

Abstract
The global spread of SARS‐CoV‐2 is fast moving and has caused a worldwide public health crisis. In the present article, we analyzed spike protein sequences of SARS‐CoV‐2 genomes to assess the impact of mutational diversity. We observed from amino acid usage patterns that spike proteins are associated with a diversity of mutational changes and most important underlying cause of variation of amino acid usage is the changes in hydrophobicity of spike proteins. The changing patterns of hydrophobicity of spike proteins over time and its influence on the receptor binding affinity provides crucial information on the SARS‐CoV‐2 interaction with human receptor. Our results also show that spike proteins have evolved to prefer more hydrophobic residues over time. The present study provides a comprehensive analysis of molecular sequence data to consider that mutational variants might play a crucial role in modulating the virulence and spread of the virus and has immediate implications for therapeutic strategies.
Keywords: amino acid usage, correspondence analysis, hydrophobicity, molecular docking, spike protein sequence
Abbreviations
- ACE2
Angiotensin Converting Enzyme 2
- MD
Molecular Dynamics
- PDB
Protein Data Bank
- Rg
Radius of Gyration
- RMSD
Root Mean Square Deviation
- SPLHR
Spike Proteins with Less Hydrophobic Residues
- SPMHR
Spike Proteins with More Hydrophobic Residues
1. INTRODUCTION
Coronaviruses are members of the family Coronaviridae. They are single‐stranded, positive‐sense RNA viruses that cause widespread respiratory, gastrointestinal, and neurological clinical symptoms. 1 , 2 To survive under immunological pressure within humans the virus accumulates mutations to outwit the immune system. 3 These mutations may lead to change the virulence of the virus and its infectivity. 4 Therefore, it is important to analyze the mutational pattern in order to ascertain virus evolutionary dynamics.
Gene sequence data from pathogen genome has been widely recognized as an important tool to study the infection dynamics. 5 , 6 Coronavirus replication is error prone as compared to other RNA viruses and the estimated mutation rate is 4 × 10−4 nucleotide substitutions/site/year. 7 Wang et al. 8 reported 13 mutations in SARS‐CoV‐2 genome from the genome sequences submitted till February 2020. In another study, 93 mutations were identified across the SARS‐CoV‐2 genome, which includes three mutations in RBD of S protein demanding further study to understand the impact of these mutations on antigenicity of the SARS‐CoV‐2. 9 van Dorp et al. 10 studied 7,666 public genome assemblies of SARS‐CoV‐2 and identified invariant and diversified regions of the genome. They observed 198 recurrent mutations across the genome when compared with reference genomes of Wuhan‐Hu‐1 (accession IDsC_045512.2 and EPI_ISL_402125). Earlier reports found that most (80%) of the mutations were non‐synonymous at the protein level. 9 , 11 More recent studies suggest that the D614G variant is close to reaching fixation around the world. 12 Groves et al. suggested that mutations in spike proteins, which are associated with higher viral loads may lead to a more open conformation enhancing the binding of the virus spike to the ACE2 receptor. 13
Coronavirus uses spike proteins for primary interaction with human host. Spike protein binds with the human cellular receptor angiotensin‐converting enzyme 2 (ACE2). The binding affinity of spike protein with ACE2 represents important determinant of coronavirus host range. 14 , 15 , 16 Spike proteins are trimers containing two functional subunits designated as S1 and S2. S1 is responsible for binding to the receptor and S2 is responsible for fusion of cellular membrane. 17 The interacting residues between pathogen and host proteins can be identified using molecular docking technique. It may provide the scope to characterize the more evolutionary constrained regions as target in the pathogen to avoid rapid drug and vaccine escape mutants. Various mutation sites for spike proteins from SARS‐CoV‐2 isolates have been mapped on to protein three‐dimensional structure. 18 It is reported that the spike protein of SARS‐CoV‐2 is in a highly stable state and binds to the ACE2 with the higher affinity. 19
The huge pool of genome sequence data of SARS‐CoV‐2 provides us ample opportunity to analyze the evolutionary dynamics of the virus. The rapid spread of SARS‐CoV‐2 raises many questions on the mutational diversity and its impact on evolution of the virus. The present study is designed to focus on the mutational pattern of spike protein sequences, the underlying cause of variation of mutational patterns and their significance to the binding affinity with human host ACE2 protein. Spike protein has been studied as a potential drug target and also as a virus antigen. 20 Therefore, the present study might be important for the development of therapeutic, and prevention of SARS‐CoV‐2.
