

Abstract
Hereditary angioedema (HAE) is an uncommon disorder with a global prevalence of approximately 1 in 10,000 to 1 in 50,000 population. This disease is grossly underrecognized in India because of lack of awareness and/or lack of diagnostic facilities. Clinical manifestations include swelling over face, eyes, lips, hands, feet, and genitals, abdominal pain, and life-threatening laryngeal edema. HAE should be suspected in all patients who present with angioedema without wheals and who do not respond to antihistamines and/or steroids. C1 levels, C1-INH levels, and C1-INH function should be checked in all patients suspected to have HAE. C1q levels should be assessed in patients with suspected autoimmune-mediated acquired angioedema. Management of HAE constitutes the treatment of acute attack and short-term and long-term prophylaxis. Because of lack of all first-line recommended medications, the management of HAE in India is a challenging task. Patients are managed using fresh frozen plasma (acute treatment), tranexamic acid, and attenuated androgens (prophylaxis). Even though attenuated androgens have been shown to be effective in the prevention of attacks of HAE, the side effect profile especially in children and in females is a serious concern. Hence, the treatment needs to be individualized considering the risk-benefit ratio of long-term prophylaxis. In this review, we provide an overview of diagnostic strategy for patients with HAE and the current treatment concepts with emphasis on currently available treatment options in resource-constrained settings.
Keywords: Androgens, C1-Inhibitor, fresh frozen plasma, hereditary angioedema, tranexamic acid
Introduction
Angioedema is defined as a deep-seated, ill-defined, nonpitting edema or swelling of skin or mucosae that lasts much longer (2–3 days) than an average urticarial wheal, which is superficial, well-defined swelling of skin that usually lasts for few hours. Urticaria is usually intensely pruritic, whereas angioedema is not; it can rather be painful.[1] A tendency to involve nongravitational areas and asymmetric distribution differentiates angioedema from other causes of symmetrical gravitational edema such as congestive heart failure, cirrhosis, and nephrotic syndrome.[2]
Angioedema is traditionally classified according to the presence or absence of wheals.[3] Angioedema associated with wheals is commonly encountered in patients having acute or chronic urticaria or anaphylaxis, and the chief mediator for its development is histamine, which is released from the mast cells, either through immunological pathways (IgE-mediated type-1 hypersensitivity) or by agents that act directly on the mast cells to cause their degranulation.[4]
Angioedema not associated with wheals is a different entity, mediated by agents other than histamine; bradykinin being the most important one. Hereditary angioedema (HAE) is the most important entity in this category followed by acquired angioedema (AAE). AAE may have multifactorial etiologies.
HAE is an uncommon disorder with a global prevalence of approximately 1 in 10,000 to 1 in 50,000 population. There are no epidemiological data on HAE from India. However, considering the current population and the global prevalence of HAE, it may be postulated that there are more than 30,000 patients with India at present. This suggests that HAE is grossly underdiagnosed in India. This could be related to lack of awareness or at times to the lack of diagnostic facilities. As most patients with HAE present with subcutaneous swelling episodes, they would visit a dermatologist for an initial consultation. Hence, dermatologists have an important role to play in the early diagnosis of this condition and to prevent morbidity and mortality.
In this review, we discuss the diagnostic algorithm and current treatment concepts in HAE especially with regard to a developing country perspective such as ours. We also brief about the pathophysiology and clinical features of bradykinin-mediated angioedema.
Pathogenesis of “Angioedema Without Wheals”
Bradykinin, via its action on B2 receptors, mediates vasodilation and enhances vascular permeability resulting in angioedema. Anything that interferes with the degradation of bradykinin or augments its generation can potentially cause angioedema.[5,6] High molecular weight kininogen (HMWK) is cleaved by activated factor XII and kallikrein to form bradykinin. Kallikrein and activated factor XII are formed by activation of pre-kallikrein and factor XII, respectively, through contact activation. Normally, the contact activation is inhibited by C1-esterase inhibitor or simply called C1-inhibitor (C1-INH), a multi-serine protease inhibitor that controls several other catalytic pathways including classical complement activation. The common subtypes of HAE result from either a deficiency of C1-INH (type I) or its dysfunction (type II), causing excess contact activation and generation of kallikrein that in turn generates more bradykinin from HMWK and leads to angioedema. Classical complement pathway is also overactivated simultaneously in patients with HAE. In the absence of C1-INH, C1-esterase cleaves excess of C4 leading to its low serum levels, an important surrogate marker of diagnosing HAE, especially during acute attacks. However, low C4 levels are not related to the pathogenesis of HAE per se.[5]
Both type I and type II HAE are now classified as C1-INH-HAE. Type I constitutes 85% of C1-INH-HAE cases, whereas remaining belong to type II. In type I, both levels and function of C1-INH are low, whereas in type II, the C1-INH is dysfunctional, and therefore, the levels are normal to elevated; however, function is low. Type III HAE, now classified as normal-C1-INH-HAE, is characterized by normal C4 and C1-INH levels and function and can only be diagnosed by doing the genetic testing. These patients may have mutations in factor XII, angiopoietin, kininogen, plasminogen, myoferlin, or heparan sulfate 3-O-sulfotransferase 6 gene.[5,7,8,9,10,11,12,13,14] Identification of these additional genetic defects in patients with HAE has expanded our understanding of pathogenesis of HAE and has proved that this is not limited to contact-kinin-complement pathway. Identification of genetic defects in angiopoietin, heparan sulfate 3-O-sulfotransferase 6, and myoferlin gene has highlighted that endothelium has an important role in the pathogenesis of HAE.
