

Abstract
The ability to detect and monitor infectious disease in a phylogenetically informative manner is critical for their management. Phylogenetically informative diagnostic tests enable patterns of pathogen introduction or changes in the distribution of genotypes to be measured, enabling research into the ecology of the pathogen. Batrachochytrium dendrobatidis (Bd), a causative agent of chytridiomycosis in amphibian populations, emerged worldwide in the 21st century and is composed of six lineages which are display varying levels of virulence in their hosts. Research into the distribution, ecology and pathogenicity of these lineages has been hampered by an inability to type lineage efficiently. Here, we describe a lineage‐specific TaqMan qPCR assay that differentiates the two lineages of Bd most commonly associated with chytridiomycosis: BdGPL and BdCAPE. We demonstrate how this assay can be used for the surveillance of wild populations of amphibians in Southern Africa using skin swabs, tissue samples and cultured isolates.
Keywords: Batrachochytrium dendrobatidis, chytrid, diagnostic, lineage, qPCR
Short abstract
see also the Perspective by Claudio Azat
1. INTRODUCTION
Bd, a fungal pathogen of amphibians, causes the potentially lethal disease chytridiomycosis. This fungus is a member of the Chytridiomycota, and one of only two species within the phylum known to infect vertebrates (Longcore & Pessier, 1999; Martel et al., 2013). Bd has been detected infecting almost 700 species of amphibians, with chytridiomycosis implicated as the cause of population declines in over 500 species (Olson & Ronnenberg, 2014; Scheele et al., 2019). This has led to the extinction of an estimated 90 species of amphibians globally (Scheele et al., 2019).
Within Bd there are at least six phylogenetically distinct lineages – BdCAPE, BdGPL, BdASIA‐1, BdASIA‐2/BRAZIL, BdASIA‐3 and BdCH. However, BdCAPE and BdGPL are primarily associated with mass mortality events and species declines or extinctions (Byrne et al., 2019; Doddington et al., 2013; Farrer et al., 2011; Jenkinson et al., 2016; O’Hanlon et al., 2018). While only BdGPL has undergone a rapid, recent and global range expansion, recent research indicates that the other five lineages, which had previously been considered largely spatially restricted, have achieved intercontinental distributions. This raises the possibility of lineage contact and interactions occurring. One such interaction is interlineage genetic recombination (Byrne et al., 2019; Jenkinson et al., 2016; O’Hanlon et al., 2018) which may lead to increased virulence (Greenspan et al., 2018). Given the infrequency with which recombinants are isolated, molecular tools that enable the identification and monitoring of areas where they might occur are needed in order to focus research effort effectively. To date, this has been hampered by the lack of a suitable lineage‐specific diagnostic test for Bd.
Currently, a pan‐lineage qPCR assay targeting the ITS region of the genome is the most widely‐used method for detecting Bd (Boyle et al., 2004). This method has high specificity, is sensitive enough to detect subclinical infections, and works with swabs taken from adult amphibian skin or tadpole mouthparts, so is minimally invasive (Boyle et al., 2004; Garland et al., 2011; Hyatt et al., 2007; Kriger et al., 2006; Skerratt et al., 2011). A further advantage of this assay working with swab samples is that these are light, easy to carry in difficult field settings and are easy to store (Hyatt et al., 2007; Van Sluys et al., 2008). The primary drawback to this pan‐lineage qPCR assay is its inability to type the lineage of Bd present in a sample. Although there have been attempts to design lineage‐specific diagnostics based on the targeted genomic region, the ITS is unable to resolve interspecific phylogenies within Bd owing to a breakdown in concerted evolution across this multiallelic array (O’Hanlon et al., 2018; Schoch et al., 2012).
The most accurate method for determining lineage is whole genome sequencing (WGS) from a pure culture of Bd. Although comparative genomic analyses have been extremely valuable in the field of chytrid research, the expense and requirement for a pure isolate culture make this approach impractical on the large scale required to answer fundamental questions about Bd lineage distribution and ecology. The most reliable alternative approach to WGS, multilocus sequence typing (MLST) is suitable for use with swabs but requires access to a specialised technology, the Fluidigm Access Array platform, and a reasonably high infection burden (>150 genomic equivalents [GE]). It is, therefore, not applicable for all Bd positive cases, especially in hosts with a subclinical infection burden (Byrne et al., 2017).
Recent research has shown that phylogenetic analyses based on Bd mitochondrial DNA (mtDNA) recovers the same lineages as those recovered by WGS (O’Hanlon et al., 2018). The Bd mitochondrial genome has recently been fully sequenced, has a low rate of recombination relative the nuclear genome, and is present at high copy number so is likely to be detectable at low infection burdens. There is also a precedent for using mtDNA for phylogenetic studies across a wide range of taxa (Barr et al., 2011; Li et al., 2016; Penry et al., 2018; Schreeg et al., 2016; Song et al., 2016). Here, we report a lineage‐specific qPCR assay for Bd based on mtDNA which detects the two lineages of Bd most commonly associated with chytridiomycosis in wild populations – BdGPL and BdCAPE – with high specificity and sensitivity.
