

Abstract
Bitter taste receptors (TAS2Rs) function in taste perception, but are also expressed in many extraoral tissues, presenting attractive therapeutic targets. TAS2R5s expressed on human airway smooth muscle cells can induce bronchodilation for treating asthma and other obstructive diseases. But TAS2R5s display low agonist affinity and the lack of a 3D structure has hindered efforts to design more active ligands. We report the structure of the activated TAS2R5 coupled to the Gi protein and bound to each of 19 agonists, using computational approaches. These agonists bind to two polar residues in TM3 that are unique for TAS2R5 among 25 TAS2R subtypes. Our predicted results correlate well with experimental results of agonist-receptor signaling coefficients, providing validation of the predicted structure. These results provide highly specific data on how agonists activate TAS2R5, how modifications of ligand structure alter receptor activation, and a guide to structure-based drug design.
Keywords: G protein-coupled receptors, airway smooth muscle cells, molecular dynamics, GEnSeMBLE structure prediction, DarwinDock binding site prediction
Graphical Abstract
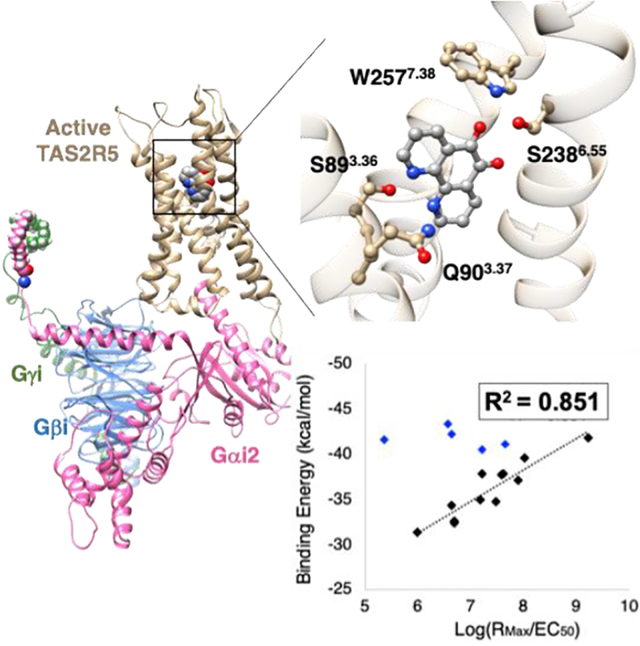
Bitter taste receptors (TAS2Rs) are responsible for bitter taste perception, which is generally considered to protect an organism from potentially toxic substances. TAS2Rs belong to the G protein-coupled receptors (GPCRs) superfamily, with 25 TAS2Rs known to be expressed in humans. In addition to the oral cavity, TAS2Rs have now been reported to be expressed in many extraoral tissues throughout the body, including airway,1 nasal epithelium,2 the gastrointestinal tract,3 heart,4 thyroid,5 and brain.6 We have proposed that these extraoral TAS2Rs represent a chemosensory system with multiple physiologic functions and may also serve as novel therapeutics targets.7,8 Indeed a number of studies have reported TAS2Rs-associated disorders and diseases including cancer,9 insulin homeostasis,10 asthma,11 upper airway disease,12 thyroid disorders,5 and cardiovascular diseases.13 However, the identity of agonist or antagonist binding sites for the 25 distinct human TAS2Rs has not been definitively established leading to high affinity (<10 nM) ligands, thus impairing drug development opportunities. In the current work, we have chosen the TAS2R5 subtype to elucidate structures and binding sites because of its potential therapeutic effect in asthma and chronic obstructive pulmonary disease (COPD), where TAS2R agonists cause marked relaxation of airway smooth muscle and dilate the airways.1,14 In addition, of the TAS2Rs expressed on human airway smooth muscle (HASM) cells, TAS2R5 is one of the few subtypes that appears to be “narrowly tuned”,15 providing the greatest opportunity to establish structure-function relationships with respect to agonist activation.
Most known TAS2R agonists display relatively low potency (~100 nM to > 10 mM) for activation of the canonical TAS2R signaling pathway (an increase in the second messenger [Ca2+]i).16 Until recently,17,18 it was not clear whether these receptors could be activated by agonists with a higher potencies or efficacies than benchmark agonists. The apparent low affinity for plant-based compounds reported for most TAS2Rs makes sense from an evolutionary standpoint for taste perception on the tongue (where the agent is in direct contact with taste cells), but this low potency/efficacy would be disadvantageous for activating receptors in other regions of the body for therapeutic purposes. Recently, we tested 1,10-phenanthroline (hereafter referred to as T5–1) as an activator of TAS2R5 in cells at the signaling and physiologic levels.17 And, in a previous screen16 T5–1 was tested against the other TAS2Rs and found not to activate them. Furthermore, none of the ~80 agonists from that screen activated TAS2R5. However, T5–1 exhibits a low apparent affinity (~30 μM), despite its specificity for TAS2R5. Consequently, 18 derivatives aimed at improving potency and efficacy were investigated.17 These studies showed that the potency and efficacy can be significantly altered by chemical modifications to the parent compound, with EC50 values varying by ~1200 fold, with the best EC50 being 120 nM. These experiments, which gave this broad range of signal transduction efficiencies from modifications of T5–1, would provide a basis for understanding the relationship between the ligand structure, receptor structure, and coupling to the G protein; but without receptor structure, further insights would at best be empirically based on serial alterations in the ligand structure without a structural basis. Indeed, this lack of receptor structural information has hampered investigations of TAS2Rs despite their apparent roles in multiple physiological functions or diseases. Three-dimensional (3D) structures of over 50 GPCRs and/or GPCR-G protein (GP) complexes have been determined, but neither X-ray crystal structures nor cryogenic electron microscopy (cryo-EM) structures are available for any TAS2R, despite numerous attempts. Moreover, TAS2Rs have little protein sequence homology with other GPCRs, so purely homology-based structures are not likely to be reliable.
Herein, we use validated computational approaches to predict the 3D structure of the fully activated TAS2R5 human bitter taste receptor coupled to the heterotrimeric Gi protein (GiP) and to predict the ligand binding site and binding poses for 19 agonists recently tested experimentally.15 The predicted binding energies correlate well with experimental data, indicating that this structural information about TAS2R5 and the ligand binding may provide new insights into the activation mechanism on TAS2R5 expressed on HASM cells. This may provide the basis for rational structure-based design of new therapeutic targets for bronchodilators targeting obstructive lung diseases such as asthma.
We used the GPCR Ensemble of Structures embedded in Membrane BiLayer Environment (GEnSeMBLE)19,20 complete sampling approach to predict the 3D structure of the fully activated TAS2R5 human bitter taste receptor coupled to the heterotrimeric GiP. As described further in Methods, we start with a template providing initial tilts for the 7 transmembrane (TM) helices based on homology to a known GPCR structure. With the tilts fixed, we examine all coupled axial rotations of 30° (BiHelix step, (12)7~35 million combinations),19 build the best 2000 into 7-TM helix bundles, and select the best structure by energy analyses. Then we examine all combinations of tilts and rotations (SuperBiHelix, 13 trillion combinations),20 select the best 2000 that we build into 7-TM helix bundles (SuperCombiHelix), from which we finally obtain the best 25 7-TM helix bundles that we use for ligand docking. This methodology has successfully predicted other GPCR structures, including class A [CCR5,21,22 AA3,23 DP prostaglandin,24 and CB125,26], class B [GLP127], and class C [TAS1R2/1R3 sweet heterodimer28].
