

Abstract
A newly reported compound, CuAgBiI5, is synthesized as powder, crystals, and thin films. The structure consists of a 3D octahedral Ag+/Bi3+ network as in spinel, but occupancy of the tetrahedral interstitials by Cu+ differs from those in spinel. The 3D octahedral network of CuAgBiI5 allows us to identify a relationship between octahedral site occupancy (composition) and octahedral motif (structure) across the whole CuI–AgI–BiI3 phase field, giving the ability to chemically control structural dimensionality. To investigate composition–structure–property relationships, we compare the basic optoelectronic properties of CuAgBiI5 with those of Cu2AgBiI6 (which has a 2D octahedral network) and reveal a surprisingly low sensitivity to the dimensionality of the octahedral network. The absorption onset of CuAgBiI5 (2.02 eV) barely changes compared with that of Cu2AgBiI6 (2.06 eV) indicating no obvious signs of an increase in charge confinement. Such behavior contrasts with that for lead halide perovskites which show clear confinement effects upon lowering dimensionality of the octahedral network from 3D to 2D. Changes in photoluminescence spectra and lifetimes between the two compounds mostly derive from the difference in extrinsic defect densities rather than intrinsic effects. While both materials show good stability, bulk CuAgBiI5 powder samples are found to be more sensitive to degradation under solar irradiation compared to Cu2AgBiI6.
Short abstract
We describe a way to chemically control the octahedral network of potentially useful photovoltaic solar absorbers in the CuI−AgI−BiI3 phase space by the synthesis of CuAgBiI5 with a 3D octahedral network. We compare the photostability of CuAgBiI5 bulk samples and the absorption coefficient and photoluminescence of solution processed thin films with those of Cu2AgBiI6, which has a 2D octahedral network. This helps to understand structure−property relationships to direct further materials optimization.
Introduction
Ternary and quaternary compounds from the CuI–AgI–BiI3 phase space show huge potential for photovoltaics due to their suitable band gaps (1.67–2.06 eV) with very high absorption coefficients exceeding 105 cm–1 and low excitonic binding energies (∼25 meV) that arise from their stable Bi3+ iodide octahedral network in a close packed iodide sublattice.1−4 In contrast, the double perovskite Cs2AgBiBr6 does not strongly absorb light at energies below its excitonic absorption peak at 2.8 eV and direct band gap at 3.03 eV,5,6 and the hypothetical compound Cs2AgBiI6, which would likely have a narrower band gap, is not a stable phase.7 Therefore, the materials from the CuI–AgI–BiI3 phase space fill an important gap in “lead-free” metal-halide materials capability. They also overcome the compromises of the wide band gap hybrid lead halide perovskites APb(BrxI1–x)3. Although the hybrid lead halide perovskites can be used to make highly efficient photovoltaic devices, they can suffer from halide segregation under illumination (due to competing phases arising from mixing of the halides)8,9 and low thermal stability (due to the presence of organic cations)10,11 and have to be carefully managed due to known toxicological issues with lead. In comparison, the CuI–AgI–BiI3 materials contain a single halide, are entirely inorganic, and are lead-free. Photovoltaic devices utilizing CuI–AgI–BiI3 materials as the solar absorbers have reached over 5% power conversion efficiencies (PCEs).12 Further improvements for devices using existing materials are expected to come from optimizing device architecture and transport layers and passivation techniques. However, further materials development is also crucial for the realization of efficient devices, and to achieve this, there is the need to understand the composition–structure–property relationships across the CuI–AgI–BiI3 phase space. The CuI–AgI–BiI3 compounds do not form perovskites. We have previously reported the structural perspective showing that the CuI–AgI–BiI3 compounds have an uninterrupted close-packed anion sublattice, whereas perovskites have a close-packed anion sublattice which is interrupted by the large A-site cations.1 Here, we report the quaternary CuAgBiI5 which shows how the widely variable composition of materials from the CuI–AgI–BiI3 phase space can be used to select the dimensionality of the octahedral motif. This is an important relationship to understand because the properties of the lead halide perovskites drastically change depending on whether the octahedral network is 2D or 3D. The 2D lead halide perovskites are more stable, but the lower dimensionality of the octahedral networks results in both wider band gaps and highly confined charge carriers leading to excitons, which are less desirable for photovoltaic applications.13,14 We compare the properties of the three-dimensional (3D) octahedral network of CuAgBiI5 with the two-dimensional (2D) octahedral network of previously reported Cu2AgBiI6.
CuI–AgI–BiI3 Phase Space
The CuI–AgI–BiI3 phase space is shown in Figure 1. It is mapped out by known binary compounds CuI, AgI, and BiI3 at the corners,15−17 ternary Ag–Bi–I (Ag3BiI6, Ag2BiI5, AgBiI4, AgBi2I7, Ag2Bi3I11)1,18−21 and Cu–Bi–I (CuBiI4, Cu2BiI5)22 compounds on the edges, and a quaternary Cu–Ag–Bi–I (Cu2AgBiI6)2 compound in the enclosed area. We have also added the new compound described in this study, CuAgBiI5. The Ag–Bi–I materials have been the most studied for photovoltaics. Low temperature synthesis and processing techniques lead to the formation of Ag2BiI5 or AgBi2I7, whereas phases Ag3BiI6, AgBiI4, and Ag2Bi3I11 are attainable via high temperature routes.4 The Ag-rich compounds Ag2BiI5 and Ag3BiI6, the latter of which is a mixture of Ag2BiI5 and AgI when solution processed into films, are reported to perform better in devices.4 The record device PCE of 5.56% uses a solar absorber with nominal composition Ag3BiI5.92S0.04—a small sulfide substitution in Ag3BiI6.12 CuBiI4 has been processed into devices with a maximum PCE of 1.1%;23,24 however, we previously found this composition to be an unstable phase, and it decomposes back into CuI and BiI3 at room temperature.2 To stabilize a Cu-containing material, we previously synthesized Cu2AgBiI6 and fabricated a preliminary device with a PCE of 0.43%.2,25 The band gap was found to be 2.06(1) eV, which was modeled to pair efficiently with a crystalline silicon solar absorber in a lead-free tandem cell. Little is known about the other Cu-containing compound Cu2BiI5, which has been reported to crystallize in a hexagonal unit cell,22 with no crystal structural solution or devices reported yet; but it has a band gap of 1.53–1.74 eV.26
Figure 1.

Reported structures in the CuI–AgI–BiI3 phase space: binaries CuI, AgI, and BiI3;15−17 ternaries Ag3BiI6, Ag2BiI5, AgBiI4, AgBi2I7, Ag2Bi3I11, CuBiI4, and Cu2BiI5;1,18−22 and quaternary Cu2AgBiI6.2 We have also added the compound we report here, CuAgBiI5. As discussed in the text, all these compounds consist of a close-packed iodide sublattice with varying arrangements of the cations filling the octahedral and tetrahedral interstitial sites to form the structures shown.
Common Structural Features
The reported compounds in the CuI–AgI–BiI3 phase space consist of Cu+, Ag+, and
Bi3+ cations occupying interstitial octahedral (Oct) or
tetrahedral
(Tet) sites in close-packed iodide sublattices (see Figure 3). In CuI, the iodide sublattice
is cubic close-packed (CCP) consisting of ABCABC stacking
(Figure S10). In the other binaries, AgI
and BiI3, the iodide sublattice is hexagonal close-packed
(HCP) with ABAB stacking. Close-packed anion sublattices
have 2 interstitial Tet sites and 1 interstitial Oct site per anion.
For example for BiI3, in which the Bi3+ is octahedrally
coordinated, the composition requires that 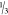 of Oct interstitial
sites are fully occupied
by Bi3+. BiI3 has layered ordering consisting
of a layer with
of Oct interstitial
sites are fully occupied
by Bi3+. BiI3 has layered ordering consisting
of a layer with 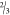 Oct interstitial sites occupied separated
by vacant layers maintaining the overall
Oct interstitial sites occupied separated
by vacant layers maintaining the overall 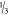 of Oct interstitial
sites occupied (Figure S11). For the room
temperature CuI and
AgI phases, in which the cations are tetrahedrally coordinated,
of Oct interstitial
sites occupied (Figure S11). For the room
temperature CuI and
AgI phases, in which the cations are tetrahedrally coordinated, 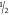 of the Tet
interstitial sites are occupied
giving a 3D network of corner-sharing tetrahedra. This concept can
be extended to the ternary and quaternary compounds but with the added
complexities of disorder and nonstoichiometric compositions. In these
compounds, the cation site occupancies are below 1. The reported crystal
structures show that Ag+, Bi3+, or vacancies
can be found on the Oct sites, and Cu+ or vacancies can
be found on the Tet sites. Unlike in room temperature AgI, Ag+ is octahedrally coordinated in the ternary and quaternary
compounds. Comparing the I–I distances in the binary and ternary
compounds suggests that Ag+ is tetrahedrally coordinated
in iodide sublattices with larger I–I distances of ∼4.6
Å, such as in AgI, but is octahedrally coordinated for shorter
I–I distances of ∼4.3 Å reported for the ternary
and quaternary systems. Atomic disorder means that we are no longer
restricted to integer ratios between the occupancy of the ions; however,
we have chosen to represent nonstoichiometric compounds with a close
stoichiometric composition for ease and report the measured or refined
composition elsewhere in the text. It should be noted that, when we
solve the structures of these systems using diffraction data, it is
required that the I– anion is considered to be ordered
with an atomic occupancy of 1. We also normalize the measured and
refined compositions to an integer amount of iodine. Here, we unravel
the complex reported crystal structures of the reported ternary and
quaternary compounds by breaking them down into three parts: the Oct
motifs (Figure 2),
the unit cells (Figure 3), and the location of the Tet interstitials
occupied by Cu+ (Figure 4). We use our introduced nomenclature to describe CuAgBiI5 and summarize all compounds in Table 1.
