

Abstract
There is an unmet need for safe and effective severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2) vaccines that are stable and can be cost‐effectively produced at large scale. Here, a biopolymer particle (BP) vaccine technology that can be quickly adapted to new and emerging variants of SARS‐CoV‐2 is used. Coronavirus antigen‐coated BPs are described as vaccines against SARS‐CoV‐2. The spike protein subunit S1 or epitopes from S and M proteins (SM) plus/minus the nucleocapsid protein (N) are selected as antigens to either coat BPs during assembly inside engineered Escherichia coli or BPs are engineered to specifically ligate glycosylated spike protein (S1‐ICC) produced by using baculovirus expression in insect cell culture (ICC). BP vaccines are safe and immunogenic in mice. BP vaccines, SM‐BP‐N and S1‐ICC‐BP induced protective immunity in the hamster SARS‐CoV‐2 infection model as shown by reduction of virus titers up to viral clearance in lungs post infection. The BP platform offers the possibility for rapid design and cost‐effective large‐scale manufacture of ambient temperature stable and globally available vaccines to combat the coronavirus disease 2019 (COVID‐19) pandemic.
Keywords: biopolyesters, COVID‐19, SARS‐CoV‐2, self‐assembly, vaccines
Ambient temperature stable, scalable coronavirus disease 2019 polymer particle vaccines are developed by engineering bacterial cell factories to assemble biopolymer particles either directly coated with severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2) antigens or to capture soluble spike protein S1 subunit produced by an insect culture. Polymer particle vaccines induce protective immunity in a hamster SARS‐CoV‐2 infection model reducing virus titers up to viral clearance in lungs post infection.
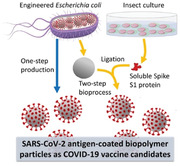
1. Introduction
There is an urgent unmet need for a globally available safe vaccine to prevent human severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2) infections to halt the worldwide pandemic that has already caused >247 million infections and >5 million deaths.[ 1 ] Despite the fact that there are >180 vaccine development projects (42 in clinical trials) internationally and more than ten emergency approved uses of vaccines there is a demand for vaccines that enable rapid global distribution and induce immunity that blocks transmission and is long lasting.[ 2 ] A further key issue is adaptability against emerging viral variants. Vaccine design is currently hampered due to our limited understanding of the complex immune response required to prevent infection and induce long‐lasting immunity.[ 3 , 4 , 5 , 6 ] Further concerns are short shelf‐life and cold‐chain requirements of vaccine candidates as well as poor manufacturability at scale. These challenges are further exacerbated by the risk of vaccine candidates inducing antibody‐dependent enhancement (ADE) of infection and/or immunopathology due to induction of a “cytokine storm” and associated inflammation,[ 7 , 8 , 9 ] and vaccine‐induced thrombosis with thrombocytopenia (VITT).[ 10 , 11 ] The cytokine storm is the major cause for severe cases of coronavirus disease 2019 (COVID‐19) and increased mortality. Hence the successful vaccine needs to be precision‐engineered to induce desired immune responses and long‐lasting immunity while avoiding ADE/immunopathology/VITT in combination with robust manufacturability at large scale.
Current emergency use approved vaccines and vaccine candidates in clinical evaluation include viral vectors (replicating, nonreplicating), virus‐like particles (VLP), protein subunits, DNA or RNA vaccines and inactivated virus vaccines mostly considering the SARS‐CoV‐2 spike glycoprotein (S) as major vaccine candidate antigen. Targeted immune responses are neutralizing antibodies that bind to the receptor binding domain (RBD) located within the S1 subunit of S and block the virus attachment to the human angiotensin converting enzyme 2 (ACE2) and hence impede virus infection. However, even in convalescent patients (CPs) these antibodies do not last longer than a few months, i.e., immunity is not long‐lasting.[ 12 ] This suggest that successful SARS‐CoV‐2 vaccines require greater immunogenicity and durability than natural infection for long‐lasting immunity.
In contrast to the mRNA and adenovirus vector vaccine types currently only emergency use approved as COVID‐19 vaccines, protein subunit vaccines against various infectious diseases have a long history of approved use demonstrating their safety and efficacy.[ 13 , 14 , 15 ] RBD and S1 have recently been demonstrated to induce neutralizing antibodies and to comprise T cell epitopes proposed to contribute to cell‐mediated immunity.[ 16 , 17 , 18 ] SARS‐CoV‐2‐specific CD8+ and CD4+ T cells were identified in ≈70% and 100% of COVID‐19 CPs, respectively. The SARS‐CoV‐2 nucleocapsid protein (N) contributed to 11%‐27% of the CD4+ T cell response in COVID‐19 CPs while CD4+ T cell responses to spike and M (membrane protein) were significant and correlated with the level of the anti‐SARS‐CoV‐2 IgG and IgA titers.[ 19 ] CD8+ T cells mostly recognized S and M. CPs also showed high antibody titers against N.[ 20 ] Hence, we decided to study immunogenicity of S1, RBD, epitopes of S1 and M in the presence or absence of N.
To improve immunogenicity, we attached these antigens to biopolymer particles (BPs) exhibiting a size of ≈200–500 nm and the core of which is composed of biocompatible polyhydroxybutyrate (PHB) surrounded by covalently linked PHB synthase used as anchoring domain for antigens.[ 15 , 21 , 22 , 23 ] BP display of protein antigens has been shown to further enhance immunogenicity by facilitating antigen uptake and processing by antigen‐presenting cells. Immune responses were specific to the displayed antigens.[ 23 , 24 , 25 ] Moreover, we recently showed that bacterial or viral antigens displayed on BPs were highly immunogenic inducing both strong cell‐mediated and long‐lasting humoral immune responses which protected mice from infection by the respective pathogen.[ 26 , 27 , 28 ] Hence, the BP platform was conceived to be suitable for SARS‐CoV‐2 vaccine development.[ 15 , 23 ] SARS‐CoV‐2 antigen coated BPs were produced either in one‐step using polymer synthesis and self‐assembly inside engineered bacteria or by using engineered BPs that display the SpyCatcher domain in order to capture Spy‐tagged S1 produced from insect culture using the baculovirus expression system. We already had shown that SpyCatcher‐coated BPs can specifically capture and ligate a Spy‐tagged target protein even when present in complex mixtures.[ 29 , 30 , 31 ]
The SARS‐CoV‐2 antigen‐coated BPs can be produced at high yields (50 g L−1 of synthetic culture media) in an endotoxin‐free mutant of E. coli, CleanColi,[ 32 ] and purified using scalable industrial bioprocess steps such as mechanical cell disruption followed by tangential flow filtration (TFF).[ 33 ] The entire process is safe and free of any animal or mammalian product derived components. Furthermore, the BP platform has intrinsic high thermal stability of up to 95 °C[ 34 , 35 ] while remaining stable after five freeze–thaw cycles.[ 29 ] Each BP is densely coated with PHB synthase genetically fused to one or more antigens or SpyCatcher for ligation of Spy‐tagged antigen. PHB synthase is linked via a thioester bond to the PHB while Spy‐tagged antigens are linked to the displayed SpyCatcher domain via a covalent isopeptide bond (Figure 1 ). Here, we show that SARS‐CoV‐2 antigen‐coated BPs are highly immunogenic in mice and hamsters inducing cell‐mediated and humoral responses contributing to protective immunity in the hamster SARS‐CoV‐2 infection model. The results of our study demonstrate the potential of the SARS‐CoV‐2 antigen‐coated BPs as a safe and effective vaccine for COVID‐19, which is very stable and can be mass‐produced as affordable vaccine in a pandemic scenario.
Figure 1.
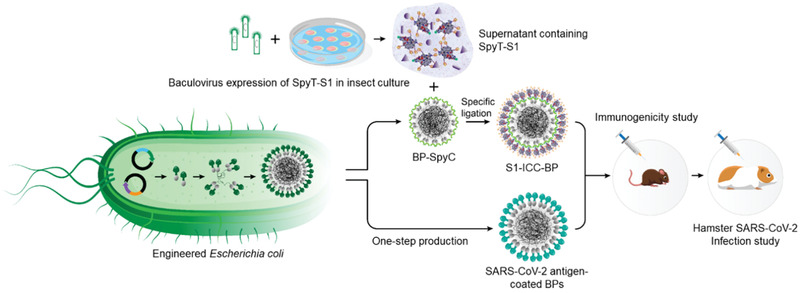
Schematic representation of the manufacture of SARS‐CoV‐2 antigen‐coated BPs. An endotoxin free production strain of Escherichia coli, ClearColi BL21(DE3), was bioengineered to produce SARS‐CoV‐2 antigen‐coated BPs in one step. BPs coated with glycosylated S1 (S1‐ICC‐BP) were generated in two steps by irreversible SpyCatcher/SpyTag (SpyC/SpyT) ligation between BP‐SpyC, produced by ClearColi BL21(DE3), and glycosylated spike protein (S1‐ICC), produced using baculovirus expression in insect cell culture (ICC). Immunogenicity of various SARS‐CoV‐2 antigen‐coated BPs was analyzed in a mouse model and the two best performing vaccine candidates were evaluated in the hamster SARS‐CoV‐2 challenge model.
2. Results
2.1. SARS‐CoV‐2 Antigens Can Be Efficiently Displayed on BPs
To directly assemble antigen‐coated BPs in E. coli (Figure 1), S1 or RBD or SM (experimentally verified and predicted B cell epitopes from S1 and M[ 36 , 37 ] are shown in Figure 2a and Table S1, Supporting Information) or N were translationally fused either to the N or C terminus of the PHB synthase (BP anchor protein) as single fusions. In dual fusions, the N protein was fused to the opposite terminus (Figure 2a). Respective hybrid genes were expressed in E coli harboring plasmid pMCS69[ 38 ] encoding the two enzymes β‐ketothiolase and acetoacetyl‐CoA reductase that convert the central metabolite acetyl‐CoA to (R)‐3‐hydroxybutyryl‐CoA, which is polymerized to PHB by the PHB synthase domain of the fusion protein resulting in antigen‐coated BPs (Figure 2b). All genetic constructs mediated formation of BPs at high yield of >8% of biomass in recombinant E. coli (Table S2, Supporting Information). Production strains could be cultured in synthetic mineral media with glucose as carbon source. Cells were harvested, mechanically disrupted to release BPs which were then purified by TFF steps.[ 33 ]
Figure 2.
