

Abstract
Patients with coronavirus disease 2019 (COVID‐19) may experience a cytokine storm with elevated interleukin‐6 (IL‐6) levels in response to severe acute respiratory syndrome‐coronavirus 2 (SARS‐CoV‐2). IL‐6 suppresses hepatic enzymes, including CYP3A; however, the effect on drug exposure and drug‐drug interaction magnitudes of the cytokine storm and resulting elevated IL‐6 levels have not been characterized in patients with COVID‐19. We used physiologically‐based pharmacokinetic (PBPK) modeling to simulate the effect of inflammation on the pharmacokinetics of CYP3A metabolized drugs. A PBPK model was developed for lopinavir boosted with ritonavir (LPV/r), using clinically observed data from people living with HIV (PLWH). The inhibition of CYPs by IL‐6 was implemented by a semimechanistic suppression model and verified against clinical data from patients with COVID‐19, treated with LPV/r. Subsequently, the verified model was used to simulate the effect of various clinically observed IL‐6 levels on the exposure of LPV/r and midazolam, a CYP3A model drug. Clinically observed LPV/r concentrations in PLWH and patients with COVID‐19 were predicted within the 95% confidence interval of the simulation results, demonstrating its predictive capability. Simulations indicated a twofold higher LPV exposure in patients with COVID‐19 compared with PLWH, whereas ritonavir exposure was predicted to be comparable. Varying IL‐6 levels under COVID‐19 had only a marginal effect on LPV/r pharmacokinetics according to our model. Simulations showed that a cytokine storm increased the exposure of the CYP3A paradigm substrate midazolam by 40%. Our simulations suggest that CYP3A metabolism is altered in patients with COVID‐19 having increased cytokine release. Caution is required when prescribing narrow therapeutic index drugs particularly in the presence of strong CYP3A inhibitors.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
☑ Coronavirus disease 2019 (COVID‐19) can cause a cytokine storm, which leads to elevated IL‐6 levels that might impact the pharmacokinetics of co‐administered drugs.
WHAT QUESTION DID THIS STUDY ADDRESS?
☑ The aim of this modeling study was to investigate the impact of IL‐6 levels in patients with COVID‐19 on drugs that are administered to treat comorbidities.
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
☑ Inflammation caused by COVID‐19 increased the exposure of drugs by a maximal twofold, which consequently can also impact the magnitude of drug‐drug interactions. Thus, caution is warranted when prescribing narrow therapeutic index drugs in patients with COVID‐19 particularly in the presence of strong CYP3A inhibitors.
HOW MIGHT THIS CHANGE CLINICAL PHARMACOLOGY AND TRANSLATIONAL SCIENCE?
☑ The physiologically‐based pharmacokinetic strategy can be effectively used to study clinical scenarios and treatment management for novel diseases, such as COVID‐19.
Patients with coronavirus disease 2019 (COVID‐19) can present a hyperinflammatory response to severe acute respiratory syndrome‐coronavirus 2 (SARS‐CoV‐2) characterized by a marked elevation of pro‐inflammatory cytokines, such as interleukin‐6 (IL‐6). This exaggerated inflammatory response, called cytokine storm, can lead to acute respiratory distress syndrome, multiple organ failure, and death.
