

Abstract
Background
Multiple myeloma (MM) is a hematological malignancy. Coronavirus disease 2019 (COVID‐19) infection correlates with MM features. This study aimed to identify MM prognostic biomarkers with potential association with COVID‐19.
Methods
Differentially expressed genes (DEGs) in five MM data sets (GSE47552, GSE16558, GSE13591, GSE6477, and GSE39754) with the same expression trends were screened out. Functional enrichment analysis and the protein‐protein interaction network were performed for all DEGs. Prognosis‐associated DEGs were screened using the stepwise Cox regression analysis in the cancer genome atlas (TCGA) MMRF‐CoMMpass cohort and the GSE24080 data set. Prognosis‐associated DEGs associated with COVID‐19 infection in the GSE164805 data set were also identified.
Results
A total of 98 DEGs with the same expression trends in five data sets were identified, and 83 DEGs were included in the protein‐protein interaction network. Cox regression analysis identified 16 DEGs were associated with MM prognosis in the TCGA cohort, and only the cytochrome c oxidase subunit 6C (COX6C) gene (HR = 1.717, 95% CI 1.231–2.428, p = .002) and the nucleotide‐binding oligomerization domain containing 2 (NOD2) gene (HR = 0.882, 95% CI 0.798–0.975, p = .014) were independent factors related to MM prognosis in the GSE24080 data set. Both of them were downregulated in patients with mild COVID‐19 infection compared with controls but were upregulated in patients with severe COVID‐19 compared with patients with mild illness.
Conclusions
The NOD2 and COX6C genes might be used as prognostic biomarkers in MM. The two genes might be associated with the development of COVID‐19 infection.
Keywords: bioinformatics analysis, coronavirus disease 2019, multiple myeloma, overall survival
1. INTRODUCTION
Multiple myeloma (MM) ranks 24th in the world in 2018, with approximately 160,000 newly diagnosed cases. 1 In hematological malignancies, the incidence of MM ranks second to non‐Hodgkin lymphoma. The incidence rate of MM is 2.1 per 100 000 persons globally and is higher (10 per 100,000 persons) in Latin American countries. 2 , 3 , 4 , 5 The survival rate of MM patients has been greatly improved over the past two decades with the introduction of new drugs, but MM remains an incurable malignancy. Also, the 5‐year and 10‐year overall survival rate of MM is still less than 60% and 40%. 2 Older MM patients have a lower survival rate and an insignificant improvement in survival from new drugs. 2
Prognostic factors can be applied for predicting individualized prognosis, making risk stratification, and treatment recommendations. 6 Factors associated with the prognosis in MM include patients' age, cytogenetics, serum creatinine, platelet count, and gene expression profile. 6 Protein coding (mRNAs) and noncoding RNAs (including miRNAs, lncRNAs, and circRNAs) with prognostic significance in MM have been proven in the past years. 7 , 8 Of note, recent evidence shows a correlation between coronavirus disease 2019 (COVID‐19) infection and MM clinical features. 9 , 10 , 11 A recent study showed that most MM patients (77%) had moderate‐severe coronavirus disease 2019 (COVID‐19) clinical features, but is lower than 89% in noncancer patients. Most patients who did not survive COVID‐19 were male with advanced tumor stages. 11 Most MM patients with a loss of functional immunoglobulins and decreased CD4+ T‐cell count, which are associated with increased infections. 12 Accordingly, MM patients are more susceptible to COVID‐19 infection. 9 , 10 However, some research studies showed that the most common MM subtypes with COVID‐19 infection were IgG and MM patients with COVID‐19 showed a longer duration to clinical improvement. 13 These studies show that there might be a potent correlation between COVID‐19 infection and MM, which might indicate the hidden and unknown mechanisms of MM.
Since pandemic COVID‐19 in 2020, more and more evidence shows that there is a correlation between gene differential expression and COVID‐19 infection. 14 However, there is limited information on the characteristics of MM patients with COVID‐19. Hence, the identification of potent prognostic biomarkers in MM that might have association with pandemic COVID‐19 might provide novel insights into the treatment strategy for MM patients hospitalized with COVID‐19.
2. MATERIALS AND METHODS
2.1. Multiple myeloma microarray data sets
Five MM gene expression microarray data sets (including GSE16558, GSE47552, GSE39754, GSE13591, and GSE6477) were selected from The National Center for Biotechnology Information (NCBI) Gene Expression Omnibus (GEO, http://www.ncbi.nlm.nih.gov/geo). The inclusion criteria of MM microarray data sets were as follows 9 : inclusive of normal bone marrow plasma cell samples from MM patients (≥40) 12 ; without restrictions on patients' gender, race, treatment response, karyotype, mutation, and pathologic stages. Forty‐one, 60, 133, 103, and 170 plasma cell samples (CD138‐positive) from MM patients were included in the data sets GSE47552, GSE16558, GSE13591, GSE6477, and GSE39754, respectively. Five (GSE13591, GSE47552, and GSE16558), six (GSE39754), or 15 (GSE6477) plasma cell samples from healthy donors were included in each data set and were used as healthy control samples. These data sets were performed based on three platforms GPL6244 ([HuGene‐1_0‐st] Affymetrix Human Gene 1.0 ST Array [transcript (gene) version]; GSE47552 and GSE16558), GPL96 ([HG‐U133A] Affymetrix Human Genome U133A Array; GSE13591 and GSE6477), and GPL5175 ([HuEx‐1_0‐st] Affymetrix Human Exon 1.0 ST Array [transcript (gene) version]; GSE39754). Samples from patients with MM and healthy donors were retained in this study.
