

Abstract
Motivation
Many high-throughput screening studies have been carried out in cancer cell lines to identify therapeutic agents and targets. Existing consistency assessment studies only examined two datasets at a time, with conclusions based on a subset of carefully selected features rather than considering global consistency of all the data. However, poor concordance can still be observed for a large part of the data even when selected features are highly consistent.
Results
In this study, we assembled nine compound screening datasets and three functional genomics datasets. We derived direct measures of consistency as well as indirect measures of consistency based on association between functional data and copy number-adjusted gene expression data. These results have been integrated into a web application—the Functional Data Consistency Explorer (FDCE), to allow users to make queries and generate interactive visualizations so that functional data consistency can be assessed for individual features of interest.
Availability and implementation
The FDCE web tool and we have developed and the functional data consistency measures we have generated are available at https://lccl.shinyapps.io/FDCE/.
Supplementary information
Supplementary data are available at Bioinformatics online.
1 Introduction
Personalized therapy has revolutionized clinical treatment of cancer. Many high-throughput functional screening studies have been performed in search of subtype-specific vulnerabilities—pharmacogenomics screenings for new agents and gene dependency screenings for new targets. The consistency among data generated from these screens has been a hot topic of interest with sometimes inconsistent findings from different assessments (Cancer Cell Line Encyclopedia and Genomics of Drug Sensitivity in Cancer, 2015; Dempster et al., 2019; Haibe-Kains et al., 2013; Haverty et al., 2016; Morgens et al., 2016; Safikhani et al., 2016). All of these reproducibility assessments performed to-date only examined two datasets at a time and conclusions of consistency were often based on a limited set of carefully selected features. It is important to realize that in the broad distribution of consistency measures, poor concordance can still be observed for a large part of the data even when selected features are highly consistent. Such discordance may arise from variations in experimental design or result from the lack of differential sensitivity among the cell lines. For researchers interested in a specific compound or gene, it has been a daunting task to identify relevant information about data consistency from the existing screens. To enable efficient re-use of these valuable datasets, we have assembled nine pharmacogenomics screening datasets and three gene essentiality screening datasets and have derived consistency measures on a per-feature basis. We also developed a user-friendly web application for result retrieval and visualization.
2 Materials and methods
2.1 Compound sensitivity data download and processing
‘NCI60’ compound sensitivity screening data was downloaded from https://discover.nci.nih.gov/cellminer/loadDownload.do, the CellMiner website (Reinhold et al., 2012) as ‘DTP_NCI60_ZSCORE.xlsx’ on August 15, 2020. Cell lines ADR-RES, MDA-N were removed due to contamination as noted by cell line database Cellosaurus (Bairoch, 2018). Some cell line names were manually corrected to match names used in other datasets. For example, ‘786O’ (last character being letter ‘O’) was changed to ‘7860’ (last character being number ‘0’). ‘CCLE’ compound sensitivity screening (Barretina et al., 2012) data was downloaded from the DepMap portal as ‘CCLE_NP24.2009_Drug_data_2015.02.24.csv’. The associated annotation file was downloaded as ‘CCLE_NP24.2009_profiling_2012.02.20.csv’. ‘GDSC’ compound sensitivity screening (Iorio et al., 2016; Picco et al., 2019) data ‘sanger-dose-response.csv’ (Release 8.1, October 2019) was downloaded on November 27, 2020 from the DepMap portal. Duplicated AUC data from GDSC1 and GDSC2 for the same cell line and same compound were averaged. CCLE_ID was mapped from DepMap_ID based on ‘sample_info.csv’ downloaded from 20Q2 data release. The associated annotation file was downloaded as ‘screened_compounds_rel_8.0.csv’ on August 12, 2019. ‘CTRP’ compound sensitivity screening (Seashore-Ludlow et al., 2015) data was downloaded from the DepMap portal as ‘v20.data.curves_post_qc.txt’ extracted from ‘http://ctrpv2.0_2015_ctd2_expandeddataset.zip’, ‘v20.meta.per_compound.txt’ was used as compound annotation. Replicate data for the same cell lines were averaged. Majority of the cell line names were replaced by matching CCLE_ID. The remaining cell lines were renamed with the CCLE_ID naming convention in NAME_DISEASE format. PRISM compound sensitivity screening (Corsello et al., 2020) datasets were downloaded from the DepMap portal 19Q4 release. For ‘PRISM_1ST’ (the primary screen), the compound sensitivity data was downloaded as ‘primary-screen-replicate-collapsed-logfold-change.csv’. Compound annotation was downloaded as ‘primary-screen-replicate-collapsed-treatment-info.csv’. For ‘PRISM_2nd’ (the secondary screen), the compound sensitivity data was downloaded as ‘secondary-screen-dose-response-curve-parameters.csv’. Compound annotation was downloaded as ‘secondary-screen-replicate-collapsed-treatment-info.csv’. ‘POPS’ data was previously published (McMillan et al., 2018) as Supplementary Table ‘table6_chemical_screen_data.xlsx’. ‘ChenNSCLC’ data was previously published (Chen et al., 2019) as Supplementary Table ‘S4_NSCLC_data’. ‘PolleySCLC’ data (Polley et al., 2016) was downloaded as ‘data_nciSclc_act.txt’ from https://discover.nci.nih.gov/SclcCellMinerCDB/ (Tlemsani et al., 2020). Compound annotation was downloaded as ‘Table_Drugs_Synonyms_cdb.txt’.
