

Abstract
Frontotemporal dementia (FTD) and amyotrophic lateral sclerosis (ALS) are neurodegenerative disorders characterized by declining motor and cognitive functions. Even though these diseases present with distinct sets of symptoms, FTD and ALS are two extremes of the same disease spectrum, as they show considerable overlap in genetic, clinical and neuropathological features. Among these overlapping features, mitochondrial dysfunction is associated with both FTD and ALS. Recent studies have shown that cells derived from patients’ induced pluripotent stem cells (iPSC)s display mitochondrial abnormalities, and similar abnormalities have been observed in a number of animal disease models. Drosophila models have been widely used to study FTD and ALS because of their rapid generation time and extensive set of genetic tools. A wide array of fly models have been developed to elucidate the molecular mechanisms of toxicity for mutations associated with FTD/ALS. Fly models have been often instrumental in understanding the role of disease associated mutations in mitochondria biology. In this review, we discuss how mutations associated with FTD/ALS disrupt mitochondrial function, and we review how the use of Drosophila models has been pivotal to our current knowledge in this field.
Keywords: Drosophila, neurodegeneration, mitochondria, ALS, FTD
Introduction
Frontotemporal dementia (FTD) is the second most common early onset dementia, with Amyotrophic lateral sclerosis (ALS) representing the most prevalent motor neuron disease. While the two diseases are diagnosed based on distinct sets of symptoms, recent evidence suggests that they can in fact be considered two ends of the same disease spectrum, sharing genetic risk factors and neuropathological hallmarks (Ferrari et al., 2011; Van Blitterswijk et al., 2012). 15% of FTD patients also present with ALS, and 50% of ALS patients go on to develop FTD. In certain familial cases, different members of the same family will go on to develop either FTD or ALS (Martinaud et al., 2005). Pathologically, both diseases are characterized by intracellular proteinaceous inclusions, most commonly formed of the DNA/RNA binding protein TDP43, although aggregates can also be composed of superoxide dismutase 1 (SOD1), fused in sarcoma (FUS) or microtubule-associated protein tau (MAPT) (Ling et al., 2013; Turner et al., 2017), amongst others.
Amyotrophic lateral sclerosis, also called Lou Gehrig’s disease, is clinically characterized by the gradual loss of upper and lower motor neurons, eventually leading to death by respiratory failure. The average life expectancy is 2–5 years after diagnosis; however, 5–10% patients can survive up to 10 years after developing clinical symptoms (Millul et al., 2005; Mateen et al., 2010). There is no cure for ALS and the drugs that have been approved so far, such as Riluzole and Edavarone (Bensimon et al., 1994; Lacomblez et al., 1996; Pandya et al., 2013; Nguyen et al., 2018; Brown et al., 2020), only prolong life by 3–6 months (Nguyen et al., 2018). The overall global incidence of ALS is 1.59 in 100,000 people whilst the prevalence is 4.42 in 100,000 (Xu et al., 2020). The average age of onset is between 55 and 65 years of age, but for familial forms earlier ages of onset have been recorded (Orsini et al., 2015). Late-onset ALS is more prevalent in males than females, but the sex ratio is quite similar at younger ages (Manjaly et al., 2010). Sporadic ALS (sALS), in which no inherited mutation directly causes disease, accounts for 95% of cases. Conversely, familial ALS (fALS), accounting for the remaining 5% of cases, is associated with a number of genetic mutations which usually show a pattern of autosomal dominant inheritance (Byrne et al., 2011). More than 40 genes have been identified as carrying mutations causing ALS including angiogenin (ANG), matrin3 (MATR3) and vesicle-associated membrane protein (VAMP)-associated protein B (VAPB) (Alsultan et al., 2016; Gregory et al., 2020). Among these, the gene most commonly associated with pure ALS is SOD1, accounting for 20% of identified fALS cases (Orsini et al., 2015). Several other ALS-associated mutations lie in genes that have also been associated with FTD, including ataxin-2 (ATXN2), cyclin F (CCNF), Coiled-Coil-Helix-Coiled-Coil-Helix Domain Containing 10 (CHCHD10), chromosome 9 opening reading frame 72 (C9orf72), Dynactin (DCTN1), FUS, Optineurin (OPTN), TAR DNA binding protein (TARDBP or TDP-43), TANK-binding Kinase (TBK1), T cell-restricted intracellular antigen-1 (TIA1), Tubulin alpha 4A (TUBA4A) and valosin-containing protein (VCP) (Orr, 2011; Orsini et al., 2015; Greaves and Rohrer, 2019; Table 1). The intronic hexanucleotide repeat expansion (GGGGCC) in the C9orf72 gene (C9), is the most common genetic cause of both ALS and FTD, despite being identified only a decade ago (DeJesus-Hernandez et al., 2011; Renton et al., 2011).
TABLE 1.
Major genes associated with ALS and FTD.
| Locus | Genes | ALS/FTD | Gene frequency | Mutations | Protein inclusions | Sources |
| 14q11.2 | ANG | ALS | <1% ALS | Missense | TDP43 | Greenway et al., 2006 |
| 5q31.2 | MATR3 | ALS | 1% sALS 1-2% fALS | Missense | TDP43 | Johnson et al., 2014; Nguyen et al., 2018 |
| 21q22.11 | SOD1 | ALS | 5% sALS 20% fALS | Missense | SOD1/p62 | Rosen et al., 1993; Shaw et al., 1998 |
| 20q13.32 | VAPB | ALS | <1% ALS | Missense | Nishimura et al., 2004; Millecamps et al., 2010 | |
| 12q24.12 | ATXN2 | ALS-FTD | 4.7% ALS | CAG repeat | TDP-43 | Elden et al., 2010; Bäumer et al., 2014; Rubino et al., 2019 |
| 16p13.3 | CCNF | ALS-FTD | <1% sALS 0.6–3.3% fALS <4% FTD | Missense | TDP43 | Williams et al., 2016; Nguyen et al., 2018 |
| 22q11.23 | CHCHD10 | ALS-FTD | <1% sALS 2% fALS <1% FTD | Missense/nonsense | TDP43 | Bannwarth et al., 2014; Nguyen et al., 2018 |
| 9p21.2 | C9orf72 | ALS-FTD | 6% sALS 37% fALS 5% sFTD 25% fFTD | G4C2 repeat expansion | TDP43/p62/dipeptides repeat/ubiquitin | DeJesus-Hernandez et al., 2011; Renton et al., 2011; Majounie et al., 2012 |
| 2p13.1 | DCTN1 | ALS-FTD | <1% sALS <1% fALS | Missense | TDP43 | Puls et al., 2003; Münch et al., 2004, 2005 |
| 16p11.2 | FUS | ALS-FTD | ∼1% sALS 3% fALS <1% FTD | Missense/nonsense | FUS/p62 | Kwiatkowski et al., 2009; Vance et al., 2009; Van Langenhove et al., 2012 |
| 10p13 | OPTN | ALS-FTD | <1% ALS 4% FTD | Missense/deletion/nonsense | TDP43/p62 | Maruyama et al., 2010; Van Langenhove et al., 2012; Pottier et al., 2015 |
| 5q35 | SQSTM1/p62 | ALS-FTD | 2–3% ALS 2–3% FTD | Missense/nonsense/deletion | TDP43/p62 | Fecto and Siddique, 2011; Elisa et al., 2012; Le Ber et al., 2013 |
| 12q14.2 | TBK1 | ALS-FTD | <1% sALS 3% fALS <1% FTD | Missense/nonsense | TDP43/p62 | Cirulli et al., 2015; Freischmidt et al., 2015; Nguyen et al., 2018; Lamb et al., 2019 |
| 2p13.3 | TIA1 | ALS-FTD | <1% sALS 2.2% fALS <1% FTD | Missense | TDP43/p62 | Mackenzie et al., 2017; Nguyen et al., 2018 |
| 2q35 | TUBA4A | ALS-FTD | <1% sALS 1% fALS <1% FTD | Missense/nonsense | TDP43 | Smith et al., 2014; Nguyen et al., 2018 |
| 9p13.3 | VCP | ALS-FTD | 1–2% fALS <1% FTD | Missense | TDP43/p62 | Watts et al., 2004; Johnson et al., 2010 |
| Xp11.21 | UBQLN2 | ALS-FTD | 2% ALS <1% FTD | Missense | TDP43/p62/ubiquitin/FUS/OPTN | Deng et al., 2011; Synofzik et al., 2012 |
| 3p11.2 | CHMP2B | FTD | <1% ALS <1% FTD | Missense/nonsense | Ubiquitin/P62 | Skibinski et al., 2005; Parkinson et al., 2006; Van Langenhove et al., 2012 |
| 17q21.31 | GRN | FTD | 1–5% sFTD 5–20% fFTD | Missense/nonsense/splice site/frameshift | TDP-43 | Baker et al., 2006; Cruts et al., 2006; Rademakers et al., 2012 |
| 17q21.2 | MAPT | FTD | 5–20% fFTD | Missense/splice site/deletion | Tau | Hutton et al., 1998; Rademakers et al., 2012 |
Characteristics of major genes associated with ALS or FTD. Table is adapted from Hardy and Rogaeva (2014).
Frontotemporal dementia, also called Frontotemporal Lobar Degeneration (FTLD) and formerly known as Pick’s disease, is a common early onset dementia (Ling et al., 2013). The prevalence is 15–22 per 100,000 individuals and the incidence is 2.7–4.1 per 100,000 individual per year (Onyike and Diehl-Schmid, 2013). The age of onset for FTD is usually below the age of 65 (Mercy et al., 2008; Seltman and Matthews, 2012). Clinically, FTD is characterized by atrophy of the frontal or temporal lobes and, depending on the initial presenting symptom, can been categorized as behavioral variant FTD (bv-FTD) if associated with changes of personality or behavior, or primary progressive aphasia (PPA) if associated with language impairment (Seltman and Matthews, 2012; Van Langenhove et al., 2012; Warren et al., 2013). There is no cure to date and no approved drugs to treat symptoms specific to this disease (Seltman and Matthews, 2012). The sporadic form of FTD accounts for 70% of cases, whilst familial forms accounts for 30% of the cases (Turner et al., 2017). The major genes linked with FTD are C9orf72, MAPT, progranulin (GRN) and chromatin modifying protein 2B (CHMP2B) (Turner et al., 2017; Casterton et al., 2020). Less common mutations have also been found in VCP, Sequestosome 1 (SQSTM1), OPTN and ubiquilin-2 (UBQLN2) (Table 1).
An increasing array of cellular processes have been linked to the development of both ALS and FTD (Mejzini et al., 2019; Liscic et al., 2020). One of the first mechanisms implicated in ALS development was excitotoxicity, caused by inappropriate elevation of intracellular sodium and calcium levels in response to sustained neuronal stimulation (reviewed in King et al., 2016). This disease mechanism is targeted by Riluzole, which blocks sodium currents and therefore reduces neuronal excitability. In the clinic, however, Riluzole only increases lifespan of ALS patients by a few months (Nguyen et al., 2018). More recent studies, fueled by the identification of a growing number of risk genes, have implicated other cellular pathways that could represent alternative therapeutic targets. For instance, many of the mutations associated with FTD/ALS fall in genes implicated in protein degradation and vesicle transport; impairments in these pathways could potentially underlie the observed accumulation of protein deposits in patients’ brains (for a review see Shahheydari et al., 2017). Similarly, impairments in RNA metabolism have been implicated by the RNA binding function of the disease-associated proteins TDP-43 and FUS. These proteins both translocate to the cytoplasm in disease, leading to disruptions in transcription and splicing (Lagier-Tourenne et al., 2010; Mackenzie et al., 2010). In addition, RNA metabolism dysfunction has also been linked to other disease-associated mutations such as C9 (Cooper-Knock et al., 2014). A number of other cellular processes are also disrupted: nucleocytoplasmic transport defects (Kim and Taylor, 2017; Fallini et al., 2020), impaired DNA repair (Sun et al., 2020), axonal transport defects, and cytoskeletal disorganization have all been described (Bilsland et al., 2010; Alami et al., 2014).
Increasingly, mitochondrial dysfunction has been implicated in disease progression: several FTD/ALS linked proteins (described in detail below) interact with mitochondria (Bannwarth et al., 2014; Tsai et al., 2020), and findings from both patient tissue and animal models link mitochondria dysfunction to FTD/ALS pathogenesis (Bannwarth et al., 2014; Nakaya and Maragkakis, 2018; Tsai et al., 2020). Moreover, mitochondrial impairments have been associated with other neurodegenerative diseases such as Alzheimer’s disease (AD) and Parkinson’s disease (PD) (Bosetti et al., 2002; Abou-Sleiman et al., 2006; Anandatheerthavarada and Devi, 2007; Cozzolino and Carrì, 2012), potentially suggesting some common pathogenicity across multiple disorders.
Neurons appear to be particularly susceptible to mitochondrial defects because of their high energetic demands (which almost exclusively have to be met by oxidative phosphorylation), their unique architecture with extremely long processes (axons can be longer than 1m in humans), and the fact they are extremely long-lived post-mitotic cells which cannot dilute out defective organelles by cell division. The high energetic demands of neurons are evident from the fact that the human brain contains about 2% of the body mass but requires about 20% of the oxygen consumption and energy needs of the whole body (Seager et al., 2020). As mitochondria are the main ATP producer within a cell (Figure 1), neurons have an abundance of mitochondria to meet their ATP requirements, which allow them to perform various cellular functions and activities (Kann and Kovács, 2007). If mitochondrial function is impaired, ATP production is not only diminished, but impaired oxidative phosphorylation (OXPHOS) can also generate inappropriately high concentrations of reactive oxygen species (ROS) that can further damage mitochondria (Bhatti et al., 2017) and eventually lead to neuronal death.
FIGURE 1.
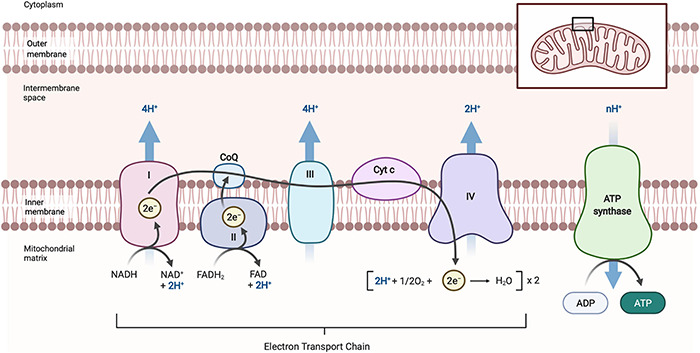
Schematic diagram of oxidative phosphorylation in mitochondria. Oxidative phosphorylation occurs at the inner mitochondrial membrane and is carried out by the electron transport chain, comprised of Complex I, II, III, IV, and ATP synthase. NADH (CI) and FADH (CII) donate electrons which then move along CoQ, CIII, cyt c and then CIV in a series of redox reaction that provide energy for the transfer of H+ ions across the intermembrane space to form an electrochemical gradient. Oxygen (O2), as the final electrons acceptor, is reduced to form water. The electrochemical gradient created in this process leads to H+ flowing back across the inner mitochondrial through ATP synthase, driving the phosphorylation of ADP to ATP. NAD, β-nicotinamide adenine dinucleotide; NADH, β-nicotinamide adenine dinucleotide 2-phosphate reduced form; FAD, flavin adenine dinucleotide; FADH, flavin adenine dinucleotide reduced from; CoQ, coenzyme Q (ubiquinone); CI, complex I; CII, complex II; CIII, complex III; CIV, complex IV; cyt c, cytochrome c; ADP, adenosine diphosphate; ATP, adenosine triphosphate. Images created with BioRender.com.
To investigate the roles of mitochondrial dysfunction and other disease mechanisms, both patient-derived cells and animal models have been essential scientific tools (Vandamme, 2014; Roberts and Speirs, 2017; Kim et al., 2020). With the advent of GWAS studies a multitude of mutations have been linked to disease development (Witte, 2010; Tam et al., 2019). Confirming that these mutations cause disease, and understanding how they lead to neurodegeneration, has often required the generation of animal models carrying these mutations. These models have allowed the field to build an initial understanding of how disease-associated mutations lead to declining organismal health generally and neurodegeneration specifically. Among animal models, the fruit fly Drosophila has proven invaluable in modeling neurodegenerative disease. Over 20 years ago, the first fly models of neurodegeneration showed, crucially, that over-expression of human disease genes in flies could not only lead to neurodegeneration (Warrick et al., 1998) but also mimic the main characteristics of the disease (Feany and Bender, 2000). Since then, a host of neurodegenerative disease models have been developed in flies, which have provided critical insights into disease pathology.
The success of flies stems from the fact that flies require little space, have short life cycles with abundant offspring, and are easy and inexpensive to breed and maintain in large numbers (providing strong statistical power) (Ashburner and Bergman, 2005; Figure 2). In this review, we will give a general overview of Drosophila as a model system to study FTD/ALS, with a focus on how Drosophila has been a powerful tool to study mitochondrial dysfunction in models of these diseases.
FIGURE 2.
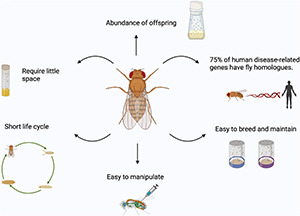
Advantages of using Drosophila as a model organism. Fruit flies are easy to breed and maintain in large numbers, easy to manipulate, require little space, have short life cycle with abundant offspring and about 75% of human disease-related genes have fly homologs. Images created with BioRender.com.
Drosophila as a Model System
Drosophila Biology
Since Thomas H. Morgan used Drosophila in his ‘Fly Room’ at Columbia University to study genetic inheritance in 1910, Drosophila has been widely used as a model organism. Thomas H. Morgan won the Nobel Prize in Physiology or Medicine in 1933 for formulating the chromosomal theory of inheritance, based on his findings looking at patterns of inheritance of the white eye pigment in flies. To date, 6 Nobel Prizes in Physiology or Medicine have been awarded to 10 researchers using flies in their research including Thomas’ proteges Allan Sturtevant, Calvin Bridges and Hermann Muller (Zhang K. et al., 2018). These trailblazing studies underscore the fundamental contribution of this unassuming model organism to our current understanding of many aspects of biology.