2.2. COVID‐19 microarray data set
One COVID‐19 gene expression data set (GSE164805) was downloaded from the GEO. It was based on the GPL26963 platform (Agilent‐085982 Arraystar human lncRNA V5 microarray). GSE164805 was composed of 15 peripheral blood mononuclear cells (PBMCs) from severe (respiratory rate ≥ 30 times/min, resting finger oxygen saturation ≤93%, and artery PaO2/FiO2 ≤ 300 mmHg) and mild COVID‐19 patients (n = 10, PCR positive) as well as healthy controls (n = 5).
2.3. Data processing and identification of differentially expressed genes
Differentially expressed genes in each data set were identified using the online program GEO2R (http://www.ncbi.nlm.nih.gov/geo/geo2r/) provided by the NCBI. DEGs in each data set were selected using the criteria of p < .05 and |log(fold change, FC)| ≥ 0.5. DEGs common to the five MM data sets were screened out using the Venn tool (http://bioinformatics.psb.ugent.be/webtools/Venn/). DEGs with the same expression trends (upregulation, logFC > 0.5; or downregulation, logFC < ‐0.5) in five data sets were retained and used for further analysis.
2.4. Gene set functional enrichment analysis
Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways and Gene Ontology (GO) biological processes associated with DEGs were obtained in the Database for Annotation, Visualization and Integrated Discovery (DAVID; version 6.8; https://david.ncifcrf.gov/). Significant themes were identified using the criteria of p < .05 and input number ≥ 1.
2.5. Construction of the protein‐protein interaction network
Search Tool for the Retrieval of Interacting Genes/Proteins (STRING; version 11.0; https://string‐db.org/cgi/input.pl) is a database of known and predicted Protein‐protein interactions. Protein interaction pairs with medium confidence (0.4) among DEGs were predicted in the STRING database. The PPI network was constructed using the Cytoscape (version 3.8.0; http://apps.cytoscape.org/), and modules with a high K‐score (>5.0) were identified using the plugin MCODE (http://apps.cytoscape.org/apps/mcode).
2.6. Selection of multiple myeloma prognosis‐associated differentially expressed genes
The association of DEGs in the PPI network with the prognosis in MM patients was used to screen the MM prognosis‐associated DEGs. The Cancer Genome Atlas (TCGA) MM RNA sequencing data set (MMRF‐CoMMpass) was obtained from the UCSC Xena (https://xenabrowser.net/datapages/). Samples (n = 784) with matched gene expression profile, overall survival time, and vital status (1 = death) were extracted and used for further analyses in this study. The association of DEG expression levels with the overall survival time in individuals with MM was analyzed using the Cox regression analysis. Also, the microarray data set GSE24080 (GPL570, [HG‐U133_Plus_2] Affymetrix Human Genome U133 Plus 2.0 Array, n = 559) was obtained to validate the association of DEGs with MM prognosis. Kaplan‐Meier plots were used to show the differences in survival percent between patients with high and low expression levels (divided by median value) of DEGs.
2.7. Identification of multiple myeloma prognosis‐associated differentially expressed genes in COVID‐19
At last, the MM prognosis‐associated DEGs which might be differentially expressed in patients with COVID‐19 infection were identified using the Venn diagram. The expression profiles of the key MM prognosis‐associated DEGs in patients with mild and severe COVID‐19 clinical features were compared.
2.8. Statistical analysis
Cox regression analysis and Kaplan‐Meier survival analysis were performed using the SPSS software (version 22.0; IBM Corporation, Somers). 95% confident interval (CI) and hazard ratio (HR) were calculated. The one‐way ANOVA test was used for the comparison in the expression level of genes among sample comparison groups in the GSE164805 data set. A statistical significance was considered when p < .05.
3. RESULTS
3.1. Differentially expressed genes identification and screening
Total of 1144, 3213, 9475, 2504, and 4428 DEGs were identified from the GSE13591, GSE16558, GSE39754, GSE47552, and GSE6477 data set, respectively. As indicated by the Venn diagram analysis, there were 109 common DEGs among the five data sets (Figure 1), including 98 DEGs with the same expression trends were identified and shown in Table S1.
FIGURE 1.