2. MATERIALS AND METHODS
2.1. Assay design and optimisation
A 175,295 kb long mtDNA alignment for 150 chytrid isolates, with 145 isolates of Bd representing BdGPL, BdCAPE, BdASIA‐1, BdASIA‐2/BRAZIL and BdCH, was manually screened for lineage‐specific single nucleotide polymorphisms (SNPs) (O’Hanlon et al., 2018). Candidate primers and probes were designed using Primer Express Software 3.0 (ThermoFisher Scientific).
We optimised primer and probe concentrations separately using a checkboard system in singleplex with DNA quantitation standards for the DNA template. We selected final primer and probe concentrations which maximised reaction efficiency compared to oligonucleotide concentration, while ensuring that the mean C t values for a given quantity of DNA remained as similar as possible between the BdCAPE and BdGPL reactions. Consistency between the reactions made it possible to multiplex the two qPCR reactions into a single assay diagnosing both BdGPL and BdCAPE. All optimisation and validation reactions were carried out on both a 7300 Real‐Time PCR system and a QuantStudio 7 Flex Real‐Time PCR system (Applied Biosystems).
The cycling conditions were identical to the pan‐lineage Bd diagnostic of Boyle et al. (2004), except that we reduced the number of cycles from 50 to 40 and increased the annealing temperature from 60 to 62°C in order to increase the specificity. We set the C t threshold for all reactions at ΔRn = 0.1, and the total reaction volume in all cases was 25 µl in total (20 µl master mix and 5 µl DNA template). The optimised multiplex reaction mix and full cycling conditions are provided in the Supporting Information. When the reactions were run in singleplex the same quantities of all reactions were used, except for an increase in the volume of filtered water commensurate with the quantity of primer and probe removed. We quantified the starting DNA present in a reaction using standard curve analysis calculated from DNA quantitation standards made from isolate IA042 (BdGPL) and isolate TF5a1 (BdCAPE) (Table S1). Both these isolates were previously typed to their respective lineages using WGS (O’Hanlon et al., 2018). The final primer and TaqMan MGB probe sequences are shown in Figure 1. We carried out pan‐lineage Bd detection using the protocol of Boyle et al. (2004).
FIGURE 1.
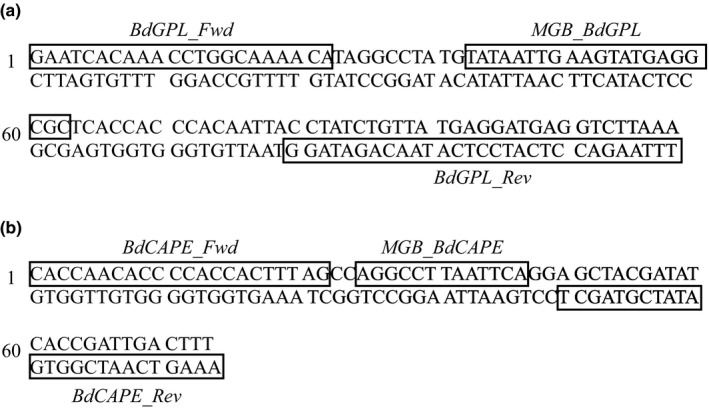
Bd mtDNA sequence sections showing the (a) BdCAPE‐specific and (b) BdGPL‐specific primer and TaqMan MGB probe sequences
2.2. Assessing lineage‐specific qPCR specificity, sensitivity and reliability
A panel of 52 isolates (38 BdGPL, six BdCAPE, four BdASIA‐1, three BdASIA‐2/BRAZIL and one BdCH) collected from 17 countries as well as from traded amphibians was used to test the specificity of the lineage‐specific qPCR (Figure 2a). All isolates had been previously assigned a lineage by WGS (O’Hanlon et al., 2018) (Figure 2b). We extracted DNA from the live cultures following RACE protocols (Supporting Information) for 50 isolates (Table S2). DNA extracts for isolates UM142 and CLFT‐065 were provided by University of Michigan. We tested all 52 isolates with both the lineage‐specific qPCR reactions in singleplex to check for cross‐reactivity. All other reactions were carried out in multiplex. To test for sensitivity, we ran the lineage‐specific qPCR assay with a 10‐fold dilution series of BdGPL and BdCAPE DNA quantitation standards in duplicate. Once a suitable range of DNA concentrations for standard curve analysis had been identified, we tested the repeatability of the assay within and between plates by comparing reaction efficiency, and R 2 and C t values.
FIGURE 2.