Drosophila development and physiology are surprisingly similar to those in more complex eukaryotes. For example, Drosophila has a similar dorsoventral patterning to vertebrates with the ventral area of Drosophila being analogous to the dorsal area in vertebrates (De Robertis and Sasai, 1996). Moreover, homologous genes control the patterning of the organism. Among these, decapentaplegic (dpp) is expressed in the dorsal region of Drosophila, whereas the vertebrate homolog, bone morphogenetic protein 4 (bmp-4), is expressed in the ventral region. Similarly, short gastrulation (sog) in Drosophila is expressed in the ventral region, whilst it vertebrate homolog, chordin (chordin), is expressed in the dorsal region (Holley et al., 1995). Drosophila also shows strong similarity to vertebrates in organ development: Hedgehog (Hh) in Drosophila, has a similar limb patterning function as Sonic hedgehog (SHH) in vertebrates (Riddle et al., 1993), and Paired Box 6 (Pax6) in both flies and vertebrates has analogous functions in eye development (Wawersik and Maas, 2000). Finally, the components of major signaling pathways, such as Wingless and Notch, are also conserved between flies and vertebrates (Baker, 1987; Fleming, 1998; Bejsovec, 2018). The conservation extends to genes and signaling pathways modulating longevity and aging, a field in which Drosophila has provided fundamental insights (Piper and Partridge, 2018). For instance, the discovery that chico (insulin-receptor substrate) and InR (insulin-like receptor) mutants extended lifespan in flies (Clancy et al., 2001; Tatar et al., 2001) prompted subsequent studies confirming similar findings in vertebrate models (Selman et al., 2008). Metabolic pathways are also highly conserved between mammals and Drosophila, making flies a valuable model system for metabolism research such as in diabetes and obesity (Baker and Thummel, 2007; Bharucha, 2009).
Flies are also the simplest model organism extensively used in research with a complex body pattern and many organs are analogous to those in humans. For example, flies have a gut, a beating heart, and a clearly defined central and peripheral nervous systems, allowing them to model multisystem phenotypes associated with disease mutations. These features have enabled Drosophila to provide numerous insights in the study of complex human diseases such as cancer, cardiovascular diseases, and neurodegenerative diseases. For instance, Drosophila has been used to identify and screen drugs for cancer therapeutics (Hirth, 2010; Bangi et al., 2019), and to study embryonic cardiac development and adult cardiac performance (Piazza and Wessells, 2011).
Drosophila has also been used extensively to study neurodegenerative diseases. Although the Drosophila brain is not as anatomically complex as the mammalian one, the cellular and molecular characteristics of the nervous system are broadly similar. At the cellular level, Drosophila and mammalian nervous systems both possess functionally distinct categories of neuronal and glial cells, and both possess a blood brain barrier (Stork et al., 2012; Kasture et al., 2018). Flies also display a range of complex behaviors such as sleep, learning and memory, courtship, and navigation. Importantly, these behaviors decline with age and in the presence of toxic genes associated with disease in humans, making flies a useful tool to study neurodegenerative diseases (Buchanan and Benzer, 1993; Jackson et al., 1998; Warrick et al., 1998).
At the genome level, Drosophila is simpler than vertebrate models, with only 4 chromosomes and 13,821 genes. However, the completed sequencing of the Drosophila genome has shown that about three-quarter of human disease-related genes have homologs in Drosophila (Reiter et al., 2001). Moreover, the Drosophila genome has relatively low genetic redundancy: classes of genes with multiple members in humans are often represented by only a single orthologous gene in flies. Drosophila research also benefits from its own comprehensive, easily searchable, database, known as FlyBase, where all published information about flies is cataloged soon after publication. This impressive repository provides researchers with access to a wide range of information, allowing them to coordinate their studies more efficiently. Because Drosophila as a model organism has been studied for over 100 years, the field has also developed a vast array of tools for manipulating and monitoring gene expression, placing Drosophila at the forefront of large-scale genetics screens. Classical X-ray radiation exposure, feeding with the mutagen ethyl methane sulfonate (EMS) (Greenspan, 1997), and the development of transposable P-element insertions have helped produce massive mutant strain collections which can be used to investigate different biological processes. Large collections of Drosophila strains have also been generated for over-expression of genes of interest (EP lines and FlyORF lines), as well as for RNAi-mediated down regulation of endogenous genes (Trip and VDRC RNAi lines) (Bellen et al., 2004; Dietzl et al., 2007; Bischof et al., 2013; Perkins et al., 2015) (Additional information about Drosophila stocks, reagents, online tools and key resources are reviewed extensively in Mohr et al., 2014). The presence of these large collections dramatically speeds up research by saving individual researchers the time and effort of designing and developing new lines. Moreover, the cell-type-specific effects of gene overexpression or knockdown can be studied using bipartite expression systems such as GAL4/UAS (upstream activating sequence) (Brand and Perrimon, 1993).
The GAL4/UAS bipartite system is an extensively used tool for controlling gene expression in flies (Brand and Perrimon, 1993). It takes advantage of the yeast transcriptional activator GAL4, which is usually cloned downstream of a tissue specific enhancer (conferring a determined expression pattern). GAL4 binds to UAS, activating the transcription of the gene of interest cloned downstream of it, in a spatiotemporal manner matching the GAL4 expression pattern (Fischer et al., 1988; Brand and Perrimon, 1993). The fly line containing the GAL4 is often called the ‘driver,’ and the transgene fly line that carries the UAS is often called the ‘responder’ or ‘reporter.’ The two elements are kept as separate fly stocks so that the expression of the UAS line is only induced in the progeny of crosses between driver and responder lines (Figure 3). There are currently nearly 8000 GAL4 and derivative lines, offering an ever increasingly precise toolkit for cell-type-specific expression of transgenes1. Importantly, most of these lines are available from public repositories. In particular, there are a plethora of GAL4 lines for different brain cell types (Pfeiffer et al., 2008; Jenett et al., 2012), allowing studies of very specific neuronal or glial sub-populations (see Doherty et al., 2009; Luan et al., 2020).
FIGURE 3.
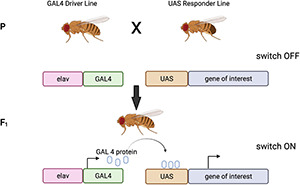
GAL4/UAS bipartite system of expression. The driver line contains a tissue specific promoter (in this case the neuronal specific elav promoter), driving the expression of yeast GAL4 protein in neurons. The responder line carries the gene of interest cloned down-stream of the UAS promoter, this, however, is not expressed. When the driver line is crossed to the responder line, the GAL4 driver and the UAS-transgene construct will come together in one animal. The GAL4 will be expressed in neurons, bind the UAS promoter and drive the expression of the gene of interest specifically in neurons. Images created with BioRender.com.
Subsequent refinements to this system have also improved temporal control: the temperature-sensitive GAL80 protein suppresses the GAL4 activity at lower temperatures (16°C) but is inactivated at higher temperatures (29°C), allowing genes to be turned on via a temperature shift (Lue et al., 1987; Duffy, 2002). More recently, Gal4-Geneswitch has been developed, where the GAL4 DNA binding domain has been fused to a progesterone inducible element which is activated upon feeding of the drug RU486 (Osterwalder et al., 2001; Nicholson et al., 2008). This system has been extensively used in neurodegenerative disease models to restrict expression of toxic constructs exclusively to adult neurons, a particularly useful feature for modeling diseases of adulthood as it avoids any confounding developmental effects (Sofola et al., 2010).
The ease of access to large libraries of stocks has enabled numerous large-scale enhancer and suppressor screens. This type of screen has been extensively used in neurodegenerative disease models to help identify downstream pathways of toxicity (Bilen and Bonini, 2007; Butzlaff et al., 2015; Zhang et al., 2015; Belfiori-Carrasco et al., 2017). The aim of these screens has been to identify factors that worsen (enhance) or ameliorate (suppress) the phenotype associated with a particular disease model. One of the most easily and commonly screened characteristics in Drosophila is the compound eye. Drosophila eyes are ideally suited as a genetic screening tool for diverse biological process, since about two-thirds of essential genes in Drosophila are involved in eye development (Thaker and Kankel, 1992). Furthermore, the complexity of the Drosophila eye, which encompasses 800 ommatidia, each in turn containing 8 photoreceptors, means that quantifiable features such as the number of ommatidia affected can precisely reflect the severity of the phenotype (Bate and Arias, 1993; St Johnston, 2002). At a macroscopic level, the size and roughness of the eye can also provide an immediate, scorable measure of cell death, which can be used as a rapid phenotypic readout. This serves as an ideal tool to conduct high throughput and unbiased screens (Lenz et al., 2013). Other screenable phenotypes linked to neurodegeneration include climbing ability, viability, and lifespan. Importantly, screens in flies have highlighted a number of pathways involved in disease development which have later been shown to be conserved in mammalian systems (reviewed in McGurk et al., 2015).
Drosophila Mitochondrial Biology
Unsurprisingly given their central role in eukaryotic biology, Drosophila and human mitochondria show a very high degree of conservation in both genetic architecture and biochemical pathways. At the genomic level, Drosophila mitochondrial DNA (mtDNA) has similar components to human mtDNA, although the human mtDNA genome is 16,559 bp, 3 kb shorter than the Drosophila mtDNA genome at 19,517 bp (Lewis et al., 1995). Human mtDNA contains 13 polypeptides, 22 tRNAs and 2 rRNAs encoding the same components as Drosophila mtDNA, albeit with a slightly different genomic order (Garesse and Kaguni, 2005). The molecular functions typically associated with mitochondria, such as OXPHOS, mitochondria transport, and biogenesis are highly conserved, as are the nuclear-encoded genes required for mitochondrial function (Chen et al., 2019). Drosophila has therefore been an excellent model system to study mitochondrial function, with relevance to mitochondrial biology broadly and to neurodegeneration specifically.
One of the earliest examples of using Drosophila to study mitochondrial biology was the characterization of phenotypes in flies mutant for the technical knockout (tko) gene (Royden et al., 1987). Tko is the nuclear-encoded mitochondrial ribosomal protein S12 (mRpS12) (Mariottini et al., 1999). The tko25t mutant has reduced complex I, II, and IV activity and reduced ATP synthesis (Toivonen et al., 2001). This disruption in the electron transport chain (ETC) leads to a number of observable phenotypes, including seizure sensitivity, larval developmental delay, hearing impairments, and defective male courtship (Toivonen et al., 2001). Remarkably, these behavioral defects resemble the key features in patients with such disorders (Jacobs et al., 2004), providing early evidence that fly models could be used to model human mitochondrial disease. Indeed, several mitochondrial diseases models have since been developed which emulate the physiological symptoms seen in patients with mitochondrial disorders (Jacobs et al., 2004; Xu et al., 2008; Fernández-Ayala et al., 2010). Beyond mitochondrial diseases specifically, Drosophila tko mutants have been used in epilepsy research as the tko bang sensitive (bs) paralytics mutant displays similar features to those seen in human seizure disorders (Song and Tanouye, 2008; Whelan et al., 2010).
Drosophila models have also proved useful in defining the phenotypes most affected by defective mitochondria. For example, Drosophila studies defined mitochondria’s key role in sperm development and male fertility. Fuzzy onions (fzo), the founding member of the Mitofusin (Mfn) family of proteins, was first characterized in flies. Mutant fzo1 mitochondria are unable to fuse in early spermatocytes, disrupting mitochondria organization and leading to male sterility (Hales and Fuller, 1997). Notably, these phenotypes have been replicated in mammalian models and observed in human patients (Zhang et al., 2016).
Finally, fly models have also proved invaluable to understanding the role of mitochondria in neurodegenerative disorders. For example, following the identification of mutations in PINK1, a PTEN-induced serine/threonine kinase 1, and in the E3 ubiquitin ligase Parkin, as causative genes in monogenic PD, flies were instrumental in identifying the role of these genes in mitochondrial biology (Clark et al., 2006; Park et al., 2006; Deng et al., 2008; Poole et al., 2008; Yang et al., 2008). Flies were used to show that both PINK and Parkin were implicated in the same biological pathway, playing a fundamental role in mitochondrial dynamics and degradation of dysfunctional mitochondria via mitophagy (Clark et al., 2006; Park et al., 2006; Deng et al., 2008; Poole et al., 2008; Yang et al., 2008).
Assays for Studying Mitochondrial Biology
The study of mitochondria in flies is bolstered by a large number of assays to characterize mitochondrial form and function in detail, both in live and fixed tissue of wildtype and mutant Drosophila (Sen and Cox, 2017).
A number of general purpose dyes have been developed to visualize mitochondria; nearly all of these can be used in Drosophila tissues. The cell-permeant mitochondrion-selective dye MitoTracker Green has been used to observe mitochondrial mass (Rana et al., 2017). Specifically, MitoTracker Green labels the mitochondria matrix by covalently binding free thiol groups on cysteine residues belonging to mitochondrial proteins (Presley et al., 2003). A number of dyes have also been developed to measure mitochondrial membrane potential (MMP), a parameter that correlates with the mitochondrial ability to generate ATP. Tetramethylrhodamine, methyl ester (TMRM) or Tetramethylrhodamine, ethyl ester (TMRE), and 5,5′, 6,6′-tetrachloro-1,1′,3,3′-tetraethyl-benzimidazolylcarbocyanine iodide (JC1) dyes are some of the most commonly used dyes used for this purpose. TMRM, TMRE, and JC-1 are all fluorescent cationic dyes that accumulate in mitochondria based on their potential, the more polarized mitochondria are, the more the dyes accumulate. For TMRM and TMRE this results in a brighter signal at high MMP (Perry et al., 2011). JC1, on the other hand, changes color according to its concentration, forming red aggregates at high concentrations in healthy mitochondria, and remaining as a green monomer in mitochondria with low MMP (Sivandzade et al., 2019). The MMP in this case is measured by the ratio of the red/green fluorescence intensity. Finally, MitoSOX dye can be applied to measure ROS production (Yarosh et al., 2008). This fluorescent cationic dye is a derivative of dihydroethidum; in the presence of superoxide, MitoSOX is selectively oxidized in the mitochondrial matrix of live cells, where it emits a red color (Roelofs et al., 2015).
In addition to dyes, a host of fluorescent genetically encoded probes and bio-sensors have also been developed, particularly in Drosophila. As the genes encoding these sensors are usually under the control of the UAS promoter, they can be driven by cell-type specific GAL4 drivers. This allows monitoring of mitochondria morphology, physiology, and key metabolites in vivo in specific cell populations (Table 2).
TABLE 2.
Genetically encoded mitochondrial biosensors/tools developed in Drosophila.
| Sensor line | Sensor type | Brief description | References |
| UAS-AT1.03NL/UAS-AT1.03RK | ATP sensor | FRET sensor for live imaging, consisting of an ATP binding sequence located between two fluorophores. Upon ATP binding the two are brought in close proximity leading to increased FRET signal. | Tsuyama et al., 2013 |
| UAS-mitoGFP UAS-mitoDSRed | Mitochondrial marker | Green or Red fluorescent protein tagged with a mitochondrial import sequence (acting as targeting signal to the mitochondrial matrix), used to visualize all mitochondria in live or fixed tissue. | Lutas et al., 2012; DeVorkin et al., 2014; Chowdhary et al., 2017; Morris et al., 2020 |
| UAS- mito-GCaMP3 | Ca2 + sensor | Circularly permutated EGFP M13/Calmodulin fusion protein, tagged with a mitochondrial import sequence. Upon Ca2 + binding the construct undergoes a conformational change, increasing fluorophore output, thus allowing to monitor Ca2 + concentration in live cells. | Lutas et al., 2012; Morris et al., 2020 |
| UAS-PercevalHR | ATP/ADP sensor | Fluorescent biosensor composed of three tandem copies of an ATP-binding protein fused to a circularly permuted monomeric Venus. It has a dual excitation spectrum, with a peak at 405 nm, enhanced by ATP binding and at 488 nm, enhanced by ADP binding. The ratiometric signal reports the occupancy and therefore ATP/ADP ration in live cells. | Morris et al., 2020; Wong et al., 2021 |
| UAS-SoNaR | NADH/NAD + sensor | Fluorescent biosensor composed of a fusion of circularly permuted yellow fluorescent protein and an NADH-binding domain. It binds to NAD and NADH with two specific conformation, associated with a different excitation spectrum. The ratiometric signal reports the occupancy and therefore NADH/NAD + ratio in live cells. | Bonnay et al., 2020; Morris et al., 2020 |
| UAS-mito-roGFP2-Grx1/UAS-mito-roGFP2-Orp1 | REDOX sensors | Mitochondrially targeted, redox sensitive GFPs, with a dithiol/disulfide switch on their surface leading to measurable ratiometric fluorescent change in response to redox changes in live mitochondria. These are fused to glutaredoxin (Grx), which increases specificity for glutathione or a microbial Orp1 H2O2 sensor, making it specific for H2O2. | Albrecht et al., 2011; Morris et al., 2020 |
| UAS-Dendra2.mito | Photoconvertible mitochondrial tag | Mitochondrially localized photoconvertible protein that can be irreversibly photoconverted from a green fluorescent form to a red fluorescent form, by UV-violet or blue light. It allows to monitor behavior of photoconverted mitochondria over time. | Hwang et al., 2014; Bertolin et al., 2018 |
| UAS-mito-PyronicSF | Pyruvate sensor | Mitochondrially targeted pyruvate FRET sensor composed of a circularly permuted GFP fused to a bacterial pyruvate sensitive transcription factor. Upon pyruvate binding a conformational change leads to increased FRET signal. | Arce-Molina et al., 2020 |
Drosophila has also been a useful model system for developing tools to assess specific molecular products of mitochondrial activity. For example, the MitoB probe, which uses a ratiometric mass spectrometry approach to measure H2O2 in the mitochondrial matrix, was developed using Drosophila (Cochemé et al., 2011). The probe contains a triphenylphosphonium (TPP) cation which is then conjugated to an arylboronic acid target in the mitochondria matrix; the final product is then measured by liquid chromatography tandem mass spectrometry.
Another important assay to study mitochondrial physiology is the measurement of oxygen consumption, which is directly linked to OXPHOS activity as the final step of the ETC involves the reduction of oxygen to water. One assay for measuring oxygen consumption is the high-resolution Oxygraph-2k, also called Oroboros, which measures oxygen consumption as well as additional parameters such as ROS production, mitochondrial membrane potential, ATP production, Ca2+, and pH2. Oroboros measures oxygen uptake and concentration after the injection of substrates for different ETC components, directly to samples immersed in a specific solution within a detection chamber. It requires a small amount of sample, with only 2 flies being sufficient for an assay (Costa et al., 2013). Advantages of this system include its flexibility to study each complex individually and its ability to perform serial measurements in the same sample, and its relatively low cost (Perry et al., 2013). Nonetheless, this system is relatively slow and low throughput.
The Seahorse XF Extracellular Flux Analyzer (Seahorse Bioscience Inc.) was introduced more recently to enable high throughput testing, albeit at extra cost. The instrument can measure the rate of mitochondrial OXPHOS (through determination of the oxygen consumption rate) and glycolysis through extracellular acidification rate (ECAR). The system achieves higher throughput by using fluorescent sensor cartridges on 24 or 96 cell-culture microplates3. In addition, the microplate-based assay has 4 ports to inject substrates and/or inhibitors to assess multiple conditions consecutively (reviewed in Horan et al., 2012).