2. MATERIALS AND METHODS
2.1. Sequence retrieval and analysis
A total number of 251,430 whole genome sequence assemblies flagged as “complete,” “high coverage,” “low coverage excl” for human host were downloaded as of January18, 2021 (8:00 GMT) from GISAID (https://www.gisaid.org/). Full‐length spike gene sequences were retrieved through BLAST search by aligning with a reference spike gene sequence (GenBank accession number: NC_045512.2). Those sequences containing unrecognized start codon, stop codon, internal stop codons, untranslatable codons, and unrecognized character (other than a, t, g, c) have been discarded from the final dataset. The final set comprises 209,148 spike gene sequences.
Correspondence analysis was performed to assess the variations in amino acid usage of spike protein dataset. 21 CoA reveals major trends of variation in the dataset by arranging them along continuous axes where consecutive axis have been arranged to have diminishing effect gradually. 22 We used CoA available in Codon W for the analysis of amino acid usage of spike gene sequences. Hydrophobicity of each spike gene sequence is calculated using the method Kyte–Doolittle available in Codon W. 23
2.2. Protein homology modeling and docking
We have followed the method proposed by Huang et al. 24 for clustering of all the spike protein sequences on the basis of sequence identity. We found 7,883 clusters of spike proteins with 100% sequence identity. Therefore, we took one spike protein sequence from each cluster for structural analysis. Out of 7,883 protein sequences, 3,676 spike proteins belong to spike proteins with more hydrophobic residues (SPMHR) and 4,207 belong to spike proteins with less hydrophobic residues (SPLHR) group.
Three‐dimensional structural models were generated for spike protein sequences through homology modeling. Spike protein structure available in Protein Data Bank (PDB) (PDB ID: 6VYB) was used as template for homology modeling with more than 99% sequence identity and 94% query coverage. The structure of angiotensin converting enzyme 2 (ACE2) was also generated through homology modeling using the protein sequence available in UniProt (UniProt ID: Q9BYF1) and then a template was used from PDB (PDB ID: 6 M18) with 100% sequence identity and 99% query coverage. 3Drefine web‐server was used for the refinement of the protein structural models generated through homology modeling. 25 Molecular interaction of viral spike protein with the human ACE2 receptor was performed using Z‐dock software. 26 Then, the resulting docking data were processed and analyzed considering binding energies and main interacting residues in each complex by using the tools of the PRODIGY software. 27 Molecular dynamics (MD) simulation of the trimeric spike protein structure with ACE2 structure was carried out using the Gromacs v5.1.4 software with Gromos53a6 force field. Root mean square deviation (RMSD) and radius of gyration (Rg) were plotted for the spike–ACE2 complexes.
3. RESULTS
3.1. Correspondence analysis on amino acid usage of spike proteins
We analyzed the variations in the amino acid usage patterns of spike gene sequences through correspondence analysis. Correspondence analyses are used to simplify rectangular matrices in which (for our purpose) the columns represent amino acid usages value and the rows represent individual genes. It creates a series of orthogonal axes to identify trends that explain the data variation, with each subsequent axis explaining a decreasing amount of the variation. Figure 1 shows the positions of the genes generated through correspondence analysis on amino acid usage along the first and second major axes. The first and second major axis accounted for 29.63 and 12.81% of the total variation in amino acid usage respectively. Since there exists a single major explanatory axis (i.e., horizontal axis with 29.63% variation), we, therefore, carried out remaining analysis in this study on the basis of the distribution of spike protein genes along the horizontal axis of correspondence analysis. Percentage of variance is expected to be low since large number of same proteins from same virus has been analyzed. Also, recent study confirms that the evolutionary rate of the spike protein remained stable throughout the 9 months of their study. Since evolutionary rate does not vary much, this also signifies the low percentage variance observed in our study. 28 We observed that the position of the genes along the horizontal axis (Figure 1) were significantly correlated with the hydrophobicity of the encoded proteins (r = .45, p < .01).The average value of the hydrophobicity of the spike proteins distributed in the negative side of the horizontal axis is −0.0767 (SD: 0.00174) and the average value of the hydrophobicity of the spike proteins distributed in the negative side of the horizontal axis is −0.0750 (SD: 0.00191).We have also observed that average hydrophobicity of the spike proteins distributed in the negative side of the horizontal axis is significantly lower (p < .0001) than the average hydrophobicity of the spike proteins distributed in the positive side of the horizontal axis. For lucidity, henceforth, spike proteins distributed in the positive side of the horizontal axis will be referred to as SPMHR and spike proteins distributed in the negative side of the horizontal axis will be referred to as SPLHR.