2.2 Compound name mapping
R package ‘webchem’ was used to query PubChem (Kim et al., 2016) for mapping compounds to PubChem IDs. For each dataset, compound names and all associated synonyms (if available) were used for mapping first, then SMILES strings were used for mapping the remaining unmapped compounds. For the NCI60 compounds that still failed to map, Substance IDs were used to query PubChem for matching PubChemIDs. After queries were made for all compound sensitivity datasets, the results were combined to identify groups of compound annotation entries with shared compound names or PubChemIDs. For each of these groups, the most frequent compound name and PubChemID were chosen to represent the other members of the group. The compound names in each dataset were then updated with the new representative compound names. Compound with more than one records has ‘(n)’ appended to its name for each record. The original names used for the compound and the associated annotations were still retained in the metadata file for each compound sensitivity datasets.
2.3 Cell line name mapping
For datasets downloaded from DepMap, RRID provided from the metadata table was used. For other datasets without RRIDs in the metadata, RRIDs and disease were retrieved from the Cellosaurus API(Bairoch, 2018) through queries and data conversion using R packages ‘httr’ and ‘jsonlite’. Disease status annotated by Cellosaurus were matched to cancer lineage defined by DepMap and appended to the cell line name. Groups of cell line synonyms matched to the same RRID were identified and replaced with a unique cell line name.
2.4 Gene dependency data download and processing
Gene dependency data were all downloaded from the DepMap portal. RNAi gene dependency data ‘demeter’ was downloaded as ‘D2_combined_gene_dep_scores.csv’ from DEMETER2 Data v6 (McFarland et al., 2018), CRISPR gene dependency data ‘achilles’ was downloaded as ‘Achilles_gene_effect.csv’ from the 20Q4 data release (2020), ‘sanger’ was downloaded as ‘gene_effect.csv’ (2019).
2.5 Generation of consistency measure ‘r.summary’
We filtered out pairwise correlations from less than 10 cell lines. For compound screening datasets that have within-study replicates, when multiple pairwise inter-study correlations are available for a specific study pair, z-score transformation is applied and only the correlation corresponding to the largest z-score is retained. After this filtering, if there is only one pair of correlation left, this is used as the r.summary; otherwise meta-analysis is performed with R package ‘metacor’ to implement the DerSimonian-Laird (DSL) random-effect meta-analytical approach with correlation coefficients as effect sizes, as described by Schulze (2004). For indirect consistency assessment, while a secondary correlation calculation was performed for two sets of compound/gene feature-versus-transcriptome correlation coefficients from the paired studies, instead of using the number of genes from the transcriptome as the sample size for meta-analysis input, we used the number of overlapping cell lines from the dataset pair with transcriptomic data available as the substitute sample size so that the weight for the random effect model would be similar to the direct assessment.
2.6 Bimodal distribution determination
To dichotomize samples into two groups, R package ‘mclust’ was use to implement Gaussian mixture modeling with assumption of two clusters with equal variance. Bimodal index (BI) is determined for the same model, following the method described by Wang et al. (2009). A higher BI value is indicative of a stronger bimodal distribution.
2.7 Determination of drug market status
We performed literature search and found WITHDRAWN—a database of withdrawn and discontinued drugs (Siramshetty et al., 2016). We downloaded lists of 270 ‘withdrawn’ compounds and 308 ‘discontinued’ compounds from with WITHDRAWN web page. According to WITHDRAWN, ‘withdrawn’ drugs are those recalled from market in at least one country due to toxic/side effects whereas ‘discontinued’ drugs are those recalled due to reasons other than safety, such as ineffectiveness of drug. 44 of the withdrawn and 45 of the discontinued compounds could be found in our summary consistency measure table. For all the remaining drugs, we used the drug group annotation from DrugBank (Wishart et al., 2006). We excluded compounds with missing annotation or with annotations as ‘vet_approved’ or ‘nutraceutical’, and we combined ‘experimental’ and ‘investigational’ into one group.
2.8 Indirect consistency assessment
RNA-seq gene expression data and copy number data from CCLE (Ghandi et al., 2019) was downloaded from the DepMap portal as ‘CCLE_depMap_19Q1_TPM.csv’ and ‘public_19Q1_gene_cn.csv’ on April 29, 2019. From the expression data, genes with variance of zero were removed from the dataset. As the standard method, only RNA-seq data was used; to apply the GRACE method as we previously described (Cai et al., 2017), for every gene we use the copy number as the predictor variable and the RNA-seq gene expression as the response variable to fit a linear regression model. The residuals from the resulting fit were saved as copy number-adjusted gene expression data. For each indirect assessment, we first computed the Pearson correlation coefficients between the functional data features and the expression data for each dataset, and then for each pair of datasets, we performed correlation of the previously computed correlation coefficients to get a single correlation coefficient for each functional data feature.
2.9 Determination of gene signature associated with vorinostat sensitivity or ZEB1 dependency
To identify mechanistically relevant gene signatures for vorinostat sensitivity or ZEB1 dependency, the top 100 genes positively or negatively associated with the functional data of interest were used to run hypergeometric tests with genesets collected in the curated Chemical and Genetic Perturbations (c2cgp) library of the molecular signatures databases (MSigDB) (Subramanian et al., 2005) collections. R package ‘fgsea’ was used for loading the geneset library. Resulting P-values were adjusted for multiple comparison by the Benjamini-Hochberg procedures and filtered by adjusted P-values. ‘LEE_DIFFERENTIATING_T_LYMPHOCYTE’ (Lee et al., 2004) (P.adj = 6.2e-36) was used as the T lymphocyte signature, whereas ‘AIGNER_ZEB1_TARGETS’ (Aigner et al., 2007) (P.adj = 1.4e-15) was selected as the ZEB1 target gene set.
