

Keywords: fibro-adipogenic progenitors, mesenchymal stromal cells, muscle stem cells, myogenesis, fibrosis, muscular disorders
Abstract
Skeletal muscle possesses a remarkable regenerative capacity that relies on the activity of muscle stem cells, also known as satellite cells. The presence of non-myogenic cells also plays a key role in the coordination of skeletal muscle regeneration. Particularly, fibro-adipogenic progenitors (FAPs) emerged as master regulators of muscle stem cell function and skeletal muscle regeneration. This population of muscle resident mesenchymal stromal cells has been initially characterized based on its bi-potent ability to differentiate into fibroblasts or adipocytes. New technologies such as single-cell RNAseq revealed the cellular heterogeneity of FAPs and their complex regulatory network during muscle regeneration. In acute injury, FAPs rapidly enter the cell cycle and secrete trophic factors that support the myogenic activity of muscle stem cells. Conversely, deregulation of FAP cell activity is associated with the accumulation of fibrofatty tissue in pathological conditions such as muscular dystrophies and ageing. Considering their central role in skeletal muscle pathophysiology, the regulatory mechanisms of FAPs and their cellular and molecular crosstalk with muscle stem cells are highly investigated in the field. In this review, we summarize the current knowledge on FAP cell characteristics, heterogeneity and the cellular crosstalk during skeletal muscle homeostasis and regeneration. We further describe their role in muscular disorders, as well as different therapeutic strategies targeting these cells to restore muscle regeneration.
1. Introduction
With more than 600 skeletal muscles and accounting for 35–45% of the body mass, striated skeletal muscle is the most abundant tissue in the human body [1]. Skeletal muscles play a role in vital functions, such as locomotion, breathing, thermoregulation, energy metabolism and endocrine signalling. Considering their role in movement and exercise, skeletal muscles are subjected to various physical stresses and traumas. To face these challenges, skeletal muscles have developed a strong adaptive capacity. Particularly, they have a remarkable regenerative capacity, owing to their population of muscle stem cells. These cells are also named satellite cells based on their anatomical location between the basal lamina and the plasma membrane of myofibres [2,3]. Muscle stem cells are quiescent in a healthy resting muscle, but they are primed for activation. Following an injury, muscle stem cells quickly enter the cell cycle to become proliferative myoblasts that transiently expand the myogenic cell pool. Thereafter, myoblasts exit the cell cycle to differentiate and fuse to form multinucleated myotubes/myofibres. A proportion of myogenic cells also resist differentiation and self-renew to replenish the muscle stem cell pool [3–5]. Animal knockout models and genetic variants in humans leading to muscle stem cell exhaustion showed that these cells are absolutely required for muscle regeneration [6,7].
Skeletal muscle injury induces the coordinated accumulation of different cell types. Single-cell RNAseq and single-cell mass cytometry have identified between nine and 15 distinct cell populations in the resting skeletal muscle and during the different phases of regeneration [8–12]. After an injury, there is a rapid accumulation of immune cells (neutrophils, pro- and anti-inflammatory macrophages, natural killer cells, B- and T-cells) and changes in the proportion of non-immune cells (endothelial cells, smooth muscle cells, glial cells, tenocytes and fibro-adipogenic progenitors). These different cell types provide molecular cues to guide muscle stem cells through myogenesis [5]. For instance, during the course of muscle regeneration, there is a switch in macrophage phenotype from pro-inflammatory macrophages to anti-inflammatory macrophages. The former secretes cytokines that promote myoblast proliferation, while the latter releases factors stimulating myoblast differentiation and fusion [13,14]. In recent years, fibro-adipogenic progenitors (FAPs), a population of muscle-specific mesenchymal stromal cells, have emerged as master regulators of skeletal muscle regeneration [15–20]. This review aims at summarizing the recent knowledge on FAP cell characteristics, cellular interactions and roles in skeletal muscle under physiological and pathological conditions.
2. FAPs developmental origin and markers
The developmental origin of FAPs has been comprehensively reviewed recently [21,22]. The seminal work of Kardon and colleagues in the early 2000s identified a population of TCF7L2/TCF4+ (transcription factor 4; also known as transcription factor 7 like 2; TCF7L2) cells arising from the mesoderm lateral plate in the chick and mouse [23]. These cells do not express myogenic markers (Pax7) and do not form myotubes. However, functional experiments using a dominant-negative form of TCF7L2/TCF4 demonstrated that these cells are critical to set up a prepattern that plays an important role for determining myogenic cell differentiation in the limb [23]. Other markers expressed by connective tissue progenitors in the mouse embryo include, T-box transcription factors (TBX) 3/4/5, HOX11 and Odd skipped-related 1 (Osr1) [24–27]. Deletion of these genes leads to limb muscle patterning defects [24–26]. These genes are not universal markers for FAPs, but are rather expressed by different subpopulations that are spatially associated with specific regions of the limb [20,24,27,28].
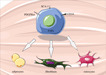
In adults, FAPs are identified by the cell surface markers stem cell antigen-1 (Sca1 or Ly6A/E) and platelet-derived growth factor receptor α (PDGFRα) [15] (figure 1). They also express CD34, like other muscle resident cells such as muscle stem cells and endothelial cells [11,29–31], but not other myogenic markers such as integrin-α7 (itga7) and syndecan-4 [11]. Notably, in adult resting muscles, FAPs express low levels of Osr1, but it is re-expressed upon injury, suggesting that FAPs reactivate a developmental programme during muscle regeneration [32].
Figure 1.

Schematic of fibro-adipogenic progenitors (FAPs). FAPs are muscle resident multipotent mesenchymal stem cells that can differentiate into adipocytes, fibroblasts or osteocytes (under specific conditions). They express key surface markers such as platelet-derived growth factor receptor α (PDGFRα), stem cell antigen-1 (Sca-1) and cluster of differentiation 34 (CD34). This figure was created with the Servier Medical Art service (https://smart.servier.com/), which is licensed under a Creative Commons Attribution 3.0 Unported License (https://creativecommons.org/licenses/by/3.0/).
3. FAPs characteristics
FAPs are located in the interstitial space of resting [21] or regenerating skeletal muscle (figure 2). They were initially described for their bi-potent ability to differentiate into fibrogenic or adipogenic cells, but not myogenic cells [15,17]. FAPs cultured in vitro can spontaneously differentiate into fibroblasts or adipocytes [15,17]. Their fibrogenic differentiation can be stimulated by adding transforming growth factor-β (TGF-β) to the medium, while their adipogenic differentiation can be promoted by a medium containing insulin, 3-isobutyl-1-methylxanthine and dexamethasone [33–35]. Lineage tracing experiments confirmed that this multipotent capacity is also observed in vivo. Experiments using PDGFRα-creERT Rosa26EYFP reporter mice confirmed that perilipin+ adipocytes observed after cardiotoxin or glycerol-induced injuries originate from PDGFRα+ FAPs [36]. Another lineage tracing study using TCF7L2/TCF4CreERT2/+ R26RmTmG/+ reporter mice confirmed that cells expressing the fibroblast marker alpha-smooth muscle actin (αSMA) were originating from TCF7L2/TCF4+ FAPs [18].
Figure 2.

Fibro-adipogenic progenitors (FAPs) in regenerating skeletal muscle. (a) Immunofluorescence of Pax7 (red), PDGFRα (orange), laminin (green) and DAPI (nuclei, blue) on regenerating skeletal muscle section (14 days post-cardiotoxin injury). Muscle stem cell (Pax7+ cell in red; identified with white arrowhead) is visible in its niche under the basal membrane (laminin; green). FAP (PDGFRα+ cell in orange) is identified with a white arrow in the interstitial space. (b) Schematic of the location of muscle stem cell and FAP in skeletal muscle.
Recent findings also indicate that FAPs can give rise to osteogenic or chondrogenic cells under specific culture conditions, such as bone morphogenic protein (BMP)-2, -7 or -9 administration [37–39]. Intramuscular transplantation of FAPs (CD31-CD45-Tie2+PDGFRα+Sca1+ cells) in combination with Matrigel and BMP2 demonstrated that these cells robustly contribute to cartilage and bone deposition in vivo [37]. Lineage tracing experiment using PDGFRα-creERT2-TdTomato mice confirmed that the vast majority of osteogenic cells came from FAPs in BMP2-induced heterotopic ossification in vivo [39]. These findings indicate that the multipotency of FAPs is broader than their name suggests.
4. FAPs in resting skeletal muscle
4.1. FAPs heterogeneity in muscle homeostasis
The emergence of single-Cell RNAseq analysis revealed cellular heterogeneity in the population of FAPs in resting muscles. A first study showed that FAPs sub-cluster in two different populations in non-injured muscles, one that is enriched for transcripts associated with ECM genes (e.g. Col4a1, Col6a1, Lum, Sparcl1, Podn, Smoc2, Mgp and Bgn) and the other one that express higher levels of genes involved in cell signalling pathways (e.g. Sfrp4, Igfbp5, Sema3c, Dpp4, Tgfrb2 and Wnt2) [40]. Similar findings were observed in another study that has subdivided the FAPs population in non-injured muscles in Cxcl14-expressing FAPs that are enriched in ECM genes (e.g. Col4a1, Col5a3, Col6a1, Smoc2 and Lum) and the Dpp4-expressing FAPs that are enriched in genes involved in different biological processes and signalling pathways (e.g. Igfbp5, Igfbp6, Wnt2, tnfaip6 and Sema3c) [9]. Furthermore, another study distinguished two cell subpopulations of FAPs based on the expression of Tek (gene encoding for Tie2) and Vcam1 [41]. Tek+ FAPs are the predominant subpopulation in non-injured muscle and they preferentially express genes implicated in Wnt and BMP signalling (e.g. Wnt5a, Wnt11, Bmp4 and Bmp6). Vcam1+ FAPs represent a smaller subset in non-injured muscle (that becomes the predominant subpopulation in response to acute injury) and display a pro-fibrotic gene signature (e.g. Acta2, Adam12, Lox and Timp1) [41]. Another recent study using unbiased scRNA-seq from healthy skeletal muscle also observed that Tek and Vcam expression are segregated in two different clusters in the FAPs population [42]. Moreover, this paper also demonstrated that the Vcam1+ population can be further subdivided to generate a third cellular subset characterized by the expression of thrombospondin-4 (Thbs4) and fibulin-7 (Fbln7) that is enriched in ECM organization and metallopeptidase activity genes [42]. Overall, while some discrepancies exist between the findings described in these independent studies, which could be attributable to different cell isolation/purification techniques and/or bioinformatic analyses, these scRNA-seq experiments reveal the cellular heterogeneity of FAPs and pinpoint common gene signatures of the different cellular subsets across the datasets.
4.2. Role of FAPs in the regulation of muscle homeostasis
FAPs are one of the predominant mononuclear cell population in non-injured muscle. These cells are quiescent in non-injured muscle, which is regulated, at least in part, by their expression of hypermethylated in cancer-1 (Hic1) [40]. Deletion of this factor leads to spontaneous cell cycle entry and expansion of the FAPs population [40]. FAPs are required for homeostatic maintenance of skeletal muscle in steady-state conditions [16,17,43]. Depletion of FAPs induced by diphtheria toxin (DTX) administration to transgenic mice containing the Fap gene (fibroblast-activation protein-a) with the insertion of the DTX receptor led to a reduction of muscle mass and myofibre size after three weeks [43]. Similarly, FAPs depletion induced by tamoxifen injection to PDGFRα.creER-DTX mice induces muscle atrophy and reduces muscle force, which can last for months, even in the absence of injury [16,44]. Depletion of FAPs does not affect the number of muscle stem cells in the first few weeks [44], but a reduction in the number of muscle stem cells is noted nine months later (other cell types such as endothelial cells and haematopoietic cells were not affected) [16]. These results indicate that FAPs are required for the long-term homeostatic maintenance of the muscle stem cell pool and myofibre growth. As FAPs do not possess myogenic properties per se, these effects are mediated by molecular crosstalk with muscle cells.