FIGURE 1.
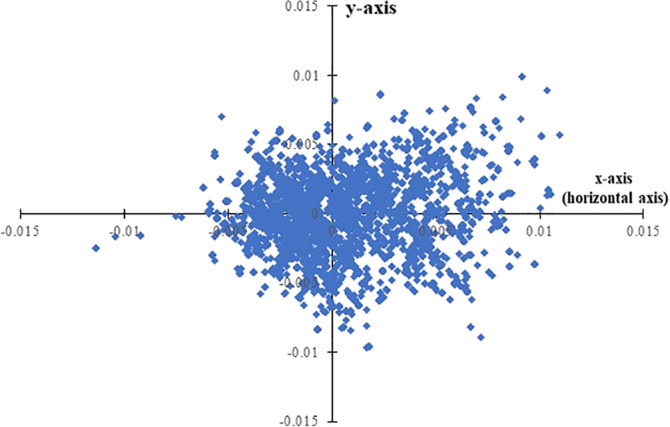
Distribution of spike(S) genes along the two major axes of correspondence analysis (COA) based on amino acid usage (AAU) data. Blue colored square boxes represent spike(S) gene sequences
The top four hydrophobic amino acids (according to Kyte–Doolittle scale) have thymine (T) in the second codon position. We have observed that average value of T composition in second codon position (T2) is significantly higher in spike proteins of SPMHR category compared to spike proteins of SPLHR category (Table S1). So, increase in T2 might be the driving force for the higher hydrophobicity of spike proteins in SPMHR category.
3.2. Distribution of date of sample collection according to differential pattern of spike protein sequences
We have compared the sequence dataset for SPMHR and SPLHR and checked the distribution of date of sequence collection for every spike protein in both the groups. We observed a skewed distribution of spike genes between SPMHR and SPLHR groups with respect to the date of collection of sequences (Figure 2). For the first couple of months (December 2019–September 2020) the percentage of sequences clustered in SPLHR group is higher than the percentage of sequences clustered in SPMHR group. However, from October 2020, the percentage of sequences clustered in SPMHR group became higher than the percentage of sequences clustered in the SPLHR group. Interestingly, as we move from December 2019, the percentage of spike protein sequences clustered in SPLHR gradually decreases with an increase in the percentage of spike protein sequences clustered in SPMHR group. To our surprise, we observed that almost 100% of spike protein sequences in December 2019 followed SPLHR pattern whereas, almost 50% of spike protein sequences followed SPMHR pattern from October 2020.
FIGURE 2.