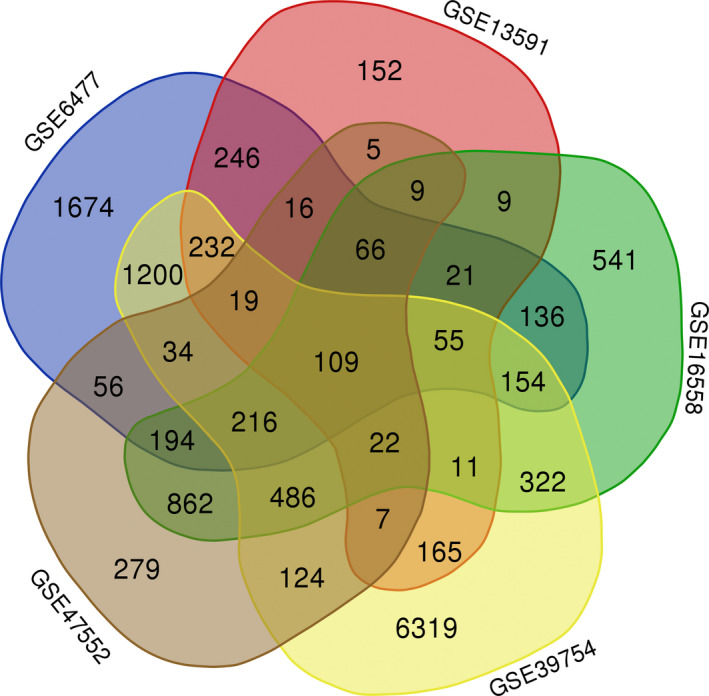
Venn diagram indicating the number of differentially expressed genes in five data sets
3.2. Functional enrichment analysis
Functional enrichment analysis showed that a cluster of upregulated genes encoding ribosomal proteins were associated with biological processes including “GO:0019083: viral transcription,” “GO: 0006364:rRNA processing,” and “GO:0002181: cytoplasmic translation,” and KEGG pathways including “hsa03010: Ribosome” (Table S2). Also, another cluster of genes were associated with biological processes such as “GO:0006123: mitochondrial electron transport, cytochrome c to oxygen” and “GO:1902600: hydrogen ion transmembrane transport.”
3.3. Construction of the protein‐protein interaction network and modules
A total of 601 interaction pairs were predicted for 83 of the above 98 DEGs, including eight downregulated DEGs and 75 upregulated DEGs. Accordingly, the PPI network was composed of 83 nodes (DEG products) and 601 edges (interactions; Figure 2). Two modules, modules 1 and 2, consisted of 27 nodes (340 edges) and 13 nodes (63 edges), respectively. All the genes in modules 1 and 2 were upregulated (Table S1 and Figure 2). Functional enrichment analysis showed that DEGs in module 1 were mainly associated with 14 biological processes including “GO:0019083: viral transcription,” “GO: 0006364:rRNA processing,” and “GO:0002181: cytoplasmic translation,” and one KEGG pathway of “hsa03010: Ribosome” (Figure S1A,B). DEGs in module 2 were associated with eight biological processes related to mitochondrial energy metabolism and seven KEGG pathways related to neurodegenerative diseases (Figure S1A,B). Those results showed that genes in modules 1 and 2 had distinct biological functions.
FIGURE 2.
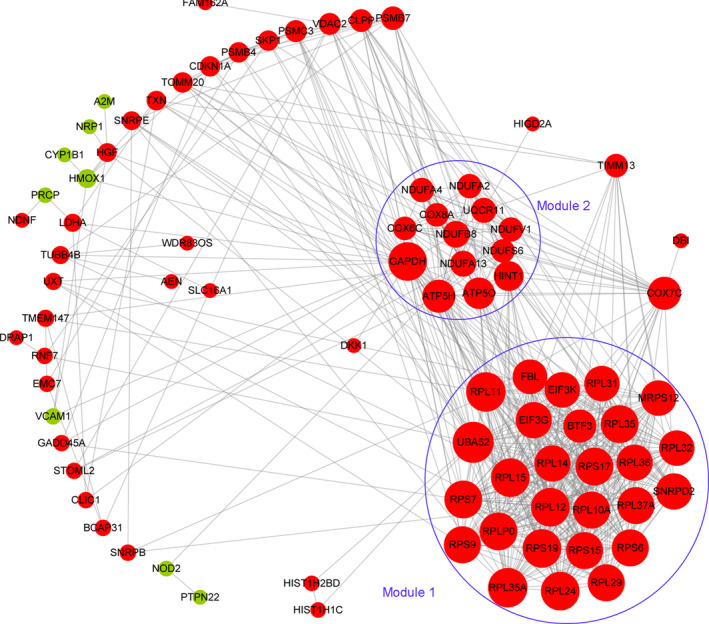
Protein‐protein interaction network. This network was constructed based on the interactions among the 98 common differentially expressed genes in five data sets. Upregulation and downregulation are indicated by red and green color, respectively. Two modules in the circle lines were identified using MCODE in the Cytoscape
3.4. Identification of differentially expressed genes associated with the prognosis of multiple myeloma
Using the TCGA MMRF‐CoMMpass cohort (n = 784) and univariate Cox regression analysis, we identified that 30 DEGs of the 83 DEGs in the PPI network were associated with the overall survival time (Table S3). Multivariate Cox regression analysis showed that 12 DEGs were confounding factors because the inclusive of them changed the other factors from protective factors (HR < 1.0) to risk factors (HR > 1.0) or vice versa. The exclusive of the 12 DEGs retained the consistent features of the other genes by univariate and multivariate Cox regression analyses (Table S4). Also, the Spearman correlation analysis showed that there were significant correlations between the expression levels of the 12 genes (Spearman correlation coefficient, r > .500, p < .05; Table S5). Four out of the other 18 DEGs were identified as prognosis‐associated DEGs by multivariate Cox regression analysis (Table S4). Also, 16 genes were considered as potent prognosis‐associated DEGs in MM patients in this study.