To predict the 7-helix transmembrane domains (TMD) of TAS2R5, we selected six starting templates for which structures are known for the GPCR in the activated configuration (Table S1): serotonin 2B (5HT2B), adenosine A2A (A2A), angiotensin II (ATII), cannabinoid 1 (CB1), muscarinic 2 (M2), and β2 adrenergic (β2AR). In addition, we included our previously predicted structure for TAS2R4 as a template,29 given its moderate homology with TAS2R5 (Table S1). Based on these 7 GPCR templates, we compared the total energies for all 7×2000 SuperCombiHelix TMDs, to select the 10 lowest energies ordered by neutralized interhelical energy (Nih), as shown in Table S2. All 10 conformations came from the TAS2R4 template, as might be expected from its high sequence and structural similarity compared to other class A GPCRs. Although, the ordering by charged interhelical (Cih) or total and charged interhelical (CNti) energies gave a different top 10, we used the Nih ordering because Coulomb interactions between charged residues can be overestimated, resulting in unfavorable conformations.
In the previous study on TAS2R4, we found that the TM1-2-7 interaction in TAS2Rs is highly conserved but quite different from other class A GPCRs, for which the well-conserved N1.50, R2.50 and S7.50 forms a hydrogen bond network,29 where the superscript is Ballesteros-Weinstein GPCR numbering.30 Moreover, we found that the polar residues at 3.36 and 6.48 positions play very important roles in ligand binding and activation processes. The best structure by Nih ordering showed a consistent result with the previous study (Fig. S1). In marked contrast, for TAS2R5, the best structures by Cih and CNti ordering show unfavorable TM1-2-7 or TM3–6 interactions due to possibly overestimation of Coulombic interactions. Thus, we selected the most energetically favorable structure by Nih ordering for the SuperCombiHelix step. The best 25 cases from SuperCombiHelix are shown in Table S3, all of which were used for the docking simulations to provide diversity.
We used DarwinDock complete sampling method31 to predict the optimum ligand structure to each of 25 7-TMD conformations of TAS2R5 predicted by GEnSeMBLE (Table S3) to understand the relation between the structure and signal transduction. DarwinDock considers ~50,000 poses spanning the possible binding regions to select 100 with optimum residue sidechains (see Methods). The experimental potencies and efficacies have been reported for 1,10-phenanthroline (compound T5–1) and 18 derivatives (denoted as T5–2 through T5–19, as in ref. 17) (Fig. 1). The derivatives were designed to examine the effects on binding of the tricyclic ring (T5–2 and T5–19), the nitrogen atoms (T5–3, T5–4, and T5–18), the addition of methyl groups (T5–5, T5–6, T5–7, and T5–12), additional polar groups (T5–8, T5–9, T5–10, T5–11, T5–13, T5–18), and large substituents (T5–14, T5–15, T5–16, T5–17). Experimental results showed that an o-quinone in T5–8 significantly improved affinity, whereas the loss of the tricyclic or both nitrogen atoms resulted in significant loss in potency.
Figure 1.
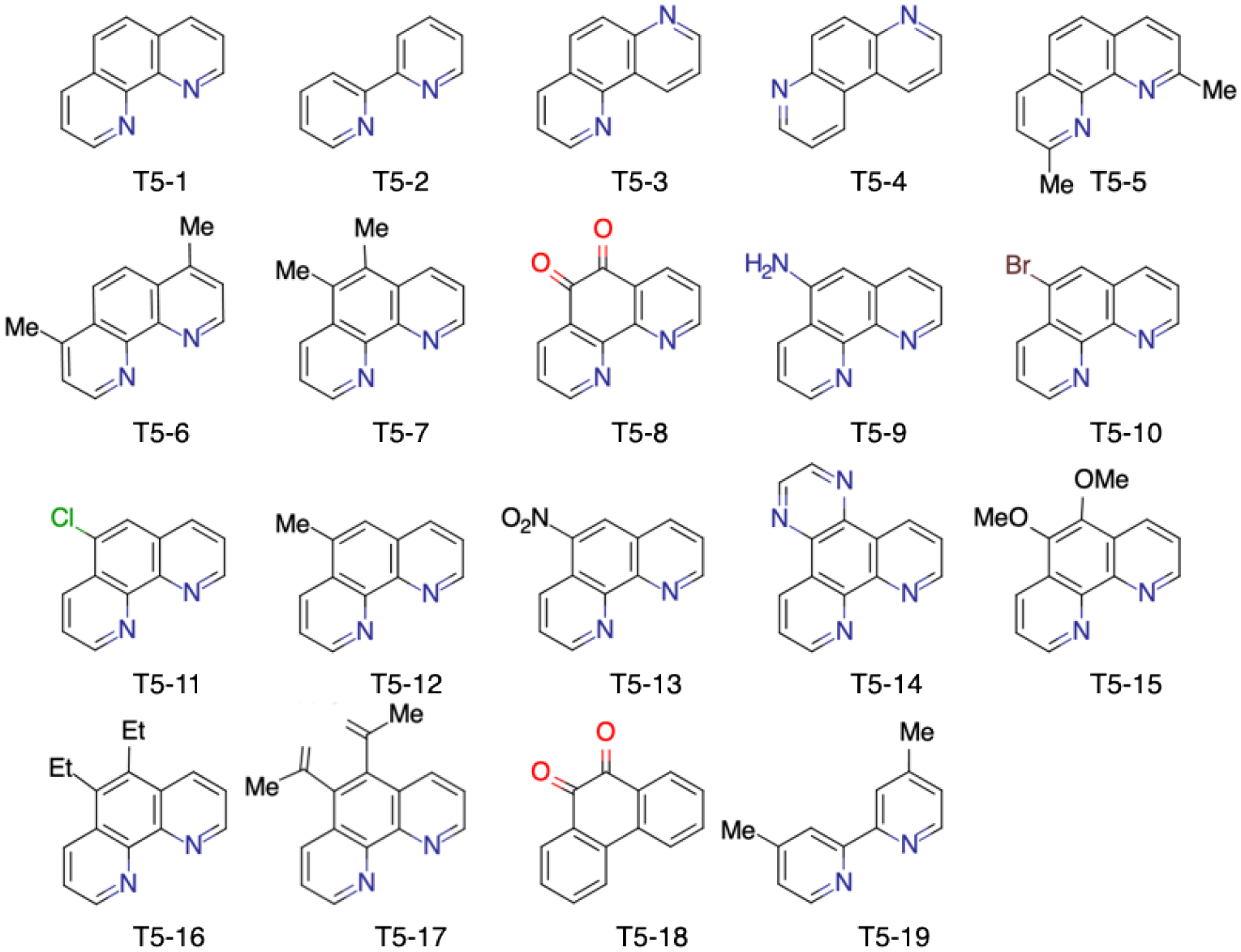
Structures of the 19 compounds examined in this study.