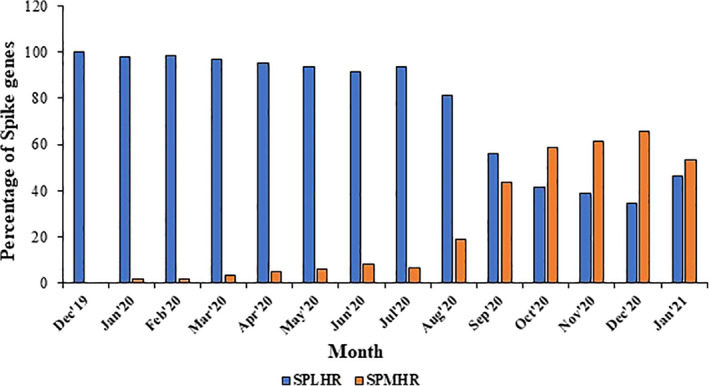
Distribution of spike genes between SPMHR and SPLHR groups with respect to the date of collection of sequences
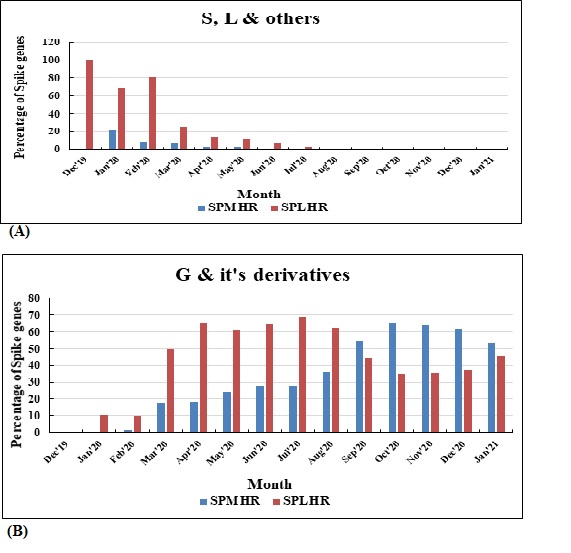
According to recent reports, several SARS‐CoV‐2 variants (S, L, O) are early diverging and G clade and its derivatives are late diverging. Therefore, in the later phase of the pandemic, G clade and its derivative are more associated with spike proteins following SPMHR pattern compared to spike proteins following SPLHR pattern. Indeed, we observed that in the early phase of pandemic when most of the spike genes followed SPLHR pattern (i.e., lower hydrophobic) mostly belongs to S, L clades. In the later part of the pandemic (September 2020 onwards) when most of the spike genes followed SPMHR pattern (i.e., higher hydrophobic) mostly belongs to G clade and its derivatives (Figure S1). 29
3.3. Interaction profile between spike protein and ACE2
Our comparison of amino acid usage underlines the differential pattern of evolution of spike protein where the hydrophobicity of the encoded protein is a significant cause of variation of amino acid usage pattern among the spike proteins. Since the receptor for SARS‐CoV‐2 has been identified as ACE2, it was very important to analyze how the differential pattern of amino acid usages of spike proteins of SARS‐CoV‐2 responded to binding to the human ACE2 receptor. Three dimensional structures of spike protein sequences from the SPMHR cluster and SPLHR cluster were constructed through homology modeling using the crystal structure of spike protein available in PDB (PDB ID: 6VYB) as template. The 3D structure of ACE2 has been generated computationally using the protein sequence available in UniProt (UniProt ID: Q9BYF1) and then a template was used from PDB (PDB ID: 6M18). Docking study was performed with ACE2 separately with all the spike proteins taken from the two groups (Figure 3a) and the average binding energy was calculated separately for SPMHR and SPLHR groups. We observed that the average binding energy for the spike–ACE2 complex taken from the SPMHR group is significantly lower than the average binding energy for the same complex taken from the SPLHR (p < .0001). A lower average binding energy for the spike–ACE2 complex taken from the SPMHR indicates its higher stability compared to the SPLHR group of complexes.
FIGURE 3.
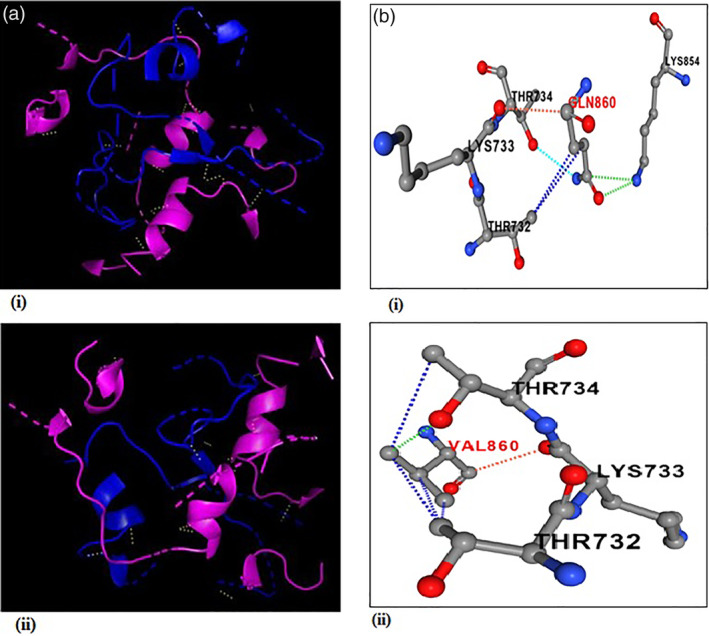
(a) Arrangement of hydrogen bonds (yellow) in spike (blue)‐ACE2 (Pink) complex. (i) Spike protein taken from the SPMHR indicates ACE2 receptor can interact with viral spike protein more effectively compared to (ii). Spike‐ACE2 complex where spike protein was taken from the SPLHR group. (b) Comparison of interaction profile of an identified mutation Q860V in spike‐protein indicating hydrophilic to hydrophobic amino acid substitution. Red and black letters represent mutant in spike protein and its receptor respectively. (i) Wild type residue type residue Q860 having one polar interaction (sky), two Van der Waals interaction (green), two hydrophobic (blue) interaction and one carbonyl interaction (orange). (ii) Mutant type residue 860 V having one Van der Waals interaction (green), four hydrophobic (blue) interaction, and one carbonyl interaction (orange)
4. DISCUSSION