Finally, a number of spectrophotometric assays have also been developed to study the activity of mitochondrial enzymes or the concentration of individual metabolites. These assays possess the advantage of being relatively cheap, rapid and reliable (Birch-Machin and Turnbull, 2001; Barrientos, 2002; Barrientos et al., 2009). They provide information on the catalytic activities of each respiratory chain complex or combination of complexes by following the production or depletion of particular substrates which absorb or transmit light at specific wavelengths. The results are then usually standardized against citrate synthase activity, frequently used as a standard in mitochondria (Frazier and Thorburn, 2012). The downsides of these assays are that few samples can be measured at one time, and they generally require a relatively large quantity of sample (Janssen and Boyle, 2019).
Assays to Assess Health and Behavior in Drosophila
As described earlier, mitochondrial defects in flies have physiological implications on the whole organism that result in alterations in behavior and lifespan. These can provide useful organism-level phenotypic readouts of mitochondrial defects. Lifespans is one of the most commonly used readouts for late onset phenotypes in flies. Because of the sensitivity of lifespan as an assay, thorough protocols have been developed to standardize the many factors that can influence lifespan (Piper and Partridge, 2016). The healthy lifespan of flies can be evaluated across ‘normal’ aging, or lifespan can be assessed in the presence of mitochondrial stressors such as paraquat or rotenone to investigate the flies’ sensitivity to oxidative stress (Menzies et al., 2005; Meulener et al., 2005).
In addition to shortened lifespan, impairments in mitochondrial function can lead to more subtle neurological defects and neurodegeneration in flies, as well as in humans. A large number of assays have been developed to monitor neuronal impairments in flies. For instance, ‘bang-sensitivity’ measures the time for flies to recover from paralysis after being strongly shaken to the base of a tube. It mimics epilepsy and seizures associated with mitochondrial disease, features found for example in mitochondrial encephalomyopathy with lactic acidosis and stroke-like episodes (MELAS) (Jacobs et al., 2004; Celotto et al., 2006; Burman et al., 2014). In addition, locomotor dysfunction can be measured in flies via a climbing or negative geotaxis assay, which quantifies the negative geotactic reflex of flies. When gently tapped to the bottom of a chamber, flies naturally climb upwards against gravity and toward light. Flies are allowed to climb for defined time, and the height climbed is scored as an easy parameter that correlates with general locomotor function (Benzer, 1967; Inagaki et al., 2010). The climbing assay is widely used in Drosophila neurodegenerative disease research as it is a rapid readout of neuromuscular function, albeit a readout that lacks information on the exact tissues and circuits involved in any observed deficits.
Locomotion, sleep, and circadian rhythms can also be monitored with the widely used Drosophila activity monitoring (DAM) system (Pfeiffenberger et al., 2010). Flies are placed in sealed tubes with food within a climate and light-controlled incubator; monitors then use infrared beams to record each time a fly walks past a defined point within the tube (an event). Events are continuously scored for 3–4 days to monitor activity. To study sleep, a significant body of Drosophila sleep research has come to the consensus that ‘sleep’ can be defined in the DAM system as zero events for a sustained period of 5 min or more (Beckwith and French, 2019). The DAM system can therefore easily record movement and sleep. Circadian rhythmicity can also be assessed by transitioning the flies to constant darkness and observing how strongly and with what period circadian activity cycles persist. All of these parameters are influenced by neuronal health, in flies as well as humans, and can be used as sensitive measures for the effects of mitochondrial mutations on brain health.
Evidence of Mitochondrial Dysfunction in Frontotemporal Dementia and Amyotrophic Lateral Sclerosis
The roles of mitochondria in FTD/ALS development have been underscored by the identification of disease mutations in the gene encoding the mitochondrial CHCHD10 protein. CHCHD10 localizes to the mitochondrial intermembrane space and is enriched at cristae junctions. Although the precise role of CHCHD10 is still unclear, muscle biopsies and fibroblasts from patients exhibit mitochondrial morphology defects, disorganization of cristae, and respiratory chain deficiencies such as abnormal complex V assembly (Bannwarth et al., 2014). CHCHD10 mutations also affect the stability of mitochondrial DNA in skeletal muscle (Bannwarth et al., 2014). Disease associated mutations can lead to impaired CHCHD10 mitochondrial import (Lehmer et al., 2018), and model systems suggest the R15L and S59L variants can display loss of function and dominant negative phenotypes, leading to mitochondria damage, accumulation of cytoplasmic TDP-43, and consequent synaptic dysfunction (Woo et al., 2017).
Other mutations linked to FTD/ALS have also been described to affect mitochondria in multiple ways, suggesting potentially a common pathogenic mechanism downstream of multiple causal mutations. We outline briefly below some of the major mitochondrial defects associated with FTD/ALS disease variants; for a more comprehensive discussion please see Smith et al. (2019).
Mitochondrial Morphology/Dynamics
Mitochondria are a highly dynamic organelles that can change shape through fission/fusion events. When mitochondrial dynamics are disrupted, mitochondrial morphology and distribution are disrupted, which impinges directly on mitochondrial function. In ALS and in disease models, a number of studies have found aberrant mitochondrial morphology. For instance, post-mortem studies of tissue from sporadic ALS patients show swollen and mis-shapen mitochondria in the spinal cord (Sasaki and Iwata, 2007). A number of transgenic models with SOD1 mutations also display aberrant dynamics and morphology: for example, SOD1 G93A SH-SY5Y and NSC-34 cell lines show disrupted mitochondrial dynamics with the fission protein dynamin-related protein (Drp1) level increased and optic atrophy 1 (Opa1), a mitochondrial fusion protein, decreased (Ferri et al., 2010). Similarly in SOD1 G93A transgenic mouse spinal cord, Drp1 and mitochondrial Fission 1 (Fis 1) proteins are increased, while levels of the fusion proteins Mfn1 and Opa1 are reduced (Liu et al., 2013). This is also recapitulated in transgenic mice over-expressing mutant SOD1 which display mitochondrial vacuolar degeneration (Kong and Xu, 1998; Jaarsma et al., 2001; Sasaki et al., 2004).
Studies focused on TDP-43 have also identified roles for this disease-associated protein in mitochondrial biology. In primary mouse neurons, overexpression of either wildtype or mutant TDP-43 decreases mitochondrial length and density in neurites; knock-down of TDP-43 exerts the opposite effect (Wang et al., 2013). Similarly, in fibroblasts derived from patients with mutations in TDP43, the mitochondria appear fragmented (Onesto et al., 2016), and EM analyses of TDP43 patient samples reveal cristae morphological defects (Wang et al., 2019).
Hexanucleotide repeat expansions in the C9orf72 gene (Figure 4), the most common genetic cause of both FTD and ALS, also appear to cause mitochondrial deficits based on recent studies. Recent studies in a mouse model expressing one of the products of the C9 repeats, the dipeptide repeat protein GR, found age dependant inner mitochondria impairment and loss of cristae formation in the prefrontal cortex (Choi et al., 2019). Mitochondria in primary cortical neurons isolated from mice expressing 80 GR repeats, are also shorter in length and less motile compared to mitochondria in wildtype neurons. At the same time, the mitochondrial network in C9 patient derived fibroblasts did not appear substantially fragmented (Onesto et al., 2016). These differences are potentially due to the different cell types, as neurons may be particularly sensitive to mitochondrial dysfunction as discussed above. In addition to morphological differences, mitochondrial dynamics appear to be disturbed in some C9orf72 models. In a C9 mouse model expressing the dipeptide repeat GR, Drp1 levels are increased and Opa1 levels are decreased, similar to the findings from SOD1 mouse models (Choi et al., 2019). Additionally, induced pluripotent stem cells (iPSC)-derived motor neurons with a C9orf72 expansion show impairments in fast axonal transport of mitochondrial cargo (Mehta et al., 2021).
FIGURE 4.
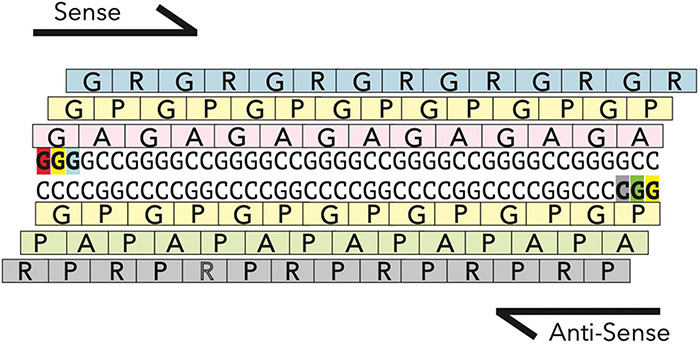
RAN translation in C9orf72 hexonucleotide repeat expansion. The hexonucleotide repeat expansion in the gene C9orf72 is transcribed both in the sense and antisense direction to generate two highly stable RNAs, which are translated via a non-canonical form of translation, called Repeat Associated non-ATG mediated (RAN) translation. RAN translation occurs in all frames along the transcript to generate five highly repetitive dipeptide repeat proteins (GR, GP, GA, AP, and PR). GR and PR being the most toxic (Mizielinska et al., 2014).
Fused in sarcoma, a gene in which mutations are associated with both FTLD and ALS, provides yet another example of a disease gene associated with mitochondrial deficits. EM analysis has shown morphological abnormalities in mitochondria from FUS patient spinal cord (Huang et al., 2010) and FTLD-FUS patient brain samples (Deng et al., 2015). In cultured neurons, expression of ALS-associated FUS mutants also leads to shortened mitochondria (Tradewell et al., 2012) and mitochondrial fragmentation (Deng et al., 2015, 2018).
Calcium Homeostasis
Mitochondria play a central role in Ca2+ homeostasis as they store calcium for cell signaling activities and neurotransmitter release. Notably, disturbed mitochondrial calcium homeostasis may be a particularly early marker of pathogenesis in FTD/ALS. For instance, brain and spinal cord samples from mice expressing G93A or G85R mutant SOD1 demonstrate a reduction of calcium-loading capacity in mitochondria very early in the disease course, long before observable symptom onset (Damiano et al., 2006).
Key to the ability of mitochondria to manage Ca2+ levels is their interaction with the endoplasmic reticulum (ER), the other main Ca2+ store. ER-mitochondria contact sites (or MAMs) regulate Ca2 + exchange between the two organelles (Romero-Garcia and Prado-Garcia, 2019) and are instrumental in maintaining appropriate intracellular Ca2+ homeostasis. As with other mitochondrial phenotypes, MAMs are disrupted in several models of FTD/ALS associated mutations. Expression of either wildtype or mutant TDP-43 or FUS in human and mouse cells disrupts MAMs by disrupting the interaction of two key mediators of ER-mitochondrial interaction: resident ER protein; VAPB and the mitochondrial protein tyrosine phosphatase-interacting protein 51 (PTPIP51) (Stoica et al., 2014). C9orf72 hexanucleotide expansions also affect Ca2+ homeostasis. An extensive characterization of iPSC derived motor neurons from patients with C9orf72 expansions showed dysfunction in Ca2+ homeostasis, reduced levels of the antiapoptotic protein Bcl-2, increased ER stress, and reduced mitochondrial membrane potential, resulting in decreased cell survival (Dafinca et al., 2016). This suggests that a disruption of Ca2+ homeostasis is a common phenotype across multiple causal mutations.
Reactive Oxygen Species
Excessive accumulation of ROS in cells can lead to cell damage and, eventually, cell death (Pizzino et al., 2017). Low levels of ROS perform essential signaling functions (Schieber and Chandel, 2014); however, excess ROS production leads to lipid peroxidation and oxidation of proteins and DNA. ROS are primarily generated in mitochondria as a by-product of oxygen and energy metabolism, making mitochondria a primary target of ROS damage. Excess ROS leads to nitration of mitochondrial proteins, mtDNA damage and mitochondrial respiratory chain dysfunction (Guo et al., 2013).
ALS and FTD, like many other neurodegenerative disorders, have been linked to excessive ROS production and consequent cellular damage. ROS has long been thought to be causally linked to ALS development as the first causal mutations linked to familial ALS were identified in SOD1, a superoxide scavenging enzyme localized to mitochondria whose primary role is to reduce ROS levels. ALS-associated mutations in SOD1 have been associated with increased ROS, with transgenic mice expressing G93A mutant human SOD1 showing oxidative damage to mitochondrial proteins and lipids (Mattiazzi et al., 2002; Vargas et al., 2011; Xiao et al., 2018; Chen et al., 2021; Méndez-López et al., 2021). In cell models as well, expression of G93A SOD1 leads to elevated ROS levels (Liu et al., 2002; Rizzardini et al., 2005) and increased lipid peroxidation level (Liu et al., 2002). Additionally, post mortem tissue from both carriers of SOD1 mutations and sporadic ALS patients display increased ROS-associated damage markers (Beal et al., 1997; Ferrante et al., 1997; Shibata et al., 2001).
Mutations in TDP43 and CHMP2B may also lead to excess ROS production by mitochondria. TDP-43 has been found to localize to mitochondria (Wang et al., 2016, 2019; Yu et al., 2020), and expression of mutant TDP-43 in fly neurons and mammalian cells lead to ROS over-production (Wang et al., 2019). Neurons with mis-localized TDP-43 showed increased ROS production relative to neurons with normal nuclear localization of TDP-43 (Wang et al., 2013). Neurons derived from FTD patients with CHMP2B mutations also show impaired cristae morphology, reduced mitochondrial respiration and increased accumulation of ROS relatively to healthy control neurons (Zhang et al., 2017).
C9 expansions can also lead to excessive ROS production in mitochondria. Motor neurons and fibroblasts derived from patients with expansions in C9orf72 show an age dependant increase in ROS production (Lopez-Gonzalez et al., 2016; Onesto et al., 2016). In fibroblasts, this is accompanied by an increase in oxygen consumption, ATP production and mitochondria hyperpolarization, possibly linked to an increase in mitochondrial biogenesis (Onesto et al., 2016). The mitochondrial dysfunction is potentially due to the highly positively charged GR, one of the repetitive dipeptides produced by the C9orf72 expansion. GR80 expressed in motor neurons has been observed to bind mitochondrial ribosomal proteins, leading to increased ROS production level and increased DNA damage; this is not the case with GA80, another C9-produced but uncharged repetitive protein (Lopez-Gonzalez et al., 2016). Reducing oxidative stress partially rescued DNA damage and partially suppressed GR80 induced cell toxicity in flies (Lopez-Gonzalez et al., 2016). Similarly, restoring the correct redox environment in SOD1 cellular models, also rescued cell death (Ferri et al., 2010), suggesting that ROS production is a driver of neurodegeneration for this model.
Metabolism
Mitochondria are responsible for the majority of ATP production. When mitochondrial metabolism is disrupted, it affects the respiratory chain and ATP synthesis, which can be yet another mechanism of toxicity for mutations linked to FTD and ALS.
Studies from both patient tissue and disease models suggest that neuronal mitochondrial ATP production is reduced in FTD/ALS. In samples of ALS patients spinal cord, citrate synthase activity and respiration chain function are reduced (Wiedemann et al., 2002). In SOD1-G39A transgenic mice, the ETC is similarly disrupted, with mitochondrial respiration and ATP synthesis reduced at the time of symptom onset but not at earlier stages (Mattiazzi et al., 2002). C9orf72 expansions can also lead to reduced ATP generation: Complex I levels are reduced in C9 patients spinal cord and in C9 patient-derived iPSC cells, leading to a reduction in Complex I activity (Wang et al., 2021). Expression of poly(GR)80 in mice also leads to mitochondrial dysfunction by binding to the ATP5A1 (complex V), thus enhancing its ubiquitination and degradation (Choi et al., 2019). Moreover, studies using cultured neurons have shown mutant TDP43 binds and inhibits translation of mRNA of mitochondrially encoded Complex I subunits (Salvatori et al., 2018). Cells derived from patients carrying CHCHD10 mutations are also susceptible to energetic stress and display Complex I deficiency, leading to increased NADH/NAD + ratio and diminished tricarboxylic acid (TCA) cycle activity (Straub et al., 2021). Taken together, these studies show that mutations in several, different FTD/ALS-associated genes lead to similar metabolic energy deficiencies, again suggesting that specific mitochondrial impairments might be a common downstream pathogenic mechanisms.
Fly Models of Frontotemporal Dementia and Amyotrophic Lateral Sclerosis and Their Role in Mitochondrial Dysfunction
Fly models of FTD/ALS generally use either gain-of -function (overexpression of the chosen gene) or loss-of-function (knockdown or knockout of the chosen gene) strategies to investigate disease pathogenesis. The overexpression and/or knockdown or knockout of the desired gene can exhibit a variety of phenotypes such as rough eye, climbing and crawling defects, eclosion defects, or wing defects, all of which suggest toxicity of the introduced genetic modification. These phenotypes can be used as quantifiable readouts of pathogenicity, helping researchers to investigate aspects of FTD/ALS pathogenesis with the goal of elucidating disease mechanisms. Similar to mammalian and human-derived cell models of disease, fly models of FTD/ALS generally display prominent mitochondrial defects, suggesting conserved mechanisms of toxicity and making flies a useful model system to investigate mitochondrial mechanisms of pathogenesis. We will focus here on examples from fly models of the most common disease risk factors: SOD1, TDP43, FUS, and C9orf72 hexanucleotide repeat expansions.
Superoxide Dismutase 1 Fly Models
Superoxide dismutase 1 is the most extensively investigated gene associated with ALS, especially in rodent models. Of all the FTD/ALS risk genes, SOD1 is one of the most directly related to mitochondrial function as it localizes and aggregates at the intermembrane space (IMS) and the outer mitochondrial membrane (OMM) (Tafuri et al., 2015). SOD1 is a superoxide dismutase which binds zinc and iron and converts superoxide anions into oxygen and hydrogen peroxide (Pardo et al., 1995), thus playing a crucial role in reducing oxidative stress. To date, more than 200 variants have been found in the SOD1 gene4. The fly gene ortholog, also named SOD1, shows 67% amino acid similarity with the human sequence. A number of fly models have been generated looking at SOD1 function in disease development. Both gain and loss of function models have been generated, as it is still a matter of debate whether ALS caused by SOD1 mutations is due to a gain or loss of function of the protein (Mockett et al., 2003; Şahin et al., 2017; Agudelo et al., 2020).
Fly models expressing mutant SOD1 display motor defects (Bahadorani et al., 2013; Gallart-Palau et al., 2016), both when expressed in muscle (Gallart-Palau et al., 2016) and the brain (Bahadorani et al., 2013). In each case, locomotor dysfunction is accompanied by mitochondrial defects. Flies over-expressing zinc deficient human SOD1 also show cristae vacuolization and reduced ATP levels in fly heads but not in bodies (Bahadorani et al., 2013), and are severely sensitized to paraquat and zinc. Both paraquat and zinc act as mitochondria toxins, with paraquat inhibiting mitochondrial complex I activity and zinc interacting with nitric oxide to cause mitochondrial dysfunction. Flies expressing SOD1-G93A, an extensively modeled dominant causal mutation, also show defects in mitochondrial morphology (Gallart-Palau et al., 2016), similar to mitochondrial defects observed in mouse models of SOD1-G93A (Higgins et al., 2003). Taken together, these studies suggest the effects of SOD1 mutations on mitochondrial dysfunction are well conserved between flies and mammals.
