

Abstract
The coronavirus disease 2019 (COVID‐19) pandemic started in March 2020 and caused over 5 million confirmed deaths worldwide as far August 2021. We have been recently overwhelmed by a wide literature on how the immune system recognizes severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2) and contributes to COVID‐19 pathogenesis. Although originally considered a respiratory viral disease, COVID‐19 is now recognized as a far more complex, multi‐organ‐, immuno‐mediated‐, and mostly heterogeneous disorder. Though efficient innate and adaptive immunity may control infection, when the patient fails to mount an adequate immune response at the start, or in advanced disease, a high innate‐induced inflammation can lead to different clinical outcomes through heterogeneous compensatory mechanisms. The variability of viral load and persistence, the genetic alterations of virus‐driven receptors/signaling pathways and the plasticity of innate and adaptive responses may all account for the extreme heterogeneity of pathogenesis and clinical patterns. As recently applied to some inflammatory disorders as asthma, rhinosinusitis with polyposis, and atopic dermatitis, herein we suggest defining different endo‐types and the related phenotypes along COVID‐19. Patients should be stratified for evolving symptoms and tightly monitored for surrogate biomarkers of innate and adaptive immunity. This would allow to preventively identify each endo‐type (and its related phenotype) and to treat patients precisely with agents targeting pathogenic mechanisms.
Keywords: COVID‐19, endo‐types, innate and adaptive Immunity, pathogenesis, SARS‐CoV‐2
1. INTRODUCTION
The coronavirus disease 2019 (COVID‐19) caused by severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2) started in Wuhan (China), was declared a pandemic in 11 March 2020 by WHO and caused over 5 million confirmed deaths up to August 2021. In a paper published last year, we underscored the unknowns about virus receptors and signaling, host immune response, disease pathogenesis and therapeutic tools able to control virus entry, replication, and spread and harmful effects. 1 After over one year of pandemic, we have been overwhelmed by an enormous number of reports on SARS‐CoV‐2 infection and COVID‐19 pathogenesis. Although originally defined as a respiratory viral infection, COVID‐19 is now clearly recognized as a far more complex, multistep, multi‐organ, immuno‐mediated, and mostly heterogeneous disease.
The angiotensin‐converting enzyme‐2 (ACE2) is one of the SARS‐CoV2 receptors and in ACE2‐bearing cells (as monocytes/macrophages and epithelial cells), the virus triggers pattern recognition receptors as toll‐like receptors (TLRs) and cytosolic sensors. Their signaling essentially follows three pathways leading to the type I/III interferon (IFN) secretion, the expression of costimulatory molecules for T‐cell activation, and production of pro‐inflammatory cytokines and chemokines. 1 This contributes to the activation of innate and adaptive immune responses, in particular of NK cells and virus‐specific cytotoxic CD8+ T cells. In mild/moderate COVID‐19 patients, such CD8+T cells are clonally expanded with a high amplification rate and express tissue‐residence molecules (CXCR‐6, and XCL1). 2 In these patients, increased virus‐specific T follicular helper (Tfh) cells cooperate with B cells to mount a protective humoral response. It starts 1–2 weeks from infection, even though in some asymptomatic/mild cases antibodies (Abs) increase much later. In some asymptomatic people, the virus‐specific T cells and Abs are totally missing. 3 The virus‐specific IgA usually correlates with the IgG response, but the IgA monomers are twofold less potent than IgG, while the IgA dimers, associated with respiratory mucosa, are 7.5 times more potent than IgG. Thus, dimeric IgA response may be particularly valuable for protection against SARS‐CoV‐2. 4 In recovered individuals circulating resting memory B (mB), cells recognizing S1 proteins are consistently detectable. The anti‐S1 Abs decline rapidly in 4 months from the infection and remain detectable for at least 11 months, correlating with the number of long‐lived S1‐specific plasma cells in bone marrow. 5
Both innate and adaptive immunity control the viral infection and determine clinical recovery in the majority of infected people; however, the virus negatively impacts on the IFN signaling pathway in infected cells leading to an impaired viral control. This triggers a persistent innate response preventing adequate adaptive immune responses. These patients show reduced circulating CD4+ and CD8+ T cells expressing inflammatory genes, membrane activation markers, and altered function. 6 , 7 In severe/critical infections, CD8+ T cells do not display a clonal expansion, are phenotypically heterogeneous, and exhibit exhaustion phenotype. 2 Of note, the neutralizing anti‐virus (S1 and receptor binding domain ‐RBD) Abs are usually higher in critical as compared to the non‐critical patients. 8 , 9
Such prevalent innate‐induced inflammation can lead to a cytokine storm‐like disorder and interstitial pneumonia, contributing (together with direct action of the virus) to a diffuse organ involvement and failure. COVID‐19 pneumonia shows immunological features different from other common respiratory viral infections. For instance, it associates with higher levels of pro‐inflammatory cytokines, pro‐coagulative state, NLRP3 inflammasome (NI) activation, and altered fatty acids profiles as compared to H1N1 infection. 10 More importantly, the dysfunction of NK and T cells and the hyperactivation of macrophages and neutrophils in COVID‐19 respiratory infection are absent in H1N1 pneumonia. 11 Lastly, eosinopenia in COVID‐19 is more pronounced than in other viral infections, including influenza. 12 Although eosinophils can promote broad‐spectrum antiviral activity or serve to regulate immunity at sites of viral infection, it is at present unclear whether eosinopenia in COVID‐19 is consequent to their massive homing to tissues. The cytokine excess of severe COVID‐19 could be also responsible for eosinophils margination or apoptosis. 13 , 14 Notably, eosinophils of COVID‐19 patients overexpress the programmed death receptor ligand‐1 which is induced by IFN‐γ hyper‐production and correlates with disease severity. 15 , 16
At present, a major unanswered question is, why the viral immune evasion may predominate on the host immunity leading to hyper activation of innate response only in a proportion of patients.
2. IMMUNO‐PATHOGENESIS OF COVID‐19
Even though many hypotheses have been proposed, the causes inducing variable responses of the immune system along such a multistep disease are only partially known. 17 , 18 We will discuss such pathogenic hypotheses, taking into account that each mechanism is not mutually exclusive and underscoring the high heterogeneity of immune responses and clinical patterns.
2.1. Altered coordination between innate and adaptive immunity
The virus exploits mechanisms inhibiting components of the IFN signaling pathway in infected cells as plasmacytoid dendritic cells (pDCs). The low IFN detected in COVID‐19 patients suggests a potential defect in antiviral defenses, which is negatively associated with disease severity. 19 While SARS‐CoV‐2 is highly sensitive to IFN, it can interfere with downstream signaling at several levels, or inhibit IFN‐stimulated gene products. 20 Remarkably, ACE2 expression on epithelial cells is upregulated by IFN itself; thus, the virus can even utilize the residual anti‐viral molecule (IFN) for further entry into these cells. 21 In addition, pDCs (the main source of IFN) are decreased in COVID‐19 due to recruitment into tissues and apoptosis. 22 When the innate response lasts too long, SARS‐CoV‐2 further replicates and compromises the adaptive immune responses. 18 The reduced pDCs can be responsible for the impaired presentation of viral epitopes to T cells and for natural killer (NK) cells dysfunction. 23 Impaired NK cells can also be due to: (i) Over‐production of cytokines (mainly IL‐6) by infected cells, which induces NK exhaustion, 24 (ii) spike subunit 1 (S1) which, through GATA3 activation, mediates HLA‐E expression and CCL26 secretion by lung epithelium. CCL26 selectively favors NK cells homing into the lung; however, the interaction of HLA‐E with NKG2A‐CD94 inhibitory receptor leads to NK cell inhibition/exhaustion, 25 , 26 (iii) KLRC2 gene (encoding the activating receptor NKG2C) deletion or HLA‐E*0101/0103 variants (poorly recognized by NKG2C) with consequent reduction/absence of NKG2C+ “memory” NK cells 27 ; (iv) impaired release into bloodstream and tissues of “inflammatory” (CD34 + DNAM1brightCXCR4+) precursors from the bone marrow, rapidly differentiating into mature NK progenies. 28 , 29
The factors delaying adaptive immunity are mostly related to early impairment of innate responses. The increasing number of Myeloid‐Derived Suppressive Cells (MDSC) and of suppressive factors as endogenous corticosteroids, TGF‐β and IL‐10, may condition the timing of the adaptive cellular response. High serum IL‐10 in COVID‐19 (mainly in critical/non‐survivors) mimics systemic sepsis. 18 , 30 Delaying factors can be amplified by age, as elderly patients show few naive T cells and a limited TCR repertoire, deferring specific T and B cell responses. 31 To compensate defective T cells, innate cells may mount excessive responses leading to lung immunopathology, and multi‐organ damages. Notably, the virus can directly (via TLR2) or indirectly (via virus/anti‐virus Abs immune complexes, IC) activate the NI, favoring the overproduction of cytokines (prevalently IL‐1β, IL‐18, and IL‐23) and severe outcomes. 30 , 31 , 32 This agrees with reports in which innate cytokine/chemokine signatures of immunopathology 22 have been associated with end‐stage COVID‐19 disease. 33 The lack of temporal coordination between innate and adaptive responses allows the persistence of a sustained viral load, which can trigger subsequent waves of inflammation worsening clinical outcomes. 23
2.2. Pre‐existing immunity to the virus in unexposed individuals
SARS‐CoV‐2‐specific T cells have been detected in about 50% of unexposed individuals, suggesting T cell cross‐reactivity between SARS‐CoV‐2 and common cold coronaviruses (CCC), which affect >75% of the general population. 34 Pre‐existing memory T cells could favor a faster and stronger adaptive immune response upon exposure to SARS‐CoV‐2, limiting disease severity. Such a secondary‐like response could associate with a faster increase in memory Tfh cells, activation of B cells, and rapid humoral response. If polymerized, some viral antigens can also directly trigger mB cell proliferation leading to short‐term antibody production. 18 This looks like what happens in recovered patients, where a robust virus‐specific T cell response is maintained, preventing reinfection. 35
The pre‐existing memory T cells might, however, be harmful, eliciting an antibody‐mediated disease enhancement mechanism. 36 Sustained response of pre‐existing T cells may be detrimental if associated with a dysfunction of naive‐ and induced‐T regulatory (Treg) cells. 37 Since Treg cells limit antiviral responses and tissue immunopathology, 38 their reduction may favor a vigorous amplification of any (specific or non‐specific) T cell response including autoreactive T cells triggering autoimmunity. The direct interaction of CD147 (an inducer of metalloproteases mainly expressed on early activated memory Treg cells) and S1 protein has been suggested to be responsible for Treg cell dysfunction observed in severe COVID‐19. 23 , 32 , 38 , 39
Regarding pre‐existing humoral response, it was reported that, in naïve and, mostly, in recovered and vaccinated (mRNA) individuals, anti‐S1‐secreting plasmablasts peaked one week after the second challenge and then disappeared three weeks later 40 ; notably, B cells recognizing also S proteins of CCC strains have been observed in these subjects. 41 Furthermore, Abs produced by germinal center (GC) B cells of lymph nodes from 8‐month‐vaccinated people, recognized not only the S1/RBD and N domain of SARS‐CoV2 but also S1 epitopes shared with some CCC strains. As described in seasonal influenza virus vaccination, these cross‐reactive B cells displayed higher levels of somatic hypermutation suggesting a strong mB cell origin. 40 , 42 , 43
2.3. Super‐antigen hypothesis
The similarity between severe COVID‐19 and sepsis suggests that SARS‐CoV‐2 could contain super‐antigenic sequences. Super‐antigens (SAtgs) may enroll and activate exhaustion of a large (even if variable) proportion of polyclonal T cells. Some HLA haplotypes are more permissive in binding SAtgs and may account for the heterogeneity of immune responses and clinical outcomes. 44 Several SARS‐CoV‐2 SAtgs have been discovered: (i) A polybasic sequence of S protein with high homology to the SARS‐Cov1 18‐mer peptide with SAtgs activity, (ii) the homology of the previous sequence with SEB: notably, an anti‐SEB Ab displays the neutralizing activity of SARS‐CoV‐2 infection by inhibiting the access of TMPRSS2 to the cleavage site, 44 (iii) the homology of the SAtg motif with neurotoxin‐like sequences binding TCR. 45 The analysis of TCR repertoire from patients with severe disease indicates a TCR skewing with extensive junctional diversity, enrichment of Vβ genes, and increased J diversity, all consistent with SAtgs‐induced activation. 46 Since SAtgs bind the monomorphic sequences of TCR and MHC class II, they may polyclonally activate a proportion of T and B cells and contribute to their dysfunction. 44 Notably, the pathogenic role of SAtg of S1 needs to be further evaluated and the described mechanism remains largely speculative. It is likely that the polyclonal T and B cell activation induced by SARS‐CoV2 SAtg sequences could be crucial in the presence of some additional mechanisms as an early impairment of Treg cells or superinfection with other pathogens displaying SAtgs. Notably, no data are available on the presence of SAtgs of S1 proteins produced by COVID‐19 vaccines.