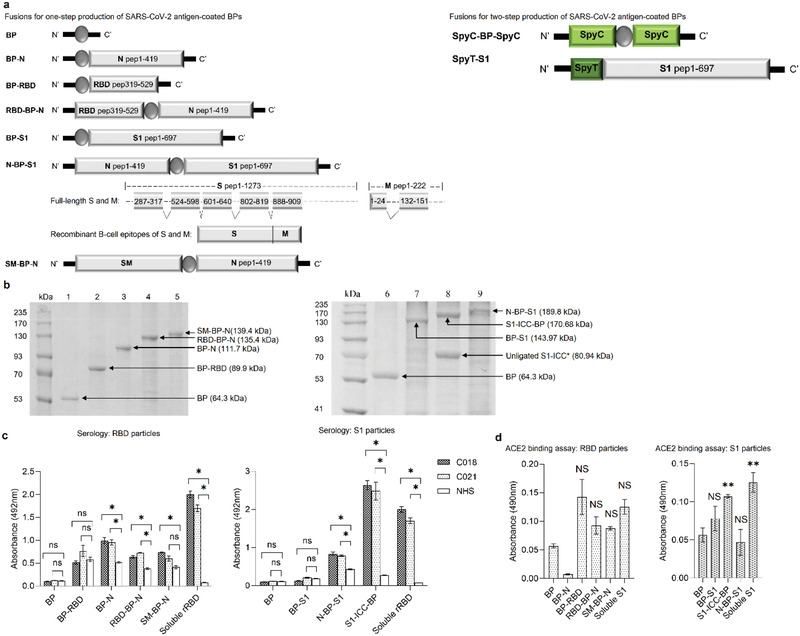
Bioengineering, production, and functional analysis of SARS‐CoV‐2 antigen‐coated BPs. a) Schematic representation of recombinant protein fusions mediated the production of SARS‐CoV‐2 antigen‐coated BPs. b) Protein profile of purified SARS‐CoV‐2 antigen‐coated BPs. kDa, molecular weight marker (GangNam‐Stain prestained protein ladder; iNtRon); lanes 1 and 6, BPs (64.3 kDa); lane 2, BP‐RBD (89.9 kDa); lane 3, BP‐N (111.7 kDa); lane 4, RBD‐BP‐N (135.4 kDa); lane 5, SM‐BP‐N (139.4 kDa); lane 7, BP‐S1 (143.97 kDa); lane 8, S1‐ICC‐BP (170.68 kDa); lane 9, N‐BP‐S1 (189.8 kDa). * The protein band with molecular weight of 80.94 kDa in lane 8 is bound, but unligated S1‐ICC. See Figure S1 in the Supporting Information for full‐length gel image. c) Evaluation of SARS‐CoV‐2 antigen‐coated BPs performance by diagnosing convalescent plasma (CP) and noninfected healthy human serum samples (NHS) using ELISA. All assays were performed in triplicates. Each data point of measurement stands for the mean ± the standard error of the mean. d) Functionality assessment of SARS‐CoV‐2 antigen‐coated BPs using an ACE2 binding assay. The assay was performed in triplicates. Each data point of measurement represents the mean ± the standard error of the mean. *, significantly higher than plain BPs (p < 0.05). *, significantly higher than NHS (p < 0.05); ns, no significant difference with NHS (p > 0.05); **, significantly higher than BP (p <0.05); NS, no significant difference with BP (p > 0.05). Statistical significance is determined by T‐test for comparison of two groups or by one‐way analysis of variance (ANOVA) with pairwise comparison of multigrouped data sets achieved using Tukey's or Dunnet's post hoc test (Prism).
To create BPs coated with glycosylated S1 (S1‐ICC (insect cell culture)) from insect culture (Figure 1), we generated a genetic fusion of the Spy‐Tag (AHIVMVDAYKPTK) with SARS‐CoV‐2 spike protein subunit S1 (Figure 2a), which was cloned into the baculovirus vector and used to infect High Five insect cells (Table S3, Supporting Information). Cells were cultured and S1‐ICC was secreted into the supernatant as shown by sodium dodecyl sulphate–polyacrylamide gel electrophoresis (SDS‐PAGE) and immunoblotting (Figure S2, Supporting Information). The supernatant was concentrated tenfold by TFF and S1‐ICC was then specifically captured and separated using the previously developed BPs densely displaying the SpyCatcher domain.[ 31 ] These BPs were coated with the dual fusion protein, SpyCatcher‐PHB synthase‐SpyCatcher, to enhance SpyCatcher density and thus Spy‐tagged antigen binding capacity. These BP‐SpyC served as an efficient capture resin, facilitating removal of impurities via sequential wash steps using diafiltration and TFF[ 33 ] while the resin with bound Spy‐Tagged S1‐ICC itself was conceived as vaccine (Figure 2b). Antigens coating BPs were confirmed as identified by SDS‐PAGE combined with tryptic peptide fingerprinting analysis using matrix‐assisted laser desorption/ionization (MALDI)‐quadrupole time of flight mass spectrometry (Q‐TOF‐MS) (Figure 2b and Table S4, Supporting Information). Spy‐tagged S1‐ICC ligated to 58% of displayed SpyCatcher domains (Figure 2b and Figure S3, Supporting Information). Densitometry was used to determine the amount of all BP associated antigens (Figure S4, Supporting Information).
2.2. SARS‐CoV‐2 Antigen Displayed on BPs Are Reactive to Sera from Convalescent Patients and Bind Human ACE2
To confirm the antigenicity of the various antigens displayed on BPs, the BPs were assessed in enzyme‐linked immunosorbent assay (ELISA) using sera from two CPs (C018, male, 63 years, mild disease, 32 d post recovery; C021, female, 27 years, mild disease, 22 d postrecovery) (Table S5, Supporting Information) and healthy donor (normal healthy serum (NHS). Purified recombinant immunogenic peptides from RBD protein (rRBD) and plain BPs without any antigen served as controls. SARS‐CoV‐2 antigen‐coated BPs were used to coat ELISA plates and IgG and IgM antibody binding was quantified (Figure 2c and Figure S6, Supporting Information). There was a significant difference between plain BPs and antigen‐coated BPs (except BP‐RBD and BP‐S1) as well as between CP sera and NHS suggesting that antigenicity of SARS‐CoV‐2 antigens, when displayed by BPs using both manufacturing routes, was retained.
To assess retention of functional conformation of S1 and RBD when displayed on BPs, the binding to ACE2 was studied. An ACE2‐Fc fusion protein was used to probe the ACE2 binding ability of S1 or S1 derived protein attached BPs. ELISA plates were coated with BPs and binding of ACE2‐Fc was measured using a protein A‐horseradish peroxidase (HRP) conjugate. S1‐ICC‐BP showed significant ACE2 binding when compared to plain BPs suggesting that functional conformation of these antigens was retained during assembly and purification of BPs (Figure 2d).
2.3. SARS‐CoV‐2 Antigen‐Coated BPs Are Immunogenic and Induce ACE2‐Blocking Antibody Responses in Mouse Models
We initially tested safety and immunogenicity of the BP vaccine candidates in mouse models. C57BL/6 mice (n = 10) were immunized intramuscularly (IM) with 20 µg of the various purified SARS‐CoV‐2 antigens displayed on BPs or BPs alone or aluminum hydroxide (alum) alone (Figure 3a). All vaccine formulations contained alum as adjuvant except for N‐BP‐S1 where we included a respective group without alum to assess how alum impacts immunogenicity. Alum is an adjuvant approved by regulators worldwide with a long history of use in vaccine formulation and is produced at large industrial scale avoiding unnecessary bottlenecks in vaccine manufacture in a pandemic situation. Alum boosts immunity toward antibody responses, which complements the BPs property to induce cell‐mediated immune responses.[ 27 , 39 , 40 ] Mice were then boosted twice with the same dose of immunogen in two weeks intervals and sera were collected one week after the first boost and 2 weeks after the last boost (Figure 3a). Sera were tested for antibodies against pure S1 and N proteins. Besides total IgG, IgG1 and IgG2c levels were determined (Figure 3b,c). The latter isotype indicates a Th1 type immune response, while a Th2 type immune response is associated with IgG1 antibodies. Th1 cells stimulate cell‐mediated immune responses desirable for prevention of viral infections as infected cells are targeted and killed interfering with the replication cycle of the virus. Th2 cells stimulate induction of antibodies that impede receptor binding and tag viruses for enhanced phagocytosis by macrophages.
Figure 3.
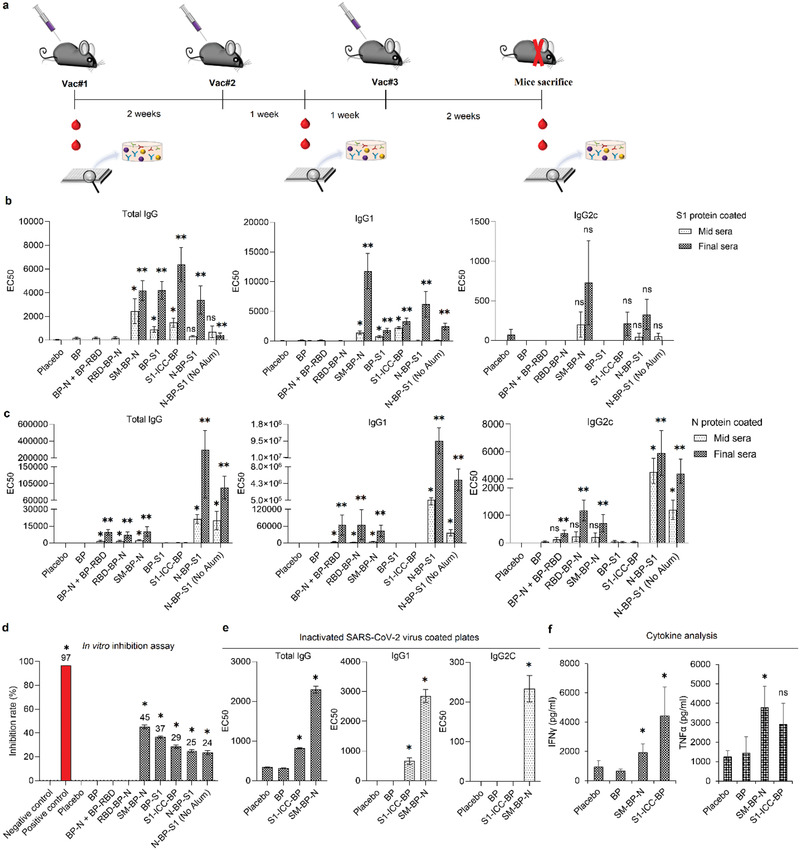
Immunogenicity study of SARS‐CoV‐2 antigen‐coated BPs in mice. a) Schematic representation of immunization schedule. There were ten female C57BL/6 mice per group. Mice were immunized three times intramuscularly at 2 weeks intervals in the thigh muscle of hind limb with freshly formulated vaccines containing 20 µg of antigen/dose, emulsified in alum (25 µL per dose). Blood collection was performed at day 0, 21, and 42, respectively, for immunogenicity analysis. b) Antibody responses to soluble S1 protein presented in EC50. Each data point stands for the results from ten mice ± the standard error of mean. c) Antibody responses to soluble N protein presented in EC50. Each data point represents the results from ten mice ± the standard error of mean. d) In vitro SARS‐CoV‐2 inhibition assay. The inhibition rate stands for the inhibition rate of postimmune sera subtracting the inhibition rate of preimmune sera. The experiment was performed in triplicates. Each data point of measurement represents the mean ± the standard error of the mean. e) Antibody response to UV‐inactivated SARS‐CoV‐2 virus presented in EC50. The experiment was done using pooled mice serum samples in triplicates. Each data point of assay represents the mean ± the standard error of the mean. f) Cytokine responses in mice vaccinated with SM‐BP‐N or S1‐ICC‐BP particles. Each data point of measurement represents the results from ten mice ± the standard error of mean. *, significantly higher than BP (p < 0.05); **, EC50 value of final serum samples is significantly higher than BP (p < 0.05); ns, no significant difference with BP (p > 0.05). Statistical significance is determined by T‐test for comparison of two groups or by one‐way ANOVA with pairwise comparison of multigrouped data sets achieved using Tukey's or Dunnet's post hoc test (Prism).
