

Abstract
Therapeutics for patients hospitalized with coronavirus disease 2019 (COVID‐19) are urgently needed during the pandemic. Bamlanivimab is a potent neutralizing monoclonal antibody that blocks severe acute respiratory syndrome‐coronavirus 2 (SARS‐CoV‐2) attachment and entry into human cells, which could potentially lead to therapeutic benefit. J2W‐MC‐PYAA was a randomized, double‐blind, sponsor unblinded, placebo‐controlled, single ascending dose first‐in‐human trial (NCT04411628) in hospitalized patients with COVID‐19. A total of 24 patients received either placebo or a single dose of bamlanivimab (700 mg, 2,800 mg, or 7,000 mg). The primary objective was assessment of safety and tolerability, including adverse events and serious adverse events, with secondary objectives of pharmacokinetic (PK) and pharmacodynamic analyses. Treatment‐emergent adverse event (TEAE) rates were identical in the placebo and pooled bamlanivimab groups (66.7%). There were no apparent dose‐related increases in the number or severity of TEAEs. There were no serious adverse events or deaths during the study, and no discontinuations due to adverse events. PKs of bamlanivimab is linear and exposure increased proportionally with dose following single i.v. administration. The half‐life was ~ 17 days. These results demonstrate the favorable safety profile of bamlanivimab, and provided the initial critical evaluation of safety, tolerability, and PKs in support of the development of bamlanivimab in several ongoing clinical trials.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
☑ As the pandemic continues, therapeutics for patients with coronavirus disease 2019 (COVID‐19) are urgently needed. Bamlanivimab is a potent neutralizing monoclonal antibody that blocks severe acute respiratory syndrome‐coronavirus 2 (SARS‐CoV‐2) attachment and entry into human cells, which could potentially lead to therapeutic benefit.
WHAT QUESTION DID THIS STUDY ADDRESS?
☑ The first human study of a fully human anti‐spike protein mAb as a potential first‐in‐human therapeutic in hospitalized patients. The study evaluated the safety, tolerability, and pharmacokinetics (PKs) of bamlanivimab at 700 mg, 2,800 mg, and 7,000 mg.
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
☑ A novel fully human mAb against SARS‐CoV‐2 S protein developed within highly accelerated timelines given as an intravenous monotherapy was found to be safe and well‐tolerated. Clinical outcome measures and viral clearance appear to have variable sensitivity to detect a change in the disease course with interventions dependent on how these are measured.
HOW MIGHT THIS CHANGE CLINICAL PHARMACOLOGY OR TRANSLATIONAL SCIENCE?
☑ These results provided the initial critical evaluation of safety, tolerability, PKs, and resistance that supported the continued development of bamlanivimab in several ongoing clinical trials.
Severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2), the causative agent of coronavirus disease 2019 (COVID‐19), has spread rapidly since its emergence in late 2019 resulting in an ongoing pandemic. Although several studies have explored the potential of antivirals (including remdesivir), 1 , 2 , 3 immunomodulators, such as baricitinib, 4 , 5 , 6 glucocorticoids, 7 , 8 and convalescent plasma, 9 , 10 for the treatment of COVID‐19, results have been mixed and outcomes vary depending upon disease severity and patient population. To date, baricitinib alone or in combination with remdesivir, 11 and three mAb therapies (sotrovimab, 12 combination of casirivimab and imdevimab, 13 and bamlanivimab and etesevimab together 14 ) have all received US Food and Drug Administration (FDA) emergency use authorization for the treatment of COVID‐19.
Bamlanivimab is a fully human neutralizing mAb that binds the SARS‐CoV‐2 spike protein, blocking interactions with the ACE2 receptor preventing viral entry into target cells. 15 , 16 This mechanism is hypothesized to result in a clinically important decrease in viral load. The reduction in viral load may also decrease the extent and duration of SARS‐CoV‐2 shedding and therefore transmission, positively impacting public health. Bamlanivimab was identified using a high‐throughput microfluidic screening of antigen‐specific B‐cells from a blood sample obtained from a patient who recovered from COVID‐19 early in the pandemic. In a non‐human primate model of COVID‐19, bamlanivimab was shown to provide protection in both the lower and upper respiratory tract against infection with SARS‐CoV‐2. 16 With a global pandemic underway, an accelerated path from selection of bamlanivimab as a clinical candidate to the first‐in‐human study was enabled in less than 2 months through innovative manufacture, nonclinical safety evaluation, and in silico model‐based approaches to clinical dose selection. These approaches were critical to the initiation of the world’s first clinical study of a neutralizing antibody for treatment of COVID‐19. This first‐in‐human study was initiated only 3 months from initiation of antibody discovery, with the timeline dramatically shortened compared with the typical duration of several years. Furthermore, the first patient was dosed on May 29, 2020, within hours of protocol approval through innovative clinical trial operational logistics.
Here, we report the safety, tolerability, pharmacokinetics (PKs), clinical course, and viral dynamic results of the first‐in‐human study of bamlanivimab in hospitalized patients with moderate to severe COVID‐19.
METHODS
Study design and participants
Study J2W‐MC‐PYAA was a randomized, investigator and patient‐blind, sponsor unblinded, placebo‐controlled, single ascending dose first‐in‐human trial (NCT04411628; see Figure S1 for study design and Figure S2 for consort diagram) conducted at five sites in the United States (see Supplementary Material for complete list of study sites). Patients were enrolled from May 29, 2020, to June 28, 2020. The study consisted of 3 cohorts recruited sequentially corresponding to ascending single i.v. doses of bamlanivimab of 700 mg, 2,800 mg, and 7,000 mg. The dose of 700 mg was selected as the maximum therapeutic dose based on 2 in silico approaches—a physiologically‐based PK model for mAbs and a viral dynamic PK/pharmacodynamic (PK/PD) model. 17 The viral dynamic model was developed based on data from patients with COVID‐19 early in the pandemic, 18 whereas the physiologically‐based PK model model was developed based on published literature incorporating mAb distribution kinetics in humans. 19 Higher doses were selected to evaluate the limit of safety and tolerability.
Eligible patients were 18–85 years of age who were hospitalized or in the process of being admitted to a hospital with laboratory confirmed COVID‐19 infection. Patients were diagnosed as having moderate to severe COVID‐19 based on FDA guidelines. Initial laboratory confirmation of COVID‐19 was required ≤ 14 days at the time of randomization.
Patients were not eligible if they had a medical condition requiring or anticipating impending need of invasive mechanical ventilation; had a saturation of peripheral oxygen < 88% while breathing room air at rest. Patients on supplemental oxygen were permitted in the trial; see Supplementary Material for complete list of inclusion and exclusion criteria.
This study was compliant with the International Conference on Harmonization guideline on Good Clinical Practice. All informed consent forms and protocols were approved by appropriate ethical review boards prior to initiation of the study. All patients gave written informed consent prior to receiving the study drug.
