

Abstract
Background
Glioblastoma multiforme (GBM) is the most common primary malignant brain tumor in adults. Amplification or overexpression of the epidermal growth factor receptor gene, part of the ErbB family, occur in approximately 40% and 60% of patients with GBM, respectively. We present data from a dose-finding study of the ErbB inhibitor afatinib in combination with radiotherapy (RT), with or without temozolomide (TMZ), in patients with GBM.
Methods
This was a phase I, open-label, 3 + 3 dose-escalation trial in patients with newly-diagnosed, histologically-confirmed grade 4 malignant glioma and proven O6-methylguanine-DNA methyltransferase gene promoter methylation status. The primary endpoint was the maximum tolerated dose (MTD) of continuous daily afatinib when given in combination with RT, with (regimen M) or without (regimen U) concomitant TMZ treatment.
Results
Fifty-five patients were enrolled; 36 received ≥ 1 dose of trial medication (regimen M, n = 20, regimen U, n = 16). Afatinib was discontinued by all patients during the study. Reasons for afatinib discontinuation (regimen M/U) included disease progression (45%/50%), dose-limiting toxicity (10%/0%), and other adverse events (AEs; 35%/38%). The most frequently reported AEs with either regimen were diarrhea and rash, with no new safety signals identified. The MTD was determined as afatinib 30 mg in combination with daily TMZ and RT, and afatinib 40 mg in combination with RT alone.
Conclusions
This study identified the MTD for afatinib in combination with RT, with and without TMZ, in patients with GBM. Further studies of afatinib in patients with GBM are warranted and should be based on appropriate biomarker-based preselection.
Trial registration
NCT00977431 (first posted September 15, 2009).
Supplementary Information
The online version contains supplementary material available at 10.1007/s11060-021-03877-6.
Keywords: Glioblastoma, Afatinib, Dose-escalation, Temozolomide, Radiotherapy
Introduction
Glioblastoma multiforme (GBM) is the most common malignant primary brain tumor in adults [1] and is associated with a poor prognosis, with a median progression-free survival (PFS) of 7.4‒10.7 months [2–4] and median overall survival (OS) of 14.6 months [5]. Limited progress has been made in improving outcomes for patients with GBM in recent decades [1]. First-line therapy for newly diagnosed patients is maximal safe surgical resection, followed by radiotherapy (RT) and temozolomide (TMZ) [6]. However, response to TMZ treatment can vary depending upon the methylation status of the methyl-guanine methyl transferase (MGMT) enzyme promoter [7, 8]. Additionally, most glioblastomas become resistant to first-line therapies, which can occur via several mechanisms, including activation of DNA repair mechanisms, evasion of apoptosis, and adaptation of the cell cycle [9, 10]. As survival rates remain low, there is a large unmet need in GBM, particularly for patients with unmethylated MGMT promoters, for whom standard treatments are less effective [1, 11]. Consequently, several biomarker-driven therapeutic targets have been investigated to date, including the ErbB family of receptors.
Dysregulation of the ErbB pathway has been reported to contribute to GBM progression [12], with mutation, rearrangement, altered splicing and/or focal amplification of the epidermal growth factor receptor (EGFR) gene observed in over half of GBM cases [13–15]. Some studies have indicated that overexpression of EGFR may be associated with worse outcomes following RT in patients with GBM [16, 17]. EGFR tyrosine kinase inhibitors (TKI) have therefore been investigated in patients with malignant glioma or GBM, but have so far shown little activity in this setting [18]. Afatinib is an ErbB-family blocker that is approved for use in patients with NSCLC [19, 20]; it irreversibly binds to and blocks EGFR (ErbB1), HER2 (ErbB2), and ErbB4. Afatinib is therefore considered to have a wider inhibitory profile than first-generation EGFR TKIs [21, 22]. Furthermore, brain penetrance is recognized as a potential hurdle in the utilization of EGFR TKIs in GBM [23, 24]. Preclinical data indicate that afatinib has a moderate capacity to penetrate the BBB, supporting its use against central nervous system (CNS) malignancies [25, 26]. Indeed, 35–82% of patients with NSCLC and CNS metastases who were treated with afatinib monotherapy experienced a CNS response [27–30]. Thus, given the wider inhibitory profile of afatinib than first-generation TKIs, and its potential for CNS penetration, afatinib represents a possible treatment for GBM.
In a phase I/II study of afatinib with or without TMZ versus TMZ alone in patients with recurrent GBM, afatinib showed a manageable safety profile and modest efficacy in this hard-to-treat population [31]. There was no difference in OS between the treatment arms in the overall trial population; however, in the small number of patients assessed by biomarker subgroup analysis, there was a non-statistically significant trend towards increased PFS in afatinib-treated patients expressing the EGFR-variant III (EGFR-vIII) mutation, an EGFR variant frequently found in GBM [14]. Given that EGFR overexpression or mutation may contribute to poor outcomes and progression of GBM, and that preclinical and clinical data have highlighted potential for afatinib to elicit antitumor activity in GBM, we hypothesized that addition of afatinib to RT and TMZ may improve tumor responses and/or delay resistance to GBM treatment. The purpose of this trial was to define the toxicity and maximum tolerated dose (MTD) of afatinib in combination with RT, with and without TMZ, for the treatment of patients with newly diagnosed GBM.
Materials and methods
Study design and patient population
The study (NCT00977431) was a phase I, open-label, 3 + 3 dose-escalation trial in patients with newly-diagnosed malignant glioma. The trial was conducted at five sites in the United Kingdom.
Eligible patients were aged ≥ 18 and < 70 years, with newly-diagnosed, histologically-confirmed World Health Organization grade 4 malignant glioma and proven MGMT gene promoter methylation status (or tumor material available for testing). Exclusion criteria included: surgery within 2 weeks prior to the start of treatment or planned during the trial; placement of a Giladel® wafer at surgery, prior radiotherapy of the cranium (including brachytherapy and/or radiosurgery for GBM); and treatment with other investigational drugs concomitantly with the study.
The trial was carried out in compliance with the clinical trial protocol, in accordance with the principles of the Declaration of Helsinki and International Conference on Harmonisation-Good Clinical Practice (ICH-GCP) guidelines, and in line with applicable regulatory requirements and Boehringer Ingelheim standard operating procedures. Prior to the initiation of any trial-related procedure, all patients were informed about the trial verbally and in writing by the investigator and provided written informed consent according to ICH-GCP and local legal requirements.
Treatment
This study included two treatment regimens: regimen M, afatinib + TMZ in combination with RT; and regimen U, afatinib in combination with RT without TMZ. During the dose-finding phase, patients with methylated MGMT status were treated with regimen M, and patients with unmethylated MGMT gene promoters were treated with regimen U. The protocol was amended following the emergence of evidence demonstrating the efficacy of TMZ in patients with GBM regardless of MGMT methylation status [32]. Once the MTD in regimen U had been determined, all new patients were assigned to regimen M regardless of methylation status.
