

Abstract
Background
Evidence is limited regarding the most effective pharmacological treatment for psychotic depression: monotherapy with an antidepressant, monotherapy with an antipsychotic, another treatment (e.g. mifepristone), or combination of an antidepressant plus an antipsychotic. This is an update of a review first published in 2005 and last updated in 2015.
Objectives
1. To compare the clinical efficacy of pharmacological treatments for patients with an acute psychotic depression: antidepressant monotherapy, antipsychotic monotherapy, mifepristone monotherapy, and the combination of an antidepressant plus an antipsychotic versus placebo and/or each other.
2. To assess whether differences in response to treatment in the current episode are related to non‐response to prior treatment.
Search methods
A search of the Cochrane Central Register of Controlled Trials (CENTRAL), in the Cochrane Library; the Cochrane Common Mental Disorders Controlled Trials Register (CCMDCTR); Ovid MEDLINE (1950‐); Embase (1974‐); and PsycINFO (1960‐) was conducted on 21 February 2020. Reference lists of all included studies and related reviews were screened and key study authors contacted.
Selection criteria
All randomised controlled trials (RCTs) that included participants with acute major depression with psychotic features, as well as RCTs consisting of participants with acute major depression with or without psychotic features, that reported separately on the subgroup of participants with psychotic features.
Data collection and analysis
Two review authors independently extracted data and assessed risk of bias in the included studies, according to criteria from the Cochrane Handbook for Systematic Reviews of Interventions. Data were entered into RevMan 5.1. We used intention‐to‐treat data. Primary outcomes were clinical response for efficacy and overall dropout rate for harm/tolerance. Secondary outcome were remission of depression, change from baseline severity score, quality of life, and dropout rate due to adverse effects.
For dichotomous efficacy outcomes (i.e. response and overall dropout), risk ratios (RRs) with 95% confidence intervals (CIs) were calculated.
Regarding the primary outcome of harm, only overall dropout rates were available for all studies. If the study did not report any of the response criteria as defined above, remission as defined here could be used as an alternative. For continuously distributed outcomes, it was not possible to extract data from the RCTs.
Main results
The search identified 3947 abstracts, but only 12 RCTs with a total of 929 participants could be included in the review. Because of clinical heterogeneity, few meta‐analyses were possible. The main outcome was reduction in severity (response) of depression, not of psychosis.
For depression response, we found no evidence of a difference between antidepressant and placebo (RR 8.40, 95% CI 0.50 to 142.27; participants = 27, studies = 1; very low‐certainty evidence) or between antipsychotic and placebo (RR 1.13, 95% CI 0.74 to 1.73; participants = 201, studies = 2; very low‐certainty evidence). Furthermore, we found no evidence of a difference in overall dropouts with antidepressant (RR 1.24, 95% CI 0.34 to 4.51; participants = 27, studies = 1; very low‐certainty evidence) or antipsychotic monotherapy (RR 0.79, 95% CI 0.57 to 1.08; participants = 201, studies = 2; very low‐certainty evidence).
No evidence suggests a difference in depression response (RR 2.09, 95% CI 0.64 to 6.82; participants = 36, studies = 1; very low‐certainty evidence) or overall dropouts (RR 1.79, 95% CI 0.18 to 18.02; participants = 36, studies = 1; very low‐certainty evidence) between antidepressant and antipsychotic.
For depression response, low‐ to very low‐certainty evidence suggests that the combination of an antidepressant plus an antipsychotic may be more effective than antipsychotic monotherapy (RR 1.83, 95% CI 1.40 to 2.38; participants = 447, studies = 4), more effective than antidepressant monotherapy (RR 1.42, 95% CI 1.11 to 1.80; participants = 245, studies = 5), and more effective than placebo (RR 1.86, 95% CI 1.23 to 2.82; participants = 148, studies = 2). Very low‐certainty evidence suggests no difference in overall dropouts between the combination of an antidepressant plus an antipsychotic versus antipsychotic monotherapy (RR 0.79, 95% CI 0.63 to 1.01; participants = 447, studies = 4), antidepressant monotherapy (RR 0.91, 95% CI 0.55 to 1.50; participants = 245, studies = 5), or placebo alone (RR 0.75, 95% CI 0.48 to 1.18; participants = 148, studies = 2).
No study measured change in depression severity from baseline, quality of life, or dropouts due to adverse events. We found no RCTs with mifepristone that fulfilled our inclusion criteria.
Risk of bias is considerable: we noted differences between studies with regards to diagnosis, uncertainties around randomisation and allocation concealment, treatment interventions (pharmacological differences between various antidepressants and antipsychotics), and outcome criteria.
Authors' conclusions
Psychotic depression is heavily under‐studied, limiting confidence in the conclusions drawn. Some evidence indicates that combination therapy with an antidepressant plus an antipsychotic is more effective than either treatment alone or placebo. Evidence is limited for treatment with an antidepressant alone or with an antipsychotic alone. Evidence for efficacy of mifepristone is lacking.
Keywords: Humans; Antidepressive Agents; Antidepressive Agents/therapeutic use; Depression; Depression/drug therapy; Depressive Disorder, Major; Depressive Disorder, Major/drug therapy; Psychotic Disorders; Psychotic Disorders/drug therapy; Systematic Reviews as Topic
Plain language summary
Pharmacological treatment for psychotic depression
Psychotic depression is a severe depression with psychotic features (i.e. delusions and/or hallucinations). Uncertainty surrounds the most effective drug treatment for psychotic depression: an antidepressant alone, an antipsychotic alone, or the combination of an antidepressant plus an antipsychotic.
The aim of this review is to compare the efficacy of the various forms of drug treatment that have been used to treat psychotic depression. We did this by analysing all randomised controlled trials (RCTs). Twelve RCTs met our inclusion criteria. These trials involved a total of 929 people.
From these trials, we found evidence that the combination of an antidepressant plus an antipsychotic provides more effective treatment for psychotic depression than either treatment alone. However, our confidence in this conclusion is limited because the information came from only a small number of RCTs, which included small numbers of people. In addition, the types of people involved varied between RCTs, and these trials differed in design, which means that we cannot confidently generalise their findings.
Summary of findings
Summary of findings 1. Antidepressant compared to placebo for psychotic depression.
| Antidepressant compared to placebo for psychotic depression | ||||||
| Patient or population: adults with psychotic depression Setting: hospital Intervention: antidepressant Comparison: placebo | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | №. of participants (studies) | Certainty of evidence (GRADE) | Comments | |
| Risk with placebo | Risk with antidepressant | |||||
| Clinical response of depression | Study population | RR 8.40 (0.50 to 142.27) | 27 (1 RCT) | ⊕⊝⊝⊝ Very lowa,b,c | Study defined depression response as HRSD‐17 < 7 | |
| 36 per 1000 | 300 per 1000 (18 to 1000) | |||||
| Overall dropouts | Study population | RR 1.24 (0.34 to 4.51) | 27 (1 RCT) | ⊕⊝⊝⊝ Very lowa,b,c | ||
| 231 per 1000 | 286 per 1000 (78 to 1000) | |||||
| Depression remission | See comment | ‐ | ‐ | ‐ | No study reported this outcome | |
| Change in depression severity from baseline | See comment | ‐ | ‐ | ‐ | No study reported this outcome | |
| Quality of life | See comment | ‐ | ‐ | ‐ | No study reported this outcome | |
| Dropouts due to adverse effects | See comment | ‐ | ‐ | ‐ | No study reported this outcome | |
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; RCT: randomised controlled trial; RR: risk ratio. | ||||||
| GRADE Working Group grades of evidence. High certainty: we are very confident that the true effect lies close to that of the estimate of the effect. Moderate certainty: we are moderately confident in the effect estimate: the true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. Low certainty: our confidence in the effect estimate is limited: the true effect may be substantially different from the estimate of the effect. Very low certainty: we have very little confidence in the effect estimate: the true effect is likely to be substantially different from the estimate of effect. | ||||||
aDowngraded one level for high risk of other bias.
bDowngraded one level for high risk of publication bias.
cDowngraded one level for imprecision due to small sample size; CIs are consistent with appreciable benefit and appreciable harm.
Summary of findings 2. Antipsychotic compared to placebo for psychotic depression.
| Antipsychotic compared to placebo for psychotic depression | ||||||
| Patient or population: adults with psychotic depression Setting: at least first week of study in hospital Intervention: antipsychotic Comparison: placebo | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | №. of participants (studies) | Certainty of evidence (GRADE) | Comments | |
| Risk with placebo | Risk with antipsychotic | |||||
| Clinical response of depression | Study population | RR 1.13 (0.74 to 1.73) | 201 (2 RCTs) | ⊕⊝⊝⊝ Very lowa,b,c | Studies defined depression response as reduction in HAMD‐24 ≥ 50% at endpoint | |
| 280 per 1000 | 316 per 1000 (207 to 484) | |||||
| Overall dropouts | Study population | RR 0.79 (0.57 to 1.08) | 201 (2 RCTs) | ⊕⊝⊝⊝ Very lowa,b,c | ||
| 470 per 1000 | 371 per 1000 (268 to 508) | |||||
| Depression remission | See comment | ‐ | ‐ | ‐ | No study reported this outcome | |
| Change in depression severity from baseline | See comment | ‐ | ‐ | ‐ | No study reported this outcome | |
| Quality of life | See comment | ‐ | ‐ | ‐ | No study reported this outcome | |
| Dropouts due to adverse effects | See comment | ‐ | ‐ | ‐ | No study reported this outcome | |
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; RCT: randomised controlled trial; RR: risk ratio. | ||||||
| GRADE Working Group grades of evidence. High certainty: we are very confident that the true effect lies close to that of the estimate of the effect. Moderate certainty: we are moderately confident in the effect estimate: the true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. Low certainty: our confidence in the effect estimate is limited: the true effect may be substantially different from the estimate of the effect. Very low certainty: we have very little confidence in the effect estimate: the true effect is likely to be substantially different from the estimate of effect. | ||||||
aDowngraded one level for high risk of other bias.
bDowngraded one level for high risk of publication bias.
cDowngraded one level for imprecision due to small sample size; CIs are consistent with appreciable benefit and appreciable harm.
Summary of findings 3. Antidepressant compared to antidepressant for psychotic depression.
| Antidepressant compared to antidepressant for psychotic depression | ||||||
| Patient or population: adults with psychotic depression Setting: hospital Intervention: antidepressant Comparison: antidepressant | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | №. of participants (studies) | Certainty of evidence (GRADE) | Comments | |
| Risk with antidepressant | Risk with antidepressant | |||||
| Clinical response | See comment | ‐ | ‐ | ⊕⊝⊝⊝ Very lowa,b,c | Meta‐analysis was not possible due to heterogeneity between the different antidepressants used van den Broek 2004a showed that imipramine may be more effective than fluvoxamine (RR 2.10, 95% CI 1.06 to 4.17) Bruijn 1996 showed that imipramine may be more effective than mirtazapine (RR 3.00, 95% CI 1.01 to 8.95) Zanardi 1996 showed that sertraline may be more effective than paroxetine (RR 3.37, 95% CI 1.19 to 9.57) Zanardi 2000 found no difference between fluvoxamine and venlafaxine (RR 1.50, 95% CI 0.82 to 2.75) Wijkstra 2010 found no difference between imipramine and venlafaxine (RR 1.57, 95% CI 0.93 to 2.67) |
|
| Overall dropouts | See comment | ‐ | ‐ | ⊕⊝⊝⊝ Very lowa,b,c |
Wijkstra 2010 found no difference between imipramine and venlafaxine (RR 0.81, 95% CI 0.33 to 2.03) Bruijn 1996 found no difference between imipramine and mirtazapine (RR 0.50, 95% CI 0.19 to 1.31) van den Broek 2004a found no difference between imipramine and fluvoxamine (RR 2.00, 95% CI 0.40 to 9.95) Zanardi 1996 found no difference between sertraline and paroxetine (RR 0.20, 95% CI 0.01 to 3.74) Zanardi 2000 found no difference between fluvoxamine and venlafaxine (RR 0.07, 95% CI 0.00 to 1.20) |
|
| Depression remission | See comment | ‐ | ‐ | ‐ | No study reported this outcome | |
| Change in depression severity from baseline | See comment | ‐ | ‐ | ‐ | No study reported this outcome | |
| Quality of life | See comment | ‐ | ‐ | ‐ | No study reported this outcome | |
| Dropouts due to adverse effects | See comment | ‐ | ‐ | ‐ | No study reported this outcome | |
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; RR: risk ratio. | ||||||
| GRADE Working Group grades of evidence. High certainty: we are very confident that the true effect lies close to that of the estimate of the effect. Moderate certainty: we are moderately confident in the effect estimate: the true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. Low certainty: our confidence in the effect estimate is limited: the true effect may be substantially different from the estimate of the effect. Very low certainty: we have very little confidence in the effect estimate: the true effect is likely to be substantially different from the estimate of effect. | ||||||
aDowngraded one level for imprecision due to small sample size.
bDowngraded one level for imprecision as CIs are consistent with appreciable benefit and appreciable harm.
cDowngraded one level for high risk of publication bias.
Summary of findings 4. Antidepressant compared to antipsychotic for psychotic depression.
| Antidepressant compared to antipsychotic for psychotic depression | ||||||
| Patient or population: adults with psychotic depression Setting: hospital Intervention: antidepressant Comparison: antipsychotic | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | №. of participants (studies) | Certainty of evidence (GRADE) | Comments | |
| Risk with antipsychotic | Risk with antidepressant | |||||
| Clinical response of depression | Study population | RR 2.09 (0.64 to 6.82) | 36 (1 RCT) | ⊕⊝⊝⊝ Very lowa,b,c | Study defined depression response as HRSD‐17 < 7 | |
| 176 per 1000 | 369 per 1000 (113 to 1000) | |||||
| Overall dropouts | Study population | RR 1.79 (0.18 to 18.02) | 36 (1 RCT) | ⊕⊝⊝⊝ Very lowa,b,c | ||
| 59 per 1000 | 105 per 1000 (11 to 1000) | |||||
| Depression remission | See comment | ‐ | ‐ | ‐ | No study reported this outcome | |
| Change in depression severity from baseline | See comment | ‐ | ‐ | ‐ | No study reported this outcome | |
| Quality of life | See comment | ‐ | ‐ | ‐ | No study reported this outcome | |
| Dropouts due to adverse effects | See comment | ‐ | ‐ | ‐ | No study reported this outcome | |
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; RCT: randomised controlled trial; RR: risk ratio. | ||||||
| GRADE Working Group grades of evidence. High certainty: we are very confident that the true effect lies close to that of the estimate of the effect. Moderate certainty: we are moderately confident in the effect estimate: the true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. Low certainty: our confidence in the effect estimate is limited: the true effect may be substantially different from the estimate of the effect. Very low certainty: we have very little confidence in the effect estimate: the true effect is likely to be substantially different from the estimate of effect. | ||||||
aDowngraded one level for imprecision due to small sample size.
bDowngraded one level for imprecision as CIs are consistent with appreciable benefit and appreciable harm.
cDowngraded one level for risk of publication bias.
Summary of findings 5. Antidepressant plus antipsychotic compared to placebo for psychotic depression.
| Antidepressant plus antipsychotic compared to placebo for psychotic depression | ||||||
| Patient or population: adults with psychotic depression Setting: at least first week of study in hospital Intervention: antidepressant plus antipsychotic Comparison: placebo | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | №. of participants (studies) | Certainty of evidence (GRADE) | Comments | |
| Risk with placebo | Risk with antidepressant plus antipsychotic | |||||
| Clinical response of depression | Study population | RR 1.86 (1.23 to 2.82) | 148 (2 RCTs) | ⊕⊝⊝⊝ Very lowa,b,c | Both studies defined response as reduction in HAMD‐24 ≥ 50% at endpoint | |
| 280 per 1000 | 521 per 1000 (344 to 790) | |||||
| Overall dropouts | Study population | RR 0.75 (0.48 to 1.18) | 148 (2 RCTs) | ⊕⊝⊝⊝ Very lowa,b,d | ||
| 470 per 1000 | 353 per 1000 (226 to 555) | |||||
| Depression remission | See comment | ‐ | ‐ | ‐ | No study reported this outcome | |
| Change in depression severity from baseline | See comment | ‐ | ‐ | ‐ | No study reported this outcome | |
| Quality of life | See comment | ‐ | ‐ | ‐ | No study reported this outcome | |
| Dropouts due to adverse effects | See comment | ‐ | ‐ | ‐ | No study reported this outcome | |
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; RCT: randomised controlled trial; RR: risk ratio. | ||||||
| GRADE Working Group grades of evidence. High certainty: we are very confident that the true effect lies close to that of the estimate of the effect. Moderate certainty: we are moderately confident in the effect estimate: the true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. Low certainty: our confidence in the effect estimate is limited: the true effect may be substantially different from the estimate of the effect. Very low certainty: we have very little confidence in the effect estimate: the true effect is likely to be substantially different from the estimate of effect. | ||||||
aDowngraded one level for for high risk of other source of bias.
bDowngraded one level for for high risk of publication bias.
cDowngraded one level for imprecision due to small sample size.
dDowngraded one level for imprecision due to small sample size; CIs are consistent with appreciable benefit and appreciable harm.
Summary of findings 6. Antidepressant plus antipsychotic compared to placebo plus antipsychotic for psychotic depression.
| Antidepressant plus antipsychotic compared to placebo plus antipsychotic for psychotic depression | ||||||
| Patient or population: adults with psychotic depression Setting: hospital (2 RCTs) or at least first week of study in hospital (2 RCTs) Intervention: antidepressant plus antipsychotic Comparison: placebo plus antipsychotic | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | №. of participants (studies) | Certainty of evidence (GRADE) | Comments | |
| Risk with placebo plus antipsychotic | Risk with antidepressant plus antipsychotic | |||||
| Clinical response of depression | Study population | RR 1.83 (1.40 to 2.38) | 447 (4 RCTs) | ⊕⊕⊝⊝ Lowa,b | 2 studies defined response as reduction in HAMD‐24 ≥ 50% at endpoint, 1 study defined response as HAMD‐17 ≦ 10, and another study defined response as HRSD‐17 < 7 | |
| 266 per 1000 | 487 per 1000 (373 to 633) | |||||
| Overall dropouts | Study population | RR 0.79 (0.63 to 1.01) | 447 (4 RCTs) | ⊕⊕⊝⊝ Very lowa,b,c | ||
| 435 per 1000 | 344 per 1000 (274 to 440) | |||||
| Depression remission | See comment | ‐ | ‐ | ‐ | No study reported this outcome | |
| Change in depression severity from baseline | See comment | ‐ | ‐ | ‐ | No study reported this outcome | |
| Quality of life | See comment | ‐ | ‐ | ‐ | No study reported this outcome | |
| Dropouts due to adverse effects | See comment | ‐ | ‐ | ‐ | No study reported this outcome | |
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; HAMD: Hamilton Depression Rating Scale; HRSD: Hamilton Rating Scale for Depression; RCT: randomised controlled trial; RR: risk ratio. | ||||||
| GRADE Working Group grades of evidence. High certainty: we are very confident that the true effect lies close to that of the estimate of the effect. Moderate certainty: we are moderately confident in the effect estimate: the true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. Low certainty: our confidence in the effect estimate is limited: the true effect may be substantially different from the estimate of the effect. Very low certainty: we have very little confidence in the effect estimate: the true effect is likely to be substantially different from the estimate of effect. | ||||||
aDowngraded one level for high risk of other source of bias.
bDowngraded one level for high risk of publication bias.
cDowngraded one level for imprecision as CIs are consistent with appreciable benefit and appreciable harm.
Summary of findings 7. Antidepressant plus antipsychotic compared to placebo plus antidepressant for psychotic depression.
| Antidepressant plus antipsychotic compared to placebo plus antidepressant for psychotic depression | ||||||
| Patient or population: adults with psychotic depression Setting: hospital Intervention: antidepressant plus antipsychotic Comparison: placebo plus antidepressant | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | №. of participants (studies) | Certainty of evidence (GRADE) | Comments | |
| Risk with placebo plus antidepressant | Risk with antidepressant plus antipsychotic | |||||
| Clinical response of depression | Study population | RR 1.42 (1.11 to 1.80) | 245 (4 RCTs) | ⊕⊕⊝⊝ Very lowa,b,c | One study defined response as HRSD‐17 < 7, another study defined response as HAMD‐17 ≦ 10, another study defined response as HAMD‐17 < 11, and a fourth study defined response as reduction in HRSD‐17 > 50% | |
| 436 per 1000 | 619 per 1000 (484 to 784) | |||||
| Overall dropouts | Study population | RR 0.91 (0.55 to 1.50) | 245 (4 RCTs) | ⊕⊝⊝⊝ Very lowa,b,d | ||
| 207 per 1,000 | 189 per 1,000 (114 to 311) | |||||
| Depression remission | See comment | ‐ | ‐ | ‐ | No study reported this outcome | |