Acquired “angioedema without wheals” may be caused by drugs, angiotensinogen converting enzyme inhibitors (ACE-i) being the most common offending drug. ACE is required for degradation of bradykinin, and its inhibition can lead to the accumulation of bradykinin causing angioedema. Other important clinical settings where one may get acquired “angioedema without wheals” are patients with lymphoma or autoimmune diseases such as systemic lupus erythematosus, where antibodies are formed against C1-INH causing its degradation.[1]
Clinical Features of HAE
HAE is characterized by recurrent episodes of ill-defined, nonpitting skin and mucosal swellings. Diagnosis of HAE should be considered in any patient who presents with angioedema in absence of wheals. Although most episodes of angioedema in patients with HAE are spontaneous in onset, a trigger such as physical trauma, mental stress, infections, and dental or other surgical procedures may be identified in a few patients. A typical episode of angioedema in patients with HAE lasts approximately 3–5 days and resolve spontaneously. It may involve lips, eyes [Figure 1], cheeks, hands, feet, genitals, gastrointestinal tract, tongue and larynx. Cutaneous swellings most frequently involve the limbs followed by face, genitals, and lips. These are nonpitting and frequently involve nondependent areas of the limbs.[15]
Figure 1.

Asymmetric left eyelid and cheek swelling in a 6-year-old girl with HAE
Abdominal colic due to angioedema of bowel wall is common and many of these patients present to the emergency settings with acute abdomen.[16] These patients may inadvertently undergo laparotomy similar to patients with acute intermittent porphyria and hereditary Mediterranean fever (HMF). However, peritoneal signs are absent in HAE and porphyria, and prominent in HMF.
Laryngeal edema is a potentially life-threatening manifestation of HAE and approximately 50% patients experience episode of laryngeal edema at least once in their lifetime.[17] Laryngeal edema in HAE or any bradykinin-mediated angioedema causes stridor; however, bronchospasm or wheezing, which is a feature of histaminergic-mediated angioedema, is characteristically absent. Facial and lip angioedema may rapidly progress to involve laryngeal edema.
Approximately 50% patients with HAE have a prodromal phase characterized by tingling, numbness, pain, and formation of faint, erythematous, serpiginous to annular, and most importantly, nonpruritic patches resembling chicken-wire on skin, called as erythema marginatum.[18,19] These skin lesions bear a striking similarity to erythema marginatum rheumaticum seen in acute rheumatic fever and may be confused with urticaria as well. Prodromal phase usually disappears by the time patients present to the physicians and dermatologists. Presence of prodromal symptoms is also more suggestive of bradykinin-mediated angioedema rather than histamine-mediated angioedema and should always be asked while evaluating a patient with suspected HAE.
Type III or normal-C1-INH-HAE is a rare entity. Clinical presentation of normal-C1-INH-HAE resembles type 1 and type 2 HAE. However, there are a few features that may help differentiate normal-C1-INH-HAE from other types of HAE. These include a higher female predominance, more frequent involvement of face, tongue, and uvula; less frequent attacks overall, less frequent abdominal attacks, and relatively later age of onset.[5] Patients with angiopoietin gene mutation may have nail fold capillary abnormalities.[10] Hemorrhages and bruising have also been reported in the skin lesions of patients who have normal-C1-INH-HAE. Patients with factor XII gene mutation (most common type of normal-C1-INH-HAE) have marked sensitivity to estrogen and most attacks develop during pregnancy and with use of estrogen-containing oral contraceptives.[14,20]
Positive family history and an autosomal dominant mode of inheritance are typical of HAE. However, approximately 25% of patients have a de novo mutation and have no family history.[21] Till date, de novo mutations have not been reported in normal-C1-INH-HAE. Most patients with AAE have onset of disease after the age of 40, whereas most patients with C1-INH-HAE have disease onset around adolescence or early adulthood.[1]
In late-onset cases, it is important to ask the history of drug intake especially ACE-i in setting of a new-onset “angioedema without wheals”, and it is useful to remember that ACE-i is contraindicated in patients having a prior diagnosis of HAE. Other drugs known to cause AAE include nonsteroidal anti-inflammatory drugs (usually leukotriene mediated), dipeptidyl peptidase inhibitors, calcium channel blockers, and tissue plasminogen activators. At last, lymphomas and autoimmune diseases should be ruled out as well on clinical and/or investigational grounds. Similar to normal-C1-INH-HAE, abdominal attacks are less frequent in AAE, whereas facial attacks are more common. In fact, normal-CI-INH-HAE is an important differential of AAE, especially in the settings of normal C1q levels.[22,23]
To summarize, unexplained, recurrent episodes of cutaneous or mucosal angioedema (recurrent abdominal colic, laryngeal swellings) in absence of pruritus and urticaria should make one think of HAE. A positive family history is an important corroborative marker; however, its absence should not deter one from making a diagnosis of HAE as up to 25% of all mutations are de-novo.