2.4. Unmasking latent auto‐inflammatory/autoimmune mechanisms
Viruses are considered the major trigger of autoimmune diseases in susceptible individuals. The hyperactivation of the immune response against SARS‐CoV2 may lead, in some patients, to unpredictable symptoms of autoimmune/auto‐inflammatory disorders (AAD), as observed in other infections. Even though they often represent transient post‐infectious epiphenomena, some COVID‐19‐related manifestations fulfill the diagnostic criteria of specific AAD. Symptoms related to autoimmune hematological diseases, autoimmune neuropathies, autoimmune coagulopathies, and Kawasaki disease‐like vasculitis have been documented during COVID‐19. 47 , 48 Histological patterns of the lung in COVID‐19 are identical to those of systemic lupus erythematosus (SLE), dermatomyositis (DM), and progressive systemic sclerosis (PSS). 49 Anti‐nuclear‐, centromere‐, PM‐Scl, SS‐B/La, Jo‐1, and Scl‐70 auto‐Abs have been described in a high proportion of severe patients, 50 while inconsistent results have been reported on anti‐phospholipid Abs. 50 , 51 , 52 Anti‐platelet factor 4 (PF4) auto‐Abs are likely responsible for the very rare post‐vaccination thromboses with thrombocytopenia. The anti‐IFN‐α auto‐Abs, further impairing the anti‐viral response, have been frequently detected in patients with life‐threatening COVID‐19. 53 The repertoire of auto‐Abs examined in multisystem inflammatory syndrome‐children (MIS‐C) patients identified 189 peptide candidates for IgG‐ and 108 for IgA autoantigens. In this library, the peptides expressed in cells of the immune system, La and Jo‐1 autoantigens (present in SLE and autoimmune myopathies), and those of tissues involved in MIS‐C are particularly abundant. 54 Furthermore, by analyzing the specificities toward more than 2500 extracellular and secreted proteins, it was found that severe COVID‐19 patients exhibited a dramatic increase in auto‐Abs compared to uninfected people. They recognize cytokines, chemokines, complement ‐C’‐ components, and cell surface proteins; thus, modulating immune responses and viral load control in a very heterogeneous manner. 55
Apparently, in odds with the ability of SARS‐CoV2 to elicit autoimmunity, many reports indicate that AAD patients, clinically stabilized with immunosuppressive drugs, display, if infected with SARS‐CoV‐2, similar morbidity rates of the general population. 56 It has been suggested that such treatment regimens likely allow to dampen some pathogenic mechanism of infection. This applies also to allergic patients infected by SARS‐CoV2, who display similar morbidity to the general population, likely due to some chronic therapeutic regimen. 57 Budesonide (an inhaled corticosteroid – ICS), largely employed in asthma, if administered at the initial phase of SARS‐CoV2 infection, has been shown to markedly reduce viral load and persistence, duration, and severity of symptoms and timing of recovery in recently infected non‐asthmatic patients. 58 It has been shown that ICS locally impair ACE2 expression on respiratory mucosa through the inhibition of IFN. 59 , 60 However, two retrospective studies on large cohorts of patients curb enthusiasm on the regular ICS use in protecting asthmatic patients against COVID‐19 infection. 61 , 62 Oral corticosteroids (OCS) also reduce ACE2 expression and virus entry, but they heavily blunt systemic immunity with the risk of increases in viral load if administered early: OCS are indicated to reduce inflammation and mortality risk only in severe patients. 63 The control of SARS‐CoV2 infection in respiratory allergy may also be due to anti‐histamine drugs. The rationale for their use is that the virus directly activates mast cells (expressing ACE2) secreting histamine in COVID‐19 pneumonia and that several anti‐histamines prevent SARS‐CoV2 entry in in vitro models. 64 , 65 In addition, patients treated with these drugs are more resistant to SARS‐CoV2, and cetirizine and famotidine alleviate pulmonary symptoms in COVID‐19. 66 These data, confirming the protective effects toward SARS‐CoV2 infection of several anti‐inflammatory drugs, underline that AAD and allergy share some pathogenic mechanisms with COVID‐19.
Besides Treg cells defect, several virus‐related mechanisms impair peripheral tolerance, and each mechanism, by itself or in association with others, may contribute to the pathogenesis of AAD‐like symptoms.
2.4.1. Molecular mimicry
S1 protein shares sequence homology with an extraordinary number of tissue proteins that, if altered, mutated, deficient, or improperly functioning by cross‐reacting Abs, may associate with a wide range of AAD. 67 An epitope mapping analysis has identified linear immunogenic epitopes from DM patients matching with the SARS‐CoV‐2 peptides. HLA‐B*15:03 which associates with Sjogren's Syndrome is able to present highly conserved SARS‐CoV‐2 peptides. SARS‐CoV‐2 shares also sequence with proteins of the brainstem respiratory nucleus and with a pulmonary surfactant, possibly explaining neurological and pulmonary damages. 68
2.4.2. Neutrophils’ extracellular traps and epitope spreading
The neutrophil extracellular traps (NETs) have been observed in COVID‐19 patients with elevated serum levels of cell‐free DNA, myeloperoxidase‐DNA complexes, and citrullinated histone H3. 33 , 69 NETs are a way to control microbial infections and this unique cell death program is called “NETosis.” NETs can be activated through many disease‐related stimuli and mediate tissue damage. Excessive spread of self‐antigens, associated with increased NETosis and/or defects of mechanisms for their elimination, leads to AAD and clotting activation. 69
2.4.3. Bystander damage
Bystander damage starts when the virus‐specific CD8+ T cells are recruited into the infected tissues where they exert cytotoxic activity. Dead cells activate macrophages to release reactive oxygen species and nitric oxide resulting in bystander killing of uninfected cells. 70 CD4+T cells may also contribute to the BD through the release of pro‐inflammatory cytokines. 70 Impaired clearance of killed cells induces spreading of autoantigens with the activation of bystander autoreactive T and B cells. BD is responsible for ARDS, myocarditis, and neurological involvement of COVID‐19.
2.4.4. Trained immunity and hyperactivation of T cells
When the virus persists, it may dysregulate NI in infected cells modifying the environmental signals (mainly IL‐1β, but also IL‐18 and IL‐23) which, in turn, promote: (i) The expression of further pro‐inflammatory molecules; (ii) the amplification of so‐called “trained innate immunity;” (iii) the activation of bystander autoreactive T and B cells. All these mechanisms, largely mediated by cytokines of the IL‐1 superfamily, contribute to an excessive immune response and to the onset of AAD. 71 The “trained Immunity” leads to an increased response of previously activated innate cells, mostly myeloid and NK cells, to subsequent triggers, defined as “innate immune memory,” responsible for the persistence of inflammation in some disorders. 72 “Trained memory” NK cells work as a rapid protective mechanism in secondary infections, but not in SARS‐CoV‐2 (primary) infection where it would contribute to the increase in the late inflammation. However, some reports indicate that trained innate immunity could overcome also primary infection due to a rapid reaction able to inhibit the virus from further dissemination. 73 No data are available regarding the onset of “trained memory” NK cells in recovered or vaccinated people.
The bystander activation of T cells, including autoreactive T cells, is favored by the excess of environment signals facilitated by Treg cell dysfunction. They contribute to ineffective virus clearance and stimulate long‐lived autoreactive B cells and auto‐Abs. Autoreactive T cells prevalently display a Th17 profile whose development is favored by IL‐1β and IL‐23 overproduced by NI‐activated macrophages. Importantly, Th17 cells have been shown in the blood and tissues of the majority of AAD and COVID‐19. Since Th17 cells are highly plastic, the environmental IL‐12 and TNF‐α usually induce their shift to a more aggressive profile (cytotoxic non‐classical Th1–ncTh1 cells) exerting further tissue injury in AAD and, likely, in COVID‐19. 74 The hyper‐production of IFN‐γ from ncTh1 and NK cells (promoted by IL‐18) improves macrophage activation; thus, starting a vicious circle that might lead to a clinical pattern known as macrophages activating syndrome (MAS). 75 The environmental conditions can also shift memory Th17 cells to produce IL‐21 and TGF‐β, but not IFN‐γ, which, in severe COVID‐19 patients, contribute to suppress T effector cells and to induce IgA2 and the egress of circulating plasmablasts. 76 Figure 1 summarizes the mechanisms leading to AAD‐related symptoms in SARS‐CoV2 infection.