2.10 Web application construction
The web application is a shiny app deployed at the shinyapps.io servers and implemented through the following R packages: ‘Cairo’, ‘data.table’, ‘dplyr’, ‘DT’, ‘GGally’, ‘ggplot2’, ‘mclust’, ‘patchwork’, ‘plotly’, ‘RColorBrewer’, ‘shiny’, ‘shinycssloaders’, ‘shinydashboardPlus’, ‘shinyjs’, ‘shinythemes’, ‘shinyTree’, ‘slickR’ and ‘tidyr’.
2.11 Other R packages used for analyses
All analyses were conducted in R (version 3.6.1). Other than the packages mentioned before, the following R packages were also used for data wrangling: ‘openxlsx’, ‘plyr’, ‘tidyverse’, ‘reshape2’. The following R packages were also used for statistical analyses: ‘stats’, ‘Hmisc’, ‘propagate’, ‘ffbase’. The following R packages were used for visualization of graphs: ‘ggrastr’, ‘ggridges’, ‘ggrepel’, ‘grid’, ‘gridGraphics’, ‘gridExtra’, ‘cowplot’.
3 Results
3.1 Harmonization of datasets
A schematic diagram that summarizes the work in this study is provided as Supplementary Figure S1. To begin, we collected nine compound screening datasets and three dependency datasets for analyses. Among the compound screening datasets, ‘CCLE’ (Barretina et al., 2012), ‘CTRP’ (Basu et al., 2013), ‘GDSC’ (Iorio et al., 2016), ‘NCI-60’ (Reinhold et al., 2012), ‘PRISM_1st’ and ‘PRISM_2nd’ (Corsello et al., 2020) are pan-cancer screens. The remaining compound screening datasets are specific for lung cancer cell lines: ‘POPS’ (McMillan et al., 2018) is a high-throughput screening (HTS) dataset for non-small cell lung cancer (NSCLC) cell lines; ‘PolleySCLC’ (Polley et al., 2016) is an HTS dataset for small cell lung cancer (SCLC) cell lines; ‘ChenNSCLC’ (Chen et al., 2019) is a dataset from manual screening of NSCLC cell lines. As summarized in Supplementary Table S1, while multiple measures are often available from a screen, we used the area under the dose response curve (AUC) whenever possible. There are generally fewer missing values with AUC data as it does not require extrapolation and can always be estimated from the dose-response curve. It has also been shown that better agreement is observed between datasets from AUC-based correlation compared to that from IC50 (Haibe-Kains et al., 2013). As indicated in Supplementary Table S1, we processed the datasets to make the lower value in each dataset always correspond to higher sensitivity. For the compound screening datasets, we identified the corresponding PubChem IDs (see material and methods) for each compound. The map rates for the nine datasets are between 80% and 100% (Fig. 1a). For dependency datasets, unique gene symbols were used as feature names. We also mapped the cell lines to Research Resource Identifiers (RRID) from cell line database Cellosaurus (Bairoch, 2018) and derived names with lineage suffix (Supplementary Table S2). These standardization procedures allow us to maximize the identification of overlapping compounds and cell lines across datasets (Fig. 1).
Fig. 1.
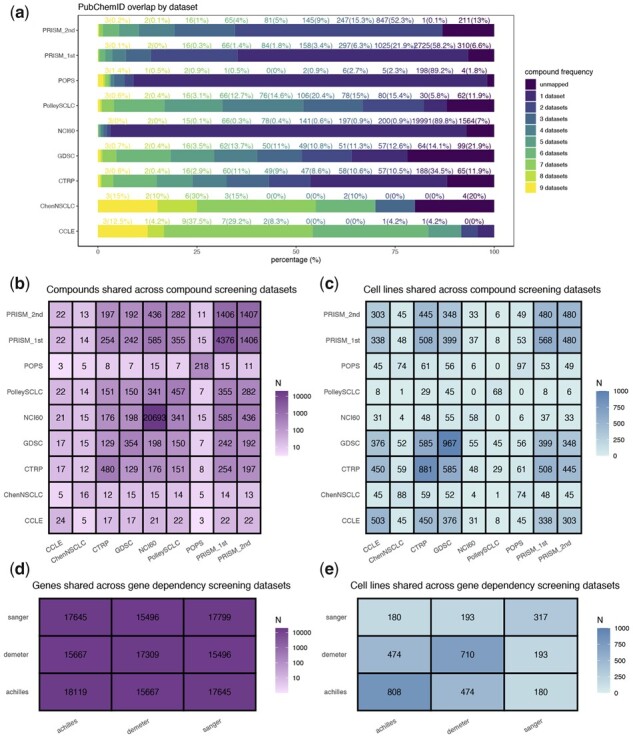
Summary of compound and dependency screening datasets. (a) Frequency of compound overlap across datasets. The percentage of overlapping compounds were colored differently by the number of overlapping datasets. Text above each color bar denotes compound frequency in counts (percentage) format. b-c, The number of overlapping compounds (b) or cell lines (c) across nine compound screening datasets. (d,e) The number of overlapping genes (d) or cell lines (e) across three gene dependency screening datasets
3.2 Compound screening data consistency is stable from intra-study and inter-study measures
Several compound screening datasets contain replicate measures for the same drug. We identified these replicates and assessed within-study consistency. As expected, the pairwise correlation from these replicates are generally more positive than all other pairwise correlations from the same dataset (Fig. 2a). We then assessed inter-study consistency by pairing all possible studies and performed correlation of functional data with the shared cell lines. Generally positive correlations were observed for all pairs of studies (Fig. 2b). However, for both intra-study and inter-study assessments, we observed that some compounds are always more consistent than others. We highlighted the EGFR inhibitor ‘Erlotinib’ as a more consistent example and c-Met inhibitor ‘PHA-665752’ as a less consistent example. For each example compound, similar degrees of consistency were seen across all pairwise assessments (Fig. 2c and d and Supplementary Fig. S2a and b). Using meta-analysis of the Pearson correlation coefficients, we generated ‘r.summary’ values for 1707 compounds that were found in at least two datasets (Fig. 2e). Erlotinib has an r.summary of 0.62, at the 98th percentile ranked by consistency, whereas PHA-665752 has an r.summary of 0.1 at the 40th percentile. We further compared the ‘r.summary’ from inter-study assessments to that from intra-study assessments on 384 compounds and observed good agreement (Fig. 2f). These findings suggest the degree of consistency is rather stable for compound screening data.