Differential Diagnosis
Hair dye-induced allergic contact dermatitis (usually associated with pain/burning/pruritus), connective tissue diseases especially dermatomyositis and systemic lupus erythematosus, trichinosis (associated eosinophilia), and granulomatous cheilitis (persistent infiltrated swelling of lips with features of granulomas on histopathology) are important differentials encountered in dermatology practice. These can be differentiated from HAE on a careful clinical evaluation. Hypereosinophilic syndrome and urticarial vasculitis may also have associated angioedema, but usually have purpuric or papular infiltrated urticarial skin lesions.[2]
In general, histamine-mediated angioedema is rapid in onset and usually resolves within 1 or 2 days, whereas bradykinin-mediated angioedema is relatively slower to begin and stays for 3 to 5 days once established. In rare cases not accompanied by classical wheals of urticaria, co-existing pruritus, flushing, flare and a positive response to antihistamines and corticosteroids also helps to distinguish mast-cell/histamine-mediated isolated idiopathic angioedema from that mediated by bradykinin.[15] As histaminergic angioedema is much more common than HAE, it is prudent to give a trial of high-dose antihistamines (double of the typical dosages) to see if the angioedema responds to antihistamines, especially in patients who have not received antihistamines before. As HAE would not respond to antihistamines, but rare causes of isolated recurrent histaminergic angioedema would, this can help to avoid extensive investigations in patients having histaminergic angioedema.
Diagnosis
Serum C4 levels, C1-INH antigenic levels (using nephelometry), and functional assay for C1-INH (using enzyme-linked immunosorbent assay, ELISA) should be carried out in all patients with suspected HAE. Testing for C4 levels alone is not a good screening test as it has a sensitivity of approximately 80% only. C4 levels are always normal in approximately 20% patients even when carried out at the time of an attack. Repeat test for C1-INH is advised if first test is normal and there is high clinical suspicion.[3]
Patients with AAE due to acquired C1-INH deficiency (secondary to antibodies generated against C1-INH) will have low C4, low or normal C1-INH levels but low C1-INH function, and low C1q levels. C4, C1-INH levels and function are normal in patients having ACE-i induced angioedema, and those with idiopathic acquired angioedema (after excluding all other causes). Patients with normal-C1-INH-HAE have normal C4, C1INH levels and C1-INH functions and at present time diagnosis of normal-C1-INH-HAE can only be established using next generation sequencing (either whole exome sequencing or targeted next generation sequencing). D-dimer levels are often elevated at the time of an acute attack in patients with HAE or during the prodrome of erythema marginatum[24] and may be a useful test in clinically suspected patients with HAE in whom other laboratory investigations are inconclusive.
Figure 2 gives a simplified algorithmic approach for laboratory diagnosis of a clinically suspected case of HAE.
Figure 2.
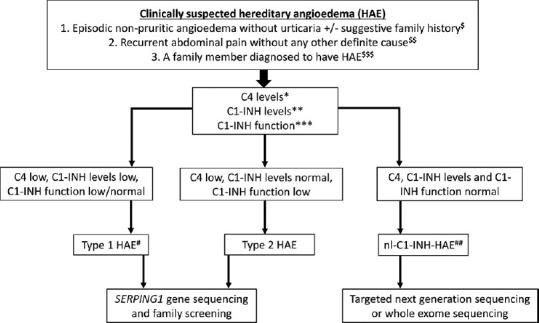
Simplified diagnostic algorithm for patients with clinically suspected HAE. $Family history of HAE may not be present in up to 20% of all patients with HAE. $$Recurrent pain abdomen may occasionally be the only clinical presentation of HAE. $$$Family members should be screened even if they are asymptomatic as late presentations and very mild presentations of HAE are known. *C4 levels are usually assessed using nephelometry, which may be normal in up to 20% of all patients even at the time of an acute attack. **C1-INH levels usually assessed using nephelometry. A repeat test is advised if the initial results are normal and there is high clinical suspicion of HAE. ***C1-INH function usually assessed using enzyme-linked immunosorbent assay (ELISA). Depending on the ease of accessibility, this test may be carried out at the time of initial presentation or after obtaining results of C4/C1-INH levels. Inappropriate storage or transport may affect the results of C1-INH functions. #A clinical possibility of acquired angioedema may be considered in patients with late-onset of symptoms (>40 years of age) and if there is no family history. Low C1q levels may be suggestive of acquired angioedema due to the presence of autoantibodies against C1-INH protein (seen in autoimmune diseases). ##At present, there are no biomarkers for diagnosis of nl-C1-INH-HAE
Management of HAE
HAE is a genetic disorder and at present there is no cure for HAE. The aim of treatment is to avoid any disease-related mortality and to improve quality of life of patients.[25,26] With availability of modern treatment options in several countries, the mortality due to HAE has reduced significantly. However, as most of the recommended first-line treatments are not available in India at present, mortality continues to be a concern for patients with HAE.[21,27]
There are 3 different principles in the management of HAE: Treatment of acute attack (on-demand therapy), prevention of long-term attacks (long-term prophylaxis), and prevention of attacks when it is anticipated (short-term prophylaxis).
Table 1 gives a summary of all drugs used in the management of HAE (including their mechanism of action, routes of administration, doses, and adverse effects). The review would, however, focus on drugs that are currently available for use in India.
Table 1.