IL‐6 has also been shown to downregulate the expression of cytochrome P450 enzymes (CYP) in vitro, particularly CYP3A, 1 due to the transcriptional suppression of CYP mRNA and the related decrease in enzyme synthesis. 2 This inhibitory effect was investigated in a clinical study including patients from orthopedic surgery. Acute inflammation showed a marked reduction in the metabolite formation of the CYP3A drug midazolam. 3 In a study including patients with rheumatoid arthritis, the exposure of the CYP3A4 substrate simvastatin was reduced by 45% by blocking the inflammation, namely after administration of the IL‐6 inhibitors tocilizumab and sarilumab. 4 , 5 Although the relation between systemic inflammation and CYP3A inhibition, and hence, increased drug exposure is underscored with these data, the direct pharmacological transition into clinical practice is challenging. Available clinical data suggest that the extent of CYP3A inhibition may depend on the degree of inflammation. Midazolam exposure was shown to be minimally altered in patients with psoriasis with C‐reactive protein (CRP) values of 2 mg/L (note: CRP is a marker of inflammation regulated by IL‐6). 6 Conversely, the trough concentration of the CYP3A4 substrate lopinavir boosted with ritonavir (LPV/r), was more than threefold higher in patients with COVID‐19 than the one observed in HIV‐infected individuals without inflammation. 7 However, despite these indicative data, the impact of cytokine storm in patients with COVID‐19 on the pharmacokinetics of CYP3A substrates has not been characterized. Furthermore, ritonavir is a strong CYP3A inhibitor which boosts the pharmacokinetics of LPV in a concentration‐dependent manner. 8 Thus, it is unclear whether high levels of inflammation can potentiate ritonavir mediated boosting of LPV and also increase the magnitude of drug‐drug interactions (DDIs) with other co‐administered drugs. A better understanding of the effect of cytokine storm on CYP3A drug exposure and on the magnitude of DDIs is pivotal for clinical practice, and hence, for supporting clinicians in prescribing to patients with COVID‐19.
Physiologically‐based pharmacokinetic (PBPK) modeling previously demonstrated its predictive power by simulating the impact of inflammation and IL‐6 levels on CYP enzymes in patients with neuromyelitis optica or neuromyelitis optica spectrum disorders in different ethnic populations. 9 A PBPK model is informed by virtual individuals, who are generated based on clinically observed organ weights, blood flows, the glomerular filtration rate, and other physiological parameters that are important to predict drug pharmacokinetics. 10 A combination of measured in vitro and clinically observed in vivo data are used to predict drug pharmacokinetics in the human body. 11 PBPK models are recommended by the health authorities to investigate clinical scenarios, which are difficult to study in clinical practice.
The aim of the present work was to investigate the impact of the cytokine storm caused by COVID‐19 on the pharmacokinetics of co‐administered drugs through our developed and verified PBPK model framework. 11
METHODS
We took three steps to understand the effect of the inflammatory cytokine release caused by COVID‐19 on the pharmacokinetics of co‐administered drugs. First, we developed and verified an LPV drug model using our previously published PBPK framework. 11 Second, clinically observed data from patients with COVID‐19 receiving LPV/r were used to verify the PBPK simulations under the consideration of IL‐6. 7 Third, the impact of different IL‐6 levels was investigated by our PBPK model.
Physiologically‐based pharmacokinetic model
A whole‐body PBPK model constructed in the mathematical programming language Matlab 2017a was used. 11 Virtual individuals aged 20 to 50 years were generated to inform the model structure, considering observed demographical (e.g., body weight), physiological (e.g., organ volume), and biological (e.g., hepatic enzyme abundance) variability. 10
Parameter of the simulated drugs
A previously published LPV model was modified and verified for our used PBPK framework. 12 LPV is metabolized by CYP3A4 (76%) and eliminated through the bile (21%) and the kidneys (2.4%), which was accounted for by a retrograde calculation. 13 The apparent permeability was estimated from the published absorption rate, 13 following the approach by Yu and Amidon. 14 The tissue distribution was predicted after Rodgers and Rowland, 11 using a scalar of two to fit the clinically observed data.
IL‐6 was implemented as a neutral compound with a molecular weight of 21,000 g/mol, a logP value of 0.01, 9 and without clearance to simulate steady‐state conditions. The suppression of CYP enzymes was implemented by a semimechanistic inhibition model based on the effect of IL‐6 on probe substrates of hepatic CYP enzymes. 1 A detailed description of the IL‐6 modeling approach can be found in Method S1. IL‐6 levels were collated from patients with COVID‐19 (Table S1 ) to simulate different steady‐state IL‐6 levels from 0.2 to 4462 pg/mL in order to investigate a realistic range of dynamically changing IL‐6 concentrations.
All other drug models used were developed and verified previously, using our PBPK framework. 15 , 16 The parameter for the simulated drugs can be found in Table S2 .
