

Abstract
Every cell in vertebrates possesses the machinery to synthesise cholesterol and to metabolise it. The major route of cholesterol metabolism is conversion to bile acids. Bile acids themselves are interesting molecules being ligands to nuclear and G protein‐coupled receptors, but perhaps the intermediates in the bile acid biosynthesis pathways are even more interesting and equally important. Here, we discuss the biological activity of the different intermediates generated in the various bile acid biosynthesis pathways. We put forward the hypothesis that the acidic pathway of bile acid biosynthesis has primary evolved to generate signalling molecules and its utilisation by hepatocytes provides an added bonus of producing bile acids to aid absorption of lipids in the intestine.
Keywords: cholestenoic acids, COVID‐19, G protein‐coupled receptors, glutamate receptors, inborn errors of metabolism, mass spectrometry, nuclear receptors, oxysterols, sterols
Have bile acid biosynthesis pathways evolved primarily to generate signalling molecules? and is bile acid formation in the hepatocyte is just a bonus? Intermediates in the conversion of cholesterol to bile acids have important roles in development, immunity, regulation of metabolism and neural homeostasis.
Abbreviations
- 22R‐HC
22R‐hydroxycholesterol, cholest‐5‐ene‐3β,22R‐diol
- 24‐HC
24‐hydroxycholesterol, cholest‐5‐ene‐3β,24‐diol
- 24S,25‐EC
24S,25‐epoxycholesterol, 3β‐hydroxycholest‐5‐en‐24S,25‐epoxide
- 25H,7O‐C
25‐hydroxy‐7‐oxocholesterol, 3β,25‐dihydroxycholest‐5‐en‐7‐one
- 25‐HC
25‐hydroxycholesterol, cholest‐5‐ene‐3β,25‐diol
- 26‐HC
(25R)26‐hydroxycholesterol, cholest‐5‐ene‐3β,(25R)26‐diol also known as 27‐hydroxycholesterol, 27‐HC
- 3β,5α‐diHC‐6O
oncosterone, 3β,5α‐dihydroxycholestan‐6‐one
- 3β,7α‐diHCA
3β,7α‐dihydroxycholest‐5‐en‐(25R)26‐oic acid
- 3β,7β‐diHCA
3β,7β‐dihydroxycholest‐5‐en‐(25R)26‐oic acid
- 3βH,7O‐CA
3β‐hydroxy‐7‐oxocholest‐5‐en‐(25R)26‐oic acid
- 3β‐HCA
3β‐hydroxycholest‐5‐en‐(25R)26‐oic acid
- 5,6‐EC
5,6‐epoxycholesterol, cholestan‐5,6‐epoxide
- 7‐DHC
7‐dehydrocholesterol, cholesta‐5,7‐dien‐3β‐ol
- 7‐OC
7‐oxocholesterol, 3β‐hydroxycholest‐5‐en‐7‐one, also known as 7‐ketocholesterol
- 7α,25‐diH,3O‐CA
7α,25‐dihydroxy‐3‐oxocholest‐4‐en‐26‐oic acid
- 7α,25‐diHC
7α,25‐dihydroxycholesterol, cholest‐5‐ene‐3β,7α,25‐triol
- 7α,25‐diHCO
7α,25‐dihydroxycholest‐4‐en‐3‐one
- 7α,26‐diHC
7α,(25R)26‐dihydroxycholesterol, cholest‐5‐ene‐3β,7α,(25R)26‐triol
- 7αH,3O‐CA
7α‐hydroxy‐3‐oxocholest‐4‐en‐(25R)26‐oic acid
- 7α‐HC
7α‐hydroxycholesterol, cholest‐5‐ene‐3β,7α‐diol
- 7β,25‐diHC
7β,25‐dihydroxycholesterol, cholest‐5‐ene‐3β,7β,25‐triol
- 7β‐HC
7β‐hydroxycholesterol, cholest‐5‐ene‐3β,7β‐diol
- ABCA1
ATP‐binding cassette subfamily A member 1
- ABCG1
ATP‐binding cassette subfamily G member 1
- AD
Alzheimer’s disease
- AIM2
absent in melanoma 2
- APOE
apolipoprotein E
- CH25H
cholesterol 25‐hydroxylase, EC:1.14.99.38
- ChEH
cholesterol epoxide hydrolase, EC:3.3.2.11
- COVID‐19
SARS‐CoV‐2
- CRD
cysteine‐rich domain
- CSF
cerebrospinal fluid
- CYP
cytochrome P450
- CYP11A1
cytochrome P450 family 11 subfamily A member 1, EC:1.14.15.6
- CYP27A1
cytochrome P450 family 27 subfamily A member 1, EC:1.14.15.15
- CYP39A1
cytochrome P450 family 39 subfamily A member 1, EC:1.14.14.26
- CYP3A11
cytochrome P450 family 3 subfamily A member 11, EC:1.14.14
- CYP3A4
cytochrome P450 family 3 subfamily A member 4, EC:1.14.14
- CYP46A1
cytochrome P450 family 46 subfamily A member 1, EC:1.14.14.25
- CYP7A1
cytochrome P450 family 7 subfamily A member 1, EC:1.14.14.23
- CYP7B1
cytochrome P450 family 7 subfamily B member 1, EC:1.14.14.29
- D8D7I
3β‐hydroxysterol‐Δ8‐Δ7‐isomerase, EC:5.3.3.5
- DDA
dendrogenin A
- DDB
dendrogenin B
- DHCR24
24‐dehydrocholesterol reductase, EC:1.3.1.72
- DHCR7
7‐dehydrocholesterol reductase, EC:1.3.1.21
- EBI2
Epstein–Barr virus‐induced gene 2, GPR183
- ER(+)BC
ER‐positive breast cancer
- ER
oestrogen receptor
- GC
gas chromatography
- Gli
glioma‐associated oncogene homolog
- GPCR
G protein‐coupled receptor
- GPR183
G protein‐coupled receptor 183
- GR
glucocorticoid receptor
- HD
Huntington’s disease
- Hh
Hedgehog
- HMGCR
3‐hydroxy‐3‐methylglutaryl‐Coenzyme A reductase, EC:1.1.1.34
- HSD
hydroxysteroid dehydrogenase
- HSD11B1
hydroxysteroid 11‐beta dehydrogenase 1, EC:1.1.1
- HSD11B2
hydroxysteroid 11‐beta dehydrogenase 2, EC:1.1.1
- HSD3B7
hydroxy‐delta‐5‐steroid dehydrogenase, 3‐beta‐ and steroid delta‐isomerase 7, EC:1.1.1.181
- IFN
interferon
- INSIG
insulin‐induced gene
- LC
liquid chromatography
- LCAT
lecithin–cholesterol acyltransferase, EC:2.3.1.43
- LIPA
lysosomal acid lipase, EC:3.1.1.13
- LSS
lanosterol synthase, EC 5.4.99.7
- LTP
long‐term potentiation
- LXR
liver X receptor
- MS
mass spectrometry
- MSI
mass spectrometry imaging
- NMDAR
N‐methyl‐D‐aspartate receptor
- NPC1
Niemann–Pick C1
- NPC2
Niemann–Pick C2
- PD
Parkinson’s disease
- Ptch1
patched‐1
- SARS‐CoV‐2
severe acute respiratory syndrome coronavirus 2
- SCAP
SREBP cleavage‐activating protein
- SERM
selective oestrogen receptor modulator
- SHH
Sonic hedgehog
- SLOS
Smith–Lemli–Opitz syndrome
- Smo
Smoothened
- SMPD1
acid sphingomyelinase, EC:3.1.4.12
- SQLE
squalene epoxidase, EC:1.14.14.17
- SREBP‐1c
sterol regulatory‐binding protein‐1c
- SREBP‐2
sterol regulatory‐binding protein‐2
- TLR
Toll‐like receptor
Introduction
Cholesterol metabolism has been studied for many decades [1, 2, 3]. In mammals, the products of cholesterol metabolism are bile acids, and steroid hormones and their metabolites [4, 5]. While bile acids and steroid hormones are of undoubted importance, in recent years interest has shifted to intermediates in their biosynthesis and to a category of molecules known as oxysterols [6, 7, 8, 9]. Oxysterols can be defined as oxidised forms of cholesterol or of its precursors. They are formed in the first steps of cholesterol metabolism, mostly by cytochrome P450 (CYP) enzymes [3, 4, 10]. They can also be formed via nonenzymatic reactions both in vivo and ex vivo [11, 12, 13]. Many oxysterols have biological activity being ligands to, for example nuclear receptors, G protein‐coupled receptors (GPCRs) and glutamate receptors [6, 8, 9, 13].
There are many areas of biology in which oxysterols play a role. At the very beginning of life, oxysterols are key molecules in embryonic development acting along with other sterols to transmit the Hedgehog (Hh) signal [14], a key pathway for fate determination of stem cells and progenitor cells. Oxysterols activate this pathway by binding to Smoothened (Smo), a GPCR found at the cell membranes of primary cilia [15]. Oxysterols also appear as key molecules for definition of neural progenitor fate by activating the liver X receptors (LXRs) [16, 17], while cholestenoic acids, downstream metabolites of oxysterols, are important for survival or death of motor neurons [18]. Oxysterols have been also linked to cancer, through overactivation of Hh signalling and via many other mechanisms [9, 15, 19]. While some oxysterols are oncogenic, others appear to be protective against cancer [9]. Perhaps unsurprisingly as oxidised forms of cholesterol, oxysterols are implicated in the atherosclerotic process being found in atherosclerotic plaques [20]. Oxysterols also appear to be involved in the immune response, having either inflammatory or anti‐inflammatory properties [21, 22, 23, 24], and are generated in response to both bacterial infection and viral infection [25, 26, 27]. There is growing evidence that certain oxysterol may inhibit infection by the SARS‐CoV‐2 virus (COVID‐19) [28, 29, 30, 31].
Accepting that oxysterols are critical biological molecules, it is important to remember that oxysterols are a family of molecules, where small changes in geometry can lead to the difference between activity and inactivity. It is also crucial to be aware that oxysterol concentrations are very often determined following a base hydrolysis step where oxysterols esterified to fatty acids are released, so what is actually being determined is the sum of the free molecules and their esterified versions. Usually, oxysterol esters are more abundant than the nonesterified molecules, but it is the nonesterified molecules that are biologically active. Today, mass spectrometry (MS) in combination with liquid chromatography (LC), that is LC‐MS, or with gas chromatography (GC), that is GC‐MS, is almost exclusively used for oxysterol measurements [32, 33, 34, 35, 36, 37]. In the following sections, we will attempt to summarise the current ‘state of play’ in oxysterol research and endeavour to highlight key unresolved questions. We arrange the review by rotating around the major primary oxysterols derived from cholesterol (Fig. 1), looking at biological activity and downstream metabolites.
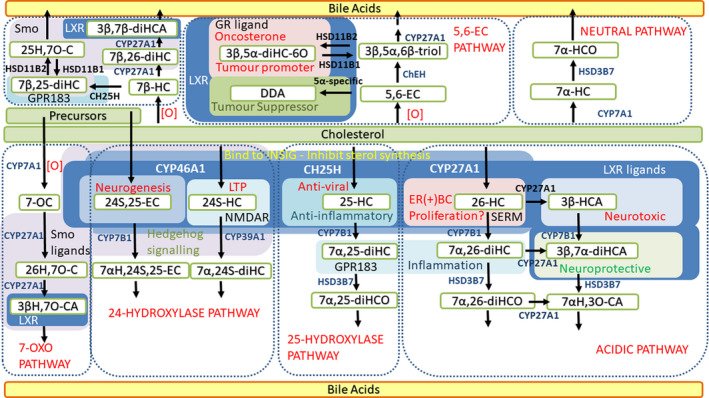
Fig. 1.