| Change in depression severity from baseline | See comment | ‐ | ‐ | ‐ | No study reported this outcome | |
| Quality of life | See comment | ‐ | ‐ | ‐ | No study reported this outcome | |
| Dropouts due to adverse effects | See comment | ‐ | ‐ | ‐ | No study reported this outcome | |
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; HAMD: Hamilton Depression Rating Scale; HRSD: Hamilton Rating Scale for Depression; RCT: randomised controlled trial; RR: risk ratio. | ||||||
| GRADE Working Group grades of evidence. High certainty: we are very confident that the true effect lies close to that of the estimate of the effect. Moderate certainty: we are moderately confident in the effect estimate: the true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. Low certainty: our confidence in the effect estimate is limited: the true effect may be substantially different from the estimate of the effect. Very low certainty: we have very little confidence in the effect estimate: the true effect is likely to be substantially different from the estimate of effect. | ||||||
aDowngraded for high risk of attrition bias and other source of bias in one study.
bDowngraded one level for high risk of publication bias.
cDowngraded one level for imprecision due to small sample size.
dDowngraded one level for imprecision due to small sample size; CIs are consistent with appreciable benefit and appreciable harm.
Summary of findings 8. Antidepressant plus antipsychotic compared to placebo plus the same antidepressant for psychotic depression.
| Antidepressant plus antipsychotic compared to placebo plus the same antidepressant for psychotic depression | ||||||
| Patient or population: adults with psychotic depression Setting: hospital Intervention: antidepressant plus antipsychotic Comparison: placebo plus the same antidepressant | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | №. of participants (studies) | Certainty of evidence (GRADE) | Comments | |
| Risk with placebo plus the same antidepressant | Risk with antidepressant plus antipsychotic | |||||
| Clinical response of depression | Study population | RR 1.70 (1.19 to 2.43) | 157 (3 RCTs) | ⊕⊕⊝⊝ Very lowa,b,c | One study defined response as HAMD‐17 < 11, another study defined response as HRSD‐17 < 7, and a third study defined response as ≧ 50% decrease in HAMD‐17 scores from baseline to study endpoint | |
| 351 per 1000 | 596 per 1000 (417 to 852) | |||||
| Overall dropouts | Study population | RR 1.04 (0.52 to 2.07) | 157 (3 RCTs) | ⊕⊝⊝⊝ Very lowa,b,d | ||
| 169 per 1000 | 176 per 1000 (88 to 349) | |||||
| Depression remission | See comment | ‐ | ‐ | ‐ | No study reported this outcome | |
| Change in depression severity from baseline | See comment | ‐ | ‐ | ‐ | No study reported this outcome | |
| Quality of life | See comment | ‐ | ‐ | ‐ | No study reported this outcome | |
| Dropouts due to adverse effects | See comment | ‐ | ‐ | ‐ | No study reported this outcome | |
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; HAMD: Hamilton Depression Rating Scale; HRSD: Hamilton Rating Scale for Depression; RCT: randomised controlled trial; RR: risk ratio. | ||||||
| GRADE Working Group grades of evidence. High certainty: we are very confident that the true effect lies close to that of the estimate of the effect. Moderate certainty: we are moderately confident in the effect estimate: the true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. Low certainty: our confidence in the effect estimate is limited: the true effect may be substantially different from the estimate of the effect. Very low certainty: we have very little confidence in the effect estimate: the true effect is likely to be substantially different from the estimate of effect. | ||||||
aDowngraded one level for high risk of other source of bias.
bDowngraded one level for high risk of publication bias.
cDowngraded one level for imprecision due to small sample size.
dDowngraded one level for imprecision due to small sample size; CIs are consistent with appreciable harm and appreciable benefit.
Background
Description of the condition
Psychotic depression is a severe condition that is defined as a depressive episode with psychotic features (i.e. delusions and/or hallucinations) in the context of a (unipolar) major depressive disorder. Psychotic depression is not uncommon. In the US Epidemiologic Catchment Area Study (Johnson 1991), 14% of participants who met the criteria for major depression had a history of episodes with psychotic features. In a European general population study, 18.5% of respondents with a major depressive episode had psychotic features; the prevalence of psychotic depression was 0.4%, and of non‐psychotic depression 2.0% (Ohayon 2002). In a US study of hospitalised patients with major depression, 25% met the criteria for psychotic depression (Coryell 1984). Compared with non‐psychotic depression, psychotic depression is marked by greater severity, increased incapacity, decreased likelihood of placebo response, longer duration of episodes, and recurrence of psychotic features in subsequent episodes (Coryell 1998).
Description of the intervention
Guidelines recommend electroconvulsive therapy (ECT) or pharmacotherapy as treatment for psychotic depression (APA 2010; NICE 2009). Pharmacotherapy for psychotic depression could consist of an antipsychotic, an antidepressant, or a combination of both. Most guidelines recommend treatment that combines an antidepressant with an antipsychotic (APA 2010; NICE 2009). However, discussion continues regarding whether the combination of an antipsychotic plus an antidepressant is more effective than monotherapy with an antidepressant or an antipsychotic (Mahli 2009; Multidisciplinaire Richtlijn Depressie 2013; Parker 1992; Wijkstra 2005; Wijkstra 2007). The intervention studied in this review is pharmacological treatment for psychotic depression, including the question of whether the combination of an antipsychotic plus an antidepressant is more effective than either treatment given as monotherapy.
How the intervention might work
All antidepressants enhance the activity of serotonin and/or noradrenaline, and some of them (also) dopamine (Sadock 2009). Most antidepressants achieve this via inhibition of reuptake of these neurotransmitters in the presynaptic neuron (tricyclic antidepressants (TCAs), selective serotonin reuptake inhibitors (SSRIs), and serotonin‐noradrenaline reuptake inhibitors (SNRIs)), although some antidepressants have other working mechanisms (e.g. blockade of postsynaptic serotonin‐2 receptors such as mirtazapine, inhibition of their breakdown via inhibition of the enzyme monoamine oxidase (MAOI)). Nevertheless, their noradrenergic and serotonergic effects do not completely explain their efficacy, as these effects occur already within hours after first intake, but it takes days to weeks before antidepressants begin to exert their effects in patients with depression or anxiety (Sadock 2009).
Almost all antipsychotics (classical as well as atypical antipsychotics, with the exception of clozapine) are blockers of the postsynaptic dopamine‐2 receptor, and their therapeutic efficacy is correlated with their affinity for dopamine‐2 receptors in vivo. However, other effects may contribute to their efficacy, such as their affinity for presynaptic serotonin‐1 receptors, postsynaptic serotonin‐2 receptors, and histamine receptors, as can be seen with some atypical antipsychotics (e.g. olanzapine, quetiapine). Similar to antidepressants, these effects do not completely explain their efficacy because they also occur already within hours after first intake, but it takes days to weeks for antipsychotics to begin to work (Sadock 2009).
The traditional view is that antidepressants work against depression and antipsychotics work against psychosis. Therefore, it seems appropriate in psychotic depression to treat the psychotic symptoms with an antipsychotic and the depressive symptoms with an antidepressant. However, when psychotic depression is considered as the most severe form of depression, and when psychosis is viewed as the distal consequence of that severity (as is the case in the Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition, Text Revision (DSM‐IV‐TR), APA 2000, and Fifth Edition (DSM‐5), APA 2013)), treatment with an antidepressant alone seems logical. On the other hand, treatment with an antipsychotic alone, especially one of the newer atypical antipsychotics with possible antidepressant effects, cannot be ruled out.
Other studies suggest that dysregulation of the hypothalamic‐pituitary‐adrenal axis found in patients with psychotic depression might be a biologically targeted treatment opportunity (Duval 2006), as abnormalities in the diurnal fluctuation of cortisol have been found (Keller 2006), and high rates of non‐suppression on the dexamethasone suppression test have been observed (Nelson 1997). Therefore, the glucocortoid receptor antagonist mifepristone has been proposed as a possible pharmacological treatment for psychotic depression (Belanoff 2001).
Why it is important to do this review
Clinical practice is characterised by uncertainty as to whether it is most appropriate to start treatment in this patient group with antidepressant monotherapy or with the combination of an antidepressant and an antipsychotic because of potential adverse effects of antipsychotics (especially extrapyramidal side effects, hyperprolactinaemia, and risk of metabolic syndrome, including weight gain). A previous meta‐analysis did not find a statistically significant difference between TCA monotherapy and combination therapy (Parker 1992). However, the findings and conclusions of that meta‐analysis were limited by inadequate methods of many included studies, which were often retrospective, uncontrolled, and/or not randomised. Some international guidelines on the pharmacological treatment of psychotic depression (in the United States ‐ Nelson 1997; in the Netherlands ‐ Multidisciplinaire Richtlijn Depressie 2013) and those presented in reviews ‐ Wheeler 2000 ‐ suggest that one may consider TCA monotherapy before adding an antipsychotic. However, in contrast, the American Psychiatric Association (APA) Practice Guideline for the Treatment of Patients with Major Depressive Disorder and the National Institute for Health and Clinical Excellence recommend initial combination therapy (APA 2010; NICE 2009). The same recommendation is made in the Coryell 1998 review. This variation between guidelines reflects the limited evidence on which these guidelines are based. In a review about evidence used in practice guidelines (Wijkstra 2007), we concluded that physicians (and patients) should be aware that guidelines for treatment recommendations may be less evidence‐based than asserted, even when it is stated that treatment recommendations are based on the highest level of evidence.
Treatment with a classical antipsychotic alone is not recommended, primarily because of findings reported by Spiker 1985, in which treatment with perphenazine alone was less effective than treatment with perphenazine plus amitriptyline. However, atypical antipsychotics may be worth reconsidering now because of reduced risk of extrapyramidal side effects and potential antidepressant properties associated with some of these agents (Rothschild 2004a).
This review is an update of our Cochrane Review first published in 2005 and updated in 2015 (Wijkstra 2005; Wijkstra 2015). The conclusion in 2015 was as follows: “psychotic depression is heavily understudied, limiting confidence in the conclusions drawn. Some evidence indicates that combination therapy with an antidepressant plus an antipsychotic is more effective than either treatment alone or placebo. Evidence is limited for treatment with an antidepressant alone or with an antipsychotic alone”.
Since 2015, a few more studies may have been conducted that might lead to a different conclusion regarding treatment with an antidepressant alone or with the combination of an antidepressant and an antipsychotic, as well as with other psychopharmacological agents, such as mifepristone.
Another important clinical issue is that differences in response to specific treatments may be explained in relation to non‐response to prior treatment(s). In generalising from observations across medical disciplines, it would be expected that patients who did not respond to adequate treatment would respond less to subsequent treatment. Some data are available on this topic with regard to pharmacological treatment of major depressive disorder (Sackeim 2001). Two studies showed that a greater degree of treatment resistance predicts an inferior response to ECT (Prudic 1990; Prudic 1996).
Objectives
To compare the clinical efficacy of pharmacological treatments for patients with an acute psychotic depression: antidepressant monotherapy, antipsychotic monotherapy, mifepristone monotherapy, and the combination of an antidepressant plus an antipsychotic versus placebo and/or each other
To assess whether differences in response to treatment in the current episode are related to non‐response to prior treatment
Methods
Criteria for considering studies for this review
Types of studies
We included randomised controlled trials (RCTs) comparing pharmacological treatments for patients with acute psychotic depression.
As we expected to identify very few RCTs assessing treatment of psychotic depression as the primary focus, we decided a priori to also include RCTs assessing treatment of major depression with or without psychotic features. Effects in the subgroup of participants with psychotic features should then be reported separately, irrespective of whether the subgroup with psychotic features was stratified before randomisation.
We applied no language restrictions for included studies.
Types of participants
Participants of any age in any setting (both inpatients and outpatients) had a major depressive disorder and a current episode with psychotic features (delusions and/or hallucinations appearing in the context of a full major depressive episode) according to the Diagnostic and Statistical Manual of Mental Disorders, Third Edition (DSM‐III)/DSM, Third Edition, Revised (DSM‐III‐R)/DSM, Fourth Edition (DSM‐IV)/DSM, Fourth Edition, Text Revision (DSM‐IV‐TR), Fifth Edition (DSM‐5) (APA 1980; APA 1987; APA 1994; APA 2000; APA 2013), or consistent with International Classification of Diseases (ICD) codes for the same.
We also included studies in which participants had comorbidities, as comorbidity was not a reason for exclusion.
As patients with a major depressive episode in the context of a bipolar disorder (bipolar depression) are at increased risk of switching to mania (Licht 2008), we intended to include only trials that assessed participants with unipolar depression. If trials had included participants with both unipolar and bipolar depression, we decided a priori to include only trials for which results in the unipolar group were reported separately, or for which the percentage of participants with bipolar depression did not exceed 20% of the total study population.
Types of interventions
We included any pharmacological treatment of a current (i.e. acute) episode. Treatment had to be given for at least four weeks with the intention of treating the current episode.
When possible, we considered the following pair‐wise comparisons.
Antidepressant versus placebo.
Antipsychotic versus placebo.
Antidepressant versus antidepressant.
Antipsychotic versus antipsychotic.
Antidepressant versus antipsychotic.
Antidepressant plus antipsychotic versus placebo.
Antidepressant plus antipsychotic versus placebo plus antidepressant.
Antidepressant plus antipsychotic versus placebo plus antipsychotic.
Mifepristone versus placebo.
Mifepristone plus antidepressant versus placebo plus antidepressant.
Other psychopharmacological agents versus placebo.
Other psychopharmacological agents plus antidepressant versus placebo plus antidepressant.
Types of outcome measures
Primary outcomes
Efficacy: clinical response of depression based on observer‐rated symptom reduction: reduction of at least 50% on the Hamilton Rating Scale for Depression (HRSD; Hamilton 1960), the Montgomery Åsberg Depression Rating Scale (MADRS; Montgomery 1979), or any other observer depression severity rating scale, or a change score on the Clinical Global Impression‐Change (CGI‐C) of 'much improved' or 'very much improved' (Guy 1976)
Harm: overall dropout rate during acute treatment as a proxy measure of overall acceptability of treatment
Secondary outcomes
Remission of depression as defined in reports and based on HRSD or MADRS or other observer depression severity rating scale. If the study did not report any of the response criteria as defined above, remission as defined here may be used as an alternative
Change from baseline in score on HRSD, MADRS, or any other observer depression severity rating scale, or change in severity on CGI‐C
Quality of life, as defined in reports
Dropout rate due to adverse effects
Search methods for identification of studies
Electronic searches
Search strategies were updated from 2013 onwards.
Previous searches, conducted up to April 2013, for earlier versions of this review ‐ Wijkstra 2005 and Wijkstra 2015 ‐ can be found in Appendix X, with a description of the Cochrane Common Mental Disorders Group (CCMD) controlled trials register (CCMDCTR) provided in Appendix X.
For this update of this review, the CCDANCTR Studies Register was searched (from 2013 to 12 February 2020) using the following terms.
Condition = (depressi* or “affective disorder*” or “affective symptoms”)
AND
Condition or Comorbidity = (psychosis or psychoses or psychotic* or delusion*)
The CCDANCTR Studies Register was searched (all years to 12 April 2013) using the following terms to identify additional untagged references.
Title/Abstract/Keywords = ((depressi* or “affective disorder*” or “affective symptoms”)
AND
Free‐Text = (psychosis or psychoses or psychotic* or delusion* or hallucin* or antipsychotic* or psychotropic*))
An information specialist with CCMD (previously known as the Cochrane Collaboration Depression, Anxiety and Neurosis Group (CCDAN)) ran an updated search (21 February 2020) of the following electronic databases, using relevant subject headings (controlled vocabularies) and search syntax, as appropriate to each resource.
Ovid MEDLINE.
Ovid Embase.
Ovid PsycINFO.
Cochrane Central Register of Controlled Trials (CENTRAL; February 2020, Issue 2 of 12).
Cochrane Common Mental Disorders Controlled Trials Register (CMDCTR).
The international trial registries (ClinicalTrials.gov and World Health Organization (WHO) International Clinical Trials Registry Platform (ICTRP)) were searched via CENTRAL, in the Cochrane Library (all years from 2013 to 12 February 2020).
We applied no restrictions on language or publication status to these searches.
Searching other resources
The reference lists of all studies, related reviews, and relevant conference proceedings were screened, and key study authors contacted.
Data collection and analysis
Selection of studies
In step 1, all abstracts of identified publications were screened independently by two review authors (JL and JW in 2005 and 2015, JK and EL in 2020), and studies were selected if they met the following criteria.
Were randomised controlled trial (RCT).
Included participants with a major depressive disorder.
Investigated the efficacy of pharmacological treatment.
Focused on acute phase treatment (minimum of four weeks' treatment), not on continuation or maintenance treatment.
If any doubt or disagreement arose between the review authors, the publication was included in step 2. Full articles were obtained for the selected abstracts.
In step 2, selected full articles were screened (JL and JW in 2005 and 2015, JK and EL in 2020) according to the following criteria.
Participants with psychotic depression were not excluded.
Results in the subgroup of psychotic depressed participants were reported separately.
If any doubt arose about an article, it was included in step 4.
In step 3, the reference lists of related reviews and of included publications, conference abstracts, and personal communications were searched.
Finally, in step 4, two review authors (JL and JW in 2005 and 2015, JK and EL in 2020) independently reviewed all identified publications according to the full inclusion criteria of the review. Any disagreement was resolved by consensus discussion with another review author (WN).
Data extraction and management
Extracted data included the following: participant characteristics (age, gender, setting: inpatient/outpatient); diagnosis (diagnostic instrument, system of classification, number of bipolar participants); prior treatment for the current episode; intervention; length of illness; suicide attempts; treatment details (treatment period, washout period, additional medication, blood levels, doses); and outcome measures. Data were extracted independently by two review authors (JL and JW in 2005 and 2015; JK and EL in 2020).
All data (from the 2013 review and recent data) were entered into RevMan 5.4 (Review Manager 2020).
Assessment of risk of bias in included studies
In the original version of this review, we assessed methodological quality of included studies using criteria set out in the Cochrane Handbook for Systematic Reviews of Interventions (Alderson 2004); however, after publication of the revised and expanded Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2021), we updated our methods accordingly. Working independently, two review authors (JL and JW in 2005 and 2015; JK and EL in 2020) assessed risk of bias of included studies using the tool described in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2021b). The following items were assessed.
Sequence generation: was the allocation sequence adequately generated?
Allocation concealment: was allocation adequately concealed?
Blinding of participants, personnel, and outcome assessors for each main outcome or class of outcomes: was knowledge of the allocated treatment adequately prevented during the study?
Incomplete outcome data for each main outcome or class of outcomes: were incomplete outcome data adequately addressed?
Selective outcome reporting: were reports of the study free of suggestion of selective outcome reporting?
Other sources of bias: was the study apparently free of other problems that could put it at high risk of bias?
We included quotations from the text of included studies and comments on how we assessed risk of bias; we judged each study to be at low, unclear, or high risk of bias.
See risk of bias figures (Figure 1; Figure 2).
1.