Workflow for simulations
The predictive performance of the used LPV model was verified against clinically observed data from people living with HIV (PLWH). These published concentration‐time profiles were digitized using GetData Graph Digitizer version 2.26, which demonstrated excellent accuracy. 17 Clinically observed in‐house data for LPV/r in patients with COVID‐19 were used for to verify the simulations in the presence of different IL‐6 levels. 7 To account for different IL‐6 levels in patients with COVID‐19 (Table S1 ), a sensitivity analysis was performed with different IL‐6 plasma concentration (1, 5, 10, 50, 100, 500, 1,000, 5,000, 10,000, and 50,000 pg/mL) with each simulated scenario (LPV/r, RTV, and midazolam). Successful predictions were judged by overlaying clinically observed data with the simulation results. Pharmacokinetic data had to be predicted within twofold of clinically observed data, which is considered to be best practice for modeling and simulation by the regulatory authorities. 18
After the successful verification, midazolam was taken as an example drug to evaluate the effect of a cytokine storm in patients with COVID‐19 on medications metabolized by CYP3A enzymes. An overview of the modeling analysis can be found in the supplementary material (Figure S1 ).
RESULTS
First, we developed a PBPK model for LPV/r, which was used to treat patients with COVID‐19 in the first instance. The verification was done against clinically observed data from PLWH, 8 , 19 which were within the 95% confidence interval of the PBPK model predictions (Figure 1 ). The area under the curve (AUCt) of LPV/r was well‐predicted with 74,019 ± 60,256 ng*h/mL vs. 73,833 ± 24,532 ng*h/mL (predicted/observed ratio: 1.00). All other pharmacokinetic parameters for LPV/r 400/100 mg twice daily were also predicted within 1.25‐fold of clinically observed data from PLWH (Table S3 ), apart from the volume of distribution with 39.7 ± 13.8 L vs. 50.4 ± 17.9 L (predicted/observed ratio: 0.79).
Figure 1.
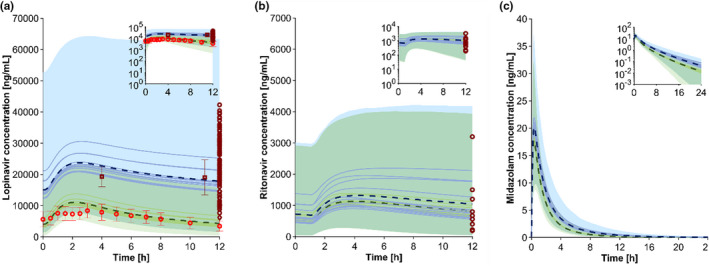
Predicted vs. observed concentration‐time profile for lopinavir/ritonavir (a), ritonavir (b), and predicted concentration‐time profile for midazolam (c) in patients with coronarivus disease 2019 (COVID‐19; blue) and people living with HIV (PLWH) a, b or healthy volunteers c (green). The red and dark red marker show clinically observed data in PLWH 8 and patients with COVID‐19 7 , 29 (mean ± SD). The solid lines, the dashed lines, and the shaded area represents the mean of each simulation with a different IL‐6 concentration (1, 5, 10, 50, 100, 500, 1,000, 5,000, 10,000, and 50,000 pg/mL; Figure S2 ), the mean, and the 95% confidence interval of all simulated scenarios to recover all possible IL‐6 concentrations in the simulations. Overlapping confidence intervals between predicted PLWH and COVID‐19 concentrations are displayed in the blue‐green shaded area. [Colour figure can be viewed at wileyonlinelibrary.com]
Second, the successfully verified PBPK model for LPV/r was used to simulate the drug concentration in patients with COVID‐19 considering the elevated cytokine concentration. 7 Patients received LPV/r twice daily, 800/200 mg on day 1 and 400/100 mg on day 2 until day 7. Pharmacokinetic parameters were taken on day 3. The clinical setting was replicated in the PBPK model simulation to verify the impact of IL‐6 on metabolizing enzymes. Figure 1a demonstrates that the PBPK model can well capture the clinically observed 12‐hour concentration of patients with COVID‐19. The AUC of LPV/r was twofold higher in patients with COVID‐19 compared with PLWH, which was predicted by our PBPK model (Table 1 ). Varying IL‐6 concentrations had only a marginal effect. Clinically observed ritonavir concentrations (Figure 1b ) were also predicted within the 95% confidence interval by our model. The difference in ritonavir exposure between patients with COVID‐19 and PLWH was negligible.