Fig. 2.

Compound screening data consistency is stable from intra-study and inter-study measures. (a) Within-study consistency of compound screening data assessed from replicates. For each of the five datasets, pairwise correlations were computed for all features. Comparison was made for correlations between replicates versus all other pairwise correlations. The number of pairwise correlations and the number of associated drugs under each type were provided on the plot as well. Density was scaled to have maximum at 1 for each plot. (b) Inter-study consistency of compound screening data. From the nine compound screening datasets, consistency was evaluated for 32 pairs of datasets (out of all 36 possible combinations, 4 pairs do not have enough overlapping cell lines for analysis). Distribution of pairwise correlation coefficients from all overlapping compounds in each pair of inter-study assessment were shown as density plots. Values for consistent example compound Erlotinib and inconsistent example compound PHA-665752 were marked as colored points. c-d, Forest plots showing meta-analysis of inter-study correlation for Erlotinib (c) or PHA-665752 (d). (e) Density plot showing distribution of inter-study consistency summary measure ‘r.summary’ for 1707 compounds. f, Scatterplot showing relationship between intra-study and inter-study ‘r.summary’ over 384 compounds. Pearson correlation coefficient is provided on the upper left corner. *, P-value < 0.05
3.3 Perturbations against functionally important targets result in more bimodally distributed data and better consistency across studies
We next examined gene dependency data. Replicates are not available from such studies, so we only examined inter-study consistency. We made the assessment separately for a group of cancer driver genes annotated as oncogenes by the COSMIC Cancer Gene Census (Sondka et al., 2018) versus all other genes. Consistency for these cancer genes is generally higher than the remaining genes (Fig. 3a). We examined the distribution of dependency scores for the top 10 most consistent cancer genes in all three datasets and observed skewed and bimodal distribution (Fig. 3b). In contrast, with the same kind of assessment for 10 inconsistent genes that do not belong to the oncogene panel, we did not observe such distribution (Fig. 3c). We reasoned that in functionally relevant subsets of cancer cell lines defined by a particular oncogenic driver, functional perturbation with drug or gene silencing against that pathway should result in biomodally distributed data. We derived the bimodal index (BI) as previously defined (Wang et al., 2009), for all genes in each datasets to represent the level of bimodal distribution. The relationship between inter-data consistency and bimodal distribution were assessed and comparison was made between cancer genes and the rest of the genes (Fig. 3d). We observed a positive correlation between the consistency measure ‘r.summary’ and bimodal indices from each of the three datasets, and these associations are more prominent for the cancer genes. Agreements among the bimodal indices from different datasets were also stronger for cancer genes (Fig. 3d). Similar relationships were observed for compound screening datasets as well (Supplementary Fig. S3a). In particular, in the dataset ‘PRISM_1st’ where compound annotations by disease area were available, we further compared the relationship in oncology drugs and non-oncology drugs. The data for oncology drugs were found to be more consistent with the other datasets and also more bimodally distributed (Supplementary Fig. S3b and c). These observations suggest that in functional screens, chemical or genetic perturbation of functionally important targets often result in bimodally distributed data and are more likely to be consistent across different studies. To see if the degree of consistency also associates with success rate of the drug, we also examined the compound consistency by market status (Supplementary Fig. S3d). Lower ‘r.summary’ was observed for compounds that were withdrawn or discontinued from the market, compared to those in the ‘approved’, ‘experimental’ or ‘investigation’ status (Siramshetty et al., 2016; Wishart et al., 2006).
Fig. 3.