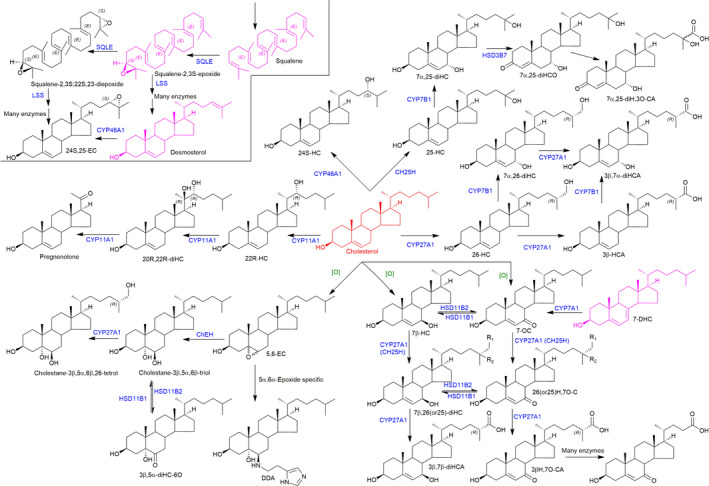
Structure of primary oxysterols and downstream metabolites. Cholesterol is shown in red, cholesterol precursors in purple and oxysterols in black. Enzymes are written in blue, and nonenzymatic oxidation is indicated by [O] in green. In 7β,26‐diHC and 26H,7O‐C, R1 = OH and R2 = H, while in 7β,25‐diHC and 25H,7O‐C, R1 = H and R2 = OH.
25‐hydroxycholesterol (25‐HC)
25‐HC is an unusual oxysterol in that cholesterol 25‐hydroxylase (CH25H, EC:1.14.99.38) is not a CYP enzyme but is a member of a small group of proteins that utilise a diiron cofactor to catalyse hydroxylation [38]. Note, 25‐HC can also be formed as a minor side product in reactions catalysed by CYP3A4 (EC:1.14.14, CYP3A11 in mouse), CYP27A1 and CYP46A1 [4, 10, 39, 40]. In normal circumstances, the level of 25‐HC is low in tissues and in the circulation [33, 41]; however, upon bacterial or viral infection CH25H (Ch25h in mouse) is upregulated in activated macrophages with the consequent enhanced formation of 25‐HC [23, 25, 26, 27, 42, 43, 44]. 25‐HC is reported to be anti‐inflammatory and antiviral [21, 26, 27, 45], and this information has stimulated much interest in 25‐HC in relation to SARS‐CoV‐2 [29, 30, 31]. CH25H is an interferon (IFN)‐stimulated gene, IFN being induced by Toll‐like receptor (TLR) 3 and TLR4 ligands upon bacterial infection [43, 44]. Upon SARS‐CoV‐2 viral infection, IFN, CH25H and other IFN‐stimulated genes are upregulated [29, 31]. Zang et al. identified 25‐HC as a potent inhibitor of SARS‐CoV‐2 replication, explaining this by 25‐HC blocking cholesterol export from the late endosome/lysosome compartment and restricting SARS‐CoV‐2 spike protein catalysed membrane fusion [29]. Interestingly, inhibition of Niemann–Pick C1 protein (NPC1), the cholesterol transporter that transports cholesterols out of late endosomes/lysosomes, also inhibited SARS‐CoV‐2 replication, supporting the theory that blocking cholesterol export from late endosomes/lysosomes inhibits viral replication (Fig. 2) [29].
Fig. 2.
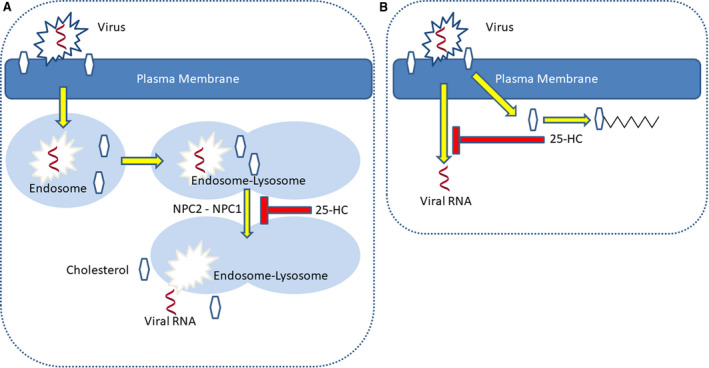
Simplified cartoon representation of the involvement of 25‐HC in protection against SARS‐CoV‐2 infection. (A) The virus enters the cell via endocytosis. Viral RNA escapes from the endosome/lysosome compartment by membrane fusion in concert with NPC2–NPC1‐mediated export of cholesterol. 25‐HC inhibits NPC1‐mediated cholesterol export and traps the virus in the late endosome/lysosome compartment [29]. Alternatively, (B) 25‐HC may activate acyl‐CoA cholesterol acyltransferase and sequester cholesterol as the ester, depleting the availability of plasma membrane nonesterified cholesterol required for membrane fusion and viral entry [31].
Deficiency in NPC1 (95% of cases) or NPC2 (5% of cases) leads to Niemann–Pick type C disease. While NPC1 protein transports cholesterol across the organelle membrane, NPC2 protein is soluble and carries nonesterified cholesterol to the NPC1 transporter [46]. Niemann–Pick type B disease shows some clinical and biochemically similarities to the type C disease [47, 48], but is genetically different, in that the type B disease results from mutations in the SMPD1 gene, and deficiency in the enzyme activity of acid sphingomyelinase (EC:3.1.4.12). It has been suggested that acid sphingomyelinase stimulates NPC2‐mediated cholesterol export by converting sphingomyelin to ceramide in the inner membranes of late endosomes [49], and the consequence of its deficiency is enhanced cholesterol content of lysosomes. In support of the hypothesis of Zang et al. [29] that blocking cholesterol export from late endosomes/lysosomes inhibits viral replication, Carpinteiro et al. [50] have recently found that inhibiting acid sphingomyelinase prevents SARS‐CoV‐2 uptake by epithelial cells, although their explanation for the involvement of acid sphingomyelinase in SARS‐CoV‐2 infection was at the level of ceramide in the outer leaflet of the plasma membrane. An alternative explanation for the antiviral activity of 25‐HC against SARS‐CoV‐2 is provided by Wang et al. [31] who found CH25H to be induced by SARS‐CoV‐2 in vitro, and suggested the antiviral activity of 25‐HC to be via inhibition of membrane fusion through depletion of plasma membrane cholesterol as a consequence of activation of acyl‐CoA cholesterol acyltransferase (EC:2.3.1.26), an enzyme that converts free cholesterol to its cholesteryl ester. Hence, one suggested mechanism of 25‐HC antiviral action is through cholesterol accumulation in the late endosomes, blocking membrane fusion and restricting the virus to this compartment [29], while a second mechanism is through depletion of plasma membrane cholesterol inhibiting viral membrane fusion and entry [31]. A combination of both mechanisms would suggest that 25‐HC can block membrane fusion and viral entry by reducing the available nonesterified cholesterol in membranes by inhibiting transport of cholesterol out of the late endosome/lysosome compartment and through activation of acyl‐CoA cholesterol acyltransferase. In Fig. 2, we present a simplified cartoon representation of the involvement of 25‐HC in preventing viral infection.
It is interesting to note that neither of the antiviral mechanisms discussed above invoked inhibition of SREBP‐2 (sterol regulatory‐binding protein‐2) processing or activation of LXRs, two key regulators of cellular cholesterol status. 25‐HC suppresses cholesterol biosynthesis by binding to the endoplasmic resident protein INSIG (insulin‐induced gene) tethering SREBP‐2 and its escort protein SCAP (SREBP cleavage‐activating protein) within the endoplasmic reticulum and preventing transport of SREBP‐2 to the Golgi for processing to its active form as the master transcription factor for the expression of genes of the cholesterol biosynthesis pathway [51]. Blanc et al. [26] proposed this mechanism to partially explain the antiviral action of macrophage produced 25‐HC towards a broad range of viruses, while Dang et al. [45] suggested inhibition of SREBP‐2 processing by 25‐HC prevents AIM2 (absent in melanoma 2) inflammasome activation in macrophages and provides an anti‐inflammatory circuit that prevents spurious AIM2 inflammasome activation. 25‐HC is also a ligand to the LXRs [52, 53], LXR activation leads to upregulation of SREBP‐1c and fatty acid synthesis [54], and also of the ABC (ATP‐binding cassette) transporters including ABCA1, ABCG1 and the cholesterol carrier protein apolipoprotein E (APOE) [55, 56]. In combination, the activation of LXR leads to removal of free cholesterol from cells by esterification, transport out of the cell by ABC transporters and its ultimate removal via apolipoproteins in the circulation. Hence, LXR activation by 25‐HC, and also 25‐HC binding to INSIG, may provide additional mechanisms for inhibition of COVID‐19 infection through depletion of membrane cholesterol.
There is now good cell‐based evidence that 25‐HC is protective against SARS‐CoV‐2 through a mechanism involving depletion of membrane cholesterol. The multiple biological activities of 25‐HC suggest that 25‐HC may have a multipronged mechanism for depleting membrane cholesterol and hence defence against the virus. It is interesting to note that total 25‐HC (sum of biologically active nonesterified 25‐HC and its inactive esterified form) is elevated in patients suffering mild SARS‐CoV‐2 [28], suggesting the successful defence against the virus by 25‐HC may be via its enhanced biosynthesis.
Key issues to resolve
Despite the availability of vaccines against SARS‐CoV‐2, which are being offered to citizens in rich countries of the developed world, it is questionable whether people in the developing world will be availed such a ‘luxury’. It is also unknown at present how effective the vaccines will be over time. Hence, alternative low‐cost strategies still require exploration. Once such alternative is treatment with the BCG vaccine, which is known to activate the TLR4 [57] and will presumably enhance the expression of IFN [58], so should theoretically enhance CH25H expression and the biosynthesis 25‐HC, leading to protection against SARS‐CoV‐2.
7α,25‐dihydroxycholesterol (7α,25‐diHC), 7β,25‐dihydroxycholesterol (7β,25‐diHC) and 25‐hydroxy‐7‐oxocholesterol (25H,7O‐C)
7α,25‐diHC is the major metabolic product of 25‐HC formed in a reaction catalysed by CYP7B1 (EC:1.14.14.29) [59]. It can also be formed from 7α‐hydroxycholesterol (7α‐HC) in a reaction catalysed by CYP3A4 (EC:1.14.14) in human and CYP3A11 (EC:1.14.14) in mouse [60]. 7α,25‐diHC does not show antiviral activity [29], and neither has it been shown to have an effect on SREBP‐2 processing or LXR activation. However, 7α,25‐diHC is a ligand towards the GPCR Epstein–Barr virus‐induced gene 2 (EBI2 or GPR 183) [22, 24] and acts as a chemoattractant to B and T cells expressing the receptor. Hence, in contrast to 25‐HC, which can be regarded as anti‐inflammatory [21, 45], 7α,25‐diHC is a proinflammatory lipokine. Further metabolism of 7α,25‐diHC leads to 7α,25‐dihydroxycholest‐4‐en‐3‐one (7α,25‐diHCO) catalysed by the enzyme hydroxysteroid dehydrogenase (HSD) 3B7 (EC:1.1.1) and further to 7α,25‐dihydroxy‐3‐oxocholest‐4‐en‐26‐oic acid (7α,25‐diH,3O‐CA), probably catalysed by CYP27A1 [61]. Interestingly, 7α,25‐diH,3O‐CA has been found to be of reduced abundance in cerebrospinal fluid (CSF) from patients with Alzheimer’s disease (AD) [62], linking the pathology with proinflammatory 7α,25‐diHC.