of the Tet
interstitial sites are occupied
giving a 3D network of corner-sharing tetrahedra. This concept can
be extended to the ternary and quaternary compounds but with the added
complexities of disorder and nonstoichiometric compositions. In these
compounds, the cation site occupancies are below 1. The reported crystal
structures show that Ag+, Bi3+, or vacancies
can be found on the Oct sites, and Cu+ or vacancies can
be found on the Tet sites. Unlike in room temperature AgI, Ag+ is octahedrally coordinated in the ternary and quaternary
compounds. Comparing the I–I distances in the binary and ternary
compounds suggests that Ag+ is tetrahedrally coordinated
in iodide sublattices with larger I–I distances of ∼4.6
Å, such as in AgI, but is octahedrally coordinated for shorter
I–I distances of ∼4.3 Å reported for the ternary
and quaternary systems. Atomic disorder means that we are no longer
restricted to integer ratios between the occupancy of the ions; however,
we have chosen to represent nonstoichiometric compounds with a close
stoichiometric composition for ease and report the measured or refined
composition elsewhere in the text. It should be noted that, when we
solve the structures of these systems using diffraction data, it is
required that the I– anion is considered to be ordered
with an atomic occupancy of 1. We also normalize the measured and
refined compositions to an integer amount of iodine. Here, we unravel
the complex reported crystal structures of the reported ternary and
quaternary compounds by breaking them down into three parts: the Oct
motifs (Figure 2),
the unit cells (Figure 3), and the location of the Tet interstitials
occupied by Cu+ (Figure 4). We use our introduced nomenclature to describe CuAgBiI5 and summarize all compounds in Table 1.
Figure 3.

Unit cell relationships, close-packed anion sublattices, and examples of octahedral (Oct) and tetrahedal (Tet) interstitials for the reported ternary and quaternary Cu–Ag–Bi–I compounds.1,2,18−22 (a) The cubic unit cell of Ag1–3xBi1+xI4 (x ≥ 0) and CuBiI4 (spinel) and (b) the small trigonal unit cell of Ag1–3xBi1+xI4, CuBiI4 (CdCl2), and Cu2AgBiI6.1,18−22 It is helpful to directly compare the structures by transforming both these cells into a large trigonal unit cell using the transformation matrices shown. (c) This large trigonal cell has a and b directions double that of the small trigonal cell with a volume four times as large and is √3/2 the volume of the cubic unit cell. The quaternary compound CuAgBiI5 crystallizes in the large trigonal unit cell. (d) Rhombohedral strain defined for the cases of the small and large trigonal cells. It can be defined as the extension or contraction of the otherwise cubic structure along the body diagonal (111)cubic, shown by the red arrows.
Figure 2.

Octahedral (Oct) motifs of ternary and quaternary Cu–Ag–Bi–I compounds.1,2,18−22 (a) The spinel Oct motif of x ≥ 0 Ag1–3xBi1+xI4 and CuBiI4 consists of a 3D edge-sharing Oct motif present in spinel (Figure S12) with half of the Oct interstitials occupied. Using the transformation matrix shown, it can be represented in a trigonal unit cell where it can be considered as alternating between layers of 75% and 25% Oct interstitials occupied, maintaining the overall half Oct site occupancy. The reduction in symmetry splits the octahedra into two different sites (depicted by the purple and yellow colors). This representation is necessary to refine the rhombohedral strain of CuAgBiI5. (b) The 2D CdCl2 Oct motif consists of alternating between layers of full Oct interstitial occupancy and vacant layers, giving overall half Oct interstitial occupancy. (c) The NaVO2 Oct motif consists of every possible Oct interstitial being occupied, with layered ordering. The layered ordering means that the layers alternate between two different Oct sites. Unit cells are drawn with solid black lines.
Figure 4.

(a) The three Cu+ sites in CuBiI4 (spinel)
color coded as yellow (site 1), orange (site 2), and red (site 3).22 A channel in the spinel octahedral (Oct) motif
is highlighted in light blue, which we take a cross section of to
show the tetrahedral (Tet) sites inside (sites located behind the
channel, which appear to be inside due to the 2D representation of
the 3D structure, have been crossed out.). The red site (site 3) is
the same in spinel.28 Some, but not all,
of the orange sites (site 2) are occupied in CuAgBiI5.
The Tet sites in CuAgBiI5 can be considered as a reflection
of the spinel sites with the mirror plane down the center of the channel
(blue dashed line). (b) The two Cu+ sites in the small
trigonal unit cell (Cu2AgBiI6 and CuBiI4 (CdCl2)) showing layered ordering.2 Also shown are the connectivities of the Tet sites, which
give a 3D Tet network. (c) The layered ordering of Cu+ sites
in CuAgBiI5 means they are only in layers with 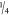 Oct interstitial
occupancy and do not occupy
all the sites associated with tetrahedral site 2 in CuBiI4 (spinel). (d) The connectivity of tetrahedra in CuAgBiI5 and spinel, which give 2D and 0D Tet networks, respectively.
Oct interstitial
occupancy and do not occupy
all the sites associated with tetrahedral site 2 in CuBiI4 (spinel). (d) The connectivity of tetrahedra in CuAgBiI5 and spinel, which give 2D and 0D Tet networks, respectively.
Table 1. Summary of the Structural Features of the Binary, Ternary, and Quaternary Compounds from the CuI–AgI–BiI3 Phase Spacea.
| system | close stoichiometric composition | unit cell (Figure 3) | space group | octahedral motif (Figure 2) | tetrahedral sites (Figure 4) | iodide sublattice | structure type | refs |
|---|---|---|---|---|---|---|---|---|
| binary | BiI3 | trigonal | R3 | BiI3-type (2D) | none | HCP | BiI3 | (17) |
| CuI | cubic | F43m | none | zinc blende (3D) | CCP | zinc blende | (15) | |
| AgI | hexagonal | P63mc | none | wurtzite (3D) | HCP | wurtzite | (16) | |
| ternary | Ag1–3xBi1+xI4x < 0 (Ag-rich) | small trigonal | R3m | NaVO2 (3D) | none | CCP | NaVO2 | (1, 18−21, 27) |
| Ag1–3xBi1+xI4x ≥ 0 (Bi-rich) | cubic | Fd3m | spinel (3D) | none | CCP | defect spinel | (1, 18−21) | |
| Ag1–3xBi1+xI4x ≥ 0 (Bi-rich) | small trigonal | R3m | CdCl2 (2D) | none | CCP | CdCl2 | (1) | |
| CuBiI4 | small trigonal | R3m | CdCl2 (2D) | antifluorite (3D, layered ordering) | CCP | CuBiI4 (CdCl2) | this work | |
| CuBiI4 | cubic | Fd3m | spinel (3D) | antifluorite (3D) | CCP | CuBiI4 (spinel) | (22) | |
| quaternary | CuAgBiI5 | large trigonal | R3m | spinel (3D) | CuAgBiI5 (2D) | CCP | CuAgBiI5 | this work |
| Cu2AgBiI6 | small trigonal | R3m | CdCl2 (2D) | antifluorite (3D, layered ordering) | CCP | Cu2AgBiI6 | (2) |
The nomenclature referred to is described in the main text and corresponding figures. Previously reported Cu2BiI5 has been omitted from the table, as its structure has not been solved.
For the CuI–AgI–BiI3 family
of materials,
the Ag+ and Bi3+ iodide octahedra are edge-sharing,
as opposed to corner-sharing in the hybrid lead perovskites. Thus
far, three different Oct motifs have been reported, corresponding
to those found in spinel,28 CdCl2,29 and NaVO2 (Figure S12).30 The
spinel Oct motif has been reported for AgBiI4, AgBi2I7, Ag2Bi3I11,
and CuBiI4.1,18,19,22 In spinel, 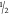 of the Oct
interstitial sites are fully
occupied to make a 3D network shown in Figure 2a,28 as they are
for the composition AgBiI4, for which Ag+ and
Bi3+ equally occupy the Oct site, and there are no vacancies.
When the compositions are Bi-rich (AgBi2I7,
Ag2Bi3I11), the materials start to
exhibit vacancies on these Oct sites. This is because for each Bi3+ added, three times as much Ag+ is removed for
charge balance, which reduces the total Oct site occupancy to less
than 50%, according to the formula Ag1–3xBi1+xI4. Therefore,
we refer to this Oct motif as spinel but highlight the fact that it
can exhibit vacancies. CuBiI4 also has the spinel Oct motif,
with octahedra occupied by 50% Bi3+ and 50% vacant, with
a total Oct site occupancy of
of the Oct
interstitial sites are fully
occupied to make a 3D network shown in Figure 2a,28 as they are
for the composition AgBiI4, for which Ag+ and
Bi3+ equally occupy the Oct site, and there are no vacancies.