In both regimens, RT was administered to patients at a dose of 2 Grays (Gy) per fraction on 5 days per week for 6 weeks (total dose of 60 Gy) in an initial RT phase. Afatinib was administered in dose escalation cohorts of 20, 30, and 40 mg/day (single oral dose) during the RT phase (i.e., days 1–42), and then at 40 mg/day following RT (maintenance phase) until investigator-assessed disease progression or undue adverse reaction, whichever occurred first. For regimen M, patients received TMZ 75 mg/m2 daily (single oral dose) during the RT phase. A 4-week TMZ-free phase followed the RT phase, after which TMZ was administered for up to six 28-day cycles (maintenance phase: TMZ single oral dose once daily on days 1–5; 150 mg/m2 in cycle 1 and 200 mg/m2 in cycles 2–6).
Afatinib treatment was paused whenever a patient experienced an adverse event (AE) that met the criteria for dose-limiting toxicity (DLT), regardless of the cycle. DLT was defined as an AE or laboratory abnormality considered to be related to afatinib and meeting pre-specified criteria (see Supplementary Methods). Upon recovery of the AE to baseline or National Cancer Institute Common Terminology Criteria for Adverse Events (CTCAE) grade 1 (whichever was higher) within 14 days, treatment could be continued at a reduced dose. Otherwise, the patient was discontinued from trial medication, except for patients with obvious clinical benefit according to the investigator’s judgment.
Endpoints and assessments
The primary endpoint was the MTD of continuous daily afatinib when given in combination with RT in patients with newly diagnosed GBM, with or without concomitant TMZ treatment. Secondary endpoints were the incidence and intensity of AEs, objective tumor response rate, and pharmacokinetics of afatinib (afatinib concentration at steady state, pre-dose: Days 8, 15, and 29; please see Supplemental Methods).
MTD was defined as the highest afatinib dose level at which no more than one of six patients experienced DLT, i.e., the highest afatinib dose with a DLT incidence ≤ 17%, during the 6-week RT phase. Patients who, for any reasons other than DLT, did not receive trial medication during the RT phase, for more than 5 consecutive days or more than 8 non-consecutive days, could stay in the trial, but were removed from the MTD assessment and replaced by additional patients.
Safety was assessed by physical examination, hematologic and chemistry laboratory values, vital signs, and electrocardiography scans. AEs were graded by CTCAE version 3.0. Serious AEs (SAEs) were defined as any AE that resulted in death, was immediately life threatening, resulted in persistent or significant disability, required or prolonged patient hospitalization, was a congenital anomaly/birth defect, or was deemed serious for any other reason.
Objective tumor response rate was assessed by the investigator according to the Macdonald criteria [33], as measured by cerebral gadolinium-enhanced MRI. Assessment of objective response was conducted during the maintenance phase, (i.e., following completion of radiotherapy). MRIs were performed between days 21–28 of cycles 1, 3, 5, 8, 10, and 12 for regimen M, and of cycles 2, 4, 6, 8, 10, and 12 for regimen U. In the second year, MRIs were performed every 3 months (cycles 15, 18, 21 and 24), and every 6 months thereafter. Objective response was defined as the best overall response [complete response (CR) or partial response] recorded since the first administration of treatment until disease progression, death, or treatment discontinuation. Unplanned post hoc analysis was performed to determine time to disease progression (TTP). TTP was calculated as the time between the first treatment date to the day following the first date with recorded progressive disease. Patients without progressive disease were censored at their most recent imaging date. The median and 95% confidence interval (CI) were calculated using Kaplan–Meier methodology.
Statistical analyses
Safety, pharmacokinetic, and efficacy parameters were summarized descriptively; no formal statistical hypothesis testing was conducted. All patients who were administered at least one dose of any study treatment were included in the efficacy and safety analyses.
Results
Patient disposition and characteristics
Between November 2009 and October 2012, 55 patients were enrolled onto the trial. Of these, 36 patients received at least one dose of trial medication; 20 and 16 patients were treated with regimens M and U, respectively. Key baseline characteristics were similar between the two treatment arms, except for median tumor size, which was greater in patients receiving regimen U (Table 1). Patients who received regimen M were predominantly male (70%) and white (95%), with a median time from first histological diagnosis of 38 days (range 28–67) and a median tumor size (sum of largest cross-section post-surgery) of 89 mm2 (range 0–2088). Patients who received regimen U were also predominantly male (69%) and white (100%), with a median time from first histological diagnosis of 36 days (range 28–51) and median tumor size of 978 mm2 (range 0–2331).
Table 1.
Patient baseline demographics and clinical characteristics
| Regimen M Afatinib + TMZ + RT N = 20 | Regimen U Afatinib + RT N = 16 | |
|---|---|---|
| Male, n (%) | 14 (70) | 11 (69) |
| Race, n (%) | ||
| White | 19 (95) | 16 (100) |
| Asian | 1 (5) | 0 |
| Age in years, median (range) | 52.5 (25–66) | 53.5 (34–68) |
| BMI in kg/m2, median (range) | 27.3 (20.6–33.8) | 28.7 (21.7–38.8) |
| Smoking history, n (%) | ||
| Never smoked | 15 (75) | 13 (81) |
| Ex-smoker | 4 (20) | 2 (13) |
| Currently smokes | 1 (5) | 1 (6) |
| Time from first histological diagnosis in days, median (range) | 38.0 (28–67) | 36.0 (28–51) |
| Karnofsky performance score, median (range) | − 10.0 (− 50–0) | − 20.0 (− 50–0) |
| Sum of largest cross-section post-surgery in mm2, median (range) | 89.0 (0–2088) | 978.3 (0–2331) |
| Unilocular, n (%) | 18 (90) | 15 (94) |
BMI body mass index, RT radiotherapy, TMZ temozolomide
With regimen M, 20 patients were treated and 15 patients continued afatinib beyond the RT phase. With regimen U, 16 patients were treated and 13 continued afatinib beyond the RT phase (Fig. 1). The median (range) durations of afatinib treatment for regimens M and U were 150 (6–2340) days and 167 (1–397) days, respectively.
Fig. 1.

Patient disposition. a Regimen M: afatinib + TMZ + RT; b regimen U: afatinib + RT. AE adverse event, DLT dose-limiting toxicity, PD progressive disease, RT radiotherapy, TMZ temozolomide. *Due to trial completion, all patients are reported as having discontinued afatinib; however, one patient continued to receive afatinib outside of the clinical trial
Afatinib was discontinued by all patients during the study. Reasons for afatinib discontinuation with regimen M included disease progression (45%), DLT (10%), and other AEs (35%) (Fig. 1). One patient who received regimen M had not experienced disease progression at data cut-off and was switched to commercially supplied afatinib; he remained on treatment for more than 6 years. With regimen U, reasons for afatinib discontinuation included disease progression (50%), and AEs other than DLTs (38%); no patients receiving regimen U discontinued afatinib treatment due to DLTs.