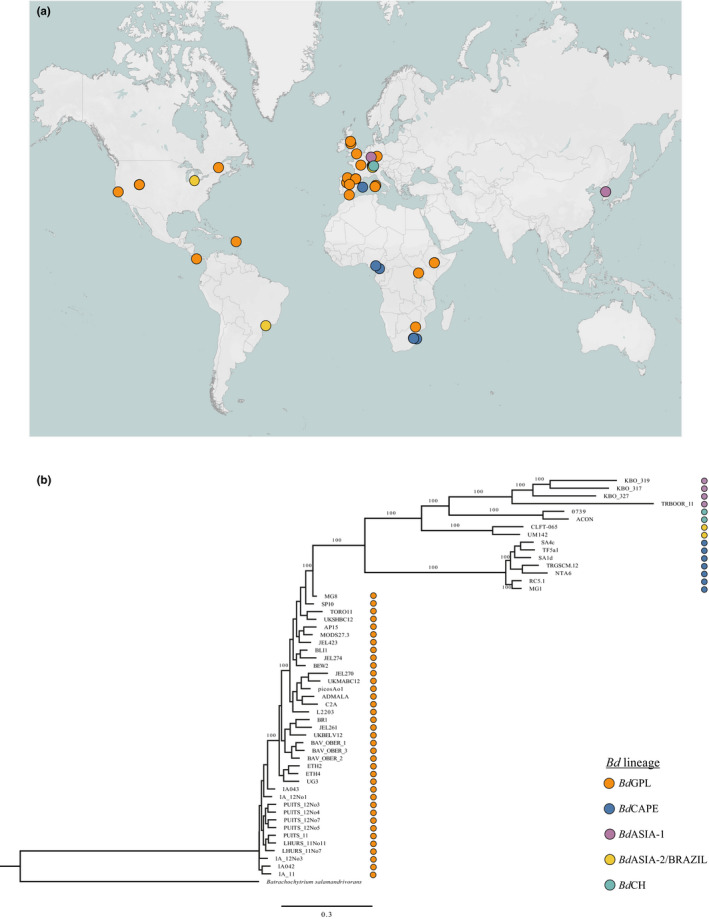
(a) Map showing source locations of Bd isolates used for assay specificity testing; (b) phylogeny of isolates used for assay specificity testing, generated from WGS analysis. Lineage is indicated by dot colour. Map generated in Tableau and formatted in Adobe Illustrator
2.3. Field sample collection and processing
WGS analysis has revealed that BdGPL and BdCAPE are both present in wild populations in South Africa in the Orange River Basin (Figure 3a). This region was therefore selected as a study system to see if the lineage‐specific qPCR assay diagnosed the same lineages from field sites as WGS. Sites were surveyed along the Orange River at approximately 100 km apart from the source in the Drakensburg Mountains to near the estuary on the border with Namibia. At each site, we attempted to culture and isolate Bd using nonlethal tissue sampling from adult amphibians and lethal mouthpart sampling from tadpoles (Fisher et al., 2018). Any resulting chytrids were lineage‐typed by WGS using the protocol described by O’Hanlon et al. (2018). All adult amphibians were also swabbed following “Risk Assessment of Chytridiomycosis to European Amphibian biodiversity (RACE)” protocols (Biodiversa, 2013; Fisher et al., 2018). DNA extraction also followed the RACE protocol (Supporting Information), following which swab samples were tested for Bd using the pan‐lineage qPCR diagnostic. Any samples which were positive in duplicate with an infection load of more than one genomic equivalent (GE) were tested with the lineage‐specific qPCR diagnostic in duplicate with DNA quantitation standards and two no‐template controls.
FIGURE 3.