Like 7α,25‐diHC, 7β,25‐diHC is also a ligand to GPR183 [22]. However, until recently pathways for the formation of 7β,25‐diHC were unknown [10, 48, 63, 64]. Low levels of both 7β‐hydroxycholesterol (7β‐HC) and 7‐oxocholesterol (7‐OC) are always present in analysis of cholesterol‐derived oxysterols from biological samples [65]. However, the fact that both molecules can be formed from cholesterol via ex vivo autoxidation reactions makes interpretation regarding their formation difficult, as similar reactions can also occur endogenously [11]. Convincing evidence for the endogenous nature of 7β‐hydroxy and 7‐oxo metabolites of cholesterol has come from analysis of plasma from people with Niemann–Pick type C disease [47, 48, 66, 67, 68, 69, 70]. Jiang et al. provided data showing elevation of 7‐OC, and also of cholestane‐3β,5α,6β‐triol, in plasma from Niemann–Pick type C patients [68]. While more recently, we found elevated levels of both these two cholesterol derivatives and also 7β‐HC in plasma of patients suffering from both Niemann–Pick type C and type B disease [48]. How can we be sure that these are endogenous molecules, not artefacts generated by ex vivo autoxidation of cholesterol? Strong evidence for their endogenous nature would be downstream enzymatic products also evident in plasma or urine from Niemann–Pick patients. In fact, Alvelius et al. found unusual 7‐oxo‐ and 7β‐hydroxy bile acids in serum and urine from a Niemann–Pick type C patient in 2001 [66], and these identifications were confirmed by Maekawa et al. [71], and by Clayton and colleagues, who also identified a bile acid derived from cholestane‐3β,5α,6β‐triol [70]. Further evidence for the in vivo nature of 7β‐HC and 7‐OC was the discovery of an entire metabolic pathway from these molecules to 3β,7β‐dihydroxychol‐5‐enoic and 3β‐hydroxy‐7‐oxochol‐5‐enoic acids [48]. In patients with Niemann–Pick disease, it is likely that 7β‐HC and 7‐OC are derived by free radical oxidation of cholesterol [48, 66]. Once formed, 7‐OC and 7β‐HC are interconvertible through the HSD11B enzymes [72, 73]. HSD11B1 (EC:1.1.1) is the 7‐OC reductase, and HSD11B2 (EC:1.1.1), the 7β‐HC dehydrogenase. Both 7β‐HC and 7‐OC are substrates for CH25H, giving 7β,25‐diHC and 25H,7O‐C, respectively, and the two products can be interconverted by HSD11B enzymes [63]. Importantly, 7β,25‐diHC will act as a chemoattractant and activator of GPR183 but 25H,7O‐C will not [22, 63].
However, 25H,7O‐C itself is a biologically active molecule, binding and activating the GPCR protein Smo [64], a member of the Frizzled class of GPCRs. Smo plays a part in the Hh signalling pathway, its activation leading to Hh signalling through Gli (glioma‐associated oncogene homolog) transcription factors. The Hh pathway is essential for proper cell differentiation, and defects in the pathway lead to dysmorphology and cancer. Other key proteins in the Hh pathway are patched‐1 (Ptch1), a sterol transport protein structurally related to NPC1 [74], and the Hh ligand, for example Sonic hedgehog (SHH) post‐translationally modified with cholesterol [75]. Like Niemann–Pick disease, Smith–Lemli–Opitz syndrome (SLOS) is an autosomal recessive monogenetic disorder presenting with elevated 7β‐HC and 7‐OC in plasma and tissues [64, 76, 77]. SLOS also presents with dysmorphology and phenocopies defective Hh signalling [78]. 25H,7O‐C is present at elevated levels in plasma from SLOS patients [64], perhaps acting as a modulator of Smo in competition with other sterol activators. 7β,25‐diHC will also activate Smo and is also found in plasma from SLOS patients [64]. It is likely that the mechanisms behind the biosynthesis of 7β,25‐diHC and 25H,7O‐C in Niemann–Pick disease and SLOS are different. In SLOS, there is a deficiency in 7‐dehydrocholesterol reductase (DHCR7, EC:1.3.1.21), one of the final enzymes in the cholesterol biosynthesis pathways [79], and the consequence of this is a build‐up in 7‐dehydrocholesterol (7‐DHC). Like cholesterol, 7‐DHC is a substrate for CYP7A1 (EC:1.14.14); however, the enzyme products are different, in that the product of CYP7A1 oxidation of 7‐DHC is 7‐OC rather than 7α‐HC, which is formed from cholesterol [76, 80]. Hence, elevated levels of 7‐OC in SLOS are a likely consequence of CYP7A1 oxidation of 7‐DHC. As discussed above, 7‐OC can be reduced to 7β‐HC by HSD11B1 and both can be oxidised to give a 25‐hydroxy product, that is 25H,7O‐C and 7β,25‐diHC, respectively. We have proposed pathways by which 25H,7O‐C and 7β,25‐diHC can be metabolised further to 3β,7β,25‐trihydroxycholest‐5‐enoic acid and ultimately the C24 bile acid 3β,7β‐dihydroxychol‐5‐enoic acid [64].
Key issues to resolve and new ideas
GPR183 has been shown to direct the movement of activated B cells expressing this receptor to outer follicle regions of secondary lymphoid organs as required for mounting a normal B‐cell response to immune challenge. 7α,25‐diHC, 7β,25‐diHC and also 7α,(25R)26‐dihydroxycholesterol (7α,26‐diHC) all act as chemoattractants to B and T cells expressing GPR183. However, the gradient of 7α/β,25‐diHC or 7α,26‐diHC has yet to be measured across lymph nodes to add further evidence to the involvement of these oxysterols in the immune response. One attractive concept is that high levels of 7α,25‐diHC in the lymph node outer follicle attract B cells to mount the inflammatory response, while 7α,26‐diHC, derived from the circulation, reverses the motion, thereby switching off the immune response. Measurements of these oxysterols in tissue should now be possible with the advent of oxysterol mass spectrometry imaging (MSI) [81]. With respect to Hh signalling, Smo and oxysterols, it is unclear how in vitro activity of oxysterols translates to the situation in vivo, as besides oxysterols, cholesterol will also bind to and activate Smo [75]. If cholesterol rather than oxysterols is the true regulator of the Hh signal, the question is how can such an abundant sterol have signalling functions? Perhaps the answer lies in measuring cholesterol and oxysterol levels in primary cilia, the locality of Smo during the signalling event. Such experiments should now be possible with the advent of sterol‐MSI and will answer the question of whether oxysterols and/or cholesterol dictate Hh signalling [81].
24‐hydroxycholesterol (24‐HC)
There are two isomers of 24‐HC, 24S‐HC and 24R‐HC. The 24S‐HC epimer is dominant in man and mouse with 24R‐HC normally constituting of only about 10% of the total in the circulation [82]. 24S‐HC, or cerebrosterol, is as the name suggests mostly synthesised in brain [36, 83]. The enzyme responsible for 24S‐hydroxylation of cholesterol is CYP46A1 (EC:1.14.14.25), which is mostly expressed in neurons [40]. 24S‐HC acts as a transport form of cholesterol providing a route for removal of excess cholesterol from brain by crossing the blood–brain barrier, something that cholesterol itself cannot do [36]. Once extracerebral, 24S‐HC can be sulfated, glucuronidated or converted to bile acids [84, 85, 86]. The 24‐hydroxycholesterol 7α‐hydroxylase is CYP39A1 (EC:1.14.14.26), required to synthesise primary bile acids from 24S‐HC, and is mostly expressed in liver [87].
No inborn error of metabolism has been found resulting from a deficiency in CYP46A1 activity, and the Cyp46a1 −/− mouse is viable, showing a comparatively mild phenotype with deficiencies in spatial, associative and motor learning, and in hippocampal long‐term potentiation (LTP) [88, 89]. Interestingly, in these mice the defect in cholesterol metabolism in brain is compensated by its reduced biosynthesis, the overall level of cholesterol in brain being unchanged in the Cyp46a1 −/− mouse compared with control [90, 91]. 24S‐HC, like 25‐HC, is a ligand to the LXRs [52, 53] and to INSIG [51], and it is also a modulator of the N‐methyl‐D‐aspartate receptors (NMDARs) [92] and of Smo [93]. It is perhaps significant that 24S‐HC, via NMDARs, enhances the ability of subthreshold stimuli to induce LTP [92], considering that the absence of 24S‐HC biosynthesis in the Cyp46a1 −/− mouse is linked with a defect in LTP [89].
As 24S‐HC is generated almost exclusively by neurons in brain, its concentration in CSF and plasma has been explored as a marker of neurodegeneration [94]. In early stage disease, one might predict an initial rise in 24S‐HC, as neuronal loss leads to enhanced availability of cholesterol, the substrate for CYP46A1, but at later stages a decay in 24S‐HC as ever‐increasing numbers of neurons, and hence CYP46A1 enzymes, is lost from brain. This can make data interpretation challenging unless samples are well‐stratified. This is illustrated below.
In a recent study, Björkhem et al. [95] found 24S‐HC to be elevated in CSF from early Parkinson’s disease (PD) patients in comparison with controls. The same investigators had previously found that CSF 24S‐HC levels correlate with PD disease progression [96]. In contrast to the situation in CSF, the level of 24S‐HC in plasma was not found to differ between PD patients and controls [96]. These data suggest that elevated 24S‐HC in CSF is a marker of neurodegeneration. In patients with AD, 24S‐HC is again elevated in CSF, and this is also true of patients with mild cognitive impairment, but as with PD no differences were found in plasma levels of 24S‐HC [97]. Interestingly, 24S‐HC in CSF was found to increase according to APOE4 (apolipoprotein E 4) status, patients with two APOE4 alleles having the highest 24S‐HC content of CSF [97]. However, in plasma from severely affected AD patients the same investigators found the 24S‐HC to cholesterol ratio to be decreased in AD [98], presumably as a consequence of loss of CYP46A1 expressing neurons. In a separate study, 24S‐HC was found to be increased in plasma of AD patients, but the levels to negatively correlate with the severity of dementia [99]. Clearly, care must be exercised in stratifying patient samples to maximise the mechanistic insight provided by analytical data. Note, in these studies total 24S‐HC was measured, that is the sum of esterified and nonesterified 24S‐HC.
Levels of 24S‐HC have also been measured in plasma of patients with Huntington’s disease (HD), and concentrations found to vary according to disease severity. Leoni et al. [100] measured 24S‐HC in a major study of 150 samples and found that 24S‐HC was elevated in an early progression HD group compared with controls, but reduced compared with controls in a latter progression HD group. These data were at variance with an earlier study performed by Leoni et al. who found 24S‐HC to be reduced in HD plasma at all disease states [101].