Risk of bias graph: review authors' judgements about each risk of bias item presented as percentages across all included studies.
2.

Risk of bias summary: review authors' judgements about each risk of bias item for each included study.
If disputes arose as to which judgement should be given, resolution was achieved after consultation with the third review author (WN).
Measures of treatment effect
Binary outcomes
For binary efficacy outcomes, such as response, remission, and dropouts, we calculated the risk ratio (RR) (with 95% confidence intervals (CI)) for each comparison using numbers randomly assigned and numbers of events.
Continuous outcomes
For continuously distributed outcomes, we calculated the standardised mean difference (SMD).
We presented skewed and non‐quantitative outcome data descriptively.
Unit of analysis issues
We identified neither cluster‐randomised nor cross‐over trials. However, if found, we would not have included them in our review, as we were interested in differences not between clusters (e.g. clinics) but between drugs or classes of drugs; nor were we interested in the results of a cross‐over phase, as the outcome of the first phase might have had an impact on the outcome of the second (i.e. cross‐over) phase.
In case a study had multiple intervention groups, we included only data for the pair‐wise comparison in question. Further, if a study compared two or more medications of the same type (e.g. venlafaxine, imipramine), we combined data into a single category, for example, the category 'antidepressant'.
Dealing with missing data
We used intention‐to‐treat (ITT) response data in the analyses, as ITT data are less biased, and because they address a more pragmatic and clinically relevant question (Higgins 2021). When necessary, we converted response data from trials into ITT response data by using the total number of randomly assigned participants per group who had started with treatment as the denominator. So participants who had started with medication but were withdrawn before endpoint were assumed not to have experienced response.
When data were missing, we contacted study authors to request the required data.
We used no other imputation techniques to deal with missing data.
Assessment of heterogeneity
First, we evaluated whether clinical homogeneity could be assumed by evaluating any between‐study dissimilarities regarding participants, interventions, and outcome measures. We excluded from the meta‐analysis studies that were considered to threaten the clinical homogeneity assumption.
We used the I² statistic supplied with a 95% CI to assess the magnitude of statistical heterogeneity when values exceeding 0.40 were considered possibly relevant. We did not perform the Q test to determine heterogeneity because of its low power in our meta‐analysis resulting from low numbers of studies in all of our comparisons.
We planned to conduct re‐stated subgroup analyses to explore sources of heterogeneity. We performed no meta‐regression.
Assessment of reporting biases
When data from at least 10 studies became available, we assessed the presence of publication bias by using contour‐enhanced funnel plots in which treatment effects expressed as RR (risk ratio) from individual studies were plotted against each study's sample size. We did not perform Egger's regression test in view of low statistical power in our meta‐analyses, again resulting from the low number of included studies.
It must be noted that asymmetry of funnel plots does not necessarily indicate publication bias but may result from other biases such as inflated results in smaller studies due to poorer methodological quality, or true heterogeneity.
Data synthesis
We used the Mantel‐Haenszel fixed‐effect method to calculate pooled risk ratios with 95% confidence intervals. Risk ratios are preferred over odds ratios because of their more straightforward interpretation (i.e. the number of times the outcome is more likely to occur given one treatment over another). We used fixed‐effect models as most meta‐analyses consisted of a small number of studies, therefore there were insufficient data to estimate random‐effects models.
Subgroup analysis and investigation of heterogeneity
If sufficient data were available, we planned to conduct the following subgroup analyses.
Participants who were non‐refractory to prior treatment(s) during the current episode.
Participants with mood congruent psychotic features only.
Because all psychotically depressed patients are considered to be severely depressed, it was not considered appropriate to evaluate baseline severity in a subgroup analysis.
Sensitivity analysis
If sufficient data were available, we planned to perform the following sensitivity analyses.
Studies focusing on psychotic depression only.
Studies in which participants with psychotic depression were separately randomised.
Studies of lower methodological quality to assess robustness of results.
Smaller versus larger studies.
Summary of findings and assessment of the certainty of the evidence
We created 'Summary of findings' tables, in which we summarised findings of studies comparing:
antidepressant versus placebo;
antipsychotic versus placebo;
antidepressant versus antidepressant;
antipsychotic versus antipsychotic;
antidepressant versus antipsychotic;
antidepressant plus antipsychotic versus placebo;
antidepressant plus antipsychotic versus placebo plus antidepressant; and
antidepressant plus antipsychotic versus placebo plus antipsychotic.
We have presented a separate 'Summary of findings' table for each comparison group. We included the following outcomes: depression response, overall dropout, depression remission, change in depression severity, quality of life, and dropout due to adverse effects as measured between baseline and end of study.
We used the five GRADE considerations (study limitations, consistency of effect, imprecision, indirectness, and publication bias) to assess the certainty of a body of evidence as it relates to studies that contributed data to meta‐analyses for prespecified outcomes. We used methods and recommendations described in Chapter 14 of the Cochrane Handbook for Systematic Reviews of Interventions (Schünemann 2020), along with GRADEpro software (GRADEpro GDT). We justified all decisions to downgrade the certainty of evidence by using footnotes, and we made comments to aid the reader's understanding of the review when necessary.
Two review authors (BV, LR) independently assessed the certainty of evidence and resolved disagreements through discussion or by consultation with a third review author (WN). We justified, documented, and incorporated judgements into reporting of results for each outcome.
Reaching conclusions
We based our conclusions only on findings from the quantitative or narrative synthesis of included studies for this review. We avoided making recommendations for practice, and our implications for research suggest priorities for future research and outline what remaining uncertainties in this area of research.
Results
Description of studies
Results of the search
From the Cochrane Central Register of Controlled Trials (CENTRAL) search conducted in 2004, we identified 1782 publications: MEDLINE 720, Embase 831; total, 3333. With some overlap from 2004, we have since found an additional 326 publications. The first step of screening abstracts of these publications in 2004 resulted in 798 relevant publications (749 from CENTRAL, 38 from MEDLINE, 11 from Embase); in 2011, we found 40 additional studies (3 from CENTRAL, 32 from CCDANCTR‐References Register, 5 from CCDANCTR‐Studies Register). The second step of screening the full articles of these publications in 2004 resulted in identification of 52 publications (47, 3, and 2, respectively) and now 17 (1 from CENTRAL, 13 from CCDANCTR‐References Register, 3 from CCDANCTR‐Studies Register). In 2004, handsearching of reference lists from relevant reviews resulted in identification of one other publication (Bellini 1994), and handsearching of included publications did not lead to identification of any further relevant publications. Now, this resulted in no publications. In 2004, the final fourth step of reviewing these 53 publications resulted in 7 included studies, and in 2004, we added 2 other publications that we knew were in press: 1 by van den Broek (van den Broek 2004a), and the other reporting 2 similar trials (Rothschild 2004a; Rothschild 2004b). Thus, in 2004 in total, we included nine publications with 10 RCTs. Now, in the fourth step, by reviewing 17 publications, we found 2 additional studies to be included.
In additional searches of CCDANCTR and CENTRAL (December 2011 to April 2013), we found another 288 references. Of these 288 references, we did not find any other publication for inclusion.
So, taking together both searches, we found 3947 publications through electronic search strategies, from which we finally included 11 publications with 12 RCTs (see Figure 3).
3.
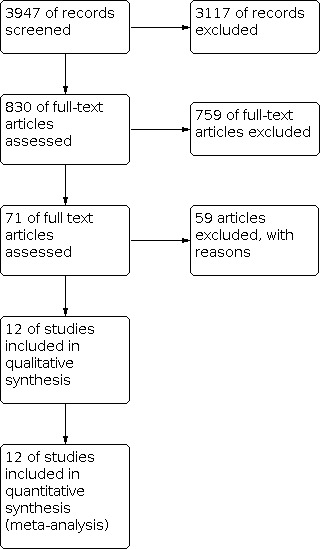
Study flow diagram.
In 2004, 13 studies needed a consensus discussion in the fourth step of the search strategy before they were excluded, and in 2013, 5 additional studies needed such a discussion before they were excluded, resulting in a total of 18 excluded studies (see Excluded studies).
In February 2020, from a search of CENTRAL and the CCMD, we identified a total of 2204 publications. After screening for duplicates, we removed 834 articles from the search and we screened a total of 1370 articles for title and abstract. In the second step, we screened 37 full‐text publications. None of these articles met the inclusion criteria of this review (see Figure 3). Thus, this update in 2021 includes the same studies that were included in the update in 2015 (Wijkstra 2015).
Included studies
See the Characteristics of included studies table for a description of the 12 included RCTs (Anton 1990; Bruijn 1996; Meyers 2009; Mulsant 2001; Rothschild 2004a; Rothschild 2004b; Spiker 1985; Spiker 1988; van den Broek 2004a; Wijkstra 2010; Zanardi 1996; Zanardi 2000).
Design
All studies were RCTs comparing the effects of pharmacological interventions for treatment of psychotic depression or for treatment of depression with and without psychotic features, but with data about participants with psychotic features published separately.
Sample size
Sample sizes were as follows: Anton 1990; 46; Bruijn 1996: 30; Meyers 2009: 259; Mulsant 2001: 54; Rothschild 2004a: 124; Rothschild 2004b: 125; Spiker 1985: 58; Spiker 1988: 27; van den Broek 2004a; 48; Wijkstra 2010: 122; Zanardi 1996: 32; and Zanardi 2000: 22, for a total of 947 participants.
Setting
Most studies included inpatients, except Meyers 2009, Rothschild 2004a, and Rothschild 2004b. In Meyers 2009, 69.1% of participants entered as inpatients. In the studies of Rothschild, participants were treated for at least one week as inpatients.
Seven studies were conducted in the United States (Anton 1990; Meyers 2009; Mulsant 2001; Rothschild 2004a; Rothschild 2004b; Spiker 1985; Spiker 1988), 3 in the Netherlands (Bruijn 1996; van den Broek 2004; Wijkstra 2010), and 2 in Italy (Zanardi 1996; Zanardi 2000).
Participants
All participants fulfilled criteria for major depressive disorder with psychotic features, classified according to a formal classification system (RDC ‐ Research Diagnostic Criteria; DSM‐III; DSM‐IV; DSM‐IV‐TR). Six studies explicitly used a semi‐structured diagnostic interview (van den Broek 2004a: Schedule for Affective Disorders and Schizophrenia (SADS); Bruijn 1996: SADS; Mulsant 2001: Brief Psychiatric Rating Scale (BPRS); Spiker 1985: SADS; Meyers 2009: Structured Clinical Interview for DSM‐IV (SCID IV); Wijkstra 2010: SCID IV). These different procedures could have led to differences in participant categories.
Seven studies included only participants with unipolar psychotic depression. In Zanardi 1996, it was possible to exclude bipolar participants from the data. In Anton 1990, 15.8% (6 out of 38) bipolar participants were included in the data that the study author used in this analysis. However, it is unclear how many of the eight dropouts, who were excluded before analysis, were bipolar participants. The study author was not able to give additional information. Therefore, we decided to assume a random dropout rate. In Spiker 1985, 15.5% of participants were bipolar (Anton 1990; Spiker 1985). In Bruijn 1996 and Zanardi 2000, we were able to exclude bipolar participants with additional information provided by the study authors. Bruijn 1996, Meyers 2009, and Wijkstra 2010 described types of psychotic symptoms in greater detail. Spiker 1985 included only participants with mood congruent delusions. This indicates some heterogeneity in diagnosis with regard to bipolarity.
Interventions
Antidepressant versus placebo was compared in one study (Spiker 1988): amitriptyline (three weeks at least 150 mg) versus placebo; treatment period was four weeks
Antipsychotic versus placebo was examined in one arm of the two identical studies of Rothschild (Rothschild 2004a; Rothschild 2004b): olanzapine (mean 11.9 mg; respectively, 14.0 mg) versus placebo; treatment period was eight weeks
Antidepressant versus antidepressant was examined in five studies. In van den Broek 2004: imipramine (plasma levels imipramine plus its metabolite desmethylimipramine 192 to 521 ng/mL) versus fluvoxamine (plasma level 109 to 325 ng/mL); treatment period was four weeks after predefined blood levels were reached; in Bruijn 1996: imipramine (plasma levels imipramine plus its metabolite desmethylimipramine 199 to 400 ng/mL) versus mirtazepine (plasma level 49 to 93 ng/mL); treatment period was four weeks after predefined blood levels were reached; in Zanardi 1996: sertraline (150 mg from day 8) versus paroxetine (50 mg from day 8); treatment period was five weeks; in Zanardi 2000: venlafaxine (300 mg from day 8) versus fluvoxamine (300 mg from day 8); treatment period was five weeks; and in one arm of Wijkstra 2010: imipramine (plasma levels imipramine plus its metabolite desmethylimipramine 200 to 300 ng/mL) versus venlafaxine (375 mg); treatment period was seven weeks
Antipsychotic versus antipsychotic was not compared in any study
Antidepressant versus antipsychotic was examined in one arm of Spiker 1985: amitriptyline (mean dose 218 mg; 130 to 500 ng/mL) versus perphenazine (mean dose 50 mg; blood level 19 to 113 ng/mL); treatment period was four weeks
The combination of antidepressant plus antipsychotic versus placebo was compared in one arm of both identical studies of Rothschild (Rothschild 2004a; Rothschild 2004b): olanzapine (12.4 mg; respectively, 13.9 mg) plus fluoxetine (23.5 mg; respectively, 22.6 mg) versus placebo; treatment period was eight weeks
The combination of antidepressant plus antipsychotic versus placebo plus antidepressant was compared in four studies. In Anton 1990: amitriptyline (150 to 250 mg) plus perphenazine (24 to 40 mg) versus amoxapine (300 to 500 mg); treatment period was four weeks; in Mulsant 2001: nortriptyline (mean 63 mg) plus perphenazine (mean 19 mg) versus nortriptyline (mean dose 76 mg); treatment started with nortriptyline, and once nortriptyline blood level was between 50 and 50 ng/mL, random assignment followed; treatment period with nortriptyline plus perphenazine (or placebo) after random assignment was 2 to 16 weeks (total treatment at least four weeks); in one arm of Spiker 1985: amitriptyline (mean 170 mg; 18 to 128 ng/mL) plus perphenazine (mean 54 mg; 157 to 690 ng/mL) versus amitriptyline (mean 218 mg; 130 to 500 ng/mL); treatment period was four weeks; and in two arms of Wijkstra 2010: venlafaxine (375 mg) plus quetiapine (600 mg) versus imipramine (plasma levels imipramine plus its metabolite desmethylimipramine 200 to 300 ng/mL) and versus venlafaxine (375 mg); treatment period was 7 weeks
The combination of antidepressant plus antipsychotic versus placebo plus antipsychotic was examined in three studies. In one arm of both identical studies of Rothschild (Rothschild 2004a; Rothschild 2004b): olanzapine (12.4 mg; respectively, 13.9 mg) plus fluoxetine (23.5 mg; respectively, 22.6 mg) versus olanzapine (mean 11.9 mg; respectively, 14.0 mg); treatment period was eight weeks; in one arm of Spiker 1985: amitriptyline (mean 170 mg; 18 to 128 ng/mL) plus perphenazine (mean 54 mg; 157 to 690 ng/mL) versus perphenazine (mean dose 50 mg; blood level 19 to 113 ng/mL); treatment period was four weeks; and in Meyers 2009: olanzapine (15 to 20 mg) plus sertraline (150 to 200 mg) versus olanzapine (15 to 20 mg); treatment period was 12 weeks
Most of these studies had a washout period before the start of treatment or random assignment, varying from four to seven days. One study had a washout period of two weeks (Spiker 1988). Because of the design of Mulsant 2001 (all participants used nortriptyline before random assignment to additional perphenazine or placebo), this study was considered a trial without a washout period. The two trials Rothschild 2004a and Rothschild 2004b had a 'screening period' of three to nine days, leaving unclear whether this was a period in which medication was not allowed. In Meyers 2009, psychotropics were tapered before random assignment without a washout period. So, heterogeneity is seen in the medication‐free period before treatment.
The dosage of psychotropics used in different trials was considered reasonably adequate. However, differences in dosing strategies led to possible bias. In four studies of TCAs, doses were adjusted according to predefined therapeutic plasma levels (Bruijn 1996; Mulsant 2001; van den Broek 2004a; Wijkstra 2010). In Spiker 1985, dose adjustment was not based on plasma levels, but afterwards it was found that plasma levels were therapeutic in most participants, although plasma levels of participants receiving TCAs alone were lower compared with those of participants receiving TCA plus antipsychotic. In the two other trials with TCAs, no plasma levels were determined. Dosages in Spiker 1988 were at least 150 mg/d, but only during three of the four study weeks. In Anton 1990, participants received at least 150 mg/d from the third day of the four‐week study period. Amitriptyline 150 mg/d is in the low range of an adequate dosage. In van den Broek 2004 and Bruijn 1996, SSRIs (fluvoxamine and mirtazapine) were dosed according to predefined plasma levels. Fixed doses were used in the studies of Zanardi (Zanardi 1996; Zanardi 2000): sertraline 150 mg, paroxetine 50 mg, venlafaxine 300 mg, and fluvoxamine 300 mg. In the studies of Rothschild (Rothschild 2004a; Rothschild 2004b), doses were clinically adjusted: olanzapine 5 to 20 mg and fluoxetine 20 to 80 mg.
Differences in additional medication strategies were also noted. In most studies, additional medication was used, such as benzodiazepines (flurazepam up to 30 mg or lorazepam as needed) and anticholinergics. In van den Broek 2004a and Bruijn 1996, some participants were treated with additional haloperidol, if clinically needed. With information provided by the study authors, we were able to identify these participants in the results and we counted them as dropouts, as in these studies the focus was the comparison of two antidepressants.
Outcomes
The primary efficacy outcome was the response rate in each study. It was not possible to transfer study authors' defined response data into rates based on one definition (i.e. at least 50% reduction in HRSD score). Some study authors used response definitions based on what is often considered remission. In addition, some study authors included psychotic symptoms in their response definition. In the absence of a better option, we decided to use the response data as reported by study authors.
Differences in outcome measures were noted. In most trials, the HRSD was used as an outcome measure. However, different versions of the HRSD were used, and study authors used different definitions of response. In six trials, the response definition was a reduction of at least 50% in HRSD score compared with baseline (Anton 1990; Bruijn 1996; Rothschild 2004a; Rothschild 2004b; van den Broek 2004; Wijkstra 2010). In four studies, study authors' definition of response was actually comparable with a frequently used definition of remission (Spiker 1985; Spiker 1988; Zanardi 1996; Zanardi 2000).
In five studies, the response definition also included psychotic symptoms (Meyers 2009; Mulsant 2001; Spiker 1985; Zanardi 1996; Zanardi 2000). Bruijn 1996 and van den Broek 2004a used a response criterion of HRSD‐17 less than 50%; Anton 1990 50% or less; Rothschild 2004a and Rothschild 2004b HRSD‐24 less than 50%; Wijkstra 2010 less than 50% plus less than 15%; Spiker 1985 HRSD‐17 less than 7 and no delusions; Spiker 1988 HRSD‐17 less than 7 and not psychotic, or HRSD‐17 6.5 to 9.5 and not psychotic and a third less of score at entry; and Mulsant 2001 HRSD‐17 less than 11 and BPRS score for items 11, 12, and 15 of 1 or 2 (i.e. not psychotic). In Meyers 2009, remission was defined as HRSD‐17 score less than 11 and no psychosis; Zanardi 1996 HRSD‐21 less than 8 and DDERS (Dimensions of Delusional Experience Rating Scale) of 0; and Zanardi 2000 HRSD‐21 less than 9 and DDERS of 0. In Zanardi 1996 and Zanardi 2000, no minimum HRSD score was applied as an inclusion criterion.
In addition to response rates based on the above criteria used by study authors, several studies reported remission rates separately (van den Broek 2004a: HRSD‐17 < 8; Rothschild 2004a and Rothschild 2004b: Hamilton Depression Rating Scale (HAMD)‐24 < 9 for two consecutive visits; Wijkstra 2010: HRSD‐17 < 8). In two studies, trial authors' definition of response is the same as what is nowadays considered the definition of remission (Spiker 1985 and Spiker 1988: HRSD‐17 < 7).
Dropout rates
Overall dropout rates for the primary outcome were available for all studies. Dropout rates due to adverse effects were available for six studies (Anton 1990; Meyers 2009; Mulsant 2001; Spiker 1985; van den Broek 2004a; Wijkstra 2010). Dropout rates due to adverse effects were not based on ITT analysis for three studies (Bruijn 1996; Rothschild 2004a; Rothschild 2004b); were unavailable for one study (Spiker 1988); and were the same as overall dropout rates for two studies (Zanardi 1996; Zanardi 2000). Dropouts specifically due to mortality or suicide were not reported, so we decided to extract only overall dropout rates.
Overall dropout rates for included studies varied from 9% to 45%. In Bruijn 1996, the reported dropout rate was 10%. However, when haloperidol‐treated participants were included as dropouts, as was our approach, the dropout rate was 40%. In the two multi‐centre trials with olanzapine/fluoxetine (Rothschild 2004a; Rothschild 2004b), dropout rates were 41%, and even higher non‐completion rates (56%) were reported. Most often, no statistically significant differences in overall dropout rates were noted between any of the treatments, neither in individual studies nor after pooling of studies.
Prior treatment
Bruijn 1996 provided information on prior treatment in the current episode. However, these data were not available for the subgroup with psychotic depression. In Wijkstra 2010, some data about prior treatment are available. The other studies did not provide information on prior treatment of the current episode. Therefore, it was not possible to examine differences in treatment response in relation to non‐response to prior treatment(s).
Excluded studies
See Characteristics of excluded studies table.
Reasons for exclusion include open study design, problems with diagnosis (exclusion of psychotic features, unclear diagnostic procedure, no adequate data on the MDD subgroup with psychotic features, > 20% bipolar participants included, and no additional data to exclude bipolar participants), too few participants with psychotic depression (N = 3), problems with treatments (continuation of mood stabilisers, additional treatment with other psychotropics), pooled analysis of studies that were included in the previously updated review, and no possibility to extract ITT data.
In the most recent search, in 2020, we identified three new articles on the treatment of psychotic depression with mifepristone (Blasey 2011; Block 2017; Block 2018). We excluded all three studies because in addition to mifepristone or placebo, as all patients received an unspecified antidepressant in a non‐standardised way as additional medication.
In Blasey 2011, participants received mifepristone or placebo during the first seven days and "throughout the study [...] one of the following antidepressant medications at standard clinical doses: bupropion, venlafaxine, fluoxetine, citalopram, escitalopram, mirtazepine, paroxetine, or sertraline". In Block 2017, participants also received mifepristone or placebo during the first seven days and from day 8 to day 56, "a single FDA‐approved [but further unspecified] antidepressant". Block 2018 pooled data from five previous studies and reported that higher plasma levels of mifepristone were associated with better response for psychotic symptoms. In this study, participants also received mifepristone or placebo during the first seven days and "an FDA approved [but further unspecified] antidepressant for 7 or 8 weeks" during the 8‐week trial. As in none of these three studies treatment with the antidepressant was standardised (i.e. all patients receiving the same regimen with a single but also the same antidepressant for all patients at a fixed dose and during a fixed period, e.g. throughout the whole study), the different antidepressant regimens may have obscured the effect of mifepristone; therefore, these studies were not enrolled in the current review.
Risk of bias in included studies
See Figure 1 and Figure 2 and the Characteristics of included studies table.
Allocation
All included studies were RCTs. Randomisation was fully described in two studies (van den Broek 2004a; Wijkstra 2010), and it was described in part in three studies (Meyers 2009; Spiker 1985; Spiker 1988). In the other seven studies, randomisation was mentioned as such, but methods of randomisation were not delineated.
(Random sequence generation: 7 studies unclear risk and 5 low risk of bias; allocation concealment: 10 unclear risk and two low risk of bias.)
Blinding
All studies were double‐blind studies, but blinding itself was not always adequately described in the methods section of the study. Blinding was adequately described in eight studies (Anton 1990; Bruijn 1996; Meyers 2009; Mulsant 2001; Spiker 1985; Spiker 1988; van den Broek 2004a; Wijkstra 2010). The method of blinding was not explicitly described in the remaining four studies (Rothschild 2004a; Rothschild 2004b; Zanardi 1996; Zanardi 2000).However, as study authors explicitly state the double‐blind condition of their studies, we have no reason to doubt that these double‐blind conditions pertained to both investigators/assessors and participants.
(Blinding of participants: 2 studies unclear risk and 10 studies low risk of bias; blinding of personnel: 2 studies unclear risk and 10 studies low risk of bias; blinding of outcome assessors: 5 studies unclear risk and 7 studies low risk of bias.)
Incomplete outcome data
The primary efficacy outcome was the response rate (of depression). It was not possible to transfer study authors' defined response data into rates based on a single definition (i.e. 50% reduction in HRSD score). Four studies used response definitions based on what is often considered remission (e.g. HAMD < 8 or 10) (Meyers 2009; Spiker 1985; Zanardi 1996; Zanardi 2000); in the absence of a better option (no response data according to our definition), we decided to use the response data as defined by study authors. In addition, six studies included psychotic symptoms in their response definition (Meyers 2009; Mulsant 2001; Spiker 1985; Spiker 1988; Zanardi 1996; Zanardi 2000); in these cases we also followed the study authors, with the preference to use response of depression rather than response of psychosis.
In 8 of the 12 studies, we recalculated intention‐to‐treat response rates using all randomly assigned participants as the denominator. Of 46 participants in Anton 1990, 8 were dropped from the study before receiving two full weeks of active medication. These participants were excluded from analysis by the study author, but we included them in our ITT analysis. In van den Broek 2004a and Bruijn 1996, we counted participants treated with haloperidol as dropouts because haloperidol treatment for these participants was started after random assignment, in part to keep them in the study. Thus, treatment with haloperidol is considered a potential bias with regard to the effect of study medication as well as dropouts. Mulsant 2001 excluded six dropouts after random assignment to perphenazine or placebo. We included them in our ITT analysis. In Rothschild 2004a and Rothschild 2004b, 7% and 9%, respectively, of randomly assigned participants were lost before baseline plus one visit. These participants were excluded from the study analysis but were included in our ITT analysis. In both studies of Spiker (Spiker 1985; Spiker 1988), seven dropouts were excluded from their analyses, but we included them in our ITT analysis.
For the secondary outcome of change in symptom severity, we decided to refrain from using continuous data from observer depression severity scales. In two studies (Bruijn 1996; van den Broek 2004), continuous data were available for the total group (psychotic and non‐psychotic) but not for the psychotic subgroup. In four studies, continuous data were not available (Anton 1990; Mulsant 2001; Spiker 1988; Zanardi 1996). In Spiker 1985, baseline and final mean HRSD data were given, but it was impossible to exclude bipolar participants from these data and convert the data to ITT data. In the studies of Rothschild (Rothschild 2004a; Rothschild 2004b), only last observation carried forward (LOCF) continuous data were available, and in this study, dropout was very high. Pooling of data from the three remaining studies are useless because three different comparisons are studied: antidepressant versus antidepressant with no ITT data (Zanardi 2000); antidepressant + antipsychotic versus antipsychotic (Meyers 2009); and antidepressant + antipsychotic versus antidepressant (Wijkstra 2010).
(Incomplete data: 1 study high risk, 3 studies unclear risk, and 8 studies low risk of bias.)
Selective reporting
All studies used generally accepted outcomes. We judged Meyers 2009 and Wijkstra 2010 to be at low risk of reporting bias, as the protocols were available and no post‐protocol changes in outcome measures had been made (except in Wijkstra 2010, which used remission as an outcome not stated in the protocol but with reasonable argument that remission has become a generally accepted outcome measure). The remaining ten studies were at unclear risk of reporting bias.
Other potential sources of bias
The subgroup of psychotic depressed participants was not stratified before random assignment in any of these studies (Bruijn 1996; Spiker 1988; van den Broek 2004a). Subgroup analyses are more likely to be carried out if the results for primary outcomes are not significant, or if they are more likely to be reported for groups for whom a significant result was found. However, in this review, we ourselves analysed subgroup data in studies that primarily reported on participants with depression with and without psychotic features.
As described under Description of studies, clinical heterogeneity is seen within the results.
In six studies, response definition included response with regard to psychotic symptoms (Meyers 2009; Mulsant 2001; Spiker 1985; Spiker 1988; Zanardi 1996; Zanardi 2000), and in the other studies (Anton 1990; Bruijn 1996; Rothschild 2004a; Rothschild 2004b; van den Broek 2004; Wijkstra 2010), response rates concerned only change in severity of depression. In our analysis, we looked only for response of depression leading to a possible bias favouring antidepressants over antipsychotics.
(Other biases together: 6 studies high risk and 6 studies unclear risk of bias.)
Effects of interventions
See: Table 1; Table 2; Table 3; Table 4; Table 5; Table 6; Table 7; Table 8
All included studies reported effects on depressive symptoms as their primary outcome. The way these were characterised for each study is listed in the Included studies section above and in the 'Summary of findings' tables. Further details on the scales are given under Characteristics of included studies.
For the secondary outcome of change in symptom severity, extracting continuous data from observer depression severity scales was not possible because in seven studies, we were not able to convert these data according to intention‐to‐treat analysis (Anton 1990; Bruijn 1996; Rothschild 2004a; Rothschild 2004b; Spiker 1985; Spiker 1988; van den Broek 2004a), and in two other studies, no continuous data were reported (Zanardi 1996; Zanardi 2000).
Comparison 1. Antidepressant versus placebo
Primary outcomes
1.1 Efficacy response rates
Spiker 1988 compared amitriptyline with placebo. The sample size (N = 27) was very small. There is insufficient data to conclude whether there is a difference between amitryptiline and placebo (risk ratio (RR) 8.40, 95% confidence interval (CI) 0.50 to 142.27; participants = 27, studies = 1; Analysis 1.1) Certainty of the evidence is rated very low.
1.1. Analysis.