Table 1.
Median (95% CI) ratio of predicted pharmacokinetic parameters of the scenario with IL‐6 divided by the scenario without IL‐6 for lopinavir/ritonavir 400/100 mg twice daily and midazolam 5 mg as a single dose
| IL‐6 [pg/mL] | Cmax | Tmax | AUCt | CLF | VdF | t1/2 |
|---|---|---|---|---|---|---|
| Lopinavir/ritonavir | ||||||
| 1 | 1.67 (1.32; 2.24) | 1.13 (1.06; 1.22) | 2.22 (1.56; 2.76) | 0.45 (0.36; 0.64) | 0.98 (0.85; 4.22) | 2.28 (1.40; 9.06) |
| 5 | 1.72 (1.30; 2.43) | 1.15 (1.06; 3.28) | 2.28 (1.50; 3.08) | 0.44 (0.32; 0.67) | 1.02 (0.83; 4.89) | 2.37 (1.36; 11.05) |
| 10 | 1.77 (1.31; 2.38) | 1.13 (1.08; 1.27) | 2.31 (1.58; 2.98) | 0.43 (0.34; 0.63) | 1.07 (0.87; 7.50) | 2.53 (1.42; 16.98) |
| 50 | 1.78 (1.28; 2.31) | 1.14 (1.06; 1.44) | 2.32 (1.48; 2.91) | 0.43 (0.34; 0.67) | 1.04 (0.85; 8.73) | 2.49 (1.27; 20.71) |
| 100 | 1.70 (1.35; 2.30) | 1.13 (1.06; 1.25) | 2.23 (1.66; 2.96) | 0.45 (0.34; 0.60) | 0.99 (0.84; 8.89) | 2.28 (1.42; 18.24) |
| 500 | 1.75 (1.35; 2.36) | 1.13 (1.07; 1.25) | 2.34 (1.62; 2.95) | 0.43 (0.34; 0.62) | 1.01 (0.85; 3.34) | 2.40 (1.45; 9.66) |
| 1,000 | 1.73 (1.27; 2.41) | 1.13 (1.05; 2.34) | 2.21 (1.33; 3.08) | 0.45 (0.32; 0.75) | 0.99 (0.77; 10.61) | 2.30 (1.09; 29.76) |
| 5,000 | 1.75 (1.36; 2.31) | 1.13 (1.07; 1.25) | 2.29 (1.73; 2.93) | 0.44 (0.34; 0.58) | 1.01 (0.85; 11.29) | 2.41 (1.49; 27.11) |
| 10,000 | 1.76 (1.31; 2.28) | 1.14 (1.07; 1.39) | 2.26 (1.58; 2.92) | 0.44 (0.34; 0.63) | 1.04 (0.88; 7.82) | 2.47 (1.49; 16.69) |
| 50,000 | 1.74 (1.32; 2.29) | 1.13 (1.07; 2.25) | 2.35 (1.62; 2.99) | 0.43 (0.33; 0.62) | 1.00 (0.84; 3.28) | 2.38 (0.62; 35.67) |
| Cmax | Tmax | AUCt | CLF | VdF | t1/2 |
|---|---|---|---|---|---|
| Midazolam | |||||
| 1.10 (1.04; 1.18) | 1.00 (1.00; 1.33) | 1.22 (1.08; 1.43) | 0.82 (0.70; 0.93) | 0.89 (0.82; 0.96) | 1.08 (1.02; 1.21) |
| 1.10 (1.05; 1.18) | 1.00 (1.00; 1.33) | 1.21 (1.09; 1.52) | 0.82 (0.66; 0.92) | 0.89 (0.83; 0.95) | 1.07 (1.03; 1.29) |
| 1.10 (1.05; 1.19) | 1.00 (1.00; 1.33) | 1.21 (1.09; 1.52) | 0.82 (0.66; 0.92) | 0.89 (0.83; 0.95) | 1.07 (1.03; 1.29) |
| 1.10 (1.04; 1.18) | 1.00 (1.00; 1.33) | 1.21 (1.08; 1.43) | 0.83 (0.70; 0.92) | 0.89 (0.82; 0.95) | 1.07 (1.03; 1.27) |
| 1.10 (1.04; 1.18) | 1.00 (1.00; 1.33) | 1.21 (1.08; 1.44) | 0.82 (0.70; 0.92) | 0.89 (0.82; 0.95) | 1.07 (1.03; 1.27) |
| 1.10 (1.02; 1.18) | 1.00 (1.00; 1.33) | 1.19 (1.06; 1.49) | 0.84 (0.67; 0.94) | 0.90 (0.83; 0.97) | 1.07 (1.02; 1.28) |
| 1.10 (1.03; 1.19) | 1.00 (1.00; 1.33) | 1.20 (1.06; 1.50) | 0.83 (0.67; 0.94) | 0.90 (0.82; 0.96) | 1.07 (1.02; 1.30) |
| 1.13 (1.05; 1.21) | 1.00 (1.00; 1.33) | 1.27 (1.09; 1.56) | 0.79 (0.64; 0.91) | 0.87 (0.79; 0.95) | 1.09 (1.03; 1.29) |
| 1.13 (1.05; 1.24) | 1.00 (1.00; 1.33) | 1.29 (1.10; 1.66) | 0.78 (0.60; 0.91) | 0.86 (0.77; 0.95) | 1.10 (1.03; 1.31) |
| 1.16 (1.06; 1.27) | 1.00 (1.00; 1.33) | 1.33 (1.12; 1.83) | 0.75 (0.55; 0.89) | 0.85 (0.75; 0.94) | 1.12 (1.04; 1.47) |
AUCt, area under the curve over time; CI, confidence interval; CLF, clearance; Cmax, peak concentration; t1/2, elimination half‐life; VdF, volume of distribution.