An overview of various drugs used in the management of HAE
| Drug | Mechanism of action | Indications | Self-administration | Dosage | Side effects |
|---|---|---|---|---|---|
| Plasma-derived C1-INH (Berinert, Cinryze and HAEGARDA) | Replaces deficient or dysfunctional C1-INH and inhibits plasma kallikrein, factor XIIa, complement proteins, and plasmin | Prophylaxis (short-term/long-term) and management of acute episode including children and during pregnancy | Yes | Variable* | Anaphylaxis, transmission of infectious agents |
| Recombinant human C1-INH (Ruconest) | Replaces deficient or dysfunctional C1-INH and inhibits plasma kallikrein, factor XIIa, complement proteins, and plasmin | Management of acute episodes in adolescents, adults, and during pregnancy | No | 50 U/kg intravenous | Anaphylaxis, transmission of infectious agents |
| Icatibant | Inhibition of Bradykinin B2 receptor | Management of acute episodes in children, adults, and during pregnancy | Yes | 10-30 mg subcutaneous depending on weight | Injection site reactions |
| Ecallantide | Inhibition of plasma kallikrein | Management of acute episodes in adolescents, adults, and during pregnancy | No | 30 mg subcutaneous | Anaphylaxis, formation of anti-drug antibodies, and prolonged activated partial thromboplastin time |
| Fresh frozen plasma | Replaces C1-INH protein | Management of acute episodes when other treatment options are not available | No | 10-20 ml/kg or 2 units | Infusion reactions, transmission of viral agents, volume overload, theoretical risk of aggravation of attack |
| Lanadelumab | Fully humanized IgG1 monoclonal antibody directed against plasma kallikrein | Long-term prophylaxis in adolescents and adults | Yes | 300 mg every 2 weekly | Injection site reactions, dizziness, prolonged activated partial thromboplastin time, and anaphylaxis |
| Berotralstat | Inhibits plasma kallikrein | Long-term prophylaxis in adolescents and adults | Yes | 150 per day oral | Gastrointestinal discomfort, vomiting, diarrhea, headache |
| Danazol | Induces intrinsic production of C1-INH and increases catabolism of bradykinin | Long-term prophylaxis (to be avoided in children and during pregnancy and breastfeeding) | Yes | 100 mg alternate days to 600 mg/day | Virilization, precocious puberty, amenorrhea, acne, infertility, aggressive behavior, depression, accelerated or discontinued growth, hypertension, headache, weight gain, muscle cramps, polycythemia, dyslipidemia |
| Stanozolol | Induces intrinsic production of C1-INH and increases catabolism of bradykinin | Yes | 0.5 mg alternate days to 4 mg/day | ||
| Tranexamic acid | Decreases the synthesis of plasmin leading to prevention of plasmin- mediated FXII activation and subsequent bradykinin production | Long-term prophylaxis | Yes | 30-50 mg/kg/day in 2-3 divided doses (maximum dose 3 g/day) | Gastrointestinal disturbance, risk of thrombosis |
*20 IU/Kg intravenous (Berinert) for acute treatment; 1000 U every 3-4 days of Cinryze for long-term prophylaxis; 60 U/Kg twice a week for HAEGARDA
On-demand treatment
Short-term prophylaxis
Long-term prophylaxis
On demand treatment
Self-administered plasma-derived C1-INH concentrate is the drug of choice for patients with HAE in most developed countries.[25,26] However, at present this drug is not available in India. Fresh frozen plasma (FFP) contains 1 unit of C1-INH per ml and is the treatment of choice for the management of acute attacks in patients with HAE in India.[27] Even though the guidelines recommend that each attack of HAE should be treated, because of potential side effects associated with use of FFP, its use may be suggested for the treatment of acute life-threatening attacks such as laryngeal edema.[28,29,30,31,32] FFP should be used in a dose of 10–20 ml/kg (or 2 units in adults). Most patients respond within 1–12 h of administration of FFP.[30,31] There is a theoretical risk of worsening of an attack of HAE with the use of FFP as it also contacts proteins that may activate the contact system and may lead to more production of bradykinin. However, the risk is more theoretical and we have not observed the worsening of acute attacks with the use of FFP in our experience. Side effects associated with the use of FFP include infusion reactions and anaphylaxis, risk of transmission of viral infections such as hepatitis B, hepatitis C, and human immunodeficiency virus (HIV), and volume overload.[33]
Short-term prophylaxis
Short-term prophylaxis is used in situations when there is a predictable risk of development of an episode of life-threatening laryngeal edema. This is usually seen at the time of a major surgery or a dental procedure. Procedures that involve laryngeal manipulation such as for administering the general anesthesia where laryngeal intubation is needed, is a particularly high-risk situation that would merit short-term prophylaxis. Several observational studies have shown that use of short-term prophylaxis during high-risk procedures is effective in preventing an attack of angioedema.[34,35,36,37]
In countries, where C1-INH therapy or Icatibant is easily available and can be used for patients with HAE as and when they develop angioedema, it may be suggested to keep a high threshold for use of short-term prophylaxis for various surgical procedures. However, in developing countries such as ours where all first-line treatment options are not available and at times, the fresh frozen plasma (that is the only option for management of acute attacks in patients with HAE) is also not easily available, it may be suggested to keep a low threshold for use of short-term prophylaxis in moderate to high-risk situations.[37]
A combination of fresh frozen plasma and attenuated androgens may be used as short-term prophylaxis. A suggested protocol for use of short-term prophylaxis in resource-constrained settings is given in Figure 3.