Comparison 1: Antidepressant versus placebo, Outcome 1: Clinical response
1.2 Harm: overall dropout rate during acute treatment
Spiker 1988 There is insufficient date to conclude that there is no difference in dropout rates between amitriptyline and placebo (RR 1.24, 95% CI 0.34 to 4.51; participants = 27, studies = 1; Analysis 1.2) Certainty of the evidence is rated very low.
1.2. Analysis.

Comparison 1: Antidepressant versus placebo, Outcome 2: Dropouts
Secondary outcomes
1.3 Remission
No study measured this outcome.
1.4 Change from baseline
No study measured this outcome.
1.5 Quality of life
No study measured this outcome.
1.6 Dropout rate due to adverse effects
No study measured this outcome.
Comparison 2. Antipsychotic versus placebo
Primary outcomes
2.1 Efficacy response rates
Two identical studies compared an antipsychotic with placebo (Rothschild 2004a; Rothschild 2004b). Pooling these studies shows it is very uncertain to conclude that olanzapine is not more effective than placebo (RR 1.13, 95% CI 0.74 to 1.73; P = 0.57; Analysis 2.1). Certainty of the evidence is rated very low.
2.1. Analysis.

Comparison 2: Antipsychotic versus placebo, Outcome 1: Clinical response
2.2 Harm: overall dropout rate during acute treatment
Meta‐analysis of two studies (Rothschild 2004a; Rothschild 2004b) show it is very uncertain whether there is no difference in dropout rates between olanzapine and placebo groups (RR 0.79, 95% CI 0.57 to 1.08; P = 0.14; Analysis 2.2). Certainty of the evidence is rated very low.
2.2. Analysis.

Comparison 2: Antipsychotic versus placebo, Outcome 2: Dropouts
Secondary outcomes
2.3 Remission
No study measured this outcome.
2.4 Change from baseline
For the secondary outcome, change in symptom severity, it was not possible to extract continuous data from observer depression severity scales; we were not able to convert these data according to ITT analysis (Rothschild 2004a; Rothschild 2004b).
2.5 Quality of life
No study measured this outcome.
2.6 Dropout rate due to adverse effects
No study measured this outcome.
Comparison 3. Antidepressant versus antidepressant
Primary outcomes
3.1 Efficacy response rates
Five studies compared two different antidepressants directly (Bruijn 1996; van den Broek 2004a; Wijkstra 2010; Zanardi 1996; Zanardi 2000). Due to heterogeneity in the types of antidepressants, it was not feasible to perform a meta‐analysis. The evidence as a whole was rated of very low certainty. Therefore, it is uncertain whether there are any differences in effectiveness between antidepressants.
van den Broek 2004a found that imipramine may be more effective than fluvoxamine (RR 2.10, 95% CI 1.06 to 4.17; Analysis 3.1), Bruijn 1996 found that imipramine may be more effective than mirtazapine (RR 3.00, 95% CI 1.01 to 8.95; Analysis 3.1), and Zanardi 1996 found that sertraline may be more effective than paroxetine (RR 3.37, 95% CI 1.19 to 9.57; Analysis 3.1). The other two studies found no difference between the two study arms (Wijkstra 2010; Zanardi 2000). Zanardi 2000 found no difference between fluvoxamine and venlafaxine (RR 1.50, 95% CI 0.82 to 2.75; Analysis 3.1). In the largest study (N = 81), Wijkstra 2010 found no difference between imipramine and venlafaxine (RR 1.57, 95% CI 0.93 to 2.67; Analysis 3.1).
3.1. Analysis.

Comparison 3: Antidepressant versus antidepressant, Outcome 1: Clinical response
3.2 Harm: overall dropout rate during acute treatment
Two different antidepressants were compared directly with each other in five studies (Bruijn 1996; van den Broek 2004a; Wijkstra 2010; Zanardi 1996; Zanardi 2000). Due to heterogeneity between studies (i.e. in all five studies, different antidepressants were used), it was not feasible to perform a meta‐analysis.
None of the five studies found evidence of a difference between antidepressants.The evidence as a whole was rated of very low certainty. Therefore, it is uncertain whether there are any differences in drop out rate between antidepressants. In the largest study (N = 81), Wijkstra 2010 found no difference between imipramine and venlafaxine (RR 0.81, 95% CI 0.33 to 2.03; Analysis 3.2). Bruijn 1996 noted no difference between imipramine and mirtazapine (RR 0.50, 95% CI 0.19 to 1.31; Analysis 3.2). van den Broek 2004a reported no difference between imipramine and fluvoxamine (RR 2.00, 95% CI 0.40 to 9.95; Analysis 3.2), and Zanardi 1996 noted no difference between sertraline and paroxetine (RR 0.20, 95% CI 0.01 to 3.74; Analysis 3.2). In the final study (Zanardi 2000), investigators reported no difference between fluvoxamine and venlafaxine (RR 0.07, 95% CI 0.00 to 1.20; Analysis 3.2).
3.2. Analysis.

Comparison 3: Antidepressant versus antidepressant, Outcome 2: Dropouts
Secondary outcomes
3.3 Remission
No study measured this outcome.
3.4 Change from baseline
Pooling was not possible because different antidepressants were used.
3.5 Quality of life
No study measured this outcome.
3.6 Dropout rate due to adverse effects
No study measured this outcome.
Comparison 4. Antipsychotic versus antipsychotic
No study compared an antipsychotic with another antipsychotic.
Comparison 5. Antidepressant versus antipsychotic
Primary outcomes
5.1 Efficacy response rates
Spiker 1985 compared an antidepressant with an antipsychotic and found no evidence of a difference between perphenazine and amitriptyline. However, the sample size (N = 36) was very small (RR 2.09, 95% CI 0.64 to 6.82; Analysis 4.1; Certainty of the evidence is rated very low.
4.1. Analysis.

Comparison 4: Antidepressant versus antipsychotic, Outcome 1: Clinical response
5.2 Harm: overall dropout rate during acute treatment
Spiker 1985 also found no evidence of difference in dropout rates between perphenazine and amitriptyline groups but again the evidence is limited by a very small sample size(RR 1.79, 95% CI 0.18 to 18.02; ) Certainty of the evidence is rated very low.
Secondary outcomes
5.3 Remission
No study measured this outcome.
5.4 Change from baseline
For the secondary outcome, change in symptom severity, it was not possible to extract continuous data from observer depression severity scales because in the only study that performed this comparison (Spiker 1985), we were not able to convert these data according to intention‐to ‐treat analysis.
5.5 Quality of life
No study measured this outcome.
5.6 Dropout rate due to adverse effects
No study measured this outcome.
Comparison 6. Antidepressant plus antipsychotic versus placebo
Primary outcomes
6.1 Efficacy response rates
Two identical studies compared the combination of fluoxetine and olanzapine versus placebo (Rothschild 2004a; Rothschild 2004b). Pooling these studies suggests it is very uncertain to conclude whether or not the combination of an antidepressant with an antipsychotic is more efficacious than placebo alone (RR 1.86, 95% CI 1.23 to 2.82; P = 0.003; Analysis 5.1). Certainty of the evidence is rated very low.
5.1. Analysis.

Comparison 5: Antidepressant plus antipsychotic versus placebo, Outcome 1: Clinical response
6.2 Harm: overall dropout rate during acute treatment
Meta‐analysis of two studies ‐ Rothschild 2004a; Rothschild 2004b ‐ found no evidence of difference in dropout rates (RR 0.75, 95% CI 0.48 to 1.18; P = 0.21; Analysis 5.2). However, substantial heterogeneity was present (I² = 76%): in Rothschild 2004b, the dropout rate was higher for placebo, and in Rothschild 2004a, it was higher for olanzapine. Certainty of the evidence is rated very low.
5.2. Analysis.

Comparison 5: Antidepressant plus antipsychotic versus placebo, Outcome 2: Dropouts
Secondary outcomes
6.3 Remission
No study measured this outcome.
6.4 Change from baseline
For the secondary outcome, change in symptom severity, it was not possible to extract continuous data from observer depression severity scales because for these two studies (Rothschild 2004a; Rothschild 2004b), we were not able to convert these data according to intention‐to ‐treat analysis.
6.5 Quality of life
No study measured this outcome.
6.6 Dropout rate due to adverse effects
No study measured this outcome.
Comparison 7. Antidepressant plus antipsychotic versus placebo plus antipsychotic
Primary outcomes
7.1 Efficacy response rates
Four studies compared the combination of an antidepressant plus an antipsychotic with antipsychotic monotherapy (Meyers 2009; Spiker 1985; Rothschild 2004a; Rothschild 2004b). Pooling of data from all four studies suggests that the combination of an antidepressant with an antipsychotic may be more efficacious than an antipsychotic alone (RR 1.83, 95% CI 1.40 to 2.38; P = 0.00001; Analysis 6.1). Certainty of the evidence is rated low.
6.1. Analysis.

Comparison 6: Antidepressant plus antipsychotic versus placebo plus antipsychotic, Outcome 1: Clinical response
7.2 Harm: overall dropout rate during acute treatment
Pooling of all four studies ‐ Meyers 2009; Rothschild 2004a; Rothschild 2004b; Spiker 1985 ‐ did not reveal a difference in dropout rates between the combination of an antidepressant and an antipsychotic and an antipsychotic alone (RR 0.79, 95% CI 0.63 to 1.01; P = 0.06; Analysis 6.2). However, there was considerable heterogeneity (I² = 63%), likely caused by the identical Rothschild 2004a and Rothschild 2004b studies, in which dropout rates were very high but also different between the two studies. Certainty of the evidence is rated very low.
6.2. Analysis.

Comparison 6: Antidepressant plus antipsychotic versus placebo plus antipsychotic, Outcome 2: Dropouts
Secondary outcomes
7.3 Remission
No study measured this outcome.
7.4 Change from baseline
For the secondary outcome, change in symptom severity, it was not possible to extract continuous data from observer depression severity scales because in three studies (Rothschild 2004a; Rothschild 2004b; Spiker 1985), we were not able to convert these data according to ITT analysis.
7.5 Quality of life
No study measured this outcome.
7.6 Dropout rate due to adverse effects
No study measured this outcome.
Comparison 8. Antidepressant plus antipsychotic versus placebo plus antidepressant
Primary outcomes
8.1 Efficacy response rates
Four studies (five comparisons) compared the combination of an antidepressant plus an antipsychotic with antidepressant monotherapy (Anton 1990; Mulsant 2001; Spiker 1985; Wijkstra 2010). Pooling of these four studies with five analyses suggests that the combination of an antidepressant plus an antipsychotic may be more effective than an antidepressant alone (RR 1.42, 95% CI 1.11 to 1.80; P = 0.002; Analysis 7.1). Certainty of the evidence is rated low.
7.1. Analysis.

Comparison 7: Antidepressant plus antipsychotic versus placebo plus antidepressant, Outcome 1: Clinical response
The unplanned subgroup analysis of the three studies that compared the same antidepressant in both arms (Mulsant 2001; Spiker 1985; Wijkstra 2010) also suggests that the combination of an antidepressant with an antipsychotic is more effective than the antidepressant alone (RR 1.70, 95% CI 1.19 to 2.43; P = 0.003; Analysis 8.1). Certainty of the evidence is rated low.
8.1. Analysis.

Comparison 8: Antidepressant plus antipsychotic versus placebo plus the same antidepressant, Outcome 1: Clinical response
8.2 Harm: overall dropout rate during acute treatment
It is very uncertain whether there were any differences between an antidepressant plus an antipsychotic versus an antidepressant alone after pooling of all four studies (RR 0.91, 95% CI 0.55 to 1.50; P = 0.69; Analysis 7.2). Certainty of the evidence is rated very low.
7.2. Analysis.

Comparison 7: Antidepressant plus antipsychotic versus placebo plus antidepressant, Outcome 2: Dropouts
In the unplanned subgroup analysis of the three studies that compared the same antidepressant in both arms (Mulsant 2001; Spiker 1985; Wijkstra 2010), it was also every uncertain whether there were any differences between interventions (RR 1.04, 95% CI 0.52 to 0.91; P = 0.91; Analysis 8.2). Certainty of the evidence is rated very low.
8.2. Analysis.