TDP-43 Fly Models
TDP-43 is an RNA/DNA binding protein involved in RNA metabolism. Neuronal inclusion bodies of hyperphosphorylated and ubiquitinated TDP-43 deposits were first described in the brain and spinal cord of FTD/ALS patients (Arai et al., 2006; Neumann et al., 2006). In healthy conditions, TDP43 localizes to the nucleus; however, in pathological conditions, TDP-43 translocates to the cytoplasm where it forms inclusions or aggregates (Ayala et al., 2008). The presence of cytoplasmic inclusions of TDP-43 suggests a toxic gain-of-function disease mechanism; in contrast, the loss of TDP-43 from the nucleus could also indicate a loss-of-function mechanism (Neumann et al., 2006; Winton et al., 2008; Giordana et al., 2010). TBPH is the fly TDP-43 ortholog, with 53% amino acid similarity between fly and human sequences. Reduction of TBPH, either by RNAi or genetic null mutants, reduces lifespan in flies and leads to a decline in motor function (Feiguin et al., 2009; Lu et al., 2009; Lin et al., 2011; Diaper et al., 2013), conversely, expression of human TDP-43 can rescue the loss of function phenotypes (Wang et al., 2011). Loss of TBPH function also causes reduced mitochondrial axonal transport linked to severe motor impairment (Baldwin et al., 2016), adding weight to TDP-43 loss of function as a disease mechanism. However, another study over-expressing wildtype human TDP-43 or A315T-mutant TDP-43 in the fly eye showed activation of mitochondrial unfolded protein response (UPRmt) and increased mitochondrial ROS levels in motor neurons (Wang et al., 2019), suggesting that both gain and loss-of-function of TDP43 can contribute to mitochondrial defects.
A number of studies have shown that over-expression of human wildtype TDP-43 in flies, either in muscle (Altanbyek et al., 2016) or neurons (Khalil et al., 2017), leads to aberrant mitochondrial morphology. Mitochondria appear fragmented, and the expression of well-known regulators of mitochondrial dynamics, such as mitochondrial assembly regulatory factor (Marf), which promotes fusion, are decreased both in muscle (Altanbyek et al., 2016) and brain (Khalil et al., 2017). Over-expression of Marf in muscle partially rescues mitochondrial morphology (Altanbyek et al., 2016), and its over-expression in neurons partially rescues the locomotor phenotypes and neuromuscular junction (NMJ) defects associated with TDP-43 over-expression (Khalil et al., 2017). Downregulation of a dynamin-like GTPase that mediates mitochondrial fission, Drp1, similarly leads to partial rescue of TDP-43 phenotypes in brain and muscle (Altanbyek et al., 2016; Khalil et al., 2017), confirming that dysfunction in mitochondrial dynamics contributes at least partially to TDP-43 toxicity. Similar defects to those in TDP-43 over-expression have been observed in flies over-expressing human FUS and TATA box-binding protein-associated factor 15 (TAF15), two additional ALS-associated genes, suggesting conserved mechanisms of pathogenicity (Altanbyek et al., 2016). The ease of generating transgenic flies and conducting rapid phenotypic analysis in flies allows for these comparisons across a number of models of disease, which would otherwise be more costly and time consuming in mammalian models of disease.
Fused in Sarcoma Fly Models
Fused in sarcoma, similarly to TDP-43, is involved in RNA-related metabolism (Lagier-Tourenne et al., 2010) and is usually located in nucleus. Like TDP-43, abnormal cytoplasmic inclusions of FUS are formed in ALS. However, FUS and TDP-43 may act independently in ALS pathogenesis given the absence of TDP-43 pathology in FUS mutant patients (Kwiatkowski et al., 2009; Vance et al., 2009). Additionally, ALS patients with FUS mutations display a more aggressive disease form, with earlier symptom onset and shorter symptoms duration, as well as increased rate of bulbar onset (Yan et al., 2010). FUS is conserved in flies, where the orthologous gene cabeza (caz) shows 51% amino acid similarity to the human gene product (Stolow and Haynes, 1995). Importantly, expression of human FUS can rescue caz mutant phenotype (Wang et al., 2011), demonstrating functional equivalence of the orthologous genes.
As in TDP-43-mediated toxicity, FUS-mediated toxicity may result from either gain-of-function or loss-of-function mechanisms. In flies, overexpression of wildtype caz, over-expression of mutant caz P398L (carrying an equivalent mutation to the human pathogenic P525L mutation), or loss of caz can all result in inhibited mitochondrial transport, with mutant caz over-expression showing the most severe mitochondrial transport impairment and greatest effects on development and locomotor activity (Baldwin et al., 2016).
FUS fly models also display prominent mitochondrial defects: wildtype or P525L FUS over-expression in adult flies leads to mitochondrial fragmentation, as seen for TDP43 (Deng et al., 2015). Both wildtype and P525L mutant FUS interact with the catalytic subunit of mitochondrial ATP synthase (ATP5B) in cells and disrupt ATP synthase supercomplex (complex V) function and formation in flies, leading to the activation of the UPRmt (Deng et al., 2018). Down-regulation of ATP5B or genes involved in UPRmt partially rescues neurodegenerative phenotypes (Deng et al., 2018), suggesting that the impaired ETC and subsequent activation of the UPRmt can at least partially drive neurodegeneration. Downregulation of Hsp60, an ATP-dependent mitochondrial chaperone protein that modulates the localization of FUS to mitochondria in human cells, also partially rescues the rough eye phenotype and locomotor dysfunction observed in FUS transgenic flies (Deng et al., 2015).
These studies in flies suggest that the mitochondrial phenotypes associated with FUS and TDP43 fly models are very similar, pointing toward common downstream pathogenic mechanisms. Importantly, these findings suggest that therapies targeted to these mechanisms might be promising areas for study in multiple types of FTD/ALS.
C9orf72 Hexonucleotide Repeat Expansion Fly Model
A hexanucleotide repeat expansion in the gene C9orf72 is the most common genetic cause of ALS and FTD (Chiò et al., 2012). Since this expansion is a relatively recently identified cause of FTD/ALS, much of our understanding of the pathogenic mechanisms of this expansion derives from models that can be rapidly generated and characterized – namely Drosophila models (Xu et al., 2013; Mizielinska et al., 2014; Freibaum et al., 2015).
Three mechanism of C9 toxicity have been proposed: (1) loss-of-function caused by reduction of endogenous C9orf72 transcription, (2) gain-of-function by formation of nuclear RNA foci transcribed from the G4C2 repeats, and (3) gain-of-function by the accumulation of highly repetitive dipeptide repeats proteins (DPRs) translated from the expanded repeats (Ash et al., 2013; Mori et al., 2013; Mizielinska et al., 2014). These DPRs can be translated from either sense (G4C2) or antisense (G2C4) transcripts through repeat-associated non-AUG (RAN) translation (Mori et al., 2013; Gendron et al., 2014). C9orf72 does not have a fly ortholog, so fly models have relied instead on overexpression of abnormal G4C2 repeats and DPR proteins to investigate potential gain-of-function mechanisms. The lack of an orthologous genes is both a strength and limitation of Drosophila in this case: the endogenous function of C9orf72 cannot be easily investigated using fly models, but the effects of the expansion can be isolated from the downregulation of the endogenous gene.
Over-expression of a variety of repeat lengths, from 30 G4C2 repeats to over 100, has been shown to lead to retinal degeneration in the eye, shortening of lifespan and impaired motor function (Xu et al., 2013; Mizielinska et al., 2014; Wen et al., 2014; Freibaum et al., 2015; Tran et al., 2015), suggesting that the repeats are highly toxic to neurons. As additional evidence of dose-dependent effects of toxicity, increasing repeat lengths show increased toxicity (Mizielinska et al., 2014). Mizielinska et al. (2014) showed that toxicity was most directly associated with the arginine-rich DPRs, GR and PR, a finding that has been confirmed in independent fly models (Wen et al., 2014; Tao et al., 2015; Yang et al., 2015; Boeynaems et al., 2016) as well as in mammalian systems (Shi et al., 2017; Zhang Y. J. et al., 2018; Choi et al., 2019; Zhang et al., 2019; Cook et al., 2020).
As seen in other genetic causes of FTD/ALS, G4C2 expansions can produce a number of mitochondrial deficits. Expression of G4C2(36) repeats disrupts mitochondrial transport when expressed in fly motor neurons (Baldwin et al., 2016). In fly muscle cells, poly(GR), which is highly positively charged, has been shown to enter mitochondria, disrupt cristae junction structure, increase membrane potential, and increase the formation of ROS (Li et al., 2020). GR80 binds to components of the Mitochondrial Contact Site and Cristae Organizing System (MICOS) complex, which is involved in cristae junction formation; the MICOS complex thus represents a potential mechanism by which GR80 can impair the exchange of mitochondrial metabolites and osmolytes. Consistent with this mechanism, downregulation of MICOS complex components in a fly model of GR toxicity rebalances ion homeostasis, reduces GR80 levels, and improves GR80 toxicity (Li et al., 2020). Rebalancing mitochondrial matrix ion levels with the K + /H + antiporter nigericin also rescues mitochondrial cristae morphology in GR(80) expressing flies (Li et al., 2020). Maintenance of appropriate MICOS complex activity is essential to mitochondrial homeostasis both in health and in disease: a recent study has found that knockdown of Drosophila MICOS genes phenocopies many of the features observed in C9orf72 models, including cristae junction loss in muscles, low mitochondrial membrane potential, imbalanced fusion/fission machinery, increased mitophagy, limited cell death, and reduced climbing ability (Wang et al., 2020). CHCHD10 mutations have also been implicated in MICOS complex integrity (Genin et al., 2018), again suggesting common mitochondrial related impairments driving toxicity in FTD/ALS. The role of MICOS might also not be limited to FTD/ALS: Pink1, a gene implicated in autosomal recessive Parkinson’s disease, has also been shown to regulate MICOS components (Tsai et al., 2018), suggesting common elements across multiple neurodegenerative disorders.
Discussion/Conclusion
In summary, Drosophila models of FTD/ALS display conserved pathological changes, demonstrating they can be rapid engines for the discovery of pathogenic mechanisms in neurodegenerative diseases. Flies have helped address questions about mitochondrial function and dysfunction in neurodegenerative diseases, aging, mitochondrial diseases, and developmental biology. Their extensive genetic tools, potential for large unbiased screens, and cost effectiveness has allowed Drosophila to provide key insights in understanding mitochondrial biology in FTD/ALS models (Figure 5). Drosophila is uniquely placed, given its facile genetics, to study convergent mechanisms of disease across multiple mutations, allowing genetic rescue experiments at scale to rapidly identify causative pathways in disease development. Drosophila also allows for screens to identify genetic or pharmacological interventions that ameliorate organismal health in addition to cellular toxicity, a potential advantage over cellular screening platforms.
FIGURE 5.
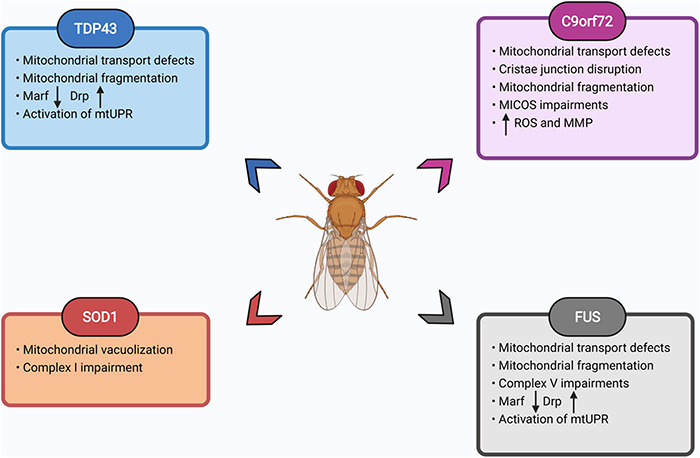
Summary of mitochondrial defects observed in fly FTD/ALS models. List of prominent mitochondrial impairments observed in fly models of TDP43, SOD1, FUS, and C9orf72 expansion. Please see text for details. Images created with BioRender.com.
In particular, flies could be used to address some of the following outstanding questions:
-
•
In addition to GR, how does concurrent expression of all possible DPRs, and combinations of DPRs, from C9 repeats affect mitochondrial biology?
-
•
Does the rescue of specific mitochondrial defects, for example MICOS integrity or Complex I activity, rescue lifespan and healthspan across models of multiple disease-associated mutations?
-
•
Flies possess unique genetic toolsets that allow precise cell-type-specific and temporal control of gene expression. This places them in an ideal setting to address questions of cell-type and age-related susceptibility to disease. Why are certain neuronal populations most susceptible to the mutations that cause disease, and why only later in life? Are mitochondrial defects the first observable impairment, or do they follow other markers of neurodegeneration?
-
•
Conversely, the same temporally controlled genetic tools can be used to answer questions related to therapeutic development. Can the rescue of a mitochondrial defect later on in disease development still rescue animal health, suggesting it could be a useful therapeutic avenue for patients already displaying symptoms?
Future research using Drosophila, particularly when complemented with confirmatory studies in mammalian or human systems, therefore has the potential to speed up and offer more thorough understanding of therapeutic strategies to address devastating neurodegenerative diseases.
Author Contributions
SA and TN wrote the manuscript. NW edited the manuscript. All authors contributed to the article and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Footnotes
Funding
This study was supported by the Alzheimer’s Society fellowship AS-JF-17b-011 (NW), ARUK Senior Fellowship ARUK-SRF2018A-003 (TN), MRC grant MR/V003585/1 (TN), and The Council of Trust for the People (MARA) (SA).