FAPs provide a supportive environment for myogenic cells as they are the main source of extracellular matrix components, such as collagens (e.g. Col6a1, Col5a1), laminin (e.g. lama2, lamb1) and fibronectin (Fn1) [45]. These extracellular matrix proteins constitute the muscle stem cell niche and play a crucial role in their self-renewal [46–49]. FAPs also express cytokines and growth factors known to regulate myogenesis and muscle growth. It was shown in vivo that FAPs specifically express growth differentiation factor 10 (GDF10; also known as bone morphogenetic protein 3b, Bmp3b), a member of the TGF-β superfamily, that stimulates myotube hypertrophy in vitro (but not myoblast differentiation/fusion) by activating the Smad1/-5/-8 and Akt pathways [44]. Deletion of this factor in Bmp3b-knockout mice induces muscle atrophy [44]. Other in vitro experiments showed that FAPs are a major source of many trophic factors, such as interleukin-6 (IL-6), IL-10 and follistatin, among others, that promote myogenesis and muscle growth [15,50–53]. The exact cocktail of cytokines secreted by FAPs and their contribution in homeostatic condition remains elusive; however, the impact of these paracrine factors on skeletal muscle regeneration has been more extensively studied, and will be discussed hereafter.
5. FAPs in skeletal muscle injury/regeneration
5.1. FAPs dynamics in regenerating skeletal muscle
Upon acute muscle injury, FAPs rapidly enter cell cycle. The rise in BrDU+ cells happens faster in FAPs than in the muscle stem cell population, which leads to an increase in the FAPs/muscle stem cell ratio during the first few days after an injury [10,15]. The total number of FAPs peaks around 3–4 days post-injury, depending on the type and severity of injury [36,54]. Thereafter, there is a strong increase in cellular apoptosis and the number of FAPs gradually returns to basal level [54]. Conditional ablation experiments revealed the importance of FAPs during muscle regeneration. Depletion of FAPs induced by tamoxifen injection in PDGFRα.creER-DTX mice does not induce myofibre necrosis in non-injured mice [44], but it prolonged necrosis and led to significant regeneration deficit after acute muscle injury (BaCl2 injection) [16]. Transplantation of FAPs into the injured muscle of ablated mice rescued muscle regenerative capacity, confirming the crucial role of FAPs in muscle regeneration.
Single-cell RNAseq analysis revealed a dynamic and heterogeneous cluster of FAPs during regeneration [8,9]. At 2 days post-injury, activated FAPs express high levels of chemokines (e.g. Ccl7, Cxcl5 and Ccl2) known to play a role in monocyte and neutrophil recruitment [8,9]. At this time point, there is also the emergence of a Tie2low Vcam1high subpopulation of FAPs expressing a pro-fibrotic gene signature [41]. At 5 days post-injury FAPs express higher levels of ECM genes such as Col3a1, Col8a1, Dcn and Fn2. After 7–10 days post-injury the expression profile of FAPs gradually returns to that observed in non-injured muscles (re-expression of markers such as CD34 and Sprouty RTK signalling antagonist 1 (Spry1)). However, at 21 days post-injury, the majority of FAPs still express high levels of Osr1, while this factor is weakly expressed in non-injured muscle, suggesting that these cells did not entirely return to quiescence at that point. Notably, Osr1+ FAPs can be divided in two subgroups, the dipeptidyl peptidase-4 (Dpp4) positive and the chemokine (C–X–C motif) ligand 4 (Cxcl4) positive cell populations indicating that they are re-acquiring cellular heterogeneity similar to what is observed in non-injured muscles [9,40].
5.2. Intrinsic regulation of FAPs in regenerating skeletal muscle
The accumulation of FAPs during muscle regeneration is tightly regulated. Yet, the intrinsic mechanisms controlling these dynamic changes are poorly characterized. One pioneer study showed that FAPs express different transcriptional variants of PDGFRα [55]. One of these variants contains a truncated kinase domain; and acts as a decoy receptor to inhibit PDGF signalling. Inhibition of this intronic polyadenylated variant increases FAPs proliferation and fibrogenic gene expression. The ratio of this intronic variant increases during muscle regeneration suggesting an intrinsic regulatory mechanism for FAPs to limit their expansion and activity. Another study showed that FAPs become ciliated upon muscle injury [36]. FAPs cilia represses Hedgehog signalling, a pathway that plays a crucial role to inhibit adipogenesis [36,56]. Consistently, deletion of FAPs cilia over-activates Hedgehog signalling and prevents adipogenesis following glycerol-induced muscle injury [36]. Another study indicated that retinoic acid receptor signalling plays an important role in the intrinsic regulation of FAPs number and cell fate decision [57]. Treatment of FAPs with retinoic acid promotes their proliferation in vitro and inhibits their adipogenic and fibrogenic differentiation both in vitro and in vivo (in obese mice fed with high-fat diet) [57]. Inversely, loss of retinoic acid receptor signalling specifically in FAPs promotes their adipogenic differentiation in vitro and in vivo; and it decreases cell apoptosis and delays their clearance following injury in vivo [57].
There is limited knowledge on the intrinsic mechanisms regulating FAPs during myogenesis; however, there is strong evidence that extrinsic factors play crucial roles in the regulation of FAPs behaviour. A transplantation experiment showed that FAPs isolated from cardiotoxin-injured muscles (which display virtually no adipogenesis) become adipocytes when transplanted into glycerol-injured muscles [17]. Conversely, FAPs isolated from glycerol-injured muscles do not become adipocytes when transplanted into cardiotoxin-injured muscles. These results suggest that the cell fate decision of FAPs is not regulated in a cell-autonomous manner, but rather by the extrinsic environment. The impact of inflammatory cells, myogenic cells and other muscle-resident cell types on FAPs will be further discussed hereafter.
6. FAPs cellular and molecular crosstalk in regenerating skeletal muscle
6.1. Interactions of FAPs with inflammatory cells
Skeletal muscle injury induces a rapid inflammatory response characterized by the subsequent accumulation of polymorphonuclear leucocytes, monocytes/macrophages and lymphocytes. The peak in inflammatory cell density coincides with the one of FAPs. Analysis of the interactome network from single-cell RNAseq data revealed that FAPs have strong receptor–ligand interactions with immune cells, especially polymorphonuclear cells and monocytes [9]. As mentioned above, in the early phase of muscle regeneration, FAPs express high levels of chemokines (e.g. Ccl7, Cxcl5 and Ccl2) and immunomodulatory cytokines that regulate the accumulation and the function of inflammatory cells such as monocytes and neutrophils (figure 3) [8,9]. Inducible depletion of FAPs by tamoxifen injection in PDGFRα.creER-DTX mice followed by muscle injury is associated with a reduction in the accumulation of CD45+ haematopoietic cells during the acute phase of muscle regeneration [16,44]. Moreover, the transcriptional profile of a subset of FAPs during muscle regeneration is enriched in canonical pathways related to dendritic cell maturation, suggesting that they might play a role in the phenotypic switch of macrophages [41]. Particularly, activated FAPs upregulate their expression of IL-10 upon muscle damage [51]. This cytokine is a central effector that triggers the change in macrophage subsets toward their anti-inflammatory phenotype, which in turn secrete factors that promote the differentiation and fusion of myogenic cells [13,14,58].
Figure 3.

Cellular interactions during muscle regeneration. Schematic of the known interactions between muscle stem cells (MuSC, pink background), fibro-adipogenic progenitors (FAPs, blue background) and immune cells (green background) throughout muscle regeneration. The blue arrows represent molecules secreted by FAPs that act on other cell types, whereas pink and green arrows indicate paracrine factors secreted by muscle stem cells and immune cells, respectively, that affect FAPs. IL, interleukin; WISP1, Wnt1-inducible-signalling pathway protein 1; IGF1, insulin growth factor-1; GDF10, growth differentiation factor 10; TNF-α, tumour necrosis factor alpha; TGF-β, transforming growth factor-β; DAMPs, damage-associated molecular pattern; CCL, C–C motif chemokine ligand; CXCL5, C–X–C motif chemokine ligand 5, PMN, polymorphonuclear cells. This figure was created with the Servier Medical Art service (https://smart.servier.com/), which is licensed under a Creative Commons Attribution 3.0 Unported License (https://creativecommons.org/licenses/by/3.0/).
Evidence also supports a role of FAPs in the modulation of regulatory T-cells (Treg) function. These cells peak late during muscle regeneration (4–7 days post-injury) and their numbers remain elevated for weeks after the injury. In skeletal muscles, FAPs are the main cellular source of IL-33, a member of the IL-1 family that has a potent immunomodulatory effect on Treg [59,60]. This alarmine is rapidly released upon muscle damage and stimulates the proliferation of Treg in vitro and in vivo [60]. Skeletal muscle injury in Il1rl1-null mice (IL-33 receptor) results in reduced accumulation of Treg during muscle regeneration and smaller size of newly formed myofibres [60]. Altogether, these findings suggest that FAPs regulate the accumulation and the function of immune cells, which in turn affect myogenesis.
The interactions between FAPs and immune cells are bilateral (figure 3). Different inflammatory cells were shown to play a key role in the regulation of FAPs accumulation, activity, cell fate decision and clearance. After an injury, the first cells to infiltrate the tissue are polymorphonuclear leucocytes. They enter the injured muscle as soon as 2 h post-injury and their numbers are peaking approximately 24 h post-injury, and rapidly decline afterward [61,62]. Among the polymorphonuclear cells, eosinophils were shown to play a central role in the regulation of FAPs [63]. Eosinophils are the main cellular source of IL-4 during muscle regeneration, and the IL-4 receptor (IL-4Rα) is highly expressed in FAPs. Lack of IL-4 signalling in Il4/Il13−/− mice and Il4rα−/− mice reduces FAPs proliferation (no effect on myogenic cell proliferation) and impairs muscle regeneration [63]. IL-4 also prevents FAPs differentiation into adipocytes in vitro, by repressing the expression of genes normally involved in adipogenesis (Pparg, Lep, Fabp4, Acaca, Cd36 and Dgat2) [63]. Moreover, FAPs showed a strong phagocytosis capacity in vitro and in vivo, which is impaired in mice lacking type 2 innate immunity (Il4/Il13−/−, Il4rα−/−) and in eosinophil-deficient mice (ΔdblGATA mice) leading to lack of clearance of necrotic debris in injured muscles. Administration of IL-4 in vivo reduced the number of necrotic cells in injured Il4/Il13−/− mice, suggesting that it rescues the phagocytotic capacity of FAPs (although a reduction in muscle degeneration through other mechanisms cannot be excluded) [63]. Altogether, these results indicate that the production of IL-4 by eosinophils regulates FAPs proliferation, phagocytosis and cell fate decision.
Monocytes/macrophages also play a key role in the regulation of FAPs. Clodronate-induced depletion of macrophages prior to muscle injury leads to a strong increase in fatty tissue infiltration [34]. Similarly, muscle injury in Ccr2-knockout mice, in which monocytes are not able to infiltrate the injured tissue, leads to the prolonged persistence of FAPs in the regenerating muscle associated with fibrosis deposition [54]. The impact of the depletion of monocytes/macrophages on the accumulation of FAPs is mediated by changes in cell apoptosis and not in cell proliferation. TNF-α, which is highly secreted by pro-inflammatory macrophages during acute injury, was shown to be the effector of FAPs apoptosis [54]. Another pro-inflammatory factor secreted by pro-inflammatory macrophages, IL-1α/β, was also shown to inhibit adipogenic differentiation of FAPs [64]. As the regeneration process progresses, there is a switch in macrophage phenotype towards anti-inflammatory macrophages, which secrete higher levels of TGF-β. This growth factor competes with TNF-α and promotes the survival of FAPs. TGF-β was also shown to downregulate the expression of the FAPs markers PDGFRα and TCF7L2/TCF4, and their downstream signalling pathways [65,66]. TGF-β influences the cell fate decision of FAPs by enhancing their survival, promoting their proliferation, inhibiting their adipogenic differentiation and favouring their differentiation into myofibroblasts [54,67,68]. In vitro experiments showed that IL-1β-polarized pro-inflammatory macrophages (expressing high levels of IL-6 and TNF-α) inhibits FAPs differentiation towards adipogenic cells, while IL-4-polarized anti-inflammatory macrophages promote the formation of adipocytes [69]. Altogether, these results indicate that the switch in macrophage phenotype needs to be fine-tuned to avoid excessive accumulation of FAPs and fibrotic tissue deposition, and to promote return to homeostasis and long-term maintenance of the cellular reserve.