The CSF and plasma measurements discussed above were all for total 24‐HC, which constitutes the sum of nonesterified and esterified 24‐HC. Usually, the nonesterified, biologically active molecules constitute only about 20% of the total [34]. In the circulation, oxysterols are esterified with fatty acids in a reaction catalysed by the enzyme lecithin–cholesterol acyltransferase (LCAT, EC:2.3.1.43) present in HDL particles. CSF lipoproteins tend to be small and spherical (≈ 10–20 nm)‐like plasma HDL [102], and human CSF contains LCAT at levels corresponding to ∼ 2.5% that of plasma LCAT [103]; however, the very minor levels of nonesterified 24‐HC (0.05 ng·mL−1 cf. 1.5 ng·mL−1esterified) in CSF indicate that this is sufficient to esterify most of the nonesterified 24S‐HC that is present [62, 104].
Major unresolved questions
In combination, the data presented above lead to the conclusion that metabolism of cholesterol to 24S‐HC is essential for brain health. However, is 24S‐HC per se an essential oxysterol? Evidence from studies on the Cyp46a1−/− mouse suggests that it is the flow through the cholesterol biosynthesis pathway that is essential rather than 24S‐HC itself [88, 89]. However, 24S‐HC is a modulator of the NMDARs, and a ligand to INSIG, LXRs and Smo, at least in vitro, and it is difficult to define its exact importance in activating these pathways as multiple other oxysterols (and sterols) have similar effects on these receptor proteins. A second important question is how good is 24S‐HC as a marker of neurodegeneration? From the studies mentioned above, it is very important to have well‐stratified groups to see a statistical effect. Is this of diagnostic value? The jury is still out.
24S,25‐epoxycholesterol (24S,25‐EC)
24S,25‐EC is one of the most efficacious endogenous LXR ligands [52, 53]. It is an unusual oxysterol in that the mechanism of its formation involves cholesterol precursors [105, 106]. There are two likely pathways: (a) 24S,25‐EC may be synthesised in parallel to the Bloch pathway of cholesterol biosynthesis but with squalene epoxidase (SQLE, also named squalene monooxygenase, EC:1.14.14.17) introducing two oxygen atoms, one to give a 2,3S‐epoxide and a second to give a 22S,23‐epoxide, to the squalene skeleton rather than just one to give the 2,3S‐epoxide. The two branches then proceed in parallel to give 24S,25‐EC and cholesterol, respectively, the only difference being that 24‐dehydrocholesterol reductase (DHCR24, EC:1.3.1.72) is absent from the pathway to generate 24S,25‐EC [6, 105, 107]. Lanosterol synthase (LSS, EC:5.4.99.7) is the enzyme that will cyclase both the squalene mono‐ and di‐epoxides, and its reduced activity will encourage di‐epoxide formation and ultimately that of 24S,25‐EC. (b) The alternative pathway to 24S,25‐EC is via CYP46A1 catalysed oxidation of desmosterol [106].
24S,25‐EC is seldom characterised in biological samples [33, 65], and this is a consequence of its comparatively low abundance and the labile nature of the 24S,25‐epoxy group. However, it has been analysed in studies, which do not include an acid or base hydrolysis step [15, 16, 17, 81, 93, 108, 109, 110]. In comparison with other oxysterols, 24S,25‐EC appears to be particularly prevalent in brain during development, perhaps a consequence of a high rate of cholesterol biosynthesis [16, 17, 109, 110, 111]. 24S,25‐EC acts as a ligand towards LXRs [52, 53], and Theofilopoulos et al. have generated compelling evidence that 24S,25‐EC, acting through LXRs, promotes midbrain dopaminergic neurogenesis [16, 17]. Besides acting as an LXR ligand, 24S,25‐EC will also bind to INSIG and repress cholesterol synthesis [51]. A more recently uncovered activity of 24S,25‐EC is as a ligand to Smo and activator of the Hh signalling pathway [15, 93, 112]. Cilia are protuberances on the outside of cells, which are required for Smo to transduce Hh signals. Smo accumulates in cilia, and cilia‐associated sterols promote this accumulation and Hh signalling. In search for sterols, which may activate the Hh pathway, Raleigh et al. [15] investigated the oxysterols enriched in cilia isolated from sea urchin. One of the oxysterols found was 24S,25‐EC. 24S,25‐EC was found to bind to the extracellular cysteine‐rich domain (CRD) of Smo and activate Smo in a dose‐dependent manner. Interestingly, 24S,25‐EC also activated Hh signalling through mutant Smo missing the CRD [15]. Molecular docking studies suggested 24S,25‐EC also bound to a cytoplasmic binding pocket and mutation studies indicated that Smo activation by 24S,25‐EC was via both binding sites [15]. Ptch1 is key protein involved in inhibition of the Hh pathway, acting as a sterol pump to deplete membranes of sterols. In an effort to identify sterols linked to Hh signalling, Qi et al. purified Ptch1 protein and identified 24S,25‐EC as one of the oxysterols co‐purified with Ptch1 [93]. They found evidence for 24S,25‐EC bound to the 7‐transmembrane region of Smo and to be more effective at activating Hh signalling than other sterols [93]. In combination, the data of Raleigh et al. and Qi et al. establish 24S,25‐EC as a ligand of Smo that can bind to multiple binding pockets and activate Hh signalling [15, 93]. The biological activities of 24S‐HC and 24S,25‐EC appear to overlap in that both activate LXRs, inhibit cholesterol biosynthesis via INSIG and repression of SREBP‐2 processing, and both are ligands to Smo. 24S‐HC and 24S,25‐EC are abundant in brain, and we speculate that brain biology has built a layer of redundancy in that CYP46A1 expressed in neurons and SQLE expressed in glia can each direct synthesis of the biologically active 24S‐oxidised sterols, that is 24S‐HC and 24S,25‐HC, respectively. Perhaps this explains the comparatively mild phenotype of the Cyp46a1−/− mouse.
Key issue
The 24S,25‐EC to cholesterol ratio is comparatively high during brain development [16, 17, 109, 110, 111]. This leads us to speculate that during brain development 24S,25‐EC acts as an in vivo ligand to Smo and controls Hh signalling and Hh‐linked development.
(25R)26‐Hydroxycholesterol (26‐HC)
26‐HC, more commonly known by the nonsystematic name 27‐hydroxycholesterol (27‐HC), is the first intermediate in the ‘acidic’, also known as the ‘alternative’, pathway of bile acid biosynthesis [2, 4, 10]. It is synthesised from cholesterol by CYP27A1 (EC:1.14.15.15) and metabolised further to 3β‐hydroxycholest‐5‐en‐(25R)26‐oic acid (3β‐HCA) or to 7α,(25R)26‐dihydroxycholesterol (7α,26‐diHC) by CYP27A1 and CYP7B1, respectively. 3β‐HCA and 7α,26‐HC are both biologically active, the former as a ligand towards LXRs [18, 113], and the latter as a ligand to GPR183 (see above) [22]. 26‐HC is in its own right an LXR ligand, although a comparatively weak agonist [53]. 26‐HC is also a selective oestrogen receptor modulator (SERM) in that it shows anti‐oestrogenic effects or pro‐oestrogenic effects that are cell type‐specific [114, 115]. Oestrogen receptors (ERs) are expressed in vascular cells and mediate cardioprotective effects of oestrogens. However, Umetani et al. [114] have shown that 26‐HC can act as a competitive antagonist of ER in the vasculature leading to a loss of oestrogen protection towards vascular disease. ERs are also expressed by breast cancer cells, and there is evidence that 26‐HC acts as a partial agonist in these cells [19, 115, 116]. Given that 26‐HC is a direct product of cholesterol metabolism, these findings have implications with respect to breast cancer and hypercholesteraemic women. In fact, Wu et al. found that in ER(+) breast cancer (ER(+)BC) patients the 26‐HC content of normal tissue was higher than that from controls, and tumour 26‐HC levels were further elevated [116]. In a study published at almost exactly the same time, Nelson et al. [19] reported that in breast tissue CYP27A1 levels correlate with tumour grade. Nelson et al. [19] showed that 26‐HC stimulated ER(+)BC proliferation through the ER and invasiveness through LXR. In a later study, Nelson [117] proposed that inhibition of CYP27A1 along with ERα and LXR antagonists could increase the efficacy of treatments towards ER(+)BC.
Surprisingly, in the light of the discussion above, a systematic review and meta‐analysis of prospective studies found a modest but statistically significant inverse association between total cholesterol, more specifically HDL cholesterol, and the risk of breast cancer [118]. In addition, in a study of almost 300 breast cancer cases no association was found between circulating 26‐HC and breast cancer risk [119], and in postmenopausal women, circulating 26‐HC was associated with a lower risk of breast cancer [120]. Unfortunately, the authors of these reports did not clarify whether they were measuring the total 26‐HC, that is the sum of the inactive ester and the active nonesterified molecule or just the active nonesterified molecule. The reader is left to guess, but in all probability a reported value of about 200 nm (80 ng·mL−1) in plasma refers to the total 26‐HC. As discussed elsewhere, there is a need for clarity in reporting of mass spectrometry data [121].
Like 25‐HC, nonesterified 26‐HC has been suggested to be an antiviral oxysterol [28]. Marcello et al., measuring total 26‐HC, found the plasma level of this oxysterol in severely affected COVID‐19 patients to be almost half that in control subjects [28]. Serum levels of cholesterol and its precursors were low in both moderate and severe COVID‐19 cases, possibly explaining reduced levels of 26‐HC. Notably, levels of antiviral 25‐HC were also reduced in severe COVID‐19 cases, although the reduction was not great (8.52 ± 2.58 ng·mL−1 in controls cf. 7.64 ± 2.49 ng·mL−1 in severe cases). Importantly, the reader should be reminded that total sterols were being measured not the nonesterified bioactive molecules.
Perhaps the reduced availability of cholesterol to cells is a key aspect in the pathophysiology of COVID‐19 leading to reduced biosynthesis of antiviral 25‐HC and 26‐HC. However, based on studies suggesting 25‐HC is antiviral through reducing cholesterol availability to membranes [29] it might be expected that reduced serum cholesterol would be beneficial in protection against COVID‐19.
Key issue
Are total oxysterol levels a good surrogate measure for concentrations of the nonesterified bioactive molecules? As LCAT is abundant in HDL particles and will esterify sterols, it not an unreasonable assumption that the levels of esterified oxysterols are reflective of bioactive nonesterified oxysterols exported from cells. However, care should be taken when relating concentrations of the total oxysterol measured in plasma to that required for an in vitro or in vivo biological activity.