3.5. Validation for the association of differentially expressed genes with multiple myeloma prognosis in GSE24080
The GSE24080 data set that contained the overall survival data of 559 patients were downloaded from the GEO. The expression profiles of the 16 potent MM prognosis‐associated DEGs were extracted. The stepwise Cox regression analysis showed that the cytochrome c oxidase subunit 6C (COX6C) gene was a risk factor for a short even‐free survival time (HR = 1.704, 95% CI 1.198–2.426, p = .003) and a short overall survival period (HR = 1.717, 95% CI 1.231–2.428, p = .002; Table 1). Besides, we confirmed that the nucleotide‐binding oligomerization domain containing 2 (NOD2) gene might be a protective factor against a poor prognosis in MM patients (even‐free survival time: HR = 0.899, 95% CI 0.811–0.996, p = .041; overall survival time: HR = 0.882, 95% CI 0.798–0.975, p = .014; Table 1).
TABLE 1.
Correlation of genes with the survival of patients with multiple myeloma in the GSE24080 dataset
| Gene symbol | Univariate | Multivariate | ||
|---|---|---|---|---|
| HR (95% CI) | P | HR (95% CI) | P | |
| Even‐free survival | ||||
| CLIC1 | 1.196 (0.897–1.595) | 0.224 | ||
| COX6C | 1.822 (1.290–2.574) | 0.001 | 1.704 (1.198–2.426) | 0.003 |
| EIF3G | 0.869 (0.700–1.079) | 0.204 | ||
| EIF3K | 0.850 (0.664–1.089) | 0.199 | ||
| HIGD2A | 0.874 (0.702–1.089) | 0.231 | ||
| MRPS12 | 1.069 (0.928–1.233) | 0.355 | ||
| NOD2 | 0.879 (0.793–0.973) | 0.013 | 0.899 (0.811–0.996) | 0.041 |
| RPL12 | 1.008 (0.803–1.264) | 0.946 | ||
| RPL14 | 0.850 (0.631–1.145) | 0.285 | ||
| RPL15 | 0.882 (0.664–1.172) | 0.387 | ||
| RPL24 | 1.006 (0.827–1.375) | 0.621 | ||
| RPS15 | 0.813 (0.573–1.152) | 0.244 | ||
| RPS17 | 1.071 (0.774–1.483) | 0.678 | ||
| RPS6 | 0.941 (0.665–1.332) | 0.732 | ||
| RPS9 | 0.825 (0.647–1.052) | 0.122 | ||
| SNRPB | 1.337 (1.027–1.741) | 0.031 | 1.177 (0.909–1.523) | 0.216 |
| Overall survival | ||||
| CLIC1 | 1.263 (0.957–1.668) | 0.099 | ||
| COX6C | 1.892 (1.349–2.653) | 2.190e–04 | 1.717 (1.213–2.428) | 0.002 |
| EIF3G | 0.906 (0.730–1.124) | 0.369 | ||
| EIF3K | 0.877 (0.683–1.127) | 0.304 | ||
| HIGD2A | 0.954 (0.765–1.190) | 0.677 | ||
| MRPS12 | 1.029 (0.894–1.184) | 0.690 | ||
| NOD2 | 0.855 (0.775–0.944) | 0.002 | 0.882 (0.798–0.975) | 0.014 |
| RPL12 | 0.980 (0.788–1.220) | 0.859 | ||
| RPL14 | 0.882 (0.663–1.174) | 0.389 | ||
| RPL15 | 0.963 (0.727–1.277) | 0.794 | ||
| RPL24 | 1.030 (0.805–1.316) | 0.816 | ||
| RPS15 | 0.841 (0.600–1.178) | 0.313 | ||
| RPS17 | 1.041 (0.761–1.424) | 0.802 | ||
| RPS6 | 0.908 (0.652–1.266) | 0.571 | ||
| RPS9 | 0.864 (0.677–1.102) | 0.238 | ||
| SNRPB | 1.421 (1.090–1.853) | 0.010 | 1.217 (0.937–1.580) | 0.141 |
3.6. Kaplan‐Meier survival analysis for COX6C and NOD2
The expression profiles of the NOD2 (downregulated) and COX6C (upregulated) genes are shown in Figure S2. The results of Kaplan‐Meier survival plot analysis GSE24080 data set and TCGA cohort showed that patients with high expression levels of COX6C had a lower overall survival ratio in both GSE24080 data set and TCGA cohort compared with patients who had low COX6C expression levels (in GSE24080: HR = 1.608, 95% CI 1.184–2.183, p = .002; in TCGA: HR = 1.392 95% CI 1.020–1.898, p = .037). However, patients with high NOD2 expression level developed a higher overall survival ratio compared with patients with low NOD2 expression level (in GSE24080: HR = 0.638, 95% CI 0.471–0.865, p = .004; in TCGA: HR = 0.725, 95% CI 0.531–0.991, p = .044; Figure 3A,B ). These results indicated that high COX6C expression and low NOD2 expression might be independent risk factors for a poor MM prognosis.
FIGURE 3.