6.2. Interaction of FAPs with myogenic cells
The formation of new muscle tissue during muscle regeneration needs to be closely coordinated with the remodelling of the ECM. Accordingly, FAPs maintain a close proximity with myofibres in resting muscle and during muscle regeneration (figure 2) [15,17]. After an injury, FAPs quickly enter the cell cycle (BrdU+ or EdU+ cells) and the ratio FAPs/MuSC increases during the first few days, suggesting that they may act upstream and help orchestrate the myogenesis process [10,15]. Cardiotoxin injury in FAPs-depleted muscles (PDGFRα.creER-DTX mice) is associated with a strong decline in the number of myogenic cells at an early time point (3 days post-injury), and a reduction in the size of the newly formed fibres at a later time point (14 days post-injury) [16,44]. Similar impairment in myogenic cell pool expansion and muscle regeneration is observed when mice are treated with the tyrosine kinase inhibitor Nilotinib (PDGFR inhibitor), which induces FAPs apoptosis and reduces the number of FAPs in the injured muscle [70,71]. In vitro co-culture experiments showed that FAPs preferentially promote the differentiation of myogenic cells and their fusion into multinucleated myotubes [15,17]. Gene expression analysis confirmed that FAPs reduce the expression of early myogenesis markers (e.g. Pax3, Pax7) and promote the expression of late myogenic differentiation markers (e.g. MyoD, Myog). Moreover, TCF7L2/TCF4+ cells (likely FAPs) [65] also regulate muscle fibre type and maturation. In vitro, transwell co-culture experiments of TCF7L2/TCF4+ FAPs and myogenic cells showed that paracrine factors secreted by FAPs enhance fusion index and slow MyHC expression. In vivo, the deletion of TCF7L2/TCF4+ using different transgenic mouse models increases the developmental myosin heavy chain (MyHC) expression and decreases the expression of slow MyHC in neonatal mice [20].
The mechanisms by which FAPs orchestrate muscle stem cell activity during myogenesis is under intense investigation in the field. FAPs are a predominant source of paracrine factors during muscle regeneration (figure 3) [15,17]. The main molecular components of the secretome of FAPs were comprehensively summarized in two recent reviews [53,72]. Briefly, activated FAPs express high levels of cytokines such as IL-6. This cytokine has been described as a myokine (i.e. secreted by myofibres in response to exercise); however, its expression in regenerating muscle is roughly 10-fold higher in FAPs than in myogenic progenitor cells [15,17]. IL-6 promotes muscle stem cell proliferation and myogenic progression [73,74]. Activated FAPs also strongly upregulate the expression of WNT1-inducible-signalling pathway protein 1 (WISP1) [75]. WISP1 promotes muscle stem cell expansion and myogenic commitment via asymmetric cell division [75]. FAPs also express high levels of IGF-1, which activates the Akt pathway that controls both protein synthesis and degradation, resulting in skeletal muscle growth [15,17,76]. In accordance with a role of FAPs in late stages of myogenesis, activated FAPs are the main mononuclear cell source of follistatin during acute muscle regeneration post-injury [50,77]. This activin-binding protein is an antagonist of the muscle growth inhibitor myostatin. FAPs-secreted follistatin also promotes myoblasts fusion into multinucleated myotubes [77,78]. Overall, FAPs secrete a cocktail of cytokines that have an impact on myogenic cells throughout the different stages of myogenesis.
Myogenic cells also contribute to the regulation of FAPs during muscle regeneration (figure 3). In vivo, the deletion of muscle stem cells using the Pax7.CreERT2-DTX mice model impaired the expansion of FAPs (TCF7L2/TCF4-expressing cells) during the early phase of muscle regeneration (5 days post-injury) but led to their prolonged accumulation and fibrosis deposition at a later time point (28 days post-injury) [18]. Muscle stem cells secrete exosomes containing miRNA, such as miR206, which inhibit collagen production by TCF7L2/TCF4+ FAPs [79]. In vitro experiments showed that myoblast conditioned medium promotes FAPs proliferation. Satellite cells were shown to secrete betabellulin and epidermal growth factor (EGF), two ligands of the EGF receptor (EGFR), which stimulate FAPs proliferation in vitro [64]. Notably, contrary to myoblast conditioned medium, myotube-conditioned medium inhibits the expression of adipogenesis genes and upregulates the expression of fibrogenesis genes, suggesting that factors secreted during late stage of myogenesis could impact on the cell fate decision of FAPs and ECM remodelling [17,80]. Other studies have suggested that inhibition of adipogenesis by differentiated myoblasts is mediated by direct contact rather than secreted factors [17,34]. The binding of delta-like ligand (DLL) expressed on the myogenic cells to the Notch receptor on the FAPs would lead to a Notch-dependent inhibition of adipogenic gene expression [34].
Among the myokines secreted by muscle cells after injury, IL-15 was shown to influence FAPs behaviour. IL-15 administration stimulates FAPs proliferation in vitro and in vivo [81]. This pro-proliferative effect is mediated trough Jak-Stat pathway, and the administration of the Jak inhibitor SAR-20347 decreased FAPs proliferation and prevented fibrosis deposition post-injury. IL-15 also influences cell fate decision of FAPs, and prevents their differentiation into adipocytes in vitro and in vivo [81–83]. This IL-15-mediated decrease in adipogenesis is associated with an upregulation of desert hedgehog (DHH) signalling pathway and its downstream effector tissue inhibitor of metalloproteinase 3 (Timp3). This pathway is regulated by primary cilia in FAPs, which has been demonstrated to repress FAPs adipogenic differentiation [36]. Conversely, IL-15 treatment increases ECM gene expression and collagen deposition in injured skeletal muscle [81]. These findings suggest that IL-15 controls the cell fate decision of FAPs to preferentially favour fibrogenesis over adipogenesis.
6.3. Interaction of FAPs with other muscle-resident cell types
Interstitial stromal cells can also have auto-regulatory effects on FAPs. These cells were identified by single-cell RNAseq analysis based on the expression of interstitial stromal cell markers such as the FAPs markers Sca1 and PDGFRa, the pericytes or mesoangioblasts marker Alpl (Alkaline phosphatase), and the PW1+ interstitial cells (PICs) marker Peg3 (paternally expressed gene 3). These interstitial stromal cells were subdivided in three subsets [42]. These subpopulations express high levels of adipogenesis genes, except one cluster expressing CD142 [42]. The CD142+ interstitial stromal cells are able to differentiate into fibrogenic cells but not adipogenic cells. Remarkably, when co-culture together, the CD142+ interstitial stromal cells inhibit the adipogenic differentiation of CD142− interstitial stromal cells. The secretion of high levels of GDF10 by these adipo-regulatory cells is at least partially responsible for the suppression of adipogenesis. FAPs are also the main mononuclear cell source of Wnt signalling protein in skeletal muscles [78]. The secretion of Wnt5a by FAPs mediate an autocrine response that activates β-catenin signalling and blocks adipogenesis. These results indicate that the different subsets of FAPs could regulate their own cell fate decision.
Schwann cells also participate in the regulation of FAPs cell fate [36]. Single-cell transcriptomics of injured muscles indicate that Schwann cells are the main source of Dhh signalling during muscle regeneration [8,9]. Notably, Schwann cells express Dhh following cardiotoxin injection, a model of injury that is devoid of adipose tissue deposition; however, Schwann cells do not express Dhh in glycerol-induced injury, which causes severe adipogenesis [36]. Dhh activates the expression of cilia-dependent Hedgehog target genes, particularly Timp3, which represses adipogenesis through inhibition of matrix metalloproteinase 14 (Mmp14).
Analysis of the putative interactome also indicates strong ligand–receptor interactions between endothelial cells and FAPs [9], which is coherent with the perivascular localization of FAPs [15,17,19,40]. Deletion of PDGFRα+ cells by tamoxifen and DTX administration to Pdgfra-MerCreMer/inducible-DTX receptor mice did not induce vascular tissue disruption in the short term in the absence of injury [19]. However, after hindlimb ischaemia, impairments in vessel size, organization and permeability were observed following PDGFRα cell ablation [19]. FAPs are a source of vascular endothelial growth factor (VEGF), which is known to promote postnatal angiogenesis [84]. Yet, the exact interplay between FAPs and endothelial cells in muscle homeostasis and regeneration needs to be further explored [72].
Overall, FAPs are central regulators of muscle homeostasis and regeneration. Their complex molecular and cellular interactions with inflammatory cells, myogenic cells and other resident cell types play a critical role in the coordination of skeletal muscle response to injury. These intrinsic and extrinsic regulatory mechanisms control the activation, proliferation, cell fate decision and clearance of FAPs to avoid the prolonged or excessive accumulation of fibrofatty tissue. However, this delicate balance is perturbed in different pathological conditions in which FAPs contribute to disease progression. The impact of FAPs in muscular dystrophies and ageing will be discussed in this next section.
7. FAPs in muscular dystrophies
7.1. Impact of FAPs in the pathogenesis of Duchenne muscular dystrophy
Loss of muscle mass and replacement with fibrous and fatty tissue is a hallmark of many muscular diseases. Considering the intrinsic ability of FAPs to differentiate into adipogenic or fibrogenic cells, they are likely to be the main effector of these pathological processes. Accumulating evidence places FAPs on the front line of several muscular diseases.
Among myopathies, DMD is the most studied. This pathology is caused by a mutation in the DMD gene which encodes for the dystrophin protein [85,86]. The dystrophin–dystroglycan complex connects the actin cytoskeleton of the muscle fibres to the extracellular matrix, and acts as a force transduction system during muscle contraction [87]. Lack of dystrophin induces muscle fibre fragility and cycle of degeneration and regeneration leading to muscle wasting, chronic inflammation and fibrosis deposition [3,88,89]. Endomysial fibrosis and fat accumulation are among the only parameters correlated with poor motor outcomes in DMD [90–92]. Therefore, a lot of effort has been invested to characterize the cells responsible for the fibrofatty accumulation and the underlying mechanisms driving fibrofatty accumulation in order to develop new therapeutic avenues [54,93].
By contrast to acute injury, which induces a transient expansion of the FAPs population, dystrophic muscles experience a persistent increase in the number of FAPs [10,35,54,94]. In dystrophin-null mdx mice, the vast majority of collagen-expressing cells (identified by Col1a1-GFP reporter) are PDGFRα+ and Sca1+ cells, suggesting that FAPs are the main source of fibrosis [95].
7.2. FAPs heterogeneity in Duchenne muscular dystrophy
An increase in cellular heterogeneity is observed in FAPs in dystrophic muscles. Two subpopulations of FAPs, the Sca1-low/CD34-low and Sca1-high/CD34-high subsets, which are predominantly found in non-injured and injured wild-type muscles, respectively, are both found alongside within mdx muscles [34]. The Sca1-high subpopulation proliferate faster than the Sca1-low subpopulation and are predisposed to differentiate into adipocytes [33]. Similarly, there is the emergence of a subpopulation of PDGFRα-low FAPs expressing high levels of fibroblast associated genes (TGFb1, Col1a1 and CTGF) in the skeletal muscles of mdx mice [66]. FAPs can also be subdivided in Vcam1− and Vcam1+ subpopulations in dystrophic muscles, the latter being absent from non-injured wild-type muscles [41]. This Vcam1+ subset has a pro-fibrotic transcriptional profile and has high proliferative state compared to other FAPs subpopulations [41]. Single-cell RNAseq analysis from different datasets suggests that there is an overlap between these different subsets, although it remains to be confirmed precisely [8,9]. Furthermore, there are also differences within the analogous subpopulations depending on the pathophysiological context. For example, Tie2-high subpopulation isolated from acute injured muscle displays changes in gene expression related to biological function such as muscle growth and dendritic cell maturation; however, these characteristics are not observed in Tie2-high subpopulation isolated from DMD muscle [41].
7.3. Regulatory network of FAPs in Duchenne muscular dystrophy
Considering the importance of the molecular and cellular crosstalk in the regulation of FAPs accumulation and cell fate decision, it is likely that the changes observed in FAPs behaviour are attributable to the degenerative microenvironment found in dystrophic muscles. Contrary to wild-type mice that display a coordinated response following injury, cardiotoxin-induced muscle injury in mdx mice does not induce the expansion of FAPs, which is associated with impairment in the myogenic cell pool growth and macrophage phenotype switching [10]. These findings suggest that the molecular signals and cellular interactions are perturbed in dystrophic muscles, which consequently deregulate the accumulation and clearance of FAPs.