Cholestenoic acids: 3β‐HCA, 3β,7α‐dihydroxycholest‐5‐en‐(25R)26‐oic (3β,7α‐diHCA), 3β,7β‐dihydroxycholest‐5‐en‐(25R)26‐oic (3β,7β‐diHCA) and 3β‐hydroxy‐7‐oxocholest‐5‐en‐(25R)26‐oic (3βH,7O‐CA) acids
3β‐HCA and 3β,7α‐diHCA are intermediates in the acidic pathway of bile acid biosynthesis [4, 10], and both these molecules, and the downstream metabolite 7α‐hydroxy‐3‐oxocholest‐4‐en‐(25R)26‐oic acid (7αH,3O‐CA), are present in CSF and/or brain [41, 62, 81]. Meaney et al. [122] have shown that 7αH,3O‐CA provides a metabolic export route for 26‐HC from brain, which itself is imported to brain, and is the first metabolite in the acidic pathway of bile acid biosynthesis [4, 10]. Both 3β‐HCA and 3β,7α‐diHCA are LXR ligands, as are 3β,7β‐diHCA and 3βH,7O‐CA, but not 7αH,3O‐CA [18, 113]. Interestingly, Theofilopoulos et al. showed that in the developing brain 3β,7α‐diHCA promoted motor neuron survival in an LXR‐dependent manner, 3βH,7O‐CA promoted maturation of precursors into motor neurons, while 3β‐HCA was toxic, showing that cholestenoic acids dictate the balance between life and death of motor neurons [18]. In a more recent study, Abdel‐Khalik et al. [64] have shown that 3β,7β‐diHCA and 3βH,7O‐CA activate Hh signalling by binding to Smo, highlighting a further potential role for cholestenoic acids in development.
New ideas
In the light of the biological activity of 26‐HC, 7α,26‐diHC and of the cholestenoic acids, we speculate that the acidic pathway evolved as more than a pathway of bile acid biosynthesis. The widespread expression of CYP27A1 contrasts to that of liver‐specific CYP7A1, the first enzyme in the neutral pathway of bile acid biosynthesis [4, 10], and it may also be significant that CYP27A1 is an inner mitochondrial enzyme, in contrast to most other CYPs involved in cholesterol metabolism, which are localised to the endoplasmic reticulum. We suggest that the acidic pathway initially evolved to generate biologically active signalling molecules and its development within hepatocytes provided the bonus of bile acid formation to remove excess cholesterol and to aid absorption in the intestine of dietary lipids. This hypothesis is supported by the activity of the acidic pathway during embryonic development [18, 109] and data that suggest that in infants the acidic pathway is more important than the neutral pathway of bile acid biosynthesis [123]. Kakiyama et al. [124] have proposed a somewhat similar evolutionary role for bile acid biosynthesis. They suggest that the acidic pathway evolved as a mechanism to remove excess cholesterol from the inner mitochondrial leaflet by CYP27A1 metabolism to 26‐HC, with further metabolism regulated by endoplasmic reticulum‐resident CYP7B1. They then proposed that an inability of the acidic pathway to increase the synthesis of bile acids without generating toxic intermediates leads to evolution of the neutral pathway starting with CYP7A1 and generating less‐toxic intermediates [124]. The two hypothesises differ in that Kakiyama and Pandak focus on toxic intermediates and a requirement for a different and neutral pathway [124], while we focus more on the positive effects of intermediates of the acidic pathway.
Cholestan‐5,6‐epoxide (5,6‐epoxycholesterol, 5,6‐EC) and cholestane‐3β,5α,6β‐triol
There are two isomers of 5,6‐EC with either 5α or 5β stereochemistry. Both are formed by free radical oxidation of cholesterol [11]. Both isomers can be hydrolysed by the enzyme cholesterol epoxide hydrolase (ChEH, EC:3.3.2.11) to cholestane‐3β,5α,6β‐triol. ChEH is an unusual enzyme in that it made up of two subunits, each of which is an enzymes in its own right, and part of the cholesterol biosynthesis pathway, that is DHCR7 and 3β‐hydroxysterol‐Δ8‐Δ7‐isomerase (D8D7I, EC:5.3.3.5) [125]. As mentioned above, cholestane‐3β,5α,6β‐triol is elevated in the circulation people with Niemann–Pick type C and type B disease and also those with lysosomal acid lipase (LIPA, EC:3.1.1.13) deficiency, known as Wolman disease when there is a complete absence of the enzyme [48, 68, 126]. The origin of cholestane‐3β,5α,6β‐triol is likely to be via hydrolysis of 5,6‐EC, which is also elevated in Niemann–Pick and in Wolman diseases [48]. Cholestane‐3β,5α,6β‐triol can be metabolised via multiple reactions to bile acids [48, 69, 70], or alternatively oxidised by HSD11B2 to 3β,5α‐dihydroxycholestan‐6‐one (oncosterone, 3β,5α‐diHC‐6O). As the trivial name oncosterone implies, 3β,5α‐diHC‐6O is a tumour promoter (Fig. 3) [127].
Fig. 3.
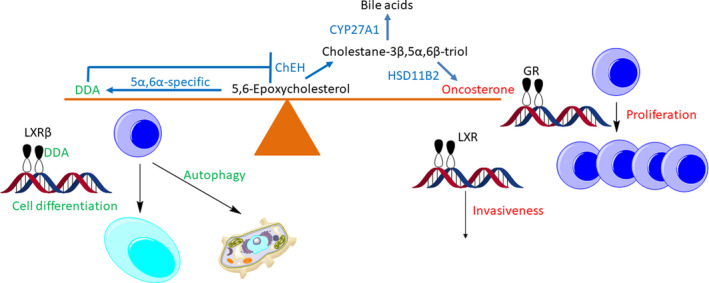
5,6‐EC at the fulcrum of protection against and the proliferation of cancer.
Oncosterone has been shown to promote proliferation of mouse and human ER(+)BC and triple‐negative breast cancer cell lines, and growth of breast cancer tumours in vivo [127]. The proliferative effects of oncosterone are through its activation of the glucocorticoid receptor (GR) [127]. Interestingly, corticosteroid ligands to GR do not share the proliferative activities of oncosterone [9]. Oncosterone also acts as a ligand to the LXR receptors, and it has been suggested that the pro‐invasive effects of oncosterone are mediated by LXR [127].
An alternative route for metabolism of 5α,6‐EC, but not 5β,6‐EC, is enzymatic conjugation with an amine nucleophile. Two such nucleophiles showed to react with 5α,6‐EC in chemically catalysed reactions are histamine and spermidine to give dendrogenin A (DDA) and dendrogenin B (DDB), respectively. Both of these metabolites have now been identified in mammalian systems [128]. In contrast to oncosterone, DDA is oncosuppressive. Importantly, DDA is not detected in cancer cell lines, and its level in breast tissue is decreased during oncogenesis [129]. The oncosuppressive effects of DDA are at least part through activation of LXRβ, inducing autophagy and cell differentiation [130], while a second pathway is through inhibition of ChEH and follow‐on inhibition of biosynthesis of oncosterone.
A Role in therapeutic development
The studies discussed above regarding metabolism of 5,6‐EC indicate three potential targets for pharmaceutical intervention:
The balance between cholestane‐3β,5α,6β‐triol and oncosterone is dependent on the enzymes HSD11B2 and HSD11B1. Inhibition of HSD11B2 will reduce the formation of oncosterone.
ChEH generates cholestane‐3β,5α,6β‐triol from 5,6‐EC. Inhibition of ChEH should reduce the formation of cholestane‐3β,5α,6β‐triol and consequently that of oncosterone. Likewise, inhibition of ChEH should shunt 5α,6‐EC towards the biosynthesis of DDA that is oncosuppressive through LXRβ.
A quite different idea is the upregulation of CYP27A1. Assuming from the studies of Le Cornet and coworkers that there is no correlation between circulating 26‐HC and breast cancer risk [119, 120], and in direct contrast to the suggestion of Nelson to inhibit CYP27A1 [117], enhancing CYP27A1 expression or activity should drive cholestane‐3β,5α,6β‐triol into the bile acid biosynthesis pathway and away from metabolism by HSD11B2 to oncosterone. These ideas are yet to be tested.
22R‐hydroxycholesterol (22R‐HC)
Cholesterol is converted to 22R‐HC by CYP11A1 (EC:1.14.15.6). Like CYP27A1, CYP11A1 is a resident of the innermitochondrial membrane. CYP11A1 can then convert 22R‐HC to 20R,22R‐dihydroxycholesterol (20R,22R‐diHC) and ultimately the C21 steroid, pregnenolone. These reactions may or may not proceed with the release of the oxysterol intermediates [131, 132]. Like other side‐chain oxysterols, 22R‐HC is a ligand to LXRs and an inhibitor of SREBP‐2 processing to its active form [51, 52]. The metabolism of 20R‐HC and 20R,22R‐diHC to bile acids rather than steroids is another story yet to be told.
Cholesterol precursors and their oxysterol derivatives
The hypothesis that the acidic pathway evolved to generate biologically active intermediates can be extended to include the Kandutsch–Russell and Bloch pathways of sterol biosynthesis. Lanosterol, the first sterol, has been shown to stimulate INSIG‐mediated degradation of HMGCoA reductase (HMGCR, EC:1.1.1.34), the enzyme catalysing the rate‐determining step of cholesterol synthesis, and remarkably, 26‐hydroxylanosterol, also called 27‐hydroxylanosterol, is 10 times more potent [133]. Other sterols in the Kandutsch–Russell and Bloch pathways with 4,4‐dimethyl groups were shown to similarly accelerate the degradation of HMGCR, as does 25‐HC [133, 134]. Lanosterol does not repress the SREBP‐2 activation of cholesterol biosynthesis, but in contrast to initial data [133], other 4,4‐dimethyl sterols are reported to inhibit SREBP‐2 activation [134]. At the other end of the cholesterol biosynthesis pathway from lanosterol, desmosterol inhibits the processing of SREBP‐2 and acts as a ligand towards LXRs [135]. As discussed above, desmosterol acts as a substrate for CYP46A1 generating biologically active 24S,25‐EC [106], which can also be formed in a shunt of the Bloch pathway [105]. 7‐DHC, the final member of the Kandutsch–Russell pathway, is also a source of bioactive metabolites, and these may be formed enzymatically, or via free radical reactions, 7‐DHC being particularly susceptible to nonenzymatic oxidation. For example, 3β,5α‐dihydroxycholest‐7‐en‐6‐one, formed by hydrolysis and oxidation of 7‐dehydrocholesterol‐5α,6‐epoxide, is a Hh pathway antagonist binding to Smo at a site distinct from the CRD and the cyclopamine pocket [136]. As discussed earlier, 7‐DHC can be converted to 7‐OC by CYP7A1 [80], and this opens a route to 7β‐HC and multiple biologically active oxysterols [22, 64, 137].
Conclusions
Oxysterols and other intermediates in the bile acid biosynthesis pathways have interested sterol chemists for decades [13]. It appears that while the neutral pathway of bile acid biosynthesis generates intermediates with comparatively little biological activity, the reverse appears to be the case with the acidic pathway. The prominence of the acidic pathway in early life highlights the importance of its intermediates, and as suggested above, it is tempting to speculate that the acidic pathway evolved to generate and regulate biologically active molecules, and its utilisation by hepatocytes provided an added bonus of generating bile acids.
Besides the acidic pathway, other pathways to bile acids, and also to steroid hormones, generate bioactive intermediates. In which case, progression towards bile acids may also be a way of regulating lipokine biosynthesis and metabolism in specific cell types and tissues, for example CYP46A1 is expressed in brain; CH25H in activated immune cells; and CYP11A1 in sex organs and the adrenal gland. In contrast to these three enzymes, CYP27A1, initiating the acidic pathway, and CYP7B1 acting as the 7α‐hydroxylase in this pathway are rather ubiquitously expressed and can be ‘lent’ to the different bile acid biosynthesis pathway to help regulate the formation/metabolism of the active intermediates (Fig. 4). Of course, an ultimate function of bile acid formation is also to remove excess cholesterol.
Fig. 4.