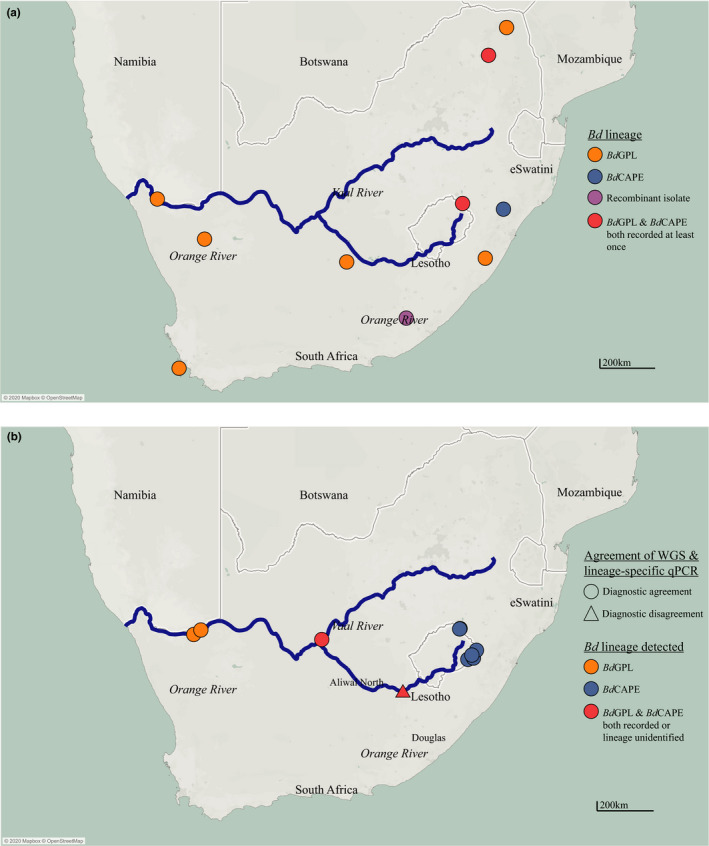
Maps showing (a) Bd lineage locations in South Africa identified by isolation and WGS analysis and (b) comparison of Bd lineage typing of sites in South Africa using WGS analysis and lineage‐specific qPCR. Agreement of diagnostics is indicated by shape of the mark; lineages identified at the site are identified by the colour. Map generated in Tableau and formatted in Adobe Illustrator, additional data downloaded from naturalearthdata.com
3. RESULTS
3.1. Lineage‐specific qPCR performance under laboratory conditions
The Bd lineage‐specific assay was able to detect all isolates assigned to the relevant lineage by WGS. The BdGPL assay also detected DNA from one isolate from the BdASIA‐2/BRAZIL lineage (UM142) but at an unrealistic concentration. This reaction also did not cross the reaction threshold during the exponential phase, as would be expected in a true positive result (Figure S1). Both lineage‐specific assays had a limit of detection of 1 GE (Figure 4). The BdCAPE‐specific qPCR recorded a plateauing of fluorescence at 10,000 GE, resulting in an underestimation of Bd load when comparing with the other quantitation standard fluorescence levels (Figure 4a). This plateauing owes to too much target DNA present in a sample quenching the PCR reaction due to the depletion of reagents. Based on these results, we recommend a DNA quantitation standard range for both lineage‐specific qPCRs of 1000 GE, 100 GE, 10 GE and 1 GE for standard curve analysis.
FIGURE 4.
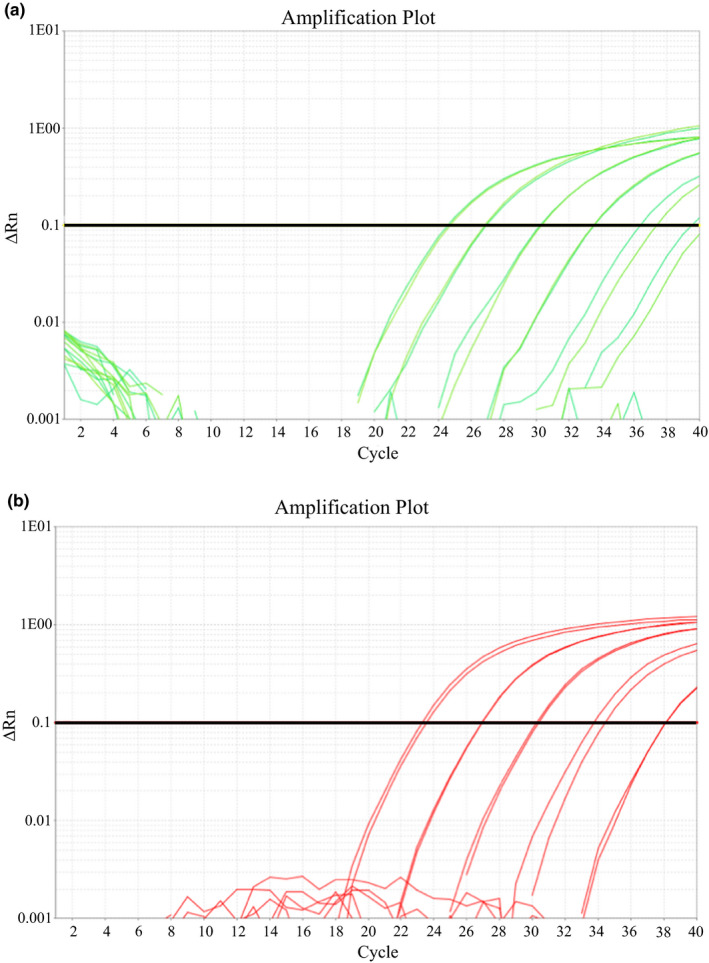
qPCR fluorescence amplification plots showing amplification of 10,000 GE, 1000 GE, 100 GE, 10 GE and 1 GE DNA quantitation standards with (a) BdCAPE‐specific primers and TaqMan MGB probe and (b) BdGPL‐specific primers and TaqMan MGB probe
Both assays performed well over three separate qPCR plates. The overall mean percentage efficiency for the BdCAPE‐specific qPCR was 108.87% (ranging from 99.35% to 126.32%), and for the BdGPL‐specific qPCR was 97.35% (ranging from 92.93% to 104.21%). All R 2 values, whether averaged across and within plates were >0.95 (ranging from 0.98 to 0.99) and the C t values of DNA quantitation standards were also consistent, all crossing the C t threshold within one cycle of each other in all cases.