References
- Abou-Sleiman P. M., Muqit M. M., Wood N. W. (2006). Expanding insights of mitochondrial dysfunction in Parkinson’s disease. Nat. Rev. Neurosci. 7 207–219. 10.1038/nrn1868 [DOI] [PubMed] [Google Scholar]
- Agudelo A., St Amand V., Grissom L., Lafond D., Achilli T., Sahin A., et al. (2020). Age-dependent degeneration of an identified adult leg motor neuron in a Drosophila SOD1 model of ALS. Biol. Open 9:bio049692. 10.1242/bio.049692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alami N. H., Smith R. B., Carrasco M. A., Williams L. A., Winborn C. S., Han S. S. W., et al. (2014). Axonal Transport of TDP-43 mRNA granules is impaired by ALS-Causing mutations. Neuron 81 536–543. 10.1016/j.neuron.2013.12.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albrecht S. C., Barata A. G., Großhans J., Teleman A. A., Dick T. P. (2011). In Vivo mapping of hydrogen peroxide and oxidized glutathione reveals chemical and regional specificity of redox homeostasis. Cell Metab. 14 819–829. 10.1016/j.cmet.2011.10.010 [DOI] [PubMed] [Google Scholar]
- Alsultan A. A., Waller R., Heath P. R., Kirby J. (2016). The genetics of amyotrophic lateral sclerosis: current insights. Degener. Neurol. Neuromuscul. Dis. 6 49–64. 10.2147/DNND.S84956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altanbyek V., Cha S. J., Kang G. U., Im D. S., Lee S., Kim H. J., et al. (2016). Imbalance of mitochondrial dynamics in Drosophila models of amyotrophic lateral sclerosis. Biochem. Biophys. Res. Commun. 481 259–264. 10.1016/j.bbrc.2016.10.134 [DOI] [PubMed] [Google Scholar]
- Anandatheerthavarada H. K., Devi L. (2007). Amyloid precursor protein and mitochondrial dysfunction in Alzheimer’s disease. Neuroscientist 13 626–638. 10.1177/1073858407303536 [DOI] [PubMed] [Google Scholar]
- Arai T., Hasegawa M., Akiyama H., Ikeda K., Nonaka T., Mori H., et al. (2006). TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem. Biophys. Res. Commun. 351 602–611. 10.1016/j.bbrc.2006.10.093 [DOI] [PubMed] [Google Scholar]
- Arce-Molina R., Cortés-Molina F., Sandoval P. Y., Galaz A., Alegría K., Schirmeier S., et al. (2020). A highly responsive pyruvate sensor reveals pathway-regulatory role of the mitochondrial pyruvate carrier MPC. Elife 9:e53917. 10.7554/eLife.53917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ash P. E., Bieniek K. F., Gendron T. F., Caulfield T., Lin W.-L., Dejesus-Hernandez M., et al. (2013). Unconventional translation of C9ORF72 GGGGCC expansion generates insoluble polypeptides specific to c9FTD/ALS. Neuron 77 639–646. 10.1016/j.neuron.2013.02.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashburner M., Bergman C. M. (2005). Drosophila melanogaster: a case study of a model genomic sequence and its consequences. Genome Res. 15 1661–1667. 10.1101/gr.3726705 [DOI] [PubMed] [Google Scholar]
- Ayala Y. M., Zago P., D’Ambrogio A., Xu Y.-F., Petrucelli L., Buratti E., et al. (2008). Structural determinants of the cellular localization and shuttling of TDP-43. J. Cell Sci. 121 3778–3785. 10.1242/jcs.038950 [DOI] [PubMed] [Google Scholar]
- Bahadorani S., Mukai S. T., Rabie J., Beckman J. S., Phillips J. P., Hilliker A. J. (2013). Expression of zinc-deficient human superoxide dismutase in Drosophila neurons produces a locomotor defect linked to mitochondrial dysfunction. Neurobiol. Aging 34 2322–2330. 10.1016/j.neurobiolaging.2013.03.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker K. D., Thummel C. S. (2007). Diabetic larvae and obese flies—emerging studies of metabolism in Drosophila. Cell Metab. 6 257–266. 10.1016/j.cmet.2007.09.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker M., Mackenzie I. R., Pickering-Brown S. M., Gass J., Rademakers R., Lindholm C., et al. (2006). Mutations in progranulin cause tau-negative frontotemporal dementia linked to chromosome 17. Nature 442 916–919. 10.1038/nature05016 [DOI] [PubMed] [Google Scholar]
- Baker N. E. (1987). Molecular cloning of sequences from wingless, a segment polarity gene in Drosophila: the spatial distribution of a transcript in embryos. EMBO J. 6 1765–1773. 10.1002/j.1460-2075.1987.tb02429.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldwin K. R., Godena V. K., Hewitt V. L., Whitworth A. J. (2016). Axonal transport defects are a common phenotype in Drosophila models of ALS. Hum. Mol. Genet. 25 2378–2392. 10.1093/hmg/ddw105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bangi E., Ang C., Smibert P., Uzilov A. V., Teague A. G., Antipin Y., et al. (2019). A personalized platform identifies trametinib plus zoledronate for a patient with KRAS-mutant metastatic colorectal cancer. Sci. Adv. 5:eaav6528. 10.1126/sciadv.aav6528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bannwarth S., Ait-El-Mkadem S., Chaussenot A., Genin E. C., Lacas-Gervais S., Fragaki K., et al. (2014). A mitochondrial origin for frontotemporal dementia and amyotrophic lateral sclerosis through CHCHD10 involvement. Brain 137 2329–2345. 10.1093/brain/awu138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrientos A. (2002). In vivo and in organello assessment of OXPHOS activities. Methods 26 307–316. 10.1016/S1046-2023(02)00036-1 [DOI] [PubMed] [Google Scholar]
- Barrientos A., Fontanesi F., Díaz F. (2009). Evaluation of the mitochondrial respiratory chain and oxidative phosphorylation system using polarography and spectrophotometric enzyme assays. Curr. Protoc. Hum. Genet. 63 19–23. 10.1002/0471142905.hg1903s63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bate M., Arias A. M. (1993). The Development of Drosophila Melanogaster. New York: Cold Spring Harbor Laboratory Press. [Google Scholar]
- Bäumer D., East S. Z., Tseu B., Zeman A., Hilton D., Talbot K., et al. (2014). FTLD-ALS of TDP-43 type and SCA2 in a family with a full ataxin-2 polyglutamine expansion. Acta Neuropathol. 128 597–604. 10.1007/s00401-014-1277-z [DOI] [PubMed] [Google Scholar]
- Beal M. F., Ferrante R. J., Browne S. E., Matthews R. T., Kowall N. W., Brown R. H., Jr. (1997). Increased 3-nitrotyrosine in both sporadic and familial amyotrophic lateral sclerosis. Ann. Neurol. 42 644–654. 10.1002/ana.410420416 [DOI] [PubMed] [Google Scholar]
- Beckwith E. J., French A. S. (2019). Sleep in drosophila and its context. Front. Physiol. 10:1167. 10.3389/fphys.2019.01167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bejsovec A. (2018). Wingless signaling: a genetic journey from morphogenesis to metastasis. Genetics 208 1311–1336. 10.1534/genetics.117.300157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belfiori-Carrasco L. F., Marcora M. S., Bocai N. I., Ceriani M. F., Morelli L., Castaño E. M. (2017). A novel genetic screen identifies modifiers of age-dependent amyloid β toxicity in the drosophila brain. Front. Aging Neurosci. 9:61. 10.3389/fnagi.2017.00061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellen H. J., Levis R. W., Liao G., He Y., Carlson J. W., Tsang G., et al. (2004). The BDGP gene disruption project: single transposon insertions associated with 40% of Drosophila genes. Genetics 167 761–781. 10.1534/genetics.104.026427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bensimon G., Lacomblez L., Meininger V. (1994). A controlled trial of riluzole in amyotrophic lateral sclerosis. ALS/RiluzoleStudy Group. N. Engl. J. Med. 330 585–591. 10.1056/NEJM199403033300901 [DOI] [PubMed] [Google Scholar]
- Benzer S. (1967). Behavioral mutants of drosophila isolated by countercurrent distribution. Proc. Natl. Acad. Sci. U. S. A. 58 1112–1119. 10.1073/pnas.58.3.1112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertolin G., Bulteau A. L., Alves-Guerra M. C., Burel A., Lavault M. T., Gavard O., et al. (2018). Aurora kinase a localises to mitochondria to control organelle dynamics and energy production. Elife 7:e38111. 10.7554/eLife.38111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bharucha K. N. (2009). The epicurean fly: using drosophila melanogaster to study metabolism. Pediatr. Res. 65 132–137. 10.1203/PDR.0b013e318191fc68 [DOI] [PubMed] [Google Scholar]
- Bhatti J. S., Bhatti G. K., Reddy P. H. (2017). Mitochondrial dysfunction and oxidative stress in metabolic disorders — A step towards mitochondria based therapeutic strategies. Biochim. Biophys. Acta Mol. Basis Dis. 1863 1066–1077. 10.1016/j.bbadis.2016.11.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bilen J., Bonini N. M. (2007). Genome-wide screen for modifiers of ataxin-3 neurodegeneration in drosophila. PLoS Genet. 3:e177. 10.1371/journal.pgen.0030177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bilsland L. G., Sahai E., Kelly G., Golding M., Greensmith L., Schiavo G. (2010). Deficits in axonal transport precede ALS symptoms in vivo. Proc. Natl. Acad. Sci. U. S. A. 107 20523–20528. 10.1073/pnas.1006869107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birch-Machin M. A., Turnbull D. M. (2001). “Chapter 5 Assaying mitochondrial respiratory complex activity in mitochondria isolated from human cells and tissues,” in Mitochondria, eds Pon L. A., Schon E. A. (Cambridge: Academic Press; ), 97–117. 10.1016/s0091-679x(01)65006-4 [DOI] [PubMed] [Google Scholar]
- Bischof J., Björklund M., Furger E., Schertel C., Taipale J., Basler K. (2013). A versatile platform for creating a comprehensive UAS-ORFeome library in Drosophila. Development 140 2434–2442. 10.1242/dev.088757 [DOI] [PubMed] [Google Scholar]
- Boeynaems S., Bogaert E., Michiels E., Gijselinck I., Sieben A., Jovièiæ A., et al. (2016). Drosophila screen connects nuclear transport genes to DPR pathology in c9ALS/FTD. Sci. Rep. 6:20877. 10.1038/srep20877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonnay F., Veloso A., Steinmann V., Köcher T., Abdusselamoglu M. D., Bajaj S., et al. (2020). Oxidative metabolism drives immortalization of neural stem cells during tumorigenesis. Cell 182 1490–1507.e19. 10.1016/j.cell.2020.07.039 [DOI] [PubMed] [Google Scholar]
- Bosetti F., Brizzi F., Barogi S., Mancuso M., Siciliano G., Tendi E. A., et al. (2002). Cytochrome c oxidase and mitochondrial F1F0-ATPase (ATP synthase) activities in platelets and brain from patients with Alzheimer’s disease. Neurobiol. Aging 23 371–376. 10.1016/s0197-4580(01)00314-1 [DOI] [PubMed] [Google Scholar]
- Brand A. H., Perrimon N. (1993). Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development 118 401–415. 10.1242/dev.118.2.401 [DOI] [PubMed] [Google Scholar]
- Brown D. G., Shorter J., Wobst H. J. (2020). Emerging small-molecule therapeutic approaches for amyotrophic lateral sclerosis and frontotemporal dementia. Bioorganic Med. Chem. Lett. 30:126942. 10.1016/j.bmcl.2019.126942. [DOI] [PubMed] [Google Scholar]
- Buchanan R. L., Benzer S. (1993). Defective Glia in the Drosophila Brain Degeneration Mutant drop-dead. Neuron 10 839–850. 10.1016/0896-6273(93)90200-B [DOI] [PubMed] [Google Scholar]
- Burman J. L., Itsara L. S., Kayser E. B., Suthammarak W., Wang A. M., Kaeberlein M., et al. (2014). A Drosophila model of mitochondrial disease caused by a complex I mutation that uncouples proton pumping from electron transfer. Dis. Model. Mech. 7 1165–1174. 10.1242/dmm.015321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butzlaff M., Hannan S. B., Karsten P., Lenz S., Ng J., Voßfeldt H., et al. (2015). Impaired retrograde transport by the Dynein/Dynactin complex contributes to Tau-induced toxicity. Hum. Mol. Genet. 24 3623–3637. 10.1093/hmg/ddv107 [DOI] [PubMed] [Google Scholar]
- Byrne S., Walsh C., Lynch C., Bede P., Elamin M., Kenna K., et al. (2011). Rate of familial amyotrophic lateral sclerosis: a systematic review and meta-analysis. J. Neurol. Neurosurg. Psychiatry 82 623–627. 10.1136/jnnp.2010.224501 [DOI] [PubMed] [Google Scholar]
- Casterton R. L., Hunt R. J., Fanto M. (2020). Pathomechanism heterogeneity in the amyotrophic lateral sclerosis and frontotemporal dementia disease spectrum: providing focus through the lens of autophagy. J. Mol. Biol. 432 2692–2713. 10.1016/j.jmb.2020.02.018 [DOI] [PubMed] [Google Scholar]
- Celotto A. M., Frank A. C., McGrath S. W., Fergestad T., Van Voorhies W. A., Buttle K. F., et al. (2006). Mitochondrial encephalomyopathy in drosophila. J. Neurosci. 26 810–820. 10.1523/JNEUROSCI.4162-05.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L., Na R., Danae McLane K., Thompson C. S., Gao J., Wang X., et al. (2021). Overexpression of ferroptosis defense enzyme Gpx4 retards motor neuron disease of SOD1G93A mice. Sci. Rep. 11:12890. 10.1038/s41598-021-92369-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z., Zhang F., Xu H. (2019). Human mitochondrial DNA diseases and Drosophila models. J. Genet. Genomics 46 201–212. 10.1016/j.jgg.2019.03.009 [DOI] [PubMed] [Google Scholar]
- Chiò A., Calvo A., Mazzini L., Cantello R., Mora G., Moglia C., et al. (2012). Extensive genetics of ALS: a population-based study in Italy. Neurology 79 1983–1989. 10.1212/WNL.0b013e3182735d36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi S. Y., Lopez-Gonzalez R., Krishnan G., Phillips H. L., Li A. N., Seeley W. W., et al. (2019). C9ORF72-ALS/FTD-associated poly(GR) binds Atp5a1 and compromises mitochondrial function in vivo. Nat. Neurosci. 22 851–862. 10.1038/s41593-019-0397-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chowdhary S., Tomer D., Dubal D., Sambre D., Rikhy R. (2017). Analysis of mitochondrial organization and function in the Drosophila blastoderm embryo. Sci. Rep. 7:5502. 10.1038/s41598-017-05679-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cirulli E. T., Lasseigne B. N., Petrovski S., Sapp P. C., Dion P. A., Leblond C. S., et al. (2015). Exome sequencing in amyotrophic lateral sclerosis identifies risk genes and pathways. Science 347 1436–1441. 10.1126/science.aaa3650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clancy D. J., Gems D., Harshman L. G., Oldham S., Stocker H., Hafen E., et al. (2001). Extension of life-span by loss of CHICO, a Drosophila insulin receptor substrate protein. Science 292 104–106. 10.1126/science.1057991 [DOI] [PubMed] [Google Scholar]
- Clark I. E., Dodson M. W., Jiang C., Cao J. H., Huh J. R., Seol J. H., et al. (2006). Drosophila pink1 is required for mitochondrial function and interacts genetically with parkin. Nature 441 1162–1166. 10.1038/nature04779 [DOI] [PubMed] [Google Scholar]
- Cochemé H. M., Quin C., McQuaker S. J., Cabreiro F., Logan A., Prime T. A., et al. (2011). Measurement of H2O2 within living drosophila during aging using a ratiometric mass spectrometry probe targeted to the mitochondrial matrix. Cell Metab. 13 340–350. 10.1016/j.cmet.2011.02.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook C. N., Wu Y., Odeh H. M., Gendron T. F., Jansen-West K., del Rosso G., et al. (2020). C9orf72 poly(GR) aggregation induces TDP-43 proteinopathy. Sci. Transl. Med. 12:eabb3774. 10.1126/scitranslmed.abb3774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper-Knock J., Walsh M. J., Higginbottom A., Robin Highley J., Dickman M. J., Edbauer D., et al. (2014). Sequestration of multiple RNA recognition motif-containing proteins by C9orf72 repeat expansions. Brain 137 2040–2051. 10.1093/brain/awu120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa A. C., Loh S. H. Y., Martins L. M. (2013). Drosophila Trap1 protects against mitochondrial dysfunction in a PINK1/parkin model of Parkinson’s disease. Cell Death Dis. 4:e467. 10.1038/cddis.2012.205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cozzolino M., Carrì M. T. (2012). Mitochondrial dysfunction in ALS. Prog. Neurobiol. 97 54–66. 10.1016/j.pneurobio.2011.06.003 [DOI] [PubMed] [Google Scholar]
- Cruts M., Gijselinck I., van der Zee J., Engelborghs S., Wils H., Pirici D., et al. (2006). Null mutations in progranulin cause ubiquitin-positive frontotemporal dementia linked to chromosome 17q21. Nature 442 920–924. 10.1038/nature05017 [DOI] [PubMed] [Google Scholar]
- Dafinca R., Scaber J., Ababneh N., Lalic T., Weir G., Christian H., et al. (2016). C9orf72 hexanucleotide expansions are associated with altered endoplasmic reticulum calcium homeostasis and stress granule formation in induced pluripotent stem cell-derived neurons from patients with amyotrophic lateral sclerosis and frontotemporal demen. Stem Cells 34 2063–2078. 10.1002/stem.2388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damiano M., Starkov A. A., Petri S., Kipiani K., Kiaei M., Mattiazzi M., et al. (2006). Neural mitochondrial Ca2+ capacity impairment precedes the onset of motor symptoms in G93A Cu/Zn-superoxide dismutase mutant mice. J. Neurochem. 96 1349–1361. 10.1111/j.1471-4159.2006.03619.x [DOI] [PubMed] [Google Scholar]
- De Robertis E. M., Sasai Y. (1996). A common plan for dorsoventral patterning in Bilateria. Nature 380 37–40. 10.1038/380037a0 [DOI] [PubMed] [Google Scholar]
- DeJesus-Hernandez M., Mackenzie I. R., Boeve B. F., Boxer A. L., Baker M., Rutherford N. J., et al. (2011). Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-Linked FTD and ALS. Neuron 72 245–256. 10.1016/j.neuron.2011.09.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng H., Dodson M. W., Huang H., Guo M. (2008). The Parkinson’s disease genes pink1 and parkin promote mitochondrial fission and/or inhibit fusion in Drosophila. Proc. Natl. Acad. Sci. U. S. A. 105 14503–14508. 10.1073/pnas.0803998105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng H.-X., Chen W., Hong S.-T., Boycott K. M., Gorrie G. H., Siddique N., et al. (2011). Mutations in UBQLN2 cause dominant X-linked juvenile and adult-onset ALS and ALS/dementia. Nature 477 211–215. 10.1038/nature10353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng J., Wang P., Chen X., Cheng H., Liu J., Fushimi K., et al. (2018). FUS interacts with ATP synthase beta subunit and induces mitochondrial unfolded protein response in cellular and animal models. Proc. Natl. Acad. Sci. U. S. A. 115 E9678–E9686. 10.1073/pnas.1806655115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng J., Yang M., Chen Y., Chen X., Liu J., Sun S., et al. (2015). FUS interacts with HSP60 to promote mitochondrial damage. PLoS Genet. 11:e1005357. 10.1371/journal.pgen.1005357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeVorkin L., Go N. E., Hou Y.-C. C., Moradian A., Morin G. B., Gorski S. M. (2014). The Drosophila effector caspase Dcp-1 regulates mitochondrial dynamics and autophagic flux via SesB. J. Cell Biol. 205 477–492. 10.1083/jcb.201303144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaper D. C., Adachi Y., Lazarou L., Greenstein M., Simoes F. A., Di Domenico A., et al. (2013). Drosophila TDP-43 dysfunction in glia and muscle cells cause cytological and behavioural phenotypes that characterize ALS and FTLD. Hum. Mol. Genet. 22 3883–3893. 10.1093/hmg/ddt243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietzl G., Chen D., Schnorrer F., Su K.-C., Barinova Y., Fellner M., et al. (2007). A genome-wide transgenic RNAi library for conditional gene inactivation in Drosophila. Nature 448 151–156. 10.1038/nature05954 [DOI] [PubMed] [Google Scholar]