Among the different molecules upregulated in DMD, TGF-β is a key regulator of FAPs functioning. A positive correlation between TGF-β levels, FAPs content and fibrosis has been observed in different muscle injuries and diseases such as denervation [66], glycerol-induced injury [66], amyotrophic lateral sclerosis (hSOD1G93A mice) [96] and DMD (mdx mice and DMD patients) [97–99]. In DMD, the chronic presence of macrophages expressing a hybrid phenotype (secreting both TNF-α and TGF-β) leads to conflicting signals to FAPs that fail to induce apoptosis [54]. Moreover, FAPs are the main producers of TGF-β-activating enzymes such as BMP1 and MMP14 that activate the latent-TGF-β secreted by macrophages [100]. High levels of TGF-β reduce the expression of TCF7L2/TCF4, as well as PDGFRα and its target genes, leading to the emergence of a TCF7L2/TCF4-low PDGFRα-low subpopulation of FAPs exhibiting a pro-fibrotic profile [65,66,101]. Corroborating with those findings, D2-mdx mice, which carry a genetic variant in the latent transforming growth factor binding protein 4 (LTBP4) that reduces its capacity to sequester TGF-β (consequently increasing the levels of active TGF-β), display increased FAPs accumulation and fibro-calcification deposition [101]. Noteworthy, chronic exposure to high levels of TGF-β in dystrophic muscles induces changes in a subset of endothelial cells and satellite cells that lose their cell identity and acquire a mesenchymal-like phenotype [97]. These cells acquire the expression of PDGFRα, produce higher levels of fibrogenic genes (e.g. Col1a1, Fn1 and Acta2) and gain the capacity to transdifferentiate into fibroblasts in vitro, suggesting that cell types other than FAPs could also contribute to fibrogenesis in DMD [97].
Evidence also indicates that the molecular signals provided by myogenic cells to FAPs through direct contact or by secretion of paracrine factors are also perturbed in DMD. Conditioned medium from myogenic cells isolated from DMD patients failed to promote FAPs proliferation and fibrogenic differentiation [80]. Moreover, while in healthy regenerating muscle the expression of the Notch ligand on the cell surface of differentiated myoblasts/myotubes inhibits adipogenic differentiation of FAPs by direct cellular contact, FAPs from mdx mice are insensitive to NOTCH-induced adipogenic inhibition [34].
Cilia-mediated signalling could also regulate FAPs adipogenic cell fate decision in dystrophic muscles. Although there is not a significant increase in the proportion of ciliated FAPs in mdx mice, the deletion of cilia specifically in FAPs reduces the number of adipocytes and enhances muscle regeneration in dystrophic muscles [36]. The impact of FAPs cilia on the regulation of adipogenesis is non-cell autonomous, suggesting a lack in hedgehog protein secreted from neighbouring cells.
Impairment in the secretion of autocrine factors by FAPs in dystrophic muscles could also have an impact on their cell fate decision. Analysis from bulk RNAseq datasets revealed downregulation of WNT ligands and receptors in FAPs from mdx mice, which could affect their cell fate considering the importance of the WNT5/GSK3/β-catenin pathway in the regulation of adipogenesis [78]. Consistently, treatment of mdx FAPs with WNT5a prevents β-catenin downregulation, which inhibits PPARγ expression and adipogenesis.
Overall, intrinsic and extrinsic factors contribute to the persistence of FAPs and to the dysregulation of their activity and cell fate decision in DMD.
7.4. FAPs in other muscular dystrophies
Apart from their role in DMD, FAPs are also implicated in the pathogenesis of many other muscular diseases. In limb girdle muscular dystrophy type 2 (LGMD2B), adipogenic replacement in dysferlin-deficient skeletal muscle is correlated with the severity of the disease [102]. Sustained muscle injury induces the extracellular release of annexin-A2, which causes FAPs accumulation and adipogenic differentiation [102]. In a model of LGMD2E (sarcoglycan-β deficient mice), it was shown that chronic muscle degeneration impairs the cellular heterogeneity of the different subsets of interstitial stromal cells (analogous to FAPs). Particularly, there is a reduction in the population of CD142+ interstitial stromal cells, which act as adipo-regulatory cells that inhibits adipocyte differentiation through GDF10 secretion [42]. Other congenital muscular dystrophies such as merosin-deficient congenital muscular dystrophy type 1A (LAMA2 deficiency) or Ullrich congenital muscular dystrophy (mutations in COL6A1, 2 or 3) are also characterized by muscle degeneration and fibrosis accumulation [103–105]. LAMA2 deficiency in skeletal muscle is associated with an over-activation of the TGF-β pathway and fibrotic tissue deposition that correlates with the severity of the disease [106–108]. Losartan treatment (an angiotensin II receptor blocker) in LAMA2-mutant mice reduces TGF-β expression and fibrosis deposition [109]. Likewise, COL6A1 myopathies are characterized by an excessive accumulation of PDGFRα+ cells and fibrosis in mouse model and in patients affected by Ullrich congenital muscular dystrophy [105]. The exact contribution of FAPs to the pathogenesis of these diseases remains to be determined.
8. Therapeutic avenues targeting FAPs in muscular dystrophies
8.1. Glucocorticoids
Glucocorticoids are the current gold-standard treatment for DMD. These drugs dampen inflammation and temporarily prevent the loss in muscle force and physical function [110]. However, glucocorticoids have numerous side effects. Glucocorticoids administration to fibroblasts or mesenchymal stromal cells in vitro increases the expression of adipocyte-associated gene (e.g. Pparg) and promotes adipocyte differentiation [111–113]. Inhibition of glucocorticoid receptor signalling using pharmacological antagonists or knockout models blocks the adipogenic differentiation of these cells in vitro [113]. In vivo, the administration of dexamethasone, a glucocorticoid, stimulates FAPs proliferation and adipogenic differentiation in injured muscle by inhibiting IL-4 expression [111]. A similar increase in adipogenesis was also observed in vivo following daily injection of prednisone, another glucocorticoid, in dystrophic mice [114]. Recent findings also suggest that glucocorticoids can have pro- or anti-adipogenic effects on FAPs depending on the type of glucocorticoids used and on the culture conditions [115]. For instance, glucocorticoids such as halcinonide and clobetasol have no effect on adipogenesis, while budesonide has anti-adipogenic effect in vitro [115]. This anti-adipogenic effect of budesonide was only observed when FAPs were actively proliferating in vitro. Conversely, budesonide has a pro-adipogenic effect when FAPs are confluent and are incubated in adipogenic induction medium [115]. Overall, while glucocorticoids preserve muscle function by reducing inflammation, there is room for improvement to develop therapeutic drugs targeting FAPs, directly or indirectly, to promote their beneficial effects (e.g. stimulation of myogenesis) and limit their harmful side effects (e.g. excessive fibrofatty accumulation) (figure 4).
Figure 4.

Molecular approaches targeting fibro-adipogenic progenitors in muscular dystrophies. Schematic of different drugs tested to inhibit fibro-adipogenic progenitor (FAP) activity, proliferation and/or their differentiation into adipocytes or fibroblasts. Arrows and T-shaped lines indicate stimulatory and inhibitory effects, respectively. DMD, Duchenne muscular dystrophy; HDAC, histone deacetylase; Vcam1, vascular cell adhesion molecule 1; PDGFRα, platelet-derived growth factor receptor α; TCF7L2/TCF4, transcription factor 4; Sca1, stem cells antigen-1; TGF-β, transforming growth factor-β; MMP, matrix metalloproteinase; GSK3, glycogen synthase kinase-3. This figure was created with the Servier Medical Art service (https://smart.servier.com/), which is licensed under a Creative Commons Attribution 3.0 Unported License (https://creativecommons.org/licenses/by/3.0/).
8.2. TGF-β inhibitors
Considering the correlation between TGF-β levels, FAPs content and fibrosis, many studies have investigated the therapeutic potential of TGF-β inhibitors. Administration of the TGF-β inhibitor, SB525334, which blocks the ATP-binding site of TGFBR1 and inhibits the TGF-β induced signalling, on FAPs cultured in vitro prevented the decrease in PDGFRα levels induced by TGF-β [66]. Intramuscular injection of ITD-1, a small molecule enhancing TGFBR2 degradation thereby inhibiting TGF-β-induced signalling, reduced the accumulation of FAPs and muscle fibrosis in injured D2-mdx mice [101]. Inhibition of the TGF-β-activating enzymes MMP14 and BMP1 (with the compounds NSC-405020 and UK-383367, respectively) also reduced collagen deposition and increased myofibre size in a fibrotic model of mdx mice [100]. Similarly, inhibition of the TGFBR1 expression using antisense oligonucleotide intramuscular injection in mdx mice reduced the expression of fibrotic genes (e.g. Col1a1) and increased the expression of myogenic genes (e.g. Myog) [116]. Altogether, the different strategies used support the therapeutic potential of targeting TGF-β to inhibit the fibrogenic activity of FAPs and fibrosis deposition in dystrophic muscles.
8.3. GSK-3 inhibitors
Library drug screening of kinase inhibitors on FAPs isolated from mdx mice showed an enrichment in GSK3-targeting molecules having anti-fibrotic and/or anti-adipogenic effects [78]. Administration of the GSK3 inhibitor, LY2090314, represses FAPs adipogenesis in glycerol-injured muscle in vivo. Moreover, GSK3 inhibitor enhances the secretion of paracrine factors, particularly follistatin, that promotes myogenesis [78]. The same group used a similar drug screening strategy with a different small molecule library, and they identified azathioprine (AZA) as a promising therapeutic compound [117]. This immunosuppressant drug targets FAPs and blunts their adipogenic differentiation in vitro. AZA inhibits the insulin-mediated activation of the Akt-mTOR pathway in FAPs isolated from mdx mice.
8.4. Tyrosine kinase inhibitors
The tyrosin kinase inhibitors, nilotinib and imatinib, initially designed to target the BCR-ABL fusion oncogene in chronic myelogenous leukaemia, also showed significant anti-fibrosis capacity. Treatment of mdx mice with these compounds increases FAPs apoptosis, and reduces FAPs accumulation and fibrogenic differentiation leading to lower collagen deposition [54,118,119]. Nilotinib and imatinib are potent inhibitors of PDGFRα and can directly target FAPs to regulate their activity. Similarly, crenolanib, another PDGFRα/β inhibitor, was also shown to reduce ECM protein expression and fibrosis deposition in skeletal muscles of mdx mice [95]. These tyrosin kinase inhibitors could also indirectly regulate FAPs activity by acting on inflammatory cells. Imatinib treatment was shown to decrease macrophage infiltration in mdx mice [118,119]; while nilotinib was shown to block the anti-apoptotic effect of TGF-β secreted by macrophages and restore the pro-apoptotic capacity of TNF-α [54]. Furthermore, nilotinib also inhibits the phosphorylation of p38 induced by TGF-β administration to the C3H10T1/2 cell line (murine mesenchymal stromal cell line), suggesting that the effect of this drug on FAPs could also be mediated by off-target mechanisms [71,120].
8.5. Histone deacetylase inhibitors
Histone deacetylase (HDAC) inhibitors have also been investigated for the treatment of DMD, due to their ability to remove the repressive epigenetic marks and promote the expression of muscle-specific genes. Preclinical trials on mdx mice showed that HDAC inhibitors such as trichostatin A (TSA) or givinostat reduce fibrosis and promote muscle regeneration and function [121–123]. A phase 2 clinical trial also showed that givinostat induces a reduction in fibrosis in DMD boys [124]. The underlying mechanism responsible for these effects seems to be mediated by FAPs [77]. Treatment of FAPs isolated from mdx mice with TSA reduces their differentiation into adipocytes and promotes their myogenic capacity through secretion of paracrine factors such as follistatin. However, these effects are only observed in young mdx mice (1.5 months old), suggesting that long-term exposure to the detrimental microenvironment in aged mdx mice (12 months old) induces intrinsic cellular changes in FAPs [77,125]. Noteworthy, while TSA inhibits adipogenesis, it has also been shown to promote fibrogenic differentiation of FAPs [78].