The binding poses for the 19 ligands deviate from each other depending on their sizes and functional groups, but most ligands share a similar binding site (Fig. S2A and S2B), with ~20 residues frequently involved in agonist binding (Fig. S2C and S2D). Since the parent agonist T5–1 activates only TAS2R5,16 we suspected that some important binding residues would be unique for TAS2R5. Fig. S3 compares those residues with sequence alignment across all 25 TAS2Rs. The result shows that S3.36, H6.48, and I6.52 are unique to TAS2R5, while Y5.39 appears twice, Q3.37 and S6.55 appear 3 times, and W7.38 and M7.42 appear 4 times, respectively, within the family. Indeed, we find that several TAS2R5-specific residues in TM3, such as the polar residues S893.36 and Q903.37, play an important role in the ligand recognition. Interestingly, D3.32 is particularly important for all biogenic amine receptors including β2AR and all opioid receptor agonists, and our previous study29 found that D923.36 plays an important role in ligand binding to TAS2R4. But, serine at the 3.36 position is unique to TAS2R5 and only 3 out of 25 TAS2Rs have glutamine at the 3.37 position, suggesting that these residues may be important for providing subtype specificity to TAS2R5, beyond the current family of agonists. The docking simulations find that S893.36 and Q903.37 interact with nitrogen in the pharmacophore (Fig. S4A) or with an additional polar group (Fig. S4B) through hydrogen bonds (HBs).
The calculated interaction energies from docking simulations for all 19 ligands are summarized in Table S4, and were compared with the experimentally obtained signal transduction coefficients, expressed as log(RMax/EC50),32 in Fig. S5. For this correlation, T5–2 and T5–4 were excluded because the responses to these compounds were minimal and the dose-response could not be fit to a sigmoid curve (Table S5).17
Fig. 2A shows the correlation between the average binding energies from MD simulations and experimentally obtained signal transduction coefficients for 17 cases, where the details of the MD simulations are described in the following sections and Methods. We find a remarkable correlation between binding energies and signaling for 12 ligands. Five ligands were excluded a priori from the final analysis for the following reasons:
T5–13 has a significantly higher binding energy than would be expected from the activation coefficient (3 orders of magnitude left-shifted). It contains a strongly electron-withdrawing nitro group on the convex side, which we find leads to strong polar interactions with positively charged residues near the binding site R552.50 or R793.26 from docking simulation (Fig. S6), or possibly residues in the ECLs (R76, R159, and K245). However, R552.50 forms a stable HB network with N1.50 and S7.50, providing the TM1-2-7 interaction in the active state.27 The interaction with R793.26 is located significantly higher than the binding site for the other agonists, which may make it less favorable for activation. Thus, we expect there may be an energy “cost” in repositioning to the proper binding site for activation, so that predicted binding energy may not correlate with activation.
The bulker ligands (T5–14, −15, −16, −17) lead to large energy gains mainly via hydrophobic interactions, causing them to rotate or translate significantly toward TM5 and away from TM6 compared to the other 12 ligands (Fig. 2B). Since the position relative to TM6 plays an important role in activation, we suspect that these 4 agonists must move to the location of the other 12 before activation, so that their higher binding does not translate to GiP activation. Indeed this change in the binding site leads to additional interactions with residues in TM5, increasing the predicted binding energy (> 8 kcal/mol) compared to T5–1 or T5–8 (~3 kcal/mol) (Table S6). Therefore, we expect that these bulky ligands they may lose the favorable binding in order to position themselves to activate the GiP.
Figure 2.
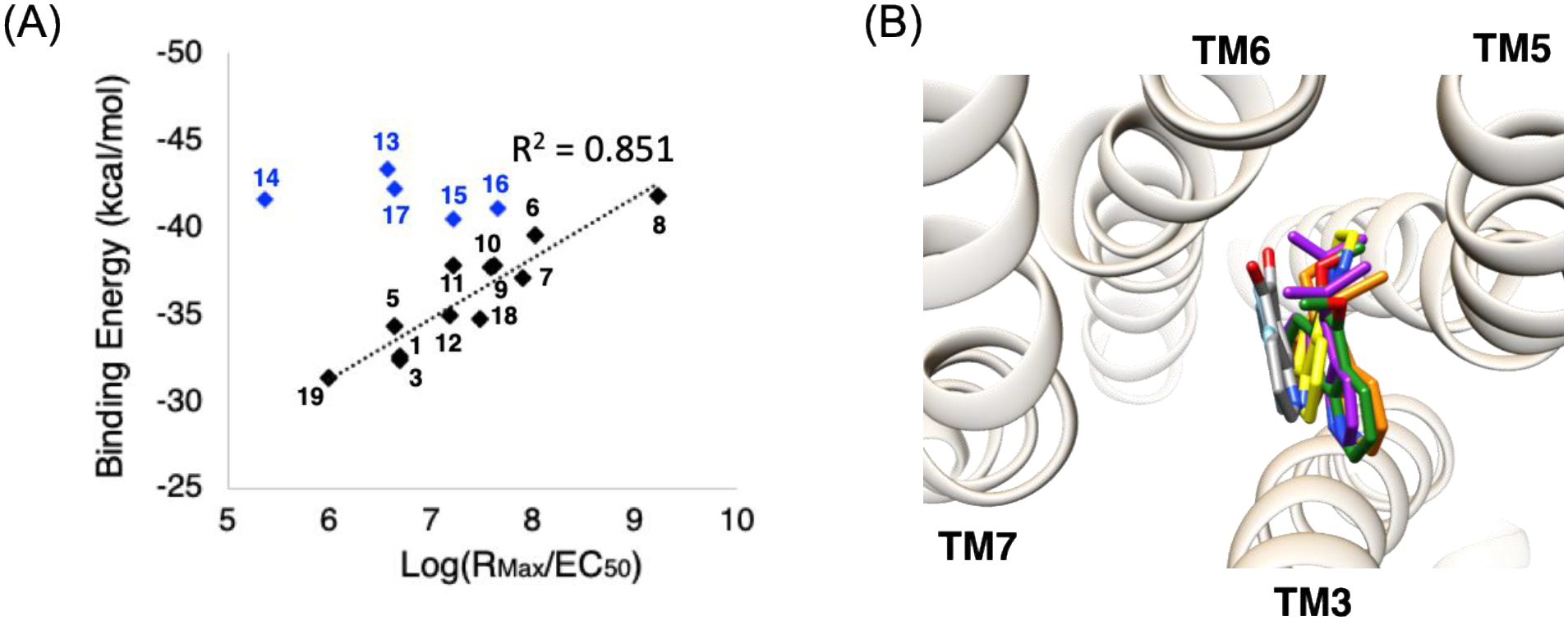
(A) The correlation between the average binding energy from 20 ns of MD simulations (average of 3 runs) and the experimental signal transduction coefficients. The black dotted trend line is to 12 cases, which excludes 5 outliers (blue symbols, see text). This fit leads to an R2=0.851. (B) Four outliers [T5–14 (yellow), T5–15 (green), T5–16 (orange), and T5–17 (purple)] are much bulkier and more hydrophobic, causing them to rotate or translate significantly toward TM5 and away from TM6, compared to the other 12 ligands, with T5–8 (gray) and T5–1 (sky blue) are shown for comparison. This modified binding site for these bulky agonists may not be suitable for activating the GP, so that the predicted binding energy fails to correlate with activity.