FIGURE 1.
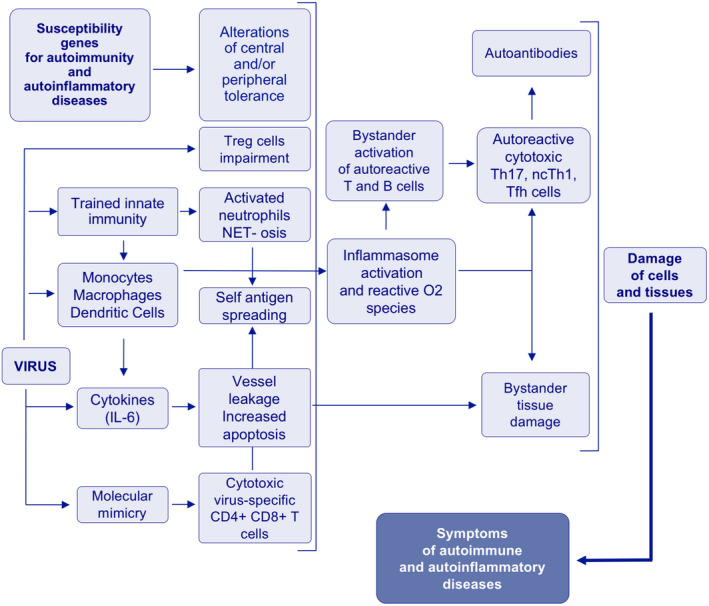
Flow chart of immuno‐mediated mechanisms leading to symptoms of autoimmune/autoinflammatory diseases in COVID‐19 patients
2.5. Prevalence of memory vs naïve T and B cells conditioning severe outcomes
Age, male‐gender, and pre‐existing comorbidities are risk factors for high morbidity and mortality of SARS‐CoV‐2 infection. They may display a higher basal pro‐inflammatory condition coupled with a progressive inability of the immune system to mount protective responses. 76 This complex status called immune‐senescence, often associated to advanced age, is characterized by: (i) Impairment of the CD4+/CD8+ T cell ratio; (ii) reduction of the TCR repertoire and clonal expansion if stimulated with novel antigens; (iii) decreased cytotoxicity of CD8+ T and NKT cells favoring ineffective response to new viruses; (iv) improved trained immunity coupled with pro‐inflammatory cytokines; and (vi) impaired development of Tfh cells, mB cells, and humoral response. 77 Of note, the humoral and mB responses induced by mRNA vaccines for COVID‐19 in naïve and recovered individuals, are consistently lower in older individuals. 78
Since immuno‐senescence is associated with higher memory and lower naive T cells, it has been speculated that such unbalance may contribute to the higher severity of the disease in adults compared with children. 79 In adults, the improved trained immunity is associated with bystander T cell activation, poor clonal T cell expansion, and low viral clearance, while, in children, the predominant naïve T cells develop a valid antiviral response with efficient clonal T cell expansion, viral clearance, and less tissue damage. 77 Besides naïve T cells, children exhibit higher levels of pre‐existing cross‐reacting IgM+mB cells. 80 Thus, they produce natural antibodies and generate most IgA+ and IgG+ switched mB cells, secreting protective Abs during early infection. 80 Neutralizing IgG from mB cells in children with COVID‐19 display none or very few somatic mutations as natural Abs; thus, suggesting that their repertoire share some SARS‐CoV‐2 specificities. 79 The abundance of naïve T and mB cells in children could contribute to explain why most pediatric patients display no or mild symptoms and early recovery. 81
Table 1 summarizes the topics of the described pathogenic hypotheses, underscoring those to be further thoroughly investigated. No data are available at the present on the proportion of patients in which each mechanism has been recognized: the next proposal to define endo‐types relating to variable clinical patterns could fill this gap.
TABLE 1.
Topics to be investigated to fully explain the proposed pathogenic hypotheses of COVID‐19
| Pathogenic hypotheses | Topics to be further investigated |
|---|---|
| Altered coordination between innate and adaptive Immunity 18 |
Factors/mechanisms delaying adaptive immunity. Early alterations in asymptomatic, mild, and severe diseases. The timing and entity of IFN impairment and of NLRP3 inflammasome activation. Macrophages activation and pDC impairment favoring NK dysfunction/exhaustion and trained NK cells response. pDC impairment and reduced antigen presentation to specific T cells. Relationship of delayed adaptive immunity and its relationship with age and other co‐morbidity with chronic inflammations. The circulating proportion of memory versus naïve T and B cells in adults compared with children. Endogeneous corticosteroids, TGF‐β and IL‐10 levels and compensatory hyperactivation of innate immunity and persistence of viral load. |
| Pre‐existing immunity to the virus in unexposed individuals 34 |
Protection given by pre‐existing immunity (memory T and B cells) toward SARS‐CoV2 infection. The permissive HLA haplotypes favoring a quick secondary‐like response. Spectrum of T cell repertoire to CCC epitopes cross‐reacting with SARS‐CoV−2 ones. The mechanism and timing of memory Treg cells impairment. Levels of Ab‐dependent endocytosis in pre‐existing immunity. |
| Super‐antigenic hypothesis 44 , 45 , 46 |
The SAtgs’ sequences of S1 protein. The timing and the degree of SAtg stimulation of T and, subsequently, of B cells. The permissive HLA‐haplotypes favoring polyclonal T cell activation till exhaustion. The interactions between chronically infected cells and polyclonal T cells activated by SAtgs. The association with Treg cell impairment and/or superinfection with other pathogens. |
| Unmasking latent autoimmune/auto‐inflammatory mechanisms 39 , 40 , 42 |
Mechanism(s) and timing of Treg cells dysfunction. Degree of inflammasome activation of infected cells leading to a prevalent type 3 (Th17, Tc17, ILC3) response. Levels of NET‐osis by activated/infected macrophages favoring epitope spreading. Bystander activation of autoreactive T cells. Phenotype and function of autoreactive T and B cells and their expansion during infection. Molecular mimicry between SARS‐CoV2 epitopes and self‐antigens. Fine specificities and pathogenic role of autoantibodies observed in COVID‐19. |
| Prevalence of memory vs naïve T and B cells 68 , 70 |
The circulating proportion of memory versus naïve T and B cells in adults compared with children. CD4+/CD8+ T cell ratio and functional Th subsets in different age ranges. Development of Tfh cells, mB cells and humoral response in different age ranges. TCR repertoire and clonal expansion to novel antigens in different age ranges. Impaired cytotoxicity of CD8+ T and NKT cells in elderly favoring not effective response to new viruses. Trained immunity coupled with pro‐inflammatory cytokines in the elderly. |
Abbreviations: CCC, common cold coronaviruses; pDC, plasmacytoid dendritic cells; IFN, interferon; ILC, innate lymphoid cells; NETs, neutrophils extracellular traps; NK, natural killer; SAtgs, superantigens; Th, T helper; Tc, T cytolytic; Tfh, T follicular helper; Treg, T regulatory.
2.6. MULTIPLE ENDO‐TYPES MAY EXPLAIN THE VARIABILITY OF COVID‐19
Despite the extraordinary amounts of reports, many unknowns on the immune response to the virus and COVID‐19 pathogenesis must be further investigated to explain the differences between severe and non‐severe COVID‐19 and why only a small fraction of patients develop severe symptoms. Although the described hypotheses contribute to shed light on multiple aspects of COVID‐19, actually none of them is sufficient to explain the variability of the disease. Indeed, a major problem of this infection is its heterogeneity. The heterogeneity concerns the viral load (varying more than 105 times among different patients) which conditions the degree and the efficacy of the immune response. 18 Heterogeneity has been observed in viral replication and persistence or tissue distribution, even though the referred long‐term PCR positivity indicates only the presence of SARS‐CoV‐2 transcripts and not of the viable virus. 18 , 22 , 31 , 33 The infection exhibits heterogeneous clinical patterns (poor or no symptoms, mild disease with recovery, severe and critical illness with acute respiratory distress syndrome (ARDS). Different biological features are prevalent in severe/critical patients: (i) The reduction of MHC class I and II on infected antigen‐presenting cells (APC) with dysregulated NK and T cells function; (ii) NI activation of macrophages is associated with high levels of IL‐1β, IL‐18, IL‐23; (iii) limited numbers of specific T cells with a prevalence of Th2/Th17 profiles; (iv) high levels of Abs with activation of C’ and clotting; v. pathogenic auto‐Abs that affect immune responses and/or tissue damages; vi variability and susceptibility of different organs to become infected by the virus; (vii) the endothelial damage directly caused by the virus, triggering clotting mechanism and multi‐organ damage. Such variability of endo‐types translates into different clinical outcomes of severe COVID‐19 (Sepsis‐like syndrome, cytokine‐released syndrome (CRS), ARDS, MAS, secondary hemophagocytic lymphohistiocytosis (HLH), disseminated intravascular coagulation (DIC), acute kidney or multi‐organ failures, and the variable symptoms of so‐called “long COVID‐19” of recovered patients, extensively described in the literature. 82 , 83 Heterogeneous response to drugs addressed to pathogenic mechanisms as those antagonizing TLR signaling (hydroxychloroquine) or cytokines (TNF‐α, IL‐1β, or IL‐6R) has been shown. 24 , 84 This has likely been the cause of the failure of clinical trials using these drugs in patients not stratified for endo‐types. Heterogeneity has been observed also in the timing of onset and intensity of immune response. 85 The size of anti‐SARS‐CoV‐2 Abs ranges more than 1000 times, and the proportions of NK and virus‐specific T cells are highly variable. 31 , 32 , 33 Single‐cell transcriptomic analysis of virus‐reactive CD4+ T cells provided evidence of heterogeneity across individual patients with different disease severity. Finally, heterogeneity of adverse events (from minimal to severe reactions, including anaphylaxis, myocarditis, and thrombosis) and of the degree of specific T and B cell responses to COVID‐19 vaccines have been documented.