MTD of afatinib with concomitant RT
Overall, 17 of 20 patients who received regimen M were evaluable for MTD determination (Fig. 2). In the first dose cohort (afatinib 20 mg/day), one of six evaluable patients had a DLT during the RT phase (grade 4 thrombocytopenia). The afatinib dose was therefore escalated to 40 mg/day; two of five patients (seven patients were treated; two were not evaluable) had DLTs: one patient had grade 4 thrombocytopenia and one patient had grade 3 vomiting. The afatinib dose was reduced and an intermediate dose level of 30 mg/day was explored; none of the six treated patients had DLTs. Accordingly, afatinib 30 mg was determined as the MTD in combination with daily TMZ and RT.
Fig. 2.
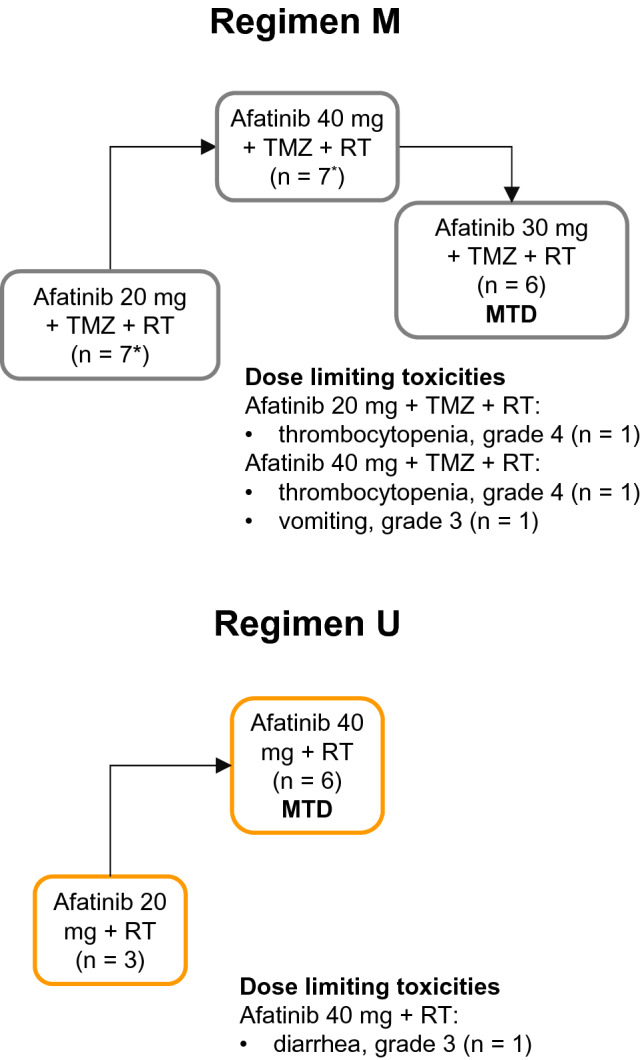
Determination of the maximum tolerated dose based on the occurrence of dose limiting toxicities during the 6-week radiotherapy phase. MTD maximum tolerated dose, RT radiotherapy, TMZ temozolomide. *In Regimen M, one patient was replaced in the 20 mg afatinib group and one patient was replaced in the 40 mg afatinib group
With regimen U, nine of 16 patients treated were evaluable for MTD determination. In the first dose cohort (afatinib 20 mg/day), three patients were treated without any DLT during the RT phase. The afatinib dose was subsequently escalated to 40 mg/day; one of six patients had a DLT (grade 3 diarrhea). Thus, afatinib 40 mg was determined as the MTD in combination with RT.
For pharmacokinetic data, please see Supplemental Results.
Safety profile of each regimen
The most frequently reported AEs with regimen M were diarrhea (85%), nausea (75%), and rash (65%). Of patients receiving regimen M, 95% had at least one drug-related AE (Table 2); the most common drug-related AEs were diarrhea (80%), rash (65%), nausea (45%), and fatigue (45%; Supplementary Table 1). Nine (45%) patients had AEs that led to discontinuation of afatinib; AEs (preferred terms) reported in more than one patient were diarrhea, fatigue, rash, and thrombocytopenia (two patients each). All other AEs leading to afatinib discontinuation were reported in single patients only; these were skin toxicity, skin ulcer, alanine aminotransferase increased, and postoperative wound infection.
Table 2.
Summary of AEs
| Regimen M Afatinib (20 mg) + TMZ + RT N = 7 | Regimen M Afatinib (30 mg) + TMZ + RT N = 6 | Regimen M Afatinib (40 mg) + TMZ + RT N = 7 | Regimen M Afatinib (total) + TMZ + RT N = 20 | |
|---|---|---|---|---|
| Any AE, n (%) | 7 (100) | 6 (100) | 7 (100) | 20 (100) |
| DLTsa, n (%) | 4 (57) | 1 (17) | 3 (43) | 8 (40) |
| Treatment-related AEs, n (%) | 7 (100) | 6 (100) | 6 (86) | 19 (95) |
| AEs leading to discontinuation of afatinib, n (%) | 4 (57) | 0 | 5 (71) | 9 (45) |
| AEs leading to discontinuation of TMZ, n (%) | 1 (14) | 0 | 2 (29) | 3 (15) |
| SAEsb, n (%) | 4 (57) | 5 (83) | 3 (43) | 12 (60) |
| Maximum CTCAE grade, n (%) | ||||
| Grade 1 | 0 | 0 | 1 (14) | 1 (5) |
| Grade 2 | 0 | 2 (33) | 1 (14) | 3 (15) |
| Grade 3 | 4 (57) | 4 (67) | 4 (57) | 12 (60) |
| Grade 4 | 3 (43) | 0 | 1 (14) | 4 (20) |
| Grade 5 | 0 | 0 | 0 | 0 |
| Regimen U afatinib (20 mg) + RT N = 3 | Regimen U afatinib (40 mg) + RT N = 13 | Regimen U afatinib (total) + RT N = 16 | |
|---|---|---|---|
| Any AE, n (%) | 3 (100) | 13 (100) | 16 (100) |
| DLTsa, n (%) | 2 (67) | 4 (31) | 6 (38) |
| Treatment-related AEs, n (%) | 3 (100) | 12 (92) | 15 (94) |
| AEs, regardless of causality, leading to discontinuation of afatinib, n (%) | 1 (33) | 9 (69) | 10 (63) |
| SAEsb, n (%) | 2 (67) | 10 (77) | 12 (75) |
| Maximum CTCAE grade, n (%) | |||
| Grade 1 | 0 | 1 (8) | 1 (6) |
| Grade 2 | 1 (33) | 2 (15) | 3 (19) |
| Grade 3 | 2 (67) | 5 (38) | 7 (44) |
| Grade 4 | 0 | 2 (15) | 2 (13) |
| Grade 5 | 0 | 3 (23) | 3 (19) |
AE adverse event, CTCAE Common Terminology Criteria for Adverse Events, DLT dose-limiting toxicity, RT radiotherapy, SAE serious adverse event, TMZ temozolomide
aDLTs occurring in the overall treatment period, as determined by the investigator
bA patient may have experienced more than one SAE
With regimen U, the most frequently reported AEs were diarrhea (94%), rash (75%), and headache (63%). Of patients receiving regimen U, 94% of patients had at least one drug-related AE (Table 2); the most frequently reported drug-related AEs were diarrhea (81%), rash (75%), and fatigue (45%; Supplementary Table 1). Ten patients (63%) had AEs that led to discontinuation of afatinib; these were diarrhea, dermatitis acneiform, rash, pneumonia, pulmonary embolism, generalized tonic–clonic seizure, increased intracranial pressure, lethargy, malignant neoplasm progression, and disease progression (one patient each).