These outliers with the nitro group or bulky functional groups may play as allosteric ligands, and possibly they are in the so-called vestibular site predicted for TAS2R46.33 After excluding the aforementioned outliers, the remaining 12 lead to an excellent correlation of predicted binding energy with experimental activation, with R2 = 0.856, indicating a structure-activity relationship that is consistent with our predicted protein structure and binding site. Thus, the predicted binding energy appears to be an adequate quantitative assessment of signal transduction for most agonists. This ligand binding is dominated by S893.36 and Q903.37 through HBs with the two nitrogen atoms plus additional functional groups on the opposite side that contribute to binding by interacting with residues in TM6 and TM7.
We note that we calculate here the binding for the fully activated agonist-receptor-GP complex, with the Gα in the open position that can release the GDP. This corresponds to the activated structures of all reported X-ray and cryo-EM structures which have an open Gα with no GDP. However, it may be that the process of agonist binding to the inactive GP to cause the Gα to open may also play a role in the overall kinetics. Here the agonist must bind to a site determined by the GPCR-GP structure with the inactive GP closed around the GDP (with the Ras-like and α-Helical domains bound tightly to the Gα). This may differ from the agonist binding energy of the fully activated agonist-receptor-GP complex.
Each TAS2R5-ligand complex obtained from GEnSeMBLE and DarwinDock was inserted into a lipid bilayer and water box after adding all loops and intracellular helix 8. Since binding of both agonist and the cognate GP is required to fully activate the GPCR, we further attached the heterotrimeric GiP complex with the open Gα subunit and the GDP removed to obtain the fully activated ligand-TAS2R5-GiP complex. Although Ggust is the GP involved for taste receptors in the oral tissue, it has been reported that Gαi1,2,3 protein couples to TAS2Rs expressed in HASM cells.14 Among these, Gαi2 protein is known to be the most highly expressed, thus we used it as the Gα subunit in our study. The constructed fully activated ligand-TAS2R5-GiP complex was then equilibrated for 100 ns.
Fig. 3A shows the equilibrated structure of TAS2R5-GiP with T5–8 agonist immersed in the lipid bilayer and solvent. The agonist binding site is based on the docked structure for T5–1 (Fig. S4A) in which S893.36 and Q903.37 made HBs with nitrogen atoms. Although these two residues also can interact with the oxygen atoms, the binding pose was energetically less favorable and unstable during the simulation. Therefore, the nitrogen atoms at 1,10 positions are likely recognized by S893.36 and Q903.37, providing the subtype specificity. The root mean square deviation (RMSD) plots for 100 ns equilibration show convergence and stabilization of the system (Fig. 3B).
Figure 3.
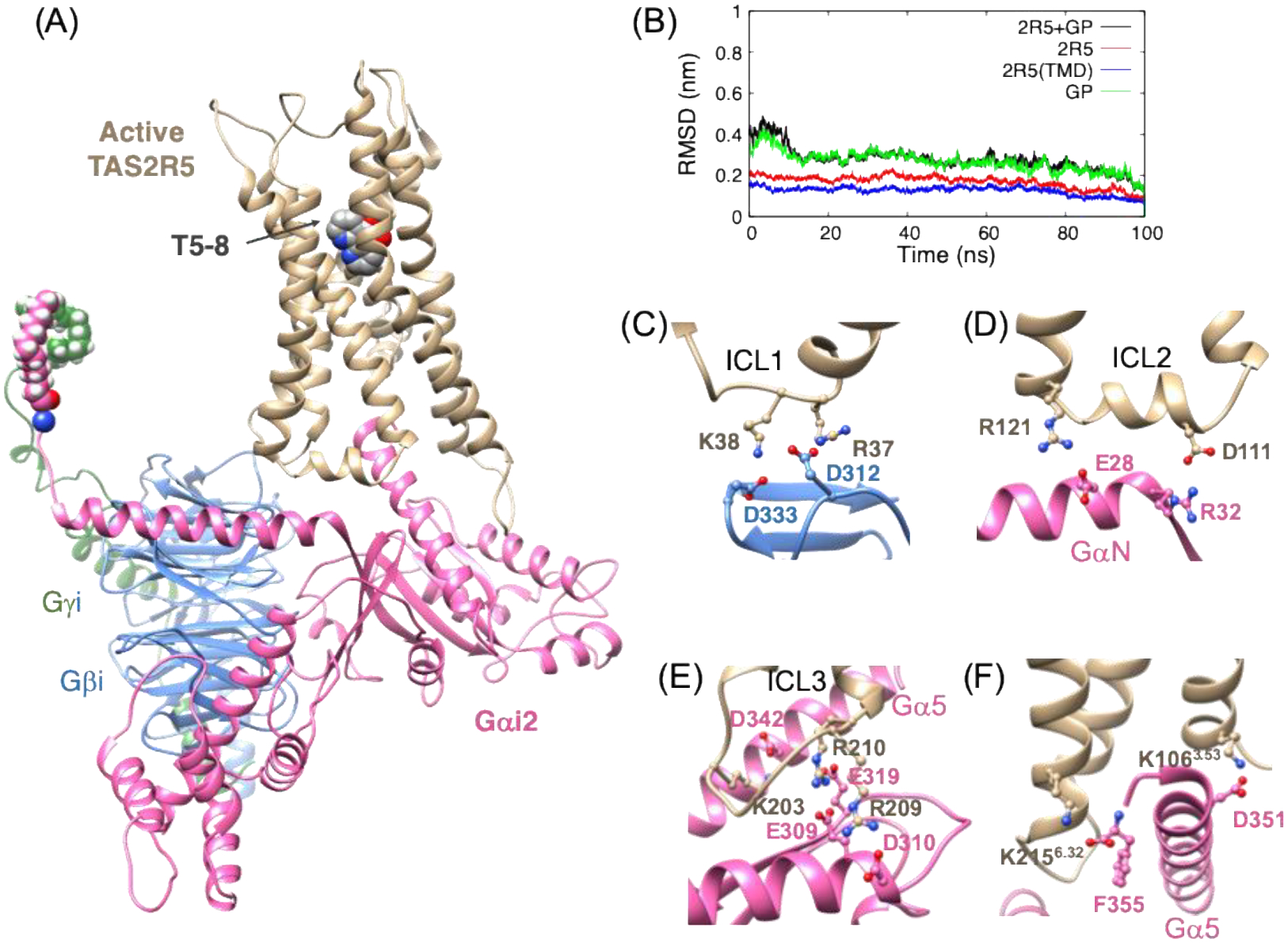
(A) The 100 ns equilibrated structure of the TAS2R5-GiP complex with bound T5–8 agonist. (B) The RMSD for backbone atoms from 100 ns MD simulations (reference to the final structure). Important polar interactions at three anchors: (C) Anchor 1: ICL1 with Gβ, (D) Anchor 2: ICL2 and GαN, and (E) Anchor 3: ICL3 and Gα. (F) Polar interactions between the Gα5 helix of the Gα subunit with the intracellular region of TAS2R5.