The current problem is, therefore, to establish the causes of this heterogeneity. Some other viral infections, such as HBV, show variable clinical outcomes owing to different balances between viral load and efficacy of antiviral immune responses. 86 As reported in the graphical abstract, many variable components can contribute to the different outcomes also in SARS‐CoV2 infection. The variability of viral load along the disease is of the utmost importance. The virus triggers a lot of co‐receptors able to start signals, which, in turn, activate several immunological mechanisms: genetic, epigenetic, pre‐existing immunity, polymorphisms of signals/receptors of the immune system, and actually may contribute to the heterogeneity of the final response. Table 2 compares the receptors targeted by S1 protein for their structure, cell, and tissue distribution, and pathophysiology, 87 , 88 , 89 , 90 , 91 , 92 , 93 , 94 , 95 , 96 , 97 , 98 , 99 , 100 , 101 whereas Figure 2 summarizes the pathways of each receptor and the antigenic activity of S1 influencing the immune response.
TABLE 2.
Comparative activities of receptors targeted by S1 protein of SARS‐CoV2
| ACE2 | TMPRSS2 | CD26 | CD147 | CD209/CD209L | TLR2/TLR7‐8 | Mannose Binding lectins | |
|---|---|---|---|---|---|---|---|
| Structure/Nomenclature | Membrane Carboxi‐peptidase | Membrane Serine‐Protease 2 (Furin) | Membrane exopeptidase (DPP‐4) | Immunoglobulin superfamily (Basiginin) EMMPRIM | Adhesion molecule (dendritic cell –3–grabbing non integrin [DC‐SIGN & L‐SIGN]) | Type I Transmembrane glycoprotein receptors (TLR2 is a surface receptor, while TLR7 and TLR8 are endosomal) | Glycan Binding receptors are divided into membrane‐bound proteins (C‐type proteins and Siglecs) and Soluble MBL (galectins) |
| Cellular/Tissue expression | Epithelium, Goblet cells, Enterocytes, Type II Pneumocytes, Secretory Mast cells, Endothelial cells, Cardiomyocytes, APC, Vessels, Respiratory mucosa, Bladder, Heart. Urothelial tract. | Epithelium, APC Enterocytes, Type II Pneumocytes, Goblet cells, Endothelial cells, Mast cells | Hepatocytes, Respiratory epithelium, Immune cells, activated leukocytes | Activated Treg cells, Some T cells subsets, Monocytes, Red blood cells, Pneumocytes, Pericytes, Fibroblasts, Resident stem cells | Dendritic cells and other APCs | T, B and NK cells, APCs, Neutrophils, Fibroblasts, Endothelial cells, Epithelial cells | C‐type receptors are a large family (60–80), expressed on APCs, NK, endothelial cells, 13 Siglec members are expressed on immune cells |
| Tissue distribution | Lungs, Arteries, Heart, Kidney, Liver, Gut, Testes, Brain | Lungs, Intestines | Liver, Lungs, kidney, intestine, prostate | Lungs | Secondary lymphoid tissues, Liver, Lungs, Kidney, Gut | Spread on different tissues, Vessels, Spleen, Lymph nodes | Vessels, Spleen, and lymphoid organs |
| Physiologic function | Regulator of the renin‐angiotensin system (degrades angiotensin II into angiotensin‐(1–7), with antioxidative, anti‐thrombotic, anti‐fibrotic, vasodilatory activity). Its gene is activated by IFN‐α | Degrades Spike protein of SARS‐CoV2 in S1 and S2 subunits essential for virus entry | Its ligand (adenosine deaminase) plays a role in glucose metabolism, T cell activation, cell adhesion, chemotaxis, apoptosis | Stimulation of Matrix metalloproteases | Adhesion molecules and Pathogen recognition receptors |
Recognition of PAMPS and DAMPS: TLR2 ligands are dyacil‐ and triacyl lipopeptides; TLR7/8 recognize ssRNA of viral origin Interaction with microRNAs and long noncoding RNAs |
Recognize a wide range of microorganisms and activates complement cascade via an antibody‐independent pathway. It functions as PPRs and intercellular signals of immune cells |
| Expression In pathologic conditions | Binds S1 protein from SARS‐CoV2, containing RBD sequence | Expressed on Activated CD4+ T effector cells in severe COVID‐19 | Receptor of MERS‐CoV | Receptor on RBC for P. Falciparum, Upregulated in Asthma (bronchi) Inflammatory processes, Tumors It mediates fibrosis | Facilitates the entry of CoVs, several viruses, and some parasites and bacteria. Soluble form is detectable in the serum | The interactions with miRNA and long‐ncRNAs have a role in several disorders including infections, autoimmune and cancer | The soluble MBL deficiency may have a role in chronic inflammation, autoimmunity, and cancer |
| Refs | 1, 7, 84, 85, 86, 87, 89, 98, 99 | 7, 86, 87 | 88, 90, 91 | 32, 38, 39 | 95, 96, 97 | 95, 96, 98, 99 | 100, 101 |
FIGURE 2.
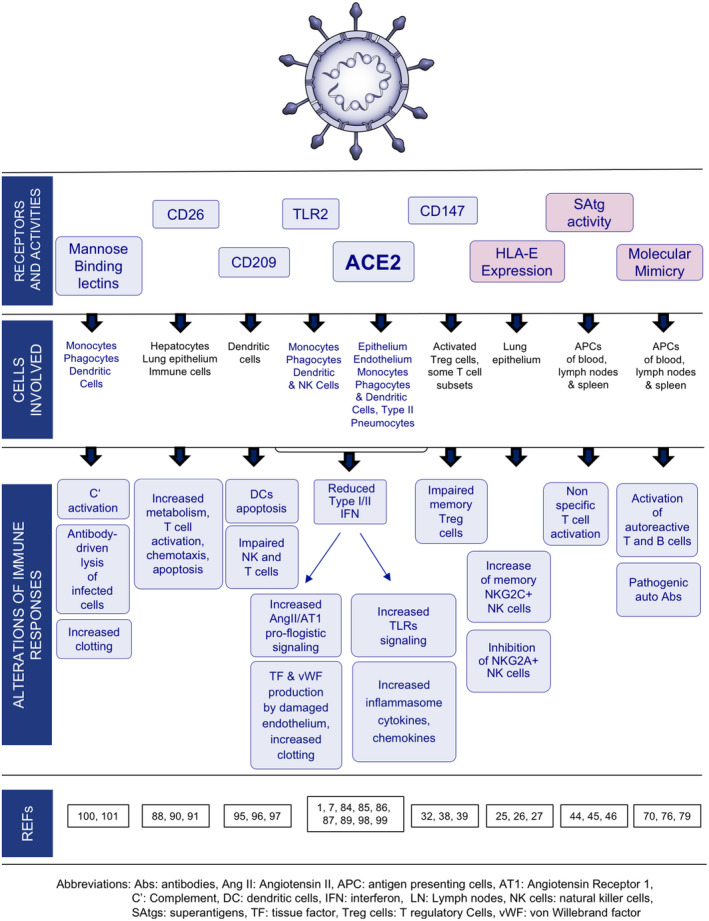
Multiple functions of SARS‐CoV‐2 S1 protein. S1 protein interacts with some active receptors/molecules expressed on many cell types, by directly or indirectly interfering with innate and adaptive immune responses. S1 protein induces also HLA‐E expression on epithelial cells mediated by GATA3 activation which may negatively affect NKG2A+ NK cells in the lung. S1 protein display also epitopes with superantigenic activity and/or cross‐reacting with self‐antigens: They may amplify polyclonal activation of not specific T cells or bystander autoreactive T and B cells
Furthermore, the immune system is diverse in different individuals who rarely respond to an infection in a similar manner. Genetic variations as TLR7 in men, 102 HLA haplotype conditioning adaptive immunity or recognizing cross‐reactive autoantigens or SAtgs of S1, molecules involved in IFN signaling, 102 susceptibility to autoimmunity, or to easy NI activation, 54 are all important to explain the heterogeneity of antiviral responses. 23 Previous and elderly‐related co‐morbidities, showing a partially controlled pro‐inflammatory condition, introduce further elements of variability. Pre‐existing factors conditioning the heterogeneity of immune response to SARS‐CoV2 are listed in Table 3. Whatever the pathway used by the immune system to counteract the virus, the timing of this multistep disease suggests a parallel underlying scenario of multistep pathogenic mechanisms. Despite the variability, it is likely that different phases of infection and timing‐related clinical outcomes (phenotypes) can be the expression of a progressive failure of precise immunological processes (detrimental endo‐types) with the reset of a novel setting. However, by exploiting the redundancy and plasticity of the immune system, this setting can recover in each clinical phase through the re‐expansion of anti‐viral immune components (neutralizing Abs, NK‐ and CD8+ T cells) modulated by an appropriate number of Treg cells (protective endo‐types). If unstable, this setting can be short‐circuited by mechanisms of the virus and immune system itself leading to a next worse phase, also subject to recovery or worsening. The virus, interacting with several cellular targets, may affect multiple immunological mechanisms, which tend to compensate for the produced alterations and damages. Protective endo‐types facilitate functional restoration while detrimental ones improve immunological alterations and favor clinical worsening. A list of the major biomarkers associable with protective or detrimental endo‐types is reported in Table 4. 103 , 104 , 105 , 106 , 107 , 108 , 109 , 110 The evolving endo‐types leading to multiple phenotypes can likely overlap or intersect in a variable way in each patient. An attempt to depict the major evolving endo‐types which, through several immune mechanisms, may lead to the major clinical patterns in severe COVID‐19 is schematically summarized in Table 5. As reported, a sepsis‐like phenotype develops when the SAtg‐driven polyclonal T cell activation in HLA‐susceptible subjects associates with Treg cell dysfunction and superinfection with other pathogens. Moreover, Treg cell impairment, molecular mimicry, and bystander autoreactive T and B cell activation are essential for the onset of AAD‐like phenotype, as described (Figure 1). DIC phenotype develops when the endothelial damage due to TNF and the virus favors the production of pro‐coagulant von Willebrand‐ and Tissue factors, added to platelets and C’ activation and the excess of anti‐phospholipids and anti‐PF4 auto‐Abs, give rise to a state of chronic clotting. In contrast, whether B cell activation leads to high levels of Abs (and/or auto‐Abs) with subsequent high ICs, these latter cause tissue damage, evolving to multi‐organ failures. 111 IFN impairment and the subsequent increased viral load strongly stimulate macrophages and neutrophils, which, in the presence of additional variable alterations, can evolve to different phenotypes (as ARDS, CRS, MAS, or secondary HLH). 112 , 113 , 114 , 115 Even though each alteration is largely described in the current literature, their sequential combination into evolving pathways has never been proposed and, therefore, is partially hypothetical.
TABLE 3.