We performed a comprehensive analysis of amino acid usage of more than 0.2 million spike proteins to understand the evolutionary dynamics of the emerging SARS‐CoV‐2 pandemic. Earlier studies have identified several mutations in the spike protein 10 , 30 , 31 ; however, the present study has categorically analyzed the underlying cause of different kinds of mutations that has shaped the evolution of spike protein during the ongoing pandemic. In the present study, variation of hydrophobicity of spike protein was observed to be a significant factor influencing the amino acid changes in spike protein. It is argued that the majority of viral mutations are harmless, however, some of these mutations may change infectivity, survival capability, pathologic property, or immunogenicity and antigenicity of the virus. 32 , 33 Previous reports pointed out several important mutations and in most of these cases directional mutational pressure changed toward higher hydrophobic/lower hydrophilic amino acid. 9 , 34 , 35 Recent studies suggest that the D614G variant is close to reaching fixation around the world. 12 The present work reinforces this observation that changes between aspartic acid to glycine contributes towards the difference in amino acid usage of spike protein associated with a change in hydrophobicity. The mutation D614G on the spike protein may also increase binding affinity between the spike protein and host ACE2 receptor, thus enhancing virus loads that lead to increased infectivity. 36 In addition, recent reports also provide evidence of this variant in viral spread associated with higher viral load. 37 , 38 Substitution of aspartic acid with glycine reduces negative charge of the protein charge, while ACE2 has a largely negative electrostatic potential on the surface, so this electrostatic effect may also facilitate corona virus infection. 39
Our results also show that compared to spike proteins collected during the earlier months, the recent sequences preferred to have more hydrophobic residues, as is evident from the higher number of sequences followed SPMHR pattern (Figure 2). The functional significance of lower binding energy for the spike–ACE2 complex for SPMHR group indicates that the ACE2 receptor can interact with viral spike protein more effectively compared to the spike–ACE2 complex from the SPLHR group. To determine the potential role of hydrophobic residues towards the binding affinity of spike‐ACE2 complex, we identified how many residues have changed between two proteins taken for our molecular docking study. We observed eight mutations (Table 1) that revealed hydrophobic to hydrophilic amino acid substitution. Figure 3b shows the change in the bonding pattern when glutamine was substituted with valine. The Gibbs free energy for unfolding was calculated to evaluate the effects of each of these mutations individually on protein complex stability. 40 The negative value of change in free energy associated with these mutations indicated stabilizing mutations. The chemical significance of the Gibbs free energy change between the two complexes comes from the molecular dynamics simulation study. RMSD and Rg plots (Figure 4a,b) illustrate that the spike–ACE2 complex from SPMHR group is comparatively more stable than the spike–ACE2 complex from SPLHR group. Further, the spike protein from SPMHR group exhibits considerable higher number of interactions with the ACE2 receptor‐binding domain, compared to the spike–ACE2 complex from SPLHR group. There are 95 interactions (1 salt bridge, 8 hydrogen bonds, and 86 non‐bonded contacts) for SPLHR category whereas, there are 103 interactions (1 salt bridge, 9 hydrogen bonds, and 93 nonbonded contacts) for SPMHR category. Higher interactions of spike protein from SPMHR group might contribute to high stable interaction with the ACE2.
TABLE 1.