When the compositions are Bi-rich (AgBi2I7,
Ag2Bi3I11), the materials start to
exhibit vacancies on these Oct sites. This is because for each Bi3+ added, three times as much Ag+ is removed for
charge balance, which reduces the total Oct site occupancy to less
than 50%, according to the formula Ag1–3xBi1+xI4. Therefore,
we refer to this Oct motif as spinel but highlight the fact that it
can exhibit vacancies. CuBiI4 also has the spinel Oct motif,
with octahedra occupied by 50% Bi3+ and 50% vacant, with
a total Oct site occupancy of 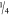 . An alternative
description of the spinel
Oct motif is to consider it as a vacancy-ordered rock salt in which
the occupied octahedra interstitial sites are arranged in the spinel
motif. Structures with the spinel Oct motif have been reported in
cubic unit cells and crystallize in space group Fd3m (Figure 3a). For AgBiI4, there is an alternative
structural description that fits single crystal and powder diffraction
data sets.1 This alternative structural
description consists of twinning of a CdCl2 Oct motif (Figure 2b). Like the spinel
motif, the CdCl2 Oct motif has
. An alternative
description of the spinel
Oct motif is to consider it as a vacancy-ordered rock salt in which
the occupied octahedra interstitial sites are arranged in the spinel
motif. Structures with the spinel Oct motif have been reported in
cubic unit cells and crystallize in space group Fd3m (Figure 3a). For AgBiI4, there is an alternative
structural description that fits single crystal and powder diffraction
data sets.1 This alternative structural
description consists of twinning of a CdCl2 Oct motif (Figure 2b). Like the spinel
motif, the CdCl2 Oct motif has 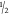 of Oct interstitial
sites occupied. The
occupied Oct sites share edges to form 2D layers which are separated
by vacant layers. In the AgBiI4 structure with the CdCl2 Oct motif, the Oct sites are occupied by 50% Ag+ and 50% Bi3+, whereas in Cu2AgBiI6, which also has the CdCl2 Oct motif, the octahedra are
occupied by 34.6% Ag+ and 30.6% Bi3+ and are
34.8% vacant. To see if the ambiguity between the spinel and CdCl2 Oct motif can exist for the CuBiI4 powder, we
fit the PXRD pattern with both. In the SI (Figure S9, Tables S1–S3), we show
that diffraction data sets collected on the CuBiI4 powder
can be alternatively fitted to a structure with a CdCl2 Oct motif. It is likely that compositions AgBi2I7 and Ag2Bi3I11 can also be
represented by a twinning of structures with the CdCl2 Oct
motif, although this has yet to be shown. Therefore, in our discussion
and Table 1, we state
that AgBiI4, CuBiI4, AgBi2I7, and Ag2Bi3I11 can all exhibit
either the spinel or CdCl2 Oct motifs. Structures with
the CdCl2 Oct motif are represented in a small trigonal
unit cell as shown in Figure 3b in space group R3m. For the special case where the structural ambiguity between
the 3D spinel and twinning of 2D CdCl2 structures exist,
the small trigonal unit cell must be metrically cubic with lattice
parameters at and ct satisfying ct/2at =
of Oct interstitial
sites occupied. The
occupied Oct sites share edges to form 2D layers which are separated
by vacant layers. In the AgBiI4 structure with the CdCl2 Oct motif, the Oct sites are occupied by 50% Ag+ and 50% Bi3+, whereas in Cu2AgBiI6, which also has the CdCl2 Oct motif, the octahedra are
occupied by 34.6% Ag+ and 30.6% Bi3+ and are
34.8% vacant. To see if the ambiguity between the spinel and CdCl2 Oct motif can exist for the CuBiI4 powder, we
fit the PXRD pattern with both. In the SI (Figure S9, Tables S1–S3), we show
that diffraction data sets collected on the CuBiI4 powder
can be alternatively fitted to a structure with a CdCl2 Oct motif. It is likely that compositions AgBi2I7 and Ag2Bi3I11 can also be
represented by a twinning of structures with the CdCl2 Oct
motif, although this has yet to be shown. Therefore, in our discussion
and Table 1, we state
that AgBiI4, CuBiI4, AgBi2I7, and Ag2Bi3I11 can all exhibit
either the spinel or CdCl2 Oct motifs. Structures with
the CdCl2 Oct motif are represented in a small trigonal
unit cell as shown in Figure 3b in space group R3m. For the special case where the structural ambiguity between
the 3D spinel and twinning of 2D CdCl2 structures exist,
the small trigonal unit cell must be metrically cubic with lattice
parameters at and ct satisfying ct/2at = 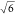 .
.
In Ag-rich compositions Ag2BiI5 and Ag3BiI6, overall Oct site occupancies are over 50%, where 50% is the maximum which can be occupied by the spinel and CdCl2 Oct motifs.18,19 In these structures, the excess Ag+ occupies Oct interstitial sites between the layers of a CdCl2 Oct motif. This means that every Oct interstitial site in the CCP iodide sublattice is occupied (Figure 2c); however, in the compositions reported so far, one Oct site has a full atomic occupancy, and the other has a very low occupancy meaning the structure alternates between layers of full and almost empty octahedra. For Ag2BiI5 (refined as Ag1.92Bi0.83I5),18 the Oct site in one of the layers is full with occupancy 67% Ag+ and 33% Bi3+, and the Oct site in the neighboring layer has a low occupancy of 9.6% Ag+. Despite this, the Oct network must be considered 3D, as every interlayer Ag+ ion connects octahedra from the adjacent layers. We refer to this Oct motif as NaVO2 (Figure S12c). Structures with the NaVO2 Oct motif can be represented in a small trigonal unit cell with space group R3m, as for the CdCl2 motif, as shown in Figure 3b. NaVO2 was first reported by Rüdorff,31 which is why Rüdorffite has been proposed to describe this family of materials,4 although the specific crystal structure it refers to was reported later;30 however, this motif only describes the ternary Ag-rich compounds. For the Cu-containing compounds CuBiI4 and Cu2AgBiI6, Cu+ occupancy is disordered over all possible Tet interstitials giving 3D corner-sharing tetrahedral connectivity. The antifluorite structure of Li2O is an example of this, in which Li+ occupies all possible Tet interstitials in the CCP O2– sublattice (Figure S12d).32 In Li2O, all of the Li+ sites are fully occupied, whereas in CuBiI4 with the spinel Oct motif, there are three Cu+ sites with low occupancies of 0.09, 0.12, and 0.18 (Figure 4a). In Figure 4, we show that one of the Tet sites is the same as that occupied in spinel. The extra occupied Tet interstitials in CuBiI4 mean it is not a spinel structure, although the Oct motif is spinel, and therefore, we refer to its structure type as CuBiI4 (spinel). In Cu2AgBiI6 and the CuBiI4 structure with the CdCl2 Oct motif (referred to as CuBiI4 (CdCl2)), there are two Cu+ sites with partial layered ordering (Figure 4b). One site is in the layer with occupied Oct interstitials, and the other site in the otherwise vacant layer. The tetrahedra are face-sharing with octahedra, edge-sharing with neighboring tetrahedra, and corner-sharing with only tetrahedra (Cu site 1), or with octahedra and tetrahedra (Cu site 2), as shown in Figure 4b. Searching for a known structure type for Cu2AgBiI6 and CuBiI4 (CdCl2), using Wyckoff positions and space group to search the ICSD and Pearson’s Crystal Data,33,34 yields heavily disordered lithium vanadates, in which disorder has been created by partial delithiation. In particular, Cu2AgBiI6 and CuBiI4 (CdCl2) are defect-versions of Li0.2V1.16O2 (Figure S12e).27 All of the Oct and Tet interstitial sites occupied in Cu2AgBiI6 and CuBiI4 with the CdCl2 Oct motif are also occupied in Li0.2V1.16O2. However, Li0.2V1.16O2 has the NaVO2 Oct motif where all Oct interstitials are occupied, as opposed to the CdCl2 Oct motif of Cu2AgBiI6 and CuBiI4 (CdCl2) where only half of these Oct interstitial sites are occupied. This demonstrates that the partial control of ordering can give different long-range average structures, all of which consist of an arrangement of cations between layers of fully anionic close-packed layers which makes them distinct from perovskite.1
CuAgBiI5 Crystal Structure
Here, we explain how we solved the crystal structure of CuAgBiI5 and describe the structural relationships to the known ternary and quaternary compounds. The final room temperature structure is shown in Figure 5, crystallographic information can be found in Table 2, bond distances and angles are reported in Table S4, and goodness of fit parameters of the Rietveld fits are reported in Table S5. We explore the CuI–AgI–BiI3 phase space and optimize synthetic parameters to isolate pure powder samples of CuAgBiI5 as described in the SI. In the SI, we describe in detail the exploratory synthesis and how compositional inhomogeneity has been overcome, as a useful guide for other researchers wanting to synthesize these materials. Multiple 0.25 g batches of powder are combined to form a sample massive enough for neutron powder diffraction measurements (Figure S13). Small crystals are picked out of the final powder (Figure S14) which are much more suitable for single crystal X-ray diffraction (SCXRD) than large crystals grown via chemical vapor transport (CVT). The CuAgBiI5 structure (Figure 5) is solved by Rietveld refinement of complementary combined room temperature high-resolution synchrotron powder X-ray diffraction (PXRD) (MAC detector, I11, Diamond Light Source, Oxfordshire, UK) and high-resolution neutron powder diffraction (NPD) (HRPD, ISIS Neutron and Muon Source, Oxfordshire, UK) data sets, with information also gathered from SCXRD data collected at 100 K.
Figure 5.

(a) The room temperature crystal structure of CuAgBiI5 from Rietveld refinement of combined PXRD and NPD data sets, including coordination environments. (b) A low d-spacing region of the fit, showing that a trigonal cell (ii) fits better than a cubic cell (i). (c) The fits to high-resolution synchrotron PXRD (MAC detector, I11, Diamond Light Source, Oxfordshire, UK) and high-resolution NPD (banks 1 and 2, HRPD, ISIS Neutron and Muon Source, Oxfordshire, UK) data sets. The goodness-of-fit parameters are presented in Table S5.