Pre‐existing factors conditioning innate and adaptive responses to SARS‐CoV‐2 infection
| Genetic (or epigenetic) polymorphisms of TLR, TLR signaling, type I/III IFN, and IFNR and their signaling, ‘C factors, Inflammasome components |
| HLA‐E haplotypes and expression of KLRC gene (encoding NKG2C) conditioning trained memory NK cells |
| HLA haplotypes binding the viral superantigens |
| HLA haplotypes presenting viral epitopes, including that cross‐reacting with self‐antigens |
| Degree of pre‐existing immunity to cross‐reacting epitopes of common cold coronaviruses |
| Naïve T/memory T cells ratio (children vs elderly people) |
| Proportion of Innate memory IgM+B cells producing natural Abs cross reacting with SARS‐CoV‐2 |
| Susceptibility genes for autoimmunity conditioning the derangement of peripheral tolerance, the proportion of autoreactive bystander T and B cells, Treg cell function and ease to produce pathogenic autoantibodies |
| Bone‐marrow reservoir of inflammatory precursors (mobilized by inflammatory molecules) able to develop rapid NK and T cell progenies |
| Bone marrow reservoir of neutrophil precursors with immunosuppressive function |
| Not stabilized co‐morbidities displaying uncontrolled inflammation |
Abbreviations: Abs, antibodies; IFN, interferon; IFNR, interferon receptor; NK, natural killer; TLRs, Toll‐like receptors; Treg, T regulatory.
TABLE 4.
Protective and detrimental biomarkers to be checked for defining COVID‐19 endotypes
| Biomarkers | Protective | Detrimental | Refs | |
|---|---|---|---|---|
| Immunologic | Cells |
High/normal proportion of CD8+ T and NK cells producing IFN‐γ High levels of Tfh and Plasmablasts Pre‐existing T cell response to common cold Coronaviruses |
Low proportion of dysfunctional NK, CD4+ and CD8+ T cells expressing PD1,TIM3 and activation or exhaustion markers Cytolytic CD4+ T cells with Tfh, Th2/Th17 profile Reduced and altered function of Treg cells Polyclonal T cells activation and exhaustion Subsets of monocytes with peculiar phenotype (CD14+, CD11b+, CD16+,CD68+, CD80+, CD163+, CD206+) producing high levels of cytokines High proportion of MDSC and NKT cells High MDSC/CD8+T cells ratio Reduction of plasmablasts |
6, 19, 22, 24, 83, 100, 135 |
| Cytokines/Chemokines |
IL‐1b, IL‐18 (for Ab production) Early IL‐12 and IFN‐γ production |
Low levels of type‐I/III IFN High levels of IL‐1β, IL‐2, IL‐4, IL‐13, IL‐6, IL‐7, IL‐8, IL‐10, IL‐15, IL‐18, IL‐23, GM‐CSF, TNF‐α, IL‐1RA, sIL‐2R, IFN‐α2 CCL2, CCL7, CCL23,CXCL10 |
22, 101, 102, 103, 106, 135 | |
| Other Molecules |
C’ activation and consumption Inflammasome's activation Presence of Immune‐complexes |
104, 105, 135 | ||
| Antibodies |
Mild IgG and IgA1 response Elevated dimeric IgA in nasal and oral mucosa |
Long IgM response High levels of IgG and IgA2 Presence of Auto‐Abs (recognizing Phospholipids, beta2 microblobulin, RO52, GP1, PF4, CCP etc) |
8, 9, 106, 135 | |
| Routinely Blood | Cells |
Trombocytopemia, Eosinopenia High Platelets‐Lymphocytes Ratios |
83, 107 | |
| Molecules | High CRP, D‐dimers, pTT, LDH, Serum Amyloid protein, NEFAs | 83, 107 |
TABLE 5.
Relationship between pathogenic mechanisms and clinical outcomes in severe/critical COVID‐19
| Evolving endotypes a | Phenotypes |
|---|---|

|
1. Sepsis‐like Syndrome 44 , 45 , 116 , 117 |

|
2. Disseminated Intravascular Coagulation 49 , 51 , 108 , 117 |

|
3. MIS‐C and Autoimmune‐ Auto‐inflammatory –like syndrome. 44 , 50 , 51 , 52 , 114 |

|
4. Acute Respiratory Distress Syndrome 11 , 107 , 112 , 118 , 123 , 124 |

|
5. Cytokine Release Syndrome 13 , 14 , 107 , 113 |

|
6 SecondaryHaemophagocytic Lymphohistiocytosis 107 , 115 |

|
7. Macrophage Activating Syndrome 107 , 112 , 117 , 123 |

|
8. Multiorgan Failure 48 , 104 , 110 , 111 , 117 |
Abbreviations: AAD: Auto‐inflammatory/autoimmune Disorders; Abs, antibodies; C’, complement; MBL, membrane binding proteins; MDSC, Myeloid‐derived suppressive cells; ncTh1, not classical Th1 cells; NETs, neutrophil extracellular traps; NK, Natural killer cells; pDC, plasmocitoid Dendritic cells; TF, tissue factor; Tfh, Follicular T helper cells; TLRs, Toll‐like receptors; vWF, von Willebrand factor.
Each pathway should not be considered one way, since conditions favoring multiple mechanisms can coexist or intersect each other. Evolving endotypes leading to Phenotypes 1, 2, 5, 6 needs further investigation.and, at presenr, must be considered hypothetical.
3. CONCLUSION
The current problems are to establish the relationship between the clinical heterogeneity (including symptoms, comorbidities, and severities, etc.) and the underlying immune responses and pathogenic mechanisms of SARS‐CoV2 infection. Recently, different sepsis endo‐types and clinical and biochemical phenotypes of ARDS have been reported in COVID‐19. 116 , 117 , 118 Several attempts to preventively identify biomarkers of endo‐types during symptom onset or at the hospital admission of patients at high risks of further clinical deterioration have been proposed. 119 , 120 , 121 , 122 , 123 , 124 , 125 Such reports propose the strategy to follow: To define precise endo‐types and the corresponding phenotypes of COVID‐19, similar to what has been done in severe asthma and other chronic allergic diseases. 126 , 127 , 128 , 129 , 130 Taking into account the mechanism of allergic inflammation and the flexibility of tissue T cells, some molecular targets shifting the bronchial immune response have been proposed as a new therapeutic strategy of asthma 131 , 132 , 133 , 134 ; biologicals targeting these molecules are used nowadays in respiratory allergy as the best example of precision medicine. 126 , 127 , 128 , 129 , 130 Based on this successful experience, we propose to stratify COVID‐19 patients for evolving symptoms and recovery along with infection and, in parallel, to monitor them tightly by defining surrogate biomarkers of innate and adaptive immunity in association with essential parameters of inflammation, coagulation, organs’ function, etc. (Table 4). 135 This approach will allow to establish a detailed guideline to preventively identify immunological alterations correlated to the symptoms trend and to exploit this tool for the most suitable therapeutic strategy in each patient (including mAbs–Sotrovimab, recognizing the virus entry pathways and biologicals targeting pathogenic molecules). Further research based on allergy pathogenesis and its therapeutic approaches is mandatory to better explore COVID‐19 heterogeneity. This strategy can also provide an outline of how we may approach emerging infections/pandemics in the future.
CONFLICT OF INTEREST
The authors declare that they have no conflicts of interest.
ACKNOWLEDGMENT
This work was supported by grants from the Ministero della Salute (grant no. RC‐2020 OPBG to L.M. and E.M.) and from Associazione Italiana per la Ricerca sul Cancro (project no. 5x1000 2018 Id 21147 and project no. IG 2017 Id 19920 to L.M.).