In our recent studies of μOR and κOR coupled to GiP, we discovered that these class A GPCRs bind strongly to the GiP via SB and HB interactions to all three ICLs.34 These interactions had not been identified in the cryo-EM experiments because of low resolution, disorder, and 140 unresolved residues. Not only we found earlier that Ggust makes similar interactions for TAS2R4,29 but also we observed similar interactions from recently solved CCR5- and 5HT1D-GiP complex structure.35,36 This suggests that these strong interactions between GPCR and GP are a common feature for class A GPCRs and TAS2Rs. For the TAS2R5-GiP complex structure, we found similar interactions:
Anchor 1: two negative residues in the Gβ subunit form SBs with ICL1: R37-ICL1 – D312-Gβ and K38-ICL1 – D333-Gβ (Fig. 3C).
Anchor 2: one negative and one positive residue in the GαN subunit form SBs with residues in ICL2: D111-ICL2 – R32-GαN and R121-ICL2 – E28-GαN (Fig. 3D).
Anchor 3: one lysine and two arginine residues in ICL3 couple with the Gα5 helix and with the Ras-like domain of the Ga subunit, K203-ICL3 – D342-Gα, R209-ICL3 – D310-Gα, and R210-ICL3 – E319-Gα (Fig. 3E).
The insertion of the Gα5 helix of the Gα subunit into the intracellular region of GPCR is a key event during the activation.37–39 Indeed, we found that the Gα5 helix forms a network of HBs and SBs to the GPCR for opioid receptors and TAS2R4.29,34 For opioid receptors, we found that key interactions are a SB from D350-Gα5 to a conserved arginine residue in ICL2 (R170 and R179 for μOR and κOR respectively). For TAS2R4, this D350 forms a SB with a K3.53 in TM3. This 3.53 position is highly conserved for all TAS2Rs being a positively charged residue, lysine or arginine. For TAS2R5, K1063.53 also forms a SB with D351-Gα5 (Fig 3F), which corresponds to D350-Gα5 for Giα1 or Ggust. For opioid receptors, moreover, we reported that the terminal carboxylate of Gα5 (F354) plays an important role in positioning of the Gα5 helix by making a SB with a R6.32 residue in TM6 while for TAS2R4 the terminal end of F354 forms a SB with K6.39. For TAS2R5, the terminal carboxylate of Gα5 (F355) forms a SB with K2156.32 (Fig. 3F).
To investigate the interaction between TAS2R5 and each of 19 ligands, we carried out 20 ns MD simulations after the 100 ns equilibration step. Fig. 4A and 4B show T5–8 (and T5–1) interacting with residues in the binding site. For T5–8, S893.36 and Q903.37 form HBs with the two nitrogen atoms of the ligand, while S2386.55 and W2577.38 form HBs with one of the ligand oxygen atoms. These HBs were very stable for the 20 ns MD simulation (Fig. 4C and 4D) and responsible for a large portion of the Coulombic contribution to the binding energy (Fig. 4E and Table S6). T5–1 with no o-quinone makes HBs only with S893.36 and Q903.37 (Fig. 4A and 4D), resulting in significantly decreased level of absolute Coulombic energy (by > 10 kcal/mol) (Table 1 and Fig. 4F). Moreover, the HB of T5–8 with W2577.38 induces significant structural changes (and rotation) in TM7 (Fig. 4B), which may promote activation (rotation of TM7 is one of the structural changes upon activation). These interaction energies and the HB with TM7 explain the experimentally observed large difference in EC50; T5–8 (0.12 mM) vs. T5–1 (30 mM).
Figure 4.
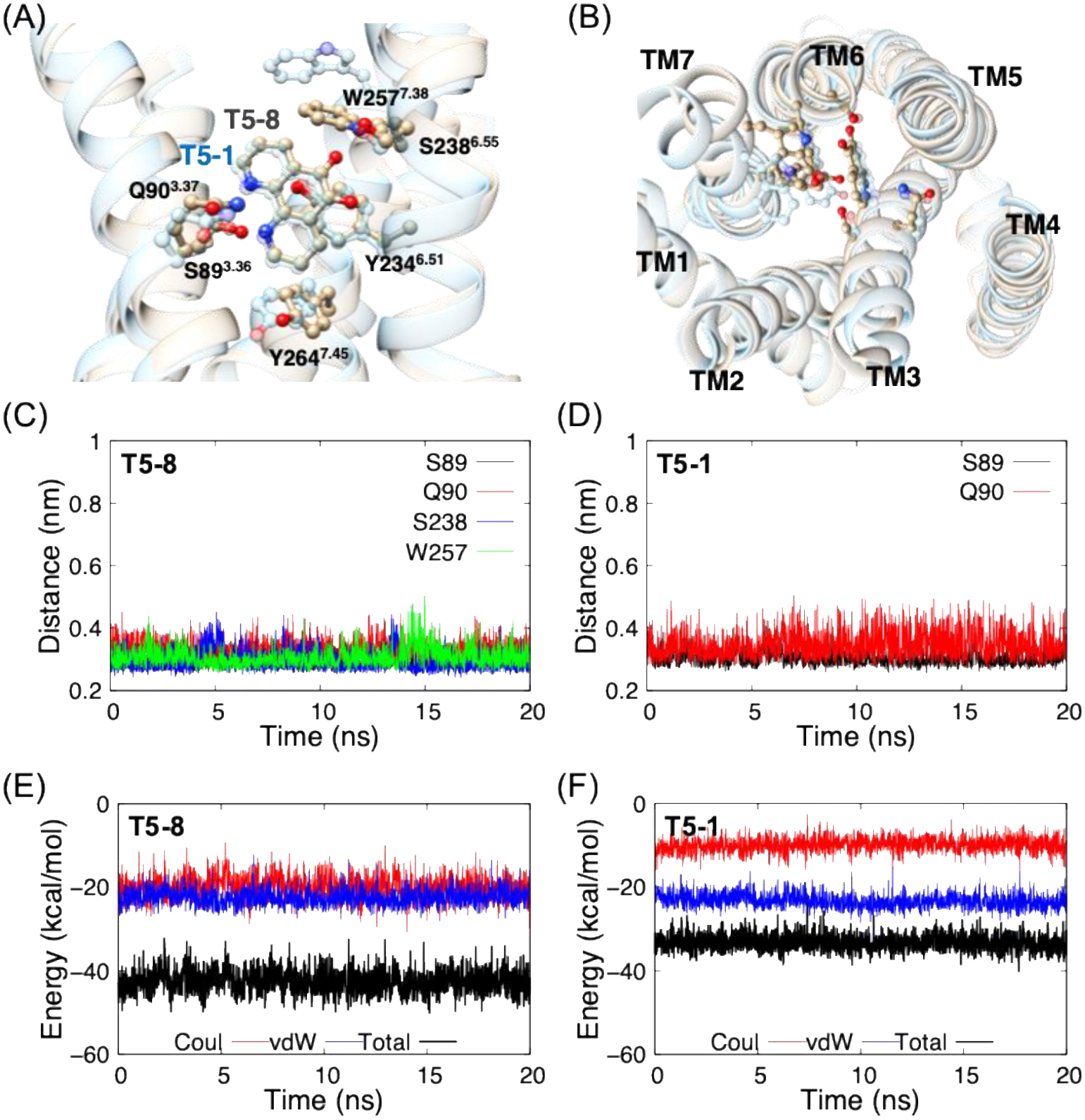
Binding site of TAS2R5 with T5–8 (and T5–1; transparent sky blue) from (A) side view and (B) extracellular view, where the ECL’s and GiP are omitted for clarity. HB distances for (C) T5–8 and (D) T5–1, and the interaction energies between TAS2R5 and (E) T5–8 and (F) T5–1.