- Doherty J., Logan M. A., Taşdemir O. E., Freeman M. R. (2009). Ensheathing glia function as phagocytes in the adult Drosophila brain. J. Neurosci. 29 4768–4781. 10.1523/JNEUROSCI.5951-08.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duffy J. B. (2002). GAL4 system in Drosophila: a fly geneticist’s Swiss army knife. Genesis 34 1–15. 10.1002/gene.10150 [DOI] [PubMed] [Google Scholar]
- Elden A. C., Kim H.-J., Hart M. P., Chen-Plotkin A. S., Johnson B. S., Fang X., et al. (2010). Ataxin-2 intermediate-length polyglutamine expansions are associated with increased risk for ALS. Nature 466 1069–1075. 10.1038/nature09320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elisa R., Rainero I., Chiò A., Rogaeva E., Galimberti D., Fenoglio P., et al. (2012). SQSTM1 mutations in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Neurology 79 1556–1562. 10.1212/WNL.0b013e31826e25df [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fallini C., Khalil B., Smith C. L., Rossoll W. (2020). Traffic jam at the nuclear pore: all roads lead to nucleocytoplasmic transport defects in ALS/FTD. Neurobiol. Dis. 140:104835. 10.1016/j.nbd.2020.104835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feany M. B., Bender W. W. (2000). A Drosophila model of Parkinson’s disease. Nature 404 394–398. 10.1038/35006074 [DOI] [PubMed] [Google Scholar]
- Fecto F., Siddique T. (2011). Making connections: pathology and genetics link amyotrophic lateral sclerosis with frontotemporal lobe dementia. J. Mol. Neurosci. 45 663–675. 10.1007/s12031-011-9637-9 [DOI] [PubMed] [Google Scholar]
- Feiguin F., Godena V. K., Romano G., D’Ambrogio A., Klima R., Baralle F. E. (2009). Depletion of TDP-43 affects Drosophila motoneurons terminal synapsis and locomotive behavior. FEBS Lett. 583 1586–1592. 10.1016/j.febslet.2009.04.019 [DOI] [PubMed] [Google Scholar]
- Fernández-Ayala D. J. M., Chen S., Kemppainen E., O’Dell K. M. C., Jacobs H. T. (2010). Gene expression in a Drosophila model of mitochondrial disease. PLoS One 5:e8549. 10.1371/journal.pone.0008549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrante R. J., Browne S. E., Shinobu L. A., Bowling A. C., Baik M. J., MacGarvey U., et al. (1997). Evidence of increased oxidative damage in both sporadic and familial amyotrophic lateral sclerosis. J. Neurochem. 69 2064–2074. 10.1046/j.1471-4159.1997.69052064.x [DOI] [PubMed] [Google Scholar]
- Ferrari R., Kapogiannis D., Huey E. D., Momeni P. (2011). FTD and ALS: a tale of two diseases. Curr. Alzheimer Res. 8 273–294. 10.2174/156720511795563700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferri A., Fiorenzo P., Nencini M., Cozzolino M., Pesaresi M. G., Valle C., et al. (2010). Glutaredoxin 2 prevents aggregation of mutant SOD1 in mitochondria and abolishes its toxicity. Hum. Mol. Genet. 19 4529–4542. 10.1093/hmg/ddq383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer J. A., Giniger E., Maniatis T., Ptashne M. (1988). GAL4 activates transcription in Drosophila. Nature 332 853–856. 10.1038/332853a0 [DOI] [PubMed] [Google Scholar]
- Fleming R. J. (1998). Structural conservation of Notch receptors and ligands. Semin. Cell Dev. Biol. 9 599–607. 10.1006/scdb.1998.0260 [DOI] [PubMed] [Google Scholar]
- Frazier A. E., Thorburn D. R. (2012). “Biochemical analyses of the electron transport chain complexes by spectrophotometry,” in Mitochondrial Disorders: Biochemical and Molecular Analysis, ed. Wong L. J. C. (Totowa: Humana Press; ), 49–62. 10.1007/978-1-61779-504-6_4 [DOI] [PubMed] [Google Scholar]
- Freibaum B. D., Lu Y., Lopez-Gonzalez R., Kim N. C., Almeida S., Lee K.-H., et al. (2015). GGGGCC repeat expansion in C9orf72 compromises nucleocytoplasmic transport. Nature 525 129–133. 10.1038/nature14974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freischmidt A., Wieland T., Richter B., Ruf W., Schaeffer V., Müller K., et al. (2015). Haploinsufficiency of TBK1 causes familial ALS and fronto-temporal dementia. Nat. Neurosci. 18 631–636. 10.1038/nn.4000 [DOI] [PubMed] [Google Scholar]
- Gallart-Palau X., Ng C. H., Ribera J., Sze S. K., Lim K. L. (2016). Drosophila expressing human SOD1 successfully recapitulates mitochondrial phenotypic features of familial amyotrophic lateral sclerosis. Neurosci. Lett. 624 47–52. 10.1016/j.neulet.2016.05.006 [DOI] [PubMed] [Google Scholar]
- Garesse R., Kaguni L. S. (2005). A drosophila model of mitochondrial DNA replication: proteins, genes and regulation. IUBMB Life 57 555–561. 10.1080/15216540500215572 [DOI] [PubMed] [Google Scholar]
- Gendron T. F., Belzil V. V., Zhang Y. J., Petrucelli L. (2014). Mechanisms of toxicity in C9FTLD/ALS. Acta Neuropathol. 127 359–376. 10.1007/s00401-013-1237-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genin E. C., Bannwarth S., Lespinasse F., Ortega-Vila B., Fragaki K., Itoh K., et al. (2018). Loss of MICOS complex integrity and mitochondrial damage, but not TDP-43 mitochondrial localisation, are likely associated with severity of CHCHD10-related diseases. Neurobiol. Dis. 119 159–171. 10.1016/j.nbd.2018.07.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giordana M. T., Piccinini M., Grifoni S., De Marco G., Vercellino M., Magistrello M., et al. (2010). TDP-43 redistribution is an early event in sporadic amyotrophic lateral sclerosis. Brain Pathol. 20 351–360. 10.1111/j.1750-3639.2009.00284.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greaves C. V., Rohrer J. D. (2019). An update on genetic frontotemporal dementia. J. Neurol. 266 2075–2086. 10.1007/s00415-019-09363-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenspan R. J. (1997). Fly Pushing: the Theory and Practice of Drosophila Genetics. New York: Cold Spring Harbour Laboratory Press. [Google Scholar]
- Greenway M. J., Andersen P. M., Russ C., Ennis S., Cashman S., Donaghy C., et al. (2006). ANG mutations segregate with familial and “sporadic” amyotrophic lateral sclerosis. Nat. Genet. 38 411–413. 10.1038/ng1742 [DOI] [PubMed] [Google Scholar]
- Gregory J. M., Fagegaltier D., Phatnani H., Harms M. B. (2020). Genetics of amyotrophic lateral sclerosis. Curr. Genet. Med. Rep. 8 121–131. 10.1007/s40142-020-00194-8 [DOI] [Google Scholar]
- Guo C., Sun L., Chen X., Zhang D. (2013). Oxidative stress, mitochondrial damage and neurodegenerative diseases. Neural Regen. Res. 8 2003–2014. 10.3969/j.issn.1673-5374.2013.21.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hales K. G., Fuller M. T. (1997). Developmentally regulated mitochondrial fusion mediated by a conserved, novel, predicted GTPase. Cell 90 121–129. 10.1016/s0092-8674(00)80319-0 [DOI] [PubMed] [Google Scholar]
- Hardy J., Rogaeva E. (2014). Motor neuron disease and frontotemporal dementia: Sometimes related, sometimes not. Exp. Neurol. 262 75–83. 10.1016/j.expneurol.2013.11.006 [DOI] [PubMed] [Google Scholar]
- Higgins C. M. J., Jung C., Xu Z. (2003). ALS-associated mutant SOD1G93A causes mitochondrial vacuolation by expansion of the intermembrane space and by involvement of SOD1 aggregation and peroxisomes. BMC Neurosci. 4:16. 10.1186/1471-2202-4-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirth F. (2010). Drosophila melanogaster in the study of human neurodegeneration. CNS Neurol. Disord. Drug Targets 9 504–523. 10.2174/187152710791556104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holley S. A., Jackson P. D., Sasai Y., Lu B., De Robertis E. M., Hoffmann F. M., et al. (1995). A conserved system for dorsal-ventral patterning in insects and vertebrates involving sog and chordin. Nature 376 249–253. 10.1038/376249a0 [DOI] [PubMed] [Google Scholar]
- Horan M. P., Pichaud N., Ballard J. W. O. (2012). Review: quantifying mitochondrial dysfunction in complex diseases of aging. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 67 1022–1035. 10.1093/gerona/glr263 [DOI] [PubMed] [Google Scholar]
- Huang E. J., Zhang J., Geser F., Trojanowski J. Q., Strober J. B., Dickson D. W., et al. (2010). Extensive FUS-immunoreactive pathology in juvenile amyotrophic lateral sclerosis with basophilic inclusions. Brain Pathol. 20 1069–1076. 10.1111/j.1750-3639.2010.00413.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutton M., Lendon C. L., Rizzu P., Baker M., Froelich S., Houlden H., et al. (1998). Association of missense and 5’-splice-site mutations in tau with the inherited dementia FTDP-17. Nature 393 702–705. 10.1038/31508 [DOI] [PubMed] [Google Scholar]
- Hwang R.-D., Wiemerslage L., LaBreck C. J., Khan M., Kannan K., Wang X., et al. (2014). The neuroprotective effect of human uncoupling protein 2 (hUCP2) requires cAMP-dependent protein kinase in a toxin model of Parkinson’s disease. Neurobiol. Dis. 69 180–191. 10.1016/j.nbd.2014.05.032 [DOI] [PubMed] [Google Scholar]
- Inagaki H. K., Kamikouchi A., Ito K. (2010). Methods for quantifying simple gravity sensing in Drosophila melanogaster. Nat. Protoc. 5 20–25. 10.1038/nprot.2009.196 [DOI] [PubMed] [Google Scholar]
- Jaarsma D., Rognoni F., Van Duijn W., Verspaget H. W., Haasdijk E. D., Holstege J. C. (2001). CuZn superoxide dismutase (SOD1) accumulates in vacuolated mitochondria in transgenic mice expressing amyotrophic lateral sclerosis-linked SOD1 mutations. Acta Neuropathol. 102 293–305. 10.1007/s004010100399 [DOI] [PubMed] [Google Scholar]
- Jackson G. R., Salecker I., Dong X., Yao X., Arnheim N., Faber P. W., et al. (1998). Polyglutamine-expanded human huntingtin transgenes induce degeneration of Drosophila photoreceptor neurons. Neuron 21 633–642. 10.1016/S0896-6273(00)80573-5 [DOI] [PubMed] [Google Scholar]
- Jacobs H. T., Fernández-Ayala D. J. M., Manjiry S., Kemppainen E., Toivonen J. M., O’Dell K. M. (2004). Mitochondrial disease in flies. Biochim. Biophys. Acta Bioenerg. 1659 190–196. 10.1016/j.bbabio.2004.07.004 [DOI] [PubMed] [Google Scholar]
- Janssen R. C., Boyle K. E. (2019). Microplate assays for spectrophotometric measurement of mitochondrial enzyme activity. Methods Mol. Biol. 1978 355–368. 10.1007/978-1-4939-9236-2_22 [DOI] [PubMed] [Google Scholar]
- Jenett A., Rubin G. M., Ngo T.-T. B., Shepherd D., Murphy C., Dionne H., et al. (2012). A GAL4-driver line resource for Drosophila neurobiology. Cell Rep. 2 991–1001. 10.1016/j.celrep.2012.09.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson J. O., Mandrioli J., Benatar M., Abramzon Y., Van Deerlin V. M., Trojanowski J. Q., et al. (2010). Exome Sequencing Reveals VCP Mutations as a Cause of Familial ALS. Neuron 68 857–864. 10.1016/j.neuron.2010.11.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson J. O., Pioro E. P., Boehringer A., Chia R., Feit H., Renton A. E., et al. (2014). Mutations in the Matrin 3 gene cause familial amyotrophic lateral sclerosis. Nat. Neurosci. 17 664–666. 10.1038/nn.3688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kann O., Kovács R. (2007). Mitochondria and neuronal activity. Am. J. Physiol. Physiol. 292 C641–C657. 10.1152/ajpcell.00222.2006 [DOI] [PubMed] [Google Scholar]
- Kasture A. S., Hummel T., Sucic S., Freissmuth M. (2018). Big lessons from tiny flies: drosophila melanogaster as a model to explore dysfunction of dopaminergic and serotonergic neurotransmitter systems. Int. J. Mol. Sci. 19:1788. 10.3390/ijms19061788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khalil B., Cabirol-Pol M. J., Miguel L., Whitworth A. J., Lecourtois M., Liévens J. C. (2017). Enhancing Mitofusin/Marf ameliorates neuromuscular dysfunction in Drosophila models of TDP-43 proteinopathies. Neurobiol. Aging 54 71–83. 10.1016/j.neurobiolaging.2017.02.016 [DOI] [PubMed] [Google Scholar]
- Kim H. J., Taylor J. P. (2017). Lost in transportation: nucleocytoplasmic transport defects in ALS and other neurodegenerative diseases. Neuron 96 285–297. 10.1016/j.neuron.2017.07.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J., Koo B.-K., Knoblich J. A. (2020). Human organoids: model systems for human biology and medicine. Nat. Rev. Mol. Cell Biol. 21 571–584. 10.1038/s41580-020-0259-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- King A. E., Woodhouse A., Kirkcaldie M. T. K., Vickers J. C. (2016). Excitotoxicity in ALS: overstimulation, or overreaction? Exp. Neurol. 275 162–171. 10.1016/j.expneurol.2015.09.019 [DOI] [PubMed] [Google Scholar]
- Kong J., Xu Z. (1998). Massive mitochondrial degeneration in motor neurons triggers the onset of amyotrophic lateral sclerosis in mice expressing a mutant SOD1. J. Neurosci. 18 3241–3250. 10.1523/jneurosci.18-09-03241.1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwiatkowski T. J., Bosco D. A., LeClerc A. L., Tamrazian E., Vanderburg C. R., Russ C., et al. (2009). Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science 323 1205–1208. 10.1126/science.1166066 [DOI] [PubMed] [Google Scholar]
- Lacomblez L., Bensimon G., Leigh P. N., Guillet P., Meininger V. (1996). Dose-ranging study of riluzole in amyotrophic lateral sclerosis. Amyotrophic lateral sclerosis/Riluzole Study Group II. Lancet 347 1425–1431. 10.1016/s0140-6736(96)91680-3 [DOI] [PubMed] [Google Scholar]
- Lagier-Tourenne C., Polymenidou M., Cleveland D. W. (2010). TDP-43 and FUS/TLS: emerging roles in RNA processing and neurodegeneration. Hum. Mol. Genet. 19 R46–R64. 10.1093/hmg/ddq137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamb R., Rohrer J. D., Real R., Lubbe S. J., Waite A. J., Blake D. J., et al. (2019). A novel TBK1 mutation in a family with diverse frontotemporal dementia spectrum disorders. Cold Spring Harb. Mol. Case Stud. 5:a003913. 10.1101/mcs.a003913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Ber I., Camuzat A., Guerreiro R., Bouya-Ahmed K., Bras J., Nicolas G., et al. (2013). SQSTM1 Mutations in french patients with frontotemporal dementia or frontotemporal dementia with amyotrophic lateral sclerosis. JAMA Neurol. 70 1403–1410. 10.1001/jamaneurol.2013.3849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehmer C., Schludi M. H., Ransom L., Greiling J., Junghänel M., Exner N., et al. (2018). A novel CHCHD10 mutation implicates a Mia40−dependent mitochondrial import deficit in ALS. EMBO Mol. Med. 10:e8558. 10.15252/emmm.201708558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenz S., Karsten P., Schulz J. B., Voigt A. (2013). Drosophila as a screening tool to study human neurodegenerative diseases. J. Neurochem. 127 453–460. 10.1111/jnc.12446 [DOI] [PubMed] [Google Scholar]
- Lewis O. L., Farr C. L., Kaguni L. S. (1995). Drosophila melanogaster mitochondrial DNA: completion of the nucleotide sequence and evolutionary comparisons. Insect Mol. Biol. 4 263–278. 10.1111/j.1365-2583.1995.tb00032.x [DOI] [PubMed] [Google Scholar]
- Li S., Wu Z., Li Y., Tantray I., De Stefani D., Mattarei A., et al. (2020). Altered MICOS Morphology and Mitochondrial Ion Homeostasis Contribute to Poly(GR) Toxicity Associated with C9-ALS/FTD. Cell Rep. 32:107989. 10.1016/j.celrep.2020.107989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin M.-J., Cheng C.-W., Shen C.-K. J. (2011). Neuronal function and dysfunction of Drosophila dTDP. PLoS One 6:e20371. 10.1371/journal.pone.0020371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling S. C., Polymenidou M., Cleveland D. W. (2013). Converging mechanisms in als and FTD: disrupted RNA and protein homeostasis. Neuron 79 416–438. 10.1016/j.neuron.2013.07.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liscic R. M., Alberici A., Cairns N. J., Romano M., Buratti E. (2020). From basic research to the clinic: innovative therapies for ALS and FTD in the pipeline. Mol. Neurodegener. 15:31. 10.1186/s13024-020-00373-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu R., Li B., Flanagan S. W., Oberley L. W., Gozal D., Qiu M. (2002). Increased mitochondrial antioxidative activity or decreased oxygen free radical propagation prevent mutant SOD1-mediated motor neuron cell death and increase amyotrophic lateral sclerosis-like transgenic mouse survival. J. Neurochem. 80 488–500. 10.1046/j.0022-3042.2001.00720.x [DOI] [PubMed] [Google Scholar]
- Liu W., Yamashita T., Tian F., Morimoto N., Ikeda Y., Deguchi K., et al. (2013). Mitochondrial fusion and fission proteins expression dynamically change in a murine model of amyotrophic lateral sclerosis. Curr. Neurovasc. Res. 10 222–230. 10.2174/15672026113109990060 [DOI] [PubMed] [Google Scholar]
- Lopez-Gonzalez R., Lu Y., Gendron T. F., Karydas A., Tran H., Yang D., et al. (2016). Poly(GR) in C9ORF72-Related ALS/FTD compromises mitochondrial function and increases oxidative stress and DNA Damage in iPSC-derived motor neurons. Neuron 92 383–391. 10.1016/j.neuron.2016.09.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y., Ferris J., Gao F. B. (2009). Frontotemporal dementia and amyotrophic lateral sclerosis-associated disease protein TDP-43 promotes dendritic branching. Mol. Brain 2 1–10. 10.1186/1756-6606-2-30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luan H., Diao F., Scott R. L., White B. H. (2020). The drosophila split Gal4 system for neural circuit mapping. Front. Neural Circ. 14:603397. 10.3389/fncir.2020.603397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lue N. F., Chasman D. I., Buchman A. R., Kornberg R. D. (1987). Interaction of GAL4 and GAL80 gene regulatory proteins in vitro. Mol. Cell. Biol. 7 3446–3451. 10.1128/mcb.7.10.3446-3451.1987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lutas A., Wahlmark C. J., Acharjee S., Kawasaki F. (2012). Genetic analysis in drosophila reveals a role for the mitochondrial protein P32 in synaptic transmission. G3 (Bethesda) 2 59–69. 10.1534/g3.111.001586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackenzie I. R., Nicholson A. M., Sarkar M., Messing J., Purice M. D., Pottier C., et al. (2017). TIA1 Mutations in amyotrophic lateral sclerosis and frontotemporal dementia promote phase separation and alter stress granule dynamics. Neuron 95 808–816.e9. 10.1016/j.neuron.2017.07.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackenzie I. R., Rademakers R., Neumann M. (2010). TDP-43 and FUS in amyotrophic lateral sclerosis and frontotemporal dementia. Lancet Neurol. 9 995–1007. 10.1016/S1474-4422(10)70195-2 [DOI] [PubMed] [Google Scholar]