Table 2. Refined Room Temperature Structural Parameters of CuAgBiI5.
| composition | refined parameters | site | atom | x | y | z | occupancy | U (Å2 × 103) | Wyckoff position | point group (Hermann-Mauguin) | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| CuAgBiI5 | formula sum | Cu3.11 Ag4.97 Bi5.05 I24 | I1 | I | 1/2 | 1/2 | 0.25040(3) | 1 | 21.6(1) | 18h | m |
| Z | 1 | I2 | I | 0 | 0 | 0.25040(3) | 1 | 21.6(1) | 6c | 3m | |
| formula weight (g/mol) | 4834.67 | Oct1 | Bi | 1/2 | 1/2 | 1/2 | 0.421(1) | 38.2(2) | 9d | 2/m | |
| crystal system | trigonal | Ag | 1/2 | 1/2 | 1/2 | 0.414(2) | 38.2(2) | 9d | 2/m | ||
| space group | R3m (166) | Oct2 | Bi | 2/3 | 1/3 | 1/3 | 0.421(1) | 38.2(2) | 3a | 3m | |
| cell parameters (Å) | a = 8.63390(5) c = 21.1398(2) | Ag | 2/3 | 1/3 | 1/3 | 0.414(2) | 38.2(2) | 3a | 3m | ||
| cell volume (Å3) | 1364.74(10) | Cu1 | Cu | 5/6 | 2/3 | 0.298(2) | 0.173(2) | 73(3) | 18h | m | |
| calcd density (g/cm3) | 5.88292 | ||||||||||
Attempts to solve the structure from SCXRD yield a structure with
the spinel Oct motif in a cubic unit cell. Unlike for AgBiI4, the structure cannot be alternatively fitted with a twinning of
structures with the 2D CdCl2 Oct motif without a large
negative peak in the residual electron density. Ultimately, however,
the structure cannot be solved by SCXRD because trying to refine Cu+ sites causes an unstable refinement. This is likely due to
correlation between the disordered occupancies and thermal parameters
of the two Cu+ sites when using a single diffraction data
set. Therefore, we take the confirmation of the spinel Oct motif and
apply it to the combined PXRD and NPD data sets. First, the high-resolution
PXRD data show that the unit cell is not cubic at lower d-spacings, whereas Cu–Ag–Bi–I materials with
spinel Oct motifs have only been reported in cubic unit cells thus
far. To account for this, we transform a structure with the spinel
Oct motif in a cubic unit cell (space group Fd3m, lattice parameter ac, volume Vc) to an equivalent
spinel Oct motif in a trigonal cell (space group R3m, lattice parameters aT and cT, and volume VT) using the transformation matrix shown in Figure 2a. This trigonal
cell is smaller in volume than the cubic cell (VT = 3Vc/4), and it is four times
larger than the trigonal cell associated with the CdCl2 and NaVO2 Oct motifs (lattice parameters at and ct and volume Vt) due to doubling of the a and b directions (VT = 4Vt). We therefore refer to the trigonal
cell with the spinel Oct motif as the large trigonal cell and subscript
lattice parameters and unit cell volumes with a capital “T”
and the small trigonal cell and lattice parameters and unit cell volumes
with subscripts of lowercase “t”. The unit cell relationships
are shown in Figure 3. The lattice parameters of CuAgBiI5 refine to aT = 8.63390(5) Å and cT = 21.1398(2) Å. The large trigonal cell of CuAgBiI5 allows us to refine rhombohedral strain, defined as how far
the value of cT/aT is from 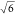 (
(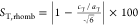 ), to a small value of 0.0132(2)%.
This
provides a much better fit to the PXRD data (Figure 5bii compared to 5bi).
This transformation and lowering of the symmetry from cubic (space
group Fd3m)
to trigonal (R3m) create two Oct sites that make up the spinel Oct motif as shown
in Figure 2a. The atomic
occupancies of these two Oct sites must be kept equal in the Rietveld
refinement, to prevent additional peaks from appearing in the calculated
PXRD pattern. The occupancy of the two Oct sites Oct1 and Oct2 refine
to 42.1(1)% Bi3+ and 41.4(2)% Ag+ leaving a
vacancy of 16.5(2)%. Transforming a spinel Oct motif into a trigonal
cell (Figure 2a) also
highlights a more precise description of the spinel motif as alternating
layers of
), to a small value of 0.0132(2)%.
This
provides a much better fit to the PXRD data (Figure 5bii compared to 5bi).
This transformation and lowering of the symmetry from cubic (space
group Fd3m)
to trigonal (R3m) create two Oct sites that make up the spinel Oct motif as shown
in Figure 2a. The atomic
occupancies of these two Oct sites must be kept equal in the Rietveld
refinement, to prevent additional peaks from appearing in the calculated
PXRD pattern. The occupancy of the two Oct sites Oct1 and Oct2 refine
to 42.1(1)% Bi3+ and 41.4(2)% Ag+ leaving a
vacancy of 16.5(2)%. Transforming a spinel Oct motif into a trigonal
cell (Figure 2a) also
highlights a more precise description of the spinel motif as alternating
layers of 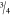 and
and 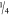 Oct interstitial occupancy. This cell and
symmetry mean there are two different interstitial Tet sites with
layered ordering so that one site belongs in the same layer as the
Oct interstitial occupancy. This cell and
symmetry mean there are two different interstitial Tet sites with
layered ordering so that one site belongs in the same layer as the 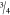 Oct interstitial
occupancy, and the other
belongs in the layers with the
Oct interstitial
occupancy, and the other
belongs in the layers with the 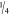 Oct interstitial
occupancy (Figure 4c). The Fourier difference
map showed that Cu+ is located only in the layers with
the
Oct interstitial
occupancy (Figure 4c). The Fourier difference
map showed that Cu+ is located only in the layers with
the 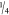 Oct site occupancy
with a refined occupancy
of 17.3(2)%. This Cu+ site is not the same as the Tet site
occupied in spinel as shown in Figure 4a but is located as if reflected in a mirror plane
running down the center of vacant octahedral channels of the spinel,
which changes their location in reference to the octahedra. The positioning
of the tetrahedral Cu+ in CuAgBiI5 means it
is not a spinel structure despite having a (rhombohedrally distorted)
spinel Oct motif. The ordering of the Tet sites in CuAgBiI5 is also distinct from those in the CuBiI4 (spinel) structure. Figure 4c shows that only
a fraction of the Cu sites of CuAgBiI5 are occupied in
comparison to CuBiI4 (spinel). As we cannot find any other
examples of a spinel Oct motif with Tet sites ordered in this way,
we refer to the tetrahedral connectivity as CuAgBiI5-type
and suggest CuAgBiI5 is a new structure type in Table 1. Tetrahedra in spinel
are isolated from other tetrahedra (0D), whereas in CuAgBiI5, a mixture of edge- and corner-sharing gives 2D tetrahedral connectivity
(Figure 4d). The refined
composition Cu0.65(1)Ag1.04(2)Bi1.05(2)I5.00 is within error of the average
powder composition Cu0.88(17)Ag1.10(6)Bi0.98(8)I5.00(11) measured by
TEM EDX, which shows some compositional inhomogeneity in the amount
of Cu remaining from the synthesis. The chemical environment of the
ions in Figure 5b shows
that the Oct sites have six equal Ag/Bi–I bond lengths of 3.0570(2)
Å with angles alternating between 89.83(3)° and 90.17(3)°,
close to 90°. The Cu+ is displaced away from the center
of the tetrahedron, toward the apex in the direction that points along
the c-axis, which leads to one Cu–I bond (2.4923(2)
Å) being shorter than the others (2.6908(3) Å). Significantly
distorted I–Cu–I angles of 112.14(4)° and 106.676(2)°
are observed. We perform X-ray photoelectron spectroscopy (XPS) on
bulk samples of CuAgBiI5 and show in the SI (Figure S15) the fitting of Cu 2p, I 3d, Bi 4f,
and Ag 3d core levels. The binding energies are associated with the
species Cu+, Ag+, and Bi3+ iodide
bonding, and no metallic species are observed.
Oct site occupancy
with a refined occupancy
of 17.3(2)%. This Cu+ site is not the same as the Tet site
occupied in spinel as shown in Figure 4a but is located as if reflected in a mirror plane
running down the center of vacant octahedral channels of the spinel,
which changes their location in reference to the octahedra. The positioning
of the tetrahedral Cu+ in CuAgBiI5 means it
is not a spinel structure despite having a (rhombohedrally distorted)
spinel Oct motif. The ordering of the Tet sites in CuAgBiI5 is also distinct from those in the CuBiI4 (spinel) structure. Figure 4c shows that only
a fraction of the Cu sites of CuAgBiI5 are occupied in
comparison to CuBiI4 (spinel). As we cannot find any other
examples of a spinel Oct motif with Tet sites ordered in this way,
we refer to the tetrahedral connectivity as CuAgBiI5-type
and suggest CuAgBiI5 is a new structure type in Table 1. Tetrahedra in spinel
are isolated from other tetrahedra (0D), whereas in CuAgBiI5, a mixture of edge- and corner-sharing gives 2D tetrahedral connectivity
(Figure 4d). The refined
composition Cu0.65(1)Ag1.04(2)Bi1.05(2)I5.00 is within error of the average
powder composition Cu0.88(17)Ag1.10(6)Bi0.98(8)I5.00(11) measured by
TEM EDX, which shows some compositional inhomogeneity in the amount
of Cu remaining from the synthesis. The chemical environment of the
ions in Figure 5b shows
that the Oct sites have six equal Ag/Bi–I bond lengths of 3.0570(2)
Å with angles alternating between 89.83(3)° and 90.17(3)°,
close to 90°. The Cu+ is displaced away from the center
of the tetrahedron, toward the apex in the direction that points along
the c-axis, which leads to one Cu–I bond (2.4923(2)
Å) being shorter than the others (2.6908(3) Å). Significantly
distorted I–Cu–I angles of 112.14(4)° and 106.676(2)°
are observed. We perform X-ray photoelectron spectroscopy (XPS) on
bulk samples of CuAgBiI5 and show in the SI (Figure S15) the fitting of Cu 2p, I 3d, Bi 4f,
and Ag 3d core levels. The binding energies are associated with the
species Cu+, Ag+, and Bi3+ iodide
bonding, and no metallic species are observed.