The alum‐only (placebo) and BP‐only groups showed low background levels of antibody against spike glycoprotein and N after the second boost as detected by ELISA (EC50 values are shown) (Figure 3b,c). Mice immunized with BPs that display S1 or SM showed high levels of antibody to S1 (EC50: 4178.67, SM‐BP‐N; 4201.86, BP‐S1; 6371.82, S1‐ICC‐BP; 3397.17, N‐BP‐S1), which strongly increased after the second boost (Figure 3b,c). Except for BP‐S1 and N‐BP‐S1, all S1 displaying BPs induced anti‐S1 IgG2c responses (Figure 3b). RBD displaying BPs did not induce significant levels of anti‐S1 antibody response. Only BPs coated with N induced strong anti‐N antibody responses which increased after the second boost and included high IgG2c levels (EC50: 1174.69, RBD‐BP‐B; 718.698, SM‐BP‐N; 5882.13, N‐BP‐S1; 4391.81, N‐BP‐S1 without alum, p < 0.05) compared to the BP‐only group (Figure 3c). To assess whether induced antibodies can block binding of SARS‐CoV‐2 to ACE2 we used the SARS‐CoV‐2 surrogate virus neutralization assay, where RBD conjugated to HRP was preincubated with the sera of interest and sera with defined neutralizing antibody titers as positive control. The antibody mediated interference of RBD binding to ACE2 was tested using ACE2 coated ELISA plates. Mice immunized with BPs that display S1, RBD or SM produced antibodies that inhibited RBD‐HRP binding to ACE2 suggesting neutralizing activity (Figure 3d).
We selected SM‐BP‐N and S1‐ICC‐BP that showed induction of specific and strong anti‐SARS‐CoV‐2 antigen antibody responses for further in‐depth analysis. We further studied whether induced antibodies could specifically bind antigens used to coat BPs by immunoblotting against whole cell lysates of production strains and purified BPs containing the antigens as well as whole UV inactivated SARS‐CoV‐2, respectively. Both BPs only induced antibodies against the antigens that were used to coat them. BPs induced the respective specific immune responses such as against the S, M, and N protein (Figure S7, Supporting Information). Further ELISA data with M protein and UV inactivated SARS‐CoV‐2 confirmed that both BPs induced high levels of antibodies directly binding to the M protein (Figure S8, Supporting Information) and the whole virus (Figure 3e).
To further evaluate the mode of immune response we analyzed sera for cytokine production. Both vaccines induced high levels of cytokines interferon‐γ and tumor necrosis fFactor alpha when compared to BP only and alum only controls (Figure 3f).
The biophysical properties of the selected SM‐BP‐N and S1‐ICC‐BP were determined (Figure 4 ). Transmission electron microscopy (Figure 4a) indicated a spherical morphology while dynamic laser scattering revealed a size distribution of 500–1200 nm with a polydispersity index of ≈0.2 (Figure 4b). The size gap observed when comparing the transmission electron microscopy (TEM) image with the dynamic light scattering (DLS) data suggests that BPs in suspension might exhibit some level of aggregation. The zeta potential revealed a negative surface charge in alignment with the isoelectric point of the fusion proteins coating the BPs (Figure 4c). The stability of the two vaccine candidates was tested by freeze‐drying and rehydration as well as by incubating BP suspension at different temperatures (4, 25, 37, and 50 °C) for 48 h plus at 4 °C for 6 months. We used the best performing mouse sera from each vaccine groups to show in ELISA that antigenicity was retained after freeze‐drying and incubation at elevated temperatures, while the antigens attached to BPs were retained after 6 months storage at 4 °C (Figure 4d–f). We also assessed the impact on the antigen stability, BP size distribution and zeta potential after incubation at the various temperatures and times showing retention of the original properties (Figures S11–S14, Supporting Information).
Figure 4.
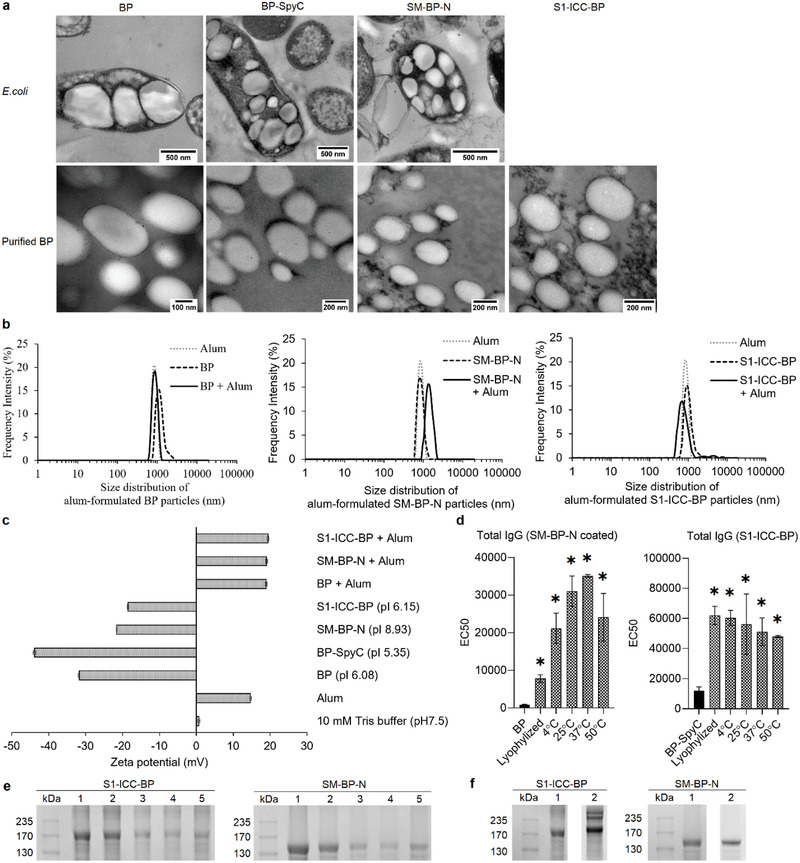
Characterization of SM‐BP‐N and S1‐ICC‐BP. a) TEM images of an endotoxin free production host ClearColi BL21(DE3) producing SARS‐CoV‐2 antigen‐coated BPs and of the purified BPs. (Scale bars: E. coli cells, 500 nm; purified particles, 100 or 200 nm). b) Size distribution of SARS‐CoV‐2 antigen‐coated BPs before and after formulation with alum. All vaccine particle sizes were measured three times consecutively using Litesizer500 (Anton Paar, Australia). Each data point of measurement represents the mean ± the standard error of the mean. c) ζ‐potential of various SARS‐CoV‐2 antigen‐coated BPs before and after formulation with alum. The ζ‐potential of all particulate vaccine samples was analyzed three times by Litesizer500 (Anton Paar, Australia). Each data point of measurement stands for the mean ± the standard error of the mean. pI of recombinant S1‐ICC‐BP fusion, 6.15; pI of recombinant SM‐BP‐N fusion, 8.93; pI of recombinant BP‐SpyC, 5.35; pI of BP protein, 6.08. d) Antigenicity analysis of SM‐BP‐N and S1‐ICC‐BP after treatment with various temperatures for 48 h. The assay was analyzed by ELISA using serum samples from mice immunized with SM‐BP‐N and S1‐ICC‐BP. The experiment was performed in triplicates. Each data point of measurement represents the mean ± the standard error of the mean. e) Protein profile of S1‐ICC‐BP and SM‐BP‐N after various temperature treatment. Lane 1, particle treated with freeze dryer; lane 2, particle treated at 4 °C for 48 h; lane 3, particle treated at 25 °C for 48 h; lane 4, particle treated at 37 °C for 48 h; lane 5, particle treated at 50 °C for 48 h. See Figure S9 in the Supporting Information for full‐length SDS‐PAGE gel image. f) Protein profile of S1‐ICC‐BP and SM‐BP‐N after stored at 4 °C for 0 and 6 months. Lane 1, particle stored at 4 °C in 10 × 10−3 m Tris buffer for 6 months; lane 2, particles stored at 4 °C in 10 × 10−3 m Tris buffer for 0 months. See Figure S10 in the Supporting Information for full protein profile gel image. *, significantly higher than BP (p < 0.05). Statistical significance is determined by T‐test for comparison of two groups or by one‐way ANOVA with pairwise comparison of multigrouped data sets achieved using Tukey's or Dunnet's post hoc test (Prism).
2.4. SM‐BP‐N and S1‐ICC‐BP Induce Protective Immunity against SARS‐CoV‐2 Infection in Hamsters
The golden Syrian hamster SARS‐CoV‐2 infection model was used to evaluate whether induced immune responses can be correlated with protection against SARS‐CoV‐2 infection. Golden Syrian hamsters had been shown to be susceptible to SARS‐CoV‐2 infection exhibiting efficient virus replication in the lung similar to level of replication in the nasal turbinates and associated with severe pathological lesions.[ 41 , 42 ] We selected SM‐BP‐N and S1‐ICC‐BP based on their immunogenicity and induction of functional immune responses in mice. These BP vaccine candidates are also representing two different manufacturing processes with SM‐BP‐N completely assembled inside engineered E. coli in one‐step, while S1‐ICC‐BP are made of BP‐SpyC that were assembled in E. coli and subsequently coated with Spy‐tagged glycosylated S1‐ICC produced from insect culture. E. coli does not enable production of glycosylated S1. The vaccines were formulated with alum and 50 µg antigen was IM administered using a prime‐boost‐boost regime with 2‐week intervals (Figure 5a). Alum‐only served as control. Groups comprised six male and six female animals. Blood samples were taken in 2‐week intervals to monitor development of the immune response. Animals were intranasally infected with 103 plaque‐forming units of SARS‐CoV‐2 viruses two weeks after the last vaccination and half of the animals were sacrificed 3 d and the other half 6 d postinfection. ELISA showed that SM‐BP‐N induced significant levels anti‐S‐ and anti‐N‐antibodies while S1‐ICC‐BP induced significant levels of anti‐S antibodies when compared to the alum‐only group (Figure 5b). Viral titers in the lung were determined (Figure 5c). Both vaccines, SM‐BP‐N and S1‐ICC‐BP, induced protective immunity resulting in a significant decrease in virus titers on day 6 postinfection when compared with the placebo group. SM‐BP‐N induced only in female hamsters a reduction of virus titers in the lung of about six‐ and tenfold measured 3 or 6 d postinfection, respectively. S1‐ICC‐BP induced immune responses mediating a >100‐fold reduction of virus titer in the lung on day 6 postinfection in both female and male hamster while no virus was observed in 2 of 6 animals.
Figure 5.