Frequencies of grade ≥ 3 drug-related AEs and SAEs by treatment regimen and dose are shown in Table 2. SAEs were reported in 12 (60%) patients who received regimen M and 12 (75%) patients who received regimen U. There were no fatal AEs reported for patients who received regimen M. Fatal AEs were reported in three patients (19%) who received regimen U. The causes of death in these patients were bacterial meningitis, pneumonia, and disease progression. For all three deaths, none were considered to be drug-related.
Response to therapy
Of 20 evaluable patients who received regimen M, five (25%) patients achieved an objective response, including one CR, and 11 (55%) patients had stable disease. With regimen U, one (6%) of 16 evaluable patients had an objective response and eight (50%) had stable disease according to the Macdonald response assessment criteria [33]. The best overall responses by afatinib dose with regimens M and U are shown in Table 3. The median time to disease progression for evaluable patients who received regimens M and U were 434 days (95% CI 205–NA, n = 18) and 211 days (95% CI 101–NA, n = 14), respectively. Insufficient data were available to calculate upper 95% CIs.
Table 3.
Summary of treatment response
| Regimen M Afatinib (20 mg) + TMZ + RT N = 7 | Regimen M Afatinib (30 mg) + TMZ + RT N = 6 | Regimen M Afatinib (40 mg) + TMZ + RT N = 7 | Regimen M Afatinib (total) + TMZ + RT N = 20 | |
|---|---|---|---|---|
| Best overall response, n (%) | ||||
| CR | 0 | 1 (17) | 0 | 1 (5) |
| PR | 2 (29) | 2 (33) | 0 | 4 (20) |
| SD | 5 (71) | 2 (33) | 4 (57) | 11 (55) |
| PD | 0 | 1 (17) | 1 (14) | 2 (10) |
| Missing | 0 | 0 | 2 (29) | 2 (10) |
| Objective response, n (%) | 2 (29) | 3 (50) | 0 | 5 (25) |
| Disease control rate, n (%) | 7 (100) | 5 (83) | 4 (57) | 16 (80) |
| Regimen U afatinib (20 mg) + RT N = 3 | Regimen U afatinib (40 mg) + RT N = 13 | Regimen U afatinib (total) + RT N = 16 | |
|---|---|---|---|
| Best overall response, n (%) | |||
| CR | 0 | 0 | 0 |
| PR | 0 | 1 (8) | 1 (6) |
| SD | 1 (33) | 7 (54) | 8 (50) |
| PR | 2 (67) | 3 (23) | 5 (31) |
| Missing | 0 | 2 (15) | 2 (13) |
| Objective response, n (%) | 0 | 1 (8) | 1 (6) |
| Disease control rate, n (%) | 1 (33) | 8 (62) | 9 (56) |
CR complete response, PD progressive disease, PR partial response, RT radiotherapy, SD stable disease, TMZ temozolomide
Discussion
In this open-label, phase I dose-escalation trial in newly diagnosed patients with GBM, the MTD of afatinib was 30 mg/day in combination with RT and TMZ (regimen M; methylated MGMT promoter), and 40 mg/day in combination with RT (regimen U; unmethylated MGMT promoter).
The most frequently reported drug-related AEs in this trial with regimens M/U were diarrhea (80/81%), rash (65/75%), and fatigue (45/38%), with nausea also reported in a high proportion of the patients who received regimen M (45%). These findings are similar to the known toxicity profile of afatinib when used as a single agent in patients with NSCLC [28, 34], and also to those observed in the phase I/II study of afatinib with or without TMZ in recurrent GBM, in which the most frequent AEs observed in afatinib-containing arms (afatinib alone/afatinib + TMZ) were rash/acne and diarrhea [31]. The AE profiles of regimens M and U were also consistent with those of the afatinib combination partners, and, aside from grade 4 thrombocytopenia reported in three patients (15%) receiving the TMZ-containing regimen M, there was no evidence that afatinib increased the incidence of TMZ-associated toxicities, compared with previous reports of TMZ with or without afatinib [5, 31].
The pharmacokinetics of afatinib in combination with RT, with or without TMZ, appeared to be consistent with those previously reported for single-agent afatinib [35]. There were no meaningful differences in afatinib trough plasma concentrations over time (Days 8, 15, and 29 from start of treatment) nor between treatment regimens, suggesting that concentrations were unaffected by RT or TMZ.
The key aim of this study was not to investigate efficacy; however, disease control observed at RT completion was indicative of modest efficacy for both treatment regimens in patients with newly diagnosed GBM. It is unclear whether treatment with afatinib was a contributing factor, as it was administered in combination with treatments with known efficacy in GBM [5, 32]. Moreover, previous studies of ErbB pathways inhibitors in GBM have shown little efficacy when given alone [31, 36]. In a phase Ib/II trial of afatinib with or without TMZ in patients with recurrent GBM, the 6-month PFS rate was significantly lower with afatinib monotherapy than with afatinib plus TMZ or TMZ alone (afatinib alone: 3%; afatinib + TMZ: 10%; TMZ alone: 23%) [31]. However, median PFS was longer in afatinib-treated patients with EGFR-overexpressing tumors (3.35 months) than those with EGFR levels within a normal range (0.99 months). Similar results have been observed with other EGFR TKIs. Gefitinib (with or without chemotherapy) was associated with response rates of up to 14% and 6-month PFS rates of 5–24% in patients with recurrent glioma [36–39]. Similarly, response rates of up to 8% have been achieved with erlotinib, with little impact on overall response or PFS in patients with recurrent malignant glioma compared with TMZ (with or without chemotherapy) [40–43]. While previous studies have indicated minimal activity of ErbB family inhibitors in GBM, a case of prolonged response to afatinib has been reported previously in a patient with recurrent GBM in this study. This patient, who had several EGFR mutations, EGFR gene amplification, and EGFR-vIII seropositivity, survived for around 5 years from recurrence, nearly sixfold longer than expected in patients with recurrent GBM [44, 45]. The patient was switched to commercial supply and was still on treatment at the time of the database lock. A further two patients with GBM who had long-term responses (> 12 months) to afatinib harbored mutations in specific combinations of alleles that are causal of EGFR addiction [44, 46]. For example, one patient had a PTPN11 mutation thought to drive EGFR addiction and, hence, response to afatinib, and another patient had a tumor that was EGFR amplified and carried an additional allele on the amplicon, potentially underlying the sustained response observed [46]. These findings suggest that afatinib may be of most benefit in patients with GBM harboring EGFR aberrations.