Randomization and blinding
Sentinel dosing was undertaken for each dose level, where the first 2 participants in each cohort were randomized 1:1 to bamlanivimab and placebo. Following a safety and tolerability review at 24 hours postdosing, subsequent participants were randomized to the remaining treatment allocations, 5 to bamlanivimab and 1 to placebo. Enrollment was planned for 24 patients in total, with additional participants added as required to ensure a minimum of 8 participants in each cohort completed 7 days of study assessments, up to a maximum of 3 additional participants per cohort.
See Supplementary Material for the determination of the sample size. The anticipated number of participants per treatment provided > 80% probability within each treatment arm of observing safety events of reasonable prevalence (≥ 25%) to support the primary objective of safety assessment.
Study pharmacists who were not involved in any other study‐related procedures were unblinded at the study site for investigational product preparation. Assignment to study intervention was determined using an interactive web‐response system, and the blinded site staff were responsible for administering the study drug to the patients.
Nonclinical safety consideration
Bamlanivimab is a fully human mAb, derived from human B‐cells. As a consequence, complementarity determining regions in bamlanivimab have undergone natural positive selection for spike protein binding, and negative selection against self‐cross‐reactivity in vivo and were not expected to have off‐target binding. This first‐in‐human study in hospitalized patients with COVID‐19 was supported by studies assessing in vitro viral neutralization combined with a single‐dose non‐human primate PK study. 16 The nonclinical safety package to support subsequent studies included tissue cross‐reactivity studies on human, rat, and monkey tissues and an in vivo toxicology study.
Procedures
A complete physical examination was conducted at the screening visit, and one of three bamlanivimab doses or placebo was administered i.v. at the baseline visit (D1) with infusion rates as follows: bamlanivimab 700 mg (50 mL) administered 100 mL/hr for 30 minutes; 2,800 mg (75 mL) administered 100 mL/hr for 45 minutes; and 7,000 mg administered 100 mL/hr for 60 minutes. Placebo infusion was 0.9% normal saline, administered at the same volume and rate as the corresponding bamlanivimab dose cohort. Safety and tolerability were reviewed for sentinel participants up to 24 hours after dosing, at which point the investigators and sponsor team determined whether safety and tolerability were acceptable to continue with dosing subsequent participants. The decision to dose the next cohort was made when all participants from the previous cohort had been dosed and safety data assessed for at least 4 days after the i.v. infusion by the investigators and sponsor team in consultation with an independent safety assessment committee.
Blood samples were obtained for assessment of exploratory biomarkers, serology, clinical laboratory assessments, and PD on days 1, 3, 7, 11, 15, 22, and 29, and every 7 days until discharge or day 60, if still an inpatient. Samples for the virology assay were collected using nasopharyngeal (preferred) or mid‐turbinate methods. Blood samples obtained for PK analyses were assessed on days 1 (pre‐infusion and just before end of infusion), 4, 15, and 29, and follow‐up visits up to day 60.
Symptom‐related physical examinations and assessments of clinical symptoms were performed on days 1, 2, 3, 4, 7, 11, 15, 22, and 29, and the day of discharge from the hospital, with additional follow‐up examinations every 7 days thereafter until day 60 if the patient was not discharged from the hospital by day 29. Participants’ clinical status and concurrent procedures of special interest were recorded, including limitation on activities due to COVID‐19 and any requirements for the following procedures of special interest, such as ongoing hospital medical care, supplemental oxygen, noninvasive ventilation or a high flow oxygen device, mechanical ventilation, extracorporeal membrane oxygenation, additional organ support, or consciousness status using alert, consciousness, verbal, pain, unresponsive scale. Hospitalization events were recorded, including dates of hospital admission and discharge, admission to the intensive care unit (ICU), discharge from the ICU, and discharge location including to an extended care facility or home.
Outcomes
The primary objective was to assess safety and tolerability, including adverse events (AEs), serious adverse events (SAEs), and discontinuations due to AEs. Other objectives included PKs (e.g., mean concentration at day 29); PD viral load, area under the response time curve (AUC, from day 1 to day 29), and change from baseline time course; total symptom score; time to symptom resolution; duration of hospitalization; National Institute of Allergy and Infectious Diseases (NIAID), World Health Organization (WHO) scales, and National Early Warning Score (NEWS2); and analysis of viral resistance. Any hypothesis tests were conducted for treatment comparisons without adjustment for multiplicity except for the evaluation of treatment effect on endogenous antibody titers. Success for the trial was claimed using a Bayesian criterion if any of the 3 bamlanivimab doses had at least 60% probability to reduce at least 30% mean AUC (28‐day viral load) over placebo.
PD assays
PD samples collected by either nasopharyngeal swab or mid‐turbinate swab were tested for presence of SARS‐CoV‐2. The SARS‐CoV‐2 viral load was derived from the cycle threshold (Ct) values determined using a reverse transcription polymerase chain reaction assay of viral genes N1 and N2 20 ; Ct values ranged between 0 and 45, where higher Ct values indicated a lower viral load. 21 Where Ct was determined (a positive test result), viral load was derived as follows:
Where Cutoff is 45 for statistical analysis (other values, such as 40 for PK/PD analysis were explored); i = 1 or 2. Ct for N1 was used as the primary measure; Ct for N2 was only used when Ct for N1 was not available. Viral load results are presented in log base 10 scale.
Viral clearance was defined as two consecutive negative SARS‐CoV‐2 results in the statistical analysis. When deriving clearance, any missing data from the nasopharyngeal or mid‐turbinate swabs was set to positive. Time to SARS‐CoV‐2 clearance was defined as the earliest timepoint of the two consecutive negative results. Another approach was also used to estimate viral clearance using a longitudinal time course model of viral dynamics, where time to viral clearance was defined as the first time a viral load of zero was predicted. 17 Potential viral resistance was determined using standard methods to sequence all viral samples and assess for genetic variants with predicted resistance to bamlanivimab. Further details are provided in the Supplementary Material .
PK assay
Serum concentrations of bamlanivimab were determined using a validated hybrid liquid chromatography‐tandem accurate mass spectrometry method. See Supplementary Material for further information.
Surrogate virus neutralization assay
The neutralization capacity of bamlanivimab in study subject serum samples was determined by SARS‐CoV‐2 Surrogate Virus Neutralization Assay (sVNA), an enzyme‐linked immunosorbent assay that uses chromogenic substrates for quantitative detection of neutralizing antibodies to SARS‐CoV‐2 in human serum. 22 Further details are in the Supplementary Material .
Serology
Endogenous antibodies directed against multiple SARS‐CoV‐2 proteins were determined in subject samples using a Luminex‐based assay. Further details are in the Supplementary Material .