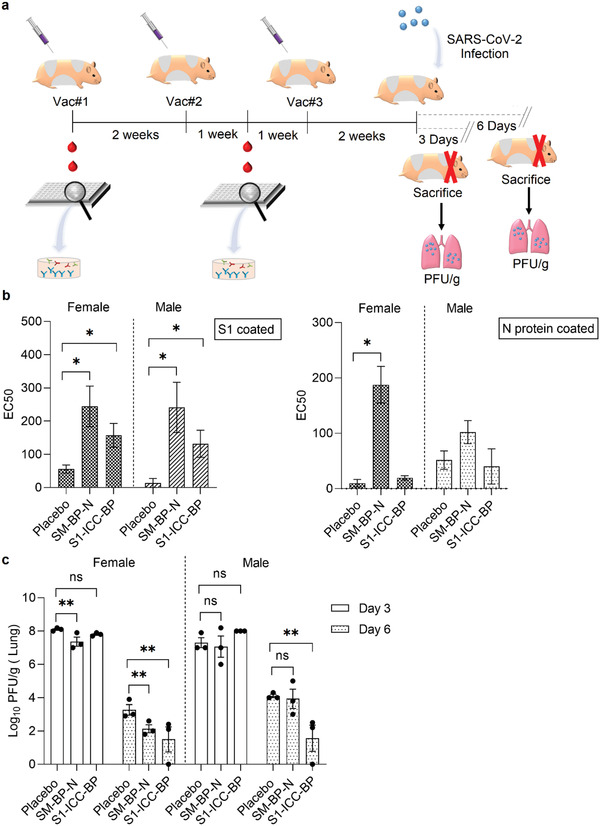
SM‐BP‐N and S1‐ICC‐BP induce protective immunity in the hamsters SARS‐CoV‐2 infections model. a) Schematic overview of hamster immunization study. There were six female and six male hamsters per group. Hamsters were vaccinated three times intramuscularly at 2 weeks intervals with freshly formulated vaccines containing 50 µg of antigen per dose, emulsified in alum (25 µL per dose). Blood collection was performed at day 0 and 21, respectively, for immunogenicity analysis. Hamsters were infected 2 weeks after the final vaccination and sacrificed 3 and 6 d later respectively for plaque‐forming unit (PFU) titer measurement. b) Antibody responses to S1 and N proteins. The serum samples were taken a week after the second immunization. Each data point represents the results from six hamsters ± the standard error of mean. *, significantly higher than the placebo group (p < 0.05). c) PFU titer per gram in the lung from the female and male hamsters immunized with SARS‐CoV‐2 antigen‐coated BPs. Each data point stands for the mean for three hamsters ± the standard error of the mean. **, significantly different with the placebo group (p < 0.05); ns, no significant difference with the placebo group (p > 0.05). Statistical significance is determined by T‐test for comparison of two groups or by one‐way ANOVA with pairwise comparison of multigrouped data sets achieved using Tukey's or Dunnet's post hoc test (Prism).
3. Discussion
In this study, we investigated whether the BP vaccine platform can be adapted for the development of a vaccine candidate for COVID‐19.[ 15 , 21 ] We previously showed that antigens attached to BPs are more immunogenic than their soluble counterparts.[ 15 , 23 ] In one study, we targeted the Hepatitis C virus (HCV) and demonstrated that Core antigen‐coated BPs induce protective immunity in a mouse model of HCV infection resulting ≈5 log10 and ≈4 log10 reduction of viral titers when compared with placebo or the soluble antigen, respectively.[ 27 ] Viral clearance in lungs was achieved in 60% of animals. Here, we showed that BPs can be coated with various SARS‐CoV‐2 antigens via direct assembly inside bacterial cells or by bacterial assembly of BPs displaying SpyC domains, which were then able to specifically bind Spy‐tagged S1 produced by using the baculovirus expression system (Figure 1). In both processes antigens are covalently linked to the BP by either using a direct translational fusion of antigen to the BP anchor, the PHB synthase, which is linked to the BP via a thioester bond or by using the SpyTag/SpyCatcher technology mediating formation of an isopeptide bond between Spy‐tagged S1 and SpyC displayed on the BP surface.[ 29 , 31 , 43 , 44 ] This study showed BP‐SpyC can specifically bind Spy‐tagged S1 in the complex mixture of the insect culture supernatant avoiding the need for laborious purification steps streamlining the downstream process that results in S1‐ICC‐coated BPs directly available for vaccination (Figures S2 and S3, Supporting Information). This is in alignment with previous studies where BP‐SpyC enabled specific immobilization and separation of Spy‐tagged target proteins present in complex bacterial lysates.[ 29 ] Prior to vaccination, we characterized all BPs confirming their composition, purity and antigenicity. All antigens and antigen combinations could be achieved as densely coating BPs (Figure 2b).
To investigate whether the protein production and assembly process retained functionality of S derived antigens, we analyzed ACE2 binding to these BPs coating ELISA plates. Except for N‐BP‐S1, all other S derived antigens attached to BPs retained ACE2 binding function (Figure 2d). The antigenicity of the various BP vaccine candidates was assessed using serum from two CPs and a healthy donor and confirmed that all N containing BPs and S1‐ICC‐BP were specifically binding antibodies from CPs (Figure 2c). Presentation of S1 fused to the C terminus of the BP anchor did not preserve antigenicity suggesting that termini to which antigen fused is critical for proper display and accessibility by antibodies. The requirement of specific linkers at C terminus that detach the fusion partner from the hydrophobic surface of BPs was previously found to be critical for functional surface display.[ 45 ] Hence, further linker designs might be required to functionally anchor C terminally fused S1. S1‐ICC‐BP showed the strongest binding of CP antibodies indicating full accessibility of multiple epitopes of S1.
All BPs were then tested for safety and immunogenicity in a mouse trial, which showed safety of BPs and that N in all BP formulations induced specific and high levels of IgG1 and IgG2c antibodies. RBD‐coated BP did not induce significant levels of anti‐S1 antibodies, which correlated with their poor antigenicity. Except for BP‐S1, all S1 and SM‐coated BPs induced high anti‐S1 antibody titers contributed by IgG1 and IgG2c isotypes (Figure 3b). We concluded that immunogenicity in mice was correlated with antigenicity as assessed by using reactivity to antibodies present in CP sera. ACE2 binding function of S1 and derived antigens did not provide an indicator for immunogenicity of BP formulations such as RBD‐coated beads bound ACE2 but failed to induce an anti‐S1 immune response. We investigated the functionality of induced antibodies to block binding of RBD to ACE2, which showed that only SM or S1 coated BPs induced neutralizing antibodies (Figure 3d). SM‐BP‐N showed the highest neutralizing antibody titers corresponding to an EC50 titer of about 245.
SM‐BP‐N and S1‐ICC‐BP were selected for further studies based on their immunogenicity and functionality of immune response. BPs biophysically characterized to determine properties such as size distribution, morphology, surface charges, functionality and antigenicity of antigens displayed on BPs (Figure 4a–d). The size of BPs of about 1 µm is suitable for efficient uptake by antigen presenting cells (APCs) (Figure 4). In general, APCs take up particles with a size range of 0.5–10 µm via phagocytosis[ 46 , 47 , 48 ] permitting antigen cross‐presentation and induction of both humoral and cell‐mediated immunity.[ 15 , 49 ] All biophysical data aligned with our previous studies.[ 15 , 23 ] Using UV‐inactivated SARS‐CoV‐2 virus in ELISA confirmed that both BPs induced antibodies that can bind to the intact virus (Figure 3e). Both vaccine candidates induced highly specific antibody responses only recognizing the respective antigens as was shown by immunoblotting using cell lysates and BPs‐only as control (Figure S7, Supporting Information). The two BPs are manufactured based on different bioprocesses employed to attach antigens including the possibility of producing post‐translationally modified glycosylated S1 from insect culture. SM‐BP‐N is solely assembled in bacterial cells enabling use of robust fermentation technology for costeffective manufacture at large scale. The S1‐ICC‐BP requires an additional insect culture step for antigen production, which might be justifiable if vaccine performance due to such as glycosylation can be improved. Nota bene, we developed BPs to efficiently and specifically bind S1‐ICC using the SpyCatcher/SpyTag interaction strongly streamlining the downstream process by combining purification of antigen with antigen coating of BPs. Insect culture based on the baculovirus expression system was chosen to produce the Spy‐tagged S1 because it is an established commercially viable manufacturing system allowing industrial scale manufacture of vaccine candidate antigens. In addition, this system enables production of viral antigens that are post‐translationally modified and generally folded in the correct conformation.[ 50 , 51 ] This expression technology was approved by regulators in the European Union and the USA for production of antigens used in vaccine products such as preventing human papilloma virus infection that causes cervical cancer or influenza.[ 50 , 51 ] The baculovirus expression systems was recently used to produce post‐translationally glycosylated RBD, which induced neutralizing antibodies and protective immunity in nonhuman primates.[ 52 ] However, the downstream process involved two chromatography steps followed by buffer exchange, which inherently increases process costs. In contrast, our process does not require chromatography.
A recent study used the SpyCatcher/SpyTag technology to attach the RBD produced from mammalian cell culture to VLPs in order to enhance immunogenicity. RBD ligated to VLP was more immunogenic than its soluble counterpart and induced neutralizing antibodies, however, protective immunity in a SARS‐CoV‐2 challenge study was not assessed.[ 53 ] The Spy‐tagged RBD was produced by mammalian cell culture requiring costly chromatography steps prior ligation to the VLP.
SM‐BP‐N and S1‐ICC‐BP were stable for at least 6 months at 4 °C and retained antigenicity, BP properties after 48h at 50 °C providing further evidence for temperature stability facilitating stockpiling and global distribution (Figure 4d–f). We provided evidence that BPs can be freeze‐dried and rehydrated for vaccine uses (Figure 4d,e). Freeze‐drying is presumably strongly enhancing shelf‐life.
It is critical for assessment of vaccine performance to investigate whether immune responses induce protective immunity in suitable infection model. The golden Syrian hamster model of SARS‐CoV‐2 infection has been widely accepted[ 42 , 54 ] to test the performance of COVID‐19 vaccine candidates’ prior further evaluation in clinical Phase I studies. Here, we vaccinated hamsters with our two vaccine candidates followed by challenge with SARS‐CoV‐2. Both vaccine candidates were safe and induced protective immunity in hamster as shown by a significant reduction of viral titers in the lung when compared to placebo. SM‐BP‐N induced immune responses appeared to already impede viral replication on day 3 postinfection which continued to reduce viral titer as assessed on day 6 (Figure 5c). S1‐ICC‐BP induced immune responses with no obvious impact on virus titers on day 3 but showed a significant reduction of >100‐fold including viral clearance in 2 of 6 animals on day 6 postinfection (Figure 5c). Since antibody responses in hamsters a week after the first boost were significant but at low levels (Figure 5b) we propose that protective immunity could be contributed by cell‐mediated immune responses, which are known to be induced by BPs.[ 15 , 27 , 55 ] This was supported by induction of IgG2c and cytokine responses in the mouse study (Figure 3b,c,f) which indicated a mixed mode Th1 and Th2 immune response desirable for prevention of SARS‐CoV‐2 infection.