Given that alterations affecting EGFR, e.g. EGFR overexpression, have been identified previously in tumors of patients with GBM, including in long-term responders to afatinib [16, 17, 44, 46], a potential limitation of the present study is that patients were not selected based on biomarker analysis. Patients were not selected in this manner as EGFR genetic testing was not routinely performed when the trial was initiated. In future trials, selection of patients based upon specific biomarkers, such as EGFR mutations or amplification, may assist in identifying patients who are more likely to benefit from EGFR-targeted therapies. Another limitation of this study is that, similar to other trials to date, it has not been possible to distinguish the efficacy of afatinib from the known effectiveness of RT and TMZ [31]. Additionally, response to therapy was evaluated using Macdonald criteria, which were in widespread use at the time of the design of this study. These criteria have largely been superseded by response assessment in neuro-oncology (RANO) criteria [47, 48].
This dose-finding study identified the MTD for afatinib in combination with RT and TMZ for patients with methylation of the MGMT promoter (30 mg/day during RT; 40 mg/day maintenance phase; regimen M), and in combination with RT for patients without methylation of the MGMT promoter (40 mg/day in RT and maintenance phases; regimen U). Treatment with both regimens was associated with a manageable AE profile that was consistent with the known safety profiles of the individual agents; the pharmacokinetic profile of afatinib was also in line with previous afatinib monotherapy studies at all dose levels. While this study only included a small number of patients, and efficacy was not the primary endpoint, antitumor activity was observed in a subset of each treatment group. Given the relationship between EGFR aberrations and poor response to treatment in GBM, the ErbB pathway remains a plausible therapeutic target in GBM. Research into the safety and pharmacokinetics of afatinib in patients with previously treated brain cancer is ongoing in a phase I study (NCT02423525). In future studies, biomarker analysis should be utilized to guide preselection of patients most likely to benefit from afatinib treatment.
Supplementary Information
Below is the link to the electronic supplementary material.
Acknowledgements
Medical writing support for the development of this manuscript, under the direction of the authors, was provided by Nadia Fowler and Jim Sinclair, PhD, of Ashfield MedComms, an Ashfield Health company, and funded by Boehringer Ingelheim. The authors are fully responsible for all content and editorial decisions, were involved at all stages of manuscript development, and have approved the final version. The authors received no direct compensation related to the development of the manuscript. Boehringer Ingelheim was given the opportunity to review the manuscript for medical and scientific accuracy as well as intellectual property considerations.
Author contributions
FS, LW, AJ, CM, SJ and FH substantially contributed to the acquisition, analysis, or interpretation of data for the work. RG and MB substantially contributed to the conception or design of the work. KP, JS, SB and AC substantially contributed to the conception or design of the work and to the acquisition, analysis, or interpretation of data for the work. All authors drafted the work/revised it critically for important intellectual content, provided final approval of the version to be published, and agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Funding
Boehringer Ingelheim International GmbH.
Data availability
To ensure independent interpretation of clinical study results, Boehringer Ingelheim grants all external authors access to relevant material, including participant-level clinical study data, as needed by them to fulfill their role and obligations as authors under the ICMJE criteria. Clinical study documents and participant clinical study data are available to be shared on request after publication of the primary manuscript in a peer-reviewed journal, and if regulatory activities are complete and other criteria met as per the BI Policy on Transparency and Publication of Clinical Study Data (see https://www.mystudywindow.com/msw/datasharing). Bona fide, qualified scientific and medical researchers are eligible to request access to the clinical study data with corresponding documentation describing the structure and content of the datasets. Upon approval, and governed by a Legal Agreement, data are shared in a secured data-access system for a limited period of 1 year, which may be extended upon request. Prior to providing access, clinical study documents and data will be examined, and, if necessary, redacted and de-identified, to protect the personal data of study participants and personnel, and to respect the boundaries of the informed consent of the study participants. Researchers should use the https://vivli.org/ link to request access to study data and visit https://www.mystudywindow.com/msw/datasharing for further information.
Code availability
(software application or custom code): not applicable.
Declarations
Conflict of interest
SJ reports having a private medical practice for CNS and Thyroid tumors, having an investment in Genesis Cancer Care at Newmarket within a Limited Company – Saunders and Jefferies Consultancy, and undertaking medicolegal work (one-two cases per year). KP declares being a Global project Manager working for Boehringer Ingelheim. JS declares employment with Boehringer Ingelheim Pharma GmbH & Co KG. SB declares employment with Boehringer Ingelheim Pharmaceuticals, Inc. AC declares employment with Boehringer Ingelheim International GmbH. All remaining authors declare no potential conflict of interest. The authors did not receive payment related to the development of the manuscript.
Ethical approval
The study was approved by the Ethics Committee of the NHS Health Research Authority London (West London and Gene Therapy Advisory Committee). All patients provided written informed consent according to International Conference on Harmonization Good Clinical Practice and local legal regulations. Patients included in the current study provided written consent for the usage of their data for research purposes.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Agnieszka Cseh passed away in May 2021.
Frank Saran and Michael Brada have contributed equally to this work.