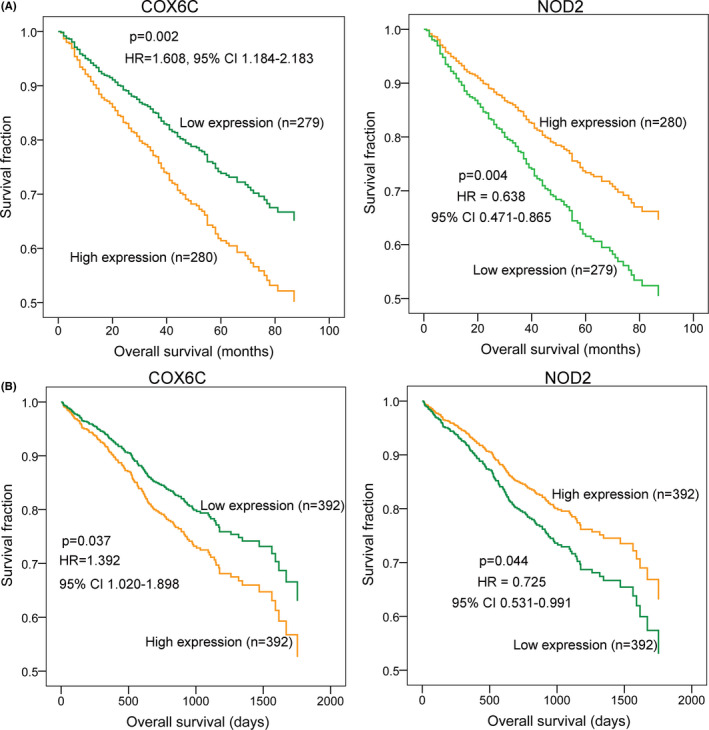
Kaplan‐Meier survival analysis for COX6C and NOD2 in multiple myeloma. A, the overall survival analyses for the COX6C and NOD2 genes in the GSE24080 data set (n = 559). B, the overall survival analyses for the COX6C and NOD2 genes in The Cancer Genome Atlas (TCGA) cohort (MMRF‐CoMMpass, n = 784)
3.7. Identification of common Differentially expressed genes in COVID‐19 patients
Based on the same criteria of |logFC| > 0.5 and p < .05, we identified 25,874 DEGs in the GSE164805 data set, including 10 of the 16 MM prognosis‐associated DEGs (Figure 4A,B). Also, the NOD2 and COX6C genes were both downregulated in patients with mild COVID‐19 infection compared with healthy controls (Figure 4B,C). However, patients with severe COVID‐19 clinical features had higher expression levels of NOD2 and COX6C compared with patients with low NOD2 (p = .0035) and COX6C (p = .0716, insignificance; Figure 4C) levels, respectively. These results suggested that the NOD2 and COX6C genes might be associated with COVID‐19 severity.
FIGURE 4.
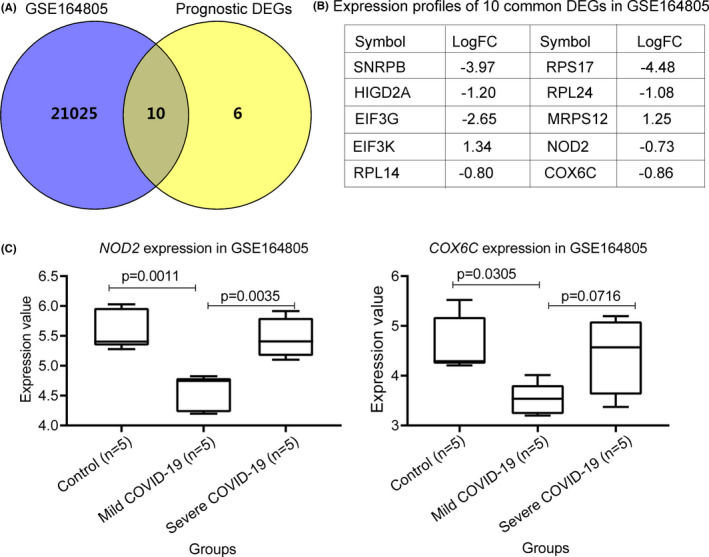
Identification and expression of differentially expressed genes in patients with COVID‐19 infection and multiple myeloma. A, the Venn diagram indicating the common DEGs between patients with COVID‐19 infection and the 16 prognostic genes in multiple myeloma. B, the expression profiles of the 10 common genes in the GSE164805 data set. C, the expression profiles of the COX6C and NOD2 genes in the peripheral blood mononuclear cells (PBMCs) from patients with (mild = 5 and severe = 5) and without (controls) COVID‐19 infection
4. DISCUSSION
Based on the integrated bioinformatics of microarray data sets from patients with MM, we identified 16 DEGs that were associated with the prognosis of patients with MM. Also, the COX6C (upregulated) and NOD2 (downregulated) genes might be independent factors associated with the prognosis in MM patients. Also, the two genes were both downregulated in patients with mild COVID‐19 infection compared with healthy controls, but were upregulated in patients with severe COVID‐19 infection compared with patients who had mile infections. These results might show that there was a potent connection between the development of MM and the expression of the two genes. Besides, the two genes might have potent association with the development of COVID‐19.