8.6. Metalloproteinase inhibitors
Considering the role of metalloproteinases in ECM remodelling, the therapeutic potential of MMP inhibitors was also investigated in muscular dystrophies. The expression of many MMPs is upregulated in the skeletal muscle of mdx mice, while the tissue inhibitors of MMP (TIMP) are downregulated [126]. Administration of the MMP inhibitor batimastat in mdx mice decreases fibrosis accumulation and increases muscle function [126]. Treatment with batimastat also blocks adipogenic differentiation of FAPs in vitro [102]. It does not affect the accumulation of FAPs in the skeletal muscle of LGMD2B mice, but it reduced their adipogenic differentiation [102]. Moreover, as mentioned above, some MMPs, such as MMP14, can convert latent-TGF-β to its active form. Thus, MMP inhibition could also reduce fibrosis by dampening TGF-β expression [100].
8.7. In vivo validation
Although these studies do not represent an exhaustive list of the different drugs tested, they illustrate that there are different strategies aiming to target FAPs. Noteworthy, drug screening on FAPs in vitro is a useful method to identify potential targets; however, the therapeutic impact of these compounds does not often transfer into benefits in vivo. A recent study screened two libraries containing 722 compounds to determine their potential to block fibrogenesis [71]. The compounds were tested on FAPs expressing the enhanced green fluorescent protein (EGFP) under the Collagen1a1 promotor. Only 21 compounds exerted a dose-dependent ability to reduce EGFP expression induced by TGF-β. Masitinib and sorafenib, two tyrosine kinase inhibitors as well as JQ1, a member of the bromodomain inhibitor family, were the most potent candidates. These lead compounds were tested in vivo, and they had mixed effects on muscle fibrosis in dystrophic mice. None of these drugs strongly and consistently reduced muscle fibrosis. Daily intra-peritoneal injections of masitinib for four weeks leads to a slight decrease in the total collagen content in the diaphragm, but its administration through osmotic minipump for eight weeks does not improve fibrosis deposition. Administration of JQ1 has no effect on the total collagen content of the diaphragm, but it reduces collagen deposition in both TA and gastrocnemius [71]. Noteworthy, the administration of JQ1 also induced muscle mass loss and myofibre atrophy. Different factors could explain the discrepancy between in vitro and in vivo results such as adverse effect on other cell types (e.g. leucocytes, myogenic cells and myofibres), wrong dosage or administration route. Therefore, the positive hits need to be carefully validated in vivo using standard operating procedures for preclinical studies on dystrophic animals. Nonetheless, these findings provide a proof-of-concept that targeting FAPs is a promising therapeutic strategy to mitigate muscular dystrophies.
9. FAPs and ageing
Ageing is characterized by a decline in several physiological functions. Particularly, ageing is associated with sarcopenia, a process characterized by a progressive and generalized loss of skeletal muscle mass and function leading to failure in the elderly [127]. It is well-characterized that, during the course of ageing, there is a reduction in the muscle stem cell pool and intrinsic cellular defects leading to impaired muscle regeneration [128–130]; even though, the contribution of these defects to the development of sarcopenia is still debated [131,132]. Defects in the secretion of paracrine factors by FAPs also contribute to this impaired regenerative response. FAPs are the main mononuclear cell source of IL-33, a cytokine associated with type 2 immunity; however, during ageing, there is a reduction in the production of this cytokine by FAPs leading to reduced accumulation of Treg and poor muscle repair [60]. Moreover, aged FAPs express lower levels of WISP, which plays an important role in the asymmetric division of muscle stem cells and muscle regeneration [75]. Aged FAPs also express lower levels of GDF10. In vitro, the addition of conditioned medium obtained from transgenic FAPs overexpressing GDF10 induces myotube hypertrophy to a higher level than conditioned medium harvested form WT or GDF10-knockout FAPs. The administration of GDF10 to aged mice reverses the loss of muscle mass and myofibre atrophy [44]. Supporting the importance of FAPs in the defect of myogenesis during ageing, the cellular transplantation of young FAPs in aged mice restores the myogenic commitment of muscle stem cells [75].
Muscle ageing is characterized by an increased amount of fibrotic tissue, suggesting impairment in the activity of FAPs [44,75]. It has been shown that during muscle ageing, there is a reduction in the number of FAPs and their proliferative capacity, as well as an increased predisposition to fibrogenic differentiation [44,75]. Different intrinsic cellular defects have been shown to contribute to these perturbations in FAPs activity during ageing. A reduction in the truncated PDGFRα variant that acts as a decoy receptor to inhibit PDGF signalling has been observed in aged FAPs [75]. Moreover, ageing is associated with an increase in cellular senescence (state of irreversible cell cycle arrest) [133,134]. In aged skeletal muscle, cellular senescence has been observed in muscle stem cells [129], but also in a population of interstitial cells [135]. Induction of cellular senescence in mesenchymal stromal cells in vitro blunted their fibrogenic and adipogenic differentiation ability. Moreover, the release of senescence-associated secretory phenotype factors by senescent mesenchymal stromal cells blocks the formation of myotubes in vitro [135]. Further studies are required to determine the contribution of FAPs cellular senescence to the pathogenesis observed in aged skeletal muscle.
The alterations in FAPs behaviour could be mediated by changes in the microenvironmental cues during muscle ageing. For instance, there are higher levels of the profibrotic factor TGF-β during ageing [136]. There are also lower levels of IL-15, a cytokine that plays a key role in the regulation of FAPs proliferation and cell fate decision [81,82]. Changes in the secretion of paracrine factors by neighbouring cells also contribute to this dysfunction. The conditioned medium of myogenic cells isolated from young individuals increases FAPs proliferation and inhibits adipogenic differentiation, while the conditioned medium of myogenic cells isolated from aged donors failed to improve FAPs proliferation and prevent adipogenic differentiation [80].
Ageing is also associated with comorbidities affecting FAPs behaviour. Particularly, there is a strong increase in the incidence of type 2 diabetes during ageing [137]. It has been shown that muscle regeneration is impaired in different mouse models of diabetes [138]. The ectopic fat deposition observed in regenerating muscle of diabetic mice was shown to originate from FAPs [138]. Insulin resistance in type 2 diabetes leads to the over-secretion of this hypoglycaemic hormone, which is a known inducer of adipogenic differentiation of FAPs in vitro [35].
Overall, intrinsic and extrinsic changes associated with ageing affect the regulation of FAPs, which, in turn, promote the accumulation of fibrofatty tissue and impair muscle regeneration. Further studies are required to evaluate the potential of therapeutic compounds targeting FAPs to rejuvenate aged skeletal muscles.
10. Conclusion
Skeletal muscle has a remarkable regenerative capacity that has been attributed to the presence and activity of muscle stem cells. The articles reviewed above clearly indicate that another type of stem cell, the FAPs, also plays a fundamental role in the regulation of skeletal muscle regeneration. This centrepiece position is attributable to their cellular crosstalks and secretion of paracrine factors that orchestrate inflammation and muscle stem cell functioning. The key to their success relies on their rapid and transient accumulation that is closely controlled by complex intrinsic and extrinsic factors. Excessive accumulation of FAPs and perturbations in their cell fate decision lead to fibrofatty deposition and impaired muscle regeneration in muscular disorders. Recent breakthroughs in single-cell transcriptomics allowed the identification of cellular heterogeneity in FAPs and their complex molecular interactome in the diverse stages of muscle regeneration and muscular disorders. These novel insights will play a key role in the development of new therapeutic avenues targeting FAPs to limit their accumulation and/or re-establish their function in order to reduce fibrofatty deposition and promote muscle regeneration in muscular disorders.
Data accessibility
This article has no additional data.
Authors' contributions
T. M. and P.F. drafted the manuscript and designed the figures. N.A.D. conceived the project and critically revised the manuscript. All authors gave final approval for publication and agreed to be held accountable for the work performed therein.
Competing interests
The authors have declared that they have no conflict of interest.
Funding
T.M. and P.F. were supported by Fonds de Recherche du Québec—Santé (FRQS) doctoral awards. N.A.D. was supported by the FRQS Junior-2 award and by research grants from the Canadian Institutes of Health Research, Natural Sciences and Engineering Research Council of Canada, Canada Foundation for Innovation, the Orphan Disease Center of the University of Pennsylvania, Muscular Dystrophy Canada, and the Quebec Cell, Tissue and Gene Therapy Network—ThéCell (a thematic network supported by the FRQS).
References
- 1.Proctor DN, O'Brien PC, Atkinson EJ, Nair KS. 1999. Comparison of techniques to estimate total body skeletal muscle mass in people of different age groups. Am. J. Physiol. 277, E489-E495. ( 10.1152/ajpendo.1999.277.3.E489) [DOI] [PubMed] [Google Scholar]
- 2.Mauro A. 1961. Satellite cell of skeletal muscle fibers. J. Biophys. Biochem. Cytol. 9, 493-495. ( 10.1083/jcb.9.2.493) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dumont NA, Bentzinger CF, Sincennes MC, Rudnicki MA. 2015. Satellite cells and skeletal muscle regeneration. Compr. Physiol. 5, 1027-1059. ( 10.1002/cphy.c140068) [DOI] [PubMed] [Google Scholar]
- 4.Dumont NA, Wang YX, Rudnicki MA. 2015. Intrinsic and extrinsic mechanisms regulating satellite cell function. Development 142, 1572-1581. ( 10.1242/dev.114223) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bentzinger CF, Wang YX, Dumont NA, Rudnicki MA. 2013. Cellular dynamics in the muscle satellite cell niche. EMBO Rep. 14, 1062-1072. ( 10.1038/embor.2013.182) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Feichtinger RG, et al. 2019. Biallelic variants in the transcription factor PAX7 are a new genetic cause of myopathy. Genet. Med. 21, 2521-2531. ( 10.1038/s41436-019-0532-z) [DOI] [PubMed] [Google Scholar]
- 7.Sambasivan R, et al. 2011. Pax7-expressing satellite cells are indispensable for adult skeletal muscle regeneration. Development 138, 3647-3656. ( 10.1242/dev.067587) [DOI] [PubMed] [Google Scholar]