Dependency inter-study consistency. (a) Density plots showing distribution of pairwise correlation coefficients from correlating gene effect scores among three dependency datasets. Comparison of correlations was made between oncogenes and all other genes (coral versus turquoise). (b) Density plots showing distribution of the 10 most consistent cancer genes in three dependency datasets. Samples were classified into low and high groups by Gaussian mixture modeling assuming equal variance, and the distribution of each group was plotted as well. (c) Density plots showing distribution of the 10 least consistent non-cancer genes in three dependency datasets. In both b and c, bimodal index (BI) was calculated and printed on the upper left corner of each plot. A higher BI value is indicative of a stronger bimodal distribution. (d) Pairwise scatterplot matrix showing the relationship between inter-study consistency measure ‘r.summary’ and BI from each of the three dependency datasets, with comparison made between oncogenes and all other genes (coral versus turquoise). Lower triangular panels show the actual datapoints with 2D density plots overlaid. Diagonal panels are distribution of each measure in the form of density plots. Upper triangular panels provide statistics from Pearson correlation. ***, P-value < 0.001
3.4 Transcriptomic association-based correlation provides a useful indirect assessment of functional data consistency
Having performed direct assessments by correlating response measurements from datasets, we also conducted indirect assessments by first deriving correlation between gene expression and functional screening data, then correlating the resulting correlation coefficients for all the genes, between pairs of datasets. This was performed with two methods, a standard approach using only the gene expression data and a copy number-adjusted gene expression data. We previously demonstrated that this ‘genomic regression analysis of coordinated expression’ (GRACE) denoising approach improves association analyses involving cancer transcriptomic data (Cai et al., 2017). In the analysis for the compound sensitivity screening datasets, we found that in some cases, indirect assessments resulted in better agreement than direct assessments, and such improvement is more dramatic by the GRACE method compared to the standard method (Fig. 4a). For example, vorinostat, a histone deacetylase (HDAC) inhibitor clinically used to treat cutaneous T-cell lymphoma has an r.summary of 0.37 from direct correlations, whereas this value increases to 0.56 from GRACE indirect correlations (Fig. 4b and c). In particular, the correlation between CTRP and GDSC datasets increased from 0.59 to 0.97. It turns out that CTRP and GDSC are the only two studies that have screened a large number of leukemia cell lines for sensitivity against vorinostat (Fig. 4d), and these cell lines of hematopoietic lineage were consistently sensitive in both screens (Fig. 4e). The association between vorinostat and gene expression from CTRP and GDSC are highly consistent, likely explained by the robust association between drug sensitivity and to lineage-specific gene expression patterns (Fig. 4f).
Fig. 4.
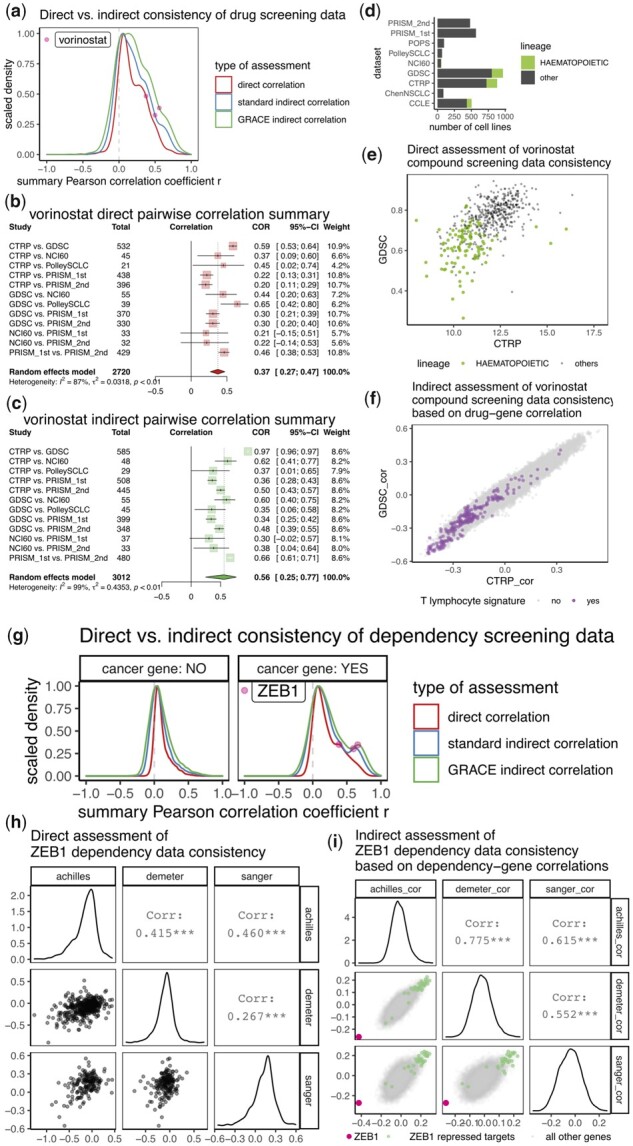
Comparison of direct and indirect assessment on functional screening datasets. (a) Density plot showing distribution of direct indirect consistency measures on 1707 compounds for inter-study consistency assessment. For indirect assessment, standard method uses RNA-seq expression data whereas GRACE method uses copy number-adjusted RNA-seq expression data. For each distribution, density was scaled to have maximum of 1. (b,c) Forest plots showing meta-analysis of inter-study consistency for vorinostat by direct (b) or indirect (c) assessment. While sample sizes for direct assessment were determined as the number of overlapping cell lines with non-missing functional data, the sample sizes for indirect assessment were determined as the number of overlapping cell lines with copy number-adjusted gene expression data. Note that pairwise correlation for CTRP versus GDSC increased from 0.59 in (b) to 0.97 in (c). (d) Number of cell lines belonging to hematopoetic or other cancer lineages for the nine compound screening datasets. (e) Scatterplot showing compound screening data (area under the dose response curve, AUC, lower value corresponds to higher sensitivity) for vorinostat from CTRP and GDSC. Cell lines with hematopoetic lineage are plotted as green dots. (f) Scatterplot showing values of correlation coefficients from association between vorinostat AUC data (from GDSC or CTRP) and copy number-adjusted transcriptomic data. Genes mapped to a T lymphocyte gene signature from ‘LEE_DIFFERENTIATING_T_LYMPHOCYTE’ (Lee et al., 2004) were plotted as purple dots. (g) Density plot showing distribution of direct indirect consistency measures on 17 966 genes for inter-study consistency assessment. Assessments were made separately for cosmic defined oncogenes and all other genes. (h) Scatterplot matrix showing relationship among gene effect data (lower value corresponds to higher lethality) for ZEB1 from all three dependency datasets. (i) scatterplot matrix showing values of correlation coefficients from association between ZEB1 deactivation effect data and copy number-adjusted transcriptomic data. Datapoints for ZEB1 and ZEB1 repressed targets were colored. In both (f) and (i), negative correlations suggest higher gene expression is associated with higher sensitivity
We also performed comparison of direct and indirect correlations for dependency screening datasets. Similar results were observed. The improvement on consistency is especially prominent for oncogenes (Fig. 4g). We provided a detailed example analysis for ZEB1, a transcriptional repressor for epithelial genes (Fig 4g–i). The r.summary from direct correlation is 0.386, and it increased to 0.661 in the GRACE indirect correlation. In all three dependency datasets, ZEB1 expression most negatively correlated with the ZEB1 dependency scores, whereas target genes of ZEB1 are mostly positively correlated with the ZEB1 dependency scores (Fig. 4i).