Relationship between Composition and Octahedral Network
The structural investigation allows suggestion of a relationship between total Oct site occupancy (composition) and the Oct motif (structure), for ternary and quaternary compositions in the CuI–AgI–BiI3 phase field. We note it will not be true when Ag+ is in tetrahedral coordination, as in room temperature AgI.16Figure 6 shows the type of Oct motif against the total atomic occupancies of the Oct sites for each reported ternary and quaternary compound. For high atomic Oct site occupancies above 50%, the NaVO2 Oct motif is obtained, which can only be obtained in the Ag-rich x < 0 Ag1–3xBi1+xI4 compositions (Ag2BiI5, Ag3BiI6) because adding more Cu+ or Bi3+ reduces Oct site occupancy. At atomic Oct site occupancies of 50%, we have the indistinguishable 3D spinel Oct motif and/or 2D CdCl2 Oct motif of AgBiI4 and CuBiI4,1 which we also expect for the Bi-rich x > 0 Ag1–3xBi1+xI4 compositions (Ag2Bi3I11, AgBi2I7). By substituting in x Bi3+ for 3x Ag+, Oct site occupancies reach as low as 45.5% and 42.9% for reported Ag2Bi3I11 and AgBi2I7, respectively. Continuing to decrease the Ag+ content will eventually lead to the BiI3 structure.
Figure 6.

(a) The relationship between occupancy of the octahedral (Oct) sites and type of Oct motif formed, giving chemical control over dimensionality of the Oct network. CuBiI4 does not fit the trend but we previously found it to be metastable at room temperature.2 (b) The same relationship shown in the CuI–AgI–BiI3 phase space, where the color map and red contour lines represent total Oct site occupancy. We note that the Oct site occupancy in (b) is not representative of materials which contain tetrahedral Ag+ such as the room temperature structure of AgI.
To obtain atomic Oct site occupancies lower than 42.9% in AgBi2I7, tetrahedral Cu+ is added. On the solid solution line between AgBiI4 and CuI, this corresponds to substituting in 4x Cu+ for every x(Ag+ + Bi3+) removed (Cu4x(AgBi)1–xI4), i.e., equal amounts of octahedral Ag+ and Bi3+ are removed. For CuAgBiI5, with an atomic Oct site occupancy of 40%, the 3D rhombohedrally distorted spinel Oct motif is obtained. This suggests that the spinel Oct motif can exist for total Oct site occupancies lower than 50%; however, the reduced Oct site induces a rhombohedral strain away from cubic. We note that this is the first unambiguously 3D Oct network due to the structural ambiguity for AgBiI4,1 CuBiI4, and also likely for Ag2Bi3I11 and AgBi2I7. For Cu2AgBiI6, with a total Oct site occupancy of 33%, the CdCl2 Oct motif is obtained showing that at some total Oct site occupancy between 40% and 33% the Oct motif becomes 2D. The outlier in the proposed relationship between total Oct site occupancy and Oct motif is CuBiI4, which has either the 3D spinel or twinned 2D CdCl2 Oct motif, with an atomic Oct site occupancy of only 25%. However, the unstable nature of the phase compared to a mixture of CuI and BiI3 at room temperature can be associated with the outlier Oct site occupancy.2 This means that CuAgBiI5 and Cu2AgBiI6 are examples of phase stable Cu-containing compounds attained by increasing total Oct site occupancy compared to CuBiI4.
Properties of 3D CuAgBiI5 Compared to 2D Cu2AgBiI6
The distinct 3D and 2D Oct motifs of CuAgBiI5 and Cu2AgBiI6, respectively, allow us to investigate structure–property relationships. To see if there is any difference in stability, we seal CuAgBiI5 and Cu2AgBiI6 powders in capillaries of laboratory air, dry synthetic air, and helium atmospheres and expose them to the solar spectrum for varying amounts of time. Measuring the stability of the powders enables us to probe the stability of the compound without solution processed induced defects, such as surface effects and grain boundaries present in thin films. After 1 week in the solar spectrum, we see that CuAgBiI5 begins to change color from dark red to yellow on the side that is irradiated. This does not correspond to any changes in the PXRD patterns (Figure S16), but we do observe an extra peak in the Raman Spectra (Figure S17). This change occurs under all atmospheres and does not happen in control samples kept in the dark and air, meaning it is a light induced change. In contrast, we do not see any sign of decomposition in Cu2AgBiI6. This may be indicative of increased phase stability of the 2D CdCl2 Oct motif of Cu2AgBiI6 compared to the strained CuAgBiI5 3D spinel Oct motif. It is unlikely to be due to the reduction of photosensitive Ag–I bonds in Cu2AgBiI6 compared to CuAgBiI5, because the more Ag-rich AgBiI4 exposed to the same conditions did not show this decomposition.2 Although we report this instability of CuAgBiI5, it does not necessarily mean that 3D Oct networks are intrinsically less stable than the 2D Oct networks, and the instability may not persist in related systems via chemical substitution. Furthermore, it should be highlighted that thermodynamically stable compositions quenched from the synthesis temperature of 350 °C (CuAgBiI5 and Cu2AgBiI6) are not necessarily the most thermodynamically stable compositions that would be obtained via low temperature solution-processing techniques or at photovoltaic device operating temperatures.
To characterize the optoelectronic properties of CuAgBiI5, we solution process films as described in the SI. It is particularly challenging to dissolve the powders in solutions concentrated enough to form films with high coverage, to prevent powder precipitating during deposition (which causes phase segregation), and to obtain a smooth, shiny surface. These challenges are overcome by using a mixed DMSO/pyridine solution, depositing from hot solutions onto a preheated substrate, and using a two-step annealing procedure, respectively. We fit the XRD pattern of thin films deposited on microscope slides to a large trigonal cell in the R3m space group with lattice parameters a = 8.724(1) Å and c = 20.800(5) Å (Figure S18a). The a and c parameters of the thin films are significantly larger and smaller than those of the CuAgBiI5 powder, respectively. To investigate this, we measure the composition of the films by SEM EDX and find them to have an average composition of Cu0.82(5)Ag0.96(9)Bi1.07(4)I3.98(13) (Figure S18b) and significantly different metal ratios compared to Cu2.52(9)Ag1.02(7)Bi0.82(11)I6.00(20) measured for Cu2AgBiI6 thin films by TEM EDX.2 While the composition of the metal cations is within 1σ of those measured for the powder, there is a large iodine deficiency of 20(3)%. It is not clear at which stage in the process the iodine is lost, and further optimization of film deposition will look to rectify this. The films show a certain level of roughness which can be seen in Figure S18c, as the film differs from a perfectly shiny black reflective surface, which arises from the morphology we show in the SEM images in Figures S18d and e; however, the films were of sufficiently high quality for spectroscopic analysis.
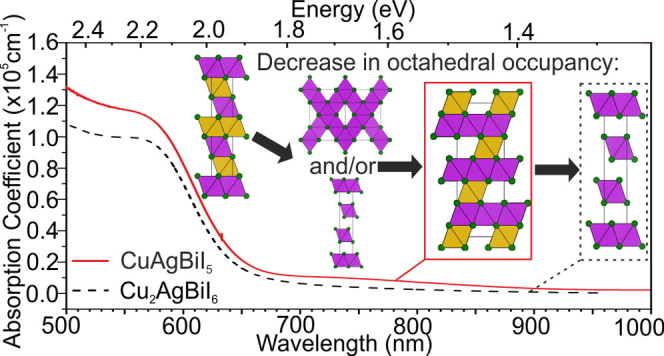
Optical properties of CuAgBiI5 thin films are measured to see how the 3D spinel Oct motif of CuAgBiI5 compares with the 2D CdCl2 Oct motif in Cu2AgBiI6. Surprisingly, the absorption spectra of CuAgBiI5 and Cu2AgBiI6 are very similar, therefore consisting of a very similar band gap and absorption coefficients (Figure 7a). The optical absorption spectra measured for CuAgBiI5 deposited on z-cut quartz substrates show a clear onset at approximately 680 nm (1.8 eV), similar to the previously reported Cu2AgBiI6,2 rising to a value of over 1 × 105 cm–1 for the absorption coefficient just above the band gap. A rough estimate of the band gap can be obtained from the inflection point of the onset of the absorption coefficient, and this gives a value of 2.02 eV, again similar to the value of the band gap reported for Cu2AgBiI6 (2.06 eV). This shows that lowering of the dimensionality of the Oct motif does not increase the band gap and charge confinement like it does for the corner-sharing networks of perovskites. We perform XPS on CuAgBiI5 and Cu2AgBiI6 powders to investigate the density of states at the top of the valence band (Figure 7b). We see an increase in density of states at the top of the valence band for Cu2AgBiI6 compared to CuAgBiI5, which, based on the composition, backs up the theoretical calculations for Cu2AgBiI6 suggesting Cu 3d states at the top of the valence band.2 In the SI, we show the energies of the valence and conduction bands with respect to vacuum for CuAgBiI5 compared to Cu2AgBiI6 (Figure S19). We point out that it is plotted by using the ionization potential measured on bulk samples, due to the surface sensitivity of solution-proceed thin films, and the optical band gap measured on thin films, due to the diffuse reflectance of powder samples broadening the optical absorption edge. The data suggest that both the valence and conduction bands of CuAgBiI5 (5.47 and 3.45 eV, respectively) are slightly lower in energy than Cu2AgBiI6 (5.21 and 3.15 eV, respectively). The lower valence band position may be due to the decreased amount of Cu+. The inclusion of Cu+ in the band edge states suggests a functionalization which contrasts with 2D perovskites; for example, calculations show that in A2PbX4 compounds, in which A cations separate layers of Pb–I octahedra, both inorganic and organic A cations have been shown not to contribute to band-edge states.35,36 The lower conduction band, theorized to be dominated by Bi 6p and I 5p states,2 may be due to the increased connectivity of the Oct network. The shift in the conduction band position of 0.3 eV is relatively small compared to the shift of 1.63 eV observed between the 3D MAPbI3 and 2D BA2PbI4 end members in the Ruddlesden–Popper (BA)2(MA)n−1PbnI3n+1 series.37 This lower sensitivity of the Cu–Ag–Bi–I materials toward the dimensionality of the octahedral network may be due to the presence of the uninterrupted CCP iodide sublattice which maximizes band dispersion and helps maintain electronic connectivity. We also note the presence of some subgap absorption between approximately 950–700 nm, as measured by Fourier transform infrared (FTIR) spectroscopy, which is possibly due to subgap defect states similar to those observed by Photo-Thermal Deflection Spectroscopy (PDS) in Cu2AgBiI6, although we cannot rule out scattering of long-wavelength light from the rough film surfaces leading to lower light transmission in this region but still allowing for the red transmitted light observed when the films are backlit (Figure S7f).