- 8.De Micheli AJ, Laurilliard EJ, Heinke CL, Ravichandran H, Fraczek P, Soueid-Baumgarten S, De Vlaminck I, Elemento O, Cosgrove BD. 2020. Single-cell analysis of the muscle stem cell hierarchy identifies heterotypic communication signals involved in skeletal muscle regeneration. Cell Rep. 30, 3583-3595.e5. ( 10.1016/j.celrep.2020.02.067) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Oprescu SN, Yue F, Qiu J, Brito LF, Kuang S. 2020. Temporal dynamics and heterogeneity of cell populations during skeletal muscle regeneration. iScience 23, 100993. ( 10.1016/j.isci.2020.100993) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Petrilli LL, Spada F, Palma A, Reggio A, Rosina M, Gargioli C, Castagnoli L, Fuoco C, Cesareni G. 2020. High-dimensional single-cell quantitative profiling of skeletal muscle cell population dynamics during regeneration. Cells 9, 1723. ( 10.3390/cells9071723) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Giordani L, et al. 2019. High-dimensional single-cell cartography reveals novel skeletal muscle-resident cell populations. Mol. Cell 74, 609-621.e6. ( 10.1016/j.molcel.2019.02.026) [DOI] [PubMed] [Google Scholar]
- 12.Dell'Orso S, Juan AH, Ko K-D, Naz F, Perovanovic J, Gutierrez-Cruz G, Feng X, Sartorelli V. 2019. Single cell analysis of adult mouse skeletal muscle stem cells in homeostatic and regenerative conditions. Development 146, dev174177. ( 10.1242/dev.174177) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Arnold L, Henry A, Poron F, Baba-Amer Y, van Rooijen N, Plonquet A, Gherardi RK, Chazaud B. 2007. Inflammatory monocytes recruited after skeletal muscle injury switch into antiinflammatory macrophages to support myogenesis. J. Exp. Med. 204, 1057-1069. ( 10.1084/jem.20070075) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dort J, Fabre P, Molina T, Dumont NA. 2019. Macrophages are key regulators of stem cells during skeletal muscle regeneration and diseases. Stem Cells Int. 2019, 4761427. ( 10.1155/2019/4761427) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Joe AWB, Yi L, Natarajan A, Le Grand F, So L, Wang J, Rudnicki MA, Rossi FMV. 2010. Muscle injury activates resident fibro/adipogenic progenitors that facilitate myogenesis. Nat. Cell Biol. 12, 153-163. ( 10.1038/ncb2015) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wosczyna MN, Konishi CT, Perez Carbajal EE, Wang TT, Walsh RA, Gan Q, Wagner MW, Rando TA. 2019. Mesenchymal stromal cells are required for regeneration and homeostatic maintenance of skeletal muscle. Cell. Rep. 27, 2029-35; e5. ( 10.1016/j.celrep.2019.04.074) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Uezumi A, Fukada S, Yamamoto N, Takeda S, Tsuchida K. 2010. Mesenchymal progenitors distinct from satellite cells contribute to ectopic fat cell formation in skeletal muscle. Nat. Cell Biol. 12, 143-152. ( 10.1038/ncb2014) [DOI] [PubMed] [Google Scholar]
- 18.Murphy MM, Lawson JA, Mathew SJ, Hutcheson DA, Kardon G. 2011. Satellite cells, connective tissue fibroblasts and their interactions are crucial for muscle regeneration. Development 138, 3625-3637. ( 10.1242/dev.064162) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Santini MP, et al. 2020. Tissue-resident PDGFRalpha(+) progenitor cells contribute to fibrosis versus healing in a context- and spatiotemporally dependent manner. Cell Rep. 30, 555-70 e7. ( 10.1016/j.celrep.2019.12.045) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mathew SJ, Hansen JM, Merrell AJ, Murphy MM, Lawson JA, Hutcheson DA, Hansen MS, Angus-Hill M, Kardon G. 2011. Connective tissue fibroblasts and Tcf4 regulate myogenesis. Development 138, 371-384. ( 10.1242/dev.057463) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Contreras O, Rossi FMV, Theret M. 2021. Origins, potency, and heterogeneity of skeletal muscle fibro-adipogenic progenitors-time for new definitions. Skelet. Muscle 11, 16. ( 10.1186/s13395-021-00265-6) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Helmbacher F, Stricker S. 2020. Tissue cross talks governing limb muscle development and regeneration. Semin. Cell Dev. Biol. 104, 14-30. ( 10.1016/j.semcdb.2020.05.005) [DOI] [PubMed] [Google Scholar]
- 23.Kardon G, Harfe BD, Tabin CJ. 2003. A Tcf4-positive mesodermal population provides a prepattern for vertebrate limb muscle patterning. Dev. Cell 5, 937-944. ( 10.1016/S1534-5807(03)00360-5) [DOI] [PubMed] [Google Scholar]
- 24.Vallecillo-Garcia P, et al. 2017. Odd skipped-related 1 identifies a population of embryonic fibro-adipogenic progenitors regulating myogenesis during limb development. Nat. Commun. 8, 1218. ( 10.1038/s41467-017-01120-3) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Swinehart IT, Schlientz AJ, Quintanilla CA, Mortlock DP, Wellik DM. 2013. Hox11 genes are required for regional patterning and integration of muscle, tendon and bone. Development 140, 4574-4582. ( 10.1242/dev.096693) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hasson P, DeLaurier A, Bennett M, Grigorieva E, Naiche LA, Papaioannou VE, Mohun TJ, Logan MPO. 2010. Tbx4 and tbx5 acting in connective tissue are required for limb muscle and tendon patterning. Dev. Cell 18, 148-156. ( 10.1016/j.devcel.2009.11.013) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Colasanto MP, Eyal S, Mohassel P, Bamshad M, Bonnemann CG, Zelzer E, Moon AM, Kardon G. 2016. Development of a subset of forelimb muscles and their attachment sites requires the ulnar-mammary syndrome gene Tbx3. Dis. Models Mech. 9, 1257-1269. ( 10.1242/dmm.025874) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stricker S, Brieske N, Haupt J, Mundlos S. 2006. Comparative expression pattern of Odd-skipped related genes Osr1 and Osr2 in chick embryonic development. Gene Expr. Patterns 6, 826-834. ( 10.1016/j.modgep.2006.02.003) [DOI] [PubMed] [Google Scholar]
- 29.Baumhueter S, Dybdal N, Kyle C, Lasky LA. 1994. Global vascular expression of murine CD34, a sialomucin-like endothelial ligand for L-selectin. Blood 84, 2554-2565. ( 10.1182/blood.V84.8.2554.2554) [DOI] [PubMed] [Google Scholar]
- 30.Dupas T, Rouaud T, Rouger K, Lieubeau B, Cario-Toumaniantz C, Fontaine-Pérus J, Gardahaut M-F, Auda-Boucher G. 2011. Fetal muscle contains different CD34+ cell subsets that distinctly differentiate into adipogenic, angiogenic and myogenic lineages. J. Stem Cell Res. 7, 230-243. ( 10.1016/j.scr.2011.06.004) [DOI] [PubMed] [Google Scholar]
- 31.Beauchamp JR, Heslop L, Yu DS, Tajbakhsh S, Kelly RG, Wernig A, Buckingham ME, Partridge TA, Zammit PS. 2000. Expression of CD34 and Myf5 defines the majority of quiescent adult skeletal muscle satellite cells. J. Cell Biol. 151, 1221-1234. ( 10.1083/jcb.151.6.1221) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Stumm J, Vallecillo-Garcia P, Vom Hofe-Schneider S, Ollitrault D, Schrewe H, Economides AN, Marazzi G, Sassoon DA, Stricker S. 2018. Odd skipped-related 1 (Osr1) identifies muscle-interstitial fibro-adipogenic progenitors (FAPs) activated by acute injury. Stem Cell Res. 32, 8-16. ( 10.1016/j.scr.2018.08.010) [DOI] [PubMed] [Google Scholar]
- 33.Giuliani G, et al. 2021. SCA-1 micro-heterogeneity in the fate decision of dystrophic fibro/adipogenic progenitors. Cell Death Dis. 12, 122. ( 10.1038/s41419-021-03408-1) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Marinkovic M, et al. 2019. Fibro-adipogenic progenitors of dystrophic mice are insensitive to NOTCH regulation of adipogenesis. Life Sci. Alliance 2, e201900437. ( 10.26508/lsa.201900437) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Uezumi A, et al. 2011. Fibrosis and adipogenesis originate from a common mesenchymal progenitor in skeletal muscle. J. Cell Sci. 124, 3654-3664. ( 10.1242/jcs.086629) [DOI] [PubMed] [Google Scholar]
- 36.Kopinke D, Roberson EC, Reiter JF. 2017. Ciliary Hedgehog signaling restricts injury-induced adipogenesis. Cell 170, 340-351.e12. ( 10.1016/j.cell.2017.06.035) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wosczyna MN, Biswas AA, Cogswell CA, Goldhamer DJ. 2012. Multipotent progenitors resident in the skeletal muscle interstitium exhibit robust BMP-dependent osteogenic activity and mediate heterotopic ossification. J. Bone Miner. Res. 27, 1004-1017. ( 10.1002/jbmr.1562) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lees-Shepard JB, et al. 2018. Activin-dependent signaling in fibro/adipogenic progenitors causes fibrodysplasia ossificans progressiva. Nat. Commun. 9, 471. ( 10.1038/s41467-018-02872-2) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Eisner C, Cummings M, Johnston G, Tung LW, Groppa E, Chang C, Rossi FMV. 2020. Murine tissue-resident PDGFRalpha+ fibro-adipogenic progenitors spontaneously acquire osteogenic phenotype in an altered inflammatory environment. J. Bone Miner. Res. 35, 1525-1534. ( 10.1002/jbmr.4020) [DOI] [PubMed] [Google Scholar]
- 40.Scott RW, Arostegui M, Schweitzer R, Rossi FMV, Underhill TM. 2019. Hic1 defines quiescent mesenchymal progenitor subpopulations with distinct functions and fates in skeletal muscle regeneration. Cell Stem Cell 25, 797-813 e9. ( 10.1016/j.stem.2019.11.004) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Malecova B, et al. 2018. Dynamics of cellular states of fibro-adipogenic progenitors during myogenesis and muscular dystrophy. Nat. Commun. 9, 3670. ( 10.1038/s41467-018-06068-6) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Camps J, et al. 2020. Interstitial cell remodeling promotes aberrant adipogenesis in dystrophic muscles. Cell Rep. 31, 107597. ( 10.1016/j.celrep.2020.107597) [DOI] [PubMed] [Google Scholar]
- 43.Roberts EW, et al. 2013. Depletion of stromal cells expressing fibroblast activation protein-alpha from skeletal muscle and bone marrow results in cachexia and anemia. J. Exp. Med. 210, 1137-1151. ( 10.1084/jem.20122344) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Uezumi A, et al. 2021. Mesenchymal Bmp3b expression maintains skeletal muscle integrity and decreases in age-related sarcopenia. J. Clin. Invest. 131, e139617. ( 10.1172/JCI139617) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chapman MA, Mukund K, Subramaniam S, Brenner D, Lieber RL. 2017. Three distinct cell populations express extracellular matrix proteins and increase in number during skeletal muscle fibrosis. Am. J. Physiol. Cell Physiol. 312, C131-C143. ( 10.1152/ajpcell.00226.2016) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rayagiri SS, Ranaldi D, Raven A, Azhar NIFM, Lefebvre O, Zammit PS, Borycki A-G. et al. 2018. Basal lamina remodeling at the skeletal muscle stem cell niche mediates stem cell self-renewal. Nat. Commun. 9, 1-12. ( 10.1038/s41467-018-03425-3) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Baghdadi MB, Castel D, Machado L, Fukada S, Birk DE, Relaix F, Tajbakhsh S, Mourikis P. 2018. Reciprocal signalling by Notch-Collagen V-CALCR retains muscle stem cells in their niche. Nature 557, 714-718. ( 10.1038/s41586-018-0144-9) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Urciuolo A, et al. 2013. Collagen VI regulates satellite cell self-renewal and muscle regeneration. Nat. Commun. 4, 1964. ( 10.1038/ncomms2964) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lukjanenko L, et al. 2016. Loss of fibronectin from the aged stem cell niche affects the regenerative capacity of skeletal muscle in mice. Nat. Med. 22, 897-905. ( 10.1038/nm.4126) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Formicola L, et al. 2018. Inhibition of the activin receptor type-2B pathway restores regenerative capacity in satellite cell-depleted skeletal muscle. Front. Physiol. 9, 515. ( 10.3389/fphys.2018.00515) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lemos DR, Paylor B, Chang C, Sampaio A, Underhill TM, Rossi FM. 2012. Functionally convergent white adipogenic progenitors of different lineages participate in a diffused system supporting tissue regeneration. Stem Cells 30, 1152-1162. ( 10.1002/stem.1082) [DOI] [PubMed] [Google Scholar]