Table 1.
Average interaction energies between TAS2R5 and agonist from 20 ns MD simulations ordered by predicted total binding energy, where the 5 cases highlighted in red [T5–13 through T5–17] are considered outliers. The binding energies are average value of 3 runs of MD simulations, and the interaction energies of each run are shown in Fig. S7, S8, and S9, respectively.
| Agonist | Binding energy (kcal/mol) | |||
|---|---|---|---|---|
| Total | Coulomb | vdW | ||
| T5–13 | 5-nitro-1,10-phenanthroline | −43.33 ± 0.3 | −17.54 ± 0.86 | −25.8 ± 0.58 |
| T5–17 | 1,10-phenanthroline-5,6-dione | −42.18 ± 0.26 | −12.98 ± 0.15 | −29.19 ± 0.41 |
| T5–8 | 5,6-bis(prop-1-en-2yl)-1,10-phenanthroline | −41.86 ± 0.91 | −19.50 ± 0.58 | −22.37 ± 0.5 |
| T5–14 | pyrazino[2,3-f] [1,10]phenanthroline | −41.52 ± 0.63 | −16.65 ± 1.19 | −24.87 ± 0.6 |
| T5–16 | 5,6-dimethoxy-1,10-phenanthroline | −41.03 ± 1.02 | −12.18 ± 0.29 | −28.85 ± 0.75 |
| T5–15 | 4,7-dimethyl-1,10-phenanthroline | −40.49 ± 0.42 | −14.18 ± 0.5 | −26.21 ± 0.1 |
| T5–6 | 5,6-diethyl-1,10-phenanthroline | −39.57 ± 0.41 | −12.65 ± 0.34 | −26.92 ± 0.57 |
| T5–11 | 5-bromo-1,10-phenanthroline | −37.88 ± 0.68 | −12.08 ± 0.23 | −25.81 ± 0.6 |
| T5–10 | 1,10-phenanthrolin-5-amine | −37.81 ± 0.43 | −12.12 ± 0.62 | −25.69 ± 0.51 |
| T5–9 | 5,6-dimethyl-1,10-phenanthroline | −37.68 ± 0.21 | −13.42 ± 0.58 | −24.27 ± 0.39 |
| T5–7 | 5-chloro-1,10-phenanthroline | −37.08 ± 0.55 | −11.96 ± 0.27 | −25.12 ± 0.4 |
| T5–12 | 5-methyl-1,10-phenanthroline | −35.08 ± 0.2 | −11.7 ± 0.13 | −23.32 ± 0.15 |
| T5–18 | 9,10-phenanthrenequinone | −34.81 ± 0.45 | −9.12 ± 0.25 | −25.69 ± 0.35 |
| T5–5 | 1,10-phenanthroline | −34.40 ± 1.91 | −6.81 ± 2.46 | −26.09 ± 0.12 |
| T5–1 | 1,7-phenanthroline | −32.64 ± 0.51 | −10.84 ± 0.8 | −21.81 ± 1.31 |
| T5–3 | 4,4′-dimethyl-2,2′-bipyridine | −32.38 ± 0.64 | −10.6 ± 0.15 | −21.78 ± 0.5 |
| T5–19 | 2,9-dimethyl-1,10-phenanthroline | −31.43 ± 0.23 | −7.95 ± 0.1 | −23.48 ± 0.13 |
| T5–4 | 4,7-phenanthroline | −26.04 ± 0.63 | −6.1 ± 0.29 | −19.94 ± 0.64 |
| T5–2 | 2,2′-bipyridine | −24.39 ± 1.66 | −4.8 ± 1.94 | −19.58 ± 0.28 |
The HB’s of S893.36 and Q903.37 to the two nitrogen atoms at the inside of the ligands provide key interactions for ligand recognition and for high affinity. The shift of these nitrogen atoms from (1, 10) to (4, 7) positions (T5–4) loses these HBs, significantly decreasing the binding energy. The tricyclic ring appears to be important for keeping those HBs, as shown by the decrease in binding (and receptor activation in cells) of T5–2 and T5–19, with only the bipyridine (Fig. S7). Additional chemical modifications at the (5, 6) position can increase the binding energy by allowing additional interactions with residues in TM6 and TM7. Thus polar groups (T5–8, −9, −10, −11) and hydrophobic groups (T5–5, −6, −7, −12) lead to energy gains from Coulombic and van der Waals (vdW) interactions, respectively. It should be noted that the modification with a bulky or a strong electron-withdrawing functional group may lead to a significant distortion of the binding site and/or inadequate binding, making it less favorable for activation despite the larger binding energy. The interaction energies for all 19 ligands were summarized in Table 1, Table S6, and Fig. S7.
We have recently reported that six TAS2Rs (TAS2Rs 4, 5, 10, 14, 19, and 31) are expressed on HASM cells and that activation of any one of these leads to marked cell relaxation, which is equal to or greater than that from β-agonists acting at β2AR on the same cell type.1,14 This suggests the use of TAS2R agonists as potential treatments for obstructive lung diseases such as asthma, since HASM cell contraction is a main mechanism of airflow restriction. To date, β-agonists are the only available direct bronchodilators for treating obstructive lung diseases. However, these agonists are associated with a number of non-trivial adverse effects (reviewed in ref. 17), and indeed about half of asthmatics fail to reach optimal control.40 Since agonists targeting TAS2Rs on airway smooth muscle cells provides high efficacy for relaxation of bronchi,41 with a distinct mechanism of action,1 it has been proposed that such agonists could be used as first-line, or add-on (with β-agonists) therapy for treating asthma and COPD.1,14 However, overcoming the typical low potency of TAS2R agonists using empirical means without the structural basis has failed to provide highly receptor-specific, low nM affinity, agonists for airway TAS2Rs.18
In this study, we predicted the 3D structure of the fully activated TAS2R5 human bitter taste receptor by using the GEnSeMBLE complete sampling of 13 trillion 7-helix packings, leading to 25 7-helix packings that we consider to be thermally accessible. Then DarwinDock complete sampling was utilized to predict the best binding sites for each of 19 agonists to each of the 25 7-helix packings to identify the best binding sites and helix-packing for each.
