GFP subcellular localization. Because MAWT-GFP lacks CA and NC Gag domains, it does not form punctae and adopts a diffuse cytosolic signal, accompanied by a homogenous plasma membrane accumulation (Figure 6C, S6). Notably MAWT-GFP was excluded from the nucleus, even though its molecular mass (~42 kDa) is below that expected to be excluded from passive diffusion through nuclear pores. However, if MA is bound to tRNA (molecular mass ~25 kDa), the resulting ~70 kDa complex would be expected to be excluded from the nucleus, especially since RNAs exhibit markedly larger hydrodynamic radii than proteins of comparable molecular mass due to the concentrated negative charge and resulting thick hydration shells. Notably, nuclear enrichment of MAK32A-GFP or MAW36A-GFP, which is absent in MAWT-GFP expressing cells, was observed (Figure 6C, S6). These data suggest that either mutation causes loss of tRNA binding, enabling the MA-GFP protein to enter the nucleus. However, to ensure that these findings were not due to the inadvertent constitution or exposure of an active nuclear localization signal, we artificially increased the size of MAK32A-GFP, by fusing it to LacZ. Importantly, MAK32A-GFP-LacZ, like MAWT-GFP was excluded from the nucleus (Figure S6D). Quantification of GFP vs DAPI fluorescence intensities along a line profile traversing central image in a Z-stack further illustrates the enrichment of MAK32A-GFP and MAW36A-GFP in the nucleus, compared to the nuclear exclusion of MAWT-GFP or MAK32A-GFP-LacZ (Figure 6D, S6D). Even though MA-GFP is not a physiological molecule, the ability of the tRNA binding site to regulate localization is consistent with the idea that a significant portion of GagWT-GFP or MAWT-GFP in cells is tRNA bound and suggests that there is sufficient free tRNA available to bind and regulate Gag, in accordance with the nanomolar binding affinities observed in vitro. Importantly, the changes in localization observed for Gag-GFP or MA-GFP mutants were not due to differential expression levels, or the formation of truncated species (Figure S6E). These findings are consistent with in vitro NMR studies (Gaines et al., 2018b) and suggest that tight tRNA-Gag binding inhibits Gag-membrane interactions, thereby controlling Gag subcellular localization.
Effects of MA:tRNA binding on HIV-1 particle assembly and replication
MA-tRNA binding may interfere with Gag-membrane association via more than one mechanism. Bound tRNA masks the HBR, occludes the PIP2 binding site, inverts the surface charge of this globular domain, and also adds substantial steric bulk, all of which may impact Gag multimerization. MA trimerization occurs in HIV-1 particles and is required for Env incorporation and viral infectivity, but MA can also form dimers, trimers, hexamers, and higher-order assemblies such as hexamers of trimers at the membrane and in virions(Alfadhli et al., 2009; Alfadhli et al., 2016; Hill et al., 1996; Murakami and Freed, 2000; Murphy et al., 2019; Tedbury et al., 2013; Tedbury et al., 2016; Tedbury et al., 2019). To understand potential impact of tRNA binding on MA multimerization, we superposed our complex structure onto the trimeric crystal structure (Figure 5D, 5E). Interestingly, when MA is bound to the tRNA elbow, the tRNAs are splayed out and present no steric conflicts with the MA trimer (Figure 5E), possibly helping to bridge neighboring MA monomers. By contrast, tRNAs that bind via the inner corner point inward and their anticodon stem loops strongly clash with each other (Figure 5D). Electron microscopy analysis of 2D crystals initially suggested that MA trimers can further assemble into hexamers of trimers on PIP2-enriched membranes (Alfadhli et al., 2009). Recently, a cryo-electron tomography (cryo-ET) analysis of HIV-1 virions revealed that MA forms distinct hexameric lattices of trimers in immature and mature virions (Qu et al., 2020). Superimposition of our complex structure shows that elbow-bound tRNAs clash with neighboring MA trimers and are incompatible with all three observed hexameric lattices (Figure 5G-I). Thus, tRNA binding does not necessarily interfere with MA trimerization but is expected to block further assembly of the larger Gag lattice at the membrane.
To investigate whether a loss of tRNA-MA binding affected HIV-1 replication, we introduced the MAK32A mutation into a set of HIV-1 infectious molecular clones based on HIV-1NL4–3 and analyzed particle formation and replication in a variety of contexts.
First, we determined whether the MAK32A mutation affected the Env protein incorporation into virions. HIV-1 particles were generated by transfection of 293T cells with a VSV-G expression plasmid and either HIV-1NL4–3, HIV-1NL/ADA, HIV-1NL/BaL or HIV-1NL/Yu2 proviral plasmids, each encoding either MAWT or MAK32A. Then, 293T cells were infected with the VSV-G-pseudotyped particles. Viral infection of 293T cells is mediated by VSV-G, while nascent particles subsequently generated during a single cycle of infection carry the HIV-1 Env protein. Western blot analysis of Env and p24 CA levels in particles revealed that the amounts of HIV-1 Env protein incorporated into nascent virions that included MAK32A did not differ appreciably from those incorporated into WT virions (Figure 7A, S7)
Figure 7. Env-incorporation assembly and replication of MAWT and MAK32A HIV-1.
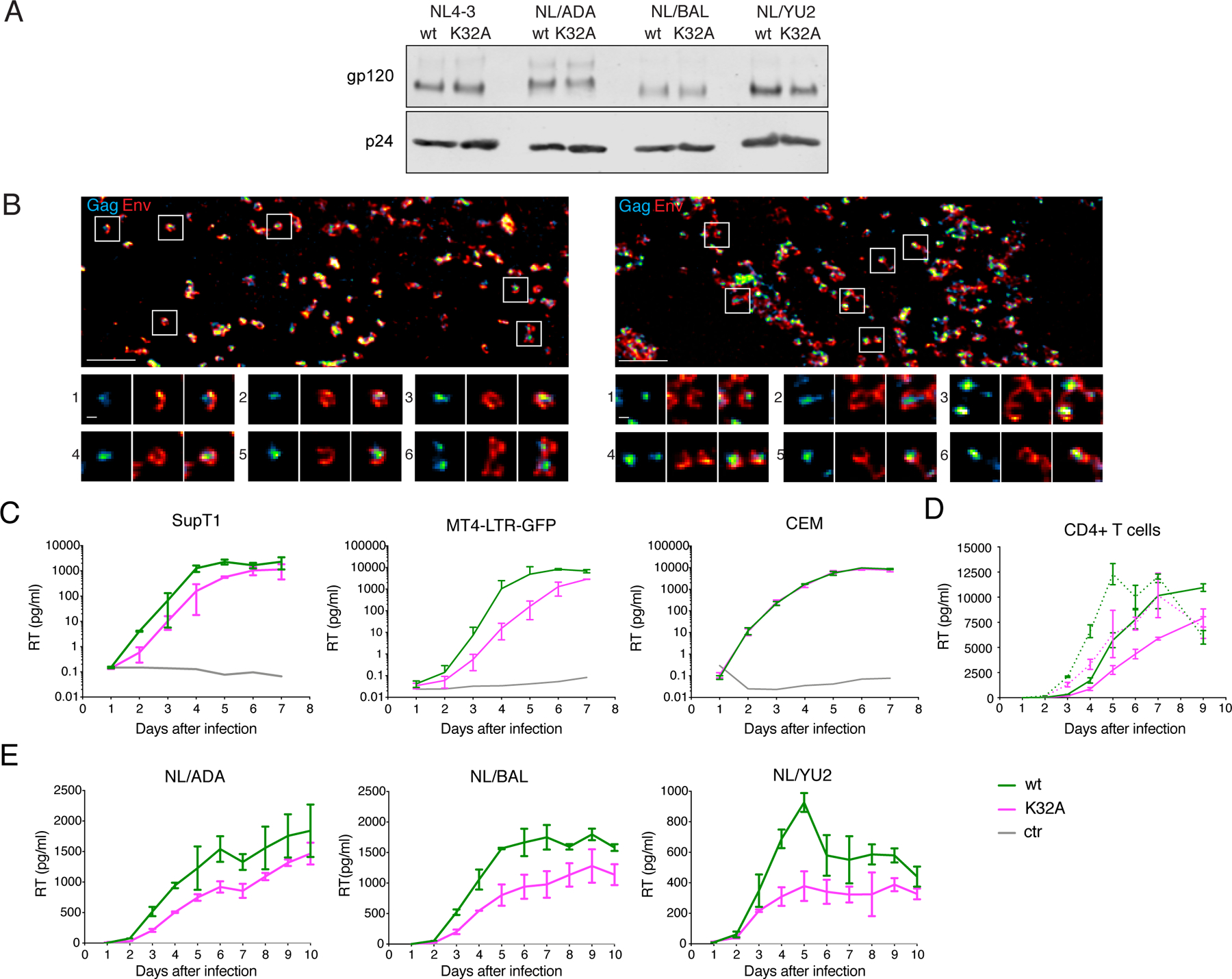
(A) 293T cells were infected with VSV-G-pseudotyped HIV-1NL4–3, HIV-1NL/ADA, HIV-1NL/BaL or HIV-1NL/Yu2 each encoding MAWT or MAK32A. Supernatants were harvested 48 h post-transfection, pelleted through a sucrose cushion and separated on SDS-PAGE. Shown are western blots probed with anti-gp120 (Env) and anti-p24 (capsid) and respective secondary antibodies. Additional western blots are in Figure S7.
(B) STED images of the ventral plasma membrane of HeLa cells transfected HIV-1NL4–3 (left) or HIV-1NL4–3(MAK32A) (right). Gag (cyan) and Env (red) were detected via indirect immunolabeling. Individual sites of particle assembly are highlighted and shown in enlargements (bottom).