In a third step, we simulated the effect of IL‐6 on another drug metabolized by CYP3A. We demonstrated an up to 40% higher exposure of the CYP3A model drug midazolam in patients with COVID‐19 compared with healthy volunteers (Figure 1c , Table 1 ). The simulation of the DDI between midazolam and the strong CYP3A inhibitor ketoconazole indicates that the magnitude of the DDI is increased by 1.6‐fold in patients with COVID‐19 compared with historical data (Figure S3 ).
DISCUSSION
Patients with COVID‐19 may experience a cytokine storm with elevated CRP and IL‐6 levels that can impact hepatic drug metabolizing enzymes. 7 In this study, we demonstrated the predictive performance of the PBPK approach to simulate the effect of elevated IL‐6 levels on LPV/r and midazolam. Our simulations indicated that a cytokine storm has a marginal effect on ritonavir exposure, which is likely explained by the fact that ritonavir inhibits its own metabolism. On the other hand, CYP3A suppression related to inflammation was apparent for LPV, as indicated by the predicted twofold increase in its exposure. Ritonavir is used at a low dose to boost LPV exposure. An inverse correlation has been reported between LPV apparent clearance and ritonavir exposure. 8 Of interest, a ritonavir plasma concentration of 360 ng/mL has been associated with a 50% maximal inhibition of LPV apparent clearance in PLWH. 20 Thus, given that LPV apparent clearance is not fully inhibited by ritonavir, it can be further inhibited by inflammation thereby leading to a higher DDI magnitude. LPV apparent clearance was predicted to be reduced by 60% in patients with COVID‐19 compared with PLWH in agreement with data from population pharmacokinetic analyses for LPV/r 21 and boosted darunavir (DRV/r) 22 in patients with COVID‐19. Despite elevated levels, these repurposed drugs failed to demonstrate a clinical benefit for the treatment of patients with COVID‐19. The population pharmacokinetic analysis of DRV/r showed an association between IL‐6 levels and DRV apparent clearance. An IL‐6 value of 18 pg/mL was found to best discriminate patients with COVID‐19 with normal versus impaired DRV apparent clearance. 22
IL‐6 was predicted to increase midazolam by 40% consistent with clinical observations reporting a 45% reduction in simvastatin exposure after administration of IL‐6 inhibitors. 4 , 5 The magnitude of DDIs between midazolam and strong CYP3A inhibitors is expected to be increased in patients with COVID‐19, as inflammation can further inhibit midazolam apparent clearance. We could demonstrate the added effect of inflammation for LPV even when combined with the strong CYP3A inhibitor ritonavir. Additionally, we simulated the DDI between midazolam and ketoconazole (Figure S3 ), which demonstrated a 1.6‐fold higher AUC‐ratio than for midazolam with ketoconazole in healthy volunteers. 23 Our assumption is further supported by real‐life clinical data reporting a more pronounced DDI between direct oral anticoagulant and LPV/r or DRV/r in patients with COVID‐19. 24 Thus, inflammation should be taken into account when prescribing for patients with COVID‐19 as both the concentrations of the victim and perpetrator drugs can be increased which can lead to a higher DDI magnitude.