Figure 7.

Absorption coefficient and PL measured on CuAgBiI5 (solid lines) and Cu2AgBiI6 (dashed line) thin films. The data for Cu2AgBiI6 is taken from Sansom et al.2 The PL spectra of CuAgBiI5 and Cu2AgBiI6 were measured in vacuum and air, respectively. (b) The density of states of the valence band measured on CuAgBiI5 (black) and Cu2AgBiI6 (red) powders, measured by XPS. (c) The shift and increase in the PL signal of CuAgBiI5 thin films exposed to air. (d) TRPL of CuAgBiI5 thin films measured in vacuum (black) and air (blue), compared to Cu2AgBiI6, measured in air (red).
We measure steady-state photoluminescence (PL) spectra on the same thin film under continuous wave excitation in vacuum, with CuAgBiI5 showing weak, broad emission peaking at approximately 760 nm (1.63 eV), giving a Stokes shift of 390 meV, similar to, though slightly larger than, that observed in Cu2AgBiI6 and characteristic of emission from localized charge-carrier states, which has been proposed as the source of PL emission in Cu2AgBiI6,25 as well as for Cs2AgBiBr6 (Figure 7a).6,38,39 Compared to Cu2AgBiI6, the PL peak of CuAgBiI5 is slightly shifted to lower energies (1.71 eV for Cu2AgBiI6) with a slightly higher Stokes shift (350 meV for Cu2AgBiI6). The PL peak of the CuAgBiI5 thin film is fitted to a Gaussian peak shape with an fwhm of 312(6) meV, slightly wider than the fwhm of 289 meV extracted for Cu2AgBiI6 films. The sharp PL peak at 800 nm marked by an asterisk is from the second diffraction of the excitation laser signal from the diffraction grating in the detection setup. The PL signal for ternary Ag–Bi–I compounds is rarely reported due to it being too low in intensity to measure, but as the intensity seems more intense for the quaternary Cu-containing compounds, we took the chance to measure it in some detail. To understand the potential impact of atmospheric and light-induced effects on CuAgBiI5, a fresh sample is left in air and darkness for 90 min, during which steady-state PL spectra are measured after 20, 60, and 90 min (Figure 7c). The thin film is only illuminated for very brief (ca. 15 s) periods during the PL measurements, during which acquisitions are taken every 3 s, after 20, 60, and 90 min, respectively. The results show PL spectra after 20 min that are similar to those measured on fresh films in vacuum but which subsequently display a clear blue-shift and large rise in PL intensity under prolonged exposure to air. A similar variation of PL spectra with atmosphere has been widely reported for conventional metal-halide perovskites.9,40−43 Exposure to air has been observed to lead to significant increases in PL intensity across lead-iodide and -bromide perovskites, and this behavior has been ascribed to the passivation of defects by oxygen.40,42,43 The behavior observed here for CuAgBiI5 films in air could follow a similar process, where deeper trap states are passivated by oxygen over time, deactivating nonradiative recombination pathways and leading to higher-energy emission and an increase in the PL intensity. This is confirmed by transient decays measured on a film after exposure to air (Figure 7c) for which the decays show a slightly stronger fluence dependence (Figure S21), with longer lifetimes at lower fluences, and a stretched exponential fit to the lowest-fluence decay gives an average lifetime of τav = 17.9 ns, much longer than for the fresh sample measured in vacuum, and approaching the values fitted for Cu2AgBiI6 in air (τav = 33 ns). This suggests that the differences seen between the PL peak position and intensity of CuAgBiI5 (measured in vacuum) and Cu2AgBiI6 (measured in air) can be accounted for by defects induced by film processing routes and exposure to atmosphere before and during measurements, rather than intrinsic material properties.
To determine whether the observed changes in CuAgBiI5 PL over time are caused by light-induced effects, PL spectra are recorded at 3 s intervals under continuous illumination by the laser after 20 and 90 min for one sample (Figure S20). When we measure under constant illumination the spectra of CuAgBiI5, it shows a drop in intensity but no change in spectral shape, a process sometimes described as “photodarkening”, implying that light-induced effects are not the source of the blue-shift of the spectrum and increase in PL intensity. Photodarkening has been observed in lead-halide perovskites, under both vacuum and nitrogen,40,41 which in one case has been ascribed to an increased density of hole traps forming under constant illumination.43 However, in our case, the very high laser excitation intensity is required to measure PL spectra, of approximately 40 Wcm–2, suggesting that the observed decrease in intensity is likely due to the degradation of the sample region under illumination leading to the creation of point defects. This is supported by the observation of small burn marks on the thin films at the end of the PL measurements. This shows that the atmosphere and light exposure of the sample before and during measurements should be carefully chosen and detailed when reporting PL spectra.
To gain an insight into the charge-carrier lifetimes in CuAgBiI5, we carry out time-resolved PL measurements in vacuum using Time-Correlated Single Photon Counting (TCSPC, see the SI for details). The transient decay shown in Figure 7d of CuAgBiI5 measured in vacuum on a fresh sample shows a very fast initial decay, on the order of 1 ns with no fluence dependence (Figure S21) to the decays across 3 orders of magnitude. The lowest-fluence decay is fitted with a stretched exponential, yielding an average lifetime of τav = 0.73 ns and a stretching exponent of β = 0.32.44 The low value of β is indicative of a highly heterogeneous decay, very similar to that observed in both Cs2AgBiBr6 and Cu2AgBiI6,2,5 and is likely due to a distribution of trap states with slightly varying trapping dynamics. The very short lifetime and lack of fluence dependence of the decays are indicative of a high trap density in the CuAgBiI5 films, leading to fast trap-mediated recombination and scarcity of radiative band-to-band recombination, consistent with the weak steady-state PL emission. This finding is further supported by time-resolved emission spectra, also measured in vacuum using TCSPC on a fresh sample and shown in Figure S21. We also observe evidence of a high-energy emission, less Stokes-shifted around 600 nm over the first 1 ns, where the emission band at 600 nm decreases in intensity relative to the main peak, and the transient decay at 600 nm is faster than that at 720 nm.
Finally, optical-pump terahertz-probe spectroscopy is used to measure the effective charge-carrier mobility for two thin films of CuAgBiI5, yielding values of 1.7(2) and 1.3(2) cm2 V–1 s–1, as shown in Figure S22. These values are comparable to charge-carrier mobilities measured for both Cs2AgBiBr6 and Cu2AgBiI62,6,45 (0.8 cm2 V–1 s–1 and 1.7(5) cm2 V–1 s–1, respectively) and are lower than values reported across conventional metal-halide perovskites.46,47 Charge-carrier mobility is influenced by intrinsic effects, such as scattering off of ionized impurities or couplings between charge carriers and the crystal lattice, and extrinsic effects, such as poor crystallinity and high energetic disorder or scattering off defects.46 Given the high trap density that is apparent from the other spectroscopic measurements, it is possible that a reduction in trap density, along with enhanced crystallinity and reduced energetic disorder, could lead to an improvement in the charge-carrier mobilities for CuAgBiI5, although the low values for charge-carrier mobilities2,48,49 and fast charge-carrier recombination39,49 reported across a variety of silver–bismuth compositions could be indicative of more fundamental limitations to charge-carrier transport in these materials that require further chemical tuning to improve.50
Conclusion
CuAgBiI5 is the first compound in the CuI–AgI–BiI3 phase field with an unambiguously 3D spinel Oct motif. The 3D Oct network has been obtained via chemical tuning, namely the total occupancy of the Ag+ and Bi3+ Oct sites, which allows selectivity between a spinel (3D), CdCl2 (2D), or NaVO2 (3D but with layered ordering) type Oct motif. We find no significant changes in band gap, absorption coefficient, PL, PL lifetimes, charge-carrier mobilities, and charge-carrier confinement (the presence of large excitonic peaks in the absorption coefficient) between the 3D Oct network of CuAgBiI5 and the 2D Oct network of Cu2AgBiI6. This could be due to the close-packed iodide sublattices or presence of tetrahedral Cu+ sites at the top of the valence band which provide enhanced electronic connectivity, thus mitigating against any changes to the electronic states due to the reduction in dimensionality of the Oct network. This contrasts with the 2D perovskites in which the cations separating the layers of Pb–I octahedra do not contribute to band-edge states. Therefore, the optoelectronic properties of Cu–Ag–Bi–I materials have a lower sensitivity toward the dimensionality of the Oct network compared to the lead halide perovskites, and thus useful materials are not restricted to 3D Oct networks. CuAgBiI5 shows a light-induced change in color and Raman spectra when exposed to the solar spectrum even under inert atmosphere, indicating that Cu2AgBiI6 may be the preferable composition with regards to long-term stability. We note that substituting Ag+ and Bi3+ for other cation pairings in the future may change the reliance of the optoelectronic properties on the dimensionality of the network. Structural understanding and initiating the discussion of composition–structure–property relationships will allow the materials community to envisage ways to further improve properties with the goal of applying these materials, and related materials via chemical substitution, into useful optoelectronic devices. Beyond materials improvement, there is future scope for advances resulting from optimization of processing, passivation, and device architectures.