We coupled the TAS2R5-agonist complex to the GiP for each of the 19 agonists in their most optimal binding state to obtain the fully activated agonist-TAS2R5-GiP complexes. Just as for TAS2R4 and the κOR, we find that GiP forms stable SBs with all three ICLs of TAS2R5 while the Gα5 helix makes extensive interactions with residues in the cytoplasmic region of the GPCR.
Based on docking and MD simulations, we identified the binding sites and binding poses for 1,10-phenanthroline and 18 derivatives. We find that TAS2R5-specific S893.36 and Q903.37 residues play important roles in the ligand recognition and binding by forming HBs with the two nitrogen atoms on the inside of the phenanthroline ring. Moreover, for the most potent agonist, T5–8, we find additional stable HBs of o-quinone with S2386.55 and W2577.38. This explains the subtype selectivity and high potency and efficacy of the most active ligand in the series. Although no study has examined these key residues experimentally to date, these positions are also known to involve in ligand binding for other TAS2Rs or class A GPCRs.42 To our knowledge the ascertainment of such highly specific binding site predictions coupled with experimentally derived activation coefficients has not been performed with an array of agonists with such similarities in structure that we have used here. This has now provided the opportunity to model agonist-receptor interactions with an emphasis on the identified binding sites using very large libraries of compounds in silico to potentially further improve potency.
Supplementary Material
ACKNOWLEDGMENT
This research was supported by grants from the NIH (1R01HL155532 and P01HL114471). We thank Dr. Amirhossein Mafi for helpful discussions. The computational resource for this research was provided by KISTI National Supercomputing Center (KSC-2018-CHA-0049).
Footnotes
Supporting Information.
The following files are available free of charge.
Details of the GEnSeMBLE calculation, Darwindock simulations, and MD simulations (PDF)
The authors declare no competing financial interests.
REFERENCES
- (1).Deshpande DA; Wang WC; McIlmoyle EL; Robinett KS; Schillinger RM; An SS; Sham JSK; Liggett SB Bitter taste receptors on airway smooth muscle bronchodilate by localized calcium signaling and reverse obstruction. Nat. Med 2010, 16, 1299–1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Finger TE; Bottger B; Hansen A; Anderson KT; Alimohammadi H; Silver WL Solitary chemoreceptor cells in the nasal cavity serve as sentinels of respiration. Proc. Natl. Acad. Sci. U. S. A 2003, 100, 8981–8986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Wu SV; Rozengurt N; Yang M; Young SH; Sinnett-Smith J; Rozengurt E Expression of bitter taste receptors of the T2R family in the gastrointestinal tract and enteroendocrine STC-1 cells. Proc. Natl. Acad. Sci. U. S. A 2002, 99, 2392–2397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Foster SR; Blank K; See Hoe LE; Behrens M; Meyerhof W; Peart JN; Thomas WG Bitter taste receptor agonists elicit G-protein-dependent negative inotropy in the murine heart. FEBS J. 2014, 28, 4497–4508. [DOI] [PubMed] [Google Scholar]
- (5).Clark AA; Dotson CD; Elson AET; Voigt A; Boehm U; Meyerhof W; Steinle NI; Munger SD TAS2R bitter taste receptors regulate thyroid function. FASEB J. 2015, 29, 164–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Singh N; Vrontakis M; Parkinson F; Chelikani P Functional bitter taste receptors are expressed in brain cells. Biochem. Biophys. Res. Commun 2011, 406, 146–151. [DOI] [PubMed] [Google Scholar]
- (7).An SS; Liggett SB Taste and smell GPCRs in the lung: Evidence for a previously unrecognized widespread chemosensory system. Cell Signal. 2018, 41, 82–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Liggett SB Bitter taste receptors on airway smooth muscle as targets for novel bronchodilators. Expert Opin. Ther. Targets 2013, 17, 721–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Martin LTP; Nachtigal MW; Selman T; Nguyen E; Sasman J; Dellaire G; Dupré DJ Bitter taste receptors are expressed in human epithelial ovarian and prostate cancers cells and noscapine stimulation impacts cell survival. Mol. Cell. Biochem 2019, 454, 203–214. [DOI] [PubMed] [Google Scholar]
- (10).Dotson CD; Zhang L; Xu H; Shin YK; Vigues S; Ott SH; Elson AE; Choi HJ; Shaw H; Egan JM; Mitchell BD; Li X; Steinle NI; Munger SD Bitter taste receptors influence glucose homeostasis. PLoS One 2008, 3, e3974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Yoon S-Y; Shin E-S; Park SY; Kim S; Kwon H-S; Cho YS; Moon H-B; Kim T-B Association between Polymorphisms in Bitter Taste Receptor Genes and Clinical Features in Korean Asthmatics. Respiration 2016, 91, 141–150. [DOI] [PubMed] [Google Scholar]
- (12).Zborowska-Piskadło K; Stachowiak M; Rusetska N; Sarnowska E; Siedlecki J; Dżaman K The expression of bitter taste receptor TAS2R38 in patients with chronic rhinosinusitis. Arch. Immunol. Ther. Exp. (Warsz) 2020, 68, 26. [DOI] [PubMed] [Google Scholar]
- (13).Shiffman D; O’Meara ES; Bare LA; Rowland CM; Louie JZ; Arellano AR; Lumley T; Rice K; Iakoubova O; Luke MM; Young BA; Malloy MJ; Kane JP; Ellis SG; Tracy RP; Devlin JJ; Psaty BM Association of gene variants with incident myocardial infarction in the cardiovascular health study. Arterioscler. Thromb. Vasc. Biol 2008, 28, 173–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Kim D; Woo JA; Geffken E; An SS; Liggett SB Coupling of airway smooth muscle bitter taste receptors to intracellular signaling and relaxation is via Gαi1,2,3. Am. J. Respir. Cell Mol. Biol 2017, 56, 762–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Behrens M; Meyerhof W Vertebrate Bitter Taste Receptors: Keys for Survival in Changing Environments, J. Agric. Food Chem 2018, 66, 2204–2213. [DOI] [PubMed] [Google Scholar]
- (16).Meyerhof W; Batram C; Kuhn C; Brockhoff A; Chudoba E; Bufe B; Appendino G; Behrens M The molecular receptive ranges of human TAS2R bitter taste receptors. Chem. Senses 2010, 35, 157–170. [DOI] [PubMed] [Google Scholar]