Statistical analysis
All participants who were randomly assigned and received study intervention (either placebo or bamlanivimab), were included in safety and tolerability assessments. The PK population consisted of all randomized participants who received study intervention (bamlanivimab) and had at least one evaluable PK sample, and the PD population consisted of all randomized participants who received study intervention (either placebo or bamlanivimab) and provided at least one baseline and one post‐baseline measure for the relevant end point.
For continuous data, summary statistics included the mean, SD, median, minimum, maximum, and number; for log‐normal data (e.g., PK parameters), the geometric mean and geometric coefficient of variation were presented. For categorical and ordinal data, frequency count and percentages were presented. Changes from baseline in SARS‐CoV‐2 viral load data (in log base 10 scale) were analyzed using a mixed model for repeated measures, which contained baseline as a covariate and treatment, day, and treatment‐by‐day interaction as fixed effects. Data listings were provided for all participants up to the point of study completion or withdrawal, with any participants excluded from the relevant population highlighted. Summary statistics and statistical analyses were only performed for participants included in the relevant analysis population. For the calculation of summary statistics and statistical analysis, unrounded data were used.
Descriptive statistics were used to summarize differences in demographic and baseline disease characteristics.
This study is registered with ClinicalTrials.gov, number NCT04411628.
RESULTS
Patients
Twenty‐six patients were randomized to receive a single i.v. dose of placebo or one of the bamlanivimab doses (700 mg, 2,800 mg, or 7,000 mg), with 24 receiving treatment, in a 1:1 ratio for the sentinel dosing followed by a 1:5 ratio for the remaining patients (8 per cohort). Overall, patient demographics and baseline disease characteristics were similar, and no formal statistical assessments were made among groups. However, the placebo group had a numerically greater number of patients with moderate COVID‐19 at baseline, shorter time since symptom development, lower mean age, and lower viral load. All 3 patients with critical COVID‐19 per FDA guidance 23 were in the 7,000 mg bamlanivimab group (Table 1 ). The median time from symptom onset ranged from 7.5 (range 5–13) days in the placebo group to 11.0 (range 5–25) days in the 7,000 mg bamlanivimab group. The average age ranged from 43.2 years in the placebo group to 66.7 years in the 7,000 mg bamlanivimab group. Two‐thirds of the patients in each group were men except for the 7,000 mg bamlanivimab group, which had 1 man. Patient demographics and comorbidities are listed in Table 1 . Weight was not collected during this study as patients were bedridden and in isolation.
Table 1.
Baseline demographics and disease characteristics
| Mean (SD) unless otherwise specified | Treatment groups | |||
|---|---|---|---|---|
| Bamlanivimab | ||||
| Placebo (N = 6) | 700 mg (N = 6) | 2,800 mg (N = 6) | 7,000 mg (N = 6) | |
| Age, years | 43.2 (8.6) | 57.2 (6.8) | 48.5 (12.1) | 66.7 (6.7) |
| Male sex, n (%) | 4 (66.7) | 4 (66.7) | 4 (66.7) | 1 (16.7) |
| Race, n (%) | ||||
| American Indian or Alaska Native | 0 | 0 | 0 | 0 |
| Asian | 0 | 0 | 0 | 1 (16.7) |
| Black or African American | 0 | 1 (16.7) | 1 (16.7) | 0 |
| Native Hawaiian or Pacific Islander | 0 | 0 | 0 | 0 |
| White | 5 (83.3) | 4 (66.7) | 5 (83.3) | 5 (83.3) |
| Unknown | 1 (16.7) | 1 (16.7) | 0 | 0 |
| Hispanic or Latino, n (%) | 5 (83.3) | 4 (66.7) | 3 (50.0) | 3 (50.0) |
| Tobacco use, n (%) | ||||
| Current | 0 | 0 | 0 | 0 |
| Former | 1 (16.7) | 2 (33.3) | 0 | 3 (50.0) |
| Never | 5 (83.3) | 4 (66.7) | 6 (100.0) | 3 (50.0) |
| Time from COVID‐19 symptom onset, median days (range) | 7.5 (5‐13) | 8.5 (5‐24) | 8.5 (2‐16) | 11.0 (5‐25) |
| Viral load, log10 scale (SD) | 3.80 (2.0) | 4.51 (2.5) | 5.83 (2.1) | 5.17 (2.3) |
| COVID‐19 severity, n (%) | ||||
| Asymptomatic | 0 | 0 | 0 | 0 |
| Mild | 0 | 0 | 0 | 0 |
| Moderate | 4 (66.7) | 1 (16.7) | 3 (50.0) | 1 (16.7) |
| Severe | 1 (16.7) | 5 (83.3) | 3 (50.0) | 2 (33.3) |
| Critical | 1 (16.7) | 0 | 0 | 3 (50.0) |
| Pre‐existing conditions, n (%) | ||||
| Asthma | 0 | 1 (16.7) | 2 (33.3) | 3 (50.0) |
| Obesity | 1 (16.7) | 0 | 0 | 1 (16.7) |
| Hyperlipidemia | 1 (16.7) | 1 (16.7) | 2 (33.3) | 2 (33.3) |
| Hypertension | 1 (16.7) | 3 (50.0) | 1 (16.7) | 3 (50.0) |
| Diabetes (type 2) | 1 (16.7) | 2 (33.3) | 2 (33.3) | 1 (16.7) |
| Coronary artery disease | 1 (16.7) | 1 (16.7) | 1 (16.7) | 0 |
| End‐stage renal disease | 1 (16.7) | 0 | 0 | 0 |
| HIV infection | 0 | 0 | 1 (16.7) | 0 |
| Concomitant medications, n (%) | ||||
| Remdesivir | 2 (33.3) | 3 (50.0) | 4 (66.7) | 3 (50.0) |
| Dexamethasone | 1 (16.7) | 0 | 1 (16.7) | 2 (33.3) |
| Antibiotics | 5 (83.3) | 5 (83.3) | 5 (83.3) | 4 (66.7) |
| All steroids | 1 (16.7) | 1 (16.7) | 2 (33.3) | 3 (50.0) |
| Pre‐specified prior medications, n (%) | ||||
| NSAIDs | 1 (16.7) | 0 | 0 | 0 |
| Antivirals (including remdesivir) | 1 (16.7) | 0 | 0 | 1 (16.7) |
| Antibiotics | 3 (50.0) | 5 (83.3) | 3 (50.0) | 3 (50.0) |
| Anti‐malarials | 0 | 0 | 0 | 0 |
| Corticosteroids (including dexamethasone) | 0 | 1 (16.7) | 1 (16.7) | 2 (33.3) |
COVID‐19, coronavirus disease 2019; NSAIDs, nonsteroidal anti‐inflammatory drugs.