Figure 3.

Suggested protocol for short-term prophylaxis in resource-constrained settings
In general, there is a risk of development of angioedema both during the procedure and 48–72 h after it. Patients who are already using attenuated androgens may double the dose of drug 2 days prior to the anticipated date of procedure and should be continued for 5 days after the procedure. If a patient is not taking attenuated androgen, then this may be initiated 2 days prior to the anticipated date of procedure and should be continued for 5 days after the procedure (Dose: stanozolol 2 mg/day). FFP may be given (dose of 10 ml/kg twice daily) 1–2 days prior to the procedure and single dose on the day of procedure. FFP may be repeated after the procedure on a case-to-case basis (especially important for patients in whom laryngeal manipulation has been carried out).
Long-term prophylaxis
Long-term prophylaxis is used to prevent an episode of angioedema in patients who have very frequent episodes that are affecting their quality of life or patients who have life-threatening episodes of angioedema. There are no definite recommendations on who should be initiated on long-term prophylaxis. However, it may be appropriate to initiate long-term prophylaxis in patients who have at least more than 1 episode of angioedema every month. In developing countries such as India where the recommended first-line on-demand therapies are not available and the only available on-demand therapy (i.e., FFP) may not be easily accessible to all patients, it is advisable to keep a low threshold for initiation of long-term prophylaxis. However, one also has to consider the risk v/s benefits of the available treatment options for long-term prophylaxis (i.e., tranexamic acid and attenuated androgens).
Table 1 enlists all drugs that are used for long-term prophylaxis. The review would, however, discuss the treatment options that are available in India.
Tranexamic acid
Tranexamic acid is an antifibrinolytic agent and inhibits the conversion of plasminogen to plasmin leading to decrease in the production of bradykinin. Tranexamic acid is less effective as compared to attenuated androgens but its better safety profile makes it an attractive option for long-term prophylaxis especially in children, adolescents and if required during pregnancy.[21,27,38,39,40] Tranexamic acid can be used in a dose of 30–50 mg/kg/day in 2–3 divided doses (maximum dose 3 g/day).
Attenuated androgens
Attenuated androgens increase the production of C1-INH in the body and are one of the most commonly used drugs for long-term prophylaxis in patients with HAE. Stanozolol (dose 05 mg every other day to 4 mg/day) and danazol (100 mg every other day to 600 mg/day) are the 2 attenuated androgens used for this indication. These drugs have been shown to reduce the frequency of attacks of angioedema including life-threatening laryngeal episodes.[41,42,43,44,45] Dose-limiting side effects of these drugs include weight gain, hypertension, acne, menstrual irregularities, virilization, hoarseness of voice, growth retardation, hirsutism, behavioural and mood alterations, and hepatic abnormalities such as adenomas.[21,43,44] Hence, these drugs should be used at a minimum possible dose that can be tolerated by patients. At least annual monitoring of growth, blood pressure, liver enzymes, and liver ultrasound should be carried out. There are no differences between stanozolol and danazol as far as the efficacy and side effects are concerned. However, in our experience, stanozolol appears to be better tolerated as compared to danazol.
Other general measures
All patients with HAE may not be able to identify a trigger for their attacks. However, a proportion of patients may be able to identify a potential trigger and they must be counseled to avoid these triggers. ACE inhibitors should preferably be avoided in all patients with HAE.[46] In addition, Helicobacter pylori infection,[47] celiac disease,[48] and other inflammatory conditions[49] may lower the threshold for swelling episodes in patients with HAE. An attempt should be made to identify and treat these potential triggers.
Role of Physician Societies and Patient Organizations
The role of physician society and patient organizations in improving awareness about HAE and bring better treatment options for patients cannot be overemphasized. India now has a dedicated physician society for HAE with the name “Hereditary Angioedema Society of India” (www.haesi.in). The society was established in February 2021 and held its first national conference on 16th May, 2021 (on the occasion of International HAE day). This meeting was attended by more than 300 delegates across the country. In addition, there is a patient support group in India with the name “HAE India” (https://haeindia.haei.org) and an international patient group “HAE International” (https://haei.org) who work in close collaboration with physicians who are interested in HAE and are willing to improve patient care. The patient groups and physician society are actively pursuing the pharmaceutical companies to bring better treatment options for HAE in India. As a result, one local pharmaceutical company is now about to launch a locally manufactured plasma-derived C1-INH preparation and another international pharmaceutical company is willing to market their plasma-derived C1-INH preparation in India. This has been a remarkable progress and is likely to have a significant impact on the quality of life of patients with HAE.