Recent studies used virus‐based spike protein vaccine candidates such as recombinant vesicular stomatitis virus or adenovirus that both induced immune responses mediating a ≈1000‐fold reduction of virus titer in the lung in hamster postchallenge with SARS‐CoV‐2.[ 56 , 57 ] Immunity was due to induction of neutralizing antibodies but no immunity leading to viral clearance in lungs post infection was observed in these studies. There is tremendous scope to improve our two BP‐based vaccines by such as, e.g., optimizing the dose, adjuvant and administration regime. There is a strong focus on neutralizing antibodies in SARS‐CoV‐2 vaccine development while underestimating the importance of T cell immunity and long‐lasting immunity based on memory T cells. The importance of adaptive T cell immunity was supported by identifying circulating SARS‐CoV‐2‐specific CD8+ and CD4+ T cells in ≈70% and 100% of COVID‐19 convalescent patients, respectively.[ 19 , 58 ] Hence, the BP vaccine candidate's bias toward induction of T cell immune responses might be advantageous in the context of SARS‐CoV‐2 infection. Overall, the data show that the BP vaccine platform can be adapted to develop robust COVID‐19 vaccine candidates. It leverages the BP platform's highly beneficial attributes of strongly facilitated costeffective manufacture at industrial scale aligned with outstanding stability ideally suited for global supply and distribution of a vaccine during a pandemic. The BP platform's design space and versatility will enable rapid development of vaccine candidates that incorporate most recent immunity and virus variant information to combat emerging pandemic threats.
4. Conclusions
Here, we demonstrated that the BP vaccine technology can be adapted to the development of safe, efficient and stable SARS‐CoV‐2 vaccine candidates. The study showed that full‐length antigens as well as selected epitopes of SARS‐CoV‐2 could be engineered to densely coat BPs. Two manufacturing processes were developed using either the bacterial cell factory to assemble the antigen‐coated BPs in one step or to assemble SpyCatcher‐coated BPs that specifically captured the glycosylated S1 protein secreted by recombinant insect cultures. Although the latter process requires two production steps, the use of the BPs as bioseparation resin and particulate vaccine strongly enhances manufacturability. All BPs retained antigenicity of SARS‐CoV‐2 derived antigens and were immunogenic in mice. The two best performing vaccine candidates SM‐BP‐N and S1‐ICC‐BP induced protective immunity in the hamster model of SARS‐CoV‐2 infection. Overall, our study proposes that the BP vaccine technology is applicable to rapidly design safe and efficient vaccine candidates against SARS‐CoV‐2 and its variants. The ambient temperature stability of BP vaccines combined with cost‐effective scalable manufacturability will strongly facilitate dissemination of vaccines in a pandemic scenario suggesting utility of the BP technology as first line rapid response vaccine technology.
5. Experimental Section
Statistical Analysis
All statistical analyses were performed using GraphPad Prism 8 (GraphPad Software). Statistical differences were analyzed using either two‐tailed Mann–Whitney U test or Kruskal–Wallis test followed by Dunn's multiple comparisons. A p‐value < 0.05 was deemed statistically significant.
One‐Step Bacterial Production of SARS‐CoV‐2 Antigen‐Coated BPs
All the plasmids used in this study are shown in Table S1 in the Supporting Information. The gene fragments encoding SARS‐CoV‐2 antigens (Table S2, Supporting Information), including N protein with amino acid sequence, RBD with amino acid sequence, spike glycoprotein S1 with amino acid sequence, and recombinant B cell epitopes from spike glycoprotein S and membrane proteins with amino acid sequence, were synthesized by Biomatik (Canada) and codon optimized for E. coli. All cloning procedures were carried out in the E. coli Top10 strain (Table S1, Supporting Information). The specific B‐cell epitopes of SARS‐CoV‐2 S and M proteins were illustrated in Table S2 in the Supporting Information. The molecular cloning techniques were performed as described in ref. [ 24 ]. The target DNA sequences of all constructed pET plasmids were confirmed by the Griffith University DNA Sequencing Facility (Griffith University, Australia). Three genes, phaA, phaB, and phaC, encoding enzymes PhaA, PhaB, and PhaC are required for BP formation. The plasmid pMCS69 containing the genes phaA and phaB encoding enzymes PhaA and PhaB which produce precursor molecules for polymer synthesis was cotransformed. Polymer synthase PhaC is required for biosynthesis of BP and is encoded by the gene phaC in the pET plasmid. The plasmid pMCS69E contains additional Erv1p encoding yeast sulfhydryl oxidase, which is able to improve the production of disulfide bonded proteins in the cytoplasm. Both pET‐14b phac‐SARS‐CoV‐2 genes and pMCS69/pMCS69E were transformed in the production strain ClearColi BL21 (DE3) (Table S1, Supporting Information) for production of SARS‐CoV‐2 antigen‐coated BPs.
Cultivation condition for production of BPs was described previously.[ 15 , 23 , 24 ] After bacterial cells were grown and accumulated BPs, respective biomass was processed for BP purification as previously described.[ 24 , 33 ] Briefly, cells were mechanically disrupted and cell debris was selectively solubilized. Purified BPs were recovered after sequential washes. The bulk BPs were stored in Tris buffer (10 × 10−3 m Tris, pH 7.5) with 20% ethanol at 4 °C until further use.
Production of S1‐ICC‐BPs
Spy‐tagged‐S1 was produced using the baculovirus expression system. In particular, a hybrid gene encoding a fusion protein composed of an N‐terminal SpyTag peptide (AHIVMVDAYKPTK) followed by a spacer (GGG) and the SARS‐CoV‐2 spike protein S1 containing a C‐terminal 10x‐histidine tag was inserted between the BamHI and SphI restriction sites in the vector pBAC‐1gp67. The vector encoded the signal peptide gp67 translationally fused to the fusion protein. The final construct was verified by DNA sequencing.
Recombinant baculovirus was obtained using the flashBAC ULTRA system (Oxford Expression Technologies) as described previously.[ 59 ] High Five cells grown in ESF921 medium (Expression Systems, USA) at 27 °C were infected at the cell density of 1.5 × 106 cells mL−1 with an estimated multiplicity of infection of 5 (infectious unit per cell). The culture supernatant was harvested at 48 h postinfection by centrifugation at 12 200 x g for 30 min at 4 °C and subsequently concentrated 10 × by tangential flow filtration using a Sartoflow Study (Sartorius Stedium Biotech, Germany) with 30 kDa molecular weight cut‐off polyethersulfone membrane (Sartorius Stedim Biotech, Germany). S1‐ICC production was confirmed by SDS‐PAGE and immunoblotting using anti‐Histidine‐HRP antibodies (Miltenyia Biotech).
To coat BPs with Spy‐tagged S1‐ICC produced by insect culture, SpyC displaying BPs (BP‐SpyC) were manufactured as previously described.[ 29 , 31 ] Initially, Spy‐tagged‐green fluorescent protein (GFP) was used to optimize the binding of target protein to BP‐SpyC as well as to determine their binding capacity. Briefly, 100 µL of 20% w/v BP‐SpyC was incubated with whole cell lysate containing Spy‐tagged‐GFP in excess. The resulting BPs were then washed three times with 50 × 10−3 m Tris buffer (pH 7.5). The binding capacity was then determined using densitometry. BP‐SpyC was mixed with the concentrated insect culture supernatant at a ratio to Spy‐tagged S1‐ICC to ensure saturation of all SpyC binding sites. Spy‐tagged‐S1‐ICC was captured by mixing 1 mL of 20% w/v BP‐SpyC with the 500 mL of concentrated supernatant containing Spy‐tagged‐S1‐ICC. After 16 h incubation at 4 °C, purified BP‐SpyC ligated with Spy‐tagged‐S1‐ICC recovered after sequential washing steps as described in ref. [ 29 ]. This innovative approach combined antigen purification with BP coating, i.e., strongly facilitated downstream processing and vaccine formulation.
BP Characterization
Protein profiles of BP vaccines was analyzed by SDS‐PAGE and proteins were quantified using densitometry (Image Lab Software (Bio‐Rad Laboratories, USA)) as previously described.[ 24 ] The recombinant target protein band was excised from the gel and subjected to protein identification using Q‐TOF/MS, performed at Mass Spectrometry Facility, University of Queensland Centre for Clinical Research (Queensland, Australia). Bovine serum albumin (BSA, Sigma‐Aldrich, USA) ranging between 62.5 and 500 ng served as standards for protein quantification. The size distribution and ζ‐potential of all particulate vaccine samples were analyzed by Litesizer500 (Anton Paar, Australia). ClearColi BL21(DE3) producing BP SARS‐CoV‐2 vaccine particles and of purified BP SARS‐CoV‐2 vaccine particles were visualized by TEM at University of Queensland in Australia.
ACE2‐Binding Assay Using SARS‐CoV‐2 Antigen‐Coated BPs
High‐binding plates (Greiner Bio‐One, Germany) were coated overnight at 4 °C with 100 µL of 5 µg mL−1 purified SARS‐CoV‐2 antigen‐coated BPs diluted in phosphate‐buffered saline (PBS) containing 0.05% (v/v) Tween 20, pH7.5 (PBST). Glycosylated soluble S1 from baculovirus expression (Protein Expression Facility, University of Queensland, Australia) and plain BPs served as positive and negative controls respectively. After three washes with PBST, plates were incubated for 1 h at 25 °C with 100 µL of angiotensin converting enzyme 2 (ACE2) (Human) Fc fusion (Aviscera Bioscience Inc, USA) diluted 1:1000 with PBST. After three washes with PBST, plates were incubated for 1 h at 25 °C with 100 µL of native protein A‐HRP (Abcam, United Kingdom) diluted 1:10 000 with PBST to bind to the Fc region of human ACE2 fusion. Plates were washed three times with PBST and 100 µL of o‐phenylenediamine substrate (OPD) (Abbott Diagnostics, IL, USA) was added to the plate for signal development. The reaction was stopped by adding 50 µL of 1 n H2SO4. The results were measured at 490 nm with an ELx808iu ultramicrotiter plate reader (Bio‐Tek Instruments Inc., USA).
Antigenicity Analysis of SARS‐CoV‐2 Antigen‐Coated BPs Using CP Sera
The antigenicity of various SARS‐CoV‐2 antigens attached to BPs was analyzed by using sera from CPs who recovered from mild SARS‐CoV‐2 (ethics number HREC/GU/2018/936) and normal human serum (NHS, Invitrogen, USA). This was done using ELISA plates coated with the various BPs and by measuring antigen specific antibody (IgG and/or IgM) levels. This experiment was done as a single blind study. Soluble rRBD and plain BPs were used as positive control and negative controls, respectively. Briefly, high‐binding plates (Nunc MaxiSorp) were coated with 100 µL of 1 µg mL−1 of SARS‐CoV‐2 antigens attached to BPs in Carbonate‐Bicarbonate Buffer (pH 9.6, Sigma‐Aldrich, USA) at 4 °C overnight. Plates were blocked with 5% (w/v) skim milk in PBST for 90 min at 37 °C. Plates were washed three times with PBST and three times with distilled water, respectively, followed by incubation with CP sera and NHS diluted in PBST containing 0.5% (w/v) skim milk at the concentration of 1/2000 for 90 min at 37 °C. After three times PBST and three times distilled water washes, plates were then incubated with the secondary goat antihuman IgG‐HRP (Bio‐Rad Laboratories, USA) at the concentration of 1/3000 and IgM‐HRP (Thermo Fisher Scientific, USA) at the concentration of 1/10 000. The signal was developed by incubating the plates with 100 µL of OPD solution for 20 min after three washes with PBST and three washes with distilled water, respectively. The reaction was stopped by adding 25 µL of 3 m HCl solution. The results were measured at 492 nm with an ELx808iu ultramicrotiter plate reader (Bio‐Tek Instruments Inc., USA).