(C) Reverse Transcriptase (RT) levels in supernatants of SupT1, MT4-LTR-GFP or CEM cells after infection with HIV-1NL4–3 MAWT (green) or HIV-1NL4–3 MAK32A (magenta) at an MOI of 0.1 (SupT1, MT4-LTR-GFP) or 0.01 (CEM). Data are represented as mean ± SEM.
(D) RT levels in supernatants of CD4+ T-cells after infection with HIV-1NL4–3 MAWT (green) or HIV-1NL4–3 MAK32A (magenta) at 0.4 (dotted lines)/ 0.05 (continuous lines) MT4-LTR-GFP infectious units/cell. Supernatants harvested from uninfected cells are in gray.
(E) RT levels in supernatants of primary monocyte-derived macrophages (MDM) after infection with the R5-tropic HIV-1NL/ADA, HIV-1NL/BaL or HIV-1NL/Yu2 each encoding MAWT (green) or MAK32A (magenta), at 2.5 MT4-LTR-GFP infectious units/cell.
Results for additional primary cell donors are in Figure S7.
Next, we applied super resolution microscopy to analyze the morphology of individual assembly sites at the ventral plasma membrane of HeLa cells transfected with HIV-1NL4–3 or HIV-1NL4–3(MAK32A) (Figure 7B). In HIV-1NL4–3 transfected cells, Gag forms clusters with a diameter of approximately 100 nm, surrounded by the Env protein, that forms a distinct donut-shaped distribution around the Gag cluster (Figure 7B). This characteristic distribution of viral proteins was also observed for the HIV-1NL4–3(MAK32A) variant, with assembly sites of similar size and morphology to those formed by HIV-1NL4–3 (Figure 7B). Thus, the absence of MA:tRNA binding did not confer any visible impairment on the formation of HIV-1 assembly sites or envelope protein colocalization and incorporation.
To determine the effects of the MAK32A on virus replication we infected T cell lines at low multiplicities and measured reverse transcriptase activity in culture supernatants for 7 days. Variable, cell-type dependent effects of the K32A mutation on replication were observed. HIV-1NL4–3(MAK32A) exhibited impeded replication in SupT1 and MT4-LTR-GFP cells as compared to HIV-1NL4–3 (Figure 7C). Conversely, in CEM cells, the levels of HIV-1NL4–3 and HIV-1NL4–3(MAK32A) replication were indistinguishable (Figure 7C). Next, we compared in HIV-1NL4–3 and HIV-1NL4–3(MAK32A) replication in primary CD4+ T-cells and found that the tRNA-binding mutant replicated at modestly reduced levels, with ~30–70 % reduction of supernatant RT at 3 to 7 days after infection (Figure 7D, S7C). In primary monocyte derived macrophages (MDM) infected with R5-tropic viruses (HIV-1NL/ADA, HIV-1NL/BaL or HIV-1NL/Yu2) a similarly modest effect of the K32A substitution was observed. Nevertheless, in each case, the mutant MAK32A viruses replicated less well than the MAWT virus (Figure 7E, S7D).
Discussion
Herein we show that (1) HIV-1 matrix protein contains structural features that enable recognition of host tRNA elbow structures, and (2) HIV-1 co-opts host tRNAs to negatively regulate Gag membrane association and optimize replication.
HIV-1 MA directs Gag to the plasma membrane through a hydrophobic interactions from its myristate group and electrostatic interactions from the HBR (Mercredi et al., 2016; Ono et al., 2004; Saad et al., 2006; Tang et al., 2004; Vlach and Saad, 2013). While MA trimerization drives myristate exposure and interaction with membranes, molecular dynamics simulations suggest that the HBR alone is sufficient to enable membrane binding, highlighting its key role (Monje-Galvan and Voth, 2020). As well as sequestration by MA, myristate was also reported to be sequestered by heme-oxygenase-2 (Zhu et al., 2017). HBR occlusion by tRNA thus represents a third independent activity that inhibits HIV-1 Gag interaction with membranes. MA globular head deletion or the introduction of mutations that facilitate myristate exposure or, as shown herein, prevent tRNA interaction, each increase Gag-membrane association and accelerate particle assembly. Interestingly, the W36A mutation which, like K32A, blocks tRNA binding was previously reported to ameliorate the effects of an L8A mutation which stabilizes the myristate-sequestered MA conformation (Paillart and Göttlinger, 1999). While the W36A mutation was originally thought to directly affect myristate exposure, it instead likely increases exposure of the HBR. These two processes may be coupled, as mutations or solution conditions that promote myristate exposure also inhibit tRNA binding in vitro (Gaines et al., 2018b).
The existence of independent virus-encoded mechanisms (myristate sequestration and HBR occlusion by tRNAs) strongly suggests that inhibited membrane binding is an acquired characteristic of MA that provides a selective advantage. However, the biological impetus for the negative regulation of Gag-membrane binding is unclear. This property delays production of viral progeny by an infected cell by a few hours, allowing the depletion of antiviral proteins such as APOBEC3G from infected cells prior to virion assembly (Holmes et al., 2015). While the deleterious effect of MAK32A mutation on HIV-1 replication is not restricted to APOBEG3G-positive cells, APOBEC3G is but one of several proteins whose depletion could increase the yield or infectiousness of progeny HIV-1 particles and maximize HIV-1 burst size. Postponing viral assembly could thus avoid futile particle assembly. Other possible consequences of delayed virion assembly, such as optimization of the stoichiometry of virion components, that has previously been suggested to be regulated at the level of mRNA degradation are also potential drivers of this phenomenon (Mu et al., 2015). Moreover, tRNA sequestration could have effects beyond virion assembly, such as regulation of viral translation.
The specific recognition of tRNA elbow by MA inducts HIV Gag into an expanding cohort of proteins, RNAs, and ribonucleoprotein particles that exploit this salient surface feature for tRNA recognition, processing and translocation (Figure S4). However, different recognition strategies are employed, and forces of distinct chemical nature are used (Kruger et al., 2018; Zhang and Ferré-D’Amaré, 2016). Protein enzymes that recognize the tRNA elbow include aminoacyl-tRNA synthetases (aaRSes; e.g., AlaRS, LeuRS, ValRS), tRNA-modifying enzymes (e.g., DusA, MTases, TruA), pre-tRNA processing enzymes (e.g., CCA-adding enzyme, RNase Z), etc. Besides basic residues, host proteins frequently employ aliphatic side chains (especially Ala, Val, Ile) to bind the elbow using Van der Waals interactions to achieve shape complementarity (Figure S4). By contrast, three independently evolved noncoding RNA elements, namely the ribosome L1 stalk, RNase P ribozyme and T-box riboswitches, use a common structural motif termed the interdigitated double T-loop motif (IDTM) (Korostelev et al., 2006; Reiter et al., 2010; Zhang, 2020; Zhang and Ferré-D’Amaré, 2013). The IDTM presents a surface-exposed base triple (T-boxes) or base pair (the other two) that stacks with the tertiary base pair of the tRNA elbow, reminiscent of how W36 of MA stacks with tC56 (Figure S4). Curiously, no known host proteins use Trp to recognize tRNA elbow as MA does. Furthermore, ValRS and AlaRS use a protruding arginine (R843 or R876) to recognize an extrahelically flipped tA20 or tG20, respectively, via cation-π interactions nearly identical to how R22 of MA binds the flipped tU20 at the same location (Figure S4) (Fukai et al., 2000; Naganuma et al., 2014). Thus, the strategies employed by MA to recognize the tRNA elbow borrows and blends key elements from both protein and RNA host machines, in a remarkable case of viral mimicry, elaboration and evolution.
MA binding to the tRNA elbow may also allow Gag to indirectly associate with host proteins that bind other regions of the tRNA, such as eukaryotic translation elongation factor 1A (eEF1A) and certain aaRSes, potentially to reprogram translation or facilitate the packaging of viral genomes and tRNA primers (Cimarelli and Luban, 1999; Guo et al., 2010; Javanbakht et al., 2003). The MA – tRNALys3 interaction may collaborate with the capsid – Lysyl-tRNA synthetase (LysRS) interaction to package the LysRS-tRNALys3 complex into virions. HARS2, the mitochondrial Histidyl-tRNA synthetase, was suggested to interact with MA in an HBR-dependent manner (Lama and Trono, 1998). It is unclear if this interaction is bridged by tRNA. For aaRSes that bind the tRNA elbow, such as AlaRS, LeuRS and ValRS, MA binding is likely competitive and could inhibit translation if present in sufficient quantities, as found in vitro (Cimarelli and Luban, 1999).
We found that distinct tRNA species interact with MA in non-identical ways and exhibit variable affinities in vitro, consistent with differential crosslinking in cells (Kutluay et al., 2014). The tRNA elbow is a major, common site that provides relatively high affinities, whilst a second MA site seems to exist elsewhere for several tRNAs. As this second site exhibits only micromolar affinities towards MA, it may not contribute to the overall tRNA regulation of Gag in vivo. In addition, given the abundance of free tRNAs available to bind MA, HIV-1 may only need to commandeer a handful of tRNAs for Gag regulation. Our comparative and chimeric analyses of six tRNA species suggest that the D-loop is a major determinant of MA selectivity. Notably, the tRNA elbow is elaborated with several posttranscriptional modifications absent from our in vitro transcribed tRNAs that could impact local structure or flexibility, and thus MA binding. Therefore, a thorough assessment of tRNA selectivity by MA awaits further experimentation using native, extensively modified tRNAs.