The acidic pathway of bile acid biosynthesis generates bioactive intermediates. Key enzymes in this pathway, CYP27A1 and CYP7B1, can be lent to different bile acid biosynthesis pathways to similarly generate and regulate other lipokines.
Bile acids can also be synthesised from oxysterols generated by nonenzymatic reactions. Incredibly, intermediates in these pathways also have biological activity. While 7‐OC and 7β‐HC can also be formed enzymatically, an enzyme to generate 5,6‐EC has yet to be isolated. Is there one, or has the pathway from 5,6‐EC evolved exclusively to remove products of cholesterol oxidation formed by reactive oxygen species?
The multiple pathways to bile acids provide redundancy in the biological system with many intermediates sharing similar activities, and this is probably the reason why most of the inborn errors of bile acid biosynthesis are not fatal, and the equivalent knockout mice are viable. However, these inborn errors do lead to disease indicating the imperfection of the back‐up system.
The huge range of bile acid intermediates, the crossover of pathways, sharing of enzymes, shuttling of oxysterols between different organelles, cell types and tissues make this a fascinating field to work in, with still many important discoveries to be made.
Conflict of interest
WJG and YW are listed as inventors on the patent ‘Kit and method for quantitative detection of steroids’ US9851368B2. WJG and YW are listed as inventors on the patent application ‘Diagnostic methods and kits’ WO2017037465A1. WJG, EY and YW are shareholders in CholesteniX Ltd.
Author contributions
WJG and YW conceived the paper. EY provided valuable insight regarding oxysterol analysis. All authors contributed to the writing of the paper, and reviewed and edited the final version.
Acknowledgements
This work was supported by the UK Biotechnology and Biological Sciences Research Council (BBSRC, Grant Numbers BB/N015932/1 to WJG and BB/L001942/1 to YW). Members of the European Network for Oxysterol Research (ENOR, https://www.oxysterols.net/) are thanked for informative discussions.
References
- 1. Bergstrom S & Norman A (1953) Metabolic products of cholesterol in bile and feces of rat; steroids and bile acids. Proc Soc Exp Biol Med 83, 71–74. [DOI] [PubMed] [Google Scholar]
- 2. Hofmann AF & Hagey LR (2014) Key discoveries in bile acid chemistry and biology and their clinical applications: history of the last eight decades. J Lipid Res 55, 1553–1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Russell DW (2018) Lucky, times ten: a career in Texas science. J Biol Chem 293, 18804–18827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Russell DW (2003) The enzymes, regulation, and genetics of bile acid synthesis. Annu Rev Biochem 72, 137–174. [DOI] [PubMed] [Google Scholar]
- 5. Shackleton CH (1986) Profiling steroid hormones and urinary steroids. J Chromatogr 379, 91–156. [DOI] [PubMed] [Google Scholar]
- 6. Brown AJ, Sharpe LJ & Rogers MJ (2020) Oxysterols: from physiological tuners to pharmacological opportunities. Br J Pharmacol. doi: 10.1111/bph.15073 [DOI] [PubMed] [Google Scholar]
- 7. Bjorkhem I (2013) Five decades with oxysterols. Biochimie 95, 448–454. [DOI] [PubMed] [Google Scholar]
- 8. Wang Y, Yutuc E & Griffiths WJ (2020) Neuro‐oxysterols and neuro‐sterols as ligands to nuclear receptors, GPCRs, ligand‐gated ion channels and other protein receptors. Br J Pharmacol. doi: 10.1111/bph.15191 [DOI] [PubMed] [Google Scholar]
- 9. de Medina P, Diallo K, Huc‐Claustre E, Attia M, Soules R, Silvente‐Poirot S & Poirot M (2020) The 5,6‐epoxycholesterol metabolic pathway in breast cancer: emergence of new pharmacological targets. Br J Pharmacol. doi: 10.1111/bph.15205 [DOI] [PubMed] [Google Scholar]
- 10. Griffiths WJ & Wang Y (2020) Oxysterols as lipid mediators: their biosynthetic genes, enzymes and metabolites. Prostaglandins Other Lipid Mediat 147, 106381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zerbinati C & Iuliano L (2017) Cholesterol and related sterols autoxidation. Free Radic Biol Med 111, 151–155. [DOI] [PubMed] [Google Scholar]
- 12. Murphy RC & Johnson KM (2008) Cholesterol, reactive oxygen species, and the formation of biologically active mediators. J Biol Chem 283, 15521–15525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Schroepfer GJ Jr (2000) Oxysterols: modulators of cholesterol metabolism and other processes. Physiol Rev 80, 361–554. [DOI] [PubMed] [Google Scholar]
- 14. Nachtergaele S, Mydock LK, Krishnan K, Rammohan J, Schlesinger PH, Covey DF & Rohatgi R (2012) Oxysterols are allosteric activators of the oncoprotein smoothened. Nat Chem Biol 8, 211–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Raleigh DR, Sever N, Choksi PK, Sigg MA, Hines KM, Thompson BM, Elnatan D, Jaishankar P, Bisignano P, Garcia‐Gonzalo FR et al. (2018) Cilia‐associated oxysterols activate smoothened. Mol Cell 72, 316–327.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Theofilopoulos S, Wang Y, Kitambi SS, Sacchetti P, Sousa KM, Bodin K, Kirk J, Salto C, Gustafsson M, Toledo EM et al. (2013) Brain endogenous liver X receptor ligands selectively promote midbrain neurogenesis. Nat Chem Biol 9, 126–133. [DOI] [PubMed] [Google Scholar]
- 17. Theofilopoulos S, Abreu de Oliveira WA, Yang S, Yutuc E, Saeed A, Abdel‐Khalik J, Ullgren A, Cedazo‐Minguez A, Bjorkhem I, Wang Y et al. (2019) 24(S),25‐Epoxycholesterol and cholesterol 24S‐hydroxylase (CYP46A1) overexpression promote midbrain dopaminergic neurogenesis in vivo. J Biol Chem 294, 4169–4176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Theofilopoulos S, Griffiths WJ, Crick PJ, Yang S, Meljon A, Ogundare M, Kitambi SS, Lockhart A, Tuschl K, Clayton PT et al. (2014) Cholestenoic acids regulate motor neuron survival via liver X receptors. J Clin Invest 124, 4829–4842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Nelson ER, Wardell SE, Jasper JS, Park S, Suchindran S, Howe MK, Carver NJ, Pillai RV, Sullivan PM, Sondhi V et al. (2013) 27‐Hydroxycholesterol links hypercholesterolemia and breast cancer pathophysiology. Science 342, 1094–1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Brown AJ & Jessup W (1999) Oxysterols and atherosclerosis. Atherosclerosis 142, 1–28. [DOI] [PubMed] [Google Scholar]
- 21. Reboldi A, Dang EV, McDonald JG, Liang G, Russell DW & Cyster JG (2014) Inflammation. 25‐Hydroxycholesterol suppresses interleukin‐1‐driven inflammation downstream of type I interferon. Science 345, 679–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hannedouche S, Zhang J, Yi T, Shen W, Nguyen D, Pereira JP, Guerini D, Baumgarten BU, Roggo S, Wen B et al. (2011) Oxysterols direct immune cell migration via EBI2. Nature 475, 524–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bauman DR, Bitmansour AD, McDonald JG, Thompson BM, Liang G & Russell DW (2009) 25‐Hydroxycholesterol secreted by macrophages in response to Toll‐like receptor activation suppresses immunoglobulin A production. Proc Natl Acad Sci USA 106, 16764–16769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Liu C, Yang XV, Wu J, Kuei C, Mani NS, Zhang L, Yu J, Sutton SW, Qin N, Banie H et al. (2011) Oxysterols direct B‐cell migration through EBI2. Nature 475, 519–523. [DOI] [PubMed] [Google Scholar]
- 25. Diczfalusy U, Olofsson KE, Carlsson AM, Gong M, Golenbock DT, Rooyackers O, Flaring U & Bjorkbacka H (2009) Marked upregulation of cholesterol 25‐hydroxylase expression by lipopolysaccharide. J Lipid Res 50, 2258–2264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Blanc M, Hsieh WY, Robertson KA, Kropp KA, Forster T, Shui G, Lacaze P, Watterson S, Griffiths SJ, Spann NJ et al. (2013) The transcription factor STAT‐1 couples macrophage synthesis of 25‐hydroxycholesterol to the interferon antiviral response. Immunity 38, 106–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Liu SY, Aliyari R, Chikere K, Li G, Marsden MD, Smith JK, Pernet O, Guo H, Nusbaum R, Zack JA et al. (2013) Interferon‐inducible cholesterol‐25‐hydroxylase broadly inhibits viral entry by production of 25‐hydroxycholesterol. Immunity 38, 92–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Marcello A, Civra A, Milan Bonotto R, Nascimento Alves L, Rajasekharan S, Giacobone C, Caccia C, Cavalli R, Adami M, Brambilla P et al. (2020) The cholesterol metabolite 27‐hydroxycholesterol inhibits SARS‐CoV‐2 and is markedly decreased in COVID‐19 patients. Redox Biol 36, 101682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zang R, Case JB, Yutuc E, Ma X, Shen S, Gomez Castro MF, Liu Z, Zeng Q, Zhao H, Son J et al. (2020) Cholesterol 25‐hydroxylase suppresses SARS‐CoV‐2 replication by blocking membrane fusion. Proc Natl Acad Sci USA 117, 32105–32113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zu S, Deng YQ, Zhou C, Li J, Li L, Chen Q, Li XF, Zhao H, Gold S, He J et al. (2020) 25‐Hydroxycholesterol is a potent SARS‐CoV‐2 inhibitor. Cell Res 30, 1043–1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wang S, Li W, Hui H, Tiwari SK, Zhang Q, Croker BA, Rawlings S, Smith D, Carlin AF & Rana TM (2020) Cholesterol 25‐Hydroxylase inhibits SARS‐CoV‐2 and other coronaviruses by depleting membrane cholesterol. EMBO J 39, e106057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. McDonald JG, Smith DD, Stiles AR & Russell DW (2012) A comprehensive method for extraction and quantitative analysis of sterols and secosteroids from human plasma. J Lipid Res 53, 1399–1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Stiles AR, Kozlitina J, Thompson BM, McDonald JG, King KS & Russell DW (2014) Genetic, anatomic, and clinical determinants of human serum sterol and vitamin D levels. Proc Natl Acad Sci USA 111, E4006–E4014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Dzeletovic S, Breuer O, Lund E & Diczfalusy U (1995) Determination of cholesterol oxidation products in human plasma by isotope dilution‐mass spectrometry. Anal Biochem 225, 73–80. [DOI] [PubMed] [Google Scholar]
- 35. Honda A, Yamashita K, Hara T, Ikegami T, Miyazaki T, Shirai M, Xu G, Numazawa M & Matsuzaki Y (2009) Highly sensitive quantification of key regulatory oxysterols in biological samples by LC‐ESI‐MS/MS. J Lipid Res 50, 350–357. [DOI] [PubMed] [Google Scholar]