- 52.Dagdeviren S, et al. 2017. IL-10 prevents aging-associated inflammation and insulin resistance in skeletal muscle. FASEB J. 31, 701-710. ( 10.1096/fj.201600832R) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Biferali B, Proietti D, Mozzetta C, Madaro L. 2019. Fibro-adipogenic progenitors cross-talk in skeletal muscle: the social network. Front. Physiol. 10, 1074. ( 10.3389/fphys.2019.01074) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lemos DR, et al. 2015. Nilotinib reduces muscle fibrosis in chronic muscle injury by promoting TNF-mediated apoptosis of fibro/adipogenic progenitors. Nat. Med. 21, 786-794. ( 10.1038/nm.3869) [DOI] [PubMed] [Google Scholar]
- 55.Mueller AA, Van Velthoven CT, Fukumoto KD, Cheung TH, Rando TA. 2016. Intronic polyadenylation of PDGFRalpha in resident stem cells attenuates muscle fibrosis. Nature 540, 276-279. ( 10.1038/nature20160) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bangs F, Anderson KV. 2017. Primary cilia and mammalian hedgehog signaling. Cold Spring Harb. Perspect. Biol. 9, a028175. ( 10.1101/cshperspect.a028175) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhao L, Son JS, Wang B, Tian Q, Chen Y, Liu X, De Avila JM, Zhu M-J, Du M. 2020. Retinoic acid signalling in fibro/adipogenic progenitors robustly enhances muscle regeneration. EBioMedicine 60, 103020. ( 10.1016/j.ebiom.2020.103020) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Deng B, Wehling-Henricks M, Villalta SA, Wang Y, Tidball JG. 2012. IL-10 triggers changes in macrophage phenotype that promote muscle growth and regeneration. J. Immunol. 189, 3669-3680. ( 10.4049/jimmunol.1103180) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cayrol C, Girard JP. 2014. IL-33: an alarmin cytokine with crucial roles in innate immunity, inflammation and allergy. Curr. Opin. Immunol. 31, 31-37. ( 10.1016/j.coi.2014.09.004) [DOI] [PubMed] [Google Scholar]
- 60.Kuswanto W, Burzyn D, Panduro M, Wang KK, Jang YC, Wagers AJ, Benoist C, Mathis D. 2016. Poor repair of skeletal muscle in aging mice reflects a defect in local, interleukin-33-dependent accumulation of regulatory T cells. Immunity 44, 355-367. ( 10.1016/j.immuni.2016.01.009) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Fielding RA, Manfredi TJ, Ding W, Fiatarone MA, Evans WJ, Cannon JG. 1993. Acute phase response in exercise. III. Neutrophil and IL-1 beta accumulation in skeletal muscle. Am. J. Physiol. 265, R166-R172. ( 10.1152/ajpregu.1993.265.1.R166) [DOI] [PubMed] [Google Scholar]
- 62.Tidball JG, Berchenko E, Frenette J. 1999. Macrophage invasion does not contribute to muscle membrane injury during inflammation. J. Leukoc. Biol. 65, 492-498. ( 10.1002/jlb.65.4.492) [DOI] [PubMed] [Google Scholar]
- 63.Heredia JE, Mukundan L, Chen FM, Mueller AA, Deo RC, Locksley RM, Rando TA, Chawla A. et al. 2013. Type 2 innate signals stimulate fibro/adipogenic progenitors to facilitate muscle regeneration. Cell 153, 376-388. ( 10.1016/j.cell.2013.02.053) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Vumbaca S, et al. 2021. Characterization of the skeletal muscle secretome reveals a role for extracellular vesicles and IL1alpha/IL1beta in restricting fibro/adipogenic progenitor adipogenesis. Biomolecules 11, 1171. ( 10.3390/biom11081171) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Contreras O, Soliman H, Theret M, Rossi FMV, Brandan E. 2020. TGF-beta-driven downregulation of the transcription factor TCF7L2 affects Wnt/beta-catenin signaling in PDGFRalpha(+) fibroblasts. J. Cell Sci. 133, jcs242297. ( 10.1242/jcs.242297) [DOI] [PubMed] [Google Scholar]
- 66.Contreras O, Cruz-Soca M, Theret M, Soliman H, Tung LW, Groppa E, Rossi FM, Brandan E. 2019. Cross-talk between TGF-β and PDGFRα signaling pathways regulates the fate of stromal fibro-adipogenic progenitors. J. Cell Sci. 132, jcs232157. ( 10.1242/jcs.232157) [DOI] [PubMed] [Google Scholar]
- 67.Contreras O, Rossi FM, Brandan E. 2019. Adherent muscle connective tissue fibroblasts are phenotypically and biochemically equivalent to stromal fibro/adipogenic progenitors. Matrix Biol. Plus. 2, 100006. ( 10.1016/j.mbplus.2019.04.003) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Davies MR, Liu X, Lee L, Laron D, Ning AY, Kim HT, Feeley BT. 2016. TGF-beta small molecule inhibitor SB431542 reduces rotator cuff muscle fibrosis and fatty infiltration by promoting fibro/adipogenic progenitor apoptosis. PLoS ONE 11, e0155486. ( 10.1371/journal.pone.0155486) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Moratal C, Raffort J, Arrighi N, Rekima S, Schaub S, Dechesne C, Chinetti G, Dani C. 2018. IL-1β- and IL-4-polarized macrophages have opposite effects on adipogenesis of intramuscular fibro-adipogenic progenitors in humans. Sci. Rep. 8, 1-13. ( 10.1038/s41598-018-35429-w) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Fiore D, et al. 2016. Pharmacological blockage of fibro/adipogenic progenitor expansion and suppression of regenerative fibrogenesis is associated with impaired skeletal muscle regeneration. Stem Cell Res. 17, 161-169. ( 10.1016/j.scr.2016.06.007) [DOI] [PubMed] [Google Scholar]
- 71.Theret M, et al. 2021. In vitro assessment of anti-fibrotic activity does not predict their in vivo efficacy in murine models of Duchenne muscular dystrophy. Life Sci. 279, 119482. ( 10.1016/j.lfs.2021.119482) [DOI] [PubMed] [Google Scholar]
- 72.Theret M, Rossi FMV, Contreras O. 2021. Evolving roles of muscle-resident fibro-adipogenic progenitors in health, regeneration, neuromuscular disorders, and aging. Front. Physiol. 12, 673404. ( 10.3389/fphys.2021.673404) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Tierney MT, Aydogdu T, Sala D, Malecova B, Gatto S, Puri PL, Latella L, Sacco A. 2014. STAT3 signaling controls satellite cell expansion and skeletal muscle repair. Nat. Med. 20, 1182-1186. ( 10.1038/nm.3656) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Serrano AL, Baeza-Raja B, Perdiguero E, Jardi M, Munoz-Cinoves P. 2008. Interleukin-6 is an essential regulator of satellite cell-mediated skeletal muscle hypertrophy. Cell Metab. 7, 33-44. ( 10.1016/j.cmet.2007.11.011) [DOI] [PubMed] [Google Scholar]
- 75.Lukjanenko L, et al. 2019. Aging disrupts muscle stem cell function by impairing matricellular WISP1 secretion from fibro-adipogenic progenitors. Cell Stem Cell 24, 433-46; e7. ( 10.1016/j.stem.2018.12.014) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Schiaffino S, Mammucari C. 2011. Regulation of skeletal muscle growth by the IGF1-Akt/PKB pathway: insights from genetic models. Skelet. Muscle 1, 4. ( 10.1186/2044-5040-1-4) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Mozzetta C, et al. 2013. Fibroadipogenic progenitors mediate the ability of HDAC inhibitors to promote regeneration in dystrophic muscles of young, but not old Mdx mice. EMBO Mol. Med. 5, 626-639. ( 10.1002/emmm.201202096) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Reggio A, et al. 2020. Adipogenesis of skeletal muscle fibro/adipogenic progenitors is affected by the WNT5a/GSK3/β-catenin axis. Cell Death Differ. 27, 2921-2941. ( 10.1038/s41418-020-0551-y) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Fry CS, Kirby TJ, Kosmac K, McCarthy JJ, Peterson CA. 2017. Myogenic progenitor cells control extracellular matrix production by fibroblasts during skeletal muscle hypertrophy. Cell Stem Cell 20, 56-69. ( 10.1016/j.stem.2016.09.010) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Moratal C, Arrighia N, Dechesnea CA, Dania C. 2019. Control of muscle fibro-adipogenic progenitors by myogenic lineage is altered in aging and Duchenne muscular dystrophy. Cell Physiol. Biochem. 53, 1029-1045. ( 10.33594/000000196) [DOI] [PubMed] [Google Scholar]
- 81.Kang X, et al. 2018. Interleukin-15 facilitates muscle regeneration through modulation of fibro/adipogenic progenitors. Cell Commun. Signal. 16, 42. ( 10.1186/s12964-018-0251-0) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Quinn LS, Strait-Bodey L, Anderson BG, Argiles JM, Havel PJ. 2005. Interleukin-15 stimulates adiponectin secretion by 3T3-L1 adipocytes: evidence for a skeletal muscle-to-fat signaling pathway. Cell Biol. Int. 29, 449-457. ( 10.1016/j.cellbi.2005.02.005) [DOI] [PubMed] [Google Scholar]
- 83.Li Y, Li F, Lin B, Kong X, Tang Y, Yin Y. 2014. Myokine IL-15 regulates the crosstalk of co-cultured porcine skeletal muscle satellite cells and preadipocytes. Mol. Biol. Rep. 41, 7543-7553. ( 10.1007/s11033-014-3646-z) [DOI] [PubMed] [Google Scholar]
- 84.Verma M, Asakura Y, Murakonda BSR, Pengo T, Latroche C, Chazaud B, Mcloon LK, Asakura A. 2018. Muscle satellite cell cross-talk with a vascular niche maintains quiescence via VEGF and notch signaling. Cell Stem Cell 23, 530-543.e9. ( 10.1016/j.stem.2018.09.007) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Hoffman EP, Brown RH Jr, Kunkel LM. 1987. Dystrophin: the protein product of the Duchenne muscular dystrophy locus. Cell 51, 919-928. ( 10.1016/0092-8674(87)90579-4) [DOI] [PubMed] [Google Scholar]
- 86.Monaco AP, Neve RL, Colletti-Feener C, Bertelson CJ, Kurnit DM, Kunkel LM. 1986. Isolation of candidate cDNAs for portions of the Duchenne muscular dystrophy gene. Nature 323, 646-650. ( 10.1038/323646a0) [DOI] [PubMed] [Google Scholar]
- 87.Constantin B. 2014. Dystrophin complex functions as a scaffold for signalling proteins. Biochim. Biophys. Acta-Biomembr. 1838, 635-642. ( 10.1016/j.bbamem.2013.08.023) [DOI] [PubMed] [Google Scholar]
- 88.Klingler W, Jurkat-Rott K, Lehmann-Horn F, Schleip R. 2012. The role of fibrosis in Duchenne muscular dystrophy. Acta Myol. 31, 184-195. [PMC free article] [PubMed] [Google Scholar]
- 89.Carpenter S, Karpati G. 1979. Duchenne muscular dystrophy: plasma membrane loss initiates muscle cell necrosis unless it is repaired. Brain 102, 147-161. ( 10.1093/brain/102.1.147) [DOI] [PubMed] [Google Scholar]
- 90.Desguerre I, Mayer M, Leturcq F, Barbet JP, Gherardi RK, Christov C. 2009. Endomysial fibrosis in Duchenne muscular dystrophy: a marker of poor outcome associated with macrophage alternative activation. J. Neuropathol. Exp. Neurol. 68, 762-773. ( 10.1097/NEN.0b013e3181aa31c2) [DOI] [PubMed] [Google Scholar]
- 91.Kim HK, Merrow AC, Shiraj S, Wong BL, Horn PS, Laor T. 2013. Analysis of fatty infiltration and inflammation of the pelvic and thigh muscles in boys with Duchenne muscular dystrophy (DMD): grading of disease involvement on MR imaging and correlation with clinical assessments. Pediatr. Radiol. 43, 1327-1335. ( 10.1007/s00247-013-2696-z) [DOI] [PubMed] [Google Scholar]
- 92.Gaeta M, et al. 2012. Muscle fat-fraction and mapping in Duchenne muscular dystrophy: evaluation of disease distribution and correlation with clinical assessments. Preliminary experience. Skeletal Radiol. 41, 955-961. ( 10.1007/s00256-011-1301-5) [DOI] [PubMed] [Google Scholar]
- 93.Andreetta F, Bernasconi P, Baggi F, Ferro P, Oliva L, Arnoldi E, Cornelio F, Mantegazza R, Confalonieri P. 2006. Immunomodulation of TGF-beta1 in mdx mouse inhibits connective tissue proliferation in diaphragm but increases inflammatory response: implications for antifibrotic therapy. J. Neuroimmunol. 175, 77-86. ( 10.1016/j.jneuroim.2006.03.005) [DOI] [PubMed] [Google Scholar]