Maggi E, Azzarone BG, Canonica GW, Moretta L. What we know and still ignore on COVID‐19 immune pathogenesis and a proposal based on the experience of allergic disorders. Allergy. 2022;77:1114–1128. doi: 10.1111/all.15112
REFERENCES
- 1. Maggi E, Canonica WG, Moretta L. COVID‐19: unanswered questions on immune response and pathogenesis. J Allergy Clin Immunol. 2020;146(1):18‐22. 10.1016/j.jaci.2020.05.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kusnadi A, Ramírez‐Suástegui C, Fajardo V, et al. Severely ill COVID‐19 patients display impaired exhaustion features in SARS‐CoV‐2‐reactive CD8+ T cells. Sci Immunol. 2021;65(55):eabe4782. 10.1126/sciimmunol.abe4782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Yang Y, Wang X, Du RH, et al. Serological investigation of asymptomatic cases of SARS‐CoV‐2 infection reveals weak and declining antibody responses. Emerg Microbes Infect. 2021;10(1):905‐912. 10.1080/22221751.2021.1919032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wang Z, Lorenzi JCC, Muecksch F, et al. Enhanced SARS‐CoV‐2 neutralization by dimeric IgA. Sci Transl Med. 2021;13(577):eabf1555. 10.1126/scitranslmed.abf1555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Turner JS, Kim W, Kalaidina E, et al. SARS‐CoV‐2 infection induces long‐lived bone marrow plasma cells in humans. Nature. 2021;595(7867):421‐425. 10.1038/s41586-021-03647-4 [DOI] [PubMed] [Google Scholar]
- 6. Wang F, Hou H, Luo Y, et al. The laboratory tests and host immunity of COVID‐19 patients with different severity of illness. JCI Insight. 2020;5(10):e137799. 10.1172/jci.insight.137799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Liao M, Liu Y, Yuan J, et al. Single‐cell landscape of bronchoalveolar immune cells in patients with COVID‐19. Nat Med. 2020;26(6):842‐844. 10.1038/s41591-020-0901-9 [DOI] [PubMed] [Google Scholar]
- 8. Zost SJ, Gilchuk P, Case JB, et al. Potently neutralizing and protective human antibodies against SARS‐CoV‐2. Nature. 2020;584:443‐449. 10.1038/s41586-020-2548-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Rogers TF, Zhao F, Huang D, et al. Isolation of potent SARS‐ CoV‐2 neutralizing antibodies and protection from disease in a small animal model. Science. 2020;369:956‐963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Nguyen M, Bourredjem A, Piroth L, et al. High plasma concentration of non‐esterified polyunsaturated fatty acids is a specific feature of severe COVID‐19 pneumonia. Sci Rep. 2021;11(1):10824. 10.1038/s41598-021-90362-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gibellini L, De Biasi S, Paolini A, et al. Altered bioenergetics and mitochondrial dysfunction of monocytes in patients with COVID‐19 pneumonia. EMBO Mol Med. 2020;12(12):e13001. 10.15252/emmm.202013001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Chen J, Pan Y, Li G, et al. Distinguishing between COVID‐19 and influenza during the early states by measurement of peripheral blood parameters. J Med Virol. 2021;93:1029‐1037. 10.1002/jmv.26384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Pum A, Ennemoser M, Adage T, Kungl AJ. Cytokines and chemokines in SARS‐CoV‐2 infections – therapeutic strategies targeting cytokine storm. Biomolecules. 2021;11(1):91. 10.3390/biom11010091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kim JS, Lee JY, Yang JW, et al. Immunopathogenesis and treatment of cytokine storm in COVID‐19. Theranostics. 2021;11:316‐319. 10.7150/thno.49713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Vitte J, Diallo AB, Boumaza A, et al. A granulocytic signature identifies COVID‐19 and its severity. J Infect Dis. 2020;222:1985‐1996. 10.1093/infdis/jiaa591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Arnold IC, Artola‐Borán M, Tallón de Lara P, et al. Eosinophils suppress Th1 responses and restrict bacterially‐induced gastrointestinal inflammation. J Exp Med. 2018;215:2055‐2072. 10.1084/jem.20172049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Yang L, Liu S, Liu J, et al. COVID‐19: immune pathogenesis and Immune therapeutics. Signal Transduct Target Ther. 2020;5:128. 10.1038/s41392-020-00243-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sette A, Crotty S. Adaptive immunity to SARS‐CoV‐2 and COVID‐19. Cell. 2021;184(4):861‐880. 10.1016/j.cell.2021.01.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hadjadj J, Yatim N, Barnabei L, et al. Impaired type I interferon activity and inflammatory responses in severe COVID‐19 patients. Science. 2020;369:718‐724. 10.1126/science.abc6027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Totura AL, Baric R. SARS coronavirus pathogenesis: host innate immune responses and viral antagonism of interferon. Curr Opin Virol. 2012;2:264‐275. 10.1016/j.coviro.2012.04.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ziegler CGK, Allon SJ, Nyquist SK, et al. SARS‐CoV‐2 Receptor ACE2 Is an Interferon‐Stimulated Gene in Human Airway Epithelial Cells and Is Detected in Specific Cell Subsets across Tissues. Cell. 2020;181(5):1016‐1035. 10.1016/j.cell.2020.04.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kuri‐Cervantes L, Pampena MB, Meng W, et al. Comprehensive mapping of immune perturbations associated with severe COVID‐19. Sci Immunol. 2020;5(49):eabd7114. 10.1126/sciimmunol.abd7114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Liu C, Martins AJ, Lau WW, et al. Time‐resolved systems immunology reveals a late juncture linked to fatal COVID‐19. Cell. 2021;184(7):1836‐1857. 10.1016/j.cell.2021.02.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Mazzoni A, Salvati L, Maggi L, et al. Impaired immune cell cytotoxicity in severe COVID‐19 is IL‐6 dependent. J Clin Invest. 2020;130(9):4694‐4703. 10.1172/JCI138554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bortolotti D, Gentili V, Rizzo S, et al. SARS‐CoV‐2 Spike 1 Protein Controls Natural Killer Cell Activation via the HLA‐E/NKG2A Pathway. Cells. 2020;9(9):1975. 10.3390/cells9091975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. El‐Shazly AE, Doloriert HC, Bisig B, et al. Novel cooperation between CX3CL1 and CCL26 inducing NK cell chemotaxis via CX3CR1: A possible mechanism for NK cell infiltration of the allergic nasal tissue. Clin Exp Allergy. 2013;43:322‐331. 10.1111/cea.12022 [DOI] [PubMed] [Google Scholar]
- 27. Vietzen H, Zoufaly A, Traugott M, et al. Deletion of the NKG2C receptor encoding KLRC2 gene and HLA‐E variants are risk factors for severe COVID‐19. Genet Med. 2021;23(5):963‐967. 10.1038/s41436-020-01077-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bozzano F, Della Chiesa M, Pelosi A, et al. HCMV‐controlling NKG2C+ NK cells originate from novel circulating inflammatory precursors. J Allergy Clin Immunol. 2021:2343‐2357. 10.1016/j.jaci.2020.12.648 [DOI] [PubMed] [Google Scholar]
- 29. Bozzano F, Dentone C, Perrone C, et al. Extensive activation, tissue trafficking, turnover and functional impairment of NK cells in COVID‐19 patients at disease onset associates with subsequent disease severity. PLoS Pathog. 2021;17(4):e1009448. 10.1371/journal.ppat.1009448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lucas C, Wong P, Klein J, et al. Longitudinal analyses reveal immunological misfiring in severe COVID‐19. Nature. 2020;584:463‐469. 10.1038/s41586-020-2588-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Moderbacher CR, Ramirez SI, Dan JM, et al. Antigen‐specific adaptive immunity to SARS‐CoV‐2 in acute COVID‐19 and associations with age and disease severity. Cell. 2020;183:996‐1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Vabret N, Britton GJ, Gruber C, et al. Sinai Immunology Review Immunology of COVID‐19: current state of the science. Immunity. 2020;52(6):910‐941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Radermecker C, Detrembleur N, Guiot J, et al. Neutrophil extracellular traps infiltrate the lung airway, interstitial, and vascular compartments in severe COVID‐19. J Exp Med. 2020;217(12):e20201012. 10.1084/jem.20201012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Grifoni AW, Weiskopf D, Ramirez SI, et al. Targets of T cell responses to SARS‐ CoV‐2 coronavirus in humans with COVID‐19 disease and unexposed individuals. Cell. 2020;181:1489‐1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Zuo J, Dowell AC, Pearce H, et al. Robust SARS‐CoV‐2‐specific T cell immunity is maintained at 6 months following primary infection. Nat Immunol. 2021;22(5):620‐626. 10.1038/s41590-021-00902-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ricke DO. Two different antibody‐dependent enhancement (ADE) risks for SARS‐CoV‐2 antibodies. Front Immunol. 2021;12:640093. 10.3389/fimmu.2021.640093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Le Bert N, Tan AT, Kunasegaran K, et al. SARS‐CoV‐2‐specific T cell immunity in cases of COVID‐19 and SARS, and uninfected controls. Nature. 2020;584(7821):457‐462. 10.1038/s41586-020-2550-z [DOI] [PubMed] [Google Scholar]
- 38. Wang K, Chen W, Zhang Z, et al. CD147‐spike protein is a novel route for SARS‐CoV‐2 infection to host cells. Signal Transduct Target Therap. 2020;5(1):283. 10.1038/s41392-020-00426-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Solstad T, Bains SJ, Landskron J, et al. CD147 (Basigin/Emmprin) identifies FoxP3+CD45RO+CTLA4+‐ activated human regulatory T cells. Blood. 2011;118:5141‐5151. 10.1182/blood-2011-02-339242. [DOI] [PubMed] [Google Scholar]
- 40. Turner JS, O'Halloran JA, Kalaidina E, et al. SARS‐CoV‐2 mRNA vaccines induce persistent human germinal centre responses. Nature. 2021;596(7870):109‐113. 10.1038/s41586-021-03738-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Amanat F, Thapa M, Lei T, et al. SARS‐CoV‐2 mRNA vaccination induces functionally diverse antibodies to NTD, RBD, and S2. Cell. 2021;184(15):3936‐3948. 10.1016/j.cell.2021.06.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Turner JS, Zhou JQ, Han J, et al. Human germinal centres engage memory and naive B cells after influenza vaccination. Nature. 2020;586(7827):127‐132. 10.1038/s41586-020-2711-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Amanat F, Thapa M, Lei T, et al. The plasmablast response to SARS‐CoV‐2 mRNA vaccination is dominated by non‐neutralizing antibodies that target both the NTD and the RBD. medRxiv. 2021. 10.1101/2021.03.07.21253098 [DOI] [Google Scholar]
- 44. Noval Rivas M, Porritt RA, Cheng MH, et al. COVID‐19‐associated multisystem inflammatory syndrome in children (MIS‐C): a novel disease that mimics toxic shock syndrome‐ the superantigen hypothesis. J Allergy Clin Immunol. 2021;147:57‐59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Cheng MH, Zhang S, Porritt RA, et al. Superantigenic character of an insert unique to SARS‐CoV‐2 spike supported by skewed TCR repertoire in patients with hyperinflammation. Proc Natl Acad Sci USA. 2020;117:25254‐25262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Porritt RA, Paschold L, Rivas MN, et al. Identification of a unique TCR repertoire, consistent with a superantigen selection process in Children with Multi‐system Inflammatory Syndrome. bioRxiv. 2020. 10.1101/2020.11.09.372169 [DOI] [Google Scholar]