NOD2 is a putative intracellular receptor for bacterial peptidoglycans and acts as a bacterial sensor, innate immune receptor, and antibacterial factor. 15 , 16 NOD2‐mediated inflammation and immunity contribute to the control of infections. 17 , 18 NOD2 is only activated by muramyl dipeptides (MDP) that are presented in bacterial peptidoglycan. 17 , 19 MDP and its analogs enhance nonspecific resistance to viral infection, including herpes simplex virus type 2 (HSV2) that is also defended by Bacillus Calmette‐Guérin (BCG) vaccination. 20 , 21 , 22 , 23 MDP presents in human peripheral blood, and its concentration is increased after BCG vaccination. 24 , 25 In the last year of the COVID‐19 pandemic, research studies showed that BCG‐vaccinated persons might have enhanced protection from infection of COVID‐19. 25 Accordingly, we presumed that the decreased NOD2 expression might enhance the risk of COVID‐19 infection. Also, the enhanced expression of NOD2 in patients with severe COVID‐19 pneumonia might be a self‐protection mechanism and adaptive immune response to COVID‐19 infection or drug‐induced boost to the immune system.
The NOD2 gene also functions as a tumor suppressor gene in multiple cancers. 26 , 27 , 28 , 29 Li et al 26 showed that the rs2111235 C allele mutation, which might result in a decreased secretion of pro‐inflammatory cytokines after H pylori infection, was associated with a decreased risk of gastric cancer progression. However, there is limited information on the characteristics of NOD2 expression and mutation in MM patients. 30 A recent study from Zmorzyński et al 30 showed that the 3020insC variant of the NOD2/CARD15 gene did not impact the MM risk, but resulted in the upregulation of pro‐inflammatory cytokines in MM patients. Our present study showed that patients with MM had a lower expression level of NOD2 in the bone marrow plasma cells compared with controls. Also, NOD2 expression was a positive prognostic biomarker in MM prognosis, as patients with high NOD2 expression levels had a higher survival ratio compared with patients who had low NOD2 expression levels. This correlation might be due to the increased innate immunity mediated by NOD2.
The negative prognostic biomarker in MM was the COX6C gene that encodes a cytochrome c oxidase subunit VIc, the terminal enzyme of the mitochondrial respiratory chain that catalyzes the electron transfer from reduced cytochrome c to oxygen. Mitochondrial respiratory chain complexes ensure the energy supply for cell proliferation, cell growth, DNA replication, and multiple biological processes. 31 These proteins had deregulated expression levels in cancer cells. 31 , 32 , 33 , 34 Patients with high expression levels of mitochondrial respiratory chain proteins have shorter survival periods compared with patients with low expression levels. 33 , 34 The inhibition of mitochondrial translation effectively sensitizes cancer cells to chemotherapy. 31 , 34 These results showed that the prognostic COX6C gene might be a therapeutic target in MM.
There is evidence showing that the initial exposure to influenza virus upregulates the expression level of COX6C expression in the host alveolar and bronchial epithelial cells. 35 COX6C is required for the initiation of apoptosis in the host cells and the activation of caspase‐9 and caspase‐3, 36 which was associated with inhibited virus infection and decreased viral numbers. 37 , 38 , 39 Besides, low expression of COX6C decreases caspase‐3 apoptotic pathway, which is highly favorable for viral replication. 35 , 40 These data at least provide evidence showing that patients with COVID‐19 infection and low COX6C expression had a longer duration of clinical improvement compared with noncancer patients. 13 The decreased expression level of COX6C in patients with COVID‐19 infection and increased COX6C expression level in MM patients might show that there was a potent negative correlation between the pathogenesis and development of MM and COVID‐19 infection.
5. CONCLUSIONS
In summary, our present study showed that patients with MM had a higher expression level of the COX6C gene and genes encoding ribosomal proteins, but a lower level of the NOD2 gene compared with controls. Both low NOD2 expression level and high COX6C expression level were related to a low survival ratio in MM patients. Also, the expression of NOD2 and COX6C was downregulated in patients with mild COVID‐19 pneumonia compared with healthy controls, but was upregulated in patients with severe COVID‐19 pneumonia compared with patients with mild COVID‐9 infection. The NOD2 and COX6C genes might be used as prognostic biomarkers in MM. The two genes might be associated with the development of COVID‐19 infection.
CONFLICT OF INTEREST
The authors declare that they have no competing interests.
AUTHOR CONTRIBUTIONS
Fei Wang and Baoan Chen involved in conception and design of the research and obtaining funding. Ran Liu, Jie Yang, and Baoan Chen involved in acquisition of data. Ran Liu and Jie Yang involved in analysis and interpretation of data. Fei Wang and Ran Liu involved in statistical analysis. Fei Wang involved in drafting the manuscript. Baoan Chen involved in revision of manuscript for important intellectual content. Fei Wang, Ran Liu, Jie Yang, and Baoan Chen involved in review and approval of the final manuscript.
ETHICAL APPROVAL
This article does not contain any studies with human participants performed by any of the authors.
Supporting information
Fig S1
Fig S2
Table S1
Table S2
Table S3
Table S4
Table S5
ACKNOWLEDGEMENTS
Not Applicable.