Safety and tolerability
The primary objective for this first‐in‐human study was to determine safety and tolerability of bamlanivimab vs. placebo. The number of patients with treatment emergent adverse events (TEAEs) was similar across all groups, with 4, 5, 5, and 2 TEAEs in the placebo, 700 mg bamlanivimab, 2,800 mg bamlanivimab, and 7,000 mg bamlanivimab groups, respectively, and the overall frequency of TEAEs in the combined bamlanivimab groups was the same as placebo (Table 2 ). The most frequent TEAEs included non‐cardiac chest pain (2 in the placebo group, and 1 in the 700 mg bamlanivimab group), fatigue (1 each in the 700 mg and 2,800 mg bamlanivimab groups), headache (1 each in the placebo and 700 mg bamlanivimab groups), leukopenia (1 each in the placebo and 700 mg bamlanivimab groups), respiratory distress (1 each in the 700 mg and 2,800 mg bamlanivimab groups), and chest discomfort (2 in the 7,000 mg bamlanivimab group). All TEAEs resolved by the end of the study, except three reported by three participants that were judged by the investigator as unrelated to study treatment: hypertension related to study disease, bronchial hyperreactivity related to study disease, and hypercholesterolemia related to other medical condition. There were no apparent dose‐related increases in the number or severity of TEAEs. No patients developed SAEs, and there were no discontinuations due to AEs (Table 2 ). There were no hypersensitivities, deaths, nor infusion site reactions observed in this study.
Table 2.
Safety results
| Treatment groups | ||||
|---|---|---|---|---|
| Placebo (N = 6) | Bamlanivimab | |||
| 700 mg (N = 6) | 2,800 mg (N = 6) | 7,000 mg (N = 6) | ||
| TEAE, a n (%) | 4 (66.7) | 5 (83.3) | 5 (83.3) | 2 (33.3) |
| SAE, b n (%) | 0 | 0 | 0 | 0 |
| Discontinuations due to AE, n (%) | 0 | 0 | 0 | 0 |
| Most common TEAEs, n (%) (decreasing frequency c ) | ||||
| Non‐cardiac chest pain | 2 (33.3) | 1 (16.7) | 0 | 0 |
| Fatigue | 0 | 1 (16.7) | 1 (16.7) | 0 |
| Headache | 1 (16.7) | 1 (16.7) | 0 | 0 |
| Leukopenia | 1 (16.7) | 1 (16.7) | 0 | 0 |
| Respiratory distress | 0 | 1 (16.7) | 1 (16.7) | 0 |
| Chest discomfort | 0 | 0 | 2 (33.3) | 0 |
AE, adverse event; SAE, serious adverse event; TEAE, treatment‐emegent adverse event.
TEAE: number (%) of patients with treatment‐emergent adverse event.
SAE: number (%) of patients with serious adverse event.
Most common TEAEs as percentage of total population; all TEAEs with total frequency across all groups ≥ 5% listed.
Pharmacokinetics
The PKs of bamlanivimab are linear and exposure increased proportionally with dose following single i.v. doses (Figure 1a ). Specifically, following an ~ 30‐minute 700 mg, 45‐minute 2,800 mg, or 60‐minute 7,000 mg i.v. infusion, the median maximum concentration at the end of infusion was estimated to be 214 µg/mL (90% confidence interval (CI): 118 to 402 µg/mL), 856 µg/mL (90% CI: 461 to 1598 µg/mL), and 2,103 µg/mL (90% CI: 1,112 to 3,903 µg/mL), respectively (Figure 1b , Table 3 ). The PK profiles of all doses remained above the 90% inhibitory concentration for all individuals up to the final PK sampling timepoint at day 29. The bamlanivimab half‐life was ~ 17 days (Table 3 ). The mean concentration of bamlanivimab by day 29 for 700 mg was 33.6 µg/mL (Figure 1a ), > 8‐fold above the 95th percentile of the in vitro estimated 90% inhibitory concentration (4.1 µg/mL) of SARS‐CoV‐2 neutralization.
Figure 1.
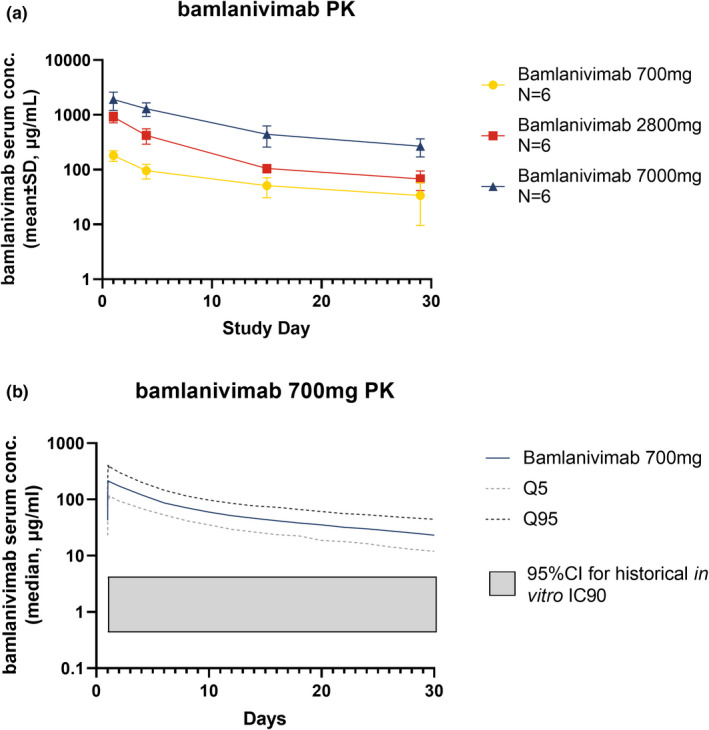
Pharmacokinetics (PKs) of bamlanivimab. (a) Mean bamlanivimab serum concentration over time after a single intravenous dose. Error bars represent SD. (b) Detailed analysis of 700 mg bamlanivimab PKs. The PK profiles of all doses remained above the 95% confidence interval (CI) for historical in vitro 90% inhibitory concentration (IC90).
Table 3.
Summary of bamlanivimab pharmacokinetic parameters
| Predicted concentration | Median | 5th and 95th prediction interval |
|---|---|---|
| End of infusion (µg/mL) | ||
| 700 mg | 214 | 118, 402 |
| 2,800 mg | 856 | 461, 1598 |
| 7,000 mg | 2103 | 1112, 3903 |
| Day 29 (µg/mL) | ||
| 700 mg | 23.2 | 12.0, 44.3 |
| 2,800 mg | 92.8 | 48.1, 177 |
| 7,000 mg | 232 | 120, 444 |
| Post hoc parameter | Geometric mean | Geometric CV% |
|---|---|---|
| CL (L/d) | 0.291 | 33.3 |
| VSS (L) | 6.01 | 11.2 |
| t1/2 (d) | 17.2 | 27.2 |
CL, systemic clearance; CV, coefficient of variation; d, days; t1/2, terminal phase half‐life; Vss, steady‐state volume of distribution.