- 36. Lutjohann D, Breuer O, Ahlborg G, Nennesmo I, Siden A, Diczfalusy U & Bjorkhem I (1996) Cholesterol homeostasis in human brain: evidence for an age‐dependent flux of 24S‐hydroxycholesterol from the brain into the circulation. Proc Natl Acad Sci USA 93, 9799–9804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Crick PJ, William Bentley T, Abdel‐Khalik J, Matthews I, Clayton PT, Morris AA, Bigger BW, Zerbinati C, Tritapepe L, Iuliano L et al. (2015) Quantitative charge‐tags for sterol and oxysterol analysis. Clin Chem 61, 400–411. [DOI] [PubMed] [Google Scholar]
- 38. Lund EG, Kerr TA, Sakai J, Li WP & Russell DW (1998) cDNA cloning of mouse and human cholesterol 25‐hydroxylases, polytopic membrane proteins that synthesize a potent oxysterol regulator of lipid metabolism. J Biol Chem 273, 34316–34327. [DOI] [PubMed] [Google Scholar]
- 39. Honda A, Miyazaki T, Ikegami T, Iwamoto J, Maeda T, Hirayama T, Saito Y, Teramoto T & Matsuzaki Y (2011) Cholesterol 25‐hydroxylation activity of CYP3A. J Lipid Res 52, 1509–1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lund EG, Guileyardo JM & Russell DW (1999) cDNA cloning of cholesterol 24‐hydroxylase, a mediator of cholesterol homeostasis in the brain. Proc Natl Acad Sci USA 96, 7238–7243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Abdel‐Khalik J, Yutuc E, Crick PJ, Gustafsson JA, Warner M, Roman G, Talbot K, Gray E, Griffiths WJ, Turner MR et al. (2017) Defective cholesterol metabolism in amyotrophic lateral sclerosis. J Lipid Res 58, 267–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Diczfalusy U (2013) On the formation and possible biological role of 25‐hydroxycholesterol. Biochimie 95, 455–460. [DOI] [PubMed] [Google Scholar]
- 43. Park K & Scott AL (2010) Cholesterol 25‐hydroxylase production by dendritic cells and macrophages is regulated by type I interferons. J Leukoc Biol 88, 1081–1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. McDonald JG & Russell DW (2010) Editorial: 25‐Hydroxycholesterol: a new life in immunology. J Leukoc Biol 88, 1071–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Dang EV, McDonald JG, Russell DW & Cyster JG (2017) Oxysterol restraint of cholesterol synthesis prevents AIM2 inflammasome activation. Cell 171, 1057–1071.e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Wang ML, Motamed M, Infante RE, Abi‐Mosleh L, Kwon HJ, Brown MS & Goldstein JL (2010) Identification of surface residues on Niemann‐Pick C2 essential for hydrophobic handoff of cholesterol to NPC1 in lysosomes. Cell Metab 12, 166–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Griffiths WJ, Gilmore I, Yutuc E, Abdel‐Khalik J, Crick PJ, Hearn T, Dickson A, Bigger BW, Wu TH, Goenka A et al. (2018) Identification of unusual oxysterols and bile acids with 7‐oxo or 3beta,5alpha,6beta‐trihydroxy functions in human plasma by charge‐tagging mass spectrometry with multistage fragmentation. J Lipid Res 59, 1058–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Griffiths WJ, Yutuc E, Abdel‐Khalik J, Crick PJ, Hearn T, Dickson A, Bigger BW, Hoi‐Yee Wu T, Goenka A, Ghosh A et al. (2019) Metabolism of non‐enzymatically derived oxysterols: clues from sterol metabolic disorders. Free Radic Biol Med 144, 124–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Gallala HD, Breiden B & Sandhoff K (2011) Regulation of the NPC2 protein‐mediated cholesterol trafficking by membrane lipids. J Neurochem 116, 702–707. [DOI] [PubMed] [Google Scholar]
- 50. Carpinteiro A, Edwards MJ, Hoffmann M, Kochs G, Gripp B, Weigang S, Adams C, Carpinteiro E, Gulbins A, Keitsch S et al. (2020) Pharmacological inhibition of acid sphingomyelinase prevents uptake of SARS‐CoV‐2 by epithelial cells. Cell Rep Med 1, 100142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Radhakrishnan A, Ikeda Y, Kwon HJ, Brown MS & Goldstein JL (2007) Sterol‐regulated transport of SREBPs from endoplasmic reticulum to Golgi: oxysterols block transport by binding to Insig. Proc Natl Acad Sci USA 104, 6511–6518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Janowski BA, Grogan MJ, Jones SA, Wisely GB, Kliewer SA, Corey EJ & Mangelsdorf DJ (1999) Structural requirements of ligands for the oxysterol liver X receptors LXRalpha and LXRbeta. Proc Natl Acad Sci USA 96, 266–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Lehmann JM, Kliewer SA, Moore LB, Smith‐Oliver TA, Oliver BB, Su JL, Sundseth SS, Winegar DA, Blanchard DE, Spencer TA et al. (1997) Activation of the nuclear receptor LXR by oxysterols defines a new hormone response pathway. J Biol Chem 272, 3137–3140. [DOI] [PubMed] [Google Scholar]
- 54. Repa JJ, Liang G, Ou J, Bashmakov Y, Lobaccaro JM, Shimomura I, Shan B, Brown MS, Goldstein JL & Mangelsdorf DJ (2000) Regulation of mouse sterol regulatory element‐binding protein‐1c gene (SREBP‐1c) by oxysterol receptors, LXRalpha and LXRbeta. Genes Dev 14, 2819–2830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Edwards PA, Kast HR & Anisfeld AM (2002) BAREing it all: the adoption of LXR and FXR and their roles in lipid homeostasis. J Lipid Res 43, 2–12. [PubMed] [Google Scholar]
- 56. Laffitte BA, Repa JJ, Joseph SB, Wilpitz DC, Kast HR, Mangelsdorf DJ & Tontonoz P (2001) LXRs control lipid‐inducible expression of the apolipoprotein E gene in macrophages and adipocytes. Proc Natl Acad Sci USA 98, 507–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Means TK, Wang S, Lien E, Yoshimura A, Golenbock DT & Fenton MJ (1999) Human toll‐like receptors mediate cellular activation by Mycobacterium tuberculosis . J Immunol 163, 3920–3927. [PubMed] [Google Scholar]
- 58. Scanga CB & Le Gros G (2000) Development of an asthma vaccine. Drugs 59, 1217–1221. [DOI] [PubMed] [Google Scholar]
- 59. Li‐Hawkins J, Lund EG, Turley SD & Russell DW (2000) Disruption of the oxysterol 7alpha‐hydroxylase gene in mice. J Biol Chem 275, 16536–16542. [DOI] [PubMed] [Google Scholar]
- 60. Griffiths WJ, Crick PJ, Meljon A, Theofilopoulos S, Abdel‐Khalik J, Yutuc E, Parker JE, Kelly DE, Kelly SL, Arenas E et al. (2019) Additional pathways of sterol metabolism: evidence from analysis of Cyp27a1‐/‐ mouse brain and plasma. Biochim Biophys Acta Mol Cell Biol Lipids 1864, 191–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Abdel‐Khalik J, Crick PJ, Yutuc E, DeBarber AE, Duell PB, Steiner RD, Laina I, Wang Y & Griffiths WJ (2018) Identification of 7alpha,24‐dihydroxy‐3‐oxocholest‐4‐en‐26‐oic and 7alpha,25‐dihydroxy‐3‐oxocholest‐4‐en‐26‐oic acids in human cerebrospinal fluid and plasma. Biochimie 153, 86–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Griffiths WJ, Abdel‐Khalik J, Yutuc E, Roman G, Warner M, Gustafsson JA & Wang Y (2019) Concentrations of bile acid precursors in cerebrospinal fluid of Alzheimer's disease patients. Free Radic Biol Med 134, 42–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Beck KR, Kanagaratnam S, Kratschmar DV, Birk J, Yamaguchi H, Sailer AW, Seuwen K & Odermatt A (2019) Enzymatic interconversion of the oxysterols 7beta,25‐dihydroxycholesterol and 7‐keto,25‐hydroxycholesterol by 11beta‐hydroxysteroid dehydrogenase type 1 and 2. J Steroid Biochem Mol Biol 190, 19–28. [DOI] [PubMed] [Google Scholar]
- 64. Abdel‐Khalik J, Hearn T, Dickson AL, Crick PJ, Yutuc E, Austin‐Muttitt K, Bigger BW, Morris AA, Shackleton CH, Clayton PT et al. (2020) Bile acid biosynthesis in Smith‐Lemli‐Opitz syndrome bypassing cholesterol: potential importance of pathway intermediates. J Steroid Biochem Mol Biol 206, 105794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Griffiths WJ, Crick PJ & Wang Y (2013) Methods for oxysterol analysis: past, present and future. Biochem Pharmacol 86, 3–14. [DOI] [PubMed] [Google Scholar]
- 66. Alvelius G, Hjalmarson O, Griffiths WJ, Bjorkhem I & Sjovall J (2001) Identification of unusual 7‐oxygenated bile acid sulfates in a patient with Niemann‐Pick disease, type C. J Lipid Res 42, 1571–1577. [PubMed] [Google Scholar]
- 67. Jiang X, Ory DS & Han X (2007) Characterization of oxysterols by electrospray ionization tandem mass spectrometry after one‐step derivatization with dimethylglycine. Rapid Commun Mass Spectrom 21, 141–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Jiang X, Sidhu R, Porter FD, Yanjanin NM, Speak AO, te Vruchte DT, Platt FM, Fujiwara H, Scherrer DE, Zhang J et al. (2011) A sensitive and specific LC‐MS/MS method for rapid diagnosis of Niemann‐Pick C1 disease from human plasma. J Lipid Res 52, 1435–1445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Jiang X, Sidhu R, Mydock‐McGrane L, Hsu FF, Covey DF, Scherrer DE, Earley B, Gale SE, Farhat NY, Porter FD et al. (2016) Development of a bile acid‐based newborn screen for Niemann‐Pick disease type C. Sci Transl Med 8, 337ra363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Mazzacuva F, Mills P, Mills K, Camuzeaux S, Gissen P, Nicoli ER, Wassif C, Te Vruchte D, Porter FD, Maekawa M et al. (2016) Identification of novel bile acids as biomarkers for the early diagnosis of Niemann‐Pick C disease. FEBS Lett 590, 1651–1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Maekawa M, Misawa Y, Sotoura A, Yamaguchi H, Togawa M, Ohno K, Nittono H, Kakiyama G, Iida T, Hofmann AF et al. (2013) LC/ESI‐MS/MS analysis of urinary 3beta‐sulfooxy‐7beta‐N‐acetylglucosaminyl‐5‐cholen‐24‐oic acid and its amides: new biomarkers for the detection of Niemann‐Pick type C disease. Steroids 78, 967–972. [DOI] [PubMed] [Google Scholar]
- 72. Hult M, Elleby B, Shafqat N, Svensson S, Rane A, Jornvall H, Abrahmsen L & Oppermann U (2004) Human and rodent type 1 11beta‐hydroxysteroid dehydrogenases are 7beta‐hydroxycholesterol dehydrogenases involved in oxysterol metabolism. Cell Mol Life Sci 61, 992–999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Schweizer RA, Zurcher M, Balazs Z, Dick B & Odermatt A (2004) Rapid hepatic metabolism of 7‐ketocholesterol by 11beta‐hydroxysteroid dehydrogenase type 1: species‐specific differences between the rat, human, and hamster enzyme. J Biol Chem 279, 18415–18424. [DOI] [PubMed] [Google Scholar]