Comparison 8: Antidepressant plus antipsychotic versus placebo plus the same antidepressant, Outcome 2: Dropouts
Secondary outcomes
8.3 Remission
No study measured this outcome.
8.4 Change from baseline
For the secondary outcome, change in symptom severity, it was not possible to extract continuous data from observer depression severity scales because we were not able to convert these data according to intention‐to‐treat analysis.
8.5 Quality of life
No study measured this outcome.
8.6 Dropout rate due to adverse effects
No study measured this outcome.
Comparison 9. Mifepristone versus placebo
No study compared mifepristone with a placebo
Comparison 10. Mifepristone plus antidepressant versus placebo plus antidepressant
No study compared mifepristone plus an antidepressant with a placebo plus an antidepressant.
Comparison 11. Other psychopharmacological agents versus placebo
No study compared other psychopharmacological agents with a placebo.
Comparison 12. Other psychopharmacological agents plus antidepressant versus placebo plus antidepressant
No study compared this.
Subgroup and sensitivity analyses
Analysis 8.1 and Analysis 8.2 (in which an antidepressant plus an antipsychotic is compared with the same antidepressant plus placebo) were unplanned sensitivity analyses.
Because of lack of data, other subgroup analyses were not possible.
Discussion
Despite our new search of the literature (screening more than 1370 abstracts and reading 37 full articles), we identified no additional randomised controlled trials (RCTs) from April 2013 up to February 2020 on pharmacological treatment with an antidepressant, an antipsychotic, or another psychopharmacological agent such as mifepristone, or the combination of any of these drugs for participants with a major depressive disorder with a current episode with psychotic features (unipolar psychotic depression).
In addition to nine studies whose main focus was treatment of participants with psychotic depression, we were able to find three studies that reported separately on effects on subgroups of participants with psychotic depression. The authors of two further studies provided us with additional information on results for the subgroup of psychotically depressed participants in their studies of both psychotic and non‐psychotic depressed patients. In our previous review, we invited authors of several studies to provide us with subgroup data if available, so we could use these data in our review, but we did not receive any data.
We also identified several studies on the efficacy of mifepristone. However, we could not include any of these studies because all study participants also received unspecified antidepressants in a non‐standardised way as additional medication.
Thus, for this update, we found no new studies fulfilling the methodological inclusion criteria of this review, which (again) illustrates that this most severe form of depression is highly under‐investigated. One probable reason for this is that it is very difficult to conduct RCTs in patients with psychotic depression. These patients often are not only psychotic but are very anxious or physically ill. Moreover, they are often offered electroconvulsive therapy (ECT) directly without a trial of pharmacological treatment. Finally, many patients with psychotic depression are not able or are reluctant to give informed consent, or they tend to withdraw their consent (Wijkstra 2015).
Summary of main results
Because of lack of included studies from April 2013 to February 2020, results of the previous update from 2015 are still valid (Wijkstra 2015)
Suggestions for the efficacy of the combination of an antidepressant plus an antipsychotic (i.e. fluoxetine plus olanzapine) was derived from two identical placebo‐controlled RCTs (RR 1.86, 95% CI 1.23 to 2.82; P = 0.003; Analysis 5.1) (Rothschild 2004a; Rothschild 2004b)
We also found suggestions for efficacy of the combination of an antidepressant plus an antipsychotic compared to antidepressant monotherapy. Pooling of four studies (with five comparisons) that compared the combination of an antidepressant plus an antipsychotic with antidepressant monotherapy showed a significant difference favouring the combination (RR 1.42, 95% CI 1.11 to 1.80; P = 0.002; Analysis 7.1) (Anton 1990; Mulsant 2001; Spiker 1988; Wijkstra 2010). When the two comparisons with a different antidepressant are left out (Anton 1990; and one comparison in Wijkstra 2010), this difference remains (RR 1.70, 95% CI 1.19 to 2.43; P = 0.003; Analysis 8.1). Thus, it can be concluded that the combination should be preferred over an antidepressant alone
There are also suggestions that the combination of an antidepressant plus an antipsychotic is more effective than antipsychotic monotherapy. Pooling of four studies that compared the combination of an antidepressant plus an antipsychotic with antipsychotic monotherapy shows a difference favouring the combination (RR 1.83, 95% CI 1.40 to 2.38; P = 0.00001; Analysis 6.1) (Meyers 2009; Rothschild 2004a; Rothschild 2004b; Spiker 1985)
No randomised controlled data are available to lead to the conclusion that monotherapy with an antidepressant is efficacious for the treatment of psychotic depression. Only one small study compared monotherapy with an antidepressant (amitriptyline) with placebo and reported no difference (RR 8.40, 95% CI 0.50 to 142.27; Analysis 1.1) (Spiker 1988)
Also no randomised controlled data are available to lead to the conclusion that monotherapy with an antipsychotic alone is efficacious. Two identical studies compared monotherapy with an antipsychotic (olanzapine) with placebo (Rothschild 2004a; Rothschild 2004b). Pooling of these two studies does not reveal a difference (RR 1.13, 95% CI 0.74 to 1.73; Analysis 3.1)
We were not able to collect data on prior treatments. So we could not address the second objective of our review: to assess whether differences in response to treatment in the current episode would be related to non‐response to prior treatment(s)
Regarding acceptability of treatment (dropout, adverse effects) and quality of life, we were only able to collect data only on overall dropout. In all studies except Meyers 2009, no differences in overall dropout rates were reported between any of the treatment groups including placebo, neither in individual studies nor after pooling of studies. With this rather rough measure, we did not find overall differences in overall acceptability of treatments. In Meyers 2009, fewer dropouts were reported in the group treated with the combination of olanzapine plus sertraline than in the group treated with olanzapine alone (RR 1.43, 95% CI 1.08 to 1.88). Study authors suggest that higher attrition rates among participants treated with olanzapine plus sertraline may be attributable to insufficient response in the olanzapine‐treated group
We found indication of heterogeneity dropout data only in instances where the two identical studies of Rothschild were included (Analysis 3.2; Analysis 5.2; Analysis 6.2) (Rothschild 2004a; Rothschild 2004b). This heterogeneity is probably due to high dropout rates, together with differences in dropout rates between the two studies (in one study (Rothschild 2004b), the dropout rate was higher for placebo, and in the other study (Rothschild 2004a), it was higher for olanzapine)
Overall completeness and applicability of evidence
Our conclusions are based on only 12 studies that fulfilled our inclusion criteria. Moreover, in the included studies, only a few different antidepressants and antipsychotics were used. Therefore, it remains unclear whether the above conclusions can be extrapolated to other antidepressants and other antipsychotics.
Strictly spoken, evidence that the combination of an antidepressant plus an antipsychotic is more effective than an antidepressant alone has been obtained in only one RCT (Wijkstra 2010), which compared the combination of venlafaxine and quetiapine with venlafaxine alone.
Evidence that the combination of an antidepressant plus an antipsychotic is more effective than an antipsychotic alone has been obtained in four RCTs for only two antipsychotics. More specifically, the combination of amitriptyline and perphenazine was reported more effective than perphenazine alone in one small study (Spiker 1985), and the three other studies involved combinations with olanzapine: in two studies with fluoxetine (Rothschild 2004a; Rothschild 2004b), and in one study with sertraline (Meyers 2009).
Nearly all participants in these studies were inpatients. This of course is a consequence of the severity of the illness and the fact that in clinical practice, most patients with psychotic depression are treated as inpatients. There is also the problem of patients who were not included in the studies: the most severely ill patients who were not able to give informed consent or who were immediately given ECT. All these points limit the generalisability of study results to all patients with psychotic depression.
These problems ans also the quality of diagnostic assessment can restrict generalisation of findings to all patients with psychotic depression, leaving aside the problem of establishing the diagnosis of psychotic depression in clinical reality.
Quality of the evidence
The strength of this review and its conclusion is that only randomised controlled studies were included, and only intention‐to‐treat (ITT) data were used in the analyses.
Several factors limit our confidence in the findings of this review. There are a number of limitations of the included studies that impact the quality of the evidence they provide. We rated the overall certainty of evidence in the meta‐analyses to be low to very low (using the GRADE criteria).
Most studies were relatively small. Only four RCTs had a more or less adequate sample size: Rothschild 2004a and Rothschild 2004b: with olanzapine 48 and 53 participants, with placebo 51 and 49 participants, but only 25 and 23 olanzapine plus fluoxetine, respectively; Meyers 2009: with olanzapine + sertraline 129 and sertraline 130 participants; and Wijkstra 2010: imipramine 42, venlafaxine 39, and venlafaxine + quetiapine 41 participants.
As with all systematic reviews, publication bias is a potentially serious source of bias. Too few studies were identified to allow further investigation into the possibility of publication bias (e.g. by making funnel plots). However, the fact that in these mostly small trials, five (50%) found a significant difference between two active treatments is suggestive of publication bias.
Allocation concealment, especially in older studies, was not explicitly described. Although we do not assume allocation concealment to be a real bias, this is of course unsure.
Dosages of antidepressants and antipsychotics used in the different trials were considered reasonably adequate. However, differences in dosing strategies were noted, leading to possible bias. Differences in additional medication strategies and differences in treatment periods were also reported (see paragraph about Other potential sources of bias for the different diagnotic procedures and different outcome measures used in the different studies).
We cannot rule out the possibility of differences in the diagnostic assessment of participants and thus in the quality of the diagnosis across studies. Although it was reported in all publications that participants included in the trials fulfilled criteria for a major depressive episode with psychotic features, according to a specified diagnostic classification system (Research Diagnostic Criteria (RDC), Diagnostic and Statistical Manual of Mental Disorders (DSM)‐III or DSM‐IV), one could doubt the reliability of the diagnoses made in some trials. Six studies used a semi‐structured diagnostic interview (Bruijn 1996; Meyers 2009; Mulsant 2001; Spiker 1985; van den Broek 2004; Wijkstra 2010), and only three studies reported types of psychotic features (Bruijn 1996; Meyers 2009; Wijkstra 2010). This leaves open the possibility that for instance the judgement that a feeling or idea of guilt was a guilt delusion was drawn differently across the studies in this review. A comparable diagnostic problem may have played a role in the judgement of whether a participant had a psychotic depression in the course of unipolar disorder or bipolar disorder. Finally, Mulsant 2001 included a geriatric sample with a mean age of 72 years, leading to the possibility that dementia or another neurological disorder was part of the diagnosis in some participants.
We could use only one outcome measure regarding efficacy: response rates as defined by study authors. It was impossible to recalculate these response rates into a standard response rate based on a single definition (e.g. reduction on the Hamilton Rating Scale for Depression (HRSD)‐17 of at least 50% compared with baseline), as many studies used other versions of the HRSD. Moreover, several studies used response definitions that are commonly used for the definition of remission.
In addition, the different studies did not use other outcome measures to assess remission rates or quality of life.
Potential biases in the review process
One problem is that there does not exist a key word (Mesh Term) for psychotic depression. Therefore, we had to search all RCTs involving depression whether included participants had depression with psychotic features, or whether such participants had been part of the group of included participants and were reported as a separate subgroup. We anticipated in the first version of this review that we might have missed one or more studies. However, we did not receive any information that we had missed any study. Therefore, in this update, we are now rather sure that we indeed have included all published studies.
In three studies (Bruijn 1996; Spiker 1988; van den Broek 2004a), the subgroup of psychotic depressed participants was part of a greater group of participants with psychotic and non‐psychotic depression, although the subgroups were not stratified before random assignment.
Another potential problem, which was not taken into account in our a priori protocol before this systematic review was performed, is that in five studies, the response definition included response with regard to psychotic symptoms (Meyers 2009; Mulsant 2001; Spiker 1985; Spiker 1988; Zanardi 2000), while in the other studies (Anton 1990; Bruijn 1996; Rothschild 2004a; Rothschild 2004b; van den Broek 2004; Wijkstra 2010; Zanardi 1996), the response definition concerned only change in severity of depression. As in our analysis, we only looked for the response of depression, and this may have lead to possible bias favouring antidepressants over antipsychotics.
Agreements and disagreements with other studies or reviews
In a review of practice guidelines regarding treatment for psychotic depression (Wijkstra 2007), we found different recommendations based on slightly different studies; most were not re‐analysed. Two guidelines were cautious in their recommendations (Multidisciplinaire Richtlijn Depressie 2013; NICE 2009). NICE 2009 recommended that "augmenting an [antidepressant] AD with an [antipsychotic] AP should be considered"; and Multidisciplinaire Richtlijn Depressie 2013 recommended "starting treatment with a [tricyclic antidepressant] TCA and if after 4 weeks there is still no response, adding an AP or starting with the combination of a TCA and an AP are reasonable options". The other reviewed guidelines, including the APA 2010 (now updated with no differences regarding treatment for psychotic depression), recommend using the combination of an AD and an AP. None of these guidelines recommended monotherapy with an antipsychotic. These recommendations were not based on a systematic review of data from all available RCTs; they were based on a few studies ‐ some randomised and some open non‐randomised.
Another review and meta‐analysis on the treatment of psychotic depression has been published (Farahani 2012), focusing on the comparison of antidepressant or antipsychotic monotherapy with combination treatment. Five studies were included (Anton 1990; Künzel 2008; Mulsant 2001; Spiker 1985; Wijkstra 2010). In our review, for this particular comparison, we excluded Künzel 2008 because it is unclear whether less or more than 20% of included participants in the ITT group had a bipolar disorder (see table Excluded studies). We included in our review the same four other studies (Anton 1990; Mulsant 2001; Spiker 1985; Wijkstra 2010), using exactly the same extracted ITT data. The conclusion of this review is consistent with ours: "Combination treatment is more effective than antidepressant monotherapy". For the comparison of an antipsychotic plus an antidepressant versus an antipsychotic, the same four studies with again exactly the same extracted ITT data were included (Meyers 2009; Rothschild 2004a; Rothschild 2004b; Spiker 1985), leading in both reviews to the same conclusion: "Combination treatment is more effective than antipsychotic monotherapy". As in our review, no differences were reported in overall dropout rates across all studies for both comparisons.
Authors' conclusions
Implications for practice.
Psychotic depression is heavily under‐studied, limiting confidence in the conclusions drawn. Evidence suggests that combination therapy with an antidepressant plus an antipsychotic is more effective than either treatment alone or placebo. Evidence for treatment with an antidepressant alone or with an antipsychotic alone is lacking.
Implications for research.
Further studies are needed:
to study the efficacy of other combinations of an antidepressant plus an antipsychotic. Regarding antidepressants: combinations with a TCA, with a selective serotonin reuptake inhibitor (SSRI), with a serotonin‐noradrenaline reuptake inhibitor (SNRI), or with other (newer) antidepressants such as mirtazapine; regarding antipsychotics: combinations with other so called atypical antipsychotics (aripiprazole, risperidone, olanzapine, etc.);
to compare the effects of the combination of an antidepressant with antipsychotics versus other pharmacological options, such as augmentation of an antidepressant with lithium or more experimental treatments such as ketamine and mifepristone;
to compare the effects of the combination of an antidepressant plus an antipsychotic versus ECT; and
to evaluate the efficacy of stepwise approaches or algorithms encompassing the previous steps after each other.
Feedback
Feedback submitted, 3 February 2015
Summary
We found a possible error in the review 'Pharmacological treatment for psychotic depression' by Wijkstra J, Lijmer J, Burger H, Geddes J, Nolen WA., which was published in issue 11 of year 2013.
When we read through the review, we found that they included 2 comparisons from a single article to calculate clinical outcome in their Analysis 7. The referenced article was Wijkstra 2010, in which 122 patients were randomized into 3 treatment groups: imipramine (n = 42), venlafaxine (n = 39), or venlafaxine + quetiapine (n = 41). In their Analysis 7, they compared the imipramine or venlafaxine group against the venlafaxine + quetiapine group independently in each subgroup. Then, when they conducted the analysis for the Total, venlafaxine + quetiapine group (n = 41) was included twice in the "antidepressant plus antipsychotic" group.
Double counting the same subjects would spuriously increase precision in the meta‐analytic estimates. Study authors should use a proper method to avoid double‐counting the same subjects.
Reply
We would like to thank Dr Matsuo and his colleagues for pointing out this mistake in the original analysis. We looked at this and we agreed that the best approach is probably to split the comparator (as described in the Cochrane Handbook for Systematic Reviews of Interventions, Chapter 16.5.4). We amended the analyses in the revised review accordingly and, given the numbers involved, it makes no material difference to the point estimates or to precision. The revised estimate for clinical response (see Analysis 7.1) was RR 1.42; 95% CI 1.11 to 1.81 (while the original pooled RR was 1.44 with 95% CI 1.15 to 1.80). The revised estimate for dropouts (see Analysis 7.2) was RR 0.91, 95% CI 0.55 to 1.50 (the original pooled RR was 0.91 with 95% CI 0.58 to 1.44).
We thank the EBMH Study Group for their interest and close reading of our review.
Contributors
Feedback submitted by Masahiro Matsuo, Aran Tajika, Toshi A. Furukawa, and Kyoto EBMH Study Group.
Response submitted by Andrea Cipriani and John Geddes.
What's new
| Date | Event | Description |
|---|---|---|
| 3 December 2021 | New search has been performed | An update search was completed in February 2020. No new studies were identified. |
| 3 December 2021 | New citation required but conclusions have not changed | This review has been updated. A 'Summary of findings' table has been added. |
History
Protocol first published: Issue 1, 2003 Review first published: Issue 4, 2005
| Date | Event | Description |
|---|---|---|
| 9 July 2015 | Feedback has been incorporated | Mistake in original analysis was corrected. This made no material difference to the results |
| 10 June 2013 | New citation required and conclusions have changed | Update of previous review. Two new studies included; conclusions slightly revised |
| 10 June 2013 | New search has been performed | Searches and methods updated |
| 2 September 2010 | Amended | Methods updated to reflect current Handbook |
| 3 November 2008 | Amended | Converted to new review format |
| 10 August 2005 | New citation required and conclusions have changed | Substantive amendments made |
Acknowledgements
The review authors extend their thanks to J Wijkstra, J Lijmer, and F Balk for important and substantive contributions to earlier versions of this review as coauthors (Wijkstra 2003; Wijkstra 2005; Wijkstra 2013; Wijkstra 2015).
We would like to thank Dr J. Bruijn, Dr W.W. van den Broek, and Dr R. Zanardi for providing additional information about their studies. A Cipriani is supported by the National Institute for Health Research (NIHR) Oxford Cognitive Health Clinical Research Facility, by an NIHR Research Professorship (grant RP‐2017‐08‐ST2‐006), by the NIHR Oxford and Thames Valley Applied Research Collaboration and by the NIHR Oxford Health Biomedical Research Centre (grant BRC‐1215‐20005) and was expert witness for Accord Healthcare for a patent issue about quetiapine extended release.
We thank the editorial team of the Cochrane Common Mental Disorders (CCMD) Group.
CCMD supported the authors in the update of this review. This update includes the same studies that were included in the update in 2015 (Wijkstra 2015), peer review was not required.
The following people conducted the editorial process for this article:
Sign‐off Editor (final editorial decision): Nick Meader, CCMD, Centre for Reviews and Dissemination, University of York.
Managing Editor (provided editorial guidance to authors, edited the article): Jessica Hendon, CCMD, Centre for Reviews and Dissemination, University of York
Information specialist (conducted search, provided editorial guidance to authors, edited the article): Sarah Dawson, CCMD & University of Bristol
Copy Editor (copy‐editing and production): Dolores Matthews, Cochrane Copy‐Editing Group, Holland, USA
The authors and the CCMD Editorial Team are grateful to Cochrane Copy Edit Support for the team's help.
Cochrane Group funding acknowledgement: The UK National Institute for Health Research (NIHR) is the largest single funder of the Cochrane Common Mental Disorders Group.
Disclaimer: The views and opinions expressed herein are those of the review authors and do not necessarily reflect those of the NIHR, National Health Service (NHS), or the Department of Health and Social Care.
Appendices
Appendix 1. Updated search strategies (all databases) 2020
Date of search: 21‐Feb‐2020 2013 onwards Ovid MEDLINE, n=332 Ovid Embase, n=621 Ovid PsycINFO, n=212 CLib: CENTRAL, n=663 International Trial Registers, ℅ CLib:CENTRAL, n=147 CCMDCTR, n=229 Total=2204 Duplicates removed, n=834 To Screen, n=1370 Ovid MEDLINE(R) and Epub Ahead of Print, In‐Process & Other Non‐Indexed Citations and Daily <1946 to February 20, 2020> Search Strategy: ‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐ 1 ((depressed or depression? or depressive?) adj5 (delusion* or psychotic or psychosis or psychoses)).ti,ab,kf. (8220) 2 (*depression/ or depressive disorder/ or depressive disorder, major/) and (psychotic disorders/ or affective disorders, psychotic/ or delusions/) (5242) 3 or/1‐2 (11662) 4 controlled clinical trial.pt. (93531) 5 randomized controlled trial.pt. (500168) 6 clinical trials as topic/ (190121) 7 (randomi#ed or randomi#ation or randomi#ing).ti,ab,kf. (618912) 8 (RCT or "at random" or (random* adj3 (administ* or allocat* or assign* or class* or cluster or crossover or cross‐over or control* or determine* or divide* or division or distribut* or expose* or fashion or number* or place* or pragmatic or quasi or recruit* or split or subsitut* or treat*))).ti,ab,kf. (543410) 9 placebo.ab,ti,kf. (210587) 10 trial.ti. (212770) 11 (control* adj3 group*).ab. (522518) 12 (control* and (trial or study or group*) and (waitlist* or wait* list* or ((treatment or care) adj2 usual))).ti,ab,kf,hw. (24123) 13 ((single or double or triple or treble) adj2 (blind* or mask* or dummy)).ti,ab,kf. (170411) 14 double‐blind method/ or random allocation/ or single‐blind method/ (275150) 15 or/4‐14 (1685135) 16 exp animals/ not humans.sh. (4670680) 17 15 not 16 (1458014) 18 3 and 17 (1185) 19 (2013* or 2014* or 2015* or 2016* or 2017* or 2018* or 2019* or 2020*).yr,dp,dt,ep,ez. (8482040) 20 18 and 19 (332) *************************** Embase <1974 to 2020 Week 07> Search Strategy: ‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐ 1 (major depression/ or *depression/) and (psychosis/ or acute psychosis/ or affective psychosis/ or brief psychotic disorder/ or delusion/) (7260) 2 depressive psychosis/ (1263) 3 ((depressed or depression? or depressive?) adj5 (delusion* or psychotic or psychosis or psychoses)).ti,ab,kw. (10743) 4 or/1‐3 (16342) 5 randomized controlled trial/ (589879) 6 randomization.de. (85791) 7 controlled clinical trial/ and drug therapy.fs. (200095) 8 placebo.de. (345862) 9 placebo.ti,ab. (300942) 10 trial.ti. (290976) 11 (randomi#ed or randomi#ation or randomi#ing).ti,ab,kw. (890491) 12 (RCT or "at random" or (random* adj3 (administ* or allocat* or assign* or class* or cluster or control* or crossover or cross‐over or determine* or divide* or division or distribut* or expose* or fashion or number* or place* or pragmatic or quasi or recruit* or split or subsitut* or treat*))).ti,ab,kw. (745781) 13 ((singl$ or doubl$ or trebl$ or tripl$) adj3 (blind$ or mask$ or dummy)).mp. (302956) 14 (control* and (study or group?) and (waitlist* or wait* list* or ((treatment or care) adj2 usual) or no? treatment)).ti,ab,kw,hw. (66667) 15 or/5‐14 (1684443) 16 ((animal or nonhuman) not (human and (animal or nonhuman))).de. (5598527) 17 15 not 16 (1525666) 18 4 and 17 (1796) 19 (2013* or 2014* or 2015* or 2016* or 2017* or 2018* or 2019* or 2020*).yr,dp,dc. (11481480) 20 18 and 19 (663) 21 (schizophrenia not ((delusion* or psychotic or psychosis or psychoses) and depress*)).ti. (80072) 22 20 not 21 (621) *************************** PsycINFO <1806 to February Week 3 2020> Search Strategy: ‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐ 1 ((depressed or depression? or depressive?) adj5 (delusion* or psychotic or psychosis or psychoses)).ti,ab,id. (8958) 2 exp major depression/ and (psychosis/ or acute psychosis/ or affective psychosis/ or delusions/) (2118) 3 1 or 2 (9616) 4 clinical trials.sh. (11573) 5 (randomi#ed or randomi#ation or randomi#ing).ti,ab,id. (83956) 6 (RCT or at random or (random* adj3 (administ* or allocat* or assign* or class* or control* or crossover or cross‐over or determine* or divide* or division or distribut* or expose* or fashion or number* or place* or recruit* or split or subsitut* or treat*))).ti,ab,id. (100141) 7 (control* and (trial or study or group) and (placebo or waitlist* or wait* list* or ((treatment or care) adj2 usual))).ti,ab,id,hw. (28575) 8 ((single or double or triple or treble) adj2 (blind* or mask* or dummy)).ti,ab,id. (25894) 9 trial.ti. (29617) 10 placebo.ti,ab,id,hw. (39738) 11 treatment outcome.md. (20165) 12 treatment effectiveness evaluation.sh. (23913) 13 or/4‐12 (190455) 14 3 and 13 (621) 15 (2013* or 2014* or 2015* or 2016* or 2017* or 2018* or 2019* or 2020*).yr,an. (1349880) 16 14 and 15 (212) *************************** Cochrane Central Register of Controlled Trials (CENTRAL), Issue 2 of 12, 2020 #1 ((depression or “depressive disorder”) and (psychosis or psychotic or delusion*)):kw #2 "depressive psychosis”:kw #3 ((depressed or depression* or depressive*) NEAR (delusion* or psychotic or psychosis or psychoses)):ti,ab #4 (#1 or #2 or #3) #5 Limit 2013 to date, n=663 Trial Registry Records ℅ CENTRAL #1 ((depression or “depressive disorder”) and (psychosis or psychotic or delusion*)):ti #2 "psychotic depression" or (depressi* near/2 (psychosis or psychoses or psychotic)) #3 (#1 or #2) #4 "clinicaltrials.gov" or “who.int" #5 (#3 and #4) n=147 *************************** Cochrane Common Mental Disorders Controlled Trials Register (CCMDCTR) (2013‐2016) [Current to June 2016 only] #1 ((depressed or depression* or depressive*) AND (delusion* or psychotic or psychosis or psychoses)):TI,EH,EMT,KW,KY,MH #2 ((depressed or depression* or depressive*) ADJ5 (delusion* or psychotic or psychosis or psychoses)):AB,SO #3 (#1 OR #2) #4 ((2013 OR 2014 OR 2015 OR 2016)):XDD AND INREGISTER #5 (#3 AND #4) n=229 ***************************
Appendix 2. Previous search strategies to 2013
The Cochrane Common Mental Disorders Controlled Trials Register (CCMDCTR) (previously known as the Cochrane Collaboration Depression, Anxiety and Neurosis Review Group's Controlled Trials Register (CCDANCTR)) was searched using the following terms. This register included relevant reports of RCTs collated from routine searches of Ovid MEDLINE (1950‐), EMBASE (1974‐), PsycINFO (1960‐) and the Cochrane Central Register of Controlled Trials (CENTRAL).
The CCMDCTR‐Studies Register was searched (all years to 12 April 2013) using the following terms: Condition = (depressi* or “affective disorder*” or “affective symptoms”) AND Condition or Comorbidity = (psychosis or psychoses or psychotic* or delusion*)
The CCMDCTR‐Studies Register was searched (all years to 12 April 2013) using the following terms to identify additional untagged references: Title/Abstract/Keywords = ((depressi* or “affective disorder*” or “affective symptoms”) AND Free‐Text=(psychosis or psychoses or psychotic* or delusion* or hallucin* or antipsychotic* or psychotropic*))
***************************
In 2010 an additional search of the Cochrane Central Register of Controlled Trials (CENTRAL) was carried out.
The Cochrane Register of Controlled Trails (CENTRAL) was searched (Issue 4, 2010) using the following terms:
#1 MeSH descriptor DEPRESSION, this term only
#2 MeSH descriptor DEPRESSIVE DISORDER, this term only
#3 MeSH descriptor DEPRESSIVE DISORDER MAJOR, this term only
#4 (depression* or depressive*):ti,ab,kw
#5 (#1 or #2 or #3 or #4)
#6 MeSH descriptor DELUSIONS, this term only
#7 delusion*:ti,ab,kw
#8 MeSH descriptor PSYCHOTIC DISORDERS, this term only
#9 MeSH AFFECTIVE DISORDERS, PSYCHOTIC, this term only
#10 (psychotic* or psychosis or psychoses) :ti,ab,kw
#11 (#6 or #7 or #8 or #9 or #10)
#12 (#5 and #11), from 2005 to 2010
#13 SR‐DEPRESSN or HS‐DEPRESSN
#14 (#12 NOT #13)
***************************
We searched the Cochrane Central Register of Controlled Trials (CENTRAL) with the terms depressive disorder and drug treatment. In addition we searched MEDLINE (1966 until April 2004) and EMBASE (1980 until April 2004) using the following terms: (“depressive disorder/drug therapy”[MESH] AND ((“delusions”[MESH Terms] OR delusions[Text Word]) OR ((“psychotic disorders”[MESH Terms] OR psychotic[Text Word]) AND features[All Fields])))) combined with a sensitive search strategy for RCTs.
***************************
Appendix 3. Description of the CCMDCTR
Specialised Register of the Cochrane Common Mental Disorders Group (CCMDCTR)
The Cochrane Common Mental Disorders Group maintains an archived, specialised register of randomised controlled trials, the CCMDCTR. This register contains over 40,000 reference records (reports of RCTs) for anxiety and depressive disorders, bipolar disorder, eating disorders, self‐harm, and other mental disorders within the scope of this Group. The CCMDCTR is a partially studies based register with > 50% of the reference records tagged to c12,600 individually PICO coded study records. Reports of trials for inclusion in the register were collated from (weekly) generic searches of key bibliographic databases to June 2016, which included MEDLINE (1950‐), Embase (1974‐), and PsycINFO (1967‐), quarterly searches of the Cochrane Central Register of Controlled Trials (CENTRAL), and review‐specific searches of additional databases. Reports of trials were also sourced from international trial registries, drug companies, handsearching of key journals, conference proceedings, and other (non‐Cochrane) systematic reviews and meta‐analyses. Details of CCMD's core search strategies (used to identify RCTs) can be found on the Group's website with an example of the core MEDLINE search displayed below.
A weekly search alert based on condition + RCT filter only 1. [MeSH Headings]: eating disorders/ or anorexia nervosa/ or binge‐eating disorder/ or bulimia nervosa/ or female athlete triad syndrome/ or pica/ or hyperphagia/ or bulimia/ or self‐injurious behavior/ or self mutilation/ or suicide/ or suicidal ideation/ or suicide, attempted/ or mood disorders/ or affective disorders, psychotic/ or bipolar disorder/ or cyclothymic disorder/ or depressive disorder/ or depression, postpartum/ or depressive disorder, major/ or depressive disorder, treatment‐resistant/ or dysthymic disorder/ or seasonal affective disorder/ or neurotic disorders/ or depression/ or adjustment disorders/ or exp antidepressive agents/ or anxiety disorders/ or agoraphobia/ or neurocirculatory asthenia/ or obsessive‐compulsive disorder/ or obsessive hoarding/ or panic disorder/ or phobic disorders/ or stress disorders, traumatic/ or combat disorders/ or stress disorders, post‐traumatic/ or stress disorders, traumatic, acute/ or anxiety/ or anxiety, castration/ or koro/ or anxiety, separation/ or panic/ or exp anti‐anxiety agents/ or somatoform disorders/ or body dysmorphic disorders/ or conversion disorder/ or hypochondriasis/ or neurasthenia/ or hysteria/ or munchausen syndrome by proxy/ or munchausen syndrome/ or fatigue syndrome, chronic/ or obsessive behavior/ or compulsive behavior/ or behavior, addictive/ or impulse control disorders/ or firesetting behavior/ or gambling/ or trichotillomania/ or stress, psychological/ or burnout, professional/ or sexual dysfunctions, psychological/ or vaginismus/ or Anhedonia/ or Affective Symptoms/ or *Mental Disorders/
2. [Title/ Author Keywords]: (eating disorder* or anorexia nervosa or bulimi* or binge eat* or (self adj (injur* or mutilat*)) or suicide* or suicidal or parasuicid* or mood disorder* or affective disorder* or bipolar i or bipolar ii or (bipolar and (affective or disorder*)) or mania or manic or cyclothymic* or depression or depressive or dysthymi* or neurotic or neurosis or adjustment disorder* or antidepress* or anxiety disorder* or agoraphobia or obsess* or compulsi* or panic or phobi* or ptsd or posttrauma* or post trauma* or combat or somatoform or somati#ation or medical* unexplained or body dysmorphi* or conversion disorder or hypochondria* or neurastheni* or hysteria or munchausen or chronic fatigue* or gambling or trichotillomania or vaginismus or anhedoni* or affective symptoms or mental disorder* or mental health).ti,kf.
3. [RCT filter]: (controlled clinical trial.pt. or randomized controlled trial.pt. or (randomi#ed or randomi#ation).ab,ti. or randomly.ab. or (random* adj3 (administ* or allocat* or assign* or class* or control* or determine* or divide* or distribut* or expose* or fashion or number* or place* or recruit* or subsitut* or treat*)).ab. or placebo*.ab,ti. or drug therapy.fs. or trial.ab,ti. or groups.ab. or (control* adj3 (trial* or study or studies)).ab,ti. or ((singl* or doubl* or tripl* or trebl*) adj3 (blind* or mask* or dummy*)).mp. or clinical trial, phase ii/ or clinical trial, phase iii/ or clinical trial, phase iv/ or randomized controlled trial/ or pragmatic clinical trial/ or (quasi adj (experimental or random*)).ti,ab. or ((waitlist* or wait* list* or treatment as usual or TAU) adj3 (control or group)).ab.)
4. (1 and 2 and 3)
Records were screened for reports of RCTs within the scope of the Cochrane Common Mental Disorders Group. Secondary reports of RCTs were tagged to the appropriate study record.
Similar weekly search alerts were also conducted on OVID Embase and PsycINFO, using relevant subject headings (controlled vocabularies) and search syntax appropriate to each resource. A quarterly search of the Cochrane Central Register of Controlled Trials (CENTRAL) was also conducted.
Data and analyses
Comparison 1. Antidepressant versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1.1 Clinical response | 1 | 27 | Risk Ratio (M‐H, Fixed, 95% CI) | 8.40 [0.50, 142.27] |
| 1.2 Dropouts | 1 | 27 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.24 [0.34, 4.51] |
Comparison 2. Antipsychotic versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 2.1 Clinical response | 2 | 201 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.13 [0.74, 1.73] |
| 2.2 Dropouts | 2 | 201 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.79 [0.57, 1.08] |
Comparison 3. Antidepressant versus antidepressant.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 3.1 Clinical response | 5 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 3.1.1 Imipramine vs venlafaxine | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 3.1.2 Imipramine vs mirtazapine | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 3.1.3 Imipramine vs fluvoxamine | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 3.1.4 Fluvoxamine vs venlafaxine | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 3.1.5 Sertraline vs paroxetine | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 3.2 Dropouts | 5 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 3.2.1 Imipramine vs venlafaxine | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 3.2.2 Imipramine vs mirtazapine | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 3.2.3 Imipramine vs fluvoxamine | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 3.2.4 Fluvoxamine vs venlafaxine | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 3.2.5 Sertraline vs paroxetine | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected |
Comparison 4. Antidepressant versus antipsychotic.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 4.1 Clinical response | 1 | 36 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.09 [0.64, 6.82] |
| 4.2 Dropouts | 1 | 36 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.79 [0.18, 18.02] |
4.2. Analysis.