Viral end points
At day 29, all bamlanivimab groups had a numerically greater change from baseline in least squares mean log base 10 viral load compared to placebo (Δ = −0.11, −0.52, and −0.10 from placebo in the 700 mg, 2,800 mg, and 7,000 mg bamlanivimab groups, respectively; Figure 2a ); however, no statistically significant difference compared with placebo was detected (P = 0.90, P = 0.55, and P = 0.91, respectively). None of the three bamlanivimab doses met the predefined Bayesian criteria: having at least 60% probability to reduce at least 30% mean AUC over placebo. At day 29, viral clearance was achieved in 2 of 6 patients (33.3%) of the placebo group, 3 of 6 patients (50%) of the 700 mg bamlanivimab group, 2 of 6 patients (33.3%) of the 2,800 mg bamlanivimab group, and 3 of 6 patients (50%) of the 7,000 mg bamlanivimab group and no statistical significance (Fisher’s exact P > 0.9) was achieved among the treatments. Based on the longitudinal viral dynamic model, developed based on a model available on a preprint server early in the pandemic, 18 66 to 83% of patients achieved viral clearance by day 29 in all 3 groups, but no difference from placebo was expected due to the small sample size (Figure 2b ).
Figure 2.
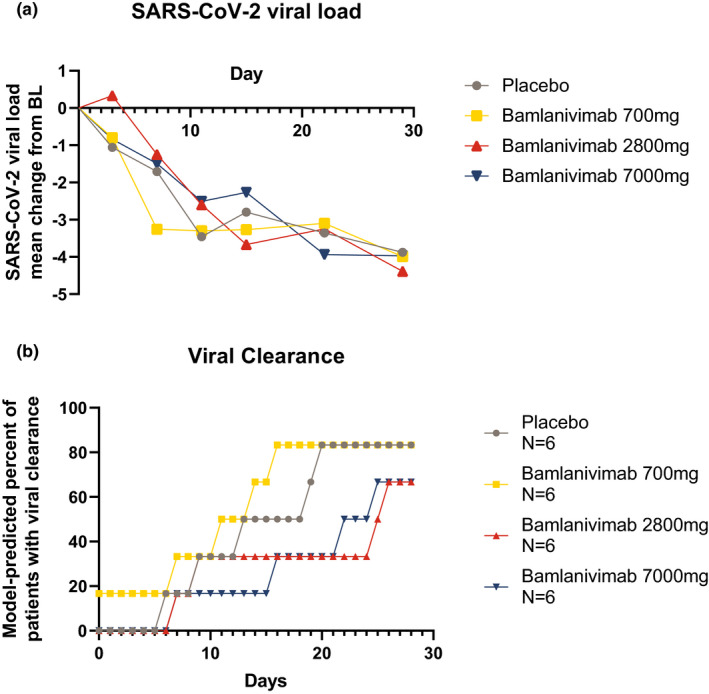
Pharmacodynamics of bamlanivimab. (a) Mean change from baseline (BL) in severe acute respiratory syndrome‐coronavirus 2 (SARS‐CoV‐2) viral load over time. (b) Model‐predicted percentage of patients with SARS‐CoV‐2 clearance.
Baseline SARS‐CoV‐2 viral loads in log base 10 scale and in Ct scale correlated with days since COVID‐19 symptom onset (Figure 2 , Figure S3 ), with higher baseline viral loads in those patients with a shorter time since symptom onset (R 2 = −0.5739, P = 0.0034). The treatment effect on change from baseline viral load also correlated to days from symptom onset, with higher reduction in patients with shorter time from symptom onset.
There was only one patient, in the 2,800 mg bamlanivimab group, who had acquired viral resistance (as defined by ≥ 20% allelic frequency compared to the Wuhan reference sequence). The infusion was well‐tolerated. Apart from a fever present predose and during day 1 peaking at 38.3ºC, the subject had unremarkable vital signs and did not require supplemental oxygen during the hospitalization. The subject was discharged 2 days after the infusion with no new concerns. The subject discontinued after withdrawing consent 38 days after study treatment infusion and did well clinically after discharge, but his withdrawal did not allow subsequent evaluation of the resistant variant.
Serology and biomarkers
All but one subject developed endogenous antibodies to SARS‐CoV‐2 (Figure 3a,b ). Antibody titers increased across the 3 treatment groups and placebo, with maximum titers reached by 7 days after baseline, or ~ 2 weeks after symptom onset. The production of these endogenous antibody titers to SARS‐CoV‐2 NTD or NCP proteins did not appear to be influenced by the administration of bamlanivimab treatment and there was no significant difference among the groups (P > 0.3). The assay for antibody IgG titers against SARS‐CoV‐2 RBD detected endogenous IgG, as shown in the placebo group, as well as bamlanivimab (Figure 3c ). Bamlanivimab at 700 mg provided a sustained IgG titer ~ 7‐fold and 3‐fold over placebo on days 7 and 29, respectively.
Figure 3.
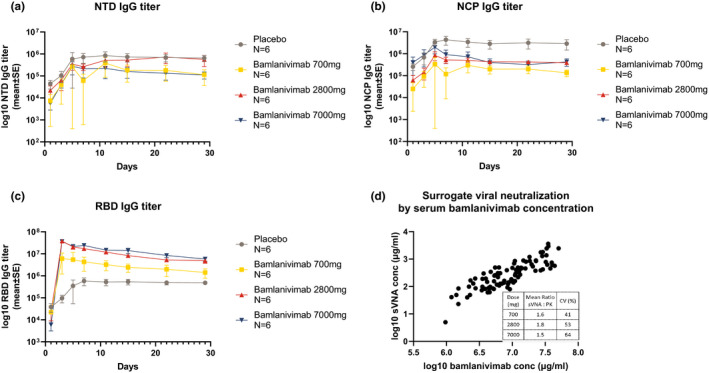
Serum biomarkers. Log10 fold change from baseline in serum concentration of antibodies against severe acute respiratory syndrome‐coronavirus 2 (SARS‐CoV‐2) (a) spike protein N‐terminal domain (NTD) and (b) nucleocapsid protein (NCP). (c) Coronavirus disease 2019 (COVID‐19) receptor binding domain (RBD) antibody titer by bamlanivimab dose. (d) Correlation of surrogate viral neutralization assay with bamlanivimab serum concentration; table inset shows ratios by bamlanivimab dose. a‐c Data presented are mean ± SE.
In an sVNA, neutralization activity of serum samples from patients treated with bamlanivimab correlated well with bamlanivimab PK concentration (R 2 = 0.831), with an sVNA:bamlanivimab concentration ratio > 1 at all doses (Figure 3d ).