Wang F, Liu R, Yang J, Chen B. New insights into genetic characteristics between multiple myeloma and COVID‐19: An integrative bioinformatics analysis of gene expression omnibus microarray and the cancer genome atlas data. Int J Lab Hematol. 2021;43:1325–1333. 10.1111/ijlh.13717
Funding information
This study is supported by the “Jiangsu Provincial Medical Youth Talent” (QNRC2016812) and the “Natural Science Foundation of Jiangsu Province for Youth” (BK20180372)
DATA AVAILABILITY STATEMENT
The original microarray data sets are available at The National Center for Biotechnology Information Gene Expression Omnibus (https://www.ncbi.nlm.nih.gov/geo/) with the accession numbers: GSE17498, GSE24080, GSE16558, GSE47552, GSE39754, GSE13591, and GSE6477. The Cancer Genome Atlas (TCGA) MM RNA sequencing data set (MMRF‐CoMMpass) was obtained from the UCSC Xena (https://xenabrowser.net/datapages/).
REFERENCES
- 1. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA‐Cancer J Clin. 2018;68(6):394–424. [DOI] [PubMed] [Google Scholar]
- 2. Costa LJ, Brill IK, Omel J, Godby K, Kumar SK, Brown EE. Recent trends in multiple myeloma incidence and survival by age, race, and ethnicity in the United States. Blood Adv. 2017;1(4):282–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Cowan AJ, Allen C, Barac A, et al. Global burden of multiple myeloma: a systematic analysis for the global burden of disease study 2016. JAMA Oncol. 2018;4(9):1221–1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Curado MP, Oliveira MM, Silva DR, Souza DL. Epidemiology of multiple myeloma in 17 Latin American countries: an update. Cancer Med. 2018;7(5):2101–2108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Terebelo HR, Abonour R, Gasparetto CJ, et al. Development of a prognostic model for overall survival in multiple myeloma using the Connect® MM patient registry. Br J Haematol. 2019;187(5):602–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hanbali A, Hassanein M, Rasheed W, Aljurf M, Alsharif F. The evolution of prognostic factors in multiple myeloma. Adv Hematol. 2017;2017:4812637. doi: 10.1155/2017/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Misiewicz‐Krzeminska I, Krzeminski P, Corchete LA, et al. Factors regulating microRNA expression and function in multiple myeloma. Non‐coding RNA. 2019;5(1):9. doi: 10.3390/ncrna5010009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Xu P, Xia T, Ling Y, Chen B. MiRNAs with prognostic significance in multiple myeloma: a systemic review and meta‐analysis. Medicine. 2019;98(33):e16711. doi: 10.1097/MD.0000000000016711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Al Saleh AS, Sher T, Gertz MA. Multiple myeloma in the time of COVID‐19. Acta Haematol. 2020;143:410–416. doi: 10.1159/000507690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Dhakal B, D'Souza A, Chhabra S, Hari P. Multiple myeloma and COVID‐19. Leukemia. 2020;34:1961–1963. [DOI] [PubMed] [Google Scholar]
- 11. Martínez‐López J, Mateos M‐V, Encinas C, et al. Multiple myeloma and SARS‐CoV‐2 infection: clinical characteristics and prognostic factors of inpatient mortality. Blood Cancer J. 2020;10(10):1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Blimark C, Holmberg E, Mellqvist U‐H, et al. Multiple myeloma and infections: a population‐based study on 9253 multiple myeloma patients. Haematologica. 2015;100(1):107–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Engelhardt M, Shoumariyeh K, Rösner A, et al. Clinical characteristics and outcome of multiple myeloma patients with concomitant COVID‐19 at comprehensive cancer centers in Germany. Haematologica. 2020;105(12):2872–2878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Pontecorvi G, Bellenghi M, Ortona E, Carè A. microRNAs as new possible actors in gender disparities of Covid‐19 pandemic. Acta Physiol. 2020;230:e13538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Girardin SE, Boneca IG, Viala J, et al. Nod2 is a general sensor of peptidoglycan through muramyl dipeptide (MDP) detection. J Biol Chem. 2003;278(11):8869–8872. [DOI] [PubMed] [Google Scholar]
- 16. Hisamatsu T, Suzuki M, Reinecker H‐C, Nadeau WJ, McCormick BA, Podolsky DK. CARD15/NOD2 functions as an antibacterial factor in human intestinal epithelial cells. Gastroenterology. 2003;124(4):993–1000. [DOI] [PubMed] [Google Scholar]
- 17. Negroni A, Pierdomenico M, Cucchiara S, Stronati L. NOD2 and inflammation: current insights. J Inflamm Res. 2018;11:49–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Santecchia I, Vernel‐Pauillac F, Rasid O, et al. Innate immune memory through TLR2 and NOD2 contributes to the control of leptospira interrogans infection. PLoS Pathog. 2019;15(5):e1007811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Mukherjee T, Hovingh ES, Foerster EG, Abdel‐Nour M, Philpott DJ, Girardin SE. NOD1 and NOD2 in inflammation, immunity and disease. Arch Biochem Biophys. 2019;670:69–81. [DOI] [PubMed] [Google Scholar]
- 20. Ikeda S, Negishi T, Nishimura C. Enhancement of non‐specific resistance to viral infection by muramyldipeptide and its analogs. Antiviral Res. 1985;5(4):207–215. [DOI] [PubMed] [Google Scholar]