References
- 1.Omuro A, DeAngelis LM. Glioblastoma and other malignant gliomas: a clinical review. JAMA. 2013;310:1842–1850. doi: 10.1001/jama.2013.280319. [DOI] [PubMed] [Google Scholar]
- 2.Gilbert MR, Dignam JJ, Armstrong TS, Wefel JS, Blumenthal DT, Vogelbaum MA, Colman H, Chakravarti A, Pugh S, Won M, Jeraj R, Brown PD, Jaeckle KA, Schiff D, Stieber VW, Brachman DG, Werner-Wasik M, Tremont-Lukats IW, Sulman EP, Aldape KD, Curran WJ, Mehta MP. A randomized trial of bevacizumab for newly diagnosed glioblastoma. N Engl J Med. 2014;370:699–708. doi: 10.1056/NEJMoa1308573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chinot OL, Wick W, Mason W, Henriksson R, Saran F, Nishikawa R, Carpentier AF, Hoang-Xuan K, Kavan P, Cernea D, Brandes AA, Hilton M, Abrey L, Cloughesy T. Bevacizumab plus radiotherapy–temozolomide for newly diagnosed glioblastoma. N Engl J Med. 2014;370:709–722. doi: 10.1056/NEJMoa1308345. [DOI] [PubMed] [Google Scholar]
- 4.Kelly C, Majewska P, Ioannidis S, Raza MH, Williams M. Estimating progression-free survival in patients with glioblastoma using routinely collected data. J Neurooncol. 2017;135:621–627. doi: 10.1007/s11060-017-2619-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJB, Belanger K, Brandes AA, Marosi C, Bogdahn U, Curschmann J, Janzer RC, Ludwin SK, Gorlia T, Allgeier A, Lacombe D, Cairncross JG, Eisenhauer E, Mirimanoff RO. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352:987–996. doi: 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- 6.Stupp R, Brada M, van den Bent MJ, Tonn JC, Pentheroudakis G, Group EGW High-grade glioma: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2014;25(Suppl 3):93–101. doi: 10.1093/annonc/mdu050. [DOI] [Google Scholar]
- 7.Hegi ME, Diserens A-C, Gorlia T, Hamou M-F, de Tribolet N, Weller M, Kros JM, Hainfellner JA, Mason W, Mariani L, Bromberg JEC, Hau P, Mirimanoff RO, Cairncross JG, Janzer RC, Stupp R. MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med. 2005;352:997–1003. doi: 10.1056/NEJMoa043331. [DOI] [PubMed] [Google Scholar]
- 8.Wick W, Platten M, Weller M. New (alternative) temozolomide regimens for the treatment of glioma. Neuro Oncol. 2009;11:69–79. doi: 10.1215/15228517-2008-078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ventero MP, Fuentes-Baile M, Quereda C, Perez-Valeciano E, Alenda C, Garcia-Morales P, Esposito D, Dorado P, Manuel Barbera V, Saceda M. Radiotherapy resistance acquisition in glioblastoma. Role of SOCS1 and SOCS3. PLoS ONE. 2019;14:e0212581–e0212581. doi: 10.1371/journal.pone.0212581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mrugala MM, Chamberlain MC. Mechanisms of disease: temozolomide and glioblastoma—look to the future. Nat Clin Pract Oncol. 2008;5:476–486. doi: 10.1038/ncponc1155. [DOI] [PubMed] [Google Scholar]
- 11.Hegi ME, Liu L, Herman JG, Stupp R, Wick W, Weller M, Mehta MP, Gilbert MR. Correlation of O6-methylguanine methyltransferase (MGMT) promoter methylation with clinical outcomes in glioblastoma and clinical strategies to modulate MGMT activity. J Clin Oncol. 2008;26:4189–4199. doi: 10.1200/JCO.2007.11.5964. [DOI] [PubMed] [Google Scholar]
- 12.Andersson U, Schwartzbaum J, Wiklund F, Sjostrom S, Liu Y, Tsavachidis S, Ahlbom A, Auvinen A, Collatz-Laier H, Feychting M, Johansen C, Kiuru A, Lonn S, Schoemaker MJ, Swerdlow AJ, Henriksson R, Bondy M, Melin B. A comprehensive study of the association between the EGFR and ERBB2 genes and glioma risk. Acta Oncol. 2010;49:767–775. doi: 10.3109/0284186x.2010.480980. [DOI] [PubMed] [Google Scholar]
- 13.Brennan CW, Verhaak RG, McKenna A, Campos B, Noushmehr H, Salama SR, Zheng S, Chakravarty D, Sanborn JZ, Berman SH, Beroukhim R, Bernard B, Wu CJ, Genovese G, Shmulevich I, Barnholtz-Sloan J, Zou L, Vegesna R, Shukla SA, Ciriello G, Yung WK, Zhang W, Sougnez C, Mikkelsen T, Aldape K, Bigner DD, Van Meir EG, Prados M, Sloan A, Black KL, Eschbacher J, Finocchiaro G, Friedman W, Andrews DW, Guha A, Iacocca M, O'Neill BP, Foltz G, Myers J, Weisenberger DJ, Penny R, Kucherlapati R, Perou CM, Hayes DN, Gibbs R, Marra M, Mills GB, Lander E, Spellman P, Wilson R, Sander C, Weinstein J, Meyerson M, Gabriel S, Laird PW, Haussler D, Getz G, Chin L. The somatic genomic landscape of glioblastoma. Cell. 2013;155:462–477. doi: 10.1016/j.cell.2013.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Frederick L, Wang XY, Eley G, James CD. Diversity and frequency of epidermal growth factor receptor mutations in human glioblastomas. Cancer Res. 2000;60:1383–1387. [PubMed] [Google Scholar]
- 15.Gonzalez-Tablas M, Arandia D, Jara-Acevedo M, Otero A, Vital AL, Prieto C, Gonzalez-Garcia N, Nieto-Librero AB, Tao H, Pascual D, Ruiz L, Sousa P, Galindo-Villardon P, Orfao A, Tabernero MD. Heterogeneous EGFR, CDK4, MDM4, and PDGFRA gene expression profiles in primary GBM: no association with patient survival. Cancers. 2020;12:231. doi: 10.3390/cancers12010231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Barker FG, Simmons ML, Chang SM, Prados MD, Larson DA, Sneed PK, Wara WM, Berger MS, Chen P, Israel MA, Aldape KD. EGFR overexpression and radiation response in glioblastoma multiforme. Int J Rad Oncol Biol Phys. 2001;51:410–418. doi: 10.1016/S0360-3016(01)01609-1. [DOI] [PubMed] [Google Scholar]
- 17.Tripathy K, Das B, Singh AK, Misra A, Misra S, Misra SS. Prognostic significance of epidermal growth factor receptor in patients of glioblastoma multiforme. J Clin Diag Res. 2017;11:EC05–EC08. doi: 10.7860/JCDR/2017/30138.10327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Raizer JJ, Abrey LE, Lassman AB, Chang SM, Lamborn KR, Kuhn JG, Yung WKA, Gilbert MR, Aldape KA, Wen PY, Fine HA, Mehta M, Deangelis LM, Lieberman F, Cloughesy TF, Robins HI, Dancey J, Prados MD, North American Brain Tumor C A phase II trial of erlotinib in patients with recurrent malignant gliomas and nonprogressive glioblastoma multiforme postradiation therapy. Neuro Oncol. 2010;12:95–103. doi: 10.1093/neuonc/nop015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sequist LV, Yang JC, Yamamoto N, O'Byrne K, Hirsh V, Mok T, Geater SL, Orlov S, Tsai CM, Boyer M, Su WC, Bennouna J, Kato T, Gorbunova V, Lee KH, Shah R, Massey D, Zazulina V, Shahidi M, Schuler M. Phase III study of afatinib or cisplatin plus pemetrexed in patients with metastatic lung adenocarcinoma with EGFR mutations. J Clin Oncol. 2013;31:3327–3334. doi: 10.1200/jco.2012.44.2806. [DOI] [PubMed] [Google Scholar]