Symptom outcomes
The median time to symptom resolution decreased with bamlanivimab treatment in a dose‐dependent manner (Figure 4a ). The mean symptom score changes from baseline decreased in the placebo group and the 700 mg bamlanivimab group initially but then plateaued approximately after day 11, whereas in the 2,800 mg bamlanivimab group, it showed a steady decrease from day 15 to day 29 and decreased in the 7,000 mg group until day 15. There was no formal statistical assessment among any of the groups (Figure 4b , Table S1 ). There were no differences or patterns in the number of days hospitalized or the time to discharge among treatment groups (Figure 4c‐d ). NIAID Ordinal Scales, WHO Ordinal Scales, and NEWS2 were similar across groups at all timepoints (Table S2‐S4 ). No patients required the use of a ventilator during the study, and one patient (2,800 mg bamlanivimab group) was admitted to the ICU; (data not shown). This patient was admitted to the ICU ~ 14 hours postdose on day 1 because of COVID‐19 and remained in the ICU for ~ 5 days. The patient had a TEAE of grade 3 (severe) respiratory distress that started ~ 11 hours postdose on day 1 and resolved after ~ 6 days.
Figure 4.
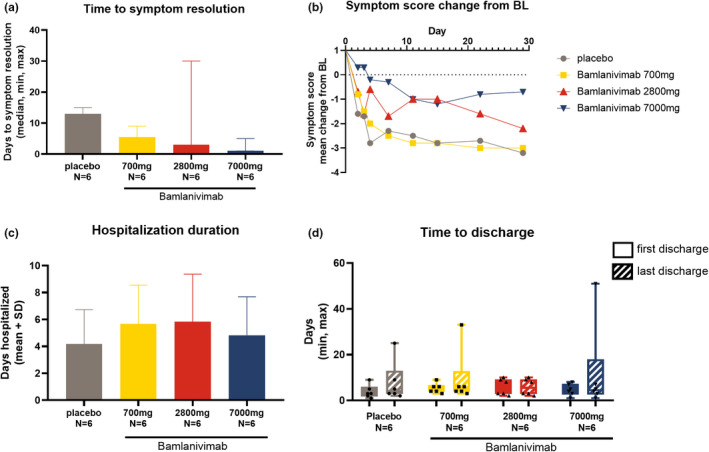
Clinical outcomes. (a) Median days to symptom resolution. Symptom resolution is defined as all symptoms on the symptom questionnaire scored as absent. Error bars represent the minimum and maximum values. (b) Change from baseline in coronavirus disease 2019 (COVID‐19) symptom score. (c) Duration of hospitalization (for COVID‐19). Data presented are mean days + SD. (d) Box plot of time to hospital discharge. Error bars represent minimum and maximum vales. Solid bars = first hospital discharge, striped bars = last hospital discharge. Patients with > 1 hospitalization are: 1 placebo, one 700 mg bamlanivimab, zero 2,800 mg bamlanivimab, and one 7,000 mg bamlanivimab. In the 2,800 mg group first discharge = last discharge as no patients had multiple hospitalizations.
DISCUSSION
In this first‐in‐human study, there were no SAEs or discontinuations due to AEs, and TEAEs in the placebo and pooled bamlanivimab groups were similar. The linear, dose proportional PKs demonstrated by bamlanivimab were consistent with a typical mAb administered at a high dose. The in vitro neutralization activity of serum bamlanivimab correlated well with serum PK concentration, indicating that circulating concentrations of bamlanivimab are active mAb. Development of natural immunity through formation of endogenous antibodies against the SARS‐CoV‐2 nucleocapsid and the N‐terminal domain of the spike protein was not obviously affected by administration of bamlanivimab in this small study. This development of natural immunity may lead to a memory response that enables the immune system to respond to future encounters with SARS‐CoV‐2. Conclusions regarding viral load reduction and efficacy were limited by the small sample size inherent to a phase I first‐in‐human trial. The study was not powered to find differences; therefore, as expected, only numerical differences were observed and results must be interpreted in this context. Therefore, in patients who were hospitalized, the lack of separation from placebo depended on three key variables: the potentially higher viral load in the “high risk” population to hospitalization, the lag to treatment administration, and the small sample size.
COVID‐19 is typically associated with viral shedding lasting 5 to 14 days, with milder cases associated with durations of viral shedding at the lower end of this range and lower viral loads than severe cases. 24 , 25 Viral load generally starts to decrease within the first week after symptom onset. 26 There is also evidence that baseline viral load may independently predict clinical outcomes of mortality and intubation. 27 However, there are also datasets showing that although severe disease tends to have higher viral load than mild disease in the COVID‐19 positive population as a whole, viral load may not differ by disease severity as much among hospitalized patients. 28 Later in the disease course, particularly with severe disease, excessive inflammatory responses or immune dysfunction have a greater impact on disease progression than viral load. 29 , 30 , 31 , 32 Moreover, molecular presence of SARS‐CoV‐2 does not guarantee infectivity, and measures of viral load and viral clearance may also be impacted by collection method and definition of clearance. Nevertheless, the observed correlation between time from symptom onset and change from baseline viral load following bamlanivimab is consistent with the expectation that the earlier the mAb administration relative to symptom onset, the greater the drug effect on viral load reduction (Figure S3 ). Patients who are hospitalized, and likely delayed in drug administration from symptom onset, may not benefit from mAb therapy.
An interim analysis of the ongoing phase II/III BLAZE‐1 study provided evidence of reduced hospitalizations in mild to moderate ambulatory patients with COVID‐19, treated with bamlanivimab, 17 resulting in an emergency use authorization for bamlanivimab alone by the FDA, 33 which was later revoked to complete transition to bamlanivimab together with etesevimab in the United States. 34 The phase II portion of BLAZE‐1 also showed a statistically significant difference in symptom improvement from baseline to day 11 in patients treated with bamlanivimab monotherapy. 35 Subsequently, data from the phase III portion of BLAZE‐1 in ambulatory patients confirmed the reduction in hospitalizations seen with bamlanivimab and etesevimab in the phase II trial. 35 , 36 In the current study, there was no difference in hospitalizations, time to discharge, or ordinal scale outcomes in individuals treated with bamlanivimab compared with those that received placebo. There are several potential reasons for these findings. First, being that this was a small phase I safety study that was not statistically powered to evaluate clinical outcome measures. Additionally, the patient population in this study, where bamlanivimab groups at baseline had COVID‐19 for a median of up to 11 days, was much further along in disease course than in BLAZE‐1 in which patients had tested positive for COVID‐19 a median of 4 days prior to treatment. Even after accounting for these factors, it may be that neutralization of viral entry into host cells in hospitalized patients does not provide clinical benefit in a population of patients hospitalized because of COVID‐19. Indeed, recent interim analyses from the National Institute of Allergy and Infectious Diseases platform trial (ACTIV‐3), of investigational treatments in hospitalized patients with COVID‐19 (without end organ failure), showed no clinical benefit with a number of mAbs, including bamlanivimab, and further evaluation for these agents, in this trial, was halted based on recommendations by the independent Data and Safety Monitoring Board. 37 , 38 , 39
Although no formal statistical analysis was conducted on symptom score change from baseline or duration of hospitalization in this small size study, a numerically faster resolution of patient‐reported symptoms in the bamlanivimab groups compared with placebo was observed. The differing responses for the 2,800 mg and 7,000 mg groups regarding mean symptom score change from baseline may be because these groups had a lower symptom score at baseline, as compared with the other groups. This highlights the challenges inherent in determining the best clinical outcome measure for treatment of COVID‐19.