Unfolding Gibbs free energies of mutations calculated using PremPS server depicting change in protein complex stability upon mutation
| Mutation position | ΔΔG | Effect of mutation |
|---|---|---|
| D215V | −0.4 | Stabilizing |
| A222V | −0.84 | Stabilizing |
| G261C | 0.06 | Mild stabilizing |
| E484G | −0.16 | Stabilizing |
| D614G | −0.31 | Stabilizing |
| Q860V | −0.94 | Stabilizing |
| K861L | −0.77 | Stabilizing |
| S970F | −1 | Stabilizing |
FIGURE 4.
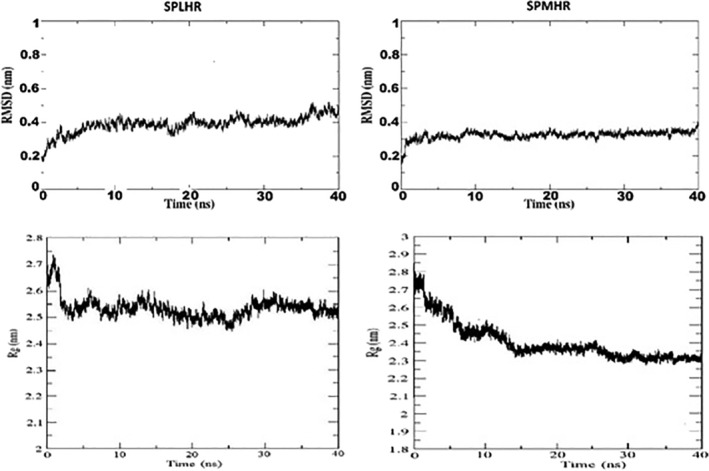
RMSD and Rg plot observed from molecular dynamics simulation study. (a) Between spike of SPLHR group with ACE2. (b) Between spike of SPMHR group with ACE2
We have found 13,064 mutations in SPMHR cluster of spike protein sequences. Then, we removed duplicate mutations and identified 1,487 unique mutations. In the set of 1,487 mutations there are 765 mutations representing conversion from hydrophilic amino acids to hydrophobic amino acids. We have checked whether these 765 mutations are associated with higher stability. 40 We found that 563 mutations out of 765 mutations are stabilizing character. So, most of the substitutions toward hydrophobic amino acids are stabilizing the spike–ACE2 docked complex. Therefore, accumulation of hydrophobic amino acids by substituting hydrophilic amino acid has a stabilizing effect on the spike–ACE2 complex. Selection of more hydrophobic amino acids in spike protein has functional significance on more effective binding with ACE2 receptor. 36
5. CONCLUSION
The present study shed light on the protein hydrophobicity as the mutational pressure for the evolution of spike protein of SARS‐CoV‐2. Here, in the present article, we have provided a comprehensive analysis of molecular evolutionary data to understand the virus infection potential which might be important for public health measures and prevent future epidemics like SARS‐CoV‐2.However, it would be judicious to consider the possibility that mutational variants might modulate the virulence and thereby might have impact on the pathogenicity of the disease. The classification of spike proteins according to the variation of hydrophobicity and thereby modulating the receptor binding affinity provides crucial information for designing treatment and, eventually, vaccines. The findings of the present study could help for the design of potential vaccine candidates/small molecular inhibitor against COVID19. Another future direction of the present study might be to undertake haplotypic/population level analysis in order to get a better view of genetic variation between SPMHR and SPLHR groups of spike proteins. The evolutionary characterization of the wide spectrum of haplotypes distributed in SPMHR and SPLHR may be used to determine the haplotype significance and its association with disease severity, various host genetic factors, and development of vaccines.
CONFLICT OF INTEREST
The authors declare that no conflicts of interest exist.
Supporting information
Figure S1 Distribution of spike genes between SPMHR and SPLHR groups with respect to respect to the date of collection of sequences in different clades as found in GISAID database. (a) Spike gene distribution in S, L & others clade. (b). Spike gene distribution in G & it's derivatives clade.
{kind=link}
Table S1 Exhaustive base composition of spike genes
ACKNOWLEDGMENT
Manisha Ghosh is supported by Senior Research Fellowship by Indian Council of Medical Research (ICMR). We also sincerely acknowledge the help of Dr. Fayaz S. M. of Department of Biotechnology, Manipal Institute of Technology in the molecular dynamics and simulation study.