- Majounie E., Renton A. E., Mok K., Dopper E. G. P., Waite A., Rollinson S., et al. (2012). Frequency of the C9orf72 hexanucleotide repeat expansion in patients with amyotrophic lateral sclerosis and frontotemporal dementia: a cross-sectional study. Lancet Neurol. 11 323–330. 10.1016/S1474-4422(12)70043-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manjaly Z. R., Scott K. M., Abhinav K., Wijesekera L., Ganesalingam J., Goldstein L. H., et al. (2010). The sex ratio in amyotrophic lateral sclerosis: a population based study. Amyotroph. Lateral Scler. 11 439–442. 10.3109/17482961003610853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mariottini P., Shah Z. H., Toivonen J. M., Bagni C., Spelbrink J. N., Amaldi F., et al. (1999). Expression of the gene for mitoribosomal protein S12 is controlled in human cells at the levels of transcription, RNA splicing, and translation. J. Biol. Chem. 274 31853–31862. 10.1074/jbc.274.45.31853 [DOI] [PubMed] [Google Scholar]
- Martinaud O., Laquerrière A., Guyant-Maréchal L., Ahtoy P., Vera P., Sergeant N., et al. (2005). Frontotemporal dementia, motor neuron disease and tauopathy: clinical and neuropathological study in a family. Acta Neuropathol. 110 84–92. 10.1007/s00401-005-1028-2 [DOI] [PubMed] [Google Scholar]
- Maruyama H., Morino H., Ito H., Izumi Y., Kato H., Watanabe Y., et al. (2010). Mutations of optineurin in amyotrophic lateral sclerosis. Nature 465 223–226. 10.1038/nature08971 [DOI] [PubMed] [Google Scholar]
- Mateen F. J., Carone M., Sorenson E. J. (2010). Patients who survive 5 years or more with ALS in Olmsted County, 1925-2004. J. Neurol. Neurosurg. Psychiatry 81 1144–1146. 10.1136/jnnp.2009.201251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattiazzi M., D’Aurelio M., Gajewski C. D., Martushova K., Kiaei M., Flint Beal M., et al. (2002). Mutated human SOD1 causes dysfunction of oxidative phosphorylation in mitochondria of transgenic mice. J. Biol. Chem. 277 29626–29633. 10.1074/jbc.M203065200 [DOI] [PubMed] [Google Scholar]
- McGurk L., Berson A., Bonini N. M. (2015). Drosophila as an in vivo model for human neurodegenerative disease. Genetics 201 377–402. 10.1534/genetics.115.179457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehta A. R., Gregory J. M., Dando O., Carter R. N., Burr K., Nanda J., et al. (2021). Mitochondrial bioenergetic deficits in C9orf72 amyotrophic lateral sclerosis motor neurons cause dysfunctional axonal homeostasis. Acta Neuropathol. 141 257–279. 10.1007/s00401-020-02252-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mejzini R., Flynn L. L., Pitout I. L., Fletcher S., Wilton S. D., Akkari P. A. (2019). ALS genetics, mechanisms, and therapeutics : where are we now? Front Neurosci. 13:1310. 10.3389/fnins.2019.01310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Méndez-López I., Sancho-Bielsa F. J., Engel T., García A. G., Padín J. F. (2021). Progressive Mitochondrial SOD1G93A accumulation causes severe structural, metabolic and functional aberrations through opa1 down-regulation in a mouse model of amyotrophic lateral sclerosis. Int. J. Mol. Sci. 22:8194. 10.3390/ijms22158194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menzies F. M., Yenisetti S. C., Min K.-T. (2005). Roles of Drosophila DJ-1 in survival of dopaminergic neurons and oxidative stress. Curr. Biol. 15 1578–1582. 10.1016/j.cub.2005.07.036 [DOI] [PubMed] [Google Scholar]
- Mercy L., Hodges J. R., Dawson K., Barker R. A., Brayne C. (2008). Incidence of early-onset dementias in Cambridgeshire, United Kingdom. Neurology 71 1496–1499. 10.1212/01.wnl.0000334277.16896.fa [DOI] [PubMed] [Google Scholar]
- Meulener M., Whitworth A. J., Armstrong-Gold C. E., Rizzu P., Heutink P., Wes P. D., et al. (2005). Drosophila DJ-1 mutants are selectively sensitive to environmental toxins associated with Parkinson’s disease. Curr. Biol. 15 1572–1577. 10.1016/j.cub.2005.07.064 [DOI] [PubMed] [Google Scholar]
- Millecamps S., Salachas F., Cazeneuve C., Gordon P., Bricka B., Camuzat A., et al. (2010). SOD1, ANG, VAPB, TARDBP, and FUS mutations in familial amyotrophic lateral sclerosis: genotype-phenotype correlations. J. Med. Genet. 47 554–560. 10.1136/jmg.2010.077180 [DOI] [PubMed] [Google Scholar]
- Millul A., Beghi E., Logroscino G., Micheli A., Vitelli E., Zardi A. (2005). Survival of patients with amyotrophic lateral sclerosis in a population-based registry. Neuroepidemiology 25 114–119. 10.1159/000086353 [DOI] [PubMed] [Google Scholar]
- Mizielinska S., Grönke S., Niccoli T., Ridler C. E., Clayton E. L., Devoy A., et al. (2014). C9orf72 repeat expansions cause neurodegeneration in Drosophila through arginine-rich proteins. Science 345 1192–1195. 10.1126/science.1256800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mockett R. J., Radyuk S. N., Benes J. J., Orr W. C., Sohal R. S. (2003). Phenotypic effects of familial amyotrophic lateral sclerosis Sod alleles in transgenic Drosophila. Proc. Natl. Acad. Sci. U. S. A. 100 301–306. 10.1073/pnas.0136976100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohr S. E., Hu Y., Kim K., Housden B. E., Perrimon N. (2014). Resources for functional genomics studies in Drosophila melanogaster. Genetics 197 1–18. 10.1534/genetics.113.154344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori K., Arzberger T., Grässer F. A., Gijselinck I., May S., Rentzsch K., et al. (2013). Bidirectional transcripts of the expanded C9orf72 hexanucleotide repeat are translated into aggregating dipeptide repeat proteins. Acta Neuropathol. 126 881–893. 10.1007/s00401-013-1189-3 [DOI] [PubMed] [Google Scholar]
- Morris O., Deng H., Tam C., Jasper H. (2020). Warburg-like metabolic reprogramming in aging intestinal stem cells contributes to tissue hyperplasia. Cell Rep. 33:108423. 10.1016/j.celrep.2020.108423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Münch C., Rosenbohm A., Sperfeld A.-D., Uttner I., Reske S., Krause B. J., et al. (2005). Heterozygous R1101K mutation of the DCTN1 gene in a family with ALS and FTD. Ann. Neurol. 58 777–780. 10.1002/ana.20631 [DOI] [PubMed] [Google Scholar]
- Münch C., Sedlmeier R., Meyer T., Homberg V., Sperfeld A. D., Kurt A., et al. (2004). Point mutations of the p150 subunit of dynactin (DCTN1) gene in ALS. Neurology 63 724–726. 10.1212/01.wnl.0000134608.83927.b1 [DOI] [PubMed] [Google Scholar]
- Nakaya T., Maragkakis M. (2018). Amyotrophic Lateral Sclerosis associated FUS mutation shortens mitochondria and induces neurotoxicity. Sci. Rep. 8:15575. 10.1038/s41598-018-33964-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann M., Sampathu D. M., Kwong L. K., Truax A. C., Micsenyi M. C., Chou T. T., et al. (2006). Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314 130–133. 10.1126/science.1134108 [DOI] [PubMed] [Google Scholar]
- Nguyen H. P., Van Broeckhoven C., van der Zee J. (2018). ALS Genes in the Genomic Era and their Implications for FTD. Trends Genet. 34 404–423. 10.1016/j.tig.2018.03.001 [DOI] [PubMed] [Google Scholar]
- Nicholson L., Singh G. K., Osterwalder T., Roman G. W., Davis R. L., Keshishian H. (2008). Spatial and temporal control of gene expression in Drosophila using the inducible GeneSwitch GAL4 system. I. Screen for larval nervous system drivers. Genetics 178 215–234. 10.1534/genetics.107.081968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimura A. L., Mitne-Neto M., Silva H. C. A., Richieri-Costa A., Middleton S., Cascio D., et al. (2004). A mutation in the vesicle-trafficking protein VAPB causes late-onset spinal muscular atrophy and amyotrophic lateral sclerosis. Am. J. Hum. Genet. 75 822–831. 10.1086/425287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onesto E., Colombrita C., Gumina V., Borghi M. O., Dusi S., Doretti A., et al. (2016). Gene-specific mitochondria dysfunctions in human TARDBP and C9ORF72 fibroblasts. Acta Neuropathol. Commun. 4:47. 10.1186/s40478-016-0316-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onyike C. U., Diehl-Schmid J. (2013). The epidemiology of frontotemporal dementia. Int. Rev. Psychiatry 25 130–137. 10.3109/09540261.2013.776523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orr H. T. (2011). FTD and ALS: genetic ties that bind. Neuron 72 189–190. 10.1016/j.neuron.2011.10.001 [DOI] [PubMed] [Google Scholar]
- Orsini M., Oliveira A. B., Nascimento O. J. M., Reis C. H. M., Leite M. A. A., de Souza J. A., et al. (2015). Amyotrophic lateral sclerosis: new perpectives and update. Neurol. Int. 7 39–47. 10.4081/ni.2015.5885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osterwalder T., Yoon K. S., White B. H., Keshishian H. (2001). A conditional tissue-specific transgene expression system using inducible GAL4. Proc. Natl. Acad. Sci. U. S. A. 98 12596–12601. 10.1073/pnas.221303298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandya R. S., Zhu H., Li W., Bowser R., Friedlander R. M., Wang X. (2013). Therapeutic neuroprotective agents for amyotrophic lateral sclerosis. Cell. Mol. Life Sci. 70 4729–4745. 10.1007/s00018-013-1415-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pardo C. A., Xu Z., Borchelt D. R., Price D. L., Sisodia S. S., Cleveland D. W. (1995). Superoxide dismutase is an abundant component in cell bodies, dendrites, and axons of motor neurons and in a subset of other neurons. Proc. Natl. Acad. Sci. U. S. A. 92 954–958. 10.1073/pnas.92.4.954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park J., Lee S. B., Lee S., Kim Y., Song S., Kim S., et al. (2006). Mitochondrial dysfunction in Drosophila PINK1 mutants is complemented by parkin. Nature 441 1157–1161. 10.1038/nature04788 [DOI] [PubMed] [Google Scholar]
- Parkinson N., Ince P. G., Smith M. O., Highley R., Skibinski G., Andersen P. M., et al. (2006). ALS phenotypes with mutations in CHMP2B (charged multivesicular body protein 2B). Neurology 67 1074–1077. 10.1212/01.wnl.0000231510.89311.8b [DOI] [PubMed] [Google Scholar]
- Perkins L. A., Holderbaum L., Tao R., Hu Y., Sopko R., McCall K., et al. (2015). The transgenic RNAi project at harvard medical school: resources and validation. Genetics 201 843–852. 10.1534/genetics.115.180208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry C. G., Kane D. A., Lanza I. R., Neufer P. D. (2013). Methods for assessing mitochondrial function in diabetes. Diabetes 62 1041–1053. 10.2337/db12-1219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry S. W., Norman J. P., Barbieri J., Brown E. B., Gelbard H. A. (2011). Mitochondrial membrane potential probes and the proton gradient: a practical usage guide. Biotechniques 50 98–115. 10.2144/000113610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeiffenberger C., Lear B. C., Keegan K. P., Allada R. (2010). Locomotor activity level monitoring using the Drosophila Activity Monitoring (DAM) System. Cold Spring Harb. Protoc. 2010:pdb.rot5518. 10.1101/pdb.prot5518 [DOI] [PubMed] [Google Scholar]
- Pfeiffer B. D., Jenett A., Hammonds A. S., Ngo T.-T. B., Misra S., Murphy C., et al. (2008). Tools for neuroanatomy and neurogenetics in Drosophila. Proc. Natl. Acad. Sci. U. S. A. 105 9715–9720. 10.1073/pnas.0803697105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piazza N., Wessells R. J. (2011). Drosophila models of cardiac disease. Prog. Mol. Biol. Transl. Sci. 100 155–210. 10.1016/B978-0-12-384878-9.00005-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piper M. D., Partridge L. (2016). Protocols to study aging in drosophila. Methods Mol. Biol. 1478 291–302. 10.1007/978-1-4939-6371-3_18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piper M. D., Partridge L. (2018). Drosophila as a model for ageing. Biochim. Biophys. Acta Mol. Basis Dis. 1864 2707–2717. 10.1016/j.bbadis.2017.09.016 [DOI] [PubMed] [Google Scholar]
- Pizzino G., Irrera N., Cucinotta M., Pallio G., Mannino F., Arcoraci V., et al. (2017). Oxidative stress: harms and benefits for human health. Oxid. Med. Cell. Longev. 2017:8416763. 10.1155/2017/8416763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poole A. C., Thomas R. E., Andrews L. A., McBride H. M., Whitworth A. J., Pallanck L. J. (2008). The PINK1/Parkin pathway regulates mitochondrial morphology. Proc. Natl. Acad. Sci. U. S. A. 105 1638–1643. 10.1073/pnas.0709336105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pottier C., Bieniek K. F., Finch N., van de Vorst M., Baker M., Perkersen R., et al. (2015). Whole-genome sequencing reveals important role for TBK1 and OPTN mutations in frontotemporal lobar degeneration without motor neuron disease. Acta Neuropathol. 130 77–92. 10.1007/s00401-015-1436-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Presley A. D., Fuller K. M., Arriaga E. A. (2003). MitoTracker Green labeling of mitochondrial proteins and their subsequent analysis by capillary electrophoresis with laser-induced fluorescence detection. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 793 141–150. 10.1016/s1570-0232(03)00371-4 [DOI] [PubMed] [Google Scholar]
- Puls I., Jonnakuty C., LaMonte B. H., Holzbaur E. L. F., Tokito M., Mann E., et al. (2003). Mutant dynactin in motor neuron disease. Nat. Genet. 33 455–456. 10.1038/ng1123 [DOI] [PubMed] [Google Scholar]
- Rademakers R., Neumann M., Mackenzie I. R. (2012). Advances in understanding the molecular basis of frontotemporal dementia. Nat. Rev. Neurol. 8 423–434. 10.1038/nrneurol.2012.117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rana A., Oliveira M. P., Khamoui A. V., Aparicio R., Rera M., Rossiter H. B., et al. (2017). Promoting Drp1-mediated mitochondrial fission in midlife prolongs healthy lifespan of Drosophila melanogaster. Nat. Commun. 8:448. 10.1038/s41467-017-00525-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiter L. T., Potocki L., Chien S., Gribskov M., Bier E. (2001). A systematic analysis of human disease-associated gene sequences in Drosophila melanogaster. Genome Res. 11 1114–1125. 10.1101/gr.169101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renton A. E., Majounie E., Waite A., Simón-Sánchez J., Rollinson S., Gibbs J. R., et al. (2011). A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 72 257–268. 10.1016/j.neuron.2011.09.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riddle R. D., Johnson R. L., Laufer E., Tabin C. (1993). Sonic hedgehog mediates the polarizing activity of the ZPA. Cell 75 1401–1416. 10.1016/0092-8674(93)90626-2 [DOI] [PubMed] [Google Scholar]
- Rizzardini M., Mangolini A., Lupi M., Ubezio P., Bendotti C., Cantoni L. (2005). Low levels of ALS-linked Cu/Zn superoxide dismutase increase the production of reactive oxygen species and cause mitochondrial damage and death in motor neuron-like cells. J. Neurol. Sci. 232 95–103. 10.1016/j.jns.2005.02.004 [DOI] [PubMed] [Google Scholar]
- Roberts S., Speirs V. (2017). Advances in the development of improved animal-free models for use in breast cancer biomedical research. Biophys. Rev. 9 321–327. 10.1007/s12551-017-0276-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roelofs B. A., Ge S. X., Studlack P. E., Polster B. M. (2015). Low micromolar concentrations of the superoxide probe MitoSOX uncouple neural mitochondria and inhibit complex IV. Free Radic. Biol. Med. 86 250–258. 10.1016/j.freeradbiomed.2015.05.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero-Garcia S., Prado-Garcia H. (2019). Mitochondrial calcium: transport and modulation of cellular processes in homeostasis and cancer (Review). Int. J. Oncol. 54 1155–1167. 10.3892/ijo.2019.4696 [DOI] [PubMed] [Google Scholar]
- Rosen D. R., Siddiquet T., Pattersont D., Figlewicz D. A., Ii P. S., Hentatit A., et al. (1993). Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 362 59–62. [DOI] [PubMed] [Google Scholar]
- Royden C. S., Pirrotta V., Jan L. Y. (1987). The tko locus, site of a behavioral mutation in D. melanogaster, codes for a protein homologous to prokaryotic ribosomal protein S12. Cell 51 165–173. 10.1016/0092-8674(87)90144-9 [DOI] [PubMed] [Google Scholar]
- Rubino E., Mancini C., Boschi S., Ferrero P., Ferrone M., Bianca S., et al. (2019). ATXN2 intermediate repeat expansions influence the clinical phenotype in frontotemporal dementia. Neurobiol. Aging 73 231.e7–231.e9. 10.1016/j.neurobiolaging.2018.09.009 [DOI] [PubMed] [Google Scholar]
- Şahin A., Held A., Bredvik K., Major P., Achilli T.-M., Kerson A. G., et al. (2017). Human SOD1 ALS mutations in a drosophila knock-in model cause severe phenotypes and reveal dosage-sensitive gain- and loss-of-function components. Genetics 205 707–723. 10.1534/genetics.116.190850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salvatori I., Ferri A., Scaricamazza S., Giovannelli I., Serrano A., Rossi S., et al. (2018). Differential toxicity of TAR DNA-binding protein 43 isoforms depends on their submitochondrial localization in neuronal cells. J. Neurochem. 146 585–597. 10.1111/jnc.14465 [DOI] [PubMed] [Google Scholar]
- Sasaki S., Iwata M. (2007). Mitochondrial alterations in the spinal cord of patients with sporadic amyotrophic lateral sclerosis. J. Neuropathol. Exp. Neurol. 66 10–16. 10.1097/nen.0b013e31802c396b [DOI] [PubMed] [Google Scholar]
- Sasaki S., Warita H., Murakami T., Abe K., Iwata M. (2004). Ultrastructural study of mitochondria in the spinal cord of transgenic mice with a G93A mutant SOD1 gene. Acta Neuropathol. 107 461–474. 10.1007/s00401-004-0837-z [DOI] [PubMed] [Google Scholar]
- Schieber M., Chandel N. S. (2014). ROS function in redox signaling and oxidative stress. Curr. Biol. 24 R453–R462. 10.1016/j.cub.2014.03.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seager R., Lee L., Henley J. M., Wilkinson K. A. (2020). Mechanisms and roles of mitochondrial localisation and dynamics in neuronal function. Neuronal Signal. 4:NS20200008. 10.1042/NS20200008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selman C., Lingard S., Choudhury A. I., Batterham R. L., Claret M., Clements M., et al. (2008). Evidence for lifespan extension and delayed age-related biomarkers in insulin receptor substrate 1 null mice. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 22 807–818. 10.1096/fj.07-9261com [DOI] [PubMed] [Google Scholar]