Acknowledgments
H.C.S. thanks EPSRC for support under EP/N004884 and for a Ph.D. studentship at the University of Liverpool and the EPSRC Prosperity Partnership EP/S004947/1 for current funding at the University of Oxford. L.R.V.B. thanks the EPSRC Centre for Doctoral Training in New and Sustainable Photovoltaics and the Oxford-Radcliffe Scholarship for financial support. We thank the STFC for access to beam time at Diamond Light Source and ISIS Spallation Source, Dr. C. Murray and Prof. C. Tang for assistance at I11, and Dr. D. Fortes for assistance at HRPD. M.J.R. is a Royal Society Research Professor.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.inorgchem.1c02773.
Sample preparation, characterization methods, and exploratory synthesis details (Figures S1–S8), figures to support main text (Figures S9–S22, Tables S1–S3), and CuAgBiI5 structural and refinement parameters (Tables S4 and S5) (PDF)
Accession Codes
CCDC 2107462–2107465 contain the supplementary crystallographic data for this paper. These data can be obtained free of charge via www.ccdc.cam.ac.uk/data_request/cif, or by emailing data_request@ccdc.cam.ac.uk, or by contacting The Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK; fax: +44 1223 336033.
Author Present Address
∥ CEMHTI CNRS UPR3079, 45071 Orléans, France
The authors declare the following competing financial interest(s): We declare that we have filed a patent protecting quaternary Cu-Ag-Bi-I phases and their use in optoelectronic devices.
Notes
The data as presented in this paper is freely available at https://datacat.liverpool.ac.uk/id/eprint/1439. The DOI is 10.17638/datacat.liverpool.ac.uk/1439.
This paper was published ASAP on November 9, 2021, with minor errors in Tables 1 and 2. The corrected version was reposted on November 10, 2021.
Supplementary Material
References
- Sansom H. C.; Whitehead G. F. S.; Dyer M. S.; Zanella M.; Manning T. D.; Pitcher M. J.; Whittles T. J.; Dhanak V. R.; Alaria J.; Claridge J. B.; Rosseinsky M. J. AgBiI4 as a Lead-Free Solar Absorber with Potential Application in Photovoltaics. Chem. Mater. 2017, 29 (4), 1538–1549. 10.1021/acs.chemmater.6b04135. [DOI] [Google Scholar]
- Sansom H. C.; Longo G.; Wright A. D.; Buizza L. R. V.; Mahesh S.; Wenger B.; Zanella M.; Abdi-Jalebi M.; Pitcher M. J.; Dyer M. S.; Manning T. D.; Friend R. H.; Herz L. M.; Snaith H. J.; Claridge J. B.; Rosseinsky M. J. Highly Absorbing Lead-Free Semiconductor Cu2AgBiI6 for Photovoltaic Applications from the Quaternary CuI-AgI-BiI3 Phase Space. J. Am. Chem. Soc. 2021, 143 (10), 3983–3992. 10.1021/jacs.1c00495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y.; Yang Z.; Jain A.; Voznyy O.; Kim G.-H.; Liu M.; Quan L. N.; García de Arquer F. P.; Comin R.; Fan J. Z.; Sargent E. H. Pure Cubic-Phase Hybrid Iodobismuthates AgBi2I7 for Thin-Film Photovoltaics. Angew. Chem., Int. Ed. 2016, 55 (33), 9586–9590. 10.1002/anie.201603608. [DOI] [PubMed] [Google Scholar]
- Turkevych I.; Kazaoui S.; Ito E.; Urano T.; Yamada K.; Tomiyasu H.; Yamagishi H.; Kondo M.; Aramaki S. Photovoltaic Rudorffites: Lead-Free Silver Bismuth Halides Alternative to Hybrid Lead Halide Perovskites. ChemSusChem 2017, 10 (19), 3754–3759. 10.1002/cssc.201700980. [DOI] [PubMed] [Google Scholar]
- Schade L.; Wright A. D.; Johnson R. D.; Dollmann M.; Wenger B.; Nayak P. K.; Prabhakaran D.; Herz L. M.; Nicholas R.; Snaith H. J.; Radaelli P. G. Structural and Optical Properties of Cs2AgBiBr6 Double Perovskite. ACS Energy Lett. 2019, 4 (1), 299–305. 10.1021/acsenergylett.8b02090. [DOI] [Google Scholar]
- Wright A. D.; Buizza L. R. V.; Savill K. J.; Longo G.; Snaith H. J.; Johnston M. B.; Herz L. M. Ultrafast Excited-State Localization in Cs2AgBiBr6 Double Perovskite. J. Phys. Chem. Lett. 2021, 12 (13), 3352–3360. 10.1021/acs.jpclett.1c00653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savory C. N.; Walsh A.; Scanlon D. O. Can Pb-Free Halide Double Perovskites Support High-Efficiency Solar Cells?. ACS Energy Lett. 2016, 1 (5), 949–955. 10.1021/acsenergylett.6b00471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knight A. J.; Borchert J.; Oliver R. D. J.; Patel J. B.; Radaelli P. G.; Snaith H. J.; Johnston M. B.; Herz L. M. Halide Segregation in Mixed-Halide Perovskites: Influence of A-Site Cations. ACS Energy Lett. 2021, 6 (2), 799–808. 10.1021/acsenergylett.0c02475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knight A. J.; Wright A. D.; Patel J. B.; McMeekin D. P.; Snaith H. J.; Johnston M. B.; Herz L. M. Electronic Traps and Phase Segregation in Lead Mixed-Halide Perovskite. ACS Energy Lett. 2019, 4 (1), 75–84. 10.1021/acsenergylett.8b02002. [DOI] [Google Scholar]
- Senocrate A.; Kim G. Y.; Grätzel M.; Maier J. Thermochemical Stability of Hybrid Halide Perovskites. ACS Energy Lett. 2019, 4 (12), 2859–2870. 10.1021/acsenergylett.9b01605. [DOI] [Google Scholar]
- Juarez-Perez E. J.; Ono L. K.; Qi Y. Thermal Degradation of Formamidinium Based Lead Halide Perovskites into Sym-Triazine and Hydrogen Cyanide Observed by Coupled Thermogravimetry-Mass Spectrometry Analysis. J. Mater. Chem. A 2019, 7 (28), 16912–16919. 10.1039/C9TA06058H. [DOI] [Google Scholar]
- Pai N.; Lu J.; Gengenbach T. R.; Seeber A.; Chesman A. S. R.; Jiang L.; Senevirathna D. C.; Andrews P. C.; Bach U.; Cheng Y.-B.; Simonov A. N. Silver Bismuth Sulfoiodide Solar Cells: Tuning Optoelectronic Properties by Sulfide Modification for Enhanced Photovoltaic Performance. Adv. Energy Mater. 2019, 9 (5), 1803396. 10.1002/aenm.201803396. [DOI] [Google Scholar]
- Marongiu D.; Saba M.; Quochi F.; Mura A.; Bongiovanni G. The Role of Excitons in 3D and 2D Lead Halide Perovskites. J. Mater. Chem. C 2019, 7 (39), 12006–12018. 10.1039/C9TC04292J. [DOI] [Google Scholar]
- Stoumpos C. C.; Cao D. H.; Clark D. J.; Young J.; Rondinelli J. M.; Jang J. I.; Hupp J. T.; Kanatzidis M. G. Ruddlesden-Popper Hybrid Lead Iodide Perovskite 2D Homologous Semiconductors. Chem. Mater. 2016, 28 (8), 2852–2867. 10.1021/acs.chemmater.6b00847. [DOI] [Google Scholar]
- Wyckoff R. W. G.; Posnjak E. The Crystal Structures of the Cuprous Halides. J. Am. Chem. Soc. 1922, 44 (1), 30–36. 10.1021/ja01422a005. [DOI] [Google Scholar]
- Aminoff G. VII. Über die Kristallstruktur von AgJ. Z. Kristallogr. - Cryst. Mater. 1922, 57 (1–6), 180. 10.1524/zkri.1922.57.1.180. [DOI] [Google Scholar]
- Trotter J.; Zobel T. The Crystal Structure of SbI3 and BiI3. Z. Kristallogr. - Cryst. Mater. 1966, 123 (1–6), 67. 10.1524/zkri.1966.123.16.67. [DOI] [Google Scholar]
- Oldag T.; Aussieker T.; Keller H.-L.; Preitschaft C.; Pfitzner A. Solvothermale Synthese und Bestimmung der Kristallstrukturen von AgBiI4 und Ag3BiI6. Z. Anorg. Allg. Chem. 2005, 631 (4), 677–682. 10.1002/zaac.200400508. [DOI] [Google Scholar]
- Mashadieva L. F.; Aliev Z. S.; Shevelkov A. V.; Babanly M. B. Experimental Investigation of the Ag-Bi-I Ternary System and Thermodynamic Properties of the Ternary Phases. J. Alloys Compd. 2013, 551, 512–520. 10.1016/j.jallcom.2012.11.033. [DOI] [Google Scholar]
- Dzeranova K. B.; Kaloev N. I.; Bukhalova G. A. The BiI3 - AgI System. Russ. J. Inorg. Chem. 1985, 30, 1700–1701. [Google Scholar]
- Fourcroy P. H.; Palazzi M.; Rivet J.; Flahaut J.; Céolin R. Etude du Systeme AgIBiI3. Mater. Res. Bull. 1979, 14 (3), 325–328. 10.1016/0025-5408(79)90096-5. [DOI] [Google Scholar]