3.2. Lineage‐specific qPCR performance under field conditions
We obtained at least one pure culture of Bd and at least one Bd‐positive skin swab that met the threshold for qPCR lineage‐testing from 10 sampling sites. At all sites except at Aliwal North, WGS analysis and the lineage‐specific qPCR agreed in their lineage typing (Figure 3b). At Aliwal North, the lineage‐specific qPCR was positive for BdCAPE, but WGS analysis revealed that the isolate recovered from the site was a CAPE/GPL recombinant. At Douglas, a single swab returned a positive result for both BdGPL and BdCAPE. Both lineages were also recovered from the same pond at Douglas by isolation and WGS.
4. DISCUSSION
We have designed a BdCAPE‐ and BdGPL‐specific qPCR diagnostic that will facilitate research into the ecology and distributions of these epidemiologically important Bd lineages. The diagnostic gap that has persisted until now resulted in an inability to gather baseline data on Bd distribution at a phylogenetically and spatially useful scale. This lack has led to difficulties investigating the ecology and epidemiological significance of lineage interactions. Our lineage‐specific qPCR complements existing diagnostics for Bd while meeting the need to decipher lineage in large numbers of samples and in animals with low infection burdens.
The qPCR assay was able to correctly detect whether BdGPL or BdCAPE was present in 51 out of 52 isolates. The most likely explanation for the low‐level cross‐reactivity of the BdGPL‐specific qPCR assay with a BdASIA‐2/BRAZIL isolate is that the DNA extract was contaminated with DNA from a BdGPL isolate at some point after WGS had been carried out. Sequencing reads from this isolate's mtDNA were screened for the presence of the BdGPL‐specific probe, which was not found. This result highlights the importance of manually examining amplification plots to assess whether they appear atypical, and to continue to utilise WGS methods where possible to corroborate results from the lineage‐specific qPCR.
The assay was sufficiently sensitive for use in cases of low infection burden, with a sensitivity of one genomic equivalent. We note, however, that mtDNA copy number has not been quantified for any Bd isolate and that ITS copy number, which is used to quantify infection burden in the majority of Bd surveillance studies, is known to be highly variable (Longo et al., 2013; Rebollar et al., 2017). We recommend therefore that in order to remain consistent with the current literature, that infection intensity is reported based on the pan‐lineage Bd diagnostic, but that this quantification should be viewed as semi‐quantitative rather than absolute. Under experimental conditions, this problem could be resolved by quantifying the copy number of the targeted region for specific isolates before carrying out any experimental work.
Novel diagnostics are challenging to test in a field setting, due to the lack of a priori knowledge of the location of the target pathogen. However, agreement of the lineage‐specific qPCR diagnostic with WGS analysis for sites in South Africa strongly supports the use of the qPCR assay under field conditions. Crucially, the results from Douglas indicate that the assay is able to identify where mixed‐lineage infections are occurring at both the population and individual host level. This indicates that the assay is appropriate for use in putative lineage contact zones where lineage interactions may occur. The lineage‐specific qPCR identified the only recombinant lineage collected as BdCAPE. This suggests uniparental inheritance of mitochondria in Bd, a new insight into the pathogen's biology.
In this study, we focussed on designing diagnostics for BdCAPE and BdGPL not only as these are the lineages most commonly associated with chytridiomycosis, but also due to knowledge barriers making similar work for the remaining lineages challenging until very recently. The Bd isolates that have been fully sequenced with published mitochondrial genomes are biased towards BdGPL and, to a lesser extent, BdCAPE. As a result, these two lineages have until recently had much more cleanly resolved internal phylogenies than the others, allowing greater confidence when identifying lineage‐specific SNPs. The comparative lack of confidence with which SNPs for the remaining lineages could be assigned made investing the time and financial costs of designing lineage‐specific assays unreasonable. However, recently the global Bd phylogeny has been more clearly resolved, and we therefore recommend that similar lineage‐specific mtDNA based diagnostics for BdASIA‐1, Bd‐ASIA‐2, BdASIA‐3 and BdCH should be designed as a research priority. This would allow the rapid and widespread generation of baseline data from field samples, allowing much finer delineation of Bd lineage distributions globally and further insight into Bd ecology.