Overall, these findings reveal a previously undescribed mechanism for the regulation of the subcellular localization of a major viral protein, and the parasitism of host molecules for the optimization of viral replication. The discovery and visualization of this selective viral protein - host RNA interface may permit therapeutic targeting by both small molecules and artificial RNA devices.
STAR★METHODS
Resource availability
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Jinwei Zhang (jinwei.zhang@nih.gov).
Materials availability
This study did not generate new unique reagents
Data and code availability
Atomic coordinates and structure factor amplitudes for tRNALys3 bound to HIV-1 MA have been deposited in the Protein Data Bank (PDB) under accession code 7MRL.
This study did not generate any original code.
Any additional information required to reanalyze the data reported in this paper is available from the Lead Contact upon request.
Experimental models and subject details
Cell lines and primary cells
HeLa, 293T, SupT1, MT4-LTR-GFP and CEM cells were cultured at 37°C and 5% CO2 in DMEM (HeLa, 293T) or RPMI (SupT1, MT4-LTR-GFP and CEM) supplemented with 10% FCS and Gentamycin. Cells are monitored regularly mycoplasma contamination. Cells used here were contamination free.
Human PBMCs were prepared from Leukopaks (Buffy coats made from human whole blood, NYBC, New York) by spinning on top of lymphocyte separation medium (Corning). CD4+ T cells were isolated from PBMCs using the EasySep Human CD4+ T cell Enrichment Kit (StemCell). Isolated CD4+ T cells were cultured in RPMI containing 2 ng/ml (20 U/ml) human recombinant IL-2 (PeproTech) and activated with 25 µl Dynabeads Human T-Activator CD3/CD28 (Thermo Scientific) per 1E6 cells. Dynabeads were removed after 48 hours and cells were plated in 96-well U bottom plates for infection. For generation of MDM, Monocytes were isolated by plastic adherence and differentiated using 100 ng/ml GM-CSF for 48 hours. Differentiation of MDM was monitored by microscopy. Upon maturation, cells were detached with StemPro Accutase Cell Dissociation Reagent (Thermo Fisher) and plated in 96-well flat bottom plates for infection.
Virus production
HIV-1 particles used for spreading replication assays were produced by transfecting 293T cells with 10 µg pNL4–3 / pNL/ADA / pNL/BAL / pNL/YU2 or their MAK32A variants (per 10 cm dish) using PEI. VSV-G-pseudotyped full-length HIV-1 particles (carrying HIV-1 Env and VSV-G on their surface) were produced by transfecting 293T cells with 10 µg pNL4–3/pNL4–3(MAK32A) (and their R5-tropic Env variants) and 1 µg pVSV-G (per 10 cm dish) using PEI. 12 h post-transfection, medium was replaced with fresh growth medium and viral supernatants were harvested and filtered 48 h post-transfection. Virus was aliquoted and stored at − 80°C.
Method details
Protein expression and purification
HIV-1 matrix domain (a.a. 1–131) was expressed in E. coli BL21(DE3)-RIL cells using a pET19b vector (Gaines et al., 2018b). Cells were grown at 37°C in LB medium until OD600 ~0.6 and induced using 1 mM isopropyl 1-thio-β-D-galactopyranoside (IPTG) and grown for an additional 4 h. Cells were resuspended in a buffer composed of 25 mM Tris-HCl (pH 7.5), 200 mM NaCl, 5% glycerol (v/v), 10 mM β-mercaptoethanol (BME) supplemented with the SIGMAFAST protease inhibitor cocktail (Sigma), lysed with a microfluidizer and centrifuged at 96, 000 g for 1 h at 4°C to remove cell debris. Nucleic acids and bound proteins were precipitated by 0.15% w/v polyethylenimine (PEI), followed by centrifuging at 61,731 g at 4°C for 30 min. The pellet was discarded and the proteins in the supernatant were precipitated by slow addition of ammonium sulfate to 90% saturation. The pellet was then dissolved in a buffer consisting of 25 mM Tris-HCl (pH 7.5), 500 mM NaCl, 10% (v/v) glycerol and 15 mM imidazole and loaded on a HisTrap Ni2+ column on an Akta Pure chromatography system. Bound MA proteins were eluted with the same buffer supplemented with 250 mM imidazole. The histag was removed by TEV protease cleavage overnight at 4°C, followed by a subtractive HisTrap Ni2+ affinity purification step to remove uncleaved histagged MA proteins. MA was further purified by gel filtration on a Superdex 200 Increase column equilibrated in 25 mM Tris-HCl pH 7.5, 150 mM NaCl and 2 mM MgCl2. All mutations were introduced using the QuikChange Lightning Site-directed Mutagenesis Kit (Agilent) and all MA variants were purified using the same procedure as described above. The purity of proteins was assessed by SDS-PAGE.
RNA preparation
RNAs used for crystallization and biophysical analyses were transcribed in vitro by T7 RNA polymerase (RNAP) or synthesized by Integrated DNA Technologies (IDT). PCR products that bear two consecutive 2′-O-methyl modifications on 5′-end of the template strand were used as templates for transcription to suppress the non-specific N+1 activity of the T7 RNAP, in the presence of 5 mM of each nucleotide triphosphate (NTP). RNAs were purified on a 8 M urea Tris-borate-EDTA denaturing preparative 10% polacrylamide (29:1 acrylamide:bisacrylamide) gel and electroeluted (Hood et al., 2019; Li et al., 2019). Extracted RNAs were washed once with 1 M KCl and desalted by washing three times with DEPC-treated water and stored in DEPC-treated water at −20°C. tRNAs were folded by heating at 90°C for 2 min in 25 mM Tris-HCl (pH 7.5) and 150 mM NaCl, followed by snap-cooling over 2 min on ice and addition of 2 mM MgCl2. Mutations in the tRNA sequence were introduced using the QuikChange Lightning kit. Oligonucleotides and RNA fragments labeled with 2AP were chemically synthesized by IDT.
Crystallization and structure determination
To facilitate crystallization, the anticodon stem of tRNALys3 was extended by 3 base pairs (bp) and the anticodon loop was replaced with a GAAA tetraloop and the flexible 3´-GCCA tail was removed. The flexible C-terminal region of MA (a.a. 109–131) was removed as well as the first six N-terminal residues. For co-crystallization, in vitro transcribed, unmodified human tRNALys3 was mixed with HIV-1 MA7−108 in equimolar amounts and 7 mg/mL of the complex was mixed 1:1 with a reservoir solution consisting of 0.1 M HEPES (pH 7.5), 20 % polyethylene glycol (PEG) 500 monomethyl ether and 10% PEG 20000. Crystallization was performed at 20°C by hanging-drop vapor diffusion. Cubic crystals were grown over 9 days to maximum dimensions of 50 X 50 X 30 μm3. The crystals were cryoprotected in a synthetic mother liquor containing 30% of PEG 500 monomethyl ether and 10% PEG 20000 before vitrification in liquid nitrogen. X-ray diffraction data were collected at the SER-CAT beamline ID-22 at the Advanced Photon Source (APS). The structure was solved by molecular replacement using PHASER (McCoy et al., 2007). Two copies of a highly similar MA structure (PDB 1HIW (Hill et al., 1996)) was first located in the asymmetric unit with a Translational Function Z (TFZ) of 20 and Log-likelihood Gain (LLG) of 300.8. The resulting initial phases enabled PHASER to locate a single copy of tRNALys3 (PDB 1FIR, (Benas et al., 2000)), producing an overall TFZ of 36.5 and LLG of 1216.9. The asymmetric unit contained one copy of tRNALys3 and two copies of MA. The structure was refined in Phenix. Refine (Liebschner et al., 2019) and iterative rounds of model building was performed using Coot (Emsley et al., 2010). The structure was optimized using ERRASER (Chou et al., 2013) and further refined. The crystals exhibited the symmetry of space group I222 and the crystallographic and refinement statistics are in summarized in Table S1.
Isothermal titration calorimetry (ITC)
tRNAs were folded by heating at 90°C for 2 min in 25 mM Tris-HCl (pH7.5) and 150 mM NaCl, snap-cooling over 2 min on ice, followed by the addition of 2 mM MgCl2. tRNAs and MA were extensively exchanged into the same buffer containing 25 mM Tris-HCl (pH 7.5), 150 mM NaCl and 2 mM MgCl2 using Amicon Ultra Filter concentrators (Millipore). All ITC measurements were performed at 25°C using a MicroCal iTC200 microcalorimeter (GE healthcare). 30 μM of tRNA was used in the cell and titrated with 350 μM of MA proteins. The raw ITC data measured in triplicates or duplicates were integrated using NITPIC and fit with SEDPHAT (Zhao et al., 2015) to obtain the dissociation constants and thermodynamic parameters reported in Table S2. For biphasic isotherms (tRNALeu, tRNASer, tRNAThr), triplicate titration data were globally fit to a two-site non-symmetric model yielding two dissociation constants.
Fluorescence spectroscopy
tRNALys3 site-specifically labelled with 2-aminopurine (2AP) at position 10 or 19 was produced by annealing two RNA fragments consisting of residues 1–33 and 34–76, respectively. 20 μM of 2AP-containing oligonucleotide was mixed with 50 μM of the unlabeled RNA fragment in 25 mM Tris-HCl and 100 mM NaCl, heated at 90°C for 2 min, cooled to 65°C over 5 min, followed by the addition of 2 mM MgCl2 and cooled to room temperature at the rate of 10C/min. 20 μM of annealed two-piece tRNAs were incubated overnight at 16°C with 10 units of T4 RNA ligase 1 (NEB) to ligate the single-stranded RNA (ssRNA) junction between positions 33 and 34. The covalent linkage was confirmed by Urea-PAGE. The ligated, 2AP-labeled tRNAs were exchanged into a buffer composed of 25 mM Tris-HCl pH 7.5, 150 mM NaCl and 2 mM MgCl2 prior to fluorescence spectroscopy measurements. The fluorescence emission spectra of 1 μM of 2AP-tRNA in the presence or absence of wild-type or mutant MA were recorded from 325 to 450 nm (maximum emission at ~ 367 nm) using a PTI QuantaMaster fluorometer with an excitation wavelength of 295 nm and a slit width of 1 mm.