3.5 A Functional data consistency explorer
We constructed a web application, ‘Functional Data Consistency Explorer’ (FDCE, at https://lccl.shinyapps.io/FDCE/), to provide tools for users to assess data consistency on a per-feature basis. Tutorial slides are provided to introduce users to the three main functions of the app—‘Overall Consistency’ for reviewing the consistency measures we derived with global and feature-specific views; ‘Pairwise Scatterplots’ for reviewing the actual pairwise relationship between datasets; and ‘Datasets’ for view and downloading data and metadata used in this study.
In the ‘Overall Consistency’ page, users can examine and download the overall consistency results for 1707 compounds across 9 compound screening datasets or 17 966 genes across 3 dependency screening datasets, with both the direct and indirect assessments we described in this paper (Fig. 5). As an example to showcase the web application, we highlight vorinostat (as examined in Fig. 4a–f). Upon submission of this feature of interest, and specification of the type of input correlation for meta-analysis, the density plots are updated to mark the vorinostat inter-study correlation coefficients. This also generates a forest plot from meta-analysis and pulls out a table to display all inter-study pairwise correlations for this feature. Along with this example, we also examined belinostat, a compound with similar mechanism of actions (moa) (Supplementary Fig. S4a). Similar degree of consistency could be found for bellinostat, and like vorinostat, between CTRP and GDSC datasets, the indirect correlation is also much higher than direct correlation. Similar analyses could also be performed for dependency screening results, as shown in Supplementary Figure S4b.
Fig. 5.

Snapshot taken from the FDCE web application for assessment of compound data consistency. Under the ‘Overall Consistency’ tab panel, users may choose to assess data consistency for compound or dependency screening data. Both distribution of direct and indirect (by GRACE method) assessment results are visualized. The top density plot panel represents distribution of ‘r.summary’, summarized from all inter-study correlations, whereas the bottom panel with multiple subplots visualizes distribution for each study pair. Upon submission of a feature to highlight and specification of the type of input pairwise correlation for meta-analysis, the datapoints corresponding to the inter-study correlation coefficients associated with the feature of interest are added to the plots and a forest plot showing results from meta-analysis will be shown below. On the right side of the web page, the top table lists the consistency summary statistics for all overlapping features across the dataset, as well as additional information about the features. Left of the table are column options for display. In this particular table, as vorinostat is a HDAC inhibitor, we checked to display the ‘moa’ and searched ‘chromatin’ as a keyword to identify additional compounds with similar moa. The bottom table contains pairwise correlation results for all study pairs specific to the feature of interest and is only rendered when user submits a feature to highlight. Annotations for column names in overall consistency table for drug screening data: ‘target’, the drug target; ‘target.source’, the source of ‘target’ annotation; ‘moa’, mechanism of drug action; ‘moa.source’, source of ‘moa’ annotation; the ‘direct’ statistics are from direct correlations of functional screening data; the ‘indirect’ statistics are from correlations of correlations between copy number-adjusted gene expression data and functional screening data; ‘pairs’ refer to the number of dataset pairs for which correlation was calculated; ‘meta-analysis’ indicates whether the feature-specific statistics is based on meta-analysis. Annotations for column names in pairwise consistency table for a specific drug: ‘cid’, PubChem ID; ‘unique_Drug_Name.1’ or ‘unique_Drug_Name.2’, compound name used for ‘dat1’ or ‘dat2’, note that when replicates exist for the same study, these names will end with ‘(n)’; ‘n.cell’, number of cell lines in the pairwise correlation; ‘n.feature’, number of features overlapping for the two datasets
From the ‘Pairwise Scatterplots’ tab panel, users may choose a compound or gene feature of interest and visualize the relationship among the different measures of this feature across different datasets in the form of a scatterplot matrix with Pearson correlation statistics. In the interactive scatterplots, cell lines are colored by the cancer lineage type. We also visualize the distribution of each measure with model-based dichotomization and calculate the associated bimodal index (Supplementary Fig. S5).
All the processed datasets used for this paper and FDCE web application can be previewed and downloaded from the ‘Datasets’ tab panel (Supplementary Fig. S6).