The consistency of any diagnostic assay across multiple laboratories is essential to its utility and ease of use. The wide application of the pan‐lineage Bd qPCR diagnostic to date means that any laboratory working on Bd is likely to have access to all the reagents and facilities required to perform the lineage‐specific assay described here, at a similar cost per sample. Lineage‐specific diagnostics also enable in vivo and in vitro experimental manipulation of Bd lineages, thus providing further insight into Bd population dynamics and structure.
AUTHOR CONTRIBUTIONS
The research was planned by P.N.G., T.W.J.G., and M.C.F. mtDNA sequences were compiled and aligned by A.R. Primer and TaqMan MGB probe design was done by P.N.G. Assay validation was carried out by P.N.G., and L.M.B., P.N.G., R.V., T.W.J.G., and C.W. carried out fieldwork in South Africa. S.J.O.H., and T.R.S. carried out whole genome sequences analyses. P.N.G. wrote the article with input from all authors.
Supporting information
Supplementary Material
ACKNOWLEDGEMENTS
PNG and MCF were supported by the Morris Animal Foundation, the Leverhulme Trust (RPG‐2014‐273), the Natural Environment Research Council (NERC NE/E006841/1) and a PhD DTP award by the ICL Grantham Institute. MCF is a fellow in the CIFAR ‘Fungal Kingdoms’ programme. The authors would like to thank Tim James, University of Michigan for providing DNA extract of isolates UM42 and CLFT‐065.
Contributor Information
Pria N. Ghosh, Email: pria.ghosh@gmail.com.
Ruhan Verster, Email: ruhan.verster@gmail.com.
Thomas R. Sewell, Email: t.sewell@imperial.ac.uk.
Simon J. O’Hanlon, Email: simon.ohanlon@imperial.ac.uk.
Lola M. Brookes, Email: Lola.Brookes@ioz.ac.uk.
Adrien Rieux, Email: adrien.rieux@cirad.fr.
Trenton W. J. Garner, Email: Trent.Garner@ioz.ac.uk.
Ché Weldon, Email: che.weldon@nwu.ac.za.
Matthew C. Fisher, Email: matthew.fisher@imperial.ac.uk.
DATA AVAILABILITY STATEMENT
The mtDNA alignment is deposited in DRYAD Data Repository https://datadryad.org/stash/dataset/doi:10.5061/dryad.r2280gbbb.
References
- Barr, A. , Premasuthan, A. , Satkoski, J. , Smith, D. G. , George, D. , & Kanthaswamy, S. (2011). A rapid quantitative real‐time PCR‐based DNA quantification assay coupled with species‐assignment capabilities for two hybridizing Macaca species. Folia Primatologica, 82, 71–80. 10.1159/000328124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biodiversa . (2013). Wildlife diseases on the increase: a serious threat for Europe’s Biodiversity. The example of the Bd fungus disease.
- Boyle, D. G. , Boyle, D. B. , Olsen, V. , Morgan, J. A. T. , & Hyatt, A. D. (2004). Rapid quantitative detection of chytridiomycosis. Diseases of Aquatic Organisms, 60, 141–148. 10.3390/s130708551 [DOI] [PubMed] [Google Scholar]
- Byrne, A. Q. , Rothstein, A. P. , Poorten, T. J. , Erens, J. , Settles, M. L. , & Rosenblum, E. B. (2017). Unlocking the story in the swab: A new genotyping assay for the amphibian chytrid fungus Batrachochytrium dendrobatidis . Molecular Ecology Resources, 17(6), 1283–1292. 10.1111/1755-0998.12675 [DOI] [PubMed] [Google Scholar]
- Byrne, A. Q. , Vredenburg, V. T. , Martel, A. , Pasmans, F. , Bell, R. C. , Blackburn, D. C. , Bletz, M. C. , Bosch, J. , Briggs, C. J. , Brown, R. M. , Catenazzi, A. , Familiar López, M. , Figueroa‐Valenzuela, R. , Ghose, S. L. , Jaeger, J. R. , Jani, A. J. , Jirku, M. , Knapp, R. A. , Muñoz, A. , … Contributed, Z. Y. (2019). Cryptic diversity of a widespread global pathogen reveals expanded threats to amphibian conservation. Proceedings of the National Academy of Sciences of the United States of America, 116(41), 20382–20387. 10.1073/pnas.1908289116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doddington, B. J. , Bosch, J. , Oliver, J. A. , Grassly, N. C. , Garcia, G. , Schmidt, B. R. , Garner, T. W. J. , & Fisher, M. C. (2013). Context‐dependent amphibian host population response to an invading pathogen. Ecology, 94(8), 1795–1804. 10.1890/12-1270.1 [DOI] [PubMed] [Google Scholar]