Sedimentation Velocity Analytical Ultracentrifugation (AUC)
Sedimentation velocity experiments on the MA, MA K32A, tRNALys3, tRNALeu, tRNASer, tRNAThr and various tRNA-MA mixtures were carried out at 50,000 rpm and 25 °C on a Beckman Coulter ProteomeLab XL-I analytical ultracentrifuge and An50-Ti rotor following standard protocols (Zhao et al., 2013a). Protein and nucleic acid stock solutions in 25 mM Tris.HCl (pH 7.5), 150 mM NaCl, and 2 mM MgCl2 were diluted in the same buffer to prepare solutions for analysis. Samples were loaded in 12 mm two-channel centerpiece cells, and sedimentation data were collected using the absorbance and interference optical detection systems. High concentration samples were analyzed in 3 mm two-channel centerpiece cells. Time corrected (Zhao et al., 2013b) data were analyzed in SEDFIT 16.2b (Schuck, 2000) in terms of a continuous c(s) distribution of sedimenting species with a maximum entropy regularization confidence interval of 0.68. The solution density, solution viscosity, and protein partial specific volumes were calculated based on their composition in SEDNTERP (Cole et al., 2008). A partial specific volume of 0.55 cm3g−1 was used for tRNA. Additivity rules were used to determine the partial specific volumes of the protein nucleic-acid complexes. Experimental sedimentation coefficients are presented because of differences in the partial specific volumes.
Nuclear magnetic resonance spectroscopy
The tRNALys3 gene encoded in a pUC57 vector was PCR amplified (EconoTaq PLUS 2X Master Mix, Lucigen) using a forward primer recognizing a region upstream of the T7 promoter site (5ʹ-GATTAAGTTGGGTAACGCCAGGGTTTTCC-3ʹ) and a reverse primer in which the first two nucleotides featured 2ʹ O-methyl modifications (5ʹ-mUmGGCGCCCGAACAGGGACTTGAACCCTGG-3ʹ). In vitro transcription was carried out in 7.5–15 mL reactions containing 80 mM Tris, pH 9.0, 2 mM DTT, 20% DMSO, 2 mM spermidine, 20 mM MgCl2, 6 mM NTPs, 0.3–0.5 mg PCR-amplified DNA template, and 0.15 mg T7 RNA polymerase (Brown, 2020; Milligan and Uhlenbeck, 1989). To quench the reaction, EDTA was added to a final concentration of 0.5 mM, the sample was boiled (100 ºC, 5 minutes) and snap cooled (0 ºC, 5 minutes) before addition of 50% glycerol and purification by denaturing polyacrylamide gel electrophoresis (SequaGel, National Diagnostics). The RNA bands were detected by UV-shadowing, eluted using the Elutrap electroelution system (Whatman) at 150 V for ~16–20 hours. The eluted RNA was loaded to a 10 kDa MWCO Amicon Centrifugal Filter Device (Millipore), concentrated, washed with 2 M NaCl to remove residual acrylamide, and desalted (Brown, 2020). The uniformly 15N-G, -U enriched tRNALys3 (100 µM) sample was titrated with various molar ratios of MA in 10 mM sodium phosphate, pH 6.5, 10 mM MgCl2, 100 mM NaCl, 5 mM BME. The 2D 1H-15N heteronuclear single quantum coherence (HSQC) data were acquired at 35 °C on a Bruker 600 MHz spectrometer equipped with a cryogenic probe. Data were processed using NMRFx (Norris et al., 2016) and analyzed with NMRViewJ (Johnson, 2004; Johnson and Blevins, 1994) using assignments reported by Puglisi et al (Puglisi and Puglisi, 2007).
Differential scanning calorimetry (DSC)
DSC experiments were performed with 20 μM of tRNA in 25 mM HEPES pH 7.5, 150 mM NaCl and 2 mM MgCl2 on a Malvern/GE VP-DSC instrument. The DSC instrument was equilibrated overnight with buffer in both sample and reference cells. The next day, the tRNA was loaded in the sample cell and the DSC scan was recorded after a 60 min equilibration. The temperature range scanned was from 25 to 120°C with a step of 1°C min-1. DSC data were corrected for instrument baselines and normalized for scan rate and RNA concentration. Data conversion and analysis was performed with Origin software (OriginLab Corporation, Northampton, MA, USA).
Electrophoretic mobility shift assays
The tRNA (4 µM) was incubated at 37 ºC for 0.5–1 h with various molar ratios of MA in 30 µL reactions carried out in 25 mM MES, pH 5.5, 2.5 mM MgCl2, 5 mM 2-mercaptoethanol. Samples were removed from the incubator and immediately supplemented with 50% glycerol. Samples were separated on 10% 29:1 polyacrylamide tris-borate gels containing 0.2 mM MgCl2 both in the gel and running buffer (Brown, 2020; Gaines et al., 2018a). Gels were resolved at room temperature at 220 V and stained using a carbocyanine dye (Stains-All, Sigma Aldrich, No. E9379).
Size-exclusion chromatography coupled with multi-angle light scattering (SEC-MALS)
To assess the binding stoichiometry of MA to tRNA, 80 μM of tRNALys3 and 90 μM or 160 μM of MA were incubated for 5 min at room temperature in a buffer consisting of 25 mM Tris-HCl pH 7.5, 150 mM NaCl, and 2 mM MgCl2, prior to injection onto a Superdex 200 Increase column on an Agilent HPLC system. For comparison, MA and tRNALys3 were also injected separately at 80 μM. The HPLC system was coupled to a DAWN HELEOSII detector equipped with a quasi-elastic light scattering module and an Optilab T-rEX refractometer (Wyatt Technology). Data were analyzed using the ASTRA 7.3 software (Wyatt Technology Europe).
Mapping tRNA interacting amino acids using CLIP
Cells were seeded onto a 10 cm dish and transfected, 24h later, with 8µg of plasmids encoding WT HIV-1 MA-3xHA or a mutant thereof. On the following day, media were replaced by culture media containing 4-thiouridine. Two days after transfection, cells were exposed to UVB light (0.15 J cm−2, λ = 365 nm, Stratalinker 2400 UV), washed in PBS and lysed in Lysis Buffer (10 mM HEPES pH 7.5, 30 mM KCl, 40 µM EDTA, 0.1% Igepal CA-630 supplemented with protease inhibitor cocktails). Lysates were clarified by centrifugation and incubated with RNAse A for 5 min at 37˚C. Anti-HA mouse antibody (BioLegend) was adsorbed to protein G agarose beads and incubated with the cell lysates for 2h at 4˚C. Beads were washed twice in NP40 Lysis Buffer, twice IP wash buffer (50 mM HEPES pH 7.5, 300 mM KCl, 2 mM EDTA, 0.5% Igepal CA-630), twice in LiCl buffer (250 mM LiCl, 10 mM Tris pH 8.0, 1 mM EDTA, 0.5% Igepal CA-630, 0.5% sodium deoxycolate), twice in NaCl Buffer (50 mM Tris pH 7.5, 1 M NaCL, 1 mM EDTA, 0.1 SDS, 0.5% sodium deoxycholate) and twice in KCl buffer (50 mM HEPES pH 7.5, 500 mM KCl, 0.05% Igepal CA-630). RNA:Protein complexes were incubated with calf intestinal phosphatase (NEB) for 13 min at 37˚C and washed in phosphatase wash buffer (50 mM Tris HCl pH 7.5, 20 mM EGTA, 0.5% NP40). Beads were resuspended in PNK buffer (50 mM Tris HCl pH 7.5, 50 mM NaCl, 10 mM MgCl2) and incubated with 5 U of PNK in the presence of 0.5 μCi/μL γ−32P ATP. Beads were washed, lysed in NuPAGE Lysis buffer and RNA:Protein complexes were resolved in a 4–12% NuPAGE gel. Complexes were transferred to nitrocellulose membrane and developed by exposing autoradiographic film.
The membranes were then blocked at room temperature and incubated with the following antibodies: anti-ZC3HAV1 (rabbit, 1:10,000, Proteintech Group), anti-TRIM25, anti-HA (rabbit, 1:5000, Rockland), anti-Tubulin (mouse, 1:10,000, Millipore Sigma), anti-gp120 HIV-1 (goat, 1:1000, American Research Products). Blots were washed and incubated with secondary antibodies: anti-Mouse IgG IR700 Dye Conjugated (Licor), Anti-Rabbit IgG IR800 Dye conjugated (Licor), Anti-Goat IgG IR800 Dye Conjugated (Licor) and Anti-Rabbit IgG horseradish peroxidase conjugated (Jackson). Blots were imaged immediately using a Licor Odyssey imager.