Ghosh M, Basak S, Dutta S. Underlying selection for the diversity of spike protein sequences of SARS‐CoV‐2. IUBMB Life. 2022;74:213–220. 10.1002/iub.2577
REFERENCES
- 1. Weiss SR, Leibowitz JL. Coronavirus pathogenesis. Adv Virus Res. 2011;81:85–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Drosten C, Günther S, Preiser W, et al. Identification of a novel coronavirus in patients with severe acute respiratory syndrome. N Engl J Med. 2003;348(20):1967–1976. [DOI] [PubMed] [Google Scholar]
- 3. Lucas M, Karrer U, Lucas A, Klenerman P. Viral escape mechanisms—Escapology taught by viruses. Int J Exp Pathol. 2001;82:269–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Berngruber TW, Froissart R, Choisy M, Gandon S. Evolution of virulence in emerging epidemics. PLoS Pathog. 2013;9:e1003209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Rambaut A, Pybus OG, Nelson MI, Viboud C, Taubenberger JK, Holmes EC. The genomic and epidemiological dynamics of human influenza A virus. Nature. 2008;453:615–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Grenfell BT, Pybus OG, Gog JR, et al. Unifying the epidemiological and evolutionary dynamics of pathogens. Science. 2004;303:327–332. [DOI] [PubMed] [Google Scholar]
- 7. Salemi M, Fitch WM, Ciccozzi M, Ruiz‐Alvarez MJ, Rezza G, Lewis MJ. Severe acute respiratory syndrome coronavirus sequence characteristics and evolutionary rate estimate from maximum likelihood analysis. J Virol. 2004;78:1602–1603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wang C, Liu Z, Chen Z, et al. The establishment of reference sequence for SARS‐CoV‐2 and variation analysis. J Med Virol. 2020;92:667–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Phan T. Genetic diversity and evolution of SARS‐CoV‐2. Infect Genet Evol. 2020. Jul;81:104260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. van Dorp L, Acman M, Richard D, et al. Emergence of genomic diversity and recurrent mutations in SARS‐CoV‐2. Infect Genet Evol. 2020;83:104351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lauring AS, Andino R. Quasispecies theory and the behavior of RNA viruses. PLoS Pathog. 2010;6(7):e1001005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Yurkovetskiy L, Wang X, Pascal KE, et al. Structural and functional analysis of the D614G SARS‐CoV‐2 spike protein variant. Cell. 2020;183:739–751.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Groves DC, Rowland‐Jones SL, Angyal A. The D614G mutations in the SARS‐CoV‐2 spike protein: Implications for viral infectivity, disease severity and vaccine design. Biochem Biophys Res Commun. 2021;538:104–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Haijema BJ, Volders H, Rottier PJ. Switching species tropism: An effective way to manipulate the feline coronavirus genome. J Virol. 2003;77:4528–4538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kuo L, Godeke GJ, Raamsman MJ, Masters PS, Rottier PJ. Retargeting of coronavirus by substitution of the spike glycoprotein ectodomain: Crossing the host cell species barrier. J Virol. 2000;74:1393–1406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Schickli JH, Thackray LB, Sawicki SG, Holmes KV. The N‐terminal region of the murine coronavirus spike glycoprotein is associated with the extended host range of viruses from persistently infected murine cells. J Virol. 2004;78:9073–9083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Tortorici MA, Walls AC, Lang Y, et al. Structural basis for human coronavirus attachment to sialic acid receptors. Nat Struct Mol Biol. 2019;26:481–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Guruprasad L. Human SARS CoV‐2 spike protein mutations. Proteins. 2021. May;89(5):569–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Priya P, Shanker A. Coevolutionary forces shaping the fitness of SARS‐CoV‐2 spike glycoprotein against human receptor ACE2. Infect Genet Evol. 2021;87:104646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ahmed SF, Quadeer AA, McKay MR. Preliminary identification of potential vaccine targets for the COVID‐19 coronavirus (SARS‐CoV‐2) based on SARS‐CoV immunological studies. Viruses. 2020;12:254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Peden JF. Analysis of codon usage. Nottingham: University of Nottingham, 2000. [Google Scholar]