Comparison 4: Antidepressant versus antipsychotic, Outcome 2: Dropouts
Comparison 5. Antidepressant plus antipsychotic versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 5.1 Clinical response | 2 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 5.1.1 Fluoxetine + olanzapine vs placebo | 2 | 148 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.86 [1.23, 2.82] |
| 5.2 Dropouts | 2 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 5.2.1 Fluoxetine + olanzapine vs placebo | 2 | 148 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.75 [0.48, 1.18] |
Comparison 6. Antidepressant plus antipsychotic versus placebo plus antipsychotic.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 6.1 Clinical response | 4 | 447 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.83 [1.40, 2.38] |
| 6.1.1 Amitriptyline + perphenazine vs perphenazine | 1 | 39 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.61 [1.23, 10.56] |
| 6.1.2 Fluoxetine + olanzapine vs olanzapine | 2 | 149 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.64 [1.10, 2.44] |
| 6.1.3 Olanzapine + sertraline vs olanzapine | 1 | 259 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.76 [1.21, 2.54] |
| 6.2 Dropouts | 4 | 447 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.79 [0.63, 1.01] |
| 6.2.1 Amitriptyline + perphenazine vs perphenazine | 1 | 39 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.09 [0.38, 25.19] |
| 6.2.2 Fluoxetine + olanzapine vs olanzapine | 2 | 149 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.95 [0.59, 1.53] |
| 6.2.3 Olanzapine + sertraline vs olanzapine | 1 | 259 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.70 [0.53, 0.92] |
Comparison 7. Antidepressant plus antipsychotic versus placebo plus antidepressant.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 7.1 Clinical response | 4 | 245 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.42 [1.11, 1.80] |
| 7.1.1 Nortriptyline + perphenazine vs nortriptyline | 1 | 36 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.12 [0.49, 2.53] |
| 7.1.2 Venlafaxine + quetiapine vs venlafaxine | 1 | 59 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.95 [1.13, 3.37] |
| 7.1.3 Amitriptyline + perphenazine vs amitriptyline | 1 | 41 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.73 [0.89, 3.37] |
| 7.1.4 Amitriptyline + perphenazine vs amoxapine | 1 | 46 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.19 [0.75, 1.88] |
| 7.1.5 Venlafaxine + quetiapine vs imipramine | 1 | 63 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.27 [0.84, 1.93] |
| 7.2 Dropouts | 4 | 245 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.91 [0.55, 1.50] |
| 7.2.1 Nortriptyline + perphenazine vs nortriptyline | 1 | 36 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.12 [0.26, 4.81] |
| 7.2.2 Venlafaxine + quetiapine vs venlafaxine | 1 | 59 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.73 [0.22, 2.46] |
| 7.2.3 Amitriptyline + perphenazine vs amitriptyline | 1 | 41 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.73 [0.35, 8.41] |
| 7.2.4 Amitriptyline + perphenazine vs amoxapine | 1 | 46 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.65 [0.29, 1.45] |
| 7.2.5 Venlafaxine + quetiapine vs imipramine | 1 | 63 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.14 [0.38, 3.47] |
Comparison 8. Antidepressant plus antipsychotic versus placebo plus the same antidepressant.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 8.1 Clinical response | 3 | 157 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.70 [1.19, 2.43] |
| 8.2 Dropouts | 3 | 157 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.04 [0.52, 2.07] |
| 8.2.1 Nortriptyline + perphenazine vs nortriptyline | 1 | 36 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.12 [0.26, 4.81] |
| 8.2.2 Venlafaxine + quetiapine vs venlafaxine | 1 | 80 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.83 [0.33, 2.08] |
| 8.2.3 Amitriptyline + perphenazine vs amitriptyline | 1 | 41 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.73 [0.35, 8.41] |
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Anton 1990.
| Study characteristics | ||
| Methods | Randomised, double‐blind comparison | |
| Participants | No explicit use of structured interview DSM‐III criteria; psychotic depressive episode HRSD‐17 > 18 Inpatients No data about prior treatment of current episode | |
| Interventions | Amoxapine vs amitriptyline + perphenazine 300 to 500 mg vs 150 to 250 mg + 24 to 40 mg No blood levels 5 days' placebo period. Additional medication in these 5 days: lorazepam or oxazepam in 'low dose' Treatment period: 4 weeks. Additional medication is not mentioned in these 4 weeks | |
| Outcomes | Dichotomous data: study author defined: response is reduction in HRSD‐17 > 50%. No remission data Continuous data: symptom reduction: no ITT data; global response: no ITT data; QOL: no data Overall dropout rate: yes Dropout due to adverse effects: yes (2 in ami + per) Mortality rate: 0 | |
| Notes | 56 participants provided informed consent. 10 dropped out in washout before receiving active medication (4 refused and six improved substantially); 46 participants were randomly assigned 46 participants: 4 dropouts in both groups (total 8). Unclear how many bipolar participants among these 8 dropouts; 38 participants were analysed, including 6 bipolar participants 6/38 bipolar = 15.8% ITT responders: amoxapine 12/21 and ami + per 17/25 (instead of 12/17 and 17/21) Dropouts after random assignment: 9/21 and 7/25 Study author had no additional data available See also 1993 J Aff Disorders 28:125‐131 (same data set) |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | As reported: "patients were randomly assigned in a double blind fashion" |
| Allocation concealment (selection bias) | Unclear risk | No information |
| Blinding (performance bias and detection bias) of participants | Low risk | As reported: "double blind treatment with identical capsules" |
| Blinding (performance bias and detection bias) of personnel | Low risk | As reported: "double blind treatment with identical capsules" |
| Blinding (performance bias and detection bias) of outcome assessors | Unclear risk | Probably yes. No explicit data |
| Incomplete outcome data (attrition bias) All outcomes | High risk | 46 participants were randomly assigned. In the publication, only those participants who completed at least 2 weeks of active medication were analysed. 4 dropouts in both groups (total 8) |
| Selective reporting (reporting bias) | Unclear risk | No protocol available. Generally accepted outcomes have been used |
| Other bias | High risk | Unclear how many bipolar participants were present among these 8 dropouts; 38 participants were analysed, including 6 bipolar participants. 6/38 bipolar = 15.8%. No additional data available to exclude bipolar participants from re‐analysis We re‐analysed the data with ITT responders (intention‐to‐treat; dropouts included): amoxapine 12/21 and ami + per 17/25 (instead of 12/17 and 17/21). ITT dropouts after random assignment: 9/21 and 7/25 |
Bruijn 1996.
| Study characteristics | ||
| Methods | Randomised, double‐blind comparison | |
| Participants | Use of checklist with DSM‐III‐R criteria SADS depression portion was performed in the presence of a second psychiatrist DSM‐III‐R depressive episode; excluded psychotic depression with hallucinations HRSD‐17 > 17 Inpatients. Subgroup psychotic depression. Probably only with delusions 51% of included participants were adequately pretreated during the current episode: adequate dose of an antidepressant during at least 4 weeks | |
| Interventions | Imipramine vs mirtazapine; 37.5 to 450 mg imipramine (blood level: 199 to 400 ng/mL) vs 40 to 100 mg mirtazapine (blood level 49 to 93 ng/mL) Washout: 3 days medication free and 4 days placebo Additional medication: 1 to 6 tablets a day containing 45 mg of an extract of valerian, lorazepam 1 to 5 mg a day, or haloperidol 1 to 15 mg a day Treatment period: 4 weeks after predefined blood levels reached (mirtazapine group: 5 to 21 days; imipramine group: 7 to 25 days) | |
| Outcomes | Dichotomous data: study author defined: response is reduction in HRSD‐17 ≥ 50%. No remission data Continuous data: symptom reduction: no ITT data; global response: no ITT data; QOL: no data Overall dropout rate: yes Dropout due to adverse effects: no ITT data in subgroup Mortality rate: 0 | |
| Notes | Worse responding in a group leads to more participants given haloperidol 107 participants included; 6 bipolar; 10 dropouts Subgroup: MDD psychotic; 30 (15 mirtazapine and 15 imipramine) Mirtazapine group: 7 haloperidol treatment (6 non‐responders, 1 responder) Imipramine group: 2 haloperidol treatment (2 non‐responders) Participants treated with haloperidol counted as dropouts Mirtazapine group: 1 dropout + 7 haloperidol treatment = 8/18; imipramine group: 2 dropouts + 2 haloperidol treatment = 4/15 Additional information from study author included | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | As reported: "patients were randomly allocated to a double blind treatment" |
| Allocation concealment (selection bias) | Unclear risk | No explicit information |
| Blinding (performance bias and detection bias) of participants | Low risk | As reported: "identical capsules. Dose adjustment by an independent psychiatrist on the basis of blood levels" |
| Blinding (performance bias and detection bias) of personnel | Low risk | As reported: "identical capsules. Dose adjustment by an independent psychiatrist" |
| Blinding (performance bias and detection bias) of outcome assessors | Low risk | Side effects were not systematically rated to prevent bias towards unblinding. After completion of the study, the research psychiatrist guessed the medication: 46 correct and 37 incorrect |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | None |
| Selective reporting (reporting bias) | Unclear risk | No protocol available. Generally accepted outcomes have been used |
| Other bias | High risk | Participants with psychotic depression with hallucinations were excluded. So only participants with psychotic depression with delusions were included in the re‐analysed subgroup We re‐analysed the data in the subgroup with psychotic depression. We counted as dropouts: 1 participant with bipolar disorder, 9 participants with haloperidol treatment (7 in mirtazapine group and 2 in imipramine group) Worse responding in psychotic depression leads in this study to more open co‐treatment with haloperidol 1 to 15 mg, especially in the mirtazapine group. Only 1 of these 9 participants (mirtazapine group) was a responder. So haloperidol probably was not instrumental in the recovery of those participants |
Meyers 2009.
| Study characteristics | ||
| Methods | Randomised double‐blind study | |
| Participants | 259 participants; DSM‐IV‐TR psychotic depression; 18 years of age or older; HAM‐D ≧ 21 and SADS delusional severity rating ≧ 3 Inpatients |
|
| Interventions | 12 weeks' treatment with olanzapine + placebo and olanzapine + sertraline | |
| Outcomes | Remission rates (HAM‐D 17 ≦ 10 and SADS delusional item score = 1) | |
| Notes | 53% dropout in olanzapine arm and 37% dropout in olanzapine + sertraline arm | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | As reported: "computer generated randomisation list" |
| Allocation concealment (selection bias) | Unclear risk | No further data |
| Blinding (performance bias and detection bias) of participants | Low risk | Study was double‐blind (reported as: "sertraline and placebo under double‐blind conditions") |
| Blinding (performance bias and detection bias) of personnel | Low risk | Well‐described double‐blinding. "Sertraline and placebo under double‐blind conditions". As reported: "investigators and raters were blind to treatment assignment" |
| Blinding (performance bias and detection bias) of outcome assessors | Low risk | Well‐described double‐blinding. "Sertraline and placebo under double‐blind conditions". As reported: "investigators and raters were blind to treatment assignment" |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | None |
| Selective reporting (reporting bias) | Low risk | Protocol available. Generally accepted outcomes have been used |
| Other bias | Unclear risk | Relatively high dropout rate with significant differences between treatment groups (53% olanzapine and 37% olanzapine + sertraline) Patients with only hallucinations excluded |
Mulsant 2001.
| Study characteristics | ||
| Methods | Randomised, double‐blind comparison | |
| Participants | Clinical interview, Brief Psychiatric Rating Scale, Global Assessment Scale, and a consensus conference were used for diagnosis DSM‐III‐R; psychotic major depressive episode (manic episode in history excluded). HAM‐D‐17 > 17 Age > 50 years Inpatients No data about prior treatment for current episode | |
| Interventions | Nortriptyline vs nortriptyline + perphenazine Open nortriptyline until therapeutic plasma level (target 100 ng/mL); once between 50 and 150 ng/mL, random assignment followed Mean doses: nortriptyline 76 mg vs nortriptyline 63 mg + perphenazine 19 mg Mean blood levels: 101 ng/mL vs 120 + 4 ng/mL Additional medication: lorazepam as needed Treatment period: after random assignment 2 to 16 weeks (total treatment at least 4 weeks) ''After a washout of other psychotropic medication except lorazepam ....'' It is unclear how long this washout has been | |
| Outcomes | Dichotomous data: study author defined: response is HAMD‐17 < 11 and BPRS (11, 12, 15) 1 or 2. No remission data Continuous data: symptom reduction: no ITT data; global response: no ITT data; QOL: no data Overall dropout rate: yes Dropout due to adverse effects: nortriptyline + perphenazine 1/17; nortriptyline + placebo 2/19 Mortality rate: 0 | |
| Notes | 54 participants included; 6 dropouts: 2 due to adverse effects and 4 for administrative reasons; 36 participants randomly assigned. This is by procedure a selected group: responders on nortriptyline and participants with adverse effects and with other reasons are excluded (28%) Open nortriptyline (8 to 21 days; median 2 weeks); once between 50 and 150 ng/mL, random assignment followed Responder somewhere between 2 and 16 weeks after randomisation (median 9 weeks); 3 dropouts in both groups after random assignment. These are excluded by the study author and were included by us for ITT analysis | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No information. As reported: "patient[s] were randomly allocated to a double blind treatment" |
| Allocation concealment (selection bias) | Unclear risk | No information. As reported: "patient[s] were randomly allocated to a double blind treatment" |
| Blinding (performance bias and detection bias) of participants | Low risk | As reported: "double blind treatment. No further data" |
| Blinding (performance bias and detection bias) of personnel | Low risk | "Dose adjustments by non blinded psychiatrists who were not involved in the care" |
| Blinding (performance bias and detection bias) of outcome assessors | Low risk | As reported: "double blind treatment" |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | None |
| Selective reporting (reporting bias) | Unclear risk | No protocol available. Generally accepted outcomes have been used |
| Other bias | High risk | No outcome data about prescription of "lorazepam as needed" EPS rating could have led to blinding bias Participants treated with only nortriptyline (+ placebo) were excluded after 4 weeks without improvement, and participants treated with nortriptyline + perphenazine after 4 + 2 weeks |
Rothschild 2004a.
| Study characteristics | ||
| Methods | Randomised, double‐blind comparison Random assignment was 2:2:1 for olanzapine, placebo, and olanzapine fluoxetine combination, respectively | |
| Participants | 124 participants; DSM‐IV diagnosis (unclear how) Major depression with psychotic features Inpatients for at least 1 week. No data about prior treatment for current episode | |
| Interventions | 2004a: olanzapine (5 to 20 mg, clinically titrated; mean 11.9 mg) vs olanzapine (5 to 20 mg, clinically titrated; mean 12.4 mg) plus fluoxetine (20 to 80 mg, clinically titrated; mean 23.5 mg) vs placebo 2004b: same procedure: olanzapine (mean 14.0 mg) vs olanzapine (mean 13.9 mg) plus fluoxetine (mean 22.6 mg) 3 to 9 days' screening; probably no washout period Treatment period: 8 weeks Additional medication: 30 mg a day diazepam equivalent for no more than 5 consecutive days or 10 cumulative days |
|
| Outcomes | Dichotomous data: study author defined: response is reduction in HAMD‐24 ≥ 50% at endpoint. Remission is HAMD‐24 ≤ 8 for 2 consecutive visits Continuous data: symptom reduction: no ITT data; global response: no ITT data; QOL: no data Overall dropout rate: yes Dropout due to adverse effects: no ITT data Mortality rate: probably 0 | |
| Notes | Washout unclear
23 investigators randomly assigned at least 1 participant. Excluded patient characteristics were not described
Dropouts in study "a": 28%; lost before baseline + 1 visit: 7% (were excluded from results; included in our data); 24% in study "a" are LOCF (last observation carried forward; in our data, not counted as dropouts); some of these LOCFs are counted as responders; total non‐completers (LOCF included) 28 + 7 + 24 = 59% Dropouts in study "b": 38%; lost before baseline + 1 visit: 9% (were excluded from results; included in our data); LOCF in study "b": 6%; total non‐completers 28 + 9 + 6 = 53% Completers in study "a": 41%; in study "b": 47% |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | As reported: "patients were randomly allocated"; no further information |
| Allocation concealment (selection bias) | Unclear risk | As reported: "patients were randomly allocated"; no further information |
| Blinding (performance bias and detection bias) of participants | Low risk | As reported: "double blind therapy. Dose adjustments in all study arms with 'capsules' (assuming identical capsules because the study is double blind)" |
| Blinding (performance bias and detection bias) of personnel | Low risk | As reported: "double blind therapy" |
| Blinding (performance bias and detection bias) of outcome assessors | Unclear risk | No explicit information |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Dropouts described in general terms. Very high dropout rate |
| Selective reporting (reporting bias) | Unclear risk | No protocol available. According to study authors, the olanzapine‐fluoxetine group was designed as an exploratory pilot arm. However, in the conclusions, it is stated that an olanzapine/fluoxetine combination was well‐tolerated treatment associated with significant and quick reduction in depressive (and psychotic) symptoms in 1 trial. With ITT data, this difference is seen in 1 study to be not statistically significant, and in the other study, barely significant. Pooling of these 2 studies would result in no significance Study authors discuss as a limitation the absence of a fluoxetine arm. They state that they cannot rule out that the effect of fluoxetine/olanzapine was due to fluoxetine. So this should have been mentioned in the conclusions |
| Other bias | High risk | High dropout rate of 34.7% reduces internal validity of the study High placebo response is contradictory to the literature |
Rothschild 2004b.
| Study characteristics | ||
| Methods | Randomised, double‐blind comparison Random assignment was 2:2:1 for olanzapine, placebo, and olanzapine fluoxetine combination, respectively | |
| Participants | 124 participants. DSM‐IV diagnosis (unclear how) Major depression with psychotic features Inpatients for at least 1 week. No data about prior treatment for current episode | |
| Interventions | 2004a: olanzapine (5 to 20 mg, clinically titrated; mean 11.9 mg) vs olanzapine (5 to 20 mg, clinically titrated; mean 12.4 mg) plus fluoxetine (20 to 80 mg, clinically titrated; mean 23.5 mg) vs placebo 2004b: same procedure: olanzapine (mean 14.0 mg) vs olanzapine (mean 13.9 mg) plus fluoxetine (mean 22.6 mg) 3 to 9 days' screening; probably no washout period Treatment period: 8 weeks Additional medication: 30 mg a day diazepam equivalent for no more than 5 consecutive days or 10 cumulative days |
|
| Outcomes | Dichotomous data: study author defined: response is reduction in HAMD‐24 ≥ 50% at endpoint. Remission is HAMD‐24 ≤ 8 for 2 consecutive visits Continuous data: symptom reduction: no ITT data; global response: no ITT data; QOL: no data Overall dropout rate: yes Dropout due to adverse effects: no ITT data Mortality rate: probably 0 | |
| Notes | Washout unclear
23 investigators randomly assigned at least 1 participant. Excluded patient characteristics not described
Dropouts in study "a": 28%; lost before baseline + 1 visit: 7% (were excluded from results; included in our data); 24% in study "a" are LOCF (last observation carried forward; in our data not counted as dropouts). Some of these LOCFs are counted as responders; total non‐completers (LOCF included) 28 + 7 + 24 = 59% Dropouts in study "b": 38%; lost before baseline + 1 visit: 9% (were excluded from results; included in our data); LOCF in study "b": 6%; total non‐completers 28 + 9 + 6 = 53% Completers in study "a": 41%; in study "b": 47% |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | As reported: "patients were randomly allocated"; no further information |
| Allocation concealment (selection bias) | Unclear risk | As reported: "patients were randomly allocated"; no further information |
| Blinding (performance bias and detection bias) of participants | Low risk | As reported: "double blind therapy. Dose adjustments in all study arms with 'capsules' (assuming identical capsules because the study is double blind)" |
| Blinding (performance bias and detection bias) of personnel | Low risk | As reported: "double blind therapy" |