Plasmids
pNL4–3 was described previously (Adachi et al., 1986). The MAK32A mutation in pNL4–3(MAK32A) was introduced using overlap extension PCR mutagenesis. MAK32A-variants of R5-tropic pNL4–3 variants were generated by introducing a PCR-fragment of pNL4–3(MAK32A) (see Resource table for primer sequences) into BssHII/SphI-digested pNL/BaL, pNL/YU2, pNL/ADA(Zhang et al., 2002) or pNL/BAL (Mariani et al., 2001) using Gibson-assembly. pCR3.1coGagGFP and pCR3.1coMA-GFP were described previously (Jouvenet et al., 2006) and the MAK32A and MAW36A mutations were introduced using overlap extension PCR mutagenesis. pCR3.1coMAK32A-GFP-LacZ was generated by amplifying the LacZ fragment from pLHCX-GFP-LacZ (Kane et al., 2016) (see Resource table for primer sequences), which was subsequently inserted into BspEI/XbaI-digested pCR3.1coMAK32A-GFP using Gibson assembly.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Goat anti-gp120 HIV-1 | ARP | Cat#12–6205 RRID:AB_1541516 |
| Anti-HIV-1 p24 (183-H12–5C) | NIH AIDS Reagent Program | Cat#3537 RRID:AB_2832923 |
| Human anti-HIV-1 gp120 Monoclonal 2G12 | Polymun Scientific | Cat#AB002 RRID:AB_2661842 |
| IRDye800CW Donkey anti-Goat IgG | LI-COR Bioscience | Cat# 926–32214 RRID:AB_621846 |
| IRDye 680RD Donkey anti-Mouse IgG | LI-COR Bioscience | Cat# 926–68072 RRID:AB_10953628 |
| Goat anti-Mouse IgG - Atto 594 | Sigma | Cat# 76085 RRID:AB_1137653 |
| Abberior STAR RED, donkey anti-human IgG | Abberior | Cat# STRED-1054 RRID:AB_2877172 |
| Bacterial and Virus Strains | ||
| BL21-CodonPlus (DE3)-RIL Competent Cells | Agilent | Cat# 230245 |
| Biological Samples | ||
| LeukoPaks – Buffy coats made from human whole blood | NYBC, New York | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| T4 RNA ligase 1 | New England BioLabs | Cat#M0204S |
| Human recombinant IL-2 | PeproTech | Cat#200–02 |
| Human recombinant GM-CSF | PeproTech | Cat#300–03 |
| Critical Commercial Assays | ||
| QuikChange Lightning | Agilent | Cat#210519 |
| Deposited Data | ||
| Atomic coordinates and structure factor amplitudes for the co-crystal structure of HIV-1 MA bound to human tRNALys3 | This paper | PDB: 7MRL |
| Experimental Models: Cell Lines | ||
| HeLa | ATCC | Cat#: CCL-2 RRID:CVCL_0030 |
| 293T | ATCC | Cat#CRL-3216 RRID: CVCL_0063 |
| SupT1 | ATCC | Cat# CRL-1942 RRID: CVCL_1714 |
| MT4-LTR-GFP | (Kane et al., 2016) | N/A |
| CEM X174 | NIH AIDS Reagent Program | Cat#234–220 RRID: CVCL_X615 |
| Oligonucleotides | ||
| 5′ RNA oligo to anneal to form tRNALys3 with 2Ap10 5′-rGrCrCrCrGrGrArUrArGrCrUrCrArGrUrCrG/i2AmPr/rUrA rGrArGrCrArUrCrArGrArCrU-3′ |
Integrated DNA Technologies | N/A |
| 3′ RNA oligo to anneal to form tRNALys3 with 2Ap10 /5′Phos/rUrUrUrArArUrCrUrGrArGrGrGrUrCrCrArGrGrGrUrUrCrArArGrUrCrCrCrUrGrUrUrCrGrGrGrCrGrCrCrA-3′ |
Integrated DNA Technologies | N/A |
| 5′ RNA oligo to anneal to form tRNALys3 with 2Ap19 5′ -rGrCrCrCrGrGrArUrA/i2AmPr/rCrUrCrArGrUrCrGrGrUrA rGrArGrCrArUrCrArGrArCrU-3′ |
Integrated DNA Technologies | N/A |
| 3′ RNA oligo to anneal to form tRNALys3 with 2Ap19 /5′Phos/rUrUrUrArArUrCrUrGrArGrGrGrUrCrCrArGrGrGrUrUrCrArArGrUrCrCrCrUrGrUrUrCrGrGrGrCrGrCrCrA-3′ |
Integrated DNA Technologies | N/A |
| Forward primer to amplify MAK32A fragment from pNL4–3(MAK32A) 5′-CCA GAG GAG ATC TCT CGA C-3’ and 5’-CCC CTT GGT TCT CTC ATC-3′ |
Integrated DNA Technologies | N/A |
| Reverse primer to amplify MAK32A fragment from pNL4–3(MAK32A) 5′-CCC CTT GGT TCT CTC ATC-3′ |
Integrated DNA Technologies | N/A |
| Forward primer to amplify the LacZ fragment from pLHCX-GFP-LacZ 5′-CTCGGCATGGACGAGCTGTACAAGTCCGGAGCCGTCGTTTTACAAC-3′ |
Integrated DNA Technologies | N/A |
| Reverse primer to amplify the LacZ fragment from pLHCX-GFP-LacZ 5′-GCTGATCAGCGGGTTTAAACGGGCCCTCTAGATTTTTGACACCAGACCAACTGGTAATGG-3′ |
Integrated DNA Technologies | N/A |
| Additional oligos for mutational analyses can be found in Supplemental Table 4 | Integrated DNA Technologies | N/A |
| Recombinant DNA | ||
| pET19b-MA | (Gaines et al., 2018b) | N/A |
| pNL4–3 | (Adachi et al., 1986) | N/A |
| pNL4–3(MAK32A) | This paper | N/A |
| pNL/YU2 | (Zhang et al., 2002) | N/A |
| pNL/ADA | (Zhang et al., 2002) | N/A |
| pNL/BAL | (Mariani et al., 2001) | N/A |
| pNL/YU2(MAK32A) | This paper | N/A |
| pNL/ADA(MAK32A) | This paper | N/A |
| pNL/BAL(MAK32A) | This paper | N/A |
| pCR3.1coGag-GFP | (Jouvenet et al., 2006) | N/A |
| pCR3.1coMAK32A-GFP | (Jouvenet et al., 2006) | N/A |
| pCR3.1coMA-3xHA | This paper | N/A |
| pCR3.1coGag(MAK32A)-GFP | This paper | N/A |
| pCR3.1coMAK32A-GFP | This paper | N/A |
| pCR3.1coMAK32A-3xHA | This paper | N/A |
| pLHCX-GFP-LacZ | (Kane et al., 2016) | N/A |
| pCR3.1coMAK32A-GFP-LacZ | This paper | N/A |
| pVSV-G | (Yee et al., 1994) | N/A |
| Software and Algorithms | ||
| SEDPHAT | (Zhao et al., 2015) | RRID:SCR_016254 http://www.analyticalultracentrifugation.com/sedphat/ |
| NITPIC | (Keller et al., 2012) | https://www.utsouthwestern.edu/labs/mbr/software/ |
| SEDFIT | (Schuck, 2000) | RRID:SCR_018365 https://sedfitsedphat.nibib.nih.gov/software/default.aspx |
| REDATE | (Zhao et al., 2013b) | https://www.utsouthwestern.edu/labs/mbr/software/ |
| SEDNTERP | (Cole et al., 2008) | RRID:SCR_016253 http://www.jphilo.mailway.com/sednterp.htm |
| ASTRA | Wyatt Technology Europe | RRID:SCR_016255 https://www.wyatt.com/products/software/astra.html |
| Phaser | (McCoy et al., 2007) | RRID:SCR_014219 https://www.phenixonline.org/documentation/reference/phaser.html |
| Phenix | (Liebschner et al., 2019) | RRID:SCR_014224 https://www.phenixonline.org |
| Coot | (Emsley et al., 2010) | RRID:SCR_014222 https://www2.mrclmb.cam.ac.uk/personal/pemsley/coot/ |
| ERRASER | (Chou et al., 2013) | https://rosie.graylab.jhu.edu/erraser |
| Other | ||
| GraphPad Prism | GraphPad Software | RRID:SCR_002798 https://www.graphpad.com |
| Origin | OriginLab Corporation | RRID:SCR_014212 https://www.originlab.com/ |
| FIJI | (Schindelin et al., 2012) | RRID:SCR_002285 https://fiji.sc |
| DeltaVision SoftWoRx | GE Healthcare | RRID:SCR_019157 https://www.gehealthcare.com |
| Imspector Image Acquisition & Analysis Software | Abberior Instruments Development Team | RRID:SCR_015249 http://www.imspector.de |
| Huygens STED Deconvolution Software, Huygens Professional | SVI | RRID:SCR_014237 https://svi.nl/Huygens-STED-Software |
Transfection experiments
For transfection experiments in HeLa cells, cells were seeded at a concentration of 25,000 cells/well in µ-Slide 8 Well Glass Bottom chambers (Ibidi) and transfected the following day using Turbofect (ThermoScientific). For widefield microscopy of Gag and MA-variants, cells were transfected with 75 ng pCR3.1coGag-GFP, pCR3.1coGag(MAK32A)-GFP, pCR3.1coGag(MAW36A)-GFP, pCR3.1coMA-GFP, pCR3.1coMAK32A-GFP, pCR3.1coMAW36A-GFP or pCR3.1coMA(K32A)-GFP-LacZ. Cells were fixed 12 hours after transfection. For STED microscopy of HIV-1 assembly sites, cells were transfected with 300 ng pNL4–3 or pNL4–3(MAK32A). Cells were fixed 20 h post-transfection.