- Seltman R. E., Matthews B. R. (2012). Frontotemporal lobar degeneration: epidemiology, pathology, diagnosis and management. CNS Drugs 26 841–870. 10.2165/11640070-000000000-00000 [DOI] [PubMed] [Google Scholar]
- Sen A., Cox R. T. (2017). “Chapter one - Fly models of human diseases: drosophila as a model for understanding human mitochondrial mutations and disease,” in Fly Models of Human Diseases, ed. Pick L. (Cambridge: Academic Press; ), 1–27. 10.1016/bs.ctdb.2016.07.001 [DOI] [PubMed] [Google Scholar]
- Shahheydari H., Ragagnin A., Walker A. K., Toth R. P., Vidal M., Jagaraj C. J., et al. (2017). Protein quality control and the amyotrophic lateral sclerosis/frontotemporal dementia continuum. Front. Mol. Neurosci. 10:119. 10.3389/fnmol.2017.00119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw C. E., Enayat Z. E., Chioza B. A., Al-Chalabi A., Radunovic A., Powell J. F., et al. (1998). Mutations in all five exons of SOD-1 may cause ALS. Ann. Neurol. 43 390–394. 10.1002/ana.410430319 [DOI] [PubMed] [Google Scholar]
- Shi K. Y., Mori E., Nizami Z. F., Lin Y., Kato M., Xiang S., et al. (2017). Toxic PR(n) poly-dipeptides encoded by the C9orf72 repeat expansion block nuclear import and export. Proc. Natl. Acad. Sci. U. S. A. 114 E1111–E1117. 10.1073/pnas.1620293114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibata N., Nagai R., Uchida K., Horiuchi S., Yamada S., Hirano A., et al. (2001). Morphological evidence for lipid peroxidation and protein glycoxidation in spinal cords from sporadic amyotrophic lateral sclerosis patients. Brain Res. 917 97–104. 10.1016/S0006-8993(01)02926-2 [DOI] [PubMed] [Google Scholar]
- Sivandzade F., Bhalerao A., Cucullo L. (2019). Analysis of the mitochondrial membrane potential using the cationic JC-1 dye as a sensitive fluorescent probe. Bio-Protocol. 9:e3128. 10.21769/bioprotoc.3128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skibinski G., Parkinson N. J., Brown J. M., Chakrabarti L., Lloyd S. L., Hummerich H., et al. (2005). Mutations in the endosomal ESCRTIII-complex subunit CHMP2B in frontotemporal dementia. Nat. Genet. 37 806–808. 10.1038/ng1609 [DOI] [PubMed] [Google Scholar]
- Smith B. N., Ticozzi N., Fallini C., Gkazi A. S., Topp S., Kenna K. P., et al. (2014). Exome-wide rare variant analysis identifies TUBA4A mutations associated with familial ALS. Neuron 84 324–331. 10.1016/j.neuron.2014.09.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith E. F., Shaw P. J., De Vos K. J. (2019). The role of mitochondria in amyotrophic lateral sclerosis. Neurosci. Lett. 710:132933. 10.1016/j.neulet.2017.06.052 [DOI] [PubMed] [Google Scholar]
- Sofola O., Kerr F., Rogers I., Killick R., Augustin H., Gandy C., et al. (2010). Inhibition of GSK-3 ameliorates Abeta pathology in an adult-onset Drosophila model of Alzheimer’s disease. PLoS Genet. 6:e1001087. 10.1371/journal.pgen.1001087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song J., Tanouye M. A. (2008). From bench to drug: human seizure modeling using Drosophila. Prog. Neurobiol. 84 182–191. 10.1016/j.pneurobio.2007.10.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- St Johnston D. (2002). The art and design of genetic screens: drosophila melanogaster. Nat. Rev. Genet. 3 176–188. 10.1038/nrg751 [DOI] [PubMed] [Google Scholar]
- Stoica R., De Vos K. J., Paillusson S., Mueller S., Sancho R. M., Lau K.-F., et al. (2014). ER-mitochondria associations are regulated by the VAPB-PTPIP51 interaction and are disrupted by ALS/FTD-associated TDP-43. Nat. Commun. 5:3996. 10.1038/ncomms4996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stolow D. T., Haynes S. R. (1995). Cabeza, a Drosophila gene encoding a novel RNA binding protein, shares homology with EWS and TLS, two genes involved in human sarcoma formation. Nucleic Acids Res. 23 835–843. 10.1093/nar/23.5.835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stork T., Bernardos R., Freeman M. R. (2012). Analysis of glial cell development and function in Drosophila. Cold Spring Harb. Protoc. 7 1–17. 10.1101/pdb.top067587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straub I. R., Weraarpachai W., Shoubridge E. A. (2021). Multi-OMICS study of a CHCHD10 variant causing ALS demonstrates metabolic rewiring and activation of endoplasmic reticulum and mitochondrial unfolded protein responses. Hum. Mol. Genet. 30 687–705. 10.1093/hmg/ddab078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y., Curle A. J., Haider A. M., Balmus G. (2020). The role of DNA damage response in amyotrophic lateral sclerosis. Essays Biochem. 64 847–861. 10.1042/EBC20200002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Synofzik M., Maetzler W., Grehl T., Prudlo J., vom Hagen J. M., Haack T., et al. (2012). Screening in ALS and FTD patients reveals 3 novel UBQLN2 mutations outside the PXX domain and a pure FTD phenotype. Neurobiol. Aging 33 2949.e13–7. 10.1016/j.neurobiolaging.2012.07.002 [DOI] [PubMed] [Google Scholar]
- Tafuri F., Ronchi D., Magri F., Comi G. P., Corti S. (2015). SOD1 misplacing and mitochondrial dysfunction in amyotrophic lateral sclerosis pathogenesis. Front. Cell. Neurosci. 9:336. 10.3389/fncel.2015.00336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tam V., Patel N., Turcotte M., Bossé Y., Paré G., Meyre D. (2019). Benefits and limitations of genome-wide association studies. Nat. Rev. Genet. 20 467–484. 10.1038/s41576-019-0127-1 [DOI] [PubMed] [Google Scholar]
- Tao Z., Wang H., Xia Q., Li K., Li K., Jiang X., et al. (2015). Nucleolar stress and impaired stress granule formation contribute to C9orf72 RAN translation-induced cytotoxicity. Hum. Mol. Genet. 24 2426–2441. 10.1093/hmg/ddv005 [DOI] [PubMed] [Google Scholar]
- Tatar M., Kopelman A., Epstein D., Tu M. P., Yin C. M., Garofalo R. S. (2001). A mutant Drosophila insulin receptor homolog that extends life-span and impairs neuroendocrine function. Science 292 107–110. 10.1126/science.1057987 [DOI] [PubMed] [Google Scholar]
- Thaker H. M., Kankel D. R. (1992). Mosaic analysis gives an estimate of the extent of genomic involvement in the development of the visual system in Drosophila melanogaster. Genetics 131 883–894. 10.1093/genetics/131.4.883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toivonen J. M., O’Dell K. M., Petit N., Irvine S. C., Knight G. K., Lehtonen M., et al. (2001). Technical knockout, a Drosophila model of mitochondrial deafness. Genetics 159 241–254. 10.1093/genetics/159.1.241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tradewell M. L., Yu Z., Tibshirani M., Boulanger M. C., Durham H. D., Richard S. (2012). Arginine methylation by prmt1 regulates nuclear-cytoplasmic localization and toxicity of FUS/TLS harbouring ALS-linked mutations. Hum. Mol. Genet. 21 136–149. 10.1093/hmg/ddr448 [DOI] [PubMed] [Google Scholar]
- Tran H., Almeida S., Moore J., Gendron T. F., Chalasani U. D., Lu Y., et al. (2015). Differential Toxicity of Nuclear RNA Foci versus Dipeptide Repeat Proteins in a Drosophila Model of C9ORF72 FTD/ALS. Neuron 87 1207–1214. 10.1016/j.neuron.2015.09.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai P.-I., Lin C.-H., Hsieh C.-H., Papakyrikos A. M., Kim M. J., Napolioni V., et al. (2018). PINK1 Phosphorylates MIC60/Mitofilin to control structural plasticity of mitochondrial crista junctions. Mol. Cell 69 744–756.e6. 10.1016/j.molcel.2018.01.026 [DOI] [PubMed] [Google Scholar]
- Tsai Y.-L., Coady T. H., Lu L., Zheng D., Alland I., Tian B., et al. (2020). ALS/FTD-associated protein FUS induces mitochondrial dysfunction by preferentially sequestering respiratory chain complex mRNAs. Genes Dev. 34 785–805. 10.1101/gad.335836.119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuyama T., Kishikawa J., Han Y.-W., Harada Y., Tsubouchi A., Noji H., et al. (2013). In Vivo Fluorescent Adenosine 5’-Triphosphate (ATP) Imaging of drosophila melanogaster and caenorhabditis elegans by using a genetically encoded fluorescent ATP biosensor optimized for low temperatures. Anal. Chem. 85 7889–7896. 10.1021/ac4015325 [DOI] [PubMed] [Google Scholar]
- Turner M. R., Al-Chalabi A., Chio A., Hardiman O., Kiernan M. C., Rohrer J. D., et al. (2017). Genetic screening in sporadic ALS and FTD. J. Neurol. Neurosurg. Psychiatry 88 1042–1044. 10.1136/jnnp-2017-315995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Blitterswijk M., Dejesus-Hernandez M., Rademakers R. (2012). How do C9ORF72 repeat expansions cause amyotrophic lateral sclerosis and frontotemporal dementia: can we learn from other noncoding repeat expansion disorders? Curr. Opin. Neurol. 25 689–700. 10.1097/WCO.0b013e32835a3efb [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Langenhove T., Van Der Zee J., Van Broeckhoven C. (2012). The molecular basis of the frontotemporal lobar degeneration-amyotrophic lateral sclerosis spectrum. Ann. Med. 44 817–828. 10.3109/07853890.2012.665471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vance C., Lehmann R., Broihier H. T., Moore L. A., Lehmann R., Lehmann R., et al. (2009). Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science 323 1208–1211. 10.1126/science.1165942.Mutations [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandamme T. (2014). Use of rodents as models of human diseases. J. Pharm. Bioallied Sci. 6 2–9. 10.4103/0975-7406.124301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vargas M. R., Johnson D. A., Johnson J. A. (2011). Decreased glutathione accelerates neurological deficit and mitochondrial pathology in familial ALS-linked hSOD1(G93A) mice model. Neurobiol. Dis. 43 543–551. 10.1016/j.nbd.2011.04.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J.-W., Brent J. R., Tomlinson A., Shneider N. A., McCabe B. D. (2011). The ALS-associated proteins FUS and TDP-43 function together to affect Drosophila locomotion and life span. J. Clin. Invest. 121 4118–4126. 10.1172/JCI57883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L. J., Hsu T., Lin H. L., Fu C. Y. (2020). Drosophila MICOS knockdown impairs mitochondrial structure and function and promotes mitophagy in muscle tissue. Biol. Open 9:bio054262. 10.1242/bio.054262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang P., Deng J., Dong J., Liu J., Bigio E. H., Mesulam M., et al. (2019). TDP-43 induces mitochondrial damage and activates the mitochondrial unfolded protein response. PLoS Genet. 15:e1007947. 10.1371/journal.pgen.1007947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T., Liu H., Itoh K., Oh S., Zhao L., Murata D., et al. (2021). C9orf72 regulates energy homeostasis by stabilizing mitochondrial complex I assembly. Cell Metab. 33 531–546.e9. 10.1016/j.cmet.2021.01.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W., Li L., Lin W. L., Dickson D. W., Petrucelli L., Zhang T., et al. (2013). The ALS disease-associated mutant TDP-43 impairs mitochondrial dynamics and function in motor neurons. Hum. Mol. Genet. 22 4706–4719. 10.1093/hmg/ddt319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W., Wang L., Lu J., Siedlak S. L., Fujioka H., Liang J., et al. (2016). The inhibition of TDP-43 mitochondrial localization blocks its neuronal toxicity. Nat. Med. 22 869–878. 10.1038/nm.4130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warren J. D., Rohrer J. D., Rossor M. N. (2013). Frontotemporal dementia. BMJ 347:f4827. 10.1136/bmj.f4827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warrick J. M., Paulson H. L., Gray-Board G. L., Bui Q. T., Fischbeck K. H., Pittman R. N., et al. (1998). Expanded polyglutamine protein forms nuclear inclusions and causes neural degeneration in Drosophila. Cell 93 939–949. 10.1016/s0092-8674(00)81200-3 [DOI] [PubMed] [Google Scholar]
- Watts G. D., Wymer J., Kovach M. J., Mehta S. G., Mumm S., Darvish D., et al. (2004). Inclusion body myopathy associated with Paget disease of bone and frontotemporal dementia is caused by mutant valosin-containing protein. Nat. Genet. 36 377–381. 10.1038/ng1332 [DOI] [PubMed] [Google Scholar]
- Wawersik S., Maas R. L. (2000). Vertebrate eye development as modeled in Drosophila. Hum. Mol. Genet. 9 917–925. 10.1093/hmg/9.6.917 [DOI] [PubMed] [Google Scholar]
- Wen X., Tan W., Westergard T., Krishnamurthy K., Markandaiah S. S., Shi Y., et al. (2014). Antisense proline-arginine RAN dipeptides linked to C9ORF72-ALS/FTD form toxic nuclear aggregates that initiate in vitro and in vivo neuronal death. Neuron 84 1213–1225. 10.1016/j.neuron.2014.12.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whelan J., Burke B., Rice A., Tong M., Kuebler D. (2010). Sensitivity to seizure-like activity in Drosophila following acute hypoxia and hypercapnia. Brain Res. 1316 120–128. 10.1016/j.brainres.2009.12.036 [DOI] [PubMed] [Google Scholar]
- Wiedemann F. R., Manfredi G., Mawrin C., Beal M. F., Schon E. A. (2002). Mitochondrial DNA and respiratory chain function in spinal cords of ALS patients. J. Neurochem. 80 616–625. 10.1046/j.0022-3042.2001.00731.x [DOI] [PubMed] [Google Scholar]
- Williams K. L., Topp S., Yang S., Smith B., Fifita J. A., Warraich S. T., et al. (2016). CCNF mutations in amyotrophic lateral sclerosis and frontotemporal dementia. Nat. Commun. 7:11253. 10.1038/ncomms11253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winton M. J., Igaz L. M., Wong M. M., Kwong L. K., Trojanowski J. Q., Lee V. M.-Y. (2008). Disturbance of nuclear and cytoplasmic TAR DNA-binding protein (TDP-43) induces disease-like redistribution, sequestration, and aggregate formation. J. Biol. Chem. 283 13302–13309. 10.1074/jbc.M800342200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witte J. S. (2010). Genome-wide association studies and beyond. Annu. Rev. Public Health 31 9–20. 10.1146/annurev.publhealth.012809.103723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong C.-O., Karagas N. E., Jung J., Wang Q., Rousseau M. A., Chao Y., et al. (2021). Regulation of longevity by depolarization-induced activation of PLC-β-IP3R signaling in neurons. Proc. Natl. Acad. Sci. U. S. A. 118:e2004253118. 10.1073/pnas.2004253118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo J. A., Liu T., Trotter C., Fang C. C., De Narvaez E., Lepochat P., et al. (2017). Loss of function CHCHD10 mutations in cytoplasmic TDP-43 accumulation and synaptic integrity. Nat. Commun. 8:15558. 10.1038/ncomms15558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao Y., Karam C., Yi J., Zhang L., Li X., Yoon D., et al. (2018). ROS-related mitochondrial dysfunction in skeletal muscle of an ALS mouse model during the disease progression. Pharmacol. Res. 138 25–36. 10.1016/j.phrs.2018.09.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H., DeLuca S. Z., O’Farrell P. H. (2008). Manipulating the metazoan mitochondrial genome with targeted restriction enzymes. Science 321 575–577. 10.1126/science.1160226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu L., Liu T., Liu L., Yao X., Chen L., Fan D., et al. (2020). Global variation in prevalence and incidence of amyotrophic lateral sclerosis: a systematic review and meta-analysis. J. Neurol. 267 944–953. 10.1007/s00415-019-09652-y [DOI] [PubMed] [Google Scholar]
- Xu Z., Poidevin M., Li X., Li Y., Shu L., Nelson D. L., et al. (2013). Expanded GGGGCC repeat RNA associated with amyotrophic lateral sclerosis and frontotemporal dementia causes neurodegeneration. Proc. Natl. Acad. Sci. U. S. A. 110 7778–7783. 10.1073/pnas.1219643110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan J., Deng H.-X., Siddique N., Fecto F., Chen W., Yang Y., et al. (2010). Frameshift and novel mutations in FUS in familial amyotrophic lateral sclerosis and ALS/dementia. Neurology 75 807–814. 10.1212/WNL.0b013e3181f07e0c [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang D., Abdallah A., Li Z., Lu Y., Almeida S., Gao F.-B. (2015). FTD/ALS-associated poly(GR) protein impairs the Notch pathway and is recruited by poly(GA) into cytoplasmic inclusions. Acta Neuropathol. 130 525–535. 10.1007/s00401-015-1448-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y., Ouyang Y., Yang L., Beal M. F., McQuibban A., Vogel H., et al. (2008). Pink1 regulates mitochondrial dynamics through interaction with the fission/fusion machinery. Proc. Natl. Acad. Sci. U. S. A. 105 7070–7075. 10.1073/pnas.0711845105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yarosh W., Monserrate J., Tong J. J., Tse S., Le P. K., Nguyen K., et al. (2008). The molecular mechanisms of OPA1-mediated optic atrophy in drosophila model and prospects for antioxidant treatment. PLoS Genet. 4:e6. 10.1371/journal.pgen.0040006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu C.-H., Davidson S., Harapas C. R., Hilton J. B., Mlodzianoski M. J., Laohamonthonkul P., et al. (2020). TDP-43 Triggers Mitochondrial DNA Release via mPTP to Activate cGAS/STING in ALS. Cell 183 636–649.e18. 10.1016/j.cell.2020.09.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J., Wang Q., Wang M., Jiang M., Wang Y., Sun Y., et al. (2016). GASZ and mitofusin-mediated mitochondrial functions are crucial for spermatogenesis. EMBO Rep. 17 220–234. 10.15252/embr.201540846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang K., Coyne A. N., Lloyd T. E. (2018). Drosophila models of amyotrophic lateral sclerosis with defects in RNA metabolism. Brain Res. 1693 109–120. 10.1016/j.brainres.2018.04.043 [DOI] [PubMed] [Google Scholar]
- Zhang Y. J., Gendron T. F., Ebbert M. T. W., O’Raw A. D., Yue M., Jansen-West K., et al. (2018). Poly(GR) impairs protein translation and stress granule dynamics in C9orf72-associated frontotemporal dementia and amyotrophic lateral sclerosis. Nat. Med. 24 1136–1142. 10.1038/s41591-018-0071-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang K., Donnelly C. J., Haeusler A. R., Grima J. C., Machamer J. B., Steinwald P., et al. (2015). The C9orf72 repeat expansion disrupts nucleocytoplasmic transport. Nature 525 56–61. 10.1038/nature14973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y., Schmid B., Nikolaisen N. K., Rasmussen M. A., Aldana B. I., Agger M., et al. (2017). Patient iPSC-derived neurons for disease modeling of frontotemporal dementia with mutation in CHMP2B. Stem Cell Rep. 8 648–658. 10.1016/j.stemcr.2017.01.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y.-J., Guo L., Gonzales P. K., Gendron T. F., Wu Y., Jansen-West K., et al. (2019). Heterochromatin anomalies and double-stranded RNA accumulation underlie C9orf72 poly(PR) toxicity. Science 363:eaav2606. 10.1126/science.aav2606 [DOI] [PMC free article] [PubMed] [Google Scholar]