- Fourcroy P. H.; Carre D.; Thevet F.; Rivet J. Structure du Tetraiodure de Cuivre(I) et de Bismuth(III), CuBiI4. Acta Crystallogr., Sect. C: Cryst. Struct. Commun. 1991, 47 (10), 2023–2025. 10.1107/S0108270191005309. [DOI] [Google Scholar]
- Zhang B.; Lei Y.; Qi R.; Yu H.; Yang X.; Cai T.; Zheng Z. An In-situ Room Temperature Route to CuBiI4 Based Bulk-Heterojunction Perovskite-like Solar Cells. Sci. China Mater. 2019, 62 (4), 519–526. 10.1007/s40843-018-9355-0. [DOI] [Google Scholar]
- Hu Z.; Wang Z.; Kapil G.; Ma T.; Iikubo S.; Minemoto T.; Yoshino K.; Toyoda T.; Shen Q.; Hayase S. Solution-Processed Air-Stable Copper Bismuth Iodide for Photovoltaics. ChemSusChem 2018, 11 (17), 2930–2935. 10.1002/cssc.201800815. [DOI] [PubMed] [Google Scholar]
- Buizza L. R. V.; Wright A. D.; Longo G.; Sansom H. C.; Xia C. Q.; Rosseinsky M. J.; Johnston M. B.; Snaith H. J.; Herz L. M. Charge-Carrier Mobility and Localization in Semiconducting Cu2AgBiI6 for Photovoltaic Applications. ACS Energy Lett. 2021, 6 (5), 1729–1739. 10.1021/acsenergylett.1c00458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adappattu Ramachandran A.; Krishnan B.; Avellaneda Avellaneda D.; Isabel Mendivil Palma M.; Amilcar Aguilar Martinez J.; Shaji S. Development of Lead-Free Cu2BiI5 Rudorffite Thin Films for Visible Light Photodetector Application. Appl. Surf. Sci. 2021, 564, 150438. 10.1016/j.apsusc.2021.150438. [DOI] [Google Scholar]
- de Picciotto L. A.; Thackeray M. M.; David W. I. F.; Bruce P. G.; Goodenough J. B. Structural Characterization of Delithiated LiVO2. Mater. Res. Bull. 1984, 19 (11), 1497–1506. 10.1016/0025-5408(84)90264-2. [DOI] [Google Scholar]
- Bragg W. H. The Structure of Magnetite and the Spinels. Nature 1915, 95 (2386), 561–561. 10.1038/095561a0. [DOI] [Google Scholar]
- Pauling L.; Hoard J. L. The Crystal Structure of Cadmium Chloride. Z. Kristallogr. - Cryst. Mater. 1930, 74 (1–6), 546–551. 10.1524/zkri.1930.74.1.546. [DOI] [Google Scholar]
- McQueen T. M.; Stephens P. W.; Huang Q.; Klimczuk T.; Ronning F.; Cava R. J. Successive Orbital Ordering Transitions in NaVO2. Phys. Rev. Lett. 2008, 101 (16), 166402. 10.1103/PhysRevLett.101.166402. [DOI] [PubMed] [Google Scholar]
- Rüdorff W.; Becker H. Notizen: Die Strukturen von LiVO2, NaVO2, LiCrO2 und NaCrO2. Z. Naturforsch. B 1954, 9 (9), 614. 10.1515/znb-1954-0911. [DOI] [Google Scholar]
- Bijvoet J.; Claassen A.; Karssen A.. The Scattering Power of Lithium and Oxygen, Determined from the Diffraction-Intensities of Powdered Lithiumoxide. In Proceedings of the Koninklijke Nederlandse Academie van Wetenschappen; 1926.
- Hellenbrandt M. The Inorganic Crystal Structure Database (ICSD)—Present and Future. Crystallogr. Rev. 2004, 10 (1), 17–22. 10.1080/08893110410001664882. [DOI] [Google Scholar]
- Villars P.; Cenzual K.. Pearson’s Crystal Data: Crystal Structure Database for Inorganic Compounds (on DVD); ASM International: Materials Park, Ohio, USA, 2020.
- Pandey M.; Jacobsen K. W.; Thygesen K. S. Band Gap Tuning and Defect Tolerance of Atomically Thin Two-Dimensional Organic-Inorganic Halide Perovskites. J. Phys. Chem. Lett. 2016, 7 (21), 4346–4352. 10.1021/acs.jpclett.6b01998. [DOI] [PubMed] [Google Scholar]
- Liu Y.; Xiao H.; Goddard W. A. Two-Dimensional Halide Perovskites: Tuning Electronic Activities of Defects. Nano Lett. 2016, 16 (5), 3335–3340. 10.1021/acs.nanolett.6b00964. [DOI] [PubMed] [Google Scholar]
- Cao D. H.; Stoumpos C. C.; Farha O. K.; Hupp J. T.; Kanatzidis M. G. 2D Homologous Perovskites as Light-Absorbing Materials for Solar Cell Applications. J. Am. Chem. Soc. 2015, 137 (24), 7843–7850. 10.1021/jacs.5b03796. [DOI] [PubMed] [Google Scholar]
- Zelewski S. J.; Urban J. M.; Surrente A.; Maude D. K.; Kuc A.; Schade L.; Johnson R. D.; Dollmann M.; Nayak P. K.; Snaith H. J.; Radaelli P.; Kudrawiec R.; Nicholas R. J.; Plochocka P.; Baranowski M. Revealing the Nature of Photoluminescence Emission in the Metal-Halide Double Perovskite Cs2AgBiBr6. J. Mater. Chem. C 2019, 7 (27), 8350–8356. 10.1039/C9TC02402F. [DOI] [Google Scholar]
- Wu B.; Ning W.; Xu Q.; Manjappa M.; Feng M.; Ye S.; Fu J.; Lie S.; Yin T.; Wang F.; Goh T. W.; Harikesh P. C.; Tay Y. K. E.; Shen Z. X.; Huang F.; Singh R.; Zhou G.; Gao F.; Sum T. C. Strong Self-Trapping by Deformation Potential Limits Photovoltaic Performance in Bismuth Double Perovskite. Science Advances 2021, 7 (8), abd3160 10.1126/sciadv.abd3160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motti S. G.; Gandini M.; Barker A. J.; Ball J. M.; Srimath Kandada A. R.; Petrozza A. Photoinduced Emissive Trap States in Lead Halide Perovskite Semiconductors. ACS Energy Lett. 2016, 1 (4), 726–730. 10.1021/acsenergylett.6b00355. [DOI] [Google Scholar]
- Brenes R.; Eames C.; Bulović V.; Islam M. S.; Stranks S. D. The Impact of Atmosphere on the Local Luminescence Properties of Metal Halide Perovskite Grains. Adv. Mater. 2018, 30 (15), 1706208. 10.1002/adma.201706208. [DOI] [PubMed] [Google Scholar]
- Galisteo-López J. F.; Anaya M.; Calvo M. E.; Míguez H. Environmental Effects on the Photophysics of Organic-Inorganic Halide Perovskites. J. Phys. Chem. Lett. 2015, 6 (12), 2200–2205. 10.1021/acs.jpclett.5b00785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motti S. G.; Meggiolaro D.; Martani S.; Sorrentino R.; Barker A. J.; De Angelis F.; Petrozza A. Defect Activity in Lead Halide Perovskites. Adv. Mater. 2019, 31 (47), 1901183. 10.1002/adma.201901183. [DOI] [PubMed] [Google Scholar]
- Johnston D. C. Stretched Exponential Relaxation Arising from a Continuous Sum of Exponential Decays. Phys. Rev. B: Condens. Matter Mater. Phys. 2006, 74 (18), 184430. 10.1103/PhysRevB.74.184430. [DOI] [Google Scholar]
- Hutter E. M.; Gélvez-Rueda M. C.; Bartesaghi D.; Grozema F. C.; Savenije T. J. Band-Like Charge Transport in Cs2AgBiBr6 and Mixed Antimony-Bismuth Cs2AgBi1-xSbxBr6 Halide Double Perovskites. ACS Omega 2018, 3 (9), 11655–11662. 10.1021/acsomega.8b01705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herz L. M. Charge-Carrier Mobilities in Metal Halide Perovskites: Fundamental Mechanisms and Limits. ACS Energy Lett. 2017, 2 (7), 1539–1548. 10.1021/acsenergylett.7b00276. [DOI] [Google Scholar]
- Herz L. M. How Lattice Dynamics Moderate the Electronic Properties of Metal-Halide Perovskites. J. Phys. Chem. Lett. 2018, 9 (23), 6853–6863. 10.1021/acs.jpclett.8b02811. [DOI] [PubMed] [Google Scholar]
- Bartesaghi D.; Slavney A. H.; Gélvez-Rueda M. C.; Connor B. A.; Grozema F. C.; Karunadasa H. I.; Savenije T. J. Charge Carrier Dynamics in Cs2AgBiBr6 Double Perovskite. J. Phys. Chem. C 2018, 122 (9), 4809–4816. 10.1021/acs.jpcc.8b00572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longo G.; Mahesh S.; Buizza L. R. V.; Wright A. D.; Ramadan A. J.; Abdi-Jalebi M.; Nayak P. K.; Herz L. M.; Snaith H. J. Understanding the Performance-Limiting Factors of Cs2AgBiBr6 Double-Perovskite Solar Cells. ACS Energy Lett. 2020, 5, 2200–2207. 10.1021/acsenergylett.0c01020. [DOI] [Google Scholar]
- Buizza L. R. V.; Herz L. M. Polarons and Charge Localization in Metal-Halide Semiconductors for Photovoltaic and Light-Emitting Devices. Adv. Mater. 2021, 33 (24), 2007057. 10.1002/adma.202007057. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Citations
- Villars P.; Cenzual K.. Pearson’s Crystal Data: Crystal Structure Database for Inorganic Compounds (on DVD); ASM International: Materials Park, Ohio, USA, 2020.