- 20.Wu YL, Zhou C, Hu CP, Feng J, Lu S, Huang Y, Li W, Hou M, Shi JH, Lee KY, Xu CR, Massey D, Kim M, Shi Y, Geater SL. Afatinib versus cisplatin plus gemcitabine for first-line treatment of Asian patients with advanced non-small-cell lung cancer harbouring EGFR mutations (LUX-Lung 6): an open-label, randomised phase 3 trial. Lancet Oncol. 2014;15:213–222. doi: 10.1016/s1470-2045(13)70604-1. [DOI] [PubMed] [Google Scholar]
- 21.Li D, Ambrogio L, Shimamura T, Kubo S, Takahashi M, Chirieac LR, Padera RF, Shapiro GI, Baum A, Himmelsbach F, Rettig WJ, Meyerson M, Solca F, Greulich H, Wong KK. BIBW2992, an irreversible EGFR/HER2 inhibitor highly effective in preclinical lung cancer models. Oncogene. 2008;27:4702–4711. doi: 10.1038/onc.2008.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Solca F, Dahl G, Zoephel A, Bader G, Sanderson M, Klein C, Kraemer O, Himmelsbach F, Haaksma E, Adolf GR. Target binding properties and cellular activity of afatinib (BIBW 2992), an irreversible ErbB family blocker. J Pharmacol Exp Ther. 2012;343:342–350. doi: 10.1124/jpet.112.197756. [DOI] [PubMed] [Google Scholar]
- 23.Westphal M, Maire CL, Lamszus K. EGFR as a target for glioblastoma treatment: an unfulfilled promise. CNS Drugs. 2017;31:723–735. doi: 10.1007/s40263-017-0456-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sarkaria JN, Hu LS, Parney IF, Pafundi DH, Brinkmann DH, Laack NN, Giannini C, Burns TC, Kizilbash SH, Laramy JK, Swanson KR, Kaufmann TJ, Brown PD, Agar NYR, Galanis E, Buckner JC, Elmquist WF. Is the blood-brain barrier really disrupted in all glioblastomas? A critical assessment of existing clinical data. Neuro Oncol. 2018;20:184–191. doi: 10.1093/neuonc/nox175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tamiya A, Tamiya M, Nishihara T, Shiroyama T, Nakao K, Tsuji T, Takeuchi N, Isa SI, Omachi N, Okamoto N, Suzuki H, Okishio K, Iwazaki A, Imai K, Hirashima T, Atagi S. Cerebrospinal fluid penetration rate and efficacy of afatinib in patients with EGFR mutation-positive non-small cell lung cancer with leptomeningeal carcinomatosis: a multicenter prospective study. Anticancer Res. 2017;37:4177–4182. doi: 10.21873/anticanres.11806. [DOI] [PubMed] [Google Scholar]
- 26.Zhang S-R, Zhu L-C, Jiang Y-P, Zhang J, Xu R-J, Xu Y-S, Xia B, Ma S-L. Efficacy of afatinib, an irreversible ErbB family blocker, in the treatment of intracerebral metastases of non-small cell lung cancer in mice. Acta Pharm Sin. 2017;38:233–240. doi: 10.1038/aps.2016.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hoffknecht P, Tufman A, Wehler T, Pelzer T, Wiewrodt R, Schutz M, Serke M, Stohlmacher-Williams J, Marten A, Maria Huber R, Dickgreber NJ. Efficacy of the irreversible ErbB family blocker afatinib in epidermal growth factor receptor (EGFR) tyrosine kinase inhibitor (TKI)-pretreated non-small-cell lung cancer patients with brain metastases or leptomeningeal disease. J Thorac Oncol. 2015;10:156–163. doi: 10.1097/jto.0000000000000380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Park K, Wan-Teck Lim D, Okamoto I, Yang JC-H. First-line afatinib for the treatment of EGFR mutation-positive non-small-cell lung cancer in the 'real-world' clinical setting. Ther Adv Med Oncol. 2019;11:1758835919836374. doi: 10.1177/1758835919836374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li SH, Liu CY, Hsu PC, Fang YF, Wang CC, Kao KC, Tseng LC, Yang CT. Response to afatinib in treatment-naive patients with advanced mutant epidermal growth factor receptor lung adenocarcinoma with brain metastases. Expert Rev Anticancer Ther. 2018;18:81–89. doi: 10.1080/14737140.2018.1409623. [DOI] [PubMed] [Google Scholar]
- 30.Schuler M, Wu YL, Hirsh V, O'Byrne K, Yamamoto N, Mok T, Popat S, Sequist LV, Massey D, Zazulina V, Yang JC. First-line afatinib versus chemotherapy in patients with non-small cell lung cancer and common epidermal growth factor receptor gene mutations and brain metastases. J Thorac Oncol. 2016;11:380–390. doi: 10.1016/j.jtho.2015.11.014. [DOI] [PubMed] [Google Scholar]
- 31.Reardon DA, Nabors LB, Mason WP, Perry JR, Shapiro W, Kavan P, Mathieu D, Phuphanich S, Cseh A, Fu Y, Cong J, Wind S, Eisenstat DD. Phase I/randomized phase II study of afatinib, an irreversible ErbB family blocker, with or without protracted temozolomide in adults with recurrent glioblastoma. Neuro Oncol. 2015;17:430–439. doi: 10.1093/neuonc/nou160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Stupp R, Hegi ME, Mason WP, van den Bent MJ, Taphoorn MJ, Janzer RC, Ludwin SK, Allgeier A, Fisher B, Belanger K, Hau P, Brandes AA, Gijtenbeek J, Marosi C, Vecht CJ, Mokhtari K, Wesseling P, Villa S, Eisenhauer E, Gorlia T, Weller M, Lacombe D, Cairncross JG, Mirimanoff RO. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009;10:459–466. doi: 10.1016/s1470-2045(09)70025-7. [DOI] [PubMed] [Google Scholar]
- 33.Macdonald DR, Cascino TL, Schold SC, Jr, Cairncross JG. Response criteria for phase II studies of supratentorial malignant glioma. J Clin Oncol. 1990;8:1277–1280. doi: 10.1200/jco.1990.8.7.1277. [DOI] [PubMed] [Google Scholar]
- 34.Park K, Tan EH, O'Byrne K, Zhang L, Boyer M, Mok T, Hirsh V, Yang JC, Lee KH, Lu S, Shi Y, Kim SW, Laskin J, Kim DW, Arvis CD, Kolbeck K, Laurie SA, Tsai CM, Shahidi M, Kim M, Massey D, Zazulina V, Paz-Ares L. Afatinib versus gefitinib as first-line treatment of patients with EGFR mutation-positive non-small-cell lung cancer (LUX-Lung 7): a phase 2B, open-label, randomised controlled trial. Lancet Oncol. 2016;17:577–589. doi: 10.1016/s1470-2045(16)30033-x. [DOI] [PubMed] [Google Scholar]