Some limitations should be acknowledged. Our model predictions demonstrated only a marginal effect of varying IL‐6 levels on drug pharmacokinetics in contrast to clinical data that suggest varying CYP3A inhibition based on the degree of inflammation. One reason could be that the data to describe the suppression of CYP3A by IL‐6 were only taken from one in vitro study. 1 The in vitro data from Dickmann et al. were used in several modeling studies with successful outcome 9 , 25 , 26 ; however, re‐analysis of the original in vitro data of Dickmann et al. led also to different half‐maximal effective concentration (EC50) and maximum effect (Emax) values. 25 Uncertainty regarding in vitro data to describe disease‐drug interaction is high, limiting translational in vitro to in vivo extrapolations. Another explanation would be the turnover of the suppressed enzyme; however, this would lead to systematic mispredictions of the used PBPK model. Our model gave realistic estimates of over 30 DDIs, involving mechanism‐based inhibition and induction of CYP3A 23 , 27 and, therefore, it appears unlikely that the implemented CYP3A half‐life is not realistic. Another approach to model the impact of inflammation on drug metabolism concluded that the used model was not sensitive to varying cytokine levels, demonstrating that more research on cytokine suppression in vitro is necessary to support more realistic model predictions. 26 However, we would like to emphasize that our model predicted the variability of LPV/r and ritonavir concentrations in patients with COVID‐19 in accordance with clinically observed data (Figure 1 ). Furthermore, it should be noted that the determination of the lowest IL‐6 cutoff associated with a significant change in pharmacokinetics may be less relevant for patients with COVID‐19, as the most part presented significantly elevated IL‐6 values which are likely to inhibit metabolism (Table S1 ). The lowest IL‐6 concentration measured in our clinical study was 13 pg/mL 7 and the resulting LPV/r concentration was predicted within the 95% confidence interval of the model (Figure 1a , Table S1 ). Furthermore, given the lag time between the elevation of IL‐6 and the downregulation of CYPs, 3 IL‐6 values may not accurately reflect CYP3A inhibition in the early phase of the inflammatory response. Taken together, the presented inflammation model might not be robust to predict the impact of very low IL‐6 levels but is verified for its use in patients with COVID‐19 presenting a high level of inflammation in the case of a severe cytokine storm.
Another limitation is that only the effect of IL‐6 was considered, but other inflammatory cytokines, such as interferons, can also impact CYP enzymes; however, IL‐6 has the most suppressive effect on CYP enzymes in vitro. 1 Furthermore, IL‐6 has an impact on other CYP enzymes 9 and transporters, 28 which were not evaluated in this study due to a lack of clinically observed data in patients with COVID‐19. In addition, other physiological changes in patients with COVID‐19 that might impact drug pharmacokinetics were not considered in the model.
In conclusion, PBPK strategy can be effectively used to study clinical scenarios and treatment management for novel diseases, such as COVID‐19. Our simulations suggest that CYP3A metabolism is altered in patients with COVID‐19 having increased cytokine release. Caution is required when prescribing narrow therapeutic index drugs (e.g., immunosuppressants, anticoagulants, and anti‐arrhythmics) particularly in the presence of strong CYP3A inhibitors.
FUNDING
C.M. was supported by the Adolf and Mary Mil Foundation. F.S. was supported by a grant from the Swiss National Science Foundation (grant number: 324730_188504).
CONFLICT OF INTEREST
The authors declared no competing interests for this work.
AUTHOR CONTRIBUTIONS
F.S. and C.M. wrote the manuscript. F.S. and C.M. designed the research. F.S. performed the research. F.S. analyzed the data. M.B. and PS. contributed new reagents/analytical tools.