| Blinding (performance bias and detection bias) of outcome assessors | Unclear risk | No explicit information |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Dropouts described in general terms. Very high dropout rate |
| Selective reporting (reporting bias) | Unclear risk | No protocol available. According to study authors, the olanzapine‐fluoxetine group was designed as a exploratory pilot arm. However, in the conclusions, it is stated that an olanzapine/fluoxetine combination was well‐tolerated treatment associated with significant and quick reduction in depressive (and psychotic) symptoms in 1 trial. With ITT data, this difference is seen in 1 study to be not statistically significant, and in the other study, to be barely significant. Pooling of these 2 studies would result in no significance. Study authors discuss as a limitation the absence of a fluoxetine arm. They state that they cannot rule out that the effect of fluoxetine/olanzapine was due to fluoxetine. So this should have been mentioned in the conclusion |
| Other bias | High risk | High dropout rate of 47.2% reduces the internal validity of the study High placebo response is contradictory to the literature |
Spiker 1985.
| Study characteristics | ||
| Methods | Randomised, double‐blind comparison Random assignment procedure described in part Blinding adequately described | |
| Participants | SADS and RDC criteria for major depressive disorder, primary subtype, and psychotic subtype (only with delusions); bipolar participants included Severity rating ≥ 4 on 6‐point scale in the SADS that rates severity of delusion HRSD‐17 > 14 Inpatients No data about prior treatment for current episode | |
| Interventions | 3 groups: perphenazine vs amitriptyline vs amitriptyline + perphenazine Doses: perphenazine mean 50 mg vs amitriptyline mean 218 mg vs amitriptyline mean 170 mg + perphenazine mean 54 mg Blood levels: perphenazine 19 to 113 ng/mL vs amitriptyline (+ nortriptyline) 130 to 500 ng/mL vs amitriptyline 157 to 690 ng/mL + perphenazine 18 to 128 ng/mL 7 days drug free Treatment period: 4 weeks Additional medication: benztropine mesylate 4 mg | |
| Outcomes | Dichotomous data: study author defined: response is HRSD‐17 < 7 and delusional rating score = 1 (6‐point scale in the SADS). No remission data (definition of response is definition of remission) Continuous data: symptom reduction: no ITT data; global response: no ITT data; QOL: no data Overall dropout rate: yes Dropout due to adverse effects: amitriptyline + perphenazine 2/22, perphenazine 1/17 Mortality rate: 0 | |
| Notes | Only participants with delusions 7 drop out in ITT (in the original data, dropouts are excluded from the analysis); response data ITT 3/17 (original 3/16); 7/19 (7/17); 14/22 (14/18) 9/58 = 15.5% bipolar participants in analysis. Because of lack of data, we were not able to exclude these bipolar participants from the analysis | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | As reported: "the hospital pharmacist assigned patients randomly" |
| Allocation concealment (selection bias) | Low risk | Probably yes: "the hospital pharmacist assigned patients randomly" |
| Blinding (performance bias and detection bias) of participants | Low risk | As reported: "the hospital pharmacist assigned patients randomly. All tablets looked identical" "All raters and floor staff and the patient were blind to the patient's drug treatment and the plasma‐level data" |
| Blinding (performance bias and detection bias) of personnel | Low risk | As reported: "the hospital pharmacist assigned patients randomly. All tablets looked identical" "All raters and floor staff and the patient were blind to the patient's drug treatment and the plasma‐level data" |
| Blinding (performance bias and detection bias) of outcome assessors | Low risk | As reported: "the hospital pharmacist assigned patients randomly. All tablets looked identical" "All raters and floor staff and the patient were blind to the patient's drug treatment and the plasma‐level data" |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | None |
| Selective reporting (reporting bias) | Unclear risk | No protocol available. Generally accepted outcomes have been used |
| Other bias | Unclear risk | Only participants with delusions are included 9/58 = 15.5% bipolar participants in analysis. Because of lack of data, we were not able to exclude these bipolar participants from the analysis We re‐analysed the data to ITT |
Spiker 1988.
| Study characteristics | ||
| Methods | Re‐analysing 2 studies (not including data from Spiker 1985) Randomised, double‐blind, placebo‐controlled Randomisation procedure not explicitly described Blinding adequately described in original studies | |
| Participants | Re‐diagnosing by using DSM‐III criteria Major depressive disorder DSM‐III HRSD‐17 > 14 (≥ 30 based on the sum of 2 raters) Inpatients. Subgroup psychotic participants No data about prior treatment for current episode | |
| Interventions | Amitriptyline vs placebo 3 days 50; 4 days 100; 7 days 150; 14 days 200 mg amitriptyline (at least 3 weeks ≥ 150 mg) Blood levels: unknown Extra medication: none 2 weeks' drug‐free washout period 1 week placebo (single‐blind); total period of 3 weeks drug free Treatment period: 4 weeks | |
| Outcomes | Dichotomous data: study author defined: response is HRSD‐17 < 7 (< 14/2) + not psychotic or HRSD‐17 = 6.5 to 9.5 (13/2 to 19 /2) + not psychotic + 1/3 or less of entering score Remission data not specified Continuous data: symptom reduction: no data; global response: no data; QOL: no data Overall dropout rate: yes Dropout due to adverse effects: no data Mortality rate: 0 (no data) | |
| Notes | 20% response in 2‐week drug‐free period (psychotic + non‐psychotic); no data about psychotic vs non‐psychotic in these 2 weeks 4 weeks' treatment; only 2 weeks 200 mg; no blood levels Subgroup of 27 participants with psychotic depression. Amitriptyline 14; placebo 13 Dropouts 4 (amitriptyline) and 3 (placebo) are excluded from analysis by study authors. Responders amitriptyline 4/10 and placebo 0/10. ITT responders: amitriptyline 4/14 and placebo 0/13 | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | As reported: "patients were randomly assigned" |
| Allocation concealment (selection bias) | Unclear risk | No data |
| Blinding (performance bias and detection bias) of participants | Low risk | As reported: "all patients received 4 identical capsules daily: patients and staff were blind" |
| Blinding (performance bias and detection bias) of personnel | Low risk | As reported: "all patients received 4 identical capsules daily: patients and staff were blind" |
| Blinding (performance bias and detection bias) of outcome assessors | Unclear risk | No data |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | None |
| Selective reporting (reporting bias) | Unclear risk | No protocol available. Generally accepted outcomes have been used |
| Other bias | High risk | Participants were retrospectively re‐diagnosed. Psychotic depression and non‐psychotic depression were included and randomly assigned. We used the data about psychotic participants 14 days' drug‐free period (20% remission with no further data) + 1 week placebo before random assignment could be due to low placebo response |
van den Broek 2004.
| Study characteristics | ||
| Methods | Randomised, double‐blind comparison | |
| Participants | DSM‐IV diagnosis major depressive disorder; assessed with the depression portion of the SADS HRSD‐17 > 16 Inpatients; subgroup of psychotic depression 39% of all included participants were pretreated with an SSRI and 22.7% with a TCA, but none as inpatients with adequate plasma level for at least 4 weeks during the present episode | |
| Interventions | Imipramine vs fluvoxamine 4 days' placebo washout Predefined blood levels. Imipramine 150 to 450 mg (blood level imipramine + desimipramine 192 to 521). Fluvoxamine 150 to 1800 mg (blood level 109 to 325 ng/mL). Treatment period: 4 weeks after reaching predefined blood levels. Additional medication: 1 to 6 tablets a day containing 45 mg of an extract of valerian, lorazepam 1 to 3 mg a day, or haloperidol 1 to 10 mg a day | |
| Outcomes | Dichotomous data: study author defined: response is reduction in HRSD‐17 ≥ 50%. Remission is HRSD‐17 < 8 | |
| Notes | Subgroup with psychotic features. Some participants in this subgroup had been treated with haloperidol (counted as dropouts). Worse responding in a group leads to more participants who were given haloperidol We used additional psychotic subgroup data from the study author | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | As reported: "a computer generated randomisation list was used" |
| Allocation concealment (selection bias) | Unclear risk | No specific data |
| Blinding (performance bias and detection bias) of participants | Low risk | As reported: "tablets identical in appearance, weight and taste were administered. Preparation of the tablets was done by the pharmacist. The treating physician received blood level data in percentages" |
| Blinding (performance bias and detection bias) of personnel | Low risk | As reported: "tablets identical in appearance, weight and taste were administered. Preparation of the tablets was done by the pharmacist. The treating physician received blood level data in percentage" |
| Blinding (performance bias and detection bias) of outcome assessors | Low risk | As reported: "the treating physicians were not involved in the ratings" |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | None |
| Selective reporting (reporting bias) | Unclear risk | No protocol available. Generally accepted outcomes have been used |
| Other bias | High risk | Worse responding in a group leads to more participants who were given haloperidol We re‐analysed the data: participants treated with haloperidol were counted as dropouts |
Wijkstra 2010.
| Study characteristics | ||
| Methods | Randomised, double‐blind study | |
| Participants |
DSM‐IV‐defined psychotic depression Inpatients |
|
| Interventions | 7 weeks of treatment with imipramine (plasma levels 200 to 300 μg/L), venlafaxine (375 mg/d), venlafaxine + quetiapine (375 mg/d + 600 mg/d) | |
| Outcomes | Dichotomous data: study author defined (response). Response is ≧ 50% decrease in HAM‐D 17 scores from baseline to study endpoint, and final HAM‐D score ≦ 14. Remission is HAMD ≦ 7 (not predefined) | |
| Notes | No quetiapine arm. Inclusion did not reach planned number (122 i.s.o. 155) Relatively low dropout rate (22/122 = 18%) |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | As reported: "randomisation was executed centrally using a computer‐generated randomisation list: randomly permuted blocks of size six" |
| Allocation concealment (selection bias) | Low risk | As reported: "randomisation was executed centrally" |
| Blinding (performance bias and detection bias) of participants | Low risk | Study was double‐blind. Blood was collected from each participant (only imipramine blood level was assessed). Treatment guesses were analysed and indicated high preservation of blindness |
| Blinding (performance bias and detection bias) of personnel | Low risk | Double‐blind study. Blood was collected from each participant (only imipramine blood level was assessed). Treatment guesses were analysed and indicated high preservation of blindness |
| Blinding (performance bias and detection bias) of outcome assessors | Low risk | As reported: "blindness was checked and high. All dose adjustments were done centrally by an independent psychiatrist" |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | None |
| Selective reporting (reporting bias) | Low risk | Protocol available. Generally accepted outcomes have been used |
| Other bias | Unclear risk | 122 participants included i.s.o. with the planned 155 resulting in loss of power Post hoc remission as secondary outcome measure |
Zanardi 1996.
| Study characteristics | ||
| Methods | Randomised, double‐blind comparison | |
| Participants | The SCID patient version was used for some participants but not for all (reply of study author to letter to editor) DSM‐III‐R criteria; psychotic depressive episode No HRSD criteria described at inclusion Inpatients No data about prior treatment for current episode | |
| Interventions | Sertraline vs paroxetine Dose: 150 mg vs 50 mg from day 8 Blood levels: unknown Additional medication: flurazepam < 30 mg (bipolar participants additional medication lithium; bipolar participants are excluded from our data) 1 week medication free (single‐blind placebo period) Treatment period: 5 weeks | |
| Outcomes | Dichotomous data: study author defined: Response is HRSD‐21 < 8 + DDERS (Dimensions of Delusional Experience RS) = 0. Remission data not specified Continuous data: symptom reduction: no data; global response: no data; QOL: no data Overall dropout rate: yes Dropout due to adverse effects: same as overall dropout Mortality rate: 0 | |
| Notes | 5/14 dropouts in paroxetine group and 0/18 in sertraline group Bipolar participants could be excluded from our analysis | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | As reported: "patients were randomly assigned to two therapy groups" |
| Allocation concealment (selection bias) | Unclear risk | No data |
| Blinding (performance bias and detection bias) of participants | Unclear risk | As reported: "patients were randomly assigned". In title and abstract: "double‐blind controlled trial. No information about methods of blinding" |
| Blinding (performance bias and detection bias) of personnel | Unclear risk | As reported: "double‐blind controlled trial" |
| Blinding (performance bias and detection bias) of outcome assessors | Unclear risk | No data |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | None |
| Selective reporting (reporting bias) | Unclear risk | No protocol available. Generally accepted outcomes have been used |
| Other bias | Unclear risk | We re‐analysed the data by excluding bipolar participants Difference in dropout is high: 5/14 vs 0/18 |
Zanardi 2000.
| Study characteristics | ||
| Methods | Randomised, double‐blind comparison | |
| Participants | Unclear diagnosing procedure DSM‐IV criteria; psychotic depressive episode No HRSD criteria described at inclusion Inpatients No data about prior treatment for current episode | |
| Interventions | Venlafaxine vs fluvoxamine Dose: 300 mg vs 300 mg from day 8 Blood levels: unknown Additional medication: flurazepam < 30 mg 1 week medication free (single‐blind placebo period) Treatment period: 5 weeks | |
| Outcomes | Dichotomous data: study author defined: response is HRSD‐21 < 9 + DDERS (Dimensions of Delusional Experience RS) = 0. Remission data not specified Continuous data: symptom reduction: no ITT data; global response: no ITT data; QOL: no data Overall dropout rate: yes Dropout due to adverse effects: same as overall dropout rate Mortality rate: 0 | |
| Notes | We used additional data from the study author to exclude bipolar participants from analysis Included 22 participants with major depressive disorder (MDD) with psychotic features. Responders in venlafaxine group 6/11 MDD. Responders fluvoxamine group 9/11 MDD. No dropouts in fluvoxamine group. 2 dropouts in venlafaxine group | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | As reported: "randomization was performed by a computer‐generated schedule" |
| Allocation concealment (selection bias) | Unclear risk | Randomisation method not described Blinding not explicitly described. No data |
| Blinding (performance bias and detection bias) of participants | Unclear risk | As reported: "patients were randomly assigned" "Double‐blind controlled study" |
| Blinding (performance bias and detection bias) of personnel | Unclear risk | "Double‐blind controlled study", but unclear whether double‐blind includes personnel |
| Blinding (performance bias and detection bias) of outcome assessors | Low risk | As reported: "raters were blind to treatment" |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | None |
| Selective reporting (reporting bias) | Unclear risk | No protocol available. Generally accepted outcomes have been used |
| Other bias | Unclear risk | We re‐analysed the data by excluding bipolar participants with additional data from study author |
DSM: Diagnostic and Statistical Manual of Mental Disorders.
HRSD/HAM‐D: Hamilton Rating Scale for Depression.
ITT: intention‐to‐treat.
LOCF: last observation carried forward.
MDD: major depressive disorder.
mg: milligram.
mL: millilitre.
ng: nanogram.
QOL: quality of life.
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Belanoff 2001 | Only 4 days of treatment |
| Bellini 1994 | 25% bipolar participants (in each group, 3 bipolar participants) The study author did not respond to our request for additional information |
| Blasey 2009 | Impossible to compare 1 defined pharmacological treatments |
| Blasey 2011 | In addition to study medication (mifepristone or placebo), all patients received an antidepressant but not as standardised treatment (i.e. not the same antidepressant for each patient and not at a fixed dose and duration). Thus, the antidepressant may have obscured the effect of mifepristone |
| Block 2017 | In addition to study medication (mifepristone or placebo), all patients received an antidepressant but not as standardised treatment (i.e. not the same antidepressant for each patient and not at a fixed dose). Thus, the antidepressant may have obscured the effect of mifepristone |
| Block 2018 | In addition to study medication (mifepristone or placebo), all patients received an antidepressant but not as standardised treatment (i.e. not the same antidepressant for each patient and not at a fixed dose and duration). Thus, the antidepressant may have obscured the effect of mifepristone |
| Cassacchia 1984 | 'Unipolar psychotic depression' is probably 'manic depressive psychosis, depressive type' (ICD9). This is not the same as 'psychotic depression' Number of bipolar participants is not clear Dropouts not in results. It is not possible to extract ITT response data Reasons for exclusion: unclear diagnosis, number of bipolar participants unclear, ITT data not available |
| Davidson 1981 | Reasons for exclusion: unclear diagnosis and short treatment period |
| DeBattista 2006 | Impossible to compare 2 defined pharmacological treatments. 48.3% + 12.9% = 61.2% HAMD response with placebo after 1 week |
| Ebert 1997 | Randomisation not adequate; open study |
| Flores 2006 | Impossible to compare 2 defined pharmacological treatments and treatment only for 7 days |
| Friedman 1966 | No comparable diagnostic procedure. No data about MDD subgroup Dropouts have been excluded |
| Künzel 2008 | No ITT data; bipolar participants 17.5% in per‐protocol data; continued treatment with lithium, valproic acid |
| Malison 1999 | Only 3 psychotic participants |
| McLaughlin 1969 | Diagnosis unclear |
| Müller 1998 | In this subgroup, no data are given about responders, bipolar participants, and dropouts The study author did not respond to our request for additional information |
| Navarro 2001 | Citalopram vs nortriptyline Subgroup with 9 psychotic depressive episodes Reason for exclusion: this subgroup was also treated with haloperidol. No data available for this subgroup are available The study author did not respond to our request for additional information |
| Nelson 1984 | Unknown from data in which group the responders are located (imipramine or ami). So comparison is impossible |
| Spiker 1982 | Pre‐published data from the 1985 study |
| Zanardi 1998 | 30.5% bipolar participants |
HAMD: Hamilton Rating Scale for Depression.
ITT: intention‐to‐treat.
MDD: major depressive disorder.
Differences between protocol and review
All studies were evaluated according to the new method used for assessing risk of bias. The background section has been updated.
Analysis 8.1 and Analysis 8.2 (where an antidepressant plus an antipsychotic is compared with the same antidepressant plus placebo) were unplanned sensitivity analyses.
Contributions of authors
JK: 2021 update of the review, literature search, screening and updating of background and other sections. EL: 2021 update of the review, literature search, screening and updating of background and other sections. HB: statistical advice. AC: statistical advice and analysis. JG: co‐author report. LR: 2021 update of the review, summary of findings tables. BV: 2021 update of the review, summary of findings tables. WN: development of protocol, data collection, analysis, overall supervision, co‐author report.
Sources of support
Internal sources
No sources of support provided
External sources
-
National Institute for Health Research (NIHR), UK
LR contribution to this review update is supported by Cochrane Infrastructure funding to the Common Mental Disorders Cochrane Review Group.
Declarations of interest
JK: no conflicts of interest. EL: no conflicts of interest. HB: no conflicts of interest. AC: has received grant funding from Johnson & Johnson and Angelini Pharma. JG: no conflicts of interest. LR: is a systematic reviewer and editor in the Cochrane Common Mental Disorders editorial team. LR was not involved in the editorial approval process for this review. BV: no conflicts of interest. WN: conducted a multi‐centre trial in participants with psychotic depression that compared treatment with imipramine, venlafaxine, and venlafaxine plus quetiapine. Wyeth and AstraZeneca financially supported this trial. Data from this trial are included in this review. To prevent bias, the data extracted from our own study explicitly have been checked by the Cochrane organisation.
New search for studies and content updated (no change to conclusions)
References
References to studies included in this review
Anton 1990 {published data only}