Immunization of Mice
The animal experiments with an ethics number GRIDD/01/20/AEC were approved by Griffith University Animal Ethics Committee (Queensland, Australia). The experiment was carried out using 6‐week‐old female C57BL/6 mice. There were ten mice per group. Each dose of formulated vaccine contained 20 µg of respective antigens and 25 µL of aluminum hydroxide (InvivoGen, USA) in a volume of 100 µL Tris‐HCl buffer pH 7.5. All BP vaccines were freshly formulated with alum before use. Mice were injected intramuscularly with 25 Gauge ½ inch needle in the thigh muscle of hind limb. Particularly, 100 µL of formulated vaccines was injected and 50 µL per injection site. Mice were vaccinated three times, 2 weeks apart.
ELISA Using Purified Soluble SARS‐CoV‐2 S1, N, and M Proteins
Mice serum samples were collected and analyzed by ELISA as previously described.[ 25 ] Briefly, 100 µL of soluble SARS‐CoV‐2 antigens, N protein or S1 (Protein Expression Facility, University of Queensland, Australia), at a concentration of 1 µg mL−1 was coated overnight at 4 °C on high‐binding ELISA plates (Greiner Bio‐One, Germany). The plates were then blocked with 3% (w/v) BSA (Sigma‐Aldrich, USA) for 1 h at 25 °C. Individual mice serum samples were used as the primary antibodies to specifically bind to N, S1, or dual M protein fusion (NativeAntigen Company, United Kingdom). After three washes with PBST, serially diluted serum samples from 1:200 to 1:25 600 were added to the plates and incubated at 25 °C for 1 h. After three washes with PBST, goat anti‐mouse IgG‐, IgG1‐, or IgG2c‐HRP (Abcam, United Kingdom) diluted 1:20 000 with PBST were added as the secondary antibodies to the wells for 1 h at 25 °C to bind to total IgG, IgG1, and IgG2c, respectively. OPD (Abbott Diagnostics, IL, USA) was added after 3 washes with PBST to the wells for signal development. The reaction was stopped by add 50 µL of 1 n H2SO4. The results were measured at 490 nm with an ELx808iu ultramicrotiter plate reader (Bio‐Tek Instruments Inc., USA). Antibody response was analyzed and presented as the half maximal inhibitory concentration (EC50). EC50‐value calculation was previously described.[ 24 ] An example of EC50‐value calculation is shown in Figure S16 in the Supporting Information.
ELISAs Using Inactivated SARS‐CoV‐2 Virus
SARS‐CoV‐2 virus (hCoV‐19/Australia/QLD02/2020; Global Initiative on Sharing Avian Influenza Data (GISAID) Accession ID; EPI_ISL_407896) was propagated in Vero E6 cells and UVC inactivated. Virus in supernatants was ultraviolet C (UVC) inactivated in six well plates (3 mL of RPMI1640 supplemented with 10% fetal calf serum (FCS) per well using a UVC Hoefer ultraviolet crosslinker (4–4.7 mW cm−2 for 5 min). The UVC dose was confirmed using a UVC Light Meter (UVC‐254A, Lutron) and inactivation confirmed by CCID50 assay. Work with live virus was conducted in a biosafety level‐3 (PC3) facility at the Queensland Institute of Medical Research (QIMR) Berghofer magnetic resonance imaging (MRI) (Australian Department of Agriculture, Water and the Environment reference number Q2326 and Office of the Gene Technology Regulator certification number, Cert‐3445).
The UV‐inactivated SARS‐CoV‐2 virus was coated on high‐binding ELISA plates (Nunc MaxiSorp) to evaluate specific antibody titers in individual serum samples from mice immunized with various SARS‐CoV‐2 antigen‐coated BPs. Briefly, 100 µL of inactivated virus diluted with Carbonate‐Bicarbonate Buffer (pH 9.6, Sigma‐Aldrich, USA) at a concentration 4.35 µg mL−1 were coated on plates overnight at 4 °C. The plates were blocked with 5% (w/v) skim milk for 1 h at 25 °C. After 3 washes with PBST, serially diluted individual mice serum samples (from 1:200 to 1:25 600) were added to plates and incubated for 1 h at 25 °C to specifically detect antibodies binding to the whole virus SARS‐CoV‐2 as opposed to individually purified antigens. Following three washes with PBST, the secondary antibody‐HRPs, goat anti‐mouse IgG‐, IgG1‐, or IgG2c‐HRP, was diluted 1:20 000 and added to plates followed by incubation for 1 h at 25 °C to detect the total IgG, IgG1, and IgG2c, respectively, in individual mice serum samples. After three washes with PBST, 100 µL of the substrate OPD (Abbott Diagnostics, IL, USA) was added to the plates. The signal development was stopped by adding 50 µL of 1 n H2SO4. The results were measured with an ELx808iu ultramicrotiter plate reader (Bio‐Tek Instruments Inc., USA) at 490 nm. Antibody response was interpreted and presented as EC50.
Cytokine Assay
The cytokine assay was carried out using AlphaLISA Mouse Interferon gamma (mIFNγ) and Mouse Tumor Necrosis Factor alpha (mTNFα) Kits (PerkinElmer, USA) according to manufacturer's instruction. Briefly, 5 µL of serum samples and diluted cytokine standards from 0.3 to 100 000 pg mL−1 was added to a white Optiplate‐384 microplate (PerkinElmer, USA). Then, 20 µL of freshly prepared mix, containing mIFNγ or mTNFα acceptor beads (25 µg mL−1) and biotinylated anti‐mIFNγ or anti‐mTNFα antibody (2.5 × 10−9 m), was added to plate and incubated for 1 h at 23 °C. Twenty‐five microliter of diluted streptavidin donor beads (80 µg mL−1) was added to the plate and incubated for 30 min at 23 °C in the dark. The result was measured on an ELx808iu ultramicrotiter plate reader (Bio‐Tek Instruments Inc., USA) with a dual filter set with excitation at 680/30nm and emission at 570/100 nm.
Antigenicity Analysis of SARS‐CoV‐2 Antigen‐Coated BPs Treated with Various Temperature
Antigenicity of various SARS‐CoV‐2 antigen‐coated BPs after different temperature treatment was evaluated by ELISA. In particular, SM‐BP‐N and S1‐ICC‐BP were freeze‐dried and reconstituted in 10 × 10−3 m Tris buffer (pH7.5). In addition, these BPs suspensions were supplemented with 10 µg mL−1 amphotericin B (ThermoFisher Scientific, USA) and 100 µg mL−1 ampicillin (ChemSupply, Australia) to avoid the risk of contaminations due to bacteria or fungi growth and then incubated at different temperatures, 4 °C, 25 °C, 37 °C, and 50 °C, for 2 d. After various temperature treatments, 100 µL of various treated SM‐BP‐N or S1‐ICC‐BP diluted with PBST at an antigen concentration at 5 µg mL−1 was coated overnight at 4 °C on high‐binding ELISA plates (Greiner Bio‐One, Germany). The plates were blocked with 3% (w/v) BSA (Sigma‐Aldrich, USA) for 1 h at 25 °C. The primary antibody was individual mouse serum sample containing the highest EC50 antibody titers from SM‐BP‐N or S1‐ICC‐BP vaccinated mouse, respectively. Following three washes with PBST, serially diluted serum samples from 1:200 to 1:25 600 were added to the plates and incubated for 1 h at 25 °C. After three washes with PBST, the secondary antibody‐HRP, goat anti‐mouse IgG‐HRP (Abcam, United Kingdom) was diluted 1:20 000 and added the plates to bind to total IgG followed by 1 h incubated at 25 °C. The substrate OPD (Abbott Diagnostics, IL, USA) was added to the plates after three washes with PBST. The reaction was stopped by adding 50 µL of 1 n H2SO4. The signal was measured with an ELx808iu ultramicrotiter plate reader (Bio‐Tek Instruments Inc., USA) at 490 nm. Antibody response was analyzed and presented as EC50.
In Vitro SARS‐CoV‐2 Inhibition Assay
The assay was performed using SARS‐CoV‐2 Surrogate Virus Neutralization Test Kit (GenScript, USA) according to the manufacturer's instructions. The kit contains a positive control with EC50 neutralizing antibody titer of 528. Briefly, 200 µL diluted serum samples and controls (20 µL samples + 180 µL sample dilution buffer) were preincubated with 200 µL HRP‐RBD for 30 min at 37 °C to allow the binding of neutralizing antibodies to HRP‐RBD. Then, 100 µL of resulting mixture was added to hACE2 precoated capture ELISA plate and incubated for 15 min at 37 °C. The unbound HRP‐RBD or any HRP‐RBD bound to non‐neutralizing antibodies will be captured by hACE2 on the plate, while the neutralizing antibodies‐HRP‐RBD complexes remained in the supernatant. The plate was washed with wash solution four times. After washing, 100 µL of 3,3′,5,5′‐tetramethylbenzidine solution was added each well and incubated in the dark for 15 min at 25 °C. The reaction was stopped by adding 50 µL of the stopping solution and the results were measured at 450 nm on an ELx808iu ultramicrotiter plate reader (Bio‐Tek Instruments Inc., USA). The inhibition rate was calculated using the following equation
| (1) |
Immunoblot Analysis to Assess Specificity of Immune Responses
To evaluate the specificity of antibody responses, pooled mice serum samples were used for immunoblotting against whole cell lysate harboring pET‐14b vector control, BP, BP‐SpyC, S1‐ICC‐BP, SM‐BP‐N, N protein, S1, M protein, and UV‐inactivated SARS‐CoV‐2 virus. First, proteins of various samples were separated by SDS‐PAGE. The amount of protein loaded for immunoblot analysis was 25–100 times less than the amount of protein loaded for protein profile analysis by SDS‐PAGE. The samples were transferred from SDS‐PAGE gel to a nitrocellulose membrane (Thermo Fisher Scientific, USA) using the iBlot 2 Dry Blotting System (Invitrogen, USA). The membrane was washed three times with PBST for 15 min, respectively. The pooled serum samples from mice immunized with alum alone (Placebo), BP, S1‐ICC‐BP, or SM‐BP‐N, were diluted 1:2000 with PBST and incubated with the nitrocellulose membranes for 1 h at 25 °C. After three washes with PBST, the nitrocellulose membranes were incubated with goat anti‐mouse IgG‐HRP at a concentration of 1/20 000 for 1 h at 25 °C. The membranes were then washed three times with PBST and incubated with SuperSignal West Pico Stable Peroxide Solution and SuperSignal West Pico Luminol/Enhancer Solution (Thermo Scientific, USA) for 5 min at 25 °C for signal development. Immunoblot images were captured using Odyssey (LI‐COR Biosciences, USA).