Supporting information
Supplementary Material
References
- 1. Dickmann, L.J. , Patel, S.K. , Rock, D.A. , Wienkers, L.C. & Slatter, J.G. Effects of interleukin‐6 (IL‐6) and an anti‐IL‐6 monoclonal antibody on drug‐metabolizing enzymes in human hepatocyte culture. Drug Metab. Dispos. 39, 1415–1422 (2011). [DOI] [PubMed] [Google Scholar]
- 2. Morgan, E. Impact of infectious and inflammatory disease on cytochrome P450–mediated drug metabolism and pharmacokinetics. Clin. Pharmacol. Ther. 85, 434–438 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lenoir, C. et al. Impact of acute inflammation on cytochromes P450 activity assessed by the Geneva cocktail. Clin. Pharmacol. Ther. 109, 1668–1676 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Schmitt, C. , Kuhn, B. , Zhang, X. , Kivitz, A. & Grange, S. Disease–drug–drug interaction involving tocilizumab and simvastatin in patients with rheumatoid arthritis. Clin. Pharmacol. Ther. 89, 735–740 (2011). [DOI] [PubMed] [Google Scholar]
- 5. Lee, E.B. et al. Disease–drug interaction of sarilumab and simvastatin in patients with rheumatoid arthritis. Clin. Pharmacokinet. 56, 607–615 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sathe, A.G. , Othman, A.A. & Mohamed, M.E.F. Therapeutic protein drug interaction potential in subjects with psoriasis: an assessment based on population pharmacokinetic analyses of sensitive cytochrome P450 probe substrates. J. Clin. Pharmacol. 61, 307–318 (2021). [DOI] [PubMed] [Google Scholar]
- 7. Marzolini, C. et al. Effect of systemic inflammatory response to SARS‐CoV‐2 on lopinavir and hydroxychloroquine plasma concentrations. Antimicrob. Agents Chemother. 64, e01177–e01220 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Crommentuyn, K.M. et al. The plasma and intracellular steady‐state pharmacokinetics of lopinavir/ritonavir in HIV‐1‐infected patients. Antivir. Ther. 9, 779–786 (2004). [PubMed] [Google Scholar]
- 9. Machavaram, K.K. et al. Simulating the impact of elevated levels of interleukin‐6 on the pharmacokinetics of various CYP450 substrates in patients with neuromyelitis optica or neuromyelitis optica spectrum disorders in different ethnic populations. AAPS J. 21, 42 (2019). [DOI] [PubMed] [Google Scholar]
- 10. Stader, F. , Siccardi, M. , Battegay, M. , Kinvig, H. , Penny, M.A. & Marzolini, C. Repository describing an aging population to inform physiologically based pharmacokinetic models considering anatomical, physiological, and biological age‐dependent changes. Clin. Pharmacokinet. 58, 483–501 (2019). [DOI] [PubMed] [Google Scholar]
- 11. Stader, F. , Penny, M.A. , Siccardi, M. & Marzolini, C. A comprehensive framework for physiologically based pharmacokinetic modelling in Matlab®. CPT Pharmacometrics Syst. Pharmacol. 8, 444–459 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Saeheng, T. , Na‐Bangchang, K. , Siccardi, M. , Rajoli, R.K. & Karbwang, J. Physiologically‐based pharmacokinetic modeling for optimal dosage prediction of quinine coadministered with ritonavir‐boosted lopinavir. Clin. Pharmacol. Ther. 107, 1209–1220 (2020). [DOI] [PubMed] [Google Scholar]
- 13. Kumar, G.N. et al. Metabolism and disposition of the HIV‐1 protease inhibitor lopinavir (ABT‐378) given in combination with ritonavir in rats, dogs, and humans. Pharm. Res. 21, 1622–1630 (2004). [DOI] [PubMed] [Google Scholar]
- 14. Yu, L.X. & Amidon, G.L. A compartmental absorption and transit model for estimating oral drug absorption. Int. J. Pharm. 186, 119–125 (1999). [DOI] [PubMed] [Google Scholar]
- 15. Stader, F. , Kinvig, H. , Penny, M.A. , Battegay, M. , Siccardi, M. & Marzolini, C. Physiologically based pharmacokinetic modelling to identify pharmacokinetic parameters driving drug exposure changes in the elderly. Clin. Pharmacokinet. 59, 383–401 (2020). [DOI] [PubMed] [Google Scholar]