HIV-1 replication assays
For analysis of HIV-1 spreading replication, T cell lines, primary CD4+ T cells or monocyte derived macrophages were used, with three donors for each (D2-D4). For T cell lines, 20,000 SupT1, MT4-LTR-GFP or CEM cells were plated in 96-well U bottom plates. For primary CD4+ T cells, 200,000 cells were plated in 96-well U bottom plates. For MDM, 50,000 cells were plated in 96-well flat bottom plates. Cells were infected with varying amounts (MOI based on MT4-LTR-GFP cells as indicated in respective Figure legends) of NL4–3 wt or NL4–3(MAK32A) virus. At 12 hpi (hours post-infection), the inoculum was removed, and cells were washed twice with PBS and incubated in 200 µl fresh growth medium. 50 µl supernatant was harvested and replaced with fresh growth medium every 24 hpi until 8 (cell lines) or 10 dpi (primary cells). RT-content in viral supernatants was assessed by SG-PERT (Pizzato et al., 2009).
Analysis of Env-incorporation into viral particles by western blot
293T cells were seeded in 6-well plates and infected with VSV-G-pseudotyped full length HIV-1 particles (containing HIV-1 Env and VSV-G on their surface) at an MOI of 0.5 (based on 293T) the following day. 16 hpi, medium was replaced by 3 ml growth medium. Viral supernatants were harvested 43 hpi, filtered and concentrated by ultracentrifugation through a 20% (w/v) sucrose cushion. Sucrose-pelleted virus was resuspended in 50 µl 1x NuPAGE sample buffer (Invitrogen) containing DTT and separated on NuPage Novex 4–12 % Bis-Tris Mini Gels (Invitrogen). Proteins were blotted onto nitrocellulose membranes. Blots were incubated with LICOR blocking buffer (LI-COR Biosciences) and probed with primary antibodies (Goat anti-gp120 HIV-1, ARP, 1:100; mouse anti-HIV-1 p24 183-H12–5C, NIH AIDS Research and Reference Reagent Program, 1:200) and secondary antibodies (IRDye800CW Donkey anti-Goat IgG; IRDye 680RD Donkey anti-Mouse IgG, both 1:10,000, LI-COR Biosciences). Blots were analyzed using the LI-COR Odyssey Imager (LI-COR Biosciences). For quantitative signal analysis, the Odyssey Image Studio software was used (LI-COR Biosciences).
Immunolabeling of Gag assembly sites for STED microscopy
HeLa cells transfected with pNL4–3 or pNL4–3(MAK32A) were fixed with 4 % paraformaldehyde 20 hpt, permeabilized with 0.1 % Triton-X-100/PBS and blocked with 2% BSA/PBS. Cells were incubated with anti-HIV-1 p24 (183-H12–5C) (NIH AIDS Research and Reference Reagent Program, 1:100) and anti-HIV-1 gp120 Monoclonal 2G12 (Polymun Scientific, 1:50), followed by incubation with respective secondary antibodies Goat anti-Mouse IgG - Atto 594 (Sigma) and Abberior STAR RED, donkey anti-human IgG (Abberior) (both 1:100).
Microscopy and image representation
Widefield-Images were captured using an DeltaVision OMX SR imaging system (GE Healthcare). Images were deconvolved using the DeltaVision SoftWoRx software (GE Healthcare). Stimulated emission depletion (STED) imaging was performed at λ = 775 nm Facility line STED system (Abberior Instruments GmbH, Göttingen, Germany), using a 100x/1.40 oil immersion objective with 561 and 640 nm excitation laser lines at room temperature. Nominal STED laser power was set to ~5–15% of the maximal power of 1200 mW and STED images were acquired with a 25 nm pixel size. Images were deconvolved using Huygens software. Representative still images were chosen. For all images shown, the camera offset value was subtracted and the contrast and brightness were adapted for optimal display of the image. Images are shown in pseudo colors. The Green lookup table (LUT) was used for widefield images, while different channels of super resolution STED images are shown with the LUTs Red Hot (referred to as ‘red’) and Green Fire Blue (referred to as ‘cyan’).
Particle analysis and line profiles of widefield microscopy data
Particle analysis of SDC microscopy data was done in FIJI (RRID:SCR_002285) (Schindelin et al., 2012) on single slices of the ventral PM. Single images were converted to 8-bit images and background was subtracted using a rolling ball. The objects of interest were automatically thresholded using the Niblack local thresholding method. A 1 px median filter was applied to all images and the watershed algorithm was applied. The region of interest was selected manually by drawing the outline of the respective cell. Finally, the size and number of particles in each cell was determined using FIJI’s Analyze Particles function. Values were further analyzed in Excel (Microsoft, Redmond, USA) and GraphPad Prism (GraphPad Software, Inc., La Jolla, USA). The number of particles detected at the ventral PM was divided by membrane area and plotted as number of Gag clusters/100 µm2. Line profiles of individual cells were generated manually in FIJI (Schindelin et al., 2012). Prior to this, the camera offset value was subtracted from the individual images and a 0.5 px median filter was applied. The line profile intensity values in the Gag and DAPI channel were exported to GraphPad Prism (GraphPad Software, Inc., La Jolla, USA). Average GFP-/DAPI-fluorescence intensity or normalized average GFP fluorescence intensity (smallest value = 0, highest value = 100) ± SEM were plotted.
Quantification and statistical analysis
Statistical analyses were performed with the Prism software (GraphPad Prism) and Origin software (OriginLab Corporation). The number of replicates for each ITC measurement and standard deviations can be found in Table S2. HIV-1 replication assays were performed on three cell donors (D2-D4) and the mean ± SEM are in Figures 7 and S7 and legends. The mean values and range for n=3 or 4 experiments of western blot analysis of Env incorporation into viral particles are in Figure S7 and legend. The error bars and the 95% confidence interval for fluorescence quantification in cells are in Figures 6 and S6 and legends, while Kruskall-Wallis test and subsequent Dunn’s multiple comparisons test were used to assess statistical significance. Statistical validation of the deposited atomic model was performed using Phenix and can be found in Table S1.
Supplementary Material
Highlights.
Co-crystal structure of HIV-1 Gag matrix (MA) domain bound to human tRNALys3
MA specifically recognizes tRNA elbow structure
K32A or W36A in MA abolishes tRNA binding and redistributes Gag to plasma membrane
Inability of Gag to bind tRNA leads to reduced HIV-1 replication
Acknowledgments
We thank I. Botos for computational support, G. Piszczek, D. Wu for support in biophysical analyses, Y. He and N. Tjandra for fermentation support, R. Levine and D.-Y. Lee for mass spectrometry support, M. Bauerle, C. Palm, J. Ejimogu, and C. Parker (UMBC) for assistance with RNA preparation and PAGE studies, and J.L. Smith and K. Suddala for discussions. We acknowledge the Rockefeller University Bio-Imaging Resource Center for support with imaging on the Abberior facility line instrument. X-ray diffraction data were collected at SER-CAT 22-ID beamline at the Advanced Photon Source of the Argonne National Laboratory, supported by the U. S. Department of Energy under Contract No. W-31–109-Eng-38. This work was supported in part by the Intramural Research Program of the NIH, The National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) (ZIADK075136 to J.Z.), a NIH Deputy Director for Intramural Research (DDIR) Challenge Award to J.Z, and grants from the National Institute of Allergy and Infectious Diseases R01AI50111 to P.D.B., R01AI150498 to M.F.S., and U54AI50470 (Center for HIV RNA studies) to P.D.B. and M.F.S., and Howard Hughes Medical Institute. C.B.N. is a recipient of an NIH Intramural AIDS Research Fellowship and a NIDDK Nancy Nossal Fellowship Award.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests
The authors declare no conflicts of interest.