- 47. Talotta R, Robertson E. Autoimmunity as the comet tail pf COVID‐19 pandemic. World J Clin Cases. 2020;8(17):3621‐3644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Belhadjer Z, Méot M, Bajolle F, et al. Acute heart failure in multisystem inflammatory syndrome in children (MIS‐C) in the context of global SARS‐CoV‐2 pandemic. Circulation. 2020;142(23):2282‐2284. 10.1161/circulationaha.120.050147 [DOI] [PubMed] [Google Scholar]
- 49. Ackermann M, Verleden SE, Kuehnel M, et al. Pulmonary Vascular Endothelialitis, Thrombosis, and Angiogenesis in Covid‐19. N Engl J Med. 2020;383(2):120‐128. 10.1056/NEJMoa2015432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Zhou Y, Han T, Chen J, et al. Clinical and Autoimmune Characteristics of Severe and Critical Cases of COVID‐19. Clin Transl Sci. 2020;13(6):1077‐1086. 10.1111/cts.12805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Borghi MO, Beltagy A, Garrafa E, et al. Anti‐Phospholipid Antibodies in COVID‐19 Are Different from Those Detectable in the Anti‐Phospholipid Syndrome. Front Immunol. 2020;15(11):584241. 10.3389/fimmu.2020.584241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Zuo Y, Estes SK, Ali RA, et al. Prothrombotic autoantibodies in serum from patients hospitalized with COVID‐19. Sci Transl Med. 2020;12(570):eabd3876. 10.1126/scitranslmed.abd3876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Bastard P, Rosen LB, Zhang Q, et al. Autoantibodies against type I IFNs in patients with life‐threatening COVID‐19. Science. 2020;370(6515):eabd4585. 10.1126/science.abd4585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Gruber CN, Patel RS, Trachtman R, et al. Mapping systemic inflammation and antibody responses in multisystem inflammatory syndrome in children (MIS‐C). Cell. 2020;183(4):982‐995. 10.1016/j.cell.2020.09.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Wang EY, Mao T, Klein J, et al. Diverse Functional Autoantibodies in Patients with COVID‐19. Nature. 2021;595(7866):283‐288. 10.1038/s41586-021-03631-y [DOI] [PubMed] [Google Scholar]
- 56. Michelena X, Borrell H, López‐Corbeto M, et al. Incidence of COVID‐19 in a cohort of adult and paediatric patients with rheumatic diseases treated with targeted biologic and synthetic disease‐modifying anti‐rheumatic drugs. Semin Arthritis Rheum. 2020;50:564‐570. 10.1016/j.semarthrit.2020.05.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Chhiba KD, Patel GB, Vu THT, et al. Prevalence and characterization of asthma in hospitalized and nonhospitalized patients with COVID‐19. J Allergy Clin Immunol. 2020;146(2):307‐314. 10.1016/j.jaci.2020.06.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Ramakrishnan S, Nicolau DV Jr, Langford B, et al. Inhaled budesonide in the treatment of early COVID‐19 (STOIC): a phase 2, open‐label, randomized controlled trial. Lancet Respir Med. 2021:763‐772. 10.1016/S2213-2600(21)00160-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Zhang Y, Hu S, Wang J, et al. Dexamethasone inhibits SARS‐CoV‐2 spike pseudotyped virus viropexis by binding to ACE2. Virology. 2021;554:83‐88. 10.1016/j.virol.2020.12.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Finney LJ, Glanville N, Farne H, et al. Inhaled corticosteroids downregulate the SARS‐CoV‐2 receptor ACE2 in COPD through suppression of type I interferon. J Allergy Clin Immunol. 2021;147(2):510‐519. 10.1016/j.jaci.2020.09.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Schultze A, Walker AJ, MacKenna B, et al. Risk of COVID‐19‐related death among patients with chronic obstructive pulmonary disease or asthma prescribed inhaled corticosteroids: an observational cohort study using the OpenSAFELY platform. Lancet Respir Med. 2020;8(11):1106‐1120. 10.1016/S2213-2600(20)30415-X [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Halpin DMG, Singh D, Hadfield RM. Inhaled corticosteroids and COVID‐19: a systematic review and clinical perspective. Eur Respir J. 2020;55(5):2001009. 10.1183/13993003.01009-2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Crisan Dabija R, Antohe I, Trofor A, Antoniu SA. Corticosteroids in SARS‐COV2 infection: certainties and uncertainties in clinical practice. Expert Rev Anti Infect Ther. 2021;1–10. 10.1080/14787210.2021.1933437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Qu C, Fuhler GM, Pan Y. Could Histamine receptor antagonists be used for treating COVID‐19? Int J Mol Sci. 2021;22:5672. 10.3390/ijms22115672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Ishola AA, Joshi T, Abdulai SI, et al. Molecular basis for the repurposing of histamine H2‐receptor antagonist to treat COVID‐19. J Biomol Struct Dyn. 2021;25:1‐18. 10.1080/07391102.2021.1873191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Reznikov LR, Norris MH, Vashisht R, et al. Identification of antiviral antihistamines for COVID‐19 repurposing. Biochem Biophys Res Commun. 2021;538:173‐179. 10.1016/j.bbrc.2020.11.095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Vojdani A, Kharrazian D. Potential antigenic cross‐reactivity between SARS‐CoV‐ 2 and human tissue with a possible link to an increase in autoimmune diseases. Clin Immunol. 2020;217:108480. 10.1016/j.clim.2020.108480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Kanduc D, Shoenfeld Y. On the molecular determinants of the SARS‐CoV‐2 attack. Clin Immunol. 2020;215:108426. 10.1016/j.clim.2020.108426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Zuo Y, Zuo M, Yalavarthi S, et al. Neutrophil extracellular traps in COVID‐19. J Thromb Thrombolysis. 2020; Nov5:1‐8. 10.1007/s11239-020-02324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Lee HG, Lee JU, Kim DH, et al. Pathogenic function of bystander‐activated memory‐like CD4(+) T cells in autoimmune encephalomyelitis. Nat Commun. 2019;10(1):709. 10.1038/s41467-019-08482-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Calado MB, da Silva Santana CE, Crovella S. Do inflammasome impact COVID‐19 severity? Virusdisease. 2021;26:1‐11. 10.1007/s13337-021-00705-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Netea MG, van der Meer JW. Trained immunity: an ancient way of remembering. Cell Host Microbe. 2017;21:297‐300. 10.1016/j.chom.2017.02.003 [DOI] [PubMed] [Google Scholar]
- 73. Sohrabi Y, Dos Santos JC, Dorenkamp M, et al. Trained immunity as a novel approach against COVID‐19 with a focus on Bacillus Calmette‐Guerin vaccine: mechanisms, challenges and perspectives. Clin Transl Immunol. 2020;9(12):e1228. 10.1002/cti2.1228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Maggi L, Cosmi L, Simonini G, et al. T cell subpopulations in juvenile idiopathic arthritis and their modifications after biotherapies. Autoimmun Rev. 2016;15(12):1141‐1144. 10.1016/j.autrev.2016.09.012 [DOI] [PubMed] [Google Scholar]
- 75. Gerfaud‐Valentin M, Jamilloux Y, Iwaz J, Sève P. Adult‐onset Still's disease. Autoimmun Rev. 2014;13:708‐722. 10.1016/j.autrev.2014.01.058 [DOI] [PubMed] [Google Scholar]
- 76. Ferreira‐Gomes M, Kruglov A, Durek P, et al. SARS‐CoV‐2 in severe COVID‐19 induces a TGF‐β‐dominated chronic immune response that does not target itself. Nat Comm. 2021;12:1961. 10.1038/s41467-021-22210-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Fulop T, Herbein G, Cossarizza A, et al. Immunosenescence and inflammatory aging as two sides of the same coin: friends or foes? Front Immunol. 2017;42:28‐46. 10.1159/000448542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Goel RR, Apostolidis SA, Painter MM, et al. Distinct antibody and memory B cell responses in SARS‐CoV‐2 naïve and recovered individuals following mRNA vaccination. Sci Immunol. 2021;6(58):eabi6950. 10.1126/sciimmunol.abi6950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. de Candia P, Prattichizzo F, Garavelli S, Matarese G. T Cells: Warriors of SARS‐CoV‐2 Infection. Trends Immunol. 2021;42(1):18‐30. 10.1016/j.it.2020.11.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Carsetti R, Di Sabatino A, Rosado MM, et al. Lack of Gut Secretory Immunoglobulin A in Memory B‐Cell Dysfunction‐Associated Disorders: A Possible Gut‐Spleen Axis. Front Immunol. 2020;10:2937. 10.3389/fimmu.2019.02937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Guo L, Ren L, Yang S, et al. Profiling Early Humoral Response to Diagnose Novel Coronavirus Disease (COVID‐19). Clin Infect Dis. 2020;71(15):778‐785. 10.1093/cid/ciaa310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Ranjeva S, Pinciroli R, Hodell E, et al. Identifying clinical and biochemical phenotypes in acute respiratory distress syndrome secondary to coronavirus disease‐2019. EClinicalMedicine. 2021;34:100829. 10.1016/j.eclinm.2021.100829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Wilfong EM, Lovly CM, Gillaspie EA, et al. Severity of illness scores at presentation predict ICU admission and mortality in COVID‐19. Emerg Crit Care Med. 2021;5:7. 10.21037/jeccm-20-92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Moutsopoulos HM. Anti‐inflammatory therapy may ameliorate the clinical picture of COVID‐19. Ann Rheum Dis. 2020;79(9):1253‐1254. 10.1136/annrheumdis-2020-217562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Mathew D, Giles JR, Baxter AE, et al. COVID Processing Unit Deep immune profiling of COVID‐19 patients reveals distinct immunotypes with therapeutic implications. Science. 2020;369(6508):eabc8511. 10.1126/science.abc8511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Liaw YF. Clinical utility of HBV surface antigen quantification in HBV e antigen‐negative chronic HBV infection. Nat Rev Gastroenterol Hepatol. 2019;16(10):631‐641. 10.1038/s41575-019-0197-8 [DOI] [PubMed] [Google Scholar]
- 87. Shang J, Wan Y, Luo C, et al. Cell entry mechanisms of SARS‐CoV‐2. Proc Natl Acad Sci U S A. 2020;117(21):11727‐11734. 10.1073/pnas.2003138117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Li F. Receptor recognition mechanisms of coronaviruses: a decade of structural studies. J Virol. 2015;89(4):1954‐1964. 10.1128/JVI.02615-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Hoffmann M, Kleine‐Weber H, Schroeder S, et al. SARS‐CoV‐2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell. 2020;181(2):271‐280. 10.1016/j.cell.2020.02.052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Kalfaoglu B, Almeida‐Santos J, Tye CA, et al. T‐Cell hyperactivation and paralysis in severe COVID‐19 infection revealed by single‐cell analysis. Front Immunol. 2020;11:589380. 10.3389/fimmu.2020.589380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Vankadari N, Wilce JA. Emerging WuHan (COVID‐19) coronavirus: glycan shield and structure prediction of spike glycoprotein and its interaction with human CD26. Emerg Microbes Infect. 2020;9(1):601–604. 10.1080/22221751.2020.1739565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Waumans Y, Baerts L, Kehoe K, et al. The Dipeptidyl Peptidase Family, Prolyl Oligopeptidase, and Prolyl Carboxypeptidase in the Immune System and Inflammatory Disease, Including Atherosclerosis. Front Immunol. 2015;6:387. 10.3389/fimmu.2015.00387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Guo H, Xun L, Zhang R, Gou X. Ratio of CD147high/CD147low in CD4+CD25+ T cells: A potential biomarker for early diagnosis and prediction of response to therapy for autoimmune diseases. Med Hypotheses. 2018;115:1–4. 10.1016/j.mehy.2018.03.005 [DOI] [PubMed] [Google Scholar]