- 16. Stader, F. et al. Effect of ageing in antiretroviral drug pharmacokinetics using clinical data combined with modelling and simulation. Br. J. Clin. Pharmacol. 87, 458–470 (2020). [DOI] [PubMed] [Google Scholar]
- 17. Wojtyniak, J.G. , Britz, H. , Selzer, D. , Schwab, M. & Lehr, T. Data digitizing: accurate and precise data extraction for quantitative systems pharmacology and physiologically‐based pharmacokinetic modeling. CPT Pharmacometrics Syst. Pharmacol. 9, 322–331 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Jones, H. et al. Physiologically based pharmacokinetic modeling in drug discovery and development: a pharmaceutical industry perspective. Clin. Pharmacol. Ther. 97, 247–262 (2015). [DOI] [PubMed] [Google Scholar]
- 19. Klein, C.E. et al. The tablet formulation of lopinavir/ritonavir provides similar bioavailability to the soft‐gelatin capsule formulation with less pharmacokinetic variability and diminished food effect. J. Acquir. Immune Defic. Syndr. 44, 401–410 (2007). [DOI] [PubMed] [Google Scholar]
- 20. Moltó, J. et al. Simultaneous population pharmacokinetic model for lopinavir and ritonavir in HIV‐infected adults. Clin. Pharmacokinet. 47, 681–692 (2008). [DOI] [PubMed] [Google Scholar]
- 21. Alvarez, J.C. et al. Population pharmacokinetics of lopinavir/ritonavir in Covid‐19 patients. Eur. J. Clin. Pharmacol. 77, 389–397 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Cojutti, P.G. et al. Comparative population pharmacokinetics of darunavir in SARS‐CoV‐2 patients vs. HIV patients: the role of interleukin‐6. Clin. Pharmacokinet. 59, 1251–1260 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Stader, F. et al. Clinical data combined with modelling and simulation indicate unchanged drug drug interaction magnitudes in the elderly. Clin. Pharmacol. Ther. 109, 471–484 (2021). [DOI] [PubMed] [Google Scholar]
- 24. Testa, S. et al. Direct oral anticoagulant plasma levels’ striking increase in severe COVID‐19 respiratory syndrome patients treated with antiviral agents: the Cremona experience. J. Thromb. Haemost. 18, 1320–1323 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Jiang, X. , Zhuang, Y. , Xu, Z. , Wang, W. & Zhou, H. Development of a physiologically based pharmacokinetic model to predict disease‐mediated therapeutic protein–drug interactions: modulation of multiple cytochrome P450 enzymes by interleukin‐6. AAPS J. 18, 767–776 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Xu, Y. , Hijazi, Y. , Wolf, A. , Wu, B. , Sun, Y.N. & Zhu, M. Physiologically based pharmacokinetic model to assess the influence of blinatumomab‐mediated cytokine elevations on cytochrome P450 enzyme activity. CPT Pharmacometrics Syst. Pharmacol. 4, 507–515 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Stader, F. , Battegay, M. & Marzolini, C. Physiologically‐based pharmacokinetic modeling to support the clinical management of drug–drug interactions with bictegravir. Clin. Pharmacol. Ther. 110, 1231–1239 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Febvre‐James, M. , Bruyère, A. , Le Vée, M. & Fardel, O. The JAK1/2 inhibitor ruxolitinib reverses interleukin‐6‐mediated suppression of drug‐detoxifying proteins in cultured human hepatocytes. Drug Metab. Dispos. 46, 131–140 (2018). [DOI] [PubMed] [Google Scholar]
- 29. Schoergenhofer, C. , Jilma, B. , Stimpfl, T. , Karolyi, M. & Zoufaly, A. Pharmacokinetics of lopinavir and ritonavir in patients hospitalized with coronavirus disease 2019 (COVID‐19). Ann. Intern. Med. 173, 670–672 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material