- 94.Contreras O, Rebolledo DL, Oyarzun JE, Olguin HC, Brandan E. 2016. Connective tissue cells expressing fibro/adipogenic progenitor markers increase under chronic damage: relevance in fibroblast–myofibroblast differentiation and skeletal muscle fibrosis. Cell Tissue Res. 364, 647-660. ( 10.1007/s00441-015-2343-0) [DOI] [PubMed] [Google Scholar]
- 95.Ieronimakis N, Hays A, Prasad A, Janebodin K, Duffield JS, Reyes M. 2016. PDGFRalpha signalling promotes fibrogenic responses in collagen-producing cells in Duchenne muscular dystrophy. J. Pathol. 240, 410-424. ( 10.1002/path.4801) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Gonzalez D, Contreras O, Rebolledo DL, Espinoza JP, Van Zundert B, Brandan E. 2017. ALS skeletal muscle shows enhanced TGF-β signaling, fibrosis and induction of fibro/adipogenic progenitor markers. PLoS ONE 12, e0177649. ( 10.1371/journal.pone.0177649) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Pessina P, Kharraz Y, Jardi M, Fukada S, Serrano AL, Perdiguero E, Muñoz-Cánoves P. 2015. Fibrogenic cell plasticity blunts tissue regeneration and aggravates muscular dystrophy. Stem Cell Rep. 4, 1046-1060. ( 10.1016/j.stemcr.2015.04.007) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Ardite E, Perdiguero E, Vidal B, Gutarra S, Serrano AL, Munoz-Canoves P. 2012. PAI-1-regulated miR-21 defines a novel age-associated fibrogenic pathway in muscular dystrophy. J. Cell Biol. 196, 163-175. ( 10.1083/jcb.201105013) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Chen YW, Nagaraju K, Bakay M, McIntyre O, Rawat R, Shi R, Hoffman EP. 2005. Early onset of inflammation and later involvement of TGFbeta in Duchenne muscular dystrophy. Neurology 65, 826-834. ( 10.1212/01.wnl.0000173836.09176.c4) [DOI] [PubMed] [Google Scholar]
- 100.Juban G, et al. 2018. AMPK Activation regulates LTBP4-dependent TGF-beta1 secretion by pro-inflammatory macrophages and controls fibrosis in Duchenne muscular dystrophy. Cell Rep. 25, 2163-2176; e6. ( 10.1016/j.celrep.2018.10.077) [DOI] [PubMed] [Google Scholar]
- 101.Mazala DA, et al. 2020. TGF-beta-driven muscle degeneration and failed regeneration underlie disease onset in a DMD mouse model. JCI Insight 5, e135703. ( 10.1172/jci.insight.135703) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Hogarth MW, Defour A, Lazarski C, Gallardo E, Diaz Manera J, Partridge TA, Nagaraju K, Jaiswal JK. 2019. Fibroadipogenic progenitors are responsible for muscle loss in limb girdle muscular dystrophy 2B. Nat. Commun. 10, 2430. ( 10.1038/s41467-019-10438-z) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Tome FM, et al. 1994. Congenital muscular dystrophy with merosin deficiency. C. R. Acad. Sci. III 317, 351-357. [PubMed] [Google Scholar]
- 104.Nevo Y, Halevy O, Genin O, Moshe I, Turgeman T, Harel M, Biton E, Reif S, Pines M. 2010. Fibrosis inhibition and muscle histopathology improvement in laminin-alpha2-deficient mice. Muscle Nerve 42, 218-229. ( 10.1002/mus.21706) [DOI] [PubMed] [Google Scholar]
- 105.Noguchi S, Ogawa M, Malicdan MC, Nonaka I, Nishino I. 2017. Muscle weakness and fibrosis due to cell autonomous and non-cell autonomous events in collagen VI deficient congenital muscular dystrophy. EBioMedicine 15, 193-202. ( 10.1016/j.ebiom.2016.12.011) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Kolbel H, Hathazi D, Jennings M, Horvath R, Roos A, Schara U. 2019. Identification of candidate protein markers in skeletal muscle of laminin-211-deficient CMD type 1A-patients. Front. Neurol. 10, 470. ( 10.3389/fneur.2019.00470) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Accorsi A, Cramer ML, Girgenrath M. 2020. Fibrogenesis in LAMA2-related muscular dystrophy is a central tenet of disease etiology. Front. Mol. Neurosci. 13, 3. ( 10.3389/fnmol.2020.00003) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Taniguchi M, et al. 2006. Expression profiling of muscles from Fukuyama-type congenital muscular dystrophy and laminin-α2 deficient congenital muscular dystrophy; is congenital muscular dystrophy a primary fibrotic disease? Biochem. Biophys. Res. Commun. 342, 489-502. ( 10.1016/j.bbrc.2005.12.224) [DOI] [PubMed] [Google Scholar]
- 109.Elbaz M, Yanay N, Aga-Mizrachi S, Brunschwig Z, Kassis I, Ettinger K, Barak V, Nevo Y. 2012. Losartan, a therapeutic candidate in congenital muscular dystrophy: studies in the dy2 J/dy2 J Mouse. Ann. Neurol. 71, 699-708. ( 10.1002/ana.22694) [DOI] [PubMed] [Google Scholar]
- 110.Matthews E, Brassington R, Kuntzer T, Jichi F, Manzur AY. 2016. Corticosteroids for the treatment of Duchenne muscular dystrophy. Cochrane Database Syst. Rev. 2016, CD003725. ( 10.1002/14651858.CD003725.pub4) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Dong Y, Silva KA, Dong Y, Zhang L. 2014. Glucocorticoids increase adipocytes in muscle by affecting IL-4 regulated FAP activity. FASEB J. 28, 4123-4132. ( 10.1096/fj.14-254011) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Wu Z, Bucher N, Farmer SR. 1996. Induction of peroxisome proliferator-activated receptor gamma during the conversion of 3T3 fibroblasts into adipocytes is mediated by C/EBPbeta, C/EBPdelta, and glucocorticoids. Mol. Cell. Biol. 16, 4128-4136. ( 10.1128/MCB.16.8.4128) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Asada M, et al. 2011. DNA binding-dependent glucocorticoid receptor activity promotes adipogenesis via Kruppel-like factor 15 gene expression. Lab Invest. 91, 203-215. ( 10.1038/labinvest.2010.170) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Quattrocelli M, Salamone IM, Page PG, Warner JL, Demonbreun AR, McNally EM. 2017. Intermittent glucocorticoid dosing improves muscle repair and function in mice with limb-girdle muscular dystrophy. Am. J. Pathol. 187, 2520-2535. ( 10.1016/j.ajpath.2017.07.017) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Perpetuini AC, et al. 2020. Janus effect of glucocorticoids on differentiation of muscle fibro/adipogenic progenitors. Sci. Rep. 10, 1-18. ( 10.1038/s41598-019-56847-4) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Kemaladewi DU, et al. 2014. Targeting TGF-beta signaling by antisense oligonucleotide-mediated knockdown of TGF-beta type I receptor. Mol. Ther. Nucleic Acids 3, e156. ( 10.1038/mtna.2014.7) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Reggio A, Spada F, Rosina M, Massacci G, Zuccotti A, Fuoco C, Gargioli C, Castagnoli L, Cesareni G. 2019. The immunosuppressant drug azathioprine restrains adipogenesis of muscle fibro/adipogenic progenitors from dystrophic mice by affecting AKT signaling. Sci. Rep. 9, 4360. ( 10.1038/s41598-019-39538-y) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Huang P, Zhao XS, Fields M, Ransohoff RM, Zhou L. 2009. Imatinib attenuates skeletal muscle dystrophy in mdx mice. FASEB J. 23, 2539-2548. ( 10.1096/fj.09-129833) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Ito T, et al. 2013. Imatinib attenuates severe mouse dystrophy and inhibits proliferation and fibrosis-marker expression in muscle mesenchymal progenitors. Neuromuscul. Disord. 23, 349-356. ( 10.1016/j.nmd.2012.10.025) [DOI] [PubMed] [Google Scholar]
- 120.Contreras O, Villarreal M, Brandan E. 2018. Nilotinib impairs skeletal myogenesis by increasing myoblast proliferation. Skelet. Muscle 8, 5. ( 10.1186/s13395-018-0150-5) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Minetti GC, et al. 2006. Functional and morphological recovery of dystrophic muscles in mice treated with deacetylase inhibitors. Nat. Med. 12, 1147-1150. ( 10.1038/nm1479) [DOI] [PubMed] [Google Scholar]
- 122.Colussi C, et al. 2010. Proteomic profile of differentially expressed plasma proteins from dystrophic mice and following suberoylanilide hydroxamic acid treatment. Proteom. Clin. Appl. 4, 71-83. ( 10.1002/prca.200900116) [DOI] [PubMed] [Google Scholar]
- 123.Consalvi S, et al. 2013. Preclinical studies in the mdx mouse model of duchenne muscular dystrophy with the histone deacetylase inhibitor givinostat. Mol. Med. 19, 79-87. ( 10.2119/molmed.2013.00011) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Bettica P, et al. 2016. Histological effects of givinostat in boys with Duchenne muscular dystrophy. Neuromuscul. Disord. 26, 643-649. ( 10.1016/j.nmd.2016.07.002) [DOI] [PubMed] [Google Scholar]
- 125.Saccone V, et al. 2014. HDAC-regulated myomiRs control BAF60 variant exchange and direct the functional phenotype of fibro-adipogenic progenitors in dystrophic muscles. Genes Dev. 28, 841-857. ( 10.1101/gad.234468.113) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Kumar A, Bhatnagar S, Kumar A. 2010. Matrix metalloproteinase inhibitor batimastat alleviates pathology and improves skeletal muscle function in dystrophin-deficient mdx mice. Am. J. Pathol. 177, 248-260. ( 10.2353/ajpath.2010.091176) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Vinel C, et al. 2018. The exerkine apelin reverses age-associated sarcopenia. Nat. Med. 24, 1360-1371. ( 10.1038/s41591-018-0131-6) [DOI] [PubMed] [Google Scholar]
- 128.Munoz-Canoves P, Neves J, Sousa-Victor P. 2020. Understanding muscle regenerative decline with aging: new approaches to bring back youthfulness to aged stem cells. FEBS J. 287, 406-416. ( 10.1111/febs.15182) [DOI] [PubMed] [Google Scholar]
- 129.Sousa-Victor P, et al. 2014. Geriatric muscle stem cells switch reversible quiescence into senescence. Nature 506, 316-321. ( 10.1038/nature13013) [DOI] [PubMed] [Google Scholar]
- 130.Sousa-Victor P, Munoz-Canoves P. 2016. Regenerative decline of stem cells in sarcopenia. Mol. Aspects Med. 50, 109-117. ( 10.1016/j.mam.2016.02.002) [DOI] [PubMed] [Google Scholar]
- 131.Snijders T, Parise G. 2017. Role of muscle stem cells in sarcopenia. Curr. Opin. Clin. Nutr. Metab. Care 20, 186-190. ( 10.1097/MCO.0000000000000360) [DOI] [PubMed] [Google Scholar]
- 132.Fry CS, et al. 2015. Inducible depletion of satellite cells in adult, sedentary mice impairs muscle regenerative capacity without affecting sarcopenia. Nat. Med. 21, 76-80. ( 10.1038/nm.3710) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Calcinotto A, Kohli J, Zagato E, Pellegrini L, Demaria M, Alimonti A. 2019. Cellular senescence: aging, cancer, and injury. Physiol. Rev. 99, 1047-1078. ( 10.1152/physrev.00020.2018) [DOI] [PubMed] [Google Scholar]
- 134.Childs BG, Durik M, Baker DJ, van Deursen JM. 2015. Cellular senescence in aging and age-related disease: from mechanisms to therapy. Nat. Med. 21, 1424-1435. ( 10.1038/nm.4000) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Sugihara H, Teramoto N, Yamanouchi K, Matsuwaki T, Nishihara M. 2018. Oxidative stress-mediated senescence in mesenchymal progenitor cells causes the loss of their fibro/adipogenic potential and abrogates myoblast fusion. Aging-Us 10, 747-763. ( 10.18632/aging.101425) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Forsey RJ, Thompson JM, Ernerudh J, Hurst TL, Strindhall J, Johansson B, Nilsson B-O, Wikby A. 2003. Plasma cytokine profiles in elderly humans. Mech. Ageing Dev. 124, 487-493. ( 10.1016/S0047-6374(03)00025-3) [DOI] [PubMed] [Google Scholar]
- 137.Fletcher B, Gulanick M, Lamendola C. 2002. Risk factors for type 2 diabetes mellitus. J. Cardiovasc. Nurs. 16, 17-23. ( 10.1097/00005082-200201000-00003) [DOI] [PubMed] [Google Scholar]
- 138.Mogi M, et al. 2016. Diabetic mice exhibited a peculiar alteration in body composition with exaggerated ectopic fat deposition after muscle injury due to anomalous cell differentiation. J. Cachexia Sarcopeni. 7, 213-223. ( 10.1002/jcsm.12044) [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
This article has no additional data.