- Farrer, R. A. , Weinert, L. A. , Bielby, J. , Garner, T. W. J. , Balloux, F. , Clare, F. , Bosch, J. , Cunningham, A. A. , Weldon, C. , du Preez, L. H. , Anderson, L. , Pond, S. L. K. , Shahar‐Golan, R. , Henk, D. A. , & Fisher, M. C. (2011). Multiple emergences of genetically diverse amphibian‐infecting chytrids include a globalized hypervirulent recombinant lineage. Proceedings of the National Academy of Sciences of the United States of America, 108(46), 18732–18736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher, M. C. , Ghosh, P. , Shelton, J. M. G. , Bates, K. , Brookes, L. , Wierzbicki, C. , Rosa, G. M. , Farrer, R. A. , Aanensen, D. M. , Alvarado‐Rybak, M. , Bataille, A. , Berger, L. , Böll, S. , Bosch, J. , Clare, F. C. , A. Courtois, E. , Crottini, A. , Cunningham, A. A. , Doherty‐Bone, T. M. , … Garner, T. W. J. (2018). Development and worldwide use of non‐lethal, and minimal population‐level impact, protocols for the isolation of amphibian chytrid fungi. Scientific Reports, 8(1), 4–11. 10.1038/s41598-018-24472-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garland, S. , Wood, J. , & Skerratt, L. F. (2011). Comparison of sensitivity between real‐time detection of a TaqMan assay for Batrachochytrium dendrobatidis and conventional detection. Diseases of Aquatic Organisms, 94(2), 101–105. 10.3354/dao02327 [DOI] [PubMed] [Google Scholar]
- Greenspan, S. E. , Lambertini, C. , Carvalho, T. , James, T. Y. , Toledo, L. F. , Haddad, C. F. B. , & Becker, C. G. (2018). Hybrids of amphibian chytrid show high virulence in native hosts. Scientific Reports, 8(1), 1–10. 10.1038/s41598-018-27828-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyatt, A. D. , Boyle, D. G. , Olsen, V. , Boyle, D. B. , Berger, L. , Obendorf, D. , Dalton, A. , Kriger, K. , Heros, M. , Hines, H. , Phillott, R. , Campbell, R. , Marantelli, G. , Gleason, F. & Colling, A. (2007). Diagnostic assays and sampling protocols for the detection of Batrachochytrium dendrobatidis . Diseases of Aquatic Organisms, 73(3), 175–192. 10.1002/adhm.201300318 [DOI] [PubMed] [Google Scholar]
- Jenkinson, T. S. , Betancourt Román, C. M. , Lambertini, C. , Valencia‐Aguilar, A. , Rodriguez, D. , Nunes‐de‐Almeida, C. H. L. , Ruggeri, J. , Belasen, A. M. , da Silva Leite, D. , Zamudio, K. R. , Longcore, J. E. , Toledo, L. F. , & James, T. Y. (2016). Amphibian‐killing chytrid in Brazil comprises both locally endemic and globally expanding populations. Molecular Ecology, 25(13), 2978–2996. 10.1111/mec.13599 [DOI] [PubMed] [Google Scholar]
- Kriger, K. M. , Hines, H. B. , Hyatt, A. D. , Boyle, D. G. , & Hero, J.‐M. (2006). Techniques for detecting chytridiomycosis in wild frogs: comparing histology with Taqman PCR. Diseases of Aquatic Organisms, 71, 141–148. Retrieved from www.int‐res.com [DOI] [PubMed] [Google Scholar]
- Li, Q. , Wei, S.‐J. , Tang, P. , Wu, Q. , Shi, M. , Sharkey, M. J. , & Chen, X.‐X. (2016). Multiple lines of evidence from mitochondrial genomes resolve phylogenetic relationships of parasitic wasps in Braconidae. Genome Biology and Evolution, 8(9), 2651–2662. 10.1093/gbe/evw184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longcore, J. , & Pessier, A. (1999). Batrachochytrium dendrobatidis gen. et sp. Nov., a chytrid pathogenic to amphibians. Mycologia, 91(2), 219–227. [Google Scholar]
- Longo, A. V. , Rodriguez, D. , da Silva Leite, D. , Toledo, L. F. , Mendoza Almeralla, C. , Burrowes, P. A. , & Zamudio, K. R. (2013). ITS1 copy number varies among Batrachochytrium dendrobatidis strains: implications for qPCR estimates of infection intensity from field‐collected amphibian skin swabs. PLoS One, 8(3), 1–10. 10.1371/journal.pone.0059499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martel, A. , Spitzen‐van der Sluijs, A. , Blooi, M. , Bert, W. , Ducatelle, R. , Fisher, M. C. , Woeltjes, A. , Bosman, W. , Chiers, K. , Bossuyt, F. , & Pasmans, F. (2013). Batrachochytrium salamandrivorans sp. nov. causes lethal chytridiomycosis in amphibians. Proceedings of the National Academy of Sciences of the United States of America, 110(38), 15325–15329. 