4 Discussion
In this study, we integrated multiple functional screening datasets for assessment of consistency. Across datasets, we observed that some features are more consistent than others and that the degree of consistency is rather stable. There could be several underlying reasons for consistent inconsistency. Variations in experimental conditions, such as cell growth conditions or types of assays, are a common concern. In the case of compound screening, drug concentration ranges and definition of output measures could affect data reproducibility (Hatzis et al., 2014). In the case of dependency screening, discordance could arise from differences in gene deactivation approach (RNAi versus CRISPR-Cas9) (Lin and Sheltzer, 2020), differences in sgRNA library and duration of the screen (Dempster et al., 2019). But we believe a key reason is that many targets are functionally unimportant, therefore many measurements are idiosyncratic within an individual experiment rather than true signals. This is supported by our observation that compound screening data are more consistent for oncology drugs and gene dependency screening data are more consistent for oncogenes. Therefore, for wet lab scientists who want to pursue a compound or gene of interest with functional studies, the degree of data consistency could help them gauge the necessity of careful assay optimization and the validity of the target itself in the in vitro setting. For dry lab scientists who devise algorithms to predict functional outcomes, the degree of data consistency could help set the expectation for prediction accuracy.
In our compound screening dataset collection, we have three lung cancer-specific datasets and six pan-cancer datasets. This predominance is a result of the successful lung cancer cell line development and research efforts (Gazdar et al., 2010) in confrontation with this most deadly cancer. In an attempt to evaluate data consistency in a lineage-specific manner, we found the consistency measures are inadvertently noisier for lineage-specific subsets compared to those from complete pan-cancer datasets, due to the much smaller sample size (Supplementary Fig. S7). For this reason, we based our analyses on the full datasets.
We also used the correlation between functional data and denoised gene expression data to derive indirect consistency measures. As gene expression data are often used for chemosensitivity prediction (Staunton et al., 2001); and association between basal gene expression data and functional screening data can also elucidate the moa (Rees et al., 2016), this indirect measure of consistency may help infer the likelihood of establishing successful predictive signatures or identification of bona fide mechanism of action. In addition, missing values are common in functional screening data. For certain features, this could result in a very small overlap of cell lines with direct measures. With gene expression data available for most of the cell lines used for functional screens, indirect assessment also partly alleviates this problem.
In conclusion, assessment of data consistency should be made on a per-feature basis. We demonstrated the importance and feasibility of such approach and devised web tools for users to make such assessments and develop further insights.
Supplementary Material
Acknowledgements
The authors thank Ms. Jessie Norris for proofreading the manuscript.
Funding
This study was supported by funding from the National Institutes of Health [P30CA142543, P50CA70907, R35CA22044901 and R35GM136375] and the Cancer Prevention and Research Institute of Texas [RP190107 and RP180805].
Conflict of Interest: J.D.M. receives licensing fees from the National Cancer Institute and UT Southwestern to distribute cell lines.
Data availability
Processed data used for this paper and the web application can be downloaded from https://lccl.shinyapps.io/FDCE/ under the ‘Datasets’ section. Source codes for the FDCE web tool are available at https://github.com/cailing20/FDCE, the input data for FDCE is available at https://doi.org/10.5061/dryad.95x69p8kq.
Contributor Information
Ling Cai, Quantitative Biomedical Research Center, Department of Population and Data Sciences, UT Southwestern Medical Center, Dallas, TX 75390, USA; Children’s Research Institute, UT Southwestern Medical Center, Dallas, TX 75390, USA; Simmons Comprehensive Cancer Center, UT Southwestern Medical Center, Dallas, TX 75390, USA.
Hongyu Liu, Quantitative Biomedical Research Center, Department of Population and Data Sciences, UT Southwestern Medical Center, Dallas, TX 75390, USA.
John D Minna, Simmons Comprehensive Cancer Center, UT Southwestern Medical Center, Dallas, TX 75390, USA; Hamon Center for Therapeutic Oncology Research, UT Southwestern Medical Center, Dallas, Dallas, TX 75390, USA; Department of Pharmacology, UT Southwestern Medical Center, Dallas, TX 75390, USA; Department of Internal Medicine, University of Texas Southwestern Medical Center, Dallas, TX 75390, USA.
Ralph J DeBerardinis, Children’s Research Institute, UT Southwestern Medical Center, Dallas, TX 75390, USA; Howard Hughes Medical Institute, University of Texas Southwestern Medical Center, Dallas, TX 75390, USA.
Guanghua Xiao, Quantitative Biomedical Research Center, Department of Population and Data Sciences, UT Southwestern Medical Center, Dallas, TX 75390, USA; Simmons Comprehensive Cancer Center, UT Southwestern Medical Center, Dallas, TX 75390, USA; Department of Bioinformatics, University of Texas Southwestern Medical Center, Dallas, TX 75390, USA.
Yang Xie, Quantitative Biomedical Research Center, Department of Population and Data Sciences, UT Southwestern Medical Center, Dallas, TX 75390, USA; Simmons Comprehensive Cancer Center, UT Southwestern Medical Center, Dallas, TX 75390, USA; Department of Bioinformatics, University of Texas Southwestern Medical Center, Dallas, TX 75390, USA.