References
- Adachi A, Gendelman HE, Koenig S, Folks T, Willey R, Rabson A, and Martin MA (1986). Production of acquired immunodeficiency syndrome-associated retrovirus in human and nonhuman cells transfected with an infectious molecular clone. J Virol 59, 284–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alfadhli A, Barklis RL, and Barklis E. (2009). HIV-1 matrix organizes as a hexamer of trimers on membranes containing phosphatidylinositol-(4,5)-bisphosphate. Virology 387, 466–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alfadhli A, Mack A, Ritchie C, Cylinder I, Harper L, Tedbury PR, Freed EO, and Barklis E. (2016). Trimer Enhancement Mutation Effects on HIV-1 Matrix Protein Binding Activities. J Virol 90, 5657–5664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benas P, Bec G, Keith G, Marquet R, Ehresmann C, Ehresmann B, and Dumas P. (2000). The crystal structure of HIV reverse-transcription primer tRNA(Lys,3) shows a canonical anticodon loop. RNA 6, 1347–1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bieniasz P, and Telesnitsky A. (2018). Multiple, Switchable Protein:RNA Interactions Regulate Human Immunodeficiency Virus Type 1 Assembly. Annual review of virology 5, 165–183. [DOI] [PubMed] [Google Scholar]
- Bou-Nader C, Barraud P, Pecqueur L, Perez J, Velours C, Shepard W, Fontecave M, Tisne C, and Hamdane D. (2019). Molecular basis for transfer RNA recognition by the double-stranded RNA-binding domain of human dihydrouridine synthase 2. Nucleic Acids Res 47, 3117–3126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bou-Nader C, Pecqueur L, Bregeon D, Kamah A, Guerineau V, Golinelli-Pimpaneau B, Guimaraes BG, Fontecave M, and Hamdane D. (2015). An extended dsRBD is required for post-transcriptional modification in human tRNAs. Nucleic Acids Res 43, 9446–9456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown JD, Kharytonchyk S, Chaudry I, Iyer AS, Carter H, Becker G, Desai Y, Glang L, Choi SH, Singh K, Lopresti MW, Orellana M, Rodriguez T, Oboh U, Hijji J, Ghinger FG, Stewart K, Frances D, Edwards B, Chen P, Case DA, Telesnitsky A, Summers MF (2020). Structural basis for transcriptional start site control of HIV-1 RNA fate. Science 368, 413–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryant M, and Ratner L. (1990). Myristoylation-dependent replication and assembly of human immunodeficiency virus 1. Proc Natl Acad Sci U S A 87, 523–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou FC, Sripakdeevong P, Dibrov SM, Hermann T, and Das R. (2013). Correcting pervasive errors in RNA crystallography through enumerative structure prediction. Nat Methods 10, 74–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chukkapalli V, Oh SJ, and Ono A. (2010). Opposing mechanisms involving RNA and lipids regulate HIV-1 Gag membrane binding through the highly basic region of the matrix domain. Proc Natl Acad Sci U S A 107, 1600–1605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cimarelli A, and Luban J. (1999). Translation elongation factor 1-alpha interacts specifically with the human immunodeficiency virus type 1 Gag polyprotein. J Virol 73, 5388–5401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole JL, Lary JW, P. Moody T, and Laue TM (2008). Analytical Ultracentrifugation: Sedimentation Velocity and Sedimentation Equilibrium. In Biophysical Tools for Biologists, Volume One: In Vitro Techniques (Elsevier; ), pp. 143–179. 10.1016/s0091-679x(07)84006-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalluge JJ, Hamamoto T, Horikoshi K, Morita RY, Stetter KO, and McCloskey JA (1997). Posttranscriptional modification of tRNA in psychrophilic bacteria. J Bacteriol 179, 1918–1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dick RA, Kamynina E, and Vogt VM (2013). Effect of multimerization on membrane association of Rous sarcoma virus and HIV-1 matrix domain proteins. J Virol 87, 13598–13608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding P, Kharytonchyk S, Waller A, Mbaekwe U, Basappa S, Kuo N, Frank HM, Quasney C, Kidane A, Swanson C, et al. (2020). Identification of the initial nucleocapsid recognition element in the HIV-1 RNA packaging signal. Proc Natl Acad Sci U S A 117, 17737–17746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, Lohkamp B, Scott WG, and Cowtan K. (2010). Features and development of Coot. Acta Crystallogr D Biol Crystallogr 66, 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukai S, Nureki O, Sekine S, Shimada A, Tao J, Vassylyev DG, and Yokoyama S. (2000). Structural basis for double-sieve discrimination of L-valine from L-isoleucine and L-threonine by the complex of tRNA(Val) and valyl-tRNA synthetase. Cell 103, 793–803. [DOI] [PubMed] [Google Scholar]
- Gaines CR, Tkacik E, Rivera-Oven A, Somani P, Achimovich A, Alabi T, Zhu A, Getachew N, Yang AL, McDonough M, et al. (2018a). HIV-1 Matrix Protein Interactions with tRNA: Implications for Membrane Targeting. J Mol Biol 430, 2113–2127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaines CR, Tkacik E, Rivera-Oven A, Somani P, Achimovich A, Alabi T, Zhu A, Getachew N, Yang AL, McDonough M, et al. (2018b). HIV-1 Matrix Protein Interactions with tRNA: Implications for Membrane Targeting. J Mol Biol 430, 2113–2127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Göttlinger HG, Sodroski JG, and Haseltine WA (1989). Role of capsid precursor processing and myristoylation in morphogenesis and infectivity of human immunodeficiency virus type 1. Proc Natl Acad Sci U S A 86, 5781–5785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo M, Shapiro R, Morris GM, Yang XL, and Schimmel P. (2010). Packaging HIV virion components through dynamic equilibria of a human tRNA synthetase. J Phys Chem B 114, 16273–16279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guy MP, Young DL, Payea MJ, Zhang X, Kon Y, Dean KM, Grayhack EJ, Mathews DH, Fields S, and Phizicky EM (2014). Identification of the determinants of tRNA function and susceptibility to rapid tRNA decay by high-throughput in vivo analysis. Genes Dev 28, 1721–1732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamann CS, and Hou YM (2000). Probing a tRNA core that contributes to aminoacylation. J Mol Biol 295, 777–789. [DOI] [PubMed] [Google Scholar]
- Heng X, Kharytonchyk S, Garcia EL, Lu K, Divakaruni SS, LaCotti C, Edme K, Telesnitsky A, and Summers MF (2012). Identification of a minimal region of the HIV-1 5’-leader required for RNA dimerization, NC binding, and packaging. J Mol Biol 417, 224–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill CP, Worthylake D, Bancroft DP, Christensen AM, and Sundquist WI (1996). Crystal structures of the trimeric human immunodeficiency virus type 1 matrix protein: implications for membrane association and assembly. Proc Natl Acad Sci U S A 93, 3099–3104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes M, Zhang F, and Bieniasz PD (2015). Single-Cell and Single-Cycle Analysis of HIV-1 Replication. PLoS Pathog 11, e1004961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hood IV, Gordon JM, Bou-Nader C, Henderson FE, Bahmanjah S, and Zhang J. (2019). Crystal structure of an adenovirus virus-associated RNA. Nat Commun 10, 2871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Javanbakht H, Halwani R, Cen S, Saadatmand J, Musier-Forsyth K, Gottlinger H, and Kleiman L. (2003). The interaction between HIV-1 Gag and human lysyl-tRNA synthetase during viral assembly. J Biol Chem 278, 27644–27651. [DOI] [PubMed] [Google Scholar]
- Jean JM, and Hall KB (2001). 2-Aminopurine fluorescence quenching and lifetimes: role of base stacking. Proc Natl Acad Sci U S A 98, 37–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson BA (2004). Using NMRView to visualize and analyze the NMR spectra of macromolecules. Methods Mol Biol 278, 313–352. [DOI] [PubMed] [Google Scholar]
- Johnson BA, and Blevins RA (1994). NMR View: A computer program for the visualization and analysis of NMR data. J Biomol NMR 4, 603–614. [DOI] [PubMed] [Google Scholar]
- Jouvenet N, Neil SJ, Bess C, Johnson MC, Virgen CA, Simon SM, and Bieniasz PD (2006). Plasma membrane is the site of productive HIV-1 particle assembly. PLoS biology 4, e435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kane M, Zang TM, Rihn SJ, Zhang F, Kueck T, Alim M, Schoggins J, Rice CM, Wilson SJ, and Bieniasz PD (2016). Identification of Interferon-Stimulated Genes with Antiretroviral Activity. Cell host & microbe 20, 392–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller S, Vargas C, Zhao H, Piszczek G, Brautigam CA, and Schuck P. (2012). High-precision isothermal titration calorimetry with automated peak-shape analysis. Anal Chem 84, 5066–5073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korostelev A, Trakhanov S, Laurberg M, and Noller HF (2006). Crystal structure of a 70S ribosome-tRNA complex reveals functional interactions and rearrangements. Cell 126, 1065–1077. [DOI] [PubMed] [Google Scholar]
- Kroupa T, Datta SAK, and Rein A. (2020). Distinct Contributions of Different Domains within the HIV-1 Gag Polyprotein to Specific and Nonspecific Interactions with RNA. Viruses 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruger DM, Neubacher S, and Grossmann TN (2018). Protein-RNA interactions: structural characteristics and hotspot amino acids. RNA 24, 1457–1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kutluay SB, Zang T, Blanco-Melo D, Powell C, Jannain D, Errando M, and Bieniasz PD (2014). Global changes in the RNA binding specificity of HIV-1 gag regulate virion genesis. Cell 159, 1096–1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuzembayeva M, Dilley K, Sardo L, and Hu WS (2014). Life of psi: how full-length HIV-1 RNAs become packaged genomes in the viral particles. Virology 454–455, 362–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lama J, and Trono D. (1998). Human immunodeficiency virus type 1 matrix protein interacts with cellular protein HO3. J Virol 72, 1671–1676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leamy KA, Yamagami R, Yennawar NH, and Bevilacqua PC (2019). Single-nucleotide control of tRNA folding cooperativity under near-cellular conditions. Proc Natl Acad Sci U S A 116, 23075–23082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leontis NB, and Westhof E. (2001). Geometric nomenclature and classification of RNA base pairs. RNA 7, 499–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Su Z, Lehmann J, Stamatopoulou V, Giarimoglou N, Henderson FE, Fan L, Pintilie GD, Zhang K, Chen M, et al. (2019). Structural basis of amino acid surveillance by higher-order tRNA-mRNA interactions. Nat Struct Mol Biol 26, 1094–1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liebschner D, Afonine PV, Baker ML, Bunkoczi G, Chen VB, Croll TI, Hintze B, Hung LW, Jain S, McCoy AJ, et al. (2019). Macromolecular structure determination using X-rays, neutrons and electrons: recent developments in Phenix. Acta Crystallogr D Struct Biol 75, 861–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lochrie MA, Waugh S, Pratt DG Jr., Clever J, Parslow TG, and Polisky B. (1997). In vitro selection of RNAs that bind to the human immunodeficiency virus type-1 gag polyprotein. Nucleic Acids Res 25, 2902–2910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu K, Heng X, and Summers MF (2011). Structural determinants and mechanism of HIV-1 genome packaging. J Mol Biol 410, 609–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mariani R, Rasala BA, Rutter G, Wiegers K, Brandt SM, Kräusslich HG, and Landau NR (2001). Mouse-human heterokaryons support efficient human immunodeficiency virus type 1 assembly. J Virol 75, 3141–3151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massiah MA, Starich MR, Paschall C, Summers MF, Christensen AM, and Sundquist WI (1994). Three-dimensional structure of the human immunodeficiency virus type 1 matrix protein. J Mol Biol 244, 198–223. [DOI] [PubMed] [Google Scholar]
- McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, and Read RJ (2007). Phaser crystallographic software. J Appl Crystallogr 40, 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercredi PY, Bucca N, Loeliger B, Gaines CR, Mehta M, Bhargava P, Tedbury PR, Charlier L, Floquet N, Muriaux D, et al. (2016). Structural and Molecular Determinants of Membrane Binding by the HIV-1 Matrix Protein. J Mol Biol 428, 1637–1655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milligan JF, and Uhlenbeck OC (1989). Synthesis of small RNAs using T7 RNA polymerase. Meth Enzymol 180, 51–62. [DOI] [PubMed] [Google Scholar]
- Monje-Galvan V, and Voth GA (2020). Binding mechanism of the matrix domain of HIV-1 gag on lipid membranes. Elife 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mu X, Fu Y, Zhu Y, Wang X, Xuan Y, Shang H, Goff SP, and Gao G. (2015). HIV-1 Exploits the Host Factor RuvB-like 2 to Balance Viral Protein Expression. Cell Host Microbe 18, 233–242. [DOI] [PubMed] [Google Scholar]
- Mücksch F, Laketa V, Müller B, Schultz C, and Kräusslich HG (2017). Synchronized HIV assembly by tunable PIP(2) changes reveals PIP(2) requirement for stable Gag anchoring. Elife 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami T, and Freed EO (2000). Genetic evidence for an interaction between human immunodeficiency virus type 1 matrix and alpha-helix 2 of the gp41 cytoplasmic tail. J Virol 74, 3548–3554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy RE, Samal AB, Vlach J, Mas V, Prevelige PE, and Saad JS (2019). Structural and biophysical characterizations of HIV-1 matrix trimer binding to lipid nanodiscs shed light on virus assembly. J Biol Chem 294, 18600–18612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naganuma M, Sekine S, Chong YE, Guo M, Yang XL, Gamper H, Hou YM, Schimmel P, and Yokoyama S. (2014). The selective tRNA aminoacylation mechanism based on a single G*U pair. Nature 510, 507–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norris M, Fetler B, Marchant J, and Johnson BA (2016). NMRFx Processor: a cross-platform NMR data processing program. J Biomol NMR 65, 205–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliva R, Cavallo L, and Tramontano A. (2006). Accurate energies of hydrogen bonded nucleic acid base pairs and triplets in tRNA tertiary interactions. Nucleic Acids Res 34, 865–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ono A, Ablan SD, Lockett SJ, Nagashima K, and Freed EO (2004). Phosphatidylinositol (4,5) bisphosphate regulates HIV-1 Gag targeting to the plasma membrane. Proc Natl Acad Sci U S A 101, 14889–14894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paillart JC, and Göttlinger HG (1999). Opposing effects of human immunodeficiency virus type 1 matrix mutations support a myristyl switch model of gag membrane targeting. J Virol 73, 2604–2612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Caballero D, Hatziioannou T, Martin-Serrano J, and Bieniasz PD (2004). Human immunodeficiency virus type 1 matrix inhibits and confers cooperativity on gag precursor-membrane interactions. J Virol 78, 9560–9563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pizzato M, Erlwein O, Bonsall D, Kaye S, Muir D, and McClure MO (2009). A one-step SYBR Green I-based product-enhanced reverse transcriptase assay for the quantitation of retroviruses in cell culture supernatants. J Virol Methods 156, 1–7. [DOI] [PubMed] [Google Scholar]
- Puglisi EV, and Puglisi JD (2007). Probing the conformation of human tRNA(3)(Lys) in solution by NMR. FEBS Lett 581, 5307–5314. [DOI] [PubMed] [Google Scholar]
- Purohit P, Dupont S, Stevenson M, and Green MR (2001). Sequence-specific interaction between HIV-1 matrix protein and viral genomic RNA revealed by in vitro genetic selection. RNA 7, 576–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu K, Ke Z, Zila V, Anders-Össwein M, Glass B, Müller B, Kräusslich H-G, and Briggs JAG (2020). Maturation of the matrix and viral membrane of HIV-1. bioRxiv, 10.1101/2020.1109.1123.309542. [DOI] [PMC free article] [PubMed]
- Reiter NJ, Osterman A, Torres-Larios A, Swinger KK, Pan T, and Mondragon A. (2010). Structure of a bacterial ribonuclease P holoenzyme in complex with tRNA. Nature 468, 784–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saad JS, Miller J, Tai J, Kim A, Ghanam RH, and Summers MF (2006). Structural basis for targeting HIV-1 Gag proteins to the plasma membrane for virus assembly. Proc Natl Acad Sci U S A 103, 11364–11369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. (2012). Fiji: an open-source platform for biological-image analysis. Nat Methods 9, 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuck P. (2000). Size-distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and lamm equation modeling. Biophys J 78, 1606–1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strulson CA, Boyer JA, Whitman EE, and Bevilacqua PC (2014). Molecular crowders and cosolutes promote folding cooperativity of RNA under physiological ionic conditions. RNA 20, 331–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundquist WI, and Kräusslich HG (2012). HIV-1 assembly, budding, and maturation. Cold Spring Harbor perspectives in medicine 2, a006924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang C, Loeliger E, Luncsford P, Kinde I, Beckett D, and Summers MF (2004). Entropic switch regulates myristate exposure in the HIV-1 matrix protein. Proc Natl Acad Sci U S A 101, 517–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang C, Ndassa Y, and Summers MF (2002). Structure of the N-terminal 283-residue fragment of the immature HIV-1 Gag polyprotein. Nature structural biology 9, 537–543. [DOI] [PubMed] [Google Scholar]
- Tedbury PR, Ablan SD, and Freed EO (2013). Global rescue of defects in HIV-1 envelope glycoprotein incorporation: implications for matrix structure. PLoS Pathog 9, e1003739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tedbury PR, Novikova M, Ablan SD, and Freed EO (2016). Biochemical evidence of a role for matrix trimerization in HIV-1 envelope glycoprotein incorporation. Proc Natl Acad Sci U S A 113, E182–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tedbury PR, Novikova M, Alfadhli A, Hikichi Y, Kagiampakis I, KewalRamani VN, Barklis E, and Freed EO (2019). HIV-1 Matrix Trimerization-Impaired Mutants Are Rescued by Matrix Substitutions That Enhance Envelope Glycoprotein Incorporation. J Virol 94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todd GC, Duchon A, Inlora J, Olson ED, Musier-Forsyth K, and Ono A. (2017). Inhibition of HIV-1 Gag-membrane interactions by specific RNAs. RNA 23, 395–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vlach J, and Saad JS (2013). Trio engagement via plasma membrane phospholipids and the myristoyl moiety governs HIV-1 matrix binding to bilayers. Proc Natl Acad Sci U S A 110, 3525–3530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westhof E, and Auffinger P. (2001). Transfer RNA Structure. In eLS (Wiley; ). 10.1002/9780470015902.a0000527.pub2. [DOI] [Google Scholar]
- Yee JK, Friedmann T, and Burns JC (1994). Generation of high-titer pseudotyped retroviral vectors with very broad host range. Methods in cell biology 43 Pt A, 99–112. [DOI] [PubMed] [Google Scholar]
- Zhang J. (2020). Unboxing the T-box riboswitches-A glimpse into multivalent and multimodal RNA-RNA interactions. Wiley Interdiscip Rev RNA 11, e1600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, and Ferré-D’Amaré AR (2013). Co-crystal structure of a T-box riboswitch stem I domain in complex with its cognate tRNA. Nature 500, 363–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, and Ferré-D’Amaré AR (2014). Direct evaluation of tRNA aminoacylation status by the T-box riboswitch using tRNA-mRNA stacking and steric readout. Mol Cell 55, 148–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, and Ferré-D’Amaré AR (2016). The tRNA Elbow in Structure, Recognition and Evolution. Life (Basel) 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang YJ, Hatziioannou T, Zang T, Braaten D, Luban J, Goff SP, and Bieniasz PD (2002). Envelope-dependent, cyclophilin-independent effects of glycosaminoglycans on human immunodeficiency virus type 1 attachment and infection. J Virol 76, 6332–6343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao H, Brautigam CA, Ghirlando R, and Schuck P. (2013a). Overview of current methods in sedimentation velocity and sedimentation equilibrium analytical ultracentrifugation. Curr Protoc Protein Sci Chapter 20, Unit20 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao H, Ghirlando R, Piszczek G, Curth U, Brautigam CA, and Schuck P. (2013b). Recorded scan times can limit the accuracy of sedimentation coefficients in analytical ultracentrifugation. Anal Biochem 437, 104–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao H, Piszczek G, and Schuck P. (2015). SEDPHAT--a platform for global ITC analysis and global multi-method analysis of molecular interactions. Methods 76, 137–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou W, Parent LJ, Wills JW, and Resh MD (1994). Identification of a membrane-binding domain within the amino-terminal region of human immunodeficiency virus type 1 Gag protein which interacts with acidic phospholipids. J Virol 68, 2556–2569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Luo S, Sabo Y, Wang C, Tong L, and Goff SP (2017). Heme Oxygenase 2 Binds Myristate to Regulate Retrovirus Assembly and TLR4 Signaling. Cell Host Microbe 21, 220–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Atomic coordinates and structure factor amplitudes for tRNALys3 bound to HIV-1 MA have been deposited in the Protein Data Bank (PDB) under accession code 7MRL.
This study did not generate any original code.
Any additional information required to reanalyze the data reported in this paper is available from the Lead Contact upon request.