Mechanisms of antiviral resistance have been widely studied in chronic infections, such as HIV, hepatitis C virus, and hepatitis B virus. The clinical significance of resistance is less clear in the setting of an acute infection, in which it is sufficient for a therapeutic to reduce viral load enough for the host immune system to effectively clear the virus. The concern with the use of single agents targeting viruses is the development of viral escape mutants that are resistant to the therapeutic; the risk of escape mutant development may be reduced if at least two antiviral agents are used, if there is adequate therapeutic exposure, or if the agents bind non‐overlapping epitopes of the target protein. 40 The current study identified a single patient that developed a potential viral escape mutation; this patient did well clinically.
These results demonstrate the favorable safety profile of bamlanivimab, and provided the initial critical evaluation of safety, tolerability, PKs, and resistance that supported the continued development of bamlanivimab in ongoing clinical trials, including BLAZE‐1.
FUNDING
This trial was fully funded by Eli Lilly and Company.
CONFLICT OF INTEREST
P.C. reports grants from Eli Lilly and Company, Gilead, and the National Institutes of Health (NIH), and scientific advisory board member for Eli Lilly and Company, Gilead, Rigel Pharmaceuticals, and Humanigen. N.R. reports grants from Eli Lilly and Company, Merck, Pfizer, Quidel, and Sanofi‐Pasteur. M.J.M. has received research grants from Eli Lilly and Company, Pfizer, and Sanofi, and serves on advisory boards for Pfizer and Meissa Vaccines. W.F. reports research funding provided to UNC from Ridgeback Biopharmaceuticals, Eli Lilly and Company, and Regeneron, consultancy fees from Merck and Roche and served on adjudication committee for Janssen and Syneos. M.D. reports personal fees from Genentech‐Roche, Partner Therapeutics, Tillots Pharma, ORIC Pharmaceuticals, Moderna, AzurRx, SQZ, and WebMD, grants from Novartis and Eli Lilly and Company, and is a scientific advisory board member for Neoleukin Therapeutics. A.S. is an advisory board member for Eli Lilly and Company. G.D., Y.G.L., J.C., K.P., E.C., P.B.A., J.P., J.F., R.J.B., P.K., A.N., and C.B. are employees and shareholders of Eli Lilly and Company. All other authors declared no competing interests for this work.
AUTHOR CONTRIBUTIONS
All authors contributed to the writing of the manuscript. A.N., P.K., C.B., G.D., J.F., M.D., K.P., R.J.B., and J.C. designed the research. G.D., P.B.A., M.M., A.S., N.R., A.K., P.C., M.D., J.P., J.F., and W.F. performed the research. P.B.A., Y.G.L., E.C., P.K., C.B., G.D., R.J.B., J.F., J.P., J.C., K.P., and M.D. analyzed the data.
DATA AVAILABILITY STATEMENT
Lilly provides access to all individual participant data collected during the trial, after anonymization, with the exception of pharmacokinetic or genetic data. Data are available to request in a timely fashion after the indication studied has been approved in the United States and the European Union and after primary publication acceptance. No expiration date of data requests is currently set once they are made available. Access is provided after a proposal has been approved by an independent review committee identified for this purpose and after receipt of a signed data sharing agreement. Data and documents, including the study protocol, statistical analysis plan, clinical study report, blank or annotated case report forms, will be provided in a secure data sharing environment for up to 2 years per proposal. For details on submitting a request, see the instructions provided at www.clinicalstudydatarequest.com.
Supporting information
Supplementary Material.
ACKNOWLEDGMENTS
The authors would like to thank the study participants and site investigators (full list provided in the Supplementary Material ); AbCellera for their early work on bamlanivimab; and the following employees of Eli Lilly and Company: Linden Green and Holly Green for providing writing and editorial support; Amanda Long, Kallin Carter, Lisa O’Brien, Lisa Ferguson‐Sells, Andrew Schade, Veavi Chang, Richard Higgs, Philip Ebert, and Stephanie Beasley for analysis support; Anthony Murphy and Timothy Holzer for bioanalytical support; David Manner for providing statistical peer review; and Daniel Skovronsky for expediting initiation of trial in the initial peak of pandemic.
- 1. Goldman, J.D. et al. Remdesivir for 5 or 10 Days in Patients with Severe Covid‐19. N. Engl. J. Med. 383, 1827–1837 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Spinner, C.D. et al. Effect of remdesivir vs standard care on clinical status at 11 days in patients with moderate COVID‐19: A randomized clinical trial. JAMA 324, 1048–1057 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Beigel, J.H. et al. Remdesivir for the treatment of covid‐19 ‐ final report. N. Engl. J. Med. 383, 1813–1826 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bronte, V. et al. Baricitinib restrains the immune dysregulation in severe COVID‐19 patients. J. Clin. Invest. 130, 6409–6416 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Stebbing, J. et al. Mechanism of baricitinib supports artificial intelligence‐predicted testing in COVID‐19 patients. EMBO Mol. Med. 12, e12697 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kalil, A.C. et al. Baricitinib plus remdesivir for hospitalized adults with covid‐19. N. Engl. J. Med. 384, 795–807 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Horby, P. et al. Dexamethasone in hospitalized patients with covid‐19 ‐ preliminary report. N. Engl. J. Med. 384, 693–704 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sterne, J.A.C. et al. Association between administration of systemic corticosteroids and mortality among critically ill patients with COVID‐19: a meta‐analysis. JAMA 324, 1330–1341 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Joyner, M.J. et al. Effect of convalescent plasma on mortality among hospitalized patients with COVID‐19. Initial Three‐Month Experience. medRxiv preprint. 10.1101/2020.08.12.20169359. [DOI]
- 10. Li, L. et al. Effect of convalescent plasma therapy on time to clinical improvement in patients with severe and life‐threatening COVID‐19: A randomized clinical trial. JAMA 324, 460–470 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. US Food and Drug Administration . EUA for emergency use of baricitinib (Olumiant), in combination with remdesivir (Veklury), for the treatment of suspected or laboratory confirmed coronavirus disease 2019 (COVID‐19) Letter of authorization <https://www.fda.gov/media/143822/download> (2020).