10.1073/pnas.1307356110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Hanlon, S. J. , Rieux, A. , Farrer, R. A. , Rosa, G. M. , Waldman, B. , Bataille, A. , Kosch, T. A. , Murray, K. A. , Brankovics, B. , Fumagalli, M. , Martin, M. D. , Wales, N. , Alvarado‐Rybak, M. , Bates, K. A. , Berger, L. , Böll, S. , Brookes, L. , Clare, F. , Courtois, E. A. , … Fisher, M. C. (2018). Recent Asian origin of chytrid fungi causing global amphibian declines. Science, 360(May), 621–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson, D. H. , & Ronnenberg, K. L. (2014). Global Bd mapping project: 2014 update. Froglog, 22(3), 17–21. 10.1371/journal [DOI] [Google Scholar]
- Penry, G. S. , Hammond, P. S. , Cockcroft, V. G. , Best, P. B. , Thornton, M. , & Graves, J. A. (2018). Phylogenetic relationships in southern African Bryde’s whales inferred from mitochondrial DNA: Further support for subspecies delineation between the two allopatric populations. Conservation Genetics, 19, 1349–1365. 10.1007/s10592-018-1105-4 [DOI] [Google Scholar]
- Rebollar, E. A. , Woodhams, D. C. , LaBumbard, B. , Kielgast, J. , & Harris, R. N. (2017). Prevalence and pathogen load estimates for the fungus Batrachochytrium dendrobatidis are impacted by ITS DNA copy number variation. Diseases of Aquatic Organisms, 123(3), 213–226. 10.3354/dao03097 [DOI] [PubMed] [Google Scholar]
- Scheele, B. C. , Pasmans, F. , Skerratt, L. F. , Berger, L. , Martel, A. , Beukema, W. , Acevedo, A. A. , Burrowes, P. A. , Carvalho, T. , Catenazzi, A. , Del la Riva, I. , Fisher, M. C. , Flechas, S. V. , Foster, C. N. , Frías‐Álvarez, P. , Garner, T. W. J. , Gratwicke, B. , Guayasamin, J. M. , Hirschfeld, M. , … Canessa, S. (2019). Amphibian fungal panzootic causes catastrophic and ongoing loss of biodiversity. Science, 363(6434), 1459–1463. Retrieved from http://science.sciencemag.org/ [DOI] [PubMed] [Google Scholar]
- Schoch, C. L. , Seifert, K. A. , Huhndorf, S. , Robert, V. , Spouge, J. L. , Levesque, C. A. , Chen, W. , Bolchacova, E. , Voigt, K. , Crous, P. W. , Miller, A. N. , Wingfield, M. J. , Aime, M. C. , An, K.‐D. , Bai, F.‐Y. , Barreto, R. W. , Begerow, D. , Bergeron, M.‐J. , Blackwell, M. , … Schindel, D. (2012). Nuclear ribosomal internal transcribed spacer (ITS) region as a universal DNA barcode marker for Fungi. Proceedings of the National Academy of Sciences of the United States of America, 109(16), 6241–6246. 10.1073/pnas.1117018109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schreeg, M. E. , Marr, H. S. , Tarigo, J. L. , Cohn, L. A. , Bird, D. M. , Scholl, E. H. , Levy, M. G. , Wiegmann, B. M. , & Birkenheuer, A. J. (2016). Mitochondrial genome sequences and structures aid in the resolution of Piroplasmida phylogeny. PLoS One, 11(11), 165702. 10.1371/journal.pone.0165702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skerratt, L. F. , Mendez, D. , McDonald, K. R. , Garland, S. , Livingstone, J. , Berger, L. , & Speare, R. (2011). Validation of diagnostic tests in wildlife: The case of Chytridiomycosis in wild amphibians. Journal of Herpetology, 45(4), 444–450. 10.1670/10-193.1 [DOI] [Google Scholar]
- Song, S.‐N. , Tang, P. , Wei, S.‐J. , & Chen, X.‐X. (2016). Comparative and phylogenetic analysis of the mitochondrial genomes in basal hymenopterans. Nature Scientific Reports, 6, 20972. 10.1038/srep20972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Sluys, M. , Kriger, K. M. , Phillott, A. D. , Campbell, R. , Skerratt, L. F. , & Hero, J.‐M. (2008). Storage of samples at high temperatures reduces the amount of amphibian chytrid fungus Batrachochytrium dendrobatidis DNA detectable by PCR assay. Diseases of Aquatic Organisms, 81, 93–97. 10.3354/dao01953 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material
Data Availability Statement
The mtDNA alignment is deposited in DRYAD Data Repository https://datadryad.org/stash/dataset/doi:10.5061/dryad.r2280gbbb.