References
- Aigner K. et al. (2007) The transcription factor ZEB1 (deltaEF1) promotes tumour cell dedifferentiation by repressing master regulators of epithelial polarity. Oncogene, 26, 6979–6988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bairoch A. (2018) The Cellosaurus, a cell-line knowledge resource. J. Biomol. Technol., 29, 25–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barretina J. et al. (2012) The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature, 483, 603–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basu A. et al. (2013) An interactive resource to identify cancer genetic and lineage dependencies targeted by small molecules. Cell, 154, 1151–1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai L. et al. (2017) Genomic regression analysis of coordinated expression. Nat. Commun., 8, 2187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Cell Line Encyclopedia and Genomics of Drug Sensitivity in Cancer. (2015) Pharmacogenomic agreement between two cancer cell line data sets. Nature, 528, 84–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen P.H. et al. (2019) Metabolic diversity in human non-small cell lung cancer cells. Mol. Cell, 76, 838–851.e835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corsello S.M. et al. (2020) Discovering the anti-cancer potential of non-oncology drugs by systematic viability profiling. Nat. Cancer, 1, 235–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dempster J.M. et al. (2019) Agreement between two large pan-cancer CRISPR-Cas9 gene dependency data sets. Nat. Commun., 10, 5817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DepMap 20Q4 Public. (2020) doi:10.6084/m9.figshare.13237076.v2.
- Gazdar A.F. et al. (2010) Lung cancer cell lines as tools for biomedical discovery and research. J. Natl. Cancer Inst., 102, 1310–1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghandi M. et al. (2019) Next-generation characterization of the Cancer Cell Line Encyclopedia. Nature, 569, 503–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haibe-Kains B. et al. (2013) Inconsistency in large pharmacogenomic studies. Nature, 504, 389–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatzis C. et al. (2014) Enhancing reproducibility in cancer drug screening: how do we move forward? Cancer Res., 74, 4016–4023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haverty P.M. et al. (2016) Reproducible pharmacogenomic profiling of cancer cell line panels. Nature, 533, 333–337. [DOI] [PubMed] [Google Scholar]
- Iorio F. et al. (2016) A landscape of pharmacogenomic interactions in cancer. Cell, 166, 740–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S. et al. (2016) PubChem substance and compound databases. Nucleic Acids Res., 44, D1202–1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee M.S. et al. (2004) Gene expression profiles during human CD4+ T cell differentiation. Int. Immunol., 16, 1109–1124. [DOI] [PubMed] [Google Scholar]
- Lin A., Sheltzer J.M. (2020) Discovering and validating cancer genetic dependencies: approaches and pitfalls. Nat. Rev. Genet., 21, 671–682. [DOI] [PubMed] [Google Scholar]
- McFarland J.M. et al. (2018) Improved estimation of cancer dependencies from large-scale RNAi screens using model-based normalization and data integration. Nat. Commun., 9, 4610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMillan E.A. et al. (2018) Chemistry-first approach for nomination of personalized treatment in lung cancer. Cell, 173, 864–878.e829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgens D.W. et al. (2016) Systematic comparison of CRISPR/Cas9 and RNAi screens for essential genes. Nat. Biotechnol., 34, 634–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picco G. et al. (2019) Functional linkage of gene fusions to cancer cell fitness assessed by pharmacological and CRISPR-Cas9 screening. Nat. Commun., 10, 2198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polley E. et al. (2016) Small Cell Lung Cancer Screen of Oncology Drugs, Investigational Agents, and Gene and microRNA Expression. J. Natl. Cancer Inst., 108, djw122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rees M.G. et al. (2016) Correlating chemical sensitivity and basal gene expression reveals mechanism of action. Nat. Chem. Biol., 12, 109–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinhold W.C. et al. (2012) CellMiner: a web-based suite of genomic and pharmacologic tools to explore transcript and drug patterns in the NCI-60 cell line set. Cancer Res., 72, 3499–3511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Safikhani Z. et al. (2016) Assessment of pharmacogenomic agreement. F1000Res, 5, 825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanger CRISPR (CERES). (2019) doi:10.6084/m9.figshare.9116732.
- Schulze R.M.-A. (2004) A Comparison of Approaches. Hogrefe Publishing, Germany. [Google Scholar]
- Seashore-Ludlow B. et al. (2015) Harnessing connectivity in a large-scale small-molecule sensitivity dataset. Cancer Discov., 5, 1210–1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siramshetty V.B. et al. (2016) WITHDRAWN–a resource for withdrawn and discontinued drugs. Nucleic Acids Res., 44, D1080–1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sondka Z. et al. (2018) The COSMIC Cancer Gene Census: describing genetic dysfunction across all human cancers. Nat. Rev. Cancer, 18, 696–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staunton J.E. et al. (2001) Chemosensitivity prediction by transcriptional profiling. Proc. Natl. Acad. Sci. USA, 98, 10787–10792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A. et al. (2005) Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA, 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tlemsani C. et al. (2020) SCLC_CellMiner: integrated genomics and therapeutics predictors of small cell lung cancer cell lines based on their genomic signatures. Cell Rep., 33, 108296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J. et al. (2009) The bimodality index: a criterion for discovering and ranking bimodal signatures from cancer gene expression profiling data. Cancer Inform., 7, 199–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wishart D.S. et al. (2006) DrugBank: a comprehensive resource for in silico drug discovery and exploration. Nucleic Acids Res., 34, D668–672. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Processed data used for this paper and the web application can be downloaded from https://lccl.shinyapps.io/FDCE/ under the ‘Datasets’ section. Source codes for the FDCE web tool are available at https://github.com/cailing20/FDCE, the input data for FDCE is available at https://doi.org/10.5061/dryad.95x69p8kq.