Conclusion
Hereditary angioedema (HAE) is an uncommon disorder characterized by episodic nonitchy indurated swellings not associated with urticaria. Because of lack of awareness, the disease often remains undiagnosed for several years. HAE should be suspected in all patients who present with episodic swelling without urticaria. C4, C1-INH levels and C1-INH function should be tested in all patients with suspected HAE. Patients with acquired angioedema and normal-C1-INH-HAE have normal levels of C4, C1-INH levels and C1-INH functions. Patients with HAE in most of the developing countries including India are managed using fresh frozen plasma, attenuated androgens, and tranexamic acid as all recommended first-line treatment options are not available at present. However, because of persistent efforts from patient groups and physician society, patients with HAE in India will soon have access to more modern treatment options.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
- 1.Patel G, Pongracic JA. Hereditary and acquired angioedema. Allergy Asthma Proc. 2019;40:441–5. doi: 10.2500/aap.2019.40.4267. [DOI] [PubMed] [Google Scholar]
- 2.Nedelea I, Deleanu D. Isolated angioedema: An overview of clinical features and etiology. Exp Ther Med. 2019;17:1068–72. doi: 10.3892/etm.2018.6982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Proper SP, Lavery WJ, Bernstein JA. Definition and classification of hereditary angioedema. Allergy Asthma Proc. 2020;41((Suppl 1)):S03–7. doi: 10.2500/aap.2020.41.200040. [DOI] [PubMed] [Google Scholar]
- 4.Maurer M, Magerl M. Differences and similarities in the mechanisms and clinical expression of bradykinin-mediated vs. mast cell-mediated angioedema. Clin Rev Allergy Immunol. 2021 doi: 10.1007/s12016-021-08841-w. doi: 10.1007/s12016-021-08841-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sharma J, Jindal AK, Banday AZ, Kaur A, Rawat A, Singh S, et al. Pathophysiology of hereditary angioedema (HAE) beyond the SERPING1 gene. Clin Rev Allergy Immunol. 2021;60:305–15. doi: 10.1007/s12016-021-08835-8. [DOI] [PubMed] [Google Scholar]
- 6.Banday AZ, Kaur A, Jindal AK, Rawat A, Singh S. An update on the genetics and pathogenesis of hereditary angioedema. Genes Dis. 2020;7:75–83. doi: 10.1016/j.gendis.2019.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Veronez CL, Csuka D, Sheikh FR, Zuraw BL, Farkas H, Bork K. The expanding spectrum of mutations in hereditary angioedema. J Allergy Clin Immunol Pract. 2021;9:2229–34. doi: 10.1016/j.jaip.2021.03.008. [DOI] [PubMed] [Google Scholar]
- 8.Bork K, Wulff K, Möhl BS, Steinmüller-Magin L, Witzke G, Hardt J, et al. Novel hereditary angioedema linked with a heparan sulfate 3-O-sulfotransferase 6 gene mutation. J Allergy Clin Immunol. 2021;S0091-6749(21):00094–4. doi: 10.1016/j.jaci.2021.01.011. doi: 10.1016/j.jaci. 2021.01.011. [DOI] [PubMed] [Google Scholar]
- 9.Bafunno V, Firinu D, D'Apolito M, Cordisco G, Loffredo S, Leccese A, et al. Mutation of the angiopoietin-1 gene (ANGPT1) associates with a new type of hereditary angioedema. J Allergy Clin Immunol. 2018;141:1009–17. doi: 10.1016/j.jaci.2017.05.020. [DOI] [PubMed] [Google Scholar]
- 10.d'Apolito M, Santacroce R, Colia AL, Cordisco G, Maffione AB, Margaglione M. Angiopoietin-1 haploinsufficiency affects the endothelial barrier and causes hereditary angioedema. Clin Exp Allergy. 2019;49:626–35. doi: 10.1111/cea.13349. [DOI] [PubMed] [Google Scholar]
- 11.Bork K, Wulff K, Steinmüller-Magin L, Braenne I, Staubach-Renz P, Witzke G, et al. Hereditary angioedema with a mutation in the plasminogen gene. Allergy. 2018;73:442–50. doi: 10.1111/all.13270. [DOI] [PubMed] [Google Scholar]
- 12.Ariano A, D'Apolito M, Bova M, Bellanti F, Loffredo S, D'Andrea G, et al. A myoferlin gain-of-function variant associates with a new type of hereditary angioedema. Allergy. 2020;75:2989–92. doi: 10.1111/all.14454. [DOI] [PubMed] [Google Scholar]
- 13.Bork K, Wulff K, Rossmann H, Steinmüller-Magin L, Braenne I, Witzke G, et al. Hereditary angioedema cosegregating with a novel kininogen 1 gene mutation changing the N-terminal cleavage site of bradykinin. Allergy. 2019;74:2479–81. doi: 10.1111/all.13869. [DOI] [PubMed] [Google Scholar]
- 14.Dewald G, Bork K. Missense mutations in the coagulation factor XII (Hageman factor) gene in hereditary angioedema with normal C1 inhibitor. Biochem Biophys Res Commun. 2006;343:1286–9. doi: 10.1016/j.bbrc.2006.03.092. [DOI] [PubMed] [Google Scholar]