- 35.Wind S, Schnell D, Ebner T, Freiwald M, Stopfer P. Clinical pharmacokinetics and pharmacodynamics of afatinib. Clin Pharmacokin. 2017;56:235–250. doi: 10.1007/s40262-016-0440-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rich JN, Reardon DA, Peery T, Dowell JM, Quinn JA, Penne KL, Wikstrand CJ, Van Duyn LB, Dancey JE, McLendon RE, Kao JC, Stenzel TT, Ahmed Rasheed BK, Tourt-Uhlig SE, Herndon JE, 2nd, Vredenburgh JJ, Sampson JH, Friedman AH, Bigner DD, Friedman HS. Phase II trial of gefitinib in recurrent glioblastoma. J Clin Oncol. 2004;22:133–142. doi: 10.1200/jco.2004.08.110. [DOI] [PubMed] [Google Scholar]
- 37.Franceschi E, Cavallo G, Lonardi S, Magrini E, Tosoni A, Grosso D, Scopece L, Blatt V, Urbini B, Pession A, Tallini G, Crino L, Brandes AA. Gefitinib in patients with progressive high-grade gliomas: a multicentre phase II study by Gruppo Italiano Cooperativo di Neuro-Oncologia (GICNO) Br J Cancer. 2007;96:1047–1051. doi: 10.1038/sj.bjc.6603669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Reardon DA, Quinn JA, Vredenburgh JJ, Gururangan S, Friedman AH, Desjardins A, Sathornsumetee S, Herndon JE, 2nd, Dowell JM, McLendon RE, Provenzale JM, Sampson JH, Smith RP, Swaisland AJ, Ochs JS, Lyons P, Tourt-Uhlig S, Bigner DD, Friedman HS, Rich JN. Phase 1 trial of gefitinib plus sirolimus in adults with recurrent malignant glioma. Clin Cancer Res. 2006;12:860–868. doi: 10.1158/1078-0432.Ccr-05-2215. [DOI] [PubMed] [Google Scholar]
- 39.Kreisl TN, Lassman AB, Mischel PS, Rosen N, Scher HI, Teruya-Feldstein J, Shaffer D, Lis E, Abrey LE. A pilot study of everolimus and gefitinib in the treatment of recurrent glioblastoma (GBM) J Neurooncol. 2009;92:99–105. doi: 10.1007/s11060-008-9741-z. [DOI] [PubMed] [Google Scholar]
- 40.Reardon DA, Desjardins A, Vredenburgh JJ, Gururangan S, Friedman AH, Herndon JE, 2nd, Marcello J, Norfleet JA, McLendon RE, Sampson JH, Friedman HS. Phase 2 trial of erlotinib plus sirolimus in adults with recurrent glioblastoma. J Neurooncol. 2010;96:219–230. doi: 10.1007/s11060-009-9950-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.de Groot JF, Gilbert MR, Aldape K, Hess KR, Hanna TA, Ictech S, Groves MD, Conrad C, Colman H, Puduvalli VK, Levin V, Yung WK. Phase II study of carboplatin and erlotinib (Tarceva, OSI-774) in patients with recurrent glioblastoma. J Neurooncol. 2008;90:89–97. doi: 10.1007/s11060-008-9637-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.van den Bent MJ, Brandes AA, Rampling R, Kouwenhoven MC, Kros JM, Carpentier AF, Clement PM, Frenay M, Campone M, Baurain JF, Armand JP, Taphoorn MJ, Tosoni A, Kletzl H, Klughammer B, Lacombe D, Gorlia T. Randomized phase II trial of erlotinib versus temozolomide or carmustine in recurrent glioblastoma: EORTC brain tumor group study 26034. J Clin Oncol. 2009;27:1268–1274. doi: 10.1200/jco.2008.17.5984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yung WK, Vredenburgh JJ, Cloughesy TF, Nghiemphu P, Klencke B, Gilbert MR, Reardon DA, Prados MD. Safety and efficacy of erlotinib in first-relapse glioblastoma: a phase II open-label study. Neuro Oncol. 2010;12:1061–1070. doi: 10.1093/neuonc/noq072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Alshami J, Guiot MC, Owen S, Kavan P, Gibson N, Solca F, Cseh A, Reardon DA, Muanza T. Afatinib, an irreversible ErbB family blocker, with protracted temozolomide in recurrent glioblastoma: a case report. Oncotarget. 2015;6:34030–34037. doi: 10.18632/oncotarget.5297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Owen S, Alshami J, Guiot M-C, Kavan P, Gibson N, Solca F, Cseh A, Reardon DA, Muanza T (2019) Afatinib in multifocal glioblastoma: a case of extended survival and follow-up (EPO2307). Presented at the 5th Congress of the European Academy of Neurology (EAN), Oslo, Norway, June 29–July 2. Available at: https://www.inoncology.com/sites/default/files/downloads/1200.36_-_ean_2019_poster.pdf
- 46.Alken S, Alshami J, Muanza T, Gibson N, Pemberton K, Solca F, Cseh A, Saran F (2015) Next generation sequencing (NGS) analysis of tumours from patients (pts) with glioblastoma (GB) and long-term response to afatinib. Presented at the 4th in the biennial series of the EORTC-EANO-ESMO Conference, Istanbul, Turkey, March 27–28
- 47.Wen PY, Macdonald DR, Reardon DA, Cloughesy TF, Sorensen AG, Galanis E, Degroot J, Wick W, Gilbert MR, Lassman AB, Tsien C, Mikkelsen T, Wong ET, Chamberlain MC, Stupp R, Lamborn KR, Vogelbaum MA, van den Bent MJ, Chang SM. Updated response assessment criteria for high-grade gliomas: response assessment in neuro-oncology working group. J Clin Oncol. 2010;28:1963–1972. doi: 10.1200/JCO.2009.26.3541. [DOI] [PubMed] [Google Scholar]
- 48.Wen PY, Chang SM, Van den Bent MJ, Vogelbaum MA, Macdonald DR, Lee EQ. Response assessment in neuro-oncology clinical trials. J Clin Oncol. 2017;35:2439–2449. doi: 10.1200/JCO.2017.72.7511. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
To ensure independent interpretation of clinical study results, Boehringer Ingelheim grants all external authors access to relevant material, including participant-level clinical study data, as needed by them to fulfill their role and obligations as authors under the ICMJE criteria. Clinical study documents and participant clinical study data are available to be shared on request after publication of the primary manuscript in a peer-reviewed journal, and if regulatory activities are complete and other criteria met as per the BI Policy on Transparency and Publication of Clinical Study Data (see https://www.mystudywindow.com/msw/datasharing). Bona fide, qualified scientific and medical researchers are eligible to request access to the clinical study data with corresponding documentation describing the structure and content of the datasets. Upon approval, and governed by a Legal Agreement, data are shared in a secured data-access system for a limited period of 1 year, which may be extended upon request. Prior to providing access, clinical study documents and data will be examined, and, if necessary, redacted and de-identified, to protect the personal data of study participants and personnel, and to respect the boundaries of the informed consent of the study participants. Researchers should use the https://vivli.org/ link to request access to study data and visit https://www.mystudywindow.com/msw/datasharing for further information.
(software application or custom code): not applicable.