- (17).Kim D; An SS; Lam H; Leahy JW; Liggett SB Identification and characterization of novel bronchodilator agonists acting at human airway smooth muscle cell TAS2R5. ACS Pharmacol. Transl. Sci 2020, 3, 1069–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Di Pizio A; Waterloo LAW; Brox R; Löber S; Weikert D; Behrens M; Gmeiner P; Niv MY Rational design of agonists for bitter taste receptor TAS2R14: from modeling to bench and back. Cell. Mol. Life Sci 2020, 77, 531–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Abrol R; Bray JK; Goddard WA III Bihelix: Towards de novo structure prediction of an ensemble of G-protein coupled receptor conformations. Proteins 2012, 80, 505–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Bray JK; Abrol R; Goddard WA III; Trzaskowski B; Scott CE SuperBiHelix method for predicting the pleiotropic ensemble of G-protein–coupled receptor conformations. Proc. Natl. Acad. Sci. U. S. A 2014, 111, E72–E78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Abrol R; Trzaskowski B; Goddard WA III; Nesterov A; Olave I; Irons C Ligand- and mutation-induced conformational selection in the CCR5 chemokine G protein-coupled receptor. Proc. Natl. Acad. Sci. U. S. A 2014, 111, 13040–13045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Berro R; Yasmeen A; Abrol R; Trzaskowski B; Abi-Habib S; Grunbeck A; Lascano D; Goddard WA III; Klasse PJ; Sakmar TP; Moore JP Use of G-protein-coupled and -uncoupled CCR5 receptors by CCR5 inhibitor-resistant and -sensitive human immunodeficiency virus type 1 variants. J. Virol 2013, 87, 6569–6581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Goddard WA III; Kim S-K; Li Y; Trzaskowski B; Griffith AR; Abrol R Predicted 3D structures for adenosine receptors bound to ligands: Comparison to the crystal structure. J. Struct. Biol 2010, 170, 10–20. [DOI] [PubMed] [Google Scholar]
- (24).Shankar V; Goddard WA III; Kim S-K; Abrol R; Liu F The 3D structure of human DP prostaglandin G-protein-coupled receptor bound to cyclopentanoindole antagonist, predicted using the DuplexBiHelix modification of the GEnSeMBLE method. J. Chem. Theory Comput 2018, 14, 1624–1642. [DOI] [PubMed] [Google Scholar]
- (25).Scott CE; Abrol R; Ahn KH; Kendall DA; Goddard WA III Molecular basis for dramatic changes in cannabinoid CB1 G protein-coupled receptor activation upon single and double point mutations. Protein Sci. 2013, 22, 101–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Ahn KH; Scott CE; Abrol R; Goddard WA III; Kendall DA Computationally-predicted CB1 cannabinoid receptor mutants show distinct patterns of salt-bridges that correlate with their level of constitutive activity reflected in G protein coupling levels, thermal stability, and ligand binding. Proteins 2013, 81, 1304–1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Kirkpatrick A; Heo J; Abrol R; Goddard WA III Predicted structure of agonist-bound glucagon-like peptide 1 receptor, a class B G protein-coupled receptor. Proc. Natl. Acad. Sc.i U. S. A 2012, 109, 19988–19993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Kim S-K; Chen Y; Abrol R; Goddard WA III; Guthrie B Activation mechanism of the G protein-coupled sweet receptor heterodimer with sweeteners and allosteric agonists. Proc. Natl. Acad. Sci. U. S. A 2017, 114, 2568–2573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Yang MY; Mafi A; Kim S-K; Goddard WA III; Guthrie B Predicted structure of fully activated human bitter taste receptor TAS2R4 complexed with G protein and agonists. QRB Discovery 2021, 2, e3 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Ballesteros JA; Weinstein H Integrated methods for the construction of three- dimensional models and computational probing of structure-function relations in G protein-coupled receptors. Methods. Neurosci 1995, 25, 366–428. [Google Scholar]
- (31).Griffith AR DarwinDock & GAG-Dock: Methods and Applications For Small Molecule Docking, PhD thesis, California Institute of Technology, 2017. [Google Scholar]
- (32).Kenakin T; Watson C; Muniz-Medina V; Christopoulos A; Novick S A simple method for quantifying functional selectivity and agonist bias. ACS Chem. Neurosci 2012, 3, 193–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Sandal M; Behrens M; Brockhoff A; Musiani F; Giorgetti A; Carloni P; Meyerhof W Evidence for a Transient Additional Ligand Binding Site in the TAS2R46 Bitter Taste Receptor. J. Chem. Theory Comput 2015, 11, 4439–4449. [DOI] [PubMed] [Google Scholar]
- (34).Mafi A; Kim S-K; Goddard WA III The atomistic level structure for the activated human κ-opioid receptor bound to the full Gi protein and the MP1104 agonist. Proc. Natl. Acad. Sci. U. S. A 2020, 117, 5836–5843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Isaikina P; Tsai C-J; Dietz N; Pamula F; Grahl A; Goldie KN; Guixà-González R; Branco C; Paolini-Bertrand M; Calo N; Cerini F; Schertler GFX; Hartley O; Stahlberg H; Maier T; Deupi X; Grzesiek S Structural basis of the activation of the CC chemokine receptor 5 by a chemokine agonist. Sci. Adv 2021, 7, eabg8685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Xu P; Huang S; Zhang H; Mao C; Zhou XE; Cheng X; Simon IA; Shen D-D; Yen H-Y; Robinson C-V; Harpsøe K; Svensson B; Guo J; Jiang H; Gloriam DE; Melcher K; Jiang Y; Zhan Y; Xu HE Structural insights into the lipid and ligand regulation of serotonin receptors. Nature 2021, 592, 469–473. [DOI] [PubMed] [Google Scholar]
- (37).Onrust R; Herzmark P; Chi P; Garcia PD; Lichtarge O; Kingsley C; Bourne HR Receptor and betagamma binding sites in the α subunit of the retinal G protein transducin. Science 1997, 275, 381–384. [DOI] [PubMed] [Google Scholar]
- (38).Oldham WM; Van Eps N; Preininger AM; Hubbell WL; Hamm HE Mechanism of the receptor-catalyzed activation of heterotrimeric G proteins. Nat. Struct. Mol. Biol 2006, 13, 772–777. [DOI] [PubMed] [Google Scholar]
- (39).Oldham WM; Hamm HE Heterotrimeric G protein activation by G-protein-coupled receptors. Nat. Rev. Mol. Cell. Biol 2008, 9, 60–71. [DOI] [PubMed] [Google Scholar]
- (40).Malmstrom K; Rodriguez-Gomez G; Guerra J; Villaran C; Pineiro A; Wei LX; Seidenberg BC; Reiss TF Oral Montelukast, inhaled beclomethasone, and placebo for chronic asthma. Ann. Intern. Med 1999, 130, 487–495. [DOI] [PubMed] [Google Scholar]
- (41).Deshpande DA; Robinett KS; Wang WC; Sham JS; An SS; Liggett SB Bronchodilator activity of bitter tastants in human tissue. Nat. Med 2011, 17, 776–778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Di Pizio A; Levitx A; Slutzki M; Behrens M; Karamanjj R; Niv MY Comparing Class A GPCRs to bitter taste receptors: Structural motifs, ligand interactions and agonist-to-antagonist ratios. Method. Cell Biol 2016, 132, 401–427. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.