- 12. US Food and Drug Administration . GSK Sotrovimab Letter of Authorization <https://wwwfdagov/media/149532/download> (2021).
- 13. US Food and Drug Administration . Casirivimab and imdevimab EUA Letter of Authorization <https://www.fda.gov/media/143891/download> (2020).
- 14. US Food and Drug Administration . Fact sheet for health care providers emergency use authorization (EUA) of Bamlanivimab and Etesevimab <https://wwwfdagov/media/145802/download> (2021).
- 15. Hoffmann, M. et al. SARS‐CoV‐2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell 181, 271–80 e8 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Jones, B.E. et al. The neutralizing antibody, LY‐CoV555, protects against SARS‐CoV‐2 infection in non‐human primates. Sci. Transl. Med. (2020). bioRxiv preprint; 10.1101/2020.09.30.318972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Chen, P. et al. SARS‐CoV‐2 neutralizing antibody LY‐CoV555 in outpatients with covid‐19. N. Engl. J. Med. 384, 229–237 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kim, K.S. et al. Modelling SARS‐CoV‐2 dynamics: implications for therapy. MedRxiv (2020). preprint. 10.1101/2020.03.23.20040493. [DOI] [Google Scholar]
- 19. Jones, H.M. et al. A physiologically‐based pharmacokinetic model for the prediction of monoclonal antibody pharmacokinetics from in vitro data. CPT Pharmacometrics Syst. Pharmacol. 8, 738–747 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. US Food and Drug Administration . Lilly SARS‐CoV‐2 Assay <https://wwwfdagov/media/140543/download> (2020).
- 21. US Food and Drug Administration . Emergency Use Authorization (EUA) Summary Lilly SARS‐CoV‐2 assay <https://wwwfdagov/media/140543/download> (2021).
- 22. Tan, C.W. et al. A SARS‐CoV‐2 surrogate virus neutralization test based on antibody‐mediated blockage of ACE2‐spike protein‐protein interaction. Nat. Biotechnol. 38, 1073–1078 (2020). [DOI] [PubMed] [Google Scholar]
- 23. US Food and Drug Administration . COVID‐19: Developing Drugs and Biological Products for Treatment or Prevention Guidance for Industry <https://www.fda.gov/regulatory‐information/search‐fda‐guidance‐documents/covid‐19‐developing‐drugs‐and‐biological‐products‐treatment‐or‐prevention> (2020).
- 24. Cevik, M. , Bamford, C.G.G. & Ho, A. COVID‐19 pandemic‐a focused review for clinicians. Clin. Microbiol. Infect. 26, 842–847 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chang, D. et al. Time kinetics of viral clearance and resolution of symptoms in novel coronavirus infection. Am. J. Respir. Crit. Care Med. 201, 1150–1152 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wölfel, R. et al. Virological assessment of hospitalized patients with COVID‐2019. Nature 581, 465–469 (2020). [DOI] [PubMed] [Google Scholar]
- 27. Magleby, R. et al. Impact of SARS‐CoV‐2 viral load on risk of intubation and mortality among hospitalized patients with coronavirus disease 2019. Clin. Infect. Dis. (2020). 10.1093/cid/ciaa851. [e‐pub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Liu, Y. et al. Viral dynamics in mild and severe cases of COVID‐19. Lancet Infect. Dis. 20, 656–657 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Shirazi, J. , Donzanti, M.J. , Nelson, K.M. , Zurakowski, R. , Fromen, C.A. & Gleghorn, J.P. Significant unresolved questions and opportunities for bioengineering in understanding and treating COVID‐19 disease progression. Cell Mol. Bioeng. 13, 259–284 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Rydyznski Moderbacher, C. et al. Antigen‐specific adaptive immunity to SARS‐CoV‐2 in acute COVID‐19 and associations with age and disease severity. Cell 183, 996–1012 e19 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hadjadj, J. et al. Impaired type I interferon activity and inflammatory responses in severe COVID‐19 patients. Science 369, 718–724 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Yao, C. et al. Cell type‐specific immune dysregulation in severely ill COVID‐19 patients. Cell Rep. 34, 108590 (2021). 10.1016/j.celrep.2020.108590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. US Food and Drug Administration . Bamlanivimab EUA Letter of Authorization <https://www.fda.gov/media/143602/download> (2020).
- 34. Eli Lilly and Company . Lilly requests revocation of emergency use authorization for bamlanivimab alone to complete transition to bamlanivimab and etesevimab together for treatment of COVID‐19 in the U.S <https://investorlillycom/news‐releases/news‐release‐details/lilly‐requests‐revocation‐emergency‐use‐authorization> (2021).
- 35. Gottlieb, R.L. et al. Effect of bamlanivimab as monotherapy or in combination with etesevimab on viral load in patients with mild to moderate COVID‐19: A randomized clinical trial. JAMA 325, 632–644 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Dougan, M. et al. Bamlanivimab plus etesevimab in mild or moderate covid‐19. N. Engl. J. Med. 10.1056/NEJMoa2102685. [e‐pub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. ACTIV‐3/TICO LY‐CoV555 Study Group . A neutralizing monoclonal antibody for hospitalized patients with covid‐19. N. Engl. J. Med. 384, 905–914 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. National Institute of Allergy and Infectious Disease . Statement—NIH‐Sponsored ACTIV‐3 Clinical Trial Closes Enrollment into Two Sub‐Studies <https://wwwnihgov/news‐events/news‐releases/nih‐sponsored‐activ‐3‐clinical‐trial‐closes‐enrollment‐into‐two‐sub‐studies> (2021).
- 39. National Institutes of Allergy and Infectious Diseases . Statement—NIH‐Sponsored ACTIV‐3 Trial Closes LY‐CoV555 Sub‐Study <https://wwwniaidnihgov/news‐events/statement‐nih‐sponsored‐activ‐3‐trial‐closes‐ly‐cov555‐sub‐study> (2020).
- 40. Mason, S. , Devincenzo, J.P. , Toovey, S. , Wu, J.Z. & Whitley, R.J. Comparison of antiviral resistance across acute and chronic viral infections. Antiviral Res. 158, 103–112 (2018). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material.
Data Availability Statement
Lilly provides access to all individual participant data collected during the trial, after anonymization, with the exception of pharmacokinetic or genetic data. Data are available to request in a timely fashion after the indication studied has been approved in the United States and the European Union and after primary publication acceptance. No expiration date of data requests is currently set once they are made available. Access is provided after a proposal has been approved by an independent review committee identified for this purpose and after receipt of a signed data sharing agreement. Data and documents, including the study protocol, statistical analysis plan, clinical study report, blank or annotated case report forms, will be provided in a secure data sharing environment for up to 2 years per proposal. For details on submitting a request, see the instructions provided at www.clinicalstudydatarequest.com.