- 15.Andrási N, Veszeli N, Kőhalmi KV, Csuka D, Temesszentandrási G, Varga L, et al. Idiopathic nonhistaminergic acquired angioedema versus hereditary angioedema. J Allergy Clin Immunol Pract. 2018;6:1205–8. doi: 10.1016/j.jaip.2018.04.018. [DOI] [PubMed] [Google Scholar]
- 16.Cao Y, Liu S, Zhi Y. Recurrent and acute abdominal pain as the main clinical manifestation in patients with hereditary angioedema. Allergy Asthma Proc. 2021;42:131–5. doi: 10.2500/aap.2021.42.210001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bork K, Siedlecki K, Bosch S, Schopf RE, Kreuz W. Asphyxiation by laryngeal edema in patients with hereditary angioedema. Mayo Clin Proc. 2000;75:349–54. doi: 10.4065/75.4.349. [DOI] [PubMed] [Google Scholar]
- 18.Leibovich-Nassi I, Reshef A. The enigma of prodromes in hereditary angioedema (HAE) Clin Rev Allergy Immunol. 2021;61:15–28. doi: 10.1007/s12016-021-08839-4. [DOI] [PubMed] [Google Scholar]
- 19.Ohsawa I, Fukunaga A, Imamura S, Iwamoto K, Tanaka A, Hide M, et al. Survey of actual conditions of erythema marginatum as a prodromal symptom in Japanese patients with hereditary angioedema. World Allergy Organ J. 2021;14:100511. doi: 10.1016/j.waojou.2021.100511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Aygören-Pürsün E, Magerl M, Maetzel A, Maurer M. Epidemiology of Bradykinin-mediated angioedema: A systematic investigation of epidemiological studies. Orphanet J Rare Dis. 2018;13:73. doi: 10.1186/s13023-018-0815-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jindal AK, Rawat A, Kaur A, Sharma D, Suri D, Gupta A, et al. Novel SERPING1 gene mutations and clinical experience of type 1 hereditary angioedema from North India. Pediatr Allergy Immunol. 2021;32:599–611. doi: 10.1111/pai.13420. [DOI] [PubMed] [Google Scholar]
- 22.Zanichelli A, Azin GM, Wu MA, Suffritti C, Maggioni L, Caccia S, et al. Diagnosis, course, and management of angioedema in patients with acquired C1-inhibitor deficiency. J Allergy Clin Immunol Pract. 2017;5:1307–13. doi: 10.1016/j.jaip.2016.12.032. [DOI] [PubMed] [Google Scholar]
- 23.Sobotkova M, Zachova R, Hakl R, Kuklinek P, Kralickova P, Krcmova I, et al. Acquired angioedema with C1 inhibitor deficiency: Occurrence, clinical features, and management: A nationwide retrospective study in the Czech Republic patients. Int Arch Allergy Immunol. 2021;182:642–9. doi: 10.1159/000512933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kőhalmi KV, Mező B, Veszeli N, Benedek S, Fehér A, Holdonner Á, et al. Changes of coagulation parameters during erythema marginatum in patients with hereditary angioedema. Int Immunopharmacol. 2020;81:106293. doi: 10.1016/j.intimp.2020.106293. [DOI] [PubMed] [Google Scholar]
- 25.Maurer M, Magerl M, Ansotegui I, Aygören-Pürsün E, Betschel S, Bork K, et al. The international WAO/EAACI guideline for the management of hereditary angioedema-The 2017 revision and update. Allergy. 2018;73:1575–96. doi: 10.1111/all.13384. [DOI] [PubMed] [Google Scholar]
- 26.Betschel S, Badiou J, Binkley K, Borici-Mazi R, Hébert J, Kanani A, et al. The International/Canadian hereditary angioedema guideline. Allergy Asthma Clin Immunol. 2019;15:72. doi: 10.1186/s13223-019-0376-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jindal AK, Reshef A, Longhurst H GEHM workgroup (Global Equity in HAE Management) Mitigating disparity in health-care resources between countries for management of hereditary angioedema. Clin Rev Allergy Immunol. 2021;61:84–97. doi: 10.1007/s12016-021-08854-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Longhurst HJ. Emergency treatment of acute attacks in hereditary angioedema due to C1 inhibitor deficiency: What is the evidence? Int J Clin Pract. 2005;59:594–9. doi: 10.1111/j.1742-1241.2005.00352.x. [DOI] [PubMed] [Google Scholar]
- 29.Pickering RJ, Good RA, Kelly JR, Gewurz H. Replacement therapy in hereditary angioedema.Successful treatment of two patients with fresh frozen plasma. Lancet. 1969;1:326–30. doi: 10.1016/s0140-6736(69)91295-1. [DOI] [PubMed] [Google Scholar]
- 30.Prematta M, Gibbs JG, Pratt EL, Stoughton TR, Craig TJ. Fresh frozen plasma for the treatment of hereditary angioedema. Ann Allergy Asthma Immunol. 2007;98:383–8. doi: 10.1016/S1081-1206(10)60886-1. [DOI] [PubMed] [Google Scholar]
- 31.Wentzel N, Panieri A, Ayazi M, Ntshalintshali SD, Pourpak Z, Hawarden D, et al. Fresh frozen plasma for on-demand hereditary angioedema treatment in South Africa and Iran. World Allergy Organ J. 2019;12:100049. doi: 10.1016/j.waojou.2019.100049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tang R, Chen S, Zhang H. Fresh frozen plasma for the treatment of hereditary angioedema acute attacks. Chin Med Sci J. 2012;27:92–5. [PubMed] [Google Scholar]