Vaccination and Challenge Study in Hamsters
The experimental study with hamsters was performed after approval by the Institutional Animal Care and Use Committee at the University of Wisconsin with an ethics number V6426. Golden Syrian hamsters (females and males; 4‐weeks old) were immunized intramuscularly three times at 2‐week intervals with freshly formulated vaccines containing 50 µg of antigen per dose, emulsified in aluminum hydroxide (25 µL per dose, InvivoGen, USA) in a volume of 100 µL Tris‐HCl buffer pH 7.5. During the course of immunization, blood samples were collected for serum isolation via the sublingual vein at day 0 and 21, respectively. Two weeks after the last immunization, hamsters were infected by intranasal inoculation with 103 plaque‐forming units of SARS‐CoV‐2 (SARS‐CoV‐2/UT‐NCGM02/Human/2020/Tokyo) while under isoflurane anesthesia. Animals were weighted daily and monitored daily for signs of illness. Animals were humanely sacrificed, and lung tissue samples were collected 3 and 6 d after infection.
Virus and Titration Assays
To determine the amount of virus in the lung samples, virus titrations were performed on Vero E6/TMPRSS2 cells that were obtained from the National Institute of Infectious Diseases, Japan.[ 60 ] Cells were maintained in Dulbecco's modified Eagle's medium (DMEM) containing 10% fatal bovine serum (FBS) and antibiotic/antimycotic solution along with G418 (1 mg mL−1). Confluent Vero E6/TMPRSS2 cells were infected with 100 µL of tenfold dilutions (10−1 to 10−6) of clarified tissue homogenates. After a 30 min incubation, the inoculum was removed, the cells were washed once, and then overlaid with 1% methylcellulose solution supplement with 5% FBS in DMEM. Plaques were counted after 3 d once the cells were fixed and stained with 20% methanol and crystal violet.
Detection of Hamster Antibodies Against SARS‐CoV‐2 Antigens
ELISAs were performed using a recombinant purified SARS‐CoV‐2 S1 (Sino Biological; 40591‐V05H1) or N (Sino Biological; 40588‐V08) as antigens. ELISA plates were coated overnight at 4 °C with 50 µL of the antigen at a concentration of 2 µg mL−1 in PBS (pH7.5). After blocking with 3% milk powder in PBST, the plates with incubated in duplicate with heat‐inactivated serum diluted in PBST with 1% milk powder. The secondary hamster IgG‐HRP (Invitrogen; 1:7000 dilution) was added to the plates. Signal was developed with SigmaFast o‐phenylenediamine dihydrochloride solution (Sigma‐Aldrich, USA), and the reaction was stopped with the addition of 3M hydrochloric acid. The absorbance was measured at a wavelength of 490 nm. Antibody response was interpreted and presented as EC50.
Biosafety Statement
Research involving SARS‐CoV‐2 at the Influenza Research Institute was approved by the University of Wisconsin‐Madison's Institutional Biosafety Committee and performed under biosafety level 3 agriculture (BSL‐3Ag) containment a designation that exceeds the standards outlined in Biosafety in Microbiological and Biomedical Laboratories (6th edition).
Conflict of Interest
B.H.A.R is inventor of the BP technology and co‐founder/shareholder of Polybatics Ltd that commercializes BP‐based products. The other authors declare no conflict of interest.
Supporting information
Supporting Information
Acknowledgements
The authors thank Dr. David Wibowo and Dr. Raisa Monteiro at Griffith Institute for Drug Discovery, Griffith University, for assisting with animal trials and editorial assistance, respectively. The authors also thank Dr. Alyssa Pyke, Queensland Health, Brisbane, for providing the QLD02 SARS‐CoV‐2 virus isolate. B.H.A.R. is funded by Fast Grants Award #2165 (Emergent Ventures at Mercatus Center, George Mason University, USA), the Australian Research Council (ARC) Discovery Project (DP200100874), the ARC Linkage Infrastructure, Equipment and Facilities (LE20010014) and Griffith University. The authors acknowledge the facilities, and the scientific and technical assistance, of the Australian Microscopy and Microanalysis Research Facility at the Centre for Microscopy and Microanalysis, as well as the Centre for Clinical Research and the Metabolomics Australia—Queensland Node at the University of Queensland. A.S. was supported by an Investigator grant from the National Health and Medical Research Council of Australia (APP1173880) and is a member of the AIDRC Global Virus Network (GVN) Center of Excellence. The authors thank Clive Berghofer and Lyn Brazil (and others) for their generous philanthropic donations that funded the establishment of the PC3 (BSL3) SARS‐CoV‐2 research facility at QIMR Berghofer MRI.
Chen S., Evert B., Adeniyi A., Salla‐Martret M., Lua L. H., Ozberk V., Pandey M., Good M. F., Suhrbier A., Halfmann P., Kawaoka Y., Rehm B. H. A., Ambient Temperature Stable, Scalable COVID‐19 Polymer Particle Vaccines Induce Protective Immunity. Adv. Healthcare Mater. 2022, 11, 2102089. 10.1002/adhm.202102089
Data Availability Statement
The data that support the findings of this study are available in the Supporting Information of this article.
References
- 1. Johns Hopkins University, Coronavirus Resource Center https://coronavirus.jhu.edu/map.html, November 2021.
- 2. Kyriakidis N. C., López‐Cortés A., González E. V., Grimaldos A. B., Prado E. O., NPJ Vaccines 2021, 6, 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Gao Q., Bao L., Mao H., Wang L., Xu K., Yang M., Li Y., Zhu L., Wang N., Lv Z., Gao H., Ge X., Kan B., Hu Y., Liu J., Cai F., Jiang D., Yin Y., Qin C., Li J., Gong X., Lou X., Shi W., Wu D., Zhang H., Zhu L., Deng W., Li Y., Lu J., Li C., Wang X., Yin W., Zhang Y., Qin C., Science 2020, 369, 77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Van Doremalen N., Lambe T., Spencer A., Belij‐Rammerstorfer S., Purushotham J. N., Port J. R., Avanzato V., Bushmaker T., Flaxman A., Ulaszewska M., Feldmann F., Allen E. R., Sharpe H., Schulz J., Holbrook M., Okumura A., Meade‐White K., Perez‐Perez L., Bissett C., Gilbride C., Williamson B. N., Rosenke R., Long D., Ishwarbhai A., Kailath R., Rose L., Morris S., Powers C., Lovaglio J., Hanley P. W., Scott D., Saturday G., de Wit E., Gilbert S. C., Munster V. J., bioRxiv 2020, 10.1101/2020.05.13.093195. [DOI] [Google Scholar]
- 5. Callaway E., Nature 2020, 581, 363. [DOI] [PubMed] [Google Scholar]
- 6. Krammer F., Nature 2020, 586, 516. [DOI] [PubMed] [Google Scholar]
- 7. Eroshenko N., Gill T., Keaveney M. K., Church G. M., Trevejo J. M., Rajaniemi H., Nat. Biotechnol. 2020, 38, 789. [DOI] [PubMed] [Google Scholar]
- 8. Kadkhoda K., mSphere 2020, 5, e00344‐20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Pedersen S. F., Ho Y.‐C., J. Clin. Invest. 2020, 130, 2202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Schultz N. H., Sørvoll I. H., Michelsen A. E., Munthe L. A., Lund‐Johansen F., Ahlen M. T., Wiedmann M., Aamodt A.‐H., Skattør T. H., Tjønnfjord G. E., Holme P. A., N. Engl. J. Med. 2021, 384, 2124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Cines D. B., Bussel J. B., N. Engl. J. Med. 2021, 384, 1.33393745 [Google Scholar]
- 12. Wheatley A. K., Juno J. A., Wang J. J., Selva K. J., Reynaldi A., Tan H.‐X., Lee W. S., Wragg K. M., Kelly H. G., Esterbauer R., Davis S. K., Kent H. E., Mordant F. L., Schlub T. E., Gordon D. L., Khoury D. S., Subbarao K., Cromer D., Gordon T. P., Chung A. W., Davenport M. P., Kent S. J., Nat. Commun. 2021, 12, 1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gomes A., Mohsen M., Bachmann M., Vaccines (Basel) 2017, 5, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Jeyanathan M., Afkhami S., Smaill F., Miller M. S., Lichty B. D., Xing Z., Nat. Rev. Immunol. 2020, 20, 615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wibowo D., Jorritsma S. H. T., Gonzaga Z. J., Evert B., Chen S., Rehm B. H. A., Biomaterials 2021, 268, 120597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ju B., Zhang Q.i, Ge J., Wang R., Sun J., Ge X., Yu J., Shan S., Zhou B., Song S., Tang X., Yu J., Lan J., Yuan J., Wang H., Zhao J., Zhang S., Wang Y., Shi X., Liu L., Zhao J., Wang X., Zhang Z., Zhang L., Nature 2020, 584, 115. [DOI] [PubMed] [Google Scholar]
- 17. Piccoli L., Park Y.‐J., Tortorici M. A., Czudnochowski N., Walls A. C., Beltramello M., Silacci‐Fregni C., Pinto D., Rosen L. E., Bowen J. E., Acton O. J., Jaconi S., Guarino B., Minola A., Zatta F., Sprugasci N., Bassi J., Peter A., De Marco A., Nix J. C., Mele F., Jovic S., Rodriguez B. F., Gupta S. V., Jin F., Piumatti G., Lo Presti G., Pellanda A. F., Biggiogero M., Tarkowski M., Pizzuto M. S., Cameroni E., Havenar‐Daughton C., Smithey M., Hong D., Lepori V., Albanese E., Ceschi A., Bernasconi E., Elzi L., Ferrari P., Garzoni C., Riva A., Snell G., Sallusto F., Fink K., Virgin H. W., Lanzavecchia A., Corti D., Veesler D., Cell 2020, 183, 1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Rogers T. F., Zhao F., Huang D., Beutler N., Burns A., He W.‐T., Limbo O., Smith C., Song G.e, Woehl J., Yang L., Abbott R. K., Callaghan S., Garcia E., Hurtado J., Parren M., Peng L., Ramirez S., Ricketts J., Ricciardi M. J., Rawlings S. A., Wu N. C., Yuan M., Smith D. M., Nemazee D., Teijaro J. R., Voss J. E., Wilson I. A., Andrabi R., Briney B., Landais E., Sok D., Jardine J. G., Burton D. R., Science 2020, 369, 956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Grifoni A., Weiskopf D., Ramirez S. I., Mateus J., Dan J. M., Moderbacher C. R., Rawlings S. A., Sutherland A., Premkumar L., Jadi R. S., Marrama D., De Silva A. M., Frazier A., Carlin A. F., Greenbaum J. A., Peters B., Krammer F., Smith D. M., Crotty S., Sette A., Cell 2020, 181, 1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. De Assis R. R., Jain A., Nakajima R., Jasinskaas A., Felgner J., Obiero J. M., Adenaiye O., Tai S., Hong F., Norris P. J., Stone M., Simmons G., Bagri A., Schreiber M., Buser A., Holbro A., Battegay M., Hosimer P., Noesen C., Milton D. K., Prometheus Study Group, Davies D. H., Contestable P., Corash L. M., Busch M. P., Felgner P. L., Khan S., bioRxiv 2020, 10.1101/2020.04.15.043364. [DOI] [Google Scholar]
- 21. Moradali M. F., Rehm B. H. A., Nat. Rev. Microbiol. 2020, 18, 195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Rehm B. H. A., Biochem. J. 2003, 376, 15.12954080 [Google Scholar]
- 23. Gonzalez‐Miro M., Chen S., Gonzaga Z. J., Evert B., Wibowo D., Rehm B. H. A., Biomacromolecules 2019, 20, 3213. [DOI] [PubMed] [Google Scholar]
- 24. Chen S., Sandford S., Kirman J. R., Rehm B. H. A., FASEB J. 2019, 33, 7505. [DOI] [PubMed] [Google Scholar]
- 25. Chen S., Sandford S., Kirman J., Rehm B. H. A., Adv. Biosyst. 2018, 2, 1800118. [Google Scholar]
- 26. Parlane N. A., Grage K., Mifune J., Basaraba R. J., Wedlock D. N., Rehm B. H. A., Buddle B. M., Clin. Vaccine Immunol. 2012, 19, 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Martínez‐Donato G., Piniella B., Aguilar D., Olivera S., Pérez A., Castañedo Y., Alvarez‐Lajonchere L., Dueñas‐Carrera S., Lee J. W., Burr N., Gonzalez‐Miro M., Rehm B. H. A., Clin. Vaccine Immunol. 2016, 23, 370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Chen S., Quan D. H., Wang X. T., Sandford S., Kirman J. R., Britton W. J., Rehm B. H. A., Nanomaterials 2021, 11, 2060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wong J. X., Gonzalez‐Miro M., Sutherland‐Smith A. J., Rehm B. H. A., Front. Bioeng. Biotechnol. 2020, 8, 44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Evert B., Vezina B., Rehm B. H. A., ACS Appl. Bio Mater. 2020, 3, 8911. [DOI] [PubMed] [Google Scholar]
- 31. Wong J. X., Rehm B. H. A., Biomacromolecules 2018, 19, 4098. [DOI] [PubMed] [Google Scholar]
- 32. Mamat U., Wilke K., Bramhill D., Schromm A. B., Lindner B., Kohl T. A., Corchero J. L., Villaverde A., Schaffer L., Head S. R., Souvignier C., Meredith T. C., Woodard R. W., Microb. Cell Fact. 2015, 14, 57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Thompson T., Rehm B. H. A., Herbert A. B., Saravolac E. G., WO/2012/077080, 2012.