- Anton RF Jr, Burch EA Jr. Amoxapine versus amitriptyline combined with perphenazine in the treatment of psychotic depression. American Journal of Psychiatry 1990;147(9):1203-8. [DOI] [PubMed] [Google Scholar]
- Anton RF Jr, Burch EA Jr. Response of psychotic depression subtypes to pharmacotherapy. Journal of Affective Disorders 1993;28(2):125-31. [DOI] [PubMed] [Google Scholar]
Bruijn 1996 {published data only}
- Bruijn JA, Moleman P, Mulder PG, den Broek WW, Hulst AM, Mast RC, et al. A double-blind, fixed blood-level study comparing mirtazapine with imipramine in depressed in-patients. Psychopharmacology 1996;127(3):231-7. [PubMed] [Google Scholar]
- Bruijn JA, Moleman P, Mulder PG, den Broek WW. Depressed in-patients respond differently to imipramine and mirtazapine. Pharmacopsychiatry 1999;32(3):87-92. [DOI] [PubMed] [Google Scholar]
Meyers 2009 {published data only}
- Andreescu C, Mulsant BH, Peasley-Miklus C, Rothschild AJ, Flint AJ, Heo M, et al. Persisting low use of antipsychotics in the treatment of major depressive disorder with psychotic features. Journal of Clinical Psychiatry 2007;68(2):194-200. [DOI] [PubMed]
- Blumberger DM, Mulsant BH, Emeremni C, Houck P, Andreescu C, Mazumdar S, et al. Impact of prior pharmacotherapy on remission of psychotic depression in a randomized controlled trial. Journal of Psychiatric Research 2011;45(7):896-901. [DOI] [PMC free article] [PubMed]
- Flint AJ. Challenges in conducting a pharmacotherapy study in psychotic depression. 157th Annual Meeting of the American Psychiatric Association; 2004 May 1-6; New York, NY. No. 63D. [Google Scholar]
- Meyers B, Rothschild A, Gabriele M, English J, McShea M, Kamara T, et al. Effectiveness of selective serotonin reuptake inhibitors combined with antipsychotic medication for the treatment of psychotic depression [STOP-PD; NCT00056472]. ClinicalTrials.gov [http://www.clinicaltrials.gov] 2004.
- Meyers BS, Flint AJ, Rothschild AJ, Mulsant BH, Whyte EM, Peasley-Miklus C, et al. A double-blind randomized controlled trial of olanzapine plus sertraline vs olanzapine plus placebo for psychotic depression: the study of pharmacotherapy of psychotic depression (STOP-PD). Archives of General Psychiatry 2009;66(8):838-47. [DOI] [PMC free article] [PubMed]
- Meyers BS, Flint AJ, Rothschild AJ, Mulsant BH, Whyte EM, Peasley-Miklus C, et al. Errors in table in: a double-blind randomized controlled trial of olanzapine plus sertraline vs olanzapine plus placebo for psychotic depression: the study of pharmacotherapy of psychotic depression (STOP-PD). Archives of General Psychiatry 2011;68(6):625-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyers BS. Diagnosis and treatment of psychosis in geriatric major depression. 46th Annual NCDEU (New Clinical Drug Evaluation Unit) Meeting; 2006 June 12-15; Boca Raton, FL. p 83. [Google Scholar]
- Schaffer A, Flint AJ, Smith E, Rothschild AJ, Mulsant BH, Szanto K, et al. Correlates of suicidality among patients with psychotic depression. Suicide and Life-Threatening Behavior 2008;38(4):403-14. [DOI] [PubMed] [Google Scholar]
- Weissman J, Flint A, Meyers B, Ghosh S, Mulsant B, Rothschild A, et al. Factors associated with non-completion in a double-blind randomized controlled trial of olanzapine plus sertraline versus olanzapine plus placebo for psychotic depression. Psychiatry Research 2012;197(3):221-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Mulsant 2001 {published data only}
- Mulsant BH, Sweet RA, Rosen J, Pollock BG, Zubenko GS, Flynn T, et al. A double-blind randomized comparison of nortriptyline plus perphenazine versus nortriptyline plus placebo in the treatment of psychotic depression in late life. Journal of Clinical Psychiatry 2001;62(8):597-604. [DOI] [PubMed] [Google Scholar]
Rothschild 2004a {published data only}
- Rothschild AJ, Williamson DJ, Tohen MF, Schatzberg A, Andersen SW, Van Campen LE, et al. A double-blind, randomized study of olanzapine and olanzapine/fluoxetine combination for major depression with psychotic features. Journal of Clinical Psychopharmacology 2004;24(4):365-73. [DOI] [PubMed]
- Williamson DJ, Andersen SW, Campen LE, Sanger TM, Paul S, Corya SA, et al. Olanzapine-fluoxetine combination for difficult-to-treat depression. 154th Annual Meeting of the American Psychiatric Association; 2001 May 5-10; New Orleans; LA:NR549. [Google Scholar]
Rothschild 2004b {published data only}
- Rothschild AJ, Williamson DJ, Tohen MF, Schatzberg A, Andersen SW, Van Campen LE, et al. A double-blind, randomized study of olanzapine and olanzapine/fluoxetine combination for major depression with psychotic features. Journal of Clinical Psychopharmacology 2004;24(4):365-73. [DOI] [PubMed] [Google Scholar]
- Williamson DJ, Andersen SW, Campen LE, Sanger TM, Paul S, et al. Olanzapine-fluoxetine combination for difficult-to-treat depression. 154th Annual Meeting of the American Psychiatric Association; 2001 May 5-10; New Orleans; LA:NR549. [Google Scholar]
Spiker 1985 {published data only}
- Spiker D, Perel JM, Hanin I, Dealy RS, Griffin SJ, Soloff PH, et al. The pharmacological treatment of delusional depression: part II. Journal of Clinical Psychopharmacology 1986;6(6):339-42. [PubMed] [Google Scholar]
- Spiker DG, Hanin I, Perel JM, Cofsky JE, Rossi AJ, Sorisio D. Pharmacological treatment of delusional depressives. Psychopharmacology Bulletin 1982;18(4):184-6. [PubMed] [Google Scholar]
- Spiker DG, Weiss JC, Dealy RS, Griffin SJ, Hanin I, Neil JF, et al. The pharmacological treatment of delusional depression. American Journal of Psychiatry 1985;142(4):430-6. [DOI] [PubMed] [Google Scholar]
Spiker 1988 {published data only}
- Spiker DG, Kupfer DJ. Placebo response rates in psychotic and nonpsychotic depression. Journal of Affective Disorders 1988;14(1):21-3. [DOI] [PubMed] [Google Scholar]
van den Broek 2004 {published data only}
Wijkstra 2010 {published data only}
- Wijkstra J, Burger H, den Broek WW, Birkenhäger TK, Janzing JG, Boks MP, et al. Treatment of unipolar psychotic depression: a randomized, double-blind study comparing imipramine, venlafaxine, and venlafaxine plus quetiapine. Acta Psychiatrica Scandinavica 2010;121(3):190-200. [DOI] [PubMed] [Google Scholar]
Zanardi 1996 {published data only}
- Zanardi R, Franchini L, Gasperini M, Perez J, Smeraldi E. Double-blind controlled trial of sertraline versus paroxetine in the treatment of delusional depression. American Journal of Psychiatry. 1996;153(12):1631-3. [DOI] [PubMed] [Google Scholar]
Zanardi 2000 {published and unpublished data}
- Zanardi R, Franchini L, Serretti A, Perez J, Smeraldi E. Venlafaxine versus fluvoxamine in the treatment of delusional depression: a pilot double-blind controlled study. Journal of Clinical Psychiatry 2000;61(1):26-9. [DOI] [PubMed] [Google Scholar]
References to studies excluded from this review
Belanoff 2001 {published data only}
- Belanoff JK, Flores BH, Kalezhan M, Sund B, Schatzberg AF. Rapid reversal of psychotic depression using mifepristone. Journal of Clinical Psychopharmacology 2001;21(5):516-21. [DOI] [PubMed] [Google Scholar]
Bellini 1994 {published data only}
- Bellini L, Gasperini M, Gatti F, Franchini L, Smeraldi E. A double blind study with fluvoxamine vs. desipramine combined with placebo or haloperidol in delusional depression. Critical Issues in the Treatment of Affective Disorders 1994;9:32-6. [Google Scholar]
Blasey 2009 {published data only}
- Blasey CM, Debattista C, Roe R, Block T, Belanoff JK. A multisite trial of mifepristone for the treatment of psychotic depression: a site-by-treatment interaction. Contemporary Clinical Trials 2009;30(4):284-8. [DOI] [PubMed] [Google Scholar]
- Blasey CM, DeBattista C, Roe R, Block T, Belanoff JK. Corrigendum to 'A multisite trial of mifepristone for the treatment of psychotic depression: a site-by-treatment interaction' [Contemp. Clin. Trials 30 (2009)284-288]. Contemporary Clinical Trials 2009;30(5):497. [DOI] [PubMed] [Google Scholar]
- Blasey CM, DeBattista C, Roe R, Block T, Belanoff JK. Corrigendum to 'A multisite trial of mifepristone for the treatment of psychotic depression: a site-by-treatment interaction' [Contemp. Clin. Trials 30 (2009) 284-288]. Contemporary Clinical Trials 2010;31(1):134. [DOI] [PubMed] [Google Scholar]
Blasey 2011 {published data only}
- Blasey CM, Block TS, Belanoff JK, Roe RL. Efficacy and safety of mifepristone for the treatment of psychotic depression. Journal of Clinical Psychopharmacology 2011;31(4):436-40. [DOI] [PubMed] [Google Scholar]
Block 2017 {published data only}
- Block T, Petrides G, Kushner H, Kalin N, Belanoff J, Schatzberg A. Mifepristone plasma level and glucocorticoid receptor antagonism associated with response in patients with psychotic depression. Journal of Clinical Psychopharmacology 2017;37(5):505-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
Block 2018 {published data only}
- Block TS, Kushner H, Kalin N, Nelson C, Belanoff J, Schatzberg A. Combined analysis of mifepristone for psychotic depression: plasma levels associated with clinical response. Biological Psychiatry 2018;84(1):46-54. [DOI] [PubMed] [Google Scholar]
Cassacchia 1984 {published data only}
- Casacchia M, Carolei A, Barba C, Frontoni M, Rossi A, Meco G, et al. A placebo-controlled study of the antidepressant activity of moclobemide, a new MAO-A inhibitor. Pharmacopsychiatry 1984;17(4):122-5. [DOI] [PubMed] [Google Scholar]
Davidson 1981 {published data only}
- Davidson JR, McLeod MN, Turnbull CD, Miller RD. A comparison of phenelzine and imipramine in depressed inpatients. Journal of Clinical Psychiatry 1981;42(10):395-7. [PubMed] [Google Scholar]
DeBattista 2006 {published data only}
- DeBattista C, Belanoff J, Glass S, Khan A, Horne RL, Blasey C, et al. Mifepristone versus placebo in the treatment of psychosis in patients with psychotic major depression. Biological Psychiatry 2006;15(12):1343-9. [DOI] [PubMed] [Google Scholar]
Ebert 1997 {published data only}
- Ebert D. Lithium-TCA combination treatment of psychotic depression: comparison with TCA-neuroleptic treatment. Journal of Clinical Psychopharmacology 1997;17(2):129-30. [DOI] [PubMed] [Google Scholar]
Flores 2006 {published data only}
- Flores BH, Kenna H, Keller J, Solvason HB, Schatzberg AF. Clinical and biological effects of mifepristone treatment for psychotic depression. Neuropsychopharmacology 2006;31(3):628-36. [DOI] [PubMed] [Google Scholar]
Friedman 1966 {published data only}
- Friedman AS, Granick S, Cohen HW, Cowitz B. Imipramine (tofranil vs. placebo in hospitalized psychotic depressives (a comparison of patients' self-ratings, psychiatrists' ratings and psychological test scores). Journal of Psychiatric Research 1966;4(1):13-36. [DOI] [PubMed] [Google Scholar]
Künzel 2008 {published data only}
- Künzel HE, Ackl N, Hatzinger M, Held K, Holsboer-Trachsler E, Ising M, et al. Outcome in delusional depression comparing trimipramine monotherapy with a combination of amitriptyline and haloperidol - a double-blind multicenter trial. Journal of Psychiatric Research 2009;43(7):702-10. [DOI] [PubMed] [Google Scholar]
Malison 1999 {published data only}
- Malison RT, Anand A, Pelton GH, Kirwin P, Carpenter L, McDougle CJ, et al. Limited efficacy of ketoconazole in treatment-refractory major depression. Journal of Clinical Psychopharmacology 1999;19(5):466-70. [DOI] [PubMed] [Google Scholar]
McLaughlin 1969 {published data only}
- McLaughlin BE. Therapy for psychotic and psychoneurotic depression with thiothixene and perphenazine-amitriptyline (a double-blind clinical study). Diseases of the Nervous System 1969;30(2):85-8. [PubMed] [Google Scholar]
Müller 1998 {published data only}
- Muller-Siecheneder F, Muller MJ, Hillert A, Szegedi A, Wetzel H, Benkert O. Risperidone versus haloperidol and amitriptyline in the treatment of patients with a combined psychotic and depressive syndrome. Journal of Clinical Psychopharmacology 1998;18(2):111-20. [DOI] [PubMed] [Google Scholar]
Navarro 2001 {published data only}
- Navarro V, Gasto C, Torres X, Marcos T, Pintor L. Citalopram versus nortriptyline in late-life depression: a 12-week randomized single-blind study. Acta Psychiatrica Scandinavica 2001;103(6):435-40. [DOI] [PubMed] [Google Scholar]
Nelson 1984 {published data only}
- Nelson WH, Kahn A, Orr WW Jr. Phenomenology, neuroendocrine function, and tricyclic antidepressant response. Journal of Affective Disorders 1984;6(3-4):297-306. [DOI] [PubMed] [Google Scholar]
Spiker 1982 {published data only}
- Spiker DG, Hanin I, Perel JM, Cofsky JE, Rossi AJ, Sorisio D. Pharmacological treatment of delusional depressives. Psychopharmacology Bulletin 1982;18(4):184-6. [PubMed] [Google Scholar]
Zanardi 1998 {published data only}
- Zanardi R, Franchini L, Gasperini M, Lucca A, Smeraldi E, Perez J. Faster onset of action of fluvoxamine in combination with pindolol in the treatment of delusional depression: a controlled study. Journal of Clinical Psychopharmacology. 1998;18(6):441-6. [DOI] [PubMed] [Google Scholar]
Additional references
Alderson 2004
- Alderson P, Green S, Higgins JPT. Cochrane Reviewer's Handbook 4.2.2. Chichester: John Wiley & Sons, Ltd, 2004. [Google Scholar]
APA 1980
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders (DSM-III). 3rd edition. Washington, DC: American Psychiatric Association, 1980. [Google Scholar]
APA 1987
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders (DSM-III-R). 3rd edition, revised. Washington, DC: American Psychiatric Association, 1987. [Google Scholar]
APA 1994
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders (DSM-IV). 4th edition. Washington, DC: American Psychiatric Association, 1994. [Google Scholar]
APA 2000
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders (DSM-IV-TR). 4th edition, text revision. Washington, DC: American Psychiatric Association, 2000. [Google Scholar]
APA 2010
- American Psychiatric Association. Practice Guideline for the Treatment of Patients With Major Depressive Disorder (revision). Washington, DC: American Psychiatric Association, 2010. [PubMed] [Google Scholar]
APA 2013
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders (DSM-V). 5th edition. Washington, DC: American Psychiatric Association, 2013. [Google Scholar]
Coryell 1984
- Coryell W, Pfohl B, Zimmerman M. The clinical and neuroendocrine features of psychotic depression. Journal of Nervous and Mental Disease 1984;172(9):521-8. [DOI] [PubMed] [Google Scholar]
Coryell 1998
- Coryell W. The treatment of psychotic depression. Journal of Clinical Psychiatry 1998;59(Suppl 1):22-9. [PubMed] [Google Scholar]
Duval 2006
- Duval F, Mokrani MC, Monreal-Ortiz JA, Fattah S, Champeval C, Schulz P, et al. Cortisol hypersecretion in unipolar major depression with melancholic and psychotic features: dopaminergic, noradrenergic and thyroid correlates. Psychoneuroendocrinology 2006;31(7):876-88. [DOI] [PubMed] [Google Scholar]
Farahani 2012
- Farahani A, Corell CU. Are antipsychotics or antidepressants needed for psychotic depression? A systematic review and meta-analysis of trials comparing antidepressant or antipsychotic monotherapy with combination treatment. Journal of Clinical Psychiatry 2012;73:486-96. [DOI] [PMC free article] [PubMed] [Google Scholar]
GRADEpro GDT [Computer program]
- GRADEpro GDT. Hamilton (ON): McMaster University (developed by Evidence Prime) (accessed prior to 3 March 2020). Available at gradepro.org.
Guy 1976
- Guy W. Clinical Global Impression. ECDEU Assessment Manual for Psychopharmacology, revised. Rockville, MD: National Institute of Mental Health, 1976.
Hamilton 1960
- Hamilton M. A rating scale for depression. Journal of Neurological and Neurosurgical Psychiatry 1960;23:56-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
Higgins 2021
- Higgins JT, Thomas J, Chandler J, Cumpston M, Li T, Page MJ, Welch VA, editor(s). Cochrane Handbook for Systematic Reviews of Interventions Version 6.2 (updated February 2021). Cochrane, 2021. Available from training.cochrane.org/handbook.
Higgins 2021b
- Higgins JT, Savović J, Page MJ, Elbers RG, Sterne JAC. Chapter 8. Assessing risk of bias in a randomized trial. In: Higgins JT, Thomas J, Chandler J, Cumpston M, Li T, Page MJ, et al (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 6.2 (updated February 2021). Cochrane, 2021. Available from www.training.cochrane.org/handbook.
Johnson 1991
- Johnson J, Horwath E, Weisman MM. The validity of major depression with psychotic features based on a community study. Archives of General Psychiatry 1991;48(12):1075-81. [DOI] [PubMed] [Google Scholar]
Keller 2006
- Keller J, Flores B, Gomez RG, Solvason HB, Kenna H, Williams GH, et al. Cortisol circadian rhythm alterations in psychotic major depression. Biological Psychiatry 2006;60(3):275-81. [DOI] [PubMed] [Google Scholar]
Licht 2008
- Licht RW, Gijsman H, Nolen WA, Angst J. Are antidepressants safe in the treatment of bipolar depression? A critical evaluation of their potential risk to induce switch into mania or cycle acceleration. Acta Psychiatrica Scandinavica 2008;118(5):337-46. [DOI] [PubMed] [Google Scholar]
Mahli 2009
- Mahli GS, Adams D, Porter R, Wignall A, Lampe L, O'Connor N, et al. Clinical practice recommendations for depression. Acta Psychiatrica Scandinavica 2009;119:8-26. [DOI] [PubMed] [Google Scholar]
Montgomery 1979
- Montgomery SA, Asberg M. A new depression scale designed to be sensitive to change. British Journal of Psychiatry 1979;134:382-9. [DOI] [PubMed] [Google Scholar]
Multidisciplinaire Richtlijn Depressie 2013
- Spijker J, Bockting CLH, Meeuwissen JAC, Vliet IM van, Emmelkamp PMG, Hermens MLM, et al (van namens de Werkgroep Multidisciplinaire richtlijnontwikkeling Angststoornissen/Depressie). Multidisciplinaire richtlijn Depressie (Derde revisie). Richtlijn voor de diagnostiek, behandeling en begeleiding van volwassen patiënten met een depressieve stoornis; 2013. Utrecht: Trimbos Institute. Available at www.nvvp.net/stream/richtlijn-depressie-2013.pdf.
Nelson 1997
- Nelson JC, Davis JM. DST studies in psychotic depression: a meta-analysis. American Journal of Psychiatry 1997;154(11):1497-503. [DOI] [PubMed] [Google Scholar]
NICE 2009
- National Institute for Health and Clinical Excellence. The treatment and management of depression in adults (updated version). National Clinical Practice Guideline 90. London: HMSO, 2009. [Google Scholar]
Ohayon 2002
- Ohayon MM, Schatzberg AF. Prevalence of depressive episodes with psychotic features in the general population. American Journal of Psychiatry 2002;159(11):1855-61. [DOI] [PubMed] [Google Scholar]
Parker 1992
- Parker G, Roy K, Hadzi-Pavlovic D, Pedic F. Psychotic (delusional) depression: a meta-analysis of physical treatments. Journal of Affective Disorders 1992;24(1):17-24. [DOI] [PubMed] [Google Scholar]
Prudic 1990
- Prudic J, Sackeim HA, Devanand DP. Medication resistance and clinical response to electroconvulsive therapy. Psychiatry Research 1990;31(3):287-96. [DOI] [PubMed] [Google Scholar]
Prudic 1996
- Prudic J, Haskett RF, Mulsant B, Malone KM, Pettinati HM, Stephens S, et al. Resistance to antidepressant medications and short-term clinical response to ECT. American Journal of Psychiatry 1996;153(8):985-92. [DOI] [PubMed] [Google Scholar]
Review Manager 2020 [Computer program]
- Review Manager 5 (RevMan). Version 5.4. Copenhagen: The Nordic Cochrane Centre, The Cochrane Collaboration, 2014.
Sackeim 2001
- Sackeim HA. The definition and meaning of treatment-resistant depression. Journal of Clinical Psychiatry 2001;62(Suppl 16):10-7. [PubMed] [Google Scholar]
Sadock 2009
- Sadock J, Sadock VA, Ruiz P (editors). Comrehensive Textbook of Psychiatry. Philadelphia: Lippincott Williams & Wilkins, 2009. [Google Scholar]
Schünemann 2020
- Schünemann HJ, Higgins JT, Vist GE, Glasziou P, Akl EA, Skoetz N, et al. Chapter 14. Completing 'Summary of findings' tables and grading the certainty of the evidence. In: Higgins JT, Thomas J, Chandler J, Cumpston MS, Li T, Page MJ, et al, editor(s), Cochrane Handbook for Systematic Reviews of Interventions Version 6.0 (updated September 2020). Cochrane, 2020. Available from www.training.cochrane.org/handbook.
van den Broek 2004a
- den Broek WW, Lely de A, Mulder PGH, Birkenhäger TK, Bruijn J. Effect of antidepressant medication resistance on short-term response to ECT. Journal of Clinical Psychopharmacology 2004;24(4):400-3. [DOI] [PubMed] [Google Scholar]
Wheeler 2000
- Wheeler Vega JA, Mortimer AM, Tyson PJ. Somatic treatment of psychotic depression: review and recommendations for practice. Journal of Clinical Psychopharmacology 2000;20(5):504-19. [DOI] [PubMed] [Google Scholar]
Wijkstra 2007
- Wijkstra J, Schubart CD, Nolen WA. Treatment of unipolar psychotic depression: the use of evidence in practice guideline. World Journal of Biological Psychiatry 2007;24:1-7. [DOI] [PubMed] [Google Scholar]
References to other published versions of this review
Wijkstra 2003
- Wijkstra J, Lijmer J, Nolen WA. Pharmacological treatment for psychotic depression. Cochrane Database of Systematic Reviews 2003, Issue 1. Art. No: CD004044. [DOI: 10.1002/14651858.CD004044] [DOI] [PubMed] [Google Scholar]
Wijkstra 2005
- Wijkstra J, Lijmer J, Balk F, Geddes J, Nolen WA. Pharmacological treatment for psychotic depression. Cochrane Database of Systematic Reviews 2005, Issue 4. Art. No: CD004044. [DOI: 10.1002/14651858.CD004044.pub2] [DOI] [PubMed] [Google Scholar]
Wijkstra 2013
- Wijkstra J, Lijmer J, Burger H, Geddes J, Nolen WA. Pharmacological treatment for psychotic depression. Cochrane Database of Systematic Reviews 2013, Issue 11. Art. No: CD004044. [DOI: 10.1002/14651858.CD004044.pub3] [DOI] [PubMed] [Google Scholar]
Wijkstra 2015
- Wijkstra J, Lijmer J, Burger H, Cipriani A, Geddes J, Nolen WA. Pharmacological treatment for psychotic depression. Cochrane Database of Systematic Reviews 2015, Issue 7. Art. No: CD004044. [DOI: 10.1002/14651858.CD004044.pub4] [DOI] [PubMed] [Google Scholar]