- 33.Kalaria S, Craig T. Assessment of hereditary angioedema treatment risks. Allergy Asthma Proc. 2013;34:519–22. doi: 10.2500/aap.2013.34.3702. [DOI] [PubMed] [Google Scholar]
- 34.Ajewole O, Lanlokun M, Dimanche S, Craig T. Short-term prophylaxis for children and adolescents with hereditary angioedema. Allergy Asthma Proc. 2021;42:205–13. doi: 10.2500/aap.2021.42.210006. [DOI] [PubMed] [Google Scholar]
- 35.Farkas H, Zotter Z, Csuka D, Szabó E, Nébenfűhrer Z, Temesszentandrási G, et al. Short-term prophylaxis in hereditary angioedema due to deficiency of the C1-inhibitor--A long-term survey. Allergy. 2012;67:1586–93. doi: 10.1111/all.12032. [DOI] [PubMed] [Google Scholar]
- 36.Zanichelli A, Ghezzi M, Santicchia I, Vacchini R, Cicardi M, Sparaco A, et al. Short-term prophylaxis in patients with angioedema due to C1-inhibitor deficiency undergoing dental procedures: An observational study. PLoS One. 2020;15:e0230128. doi: 10.1371/journal.pone.0230128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jindal AK, Singh A, Anjani G, Kaur A, Jaiswal M, Chopra S, et al. Successful perioperative management of three patients with hereditary angioedema without C1 esterase inhibitor therapy: A developing country perspective. Immunobiology. 2020;225:152022. doi: 10.1016/j.imbio.2020.152022. [DOI] [PubMed] [Google Scholar]
- 38.Sheffer AL, Austen KF, Rosen FS. Tranexamic acid therapy in hereditary angioneurotic edema. N Engl J Med. 1972;287:452–4. doi: 10.1056/NEJM197208312870907. [DOI] [PubMed] [Google Scholar]
- 39.Cicardi M, Bork K, Caballero T, Craig T, Li HH, Longhurst H, et al. Evidence-based recommendations for the therapeutic management of angioedema owing to hereditary C1 inhibitor deficiency: Consensus report of an International Working Group. Allergy. 2012;67:147–57. doi: 10.1111/j.1398-9995.2011.02751.x. [DOI] [PubMed] [Google Scholar]
- 40.Bowen T, Cicardi M, Farkas H, Bork K, Longhurst HJ, Zuraw B, et al. 2010 International consensus algorithm for the diagnosis, therapy and management of hereditary angioedema. Allergy Asthma Clin Immunol. 2010;6:24. doi: 10.1186/1710-1492-6-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Longhurst H, Zinser E. Prophylactic therapy for hereditary angioedema. Immunol Allergy Clin North Am. 2017;37:557–70. doi: 10.1016/j.iac.2017.04.003. [DOI] [PubMed] [Google Scholar]
- 42.Sheffer AL, Fearon DT, Austen KF. Clinical and biochemical effects of stanozolol therapy for hereditary angioedema. J Allergy Clin Immunol. 1981;68:181–7. doi: 10.1016/0091-6749(81)90181-0. [DOI] [PubMed] [Google Scholar]
- 43.Sheffer AL, Fearon DT, Austen KF. Methyltestosterone therapy in hereditary angioedema. Ann Intern Med. 1977;86:306–8. doi: 10.7326/0003-4819-86-3-306. [DOI] [PubMed] [Google Scholar]
- 44.Riedl MA. Critical appraisal of androgen use in hereditary angioedema: A systematic review. Ann Allergy Asthma Immunol. 2015;114:281–8. doi: 10.1016/j.anai.2015.01.003. e7. [DOI] [PubMed] [Google Scholar]
- 45.Bork K, Bygum A, Hardt J. Benefits and risks of danazol in hereditary angioedema: A long-term survey of 118 patients. Ann Allergy Asthma Immunol. 2008;100:153–61. doi: 10.1016/S1081-1206(10)60424-3. [DOI] [PubMed] [Google Scholar]
- 46.Agostoni A, Aygören-Pürsün E, Binkley KE, Blanch A, Bork K, Bouillet L, et al. Hereditary and acquired angioedema: Problems and progress: Proceedings of the third C1 esterase inhibitor deficiency workshop and beyond. J Allergy Clin Immunol. 2004;114((3 Suppl)):S51–131. doi: 10.1016/j.jaci.2004.06.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Visy B, Füst G, Bygum A, Bork K, Longhurst H, Bucher C, et al. Helicobacter pylori infection as a triggering factor of attacks in patients with hereditary angioedema. Helicobacter. 2007;12:251–7. doi: 10.1111/j.1523-5378.2007.00501.x. [DOI] [PubMed] [Google Scholar]
- 48.Csuka D, Kelemen Z, Czaller I, Molnár K, Füst G, Varga L, et al. Association of celiac disease and hereditary angioedema due to C1-inhibitor deficiency.Screening patients with hereditary angioedema for celiac disease: Is it worth the effort? Eur J Gastroenterol Hepatol. 2011;23:238–44. doi: 10.1097/MEG.0b013e328343d3b2. [DOI] [PubMed] [Google Scholar]
- 49.Farkas H, Csuka D, Gács J, Czaller I, Zotter Z, Füst G, et al. Lack of increased prevalence of immunoregulatory disorders in hereditary angioedema due to C1-inhibitor deficiency. Clin Immunol. 2011;141:58–66. doi: 10.1016/j.clim.2011.05.004. [DOI] [PubMed] [Google Scholar]