- 21. Moorlag S, Arts R, van Crevel R, Netea M. Non‐specific effects of BCG vaccine on viral infections. Clin Microbiol Infect. 2019;25(12):1473–1478. [DOI] [PubMed] [Google Scholar]
- 22. O'Neill LAJ, Netea MG. BCG‐induced trained immunity: can it offer protection against COVID‐19? Nat Rev Immunol. 2020;20(6):335‐337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Starr SE, Visintine AM, Tomeh MO, Nahmias AJ. Effects of immunostimulants on resistance of newborn mice to herpes simplex type 2 infection. Exp Biol Med. 1976;152(1):57–60. [DOI] [PubMed] [Google Scholar]
- 24. Huang Z, Wang J, Xu X, et al. Antibody neutralization of microbiota‐derived circulating peptidoglycan dampens inflammation and ameliorates autoimmunity. Nature Microbiol. 2019;4(5):766–773. [DOI] [PubMed] [Google Scholar]
- 25. Mourits VP, Koeken VA, De Bree LCJ, et al. BCG‐induced trained immunity in healthy individuals: the effect of plasma muramyl dipeptide concentrations. J Immunol Res. 2020;2020:5812743. doi: 10.1155/2020/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Li Z‐X, Wang Y‐M, Tang F‐B, et al. NOD1 and NOD2 genetic variants in association with risk of gastric cancer and its precursors in a Chinese population. PLoS One. 2015;10(5):e0124949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ma X, Qiu Y, Sun Y, et al. NOD2 inhibits tumorigenesis and increases chemosensitivity of hepatocellular carcinoma by targeting AMPK pathway. Cell Death Dis. 2020;11(3):1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Udden SN, Peng L, Gan J‐L, et al. NOD2 suppresses colorectal tumorigenesis via downregulation of the TLR pathways. Cell Rep. 2017;19(13):2756–2770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Xu D, Zhang S, Zhang S, et al. NOD2 maybe a biomarker for the survival of kidney cancer patients. Oncotarget. 2017;8(60): 101489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zmorzyński S, Popek‐Marciniec S, Styk W, et al. The impact of the NOD2/CARD15 variant (3020insC) and PSMA6 polymorphism (‐8C > G) on the development and outcome of multiple myeloma. Biomed Res Int. 2020;2020:7629456. doi: 10.1155/2020/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ripani P, Delp J, Bode K, et al. Thiazolides promote G1 cell cycle arrest in colorectal cancer cells by targeting the mitochondrial respiratory chain. Oncogene. 2020;39(11):2345–2357. [DOI] [PubMed] [Google Scholar]
- 32. Reznik E, Wang Q, La K, Schultz N, Sander C. Mitochondrial respiratory gene expression is suppressed in many cancers. Elife. 2017;6:e21592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Stein J, Tenbrock J, Kristiansen G, Müller SC, Ellinger J. Systematic expression analysis of the mitochondrial respiratory chain protein subunits identifies COX5B as a prognostic marker in clear cell renal cell carcinoma. Int J Urol. 2019;26(9):910–916. [DOI] [PubMed] [Google Scholar]
- 34. Wang B, Ao J, Yu D, Rao T, Ruan Y, Yao X. Inhibition of mitochondrial translation effectively sensitizes renal cell carcinoma to chemotherapy. Biochem Biophys Res Comm. 2017;490(3):767–773. [DOI] [PubMed] [Google Scholar]
- 35. Othumpangat S, Noti JD, Beezhold DH. Lung epithelial cells resist influenza a infection by inducing the expression of cytochrome c oxidase VIc which is modulated by miRNA 4276. Virology. 2014;468:256–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Crul T, Testelmans D, Spruit MA, et al. Gene expression profiling in vastus lateralis muscle during an acute exacerbation of COPD. Cell Physiol Biochem. 2010;25(4–5):491–500. [DOI] [PubMed] [Google Scholar]
- 37. Deng L, Adachi T, Kitayama K, et al. Hepatitis C virus infection induces apoptosis through a bax‐triggered, mitochondrion‐mediated, caspase 3‐dependent pathway. J Virol. 2008;82(21):10375–10385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kraft RM, Nguyen ML, Yang X‐H, Thor AD, Blaho JA. Caspase 3 activation during herpes simplex virus 1 infection. Virus Res. 2006;120(1‐2):163–175. [DOI] [PubMed] [Google Scholar]
- 39. Lin Z, Li Y, Gong G, et al. Restriction of H1N1 influenza virus infection by selenium nanoparticles loaded with ribavirin via resisting caspase‐3 apoptotic pathway. Int J Nanomedicine. 2018;13:5787–5797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Mohsen HA, Al‐Gayyar MM, Mohamed EES, et al. Thymoquinone therapy remediates elevated brain tissue inflammatory mediators induced by chronic administration of food preservatives. Sci Rep. 2019;9:7026. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1
Fig S2
Table S1
Table S2
Table S3
Table S4
Table S5
Data Availability Statement
The original microarray data sets are available at The National Center for Biotechnology Information Gene Expression Omnibus (https://www.ncbi.nlm.nih.gov/geo/) with the accession numbers: GSE17498, GSE24080, GSE16558, GSE47552, GSE39754, GSE13591, and GSE6477. The Cancer Genome Atlas (TCGA) MM RNA sequencing data set (MMRF‐CoMMpass) was obtained from the UCSC Xena (https://xenabrowser.net/datapages/).