- 94. Ulrich H, Pillat MM. CD147 as a Target for COVID‐19 Treatment: Suggested Effects of Azithromycin and Stem Cell Engagement. Stem Cell Rev Rep. 2020;16(3):434‐440. 10.1007/s12015-020-09976-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Amraei R, Yin W, Napoleon MA, et al. CD209L/L‐SIGN and CD209/DC‐SIGN act as receptors for SARS‐CoV‐2 and are differentially expressed in lung and kidney epithelial and endothelial cells. ACS Cent Sci. 2021;7(7):1156‐1165. 10.1021/acscentsci.0c01537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Rahimi N. C‐type Lectin CD209L/L‐SIGN and CD209/DC‐SIGN: Cell Adhesion Molecules Turned to Pathogen Recognition Receptors. Biology. 2021;10(1):1. 10.3390/biology10010001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Thépaut M, Luczkowiak J, Vivès C, et al. DC/L‐SIGN recognition of spike glycoprotein promotes SARS‐CoV‐2 trans‐infection and can be inhibited by a glycomimetic antagonist. PLoS Pathog. 2021;17(5):e1009576. 10.1371/journal.ppat.100957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Moreno‐Eutimio MA, López‐Macías C, Pastelin‐Palacios R. Bioinformatic analysis and identification of single‐stranded RNA sequences recognized by TLR7/8 in the SARS‐CoV‐2, SARS‐CoV, and MERS‐CoV genomes. Microbes Infect. 2020;22(4‐5):226‐229. 10.1016/j.micinf.2020.04.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Ghafouri‐Fard S, Abak A, Shoorei H, et al. Interaction between non‐coding RNAs and Toll‐like receptors. Biomed Pharmacother. 2021;140:111784. 10.1016/j.biopha.2021.111784 [DOI] [PubMed] [Google Scholar]
- 100. Gupta A, Gupta GS. Status of mannose‐binding lectin (MBL) and complement system in COVID‐19 patients and therapeutic applications of antiviral plant MBLs. Mol Cell Biochem. 2021;476(8):2917‐2942. 10.1007/s11010-021-04107-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. van Kooyk Y, Rabinovich GA. Protein‐glycan interactions in the control of innate and adaptive immune responses. Nat Immunol. 2008;9(6):593‐601. 10.1038/ni.f.203 [DOI] [PubMed] [Google Scholar]
- 102. Pairo‐Castineira E, Clohisey S, Klaric L, et al. Genetic mechanisms of critical illness in COVID‐19. Nature. 2021;591(7848):92‐98. 10.1038/s41586-020-03065-y [DOI] [PubMed] [Google Scholar]
- 103. Kreutmair S, Unger S, Núñez NG, et al. Distinct immunological signatures discriminate severe COVID‐19 from non‐SARS‐CoV‐2‐driven critical pneumonia. Immunity. 2021;54(7):1578‐1593. 10.1016/j.immuni.2021.05.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Gómez‐Escobar LG, Hoffman KL, Choi JJ, Cytokine signatures of end organ injury in COVID‐19. Sci Rep. 2021;11(1):12606. 10.1038/s41598-021-91859-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Vigón L, Fuertes D, García‐Pérez J, et al. Impaired Cytotoxic Response in PBMCs From Patients With COVID‐19 Admitted to the ICU: Biomarkers to Predict Disease Severity. Front Immunol. 2021;26(12):665329. 10.3389/fimmu.2021.665329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Khalil BA, Elemam NM, Maghazachi AA. Chemokines and chemokine receptors during COVID‐19 infection. Comput Struct Biotechnol J. 2021;19:976‐988. 10.1016/j.csbj.2021.01.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Zhao N, Di B, Xu LL.The NLRP3 inflammasome and COVID‐19: Activation, pathogenesis and therapeutic strategies. Cytokine Growth Factor Rev. 2021:2‐15. 10.1016/j.cytogfr.2021.06.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Ng N, Powell CA. Targeting the Complement Cascade in the Pathophysiology of COVID‐19 Disease. J Clin Med. 2021;10(10):2188. 10.3390/jcm10102188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Quast I, Tarlinton D. B cell memory: understanding COVID‐19. Immunity. 2021;54(2):205‐210. 10.1016/j.immuni.2021.01.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Laudanski K, Jihane H, Antalosky B, Unbiased Analysis of Temporal Changes in Immune Serum Markers in Acute COVID‐19 Infection With Emphasis on Organ Failure, Anti‐Viral Treatment, and Demographic Characteristics. Front Immunol. 2021;12:650465. 10.3389/fimmu.2021.650465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Guven G, Ince C, Topeli A, Caliskan K. Cardio‐pulmonary‐renal consequences of severe COVID‐19. Cardiorenal Med. 2021;11(3):133‐139. 10.1159/000516740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Kosyreva A, Dzhalilova D, Lokhonina A, et al. The Role of Macrophages in the Pathogenesis of SARS‐CoV‐2‐Associated Acute Respiratory Distress Syndrome. Front Immunol. 2021;12:682871. 10.3389/fimmu.2021.682871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Eljaaly K, Malibary H, Alsulami S, et al. Description and Analysis of Cytokine Storm in Registered COVID‐19 Clinical Trials: A Systematic Review. Pathogens. 2021;10(6):692. 10.3390/pathogens10060692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Matucci‐Cerinic C, Caorsi R, Consolaro A, et al. Multisystem inflammatory syndrome in children: unique disease or part of the kawasaki disease spectrum? Front Pediatr. 2021;9:680813. 10.3389/fped.2021.680813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Retamozo S, Brito‐Zerón P, Sisó‐Almirall A, et al. Haemophagocytic syndrome and COVID‐19. Clin Rheumatol. 2021;40(4):1233‐1244. 10.1007/s10067-020-05569-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. DeMerle KM, Angus DC, Baillie JK, et al. Sepsis Subclasses: A Framework for Development and Interpretation. Crit Care Med. 2021;49(5):748‐759. 10.1097/CCM.0000000000004842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Sweeney TE, Liesenfeld O, Wacker J, et al. Validation of inflammopathic, adaptive, and coagulopathic sepsis endotypes in coronavirus disease 2019. Crit Care Med. 2021;49(2):e170‐e178. 10.1097/CCM.0000000000004786 [DOI] [PubMed] [Google Scholar]
- 118. Ranjeva S, Pinciroli R, Hodell E, et al. Identifying clinical and biochemical phenotypes in acute respiratory distress syndrome secondary to coronavirus disease‐2019. EClinicalMedicine. 2021;3(4):100829. 10.1016/j.eclinm.2021.100829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Carsetti R, Zaffina S, Piano Mortari E, et al. Different Innate and Adaptive Immune Responses to SARS‐CoV‐2 Infection of Asymptomatic, Mild, and Severe Cases. Front Immunol. 2020;11:610300. 10.3389/fimmu.2020.610300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Vultaggio A, Vivarelli E, Virgili G, et al. Prompt Predicting of Early Clinical Deterioration of Moderate‐to‐Severe COVID‐19 Patients: Usefulness of a Combined Score Using IL‐6 in a Preliminary Study. J Allergy Clin Immunol Pract. 2020;8(8):2575‐2581.e2. 10.1016/j.jaip.2020.06.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Ye CH, Hsu WL, Peng GR, et al. Role of the Immune Microenvironment in SARS‐CoV‐2 Infection. Cell Transplant. 2021;30:9636897211010632. 10.1177/09636897211010632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Van Singer M, Brahier T, Ngai M, et al. COVID‐19 risk stratification algorithms based on sTREM‐1 and IL‐6 in emergency department. J Allergy Clin Immunol. 2021;147(1):99‐106. 10.1016/j.jaci.2020.10.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Blot M, Bour JB, Quenot JP, et al. The dysregulated innate immune response in severe COVID‐19 pneumonia that could drive poorer outcome. J Transl Med. 2020;18(1):457. 10.1186/s12967-020-02646-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Chen LD, Zhang ZY, Wei XJ, et al. Association between cytokine profiles and lung injury in COVID‐19 pneumonia. Respir Res. 2020; 21(1):201. 10.1186/s12931-020-01465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Petruccioli E, Najafi Fard S, Navarra A, et al. Exploratory analysis to identify the best antigen and the best immune biomarkers to study SARS‐CoV‐2 infection. J Transl Med. 2021;19(1):272‐287. 10.1186/s12967-021-02938-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. McDowell PJ, Heaney LG. Different endotypes and phenotypes drive the heterogeneity in severe asthma. Allergy. 2020;75(2):302‐310. 10.1111/all.13966 [DOI] [PubMed] [Google Scholar]
- 127. Baker MG, Samson HA. Phenotypes and endotypes of food allergy: A path to better understanding the pathogenesis and prognosis of food allergy. Ann Allergy Asthma Immunol. 2018;120(3):245‐253. 10.1016/j.anai.2018.01.027 [DOI] [PubMed] [Google Scholar]
- 128. Dunn JLM, Shoda T, Caldwll JM, et al. Esophageal type 2 cytokine expression heterogeneity in eosinophilic esophagitis in a multisite cohort. J Allergy Clin Immunol. 2020;145(6):1629‐1640. 10.1016/j.jaci.2020.01.051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Czamowicki T, He H, Krueger JG, Guttman‐Yassky E. Atopic dermatitis endotypes and implications for targeted therapeutics. J Allergy Clin Immunol. 2019;143(1):1‐11. 10.1016/j.jaci.2018.10.032 [DOI] [PubMed] [Google Scholar]
- 130. Staudacher AG, Peters AT, Kato A, Stevens WW. Use of endotypes, phenotypes, and inflammatory markers to guide treatment decisions in chronic rhinosinusitis. Ann Allergy Asthma Immunol. 2020;124(4):318‐325. 10.1016/j.anai.2020.01.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Holgate ST. Mechanisms of asthma and implications for its prevention and treatment: a personal journey. Allergy Asthma Immunol Res. 2013;5(6):343‐347. 10.4168/aair.2013.5.6.343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Maggi E, Parronchi P, Manetti R, et al. Reciprocal regulatory effects of IFN‐gamma and IL‐4 on the in vitro development of human Th1 and Th2 clones. J Immunol. 1992;148(7):2142‐2147. [PubMed] [Google Scholar]
- 133. Cosmi L, Maggi L, Santarlasci V, et al. Identification of a novel subset of human circulating memory CD4(+) T cells that produce both IL‐17A and IL‐4. J Allergy Clin Immunol. 2010;125(1):222‐230. 10.1016/j.jaci.2009.10.012 [DOI] [PubMed] [Google Scholar]
- 134. Assaf SM, Hanania NA. Biological treatments for severe asthma. Curr Opin Allergy Clin Immunol. 2019;19(4):379‐386. 10.1097/ACI.0000000000000549 [DOI] [PubMed] [Google Scholar]
- 135. Janssen NAF, Grondman I, de Nooijer AH, et al. Dysregulated Innate and Adaptive Immune Responses Discriminate Disease Severity in COVID‐19. J Infect Dis. 2021;223(8):1322‐1333. 10.1093/infdis/jiab [DOI] [PMC free article] [PubMed] [Google Scholar]