- 22. Roy A, Banerjee R, Basak S. HIV progression depends on codon and amino acid usage profile of envelope protein and associated host‐genetic influence. Front Microbiol. 2017;8:1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kyte J, Doolittle RF. A simple method for displaying the hydropathic character of a protein. J Mol Biol. 1982;157:105–132. [DOI] [PubMed] [Google Scholar]
- 24. Huang Y, Niu B, Gao Y, Fu L, Li W. CD‐HIT suite: A web server for clustering and comparing biological sequences. Bioinformatics. 2010;26:680–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bhattacharya D, Nowotny J, Cao R, Cheng J. 3Drefine: An interactive web server for efficient protein structure refinement. Nucleic Acids Res. 2016;44:W406–W409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Pierce BG, Wiehe K, Hwang H, Kim BH, Vreven T, Weng Z. ZDOCK server: Interactive docking prediction of protein‐protein complexes and symmetric multimers. Bioinformatics. 2014;30:1771–1773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Xue LC, Rodrigues JP, Kastritis PL, Bonvin AM, Vangone A. PRODIGY: A web server for predicting the binding affinity of protein‐protein complexes. Bioinformatics. 2016;32:3676–3678. [DOI] [PubMed] [Google Scholar]
- 28. Pereson MJ, Flichman DM, Martínez AP, Baré P, Garcia GH, Di Lello FA. Evolutionary analysis of SARS‐CoV‐2 spike protein for its different clades. J Med Virol. 2021;93:3000–3006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Matyášek R, Řehůřková K, Berta Marošiová K, Kovařík A. Mutational asymmetries in the SARS‐CoV‐2 genome may lead to increased hydrophobicity of virus proteins. Genes (Basel). 2021;12:826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Korber B, Fischer WM, Gnanakaran S, et al. Tracking changes in SARS‐CoV‐2 spike: Evidence that D614G increases infectivity of the COVID‐19 virus. Cell. 2020;182:812–827.e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Li Q, Wu J, Nie J, et al. The impact of mutations in SARS‐CoV‐2 spike on viral infectivity and antigenicity. Cell. 2020;182:1284–1294.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Portelli S, Olshansky M, Rodrigues CHM, et al. Exploring the structural distribution of genetic variation in SARS‐CoV‐2 with the COVID‐3D online resource. Nat Genet. 2020;52:999–1001. [DOI] [PubMed] [Google Scholar]
- 33. Chen J, Wang R, Wang M, Wei GW. Mutations strengthened SARS‐CoV‐2 infectivity. J Mol Biol. 2020;432:5212–5226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Forster P, Forster L, Renfrew C, Forster M. Phylogenetic network analysis of SARS‐CoV‐2 genomes. Proc Natl Acad Sci U S A. 2020;117:9241–9243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Biswas NK, Majumder PP. Analysis of RNA sequences of 3636 SARS‐CoV‐2 collected from 55 countries reveals selective sweep of one virus type. Indian J Med Res. 2020;151:450–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zeng L, Li D, Tong W, Shi T, Ning B. Biochemical features and mutations of key proteins in SARS‐CoV‐2 and their impacts on RNA therapeutics. Biochem Pharmacol. 2021;189:114424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Plante JA, Liu Y, Liu J, et al. Spike mutation D614G alters SARS‐CoV‐2 fitness. Nature. 2021;592:116–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Volz E, Hill V, McCrone JT, et al. Evaluating the effects of SARS‐CoV‐2 spike mutation D614G on transmissibility and pathogenicity. Cell. 2021;184:64–75.e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Pawłowski PH. Charged amino acids may promote coronavirus SARS‐CoV‐2 fusion with the host cell. AIMS Biophys. 2021;8:111–120. [Google Scholar]
- 40. Chen Y, Lu H, Zhang N, Zhu Z, Wang S, Li M. PremPS: Predicting the impact of missense mutations on protein stability. PLoS Comput Biol. 2020;16:e1008543. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Distribution of spike genes between SPMHR and SPLHR groups with respect to respect to the date of collection of sequences in different clades as found in GISAID database. (a) Spike gene distribution in S, L & others clade. (b). Spike gene distribution in G & it's derivatives clade.
Table S1 Exhaustive base composition of spike genes