- 34. Rasiah I. A., Rehm B. H. A., Appl. Environ. Microbiol. 2009, 75, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hooks D. O., Rehm B. H. A., Biotechnol. Lett. 2015, 37, 1415. [DOI] [PubMed] [Google Scholar]
- 36. Grifoni A., Sidney J., Zhang Y., Scheuermann R. H., Peters B., Sette A., Cell Host Microbe 2020, 27, 671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Yoshida S., Ono C., Hayashi H., Fukumoto S., Shiraishi S., Tomono K., Arase H., Matsuura Y., Nakagami H., Sci. Rep. 2021, 11, 5934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Amara A. A., Rehm B. H. A., Biochem. J. 2003, 374, 413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. González‐Miró M., Rodríguez‐Noda L. M., Fariñas‐Medina M., Cedré‐Marrero B., Madariaga‐Zarza S., Zayas‐Vignier C., Hernández‐Cedeño M., Kleffmann T., García‐Rivera D., Vérez‐Bencomo V., Rehm B. H. A., Sci. Rep. 2018, 8, 1888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. González‐Miró M., Radecker A.‐M., Rodríguez‐Noda L. M., Fariñas‐Medina M., Zayas‐Vignier C., Hernández‐Cedeño M., Serrano Y., Cardoso F., Santana‐Mederos D., García‐Rivera D., Valdés‐Balbín Y., Vérez‐Bencomo V., Rehm B. H. A., ACS Biomater. Sci. Eng. 2018, 4, 3413. [DOI] [PubMed] [Google Scholar]
- 41. Chan J. F.‐W., Zhang A. J., Yuan S., Poon V. K.‐M., Chan C. C.‐S., Lee A. C.‐Y., Chan W.‐M., Fan Z., Tsoi H.‐W., Wen L., Liang R., Cao J., Chen Y., Tang K., Luo C., Cai J.‐P., Kok K.‐H., Chu H., Chan K.‐H., Sridhar S., Chen Z., Chen H., To K. K.‐W., Yuen K.‐Y., Clin. Infect. Dis. 2020, 71, 2428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Imai H., Uchiumi T., Kodera N., Proc. Natl. Acad. Sci. USA 2020, 117, 32386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Li L., Fierer J. O., Rapoport T. A., Howarth M., J. Mol. Biol. 2014, 426, 309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Peters V., Rehm B. H. A., Appl. Environ. Microbiol. 2006, 72, 1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Jahns A. C., Rehm B. H. A., Appl. Environ. Microbiol. 2009, 75, 5461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Rehm B. H. A., Curr. Opin. Biotechnol. 2017, 48, 42. [DOI] [PubMed] [Google Scholar]
- 47. De Temmerman M.‐L., Rejman J., Demeester J., Irvine D. J., Gander B., De Smedt S. C., Drug Discovery Today 2011, 16, 569. [DOI] [PubMed] [Google Scholar]
- 48. Das A., Ali N., Expert Rev. Vaccines 2021, 20, 1273. [DOI] [PubMed] [Google Scholar]
- 49. Ackerman A. L., Kyritsis C., Tampe R., Cresswell P., Proc. Natl. Acad. Sci. USA 2003, 100, 12889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Cox M. M. J., Hashimoto Y., J. Invertebr. Pathol. 2011, 107, S31. [DOI] [PubMed] [Google Scholar]
- 51. Contreras‐Gómez A., Sánchez‐Mirón A., García‐Camacho F., Molina‐Grima E., Chisti Y., Biotechnol. Prog. 2014, 30, 1. [DOI] [PubMed] [Google Scholar]
- 52. Yang J., Wang W., Chen Z., Lu S., Yang F., Bi Z., Bao L., Mo F., Li X., Huang Y., Hong W., Yang Y., Zhao Y., Ye F., Lin S., Deng W., Chen H., Lei H., Zhang Z., Luo M., Gao H., Zheng Y., Gong Y., Jiang X., Xu Y., Lv Q., Li D., Wang M., Li F., Wang S., Wang G., Yu P., Qu Y., Yang L., Deng H., Tong A., Li J., Wang Z., Yang J., Shen G., Zhao Z., Li Y., Luo J., Liu H., Yu W., Yang M., Xu J., Wang J., Li H., Wang H., Kuang D., Lin P., Hu Z., Guo W., Cheng W., He Y., Song X., Chen C., Xue Z., Yao S., Chen L., Ma X., Chen S., Gou M., Huang W., Wang Y., Fan C., Tian Z., Shi M., Wang F.‐S., Dai L., Wu M., Li G., Wang G., Peng Y., Qian Z., Huang C., Lau J. Y.‐N., Yang Z., Wei Y., Cen X., Peng X., Qin C., Zhang K., Lu G., Wei X., Nature 2020, 586, 572. [DOI] [PubMed] [Google Scholar]
- 53. Tan T. K., Rijal P., Rahikainen R., Keeble A. H., Schimanski L., Hussain S., Harvey R., Hayes J. W. P., Edwards J. C., Mclean R. K., Martini V., Pedrera M., Thakur N., Conceicao C., Dietrich I., Shelton H., Ludi A., Wilsden G., Browning C., Zagrajek A. K., Bialy D., Bhat S., Stevenson‐Leggett P., Hollinghurst P., Tully M., Moffat K., Chiu C., Waters R., Gray A., Azhar M., Mioulet V., Newman J., Asfor A. S., Burman A., Crossley S., Hammond J. A., Tchilian E., Charleston B., Bailey D., Tuthill T. J., Graham S. P., Duyvesteyn H. M. E., Malinauskas T., Huo J., Tree J. A., Buttigieg K. R., Owens R. J., Carroll M. W., Daniels R. S., Mccauley J. W., Stuart D. I., Huang K.‐Y. A., Howarth M., Townsend A. R., Nat. Commun. 2021, 12, 542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Sia S. F., Yan L.‐M., Chin A. W. H., Fung K., Choy K.‐T., Wong A. Y. L., Kaewpreedee P., Perera R. A. P. M., Poon L. L. M., Nicholls J. M., Peiris M., Yen H.‐L., Nature 2020, 583, 834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Lee J. W., Parlane N. A., Wedlock D. N., Rehm B. H. A., Sci. Rep. 2017, 7, 41607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Tostanoski L. H., Wegmann F., Martinot A. J., Loos C., Mcmahan K., Mercado N. B., Yu J., Chan C. N., Bondoc S., Starke C. E., Nekorchuk M., Busman‐Sahay K., Piedra‐Mora C., Wrijil L. M., Ducat S., Custers J., Atyeo C., Fischinger S., Burke J. S., Feldman J., Hauser B. M., Caradonna T. M., Bondzie E. A., Dagotto G., Gebre M. S., Jacob‐Dolan C., Lin Z., Mahrokhian S. H., Nampanya F., Nityanandam R., Pessaint L., Porto M., Ali V., Benetiene D., Tevi K., Andersen H., Lewis M. G., Schmidt A. G., Lauffenburger D. A., Alter G., Estes J. D., Schuitemaker H., Zahn R., Barouch D. H., Nat. Med. 2020, 26, 1694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Yahalom‐Ronen Y., Tamir H., Melamed S., Politi B., Shifman O., Achdout H., Vitner E. B., Israeli O., Milrot E., Stein D., Cohen‐Gihon I., Lazar S., Gutman H., Glinert I., Cherry L., Vagima Y., Lazar S., Weiss S., Ben‐Shmuel A., Avraham R., Puni R., Lupu E., Bar‐David E., Sittner A., Erez N., Zichel R., Mamroud E., Mazor O., Levy H., Laskar O., Yitzhaki S., Shapira S. C., Zvi A., Beth‐Din A., Paran N., Israely T., Nat. Commun. 2020, 11, 6402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Ni L., Ye F., Cheng M.‐L., Feng Y., Deng Y.‐Q., Zhao H., Wei P., Ge J., Gou M., Li X., Sun L., Cao T., Wang P., Zhou C., Zhang R., Liang P., Guo H., Wang X., Qin C.‐F., Chen F., Dong C., Immunity 2020, 52, 971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Adeniyi A. A., Lua L. H.‐L., in Protein Nanotechnology (Eds: Gerrard J., Domigan L.), Methods in Molecular Biology Springer, New York: 2020, pp. 17–37. [Google Scholar]
- 60. Matsuyama S., Nao N., Shirato K., Kawase M., Saito S., Takayama I., Nagata N., Sekizuka T., Katoh H., Kato F., Sakata M., Tahara M., Kutsuna S., Ohmagari N., Kuroda M., Suzuki T., Kageyama T., Takeda M., Proc. Natl. Acad. Sci. USA 2020, 117, 7001. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Data Availability Statement
The data that support the findings of this study are available in the Supporting Information of this article.