- 74. Lange Y & Steck TL (1998) Four cholesterol‐sensing proteins. Curr Opin Struct Biol 8, 435–439. [DOI] [PubMed] [Google Scholar]
- 75. Kong JH, Siebold C & Rohatgi R (2019) Biochemical mechanisms of vertebrate hedgehog signaling. Development 146. doi: 10.1242/dev.166892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Bjorkhem I, Diczfalusy U, Lovgren‐Sandblom A, Starck L, Jonsson M, Tallman K, Schirmer H, Ousager LB, Crick PJ, Wang Y et al. (2014) On the formation of 7‐ketocholesterol from 7‐dehydrocholesterol in patients with CTX and SLO. J Lipid Res 55, 1165–1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Griffiths WJ, Abdel‐Khalik J, Crick PJ, Ogundare M, Shackleton CH, Tuschl K, Kwok MK, Bigger BW, Morris AA, Honda A et al. (2017) Sterols and oxysterols in plasma from Smith‐Lemli‐Opitz syndrome patients. J Steroid Biochem Mol Biol 169, 77–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Porter FD & Herman GE (2011) Malformation syndromes caused by disorders of cholesterol synthesis. J Lipid Res 52, 6–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Shackleton CH (2012) Role of a disordered steroid metabolome in the elucidation of sterol and steroid biosynthesis. Lipids 47, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Shinkyo R, Xu L, Tallman KA, Cheng Q, Porter NA & Guengerich FP (2011) Conversion of 7‐dehydrocholesterol to 7‐ketocholesterol is catalyzed by human cytochrome P450 7A1 and occurs by direct oxidation without an epoxide intermediate. J Biol Chem 286, 33021–33028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Yutuc E, Angelini R, Baumert M, Mast N, Pikuleva I, Newton J, Clench MR, Skibinski DOF, Howell OW, Wang Y et al. (2020) Localization of sterols and oxysterols in mouse brain reveals distinct spatial cholesterol metabolism. Proc Natl Acad Sci USA 117, 5749–5760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Saeed AA, Genove G, Li T, Lutjohann D, Olin M, Mast N, Pikuleva IA, Crick P, Wang Y, Griffiths W et al. (2014) Effects of a disrupted blood‐brain barrier on cholesterol homeostasis in the brain. J Biol Chem 289, 23712–23722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Bjorkhem I (2007) Rediscovery of cerebrosterol. Lipids 42, 5–14. [DOI] [PubMed] [Google Scholar]
- 84. Meng LJ, Griffiths WJ, Nazer H, Yang Y & Sjövall J (1997) High levels of (24S)‐24‐hydroxycholesterol 3‐sulfate, 24‐glucuronide in the serum and urine of children with severe cholestatic liver disease. J Lipid Res 38, 926–934. [PubMed] [Google Scholar]
- 85. Bjorkhem I, Andersson U, Ellis E, Alvelius G, Ellegard L, Diczfalusy U, Sjovall J & Einarsson C (2001) From brain to bile. Evidence that conjugation and omega‐hydroxylation are important for elimination of 24S‐hydroxycholesterol (cerebrosterol) in humans. J Biol Chem 276, 37004–37010. [DOI] [PubMed] [Google Scholar]
- 86. Autio KJ, Schmitz W, Nair RR, Selkala EM, Sormunen RT, Miinalainen IJ, Crick PJ, Wang Y, Griffiths WJ, Reddy JK et al. (2014) Role of AMACR (alpha‐methylacyl‐CoA racemase) and MFE‐1 (peroxisomal multifunctional enzyme‐1) in bile acid synthesis in mice. Biochem J 461, 125–135. [DOI] [PubMed] [Google Scholar]
- 87. Li‐Hawkins J, Lund EG, Bronson AD & Russell DW (2000) Expression cloning of an oxysterol 7alpha‐hydroxylase selective for 24‐hydroxycholesterol. J Biol Chem 275, 16543–16549. [DOI] [PubMed] [Google Scholar]
- 88. Kotti TJ, Ramirez DM, Pfeiffer BE, Huber KM & Russell DW (2006) Brain cholesterol turnover required for geranylgeraniol production and learning in mice. Proc Natl Acad Sci USA 103, 3869–3874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Kotti T, Head DD, McKenna CE & Russell DW (2008) Biphasic requirement for geranylgeraniol in hippocampal long‐term potentiation. Proc Natl Acad Sci USA 105, 11394–11399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Russell DW, Halford RW, Ramirez DM, Shah R & Kotti T (2009) Cholesterol 24‐hydroxylase: an enzyme of cholesterol turnover in the brain. Annu Rev Biochem 78, 1017–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Meljon A, Wang Y & Griffiths WJ (2014) Oxysterols in the brain of the cholesterol 24‐hydroxylase knockout mouse. Biochem Biophys Res Commun 446, 768–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Paul SM, Doherty JJ, Robichaud AJ, Belfort GM, Chow BY, Hammond RS, Crawford DC, Linsenbardt AJ, Shu HJ, Izumi Y et al. (2013) The major brain cholesterol metabolite 24(S)‐hydroxycholesterol is a potent allosteric modulator of N‐methyl‐D‐aspartate receptors. J Neurosci 33, 17290–17300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Qi X, Liu H, Thompson B, McDonald J, Zhang C & Li X (2019) Cryo‐EM structure of oxysterol‐bound human Smoothened coupled to a heterotrimeric Gi. Nature 571, 279–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Bjorkhem I, Cedazo‐Minguez A, Leoni V & Meaney S (2009) Oxysterols and neurodegenerative diseases. Mol Aspects Med 30, 171–179. [DOI] [PubMed] [Google Scholar]
- 95. Bjorkhem I, Patra K, Boxer AL & Svenningsson P (2018) 24S‐hydroxycholesterol correlates with tau and is increased in cerebrospinal fluid in Parkinson's disease and corticobasal syndrome. Front Neurol 9, 756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Bjorkhem I, Lovgren‐Sandblom A, Leoni V, Meaney S, Brodin L, Salveson L, Winge K, Palhagen S & Svenningsson P (2013) Oxysterols and Parkinson's disease: evidence that levels of 24S‐hydroxycholesterol in cerebrospinal fluid correlates with the duration of the disease. Neurosci Lett 555, 102–105. [DOI] [PubMed] [Google Scholar]
- 97. Papassotiropoulos A, Lutjohann D, Bagli M, Locatelli S, Jessen F, Buschfort R, Ptok U, Bjorkhem I, von Bergmann K & Heun R (2002) 24S‐hydroxycholesterol in cerebrospinal fluid is elevated in early stages of dementia. J Psychiatr Res 36, 27–32. [DOI] [PubMed] [Google Scholar]
- 98. Papassotiropoulos A, Lütjohann D, Bagli M, Locatelli S, Jessen F, Rao ML, Maier W, Björkhem I, von Bergmann K & Heun R (2000) Plasma 24S‐hydroxycholesterol: a peripheral indicator of neuronal degeneration and potential state marker for Alzheimer's disease. NeuroReport 11, 1959–1962. [DOI] [PubMed] [Google Scholar]
- 99. Lütjohann D, Papassotiropoulos A, Björkhem I, Locatelli S, Bagli M, Oehring RD, Schlegel U, Jessen F, Rao ML, von Bergmann K et al. (2000) Plasma 24S‐hydroxycholesterol (cerebrosterol) is increased in Alzheimer and vascular demented patients. J Lipid Res 41, 195–198. [PubMed] [Google Scholar]
- 100. Leoni V, Long JD, Mills JA, Di Donato S & Paulsen JS (2013) Plasma 24S‐hydroxycholesterol correlation with markers of Huntington disease progression. Neurobiol Dis 55, 37–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Leoni V, Mariotti C, Tabrizi SJ, Valenza M, Wild EJ, Henley SM, Hobbs NZ, Mandelli ML, Grisoli M, Bjorkhem I et al. (2008) Plasma 24S‐hydroxycholesterol and caudate MRI in pre‐manifest and early Huntington's disease. Brain 131, 2851–2859. [DOI] [PubMed] [Google Scholar]
- 102. Mahley RW (2016) Central nervous system lipoproteins: ApoE and regulation of cholesterol metabolism. Arterioscler Thromb Vasc Biol 36, 1305–1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Hirsch‐Reinshagen V, Donkin J, Stukas S, Chan J, Wilkinson A, Fan J, Parks JS, Kuivenhoven JA, Lütjohann D, Pritchard H et al. (2009) LCAT synthesized by primary astrocytes esterifies cholesterol on glia‐derived lipoproteins. J Lipid Res 50, 885–893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Sidhu R, Jiang H, Farhat NY, Carrillo‐Carrasco N, Woolery M, Ottinger E, Porter FD, Schaffer JE, Ory DS & Jiang X (2015) A validated LC‐MS/MS assay for quantification of 24(S)‐hydroxycholesterol in plasma and cerebrospinal fluid. J Lipid Res 56, 1222–1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Nelson JA, Steckbeck SR & Spencer TA (1981) Biosynthesis of 24,25‐epoxycholesterol from squalene 2,3;22,23‐dioxide. J Biol Chem 256, 1067–1068. [PubMed] [Google Scholar]
- 106. Goyal S, Xiao Y, Porter NA, Xu L & Guengerich FP (2014) Oxidation of 7‐dehydrocholesterol and desmosterol by human cytochrome P450 46A1. J Lipid Res 55, 1933–1943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Gill S, Chow R & Brown AJ (2008) Sterol regulators of cholesterol homeostasis and beyond: the oxysterol hypothesis revisited and revised. Prog Lipid Res 47, 391–404. [DOI] [PubMed] [Google Scholar]
- 108. Zhang Z, Li D, Blanchard DE, Lear SR, Erickson SK & Spencer TA (2001) Key regulatory oxysterols in liver: analysis as delta4‐3‐ketone derivatives by HPLC and response to physiological perturbations. J Lipid Res 42, 649–658. [PubMed] [Google Scholar]
- 109. Meljon A, Theofilopoulos S, Shackleton CH, Watson GL, Javitt NB, Knolker HJ, Saini R, Arenas E, Wang Y & Griffiths WJ (2012) Analysis of bioactive oxysterols in newborn mouse brain by LC/MS. J Lipid Res 53, 2469–2483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Wang Y, Sousa KM, Bodin K, Theofilopoulos S, Sacchetti P, Hornshaw M, Woffendin G, Karu K, Sjovall J, Arenas E et al. (2009) Targeted lipidomic analysis of oxysterols in the embryonic central nervous system. Mol Biosyst 5, 529–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Wang Y, Karu K, Meljon A, Turton J, Yau JL, Seckl JR, Wang Y & Griffiths WJ (2014) 24S,25‐Epoxycholesterol in mouse and rat brain. Biochem Biophys Res Commun 449, 229–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Qi X, Friedberg L, De Bose‐Boyd R, Long T & Li X (2020) Sterols in an intramolecular channel of Smoothened mediate Hedgehog signaling. Nat Chem Biol 16, 1368–1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Song C & Liao S (2000) Cholestenoic acid is a naturally occurring ligand for liver X receptor alpha. Endocrinology 141, 4180–4184. [DOI] [PubMed] [Google Scholar]
- 114. Umetani M, Domoto H, Gormley AK, Yuhanna IS, Cummins CL, Javitt NB, Korach KS, Shaul PW & Mangelsdorf DJ (2007) 27‐Hydroxycholesterol is an endogenous SERM that inhibits the cardiovascular effects of estrogen. Nat Med 13, 1185–1192. [DOI] [PubMed] [Google Scholar]
- 115. DuSell CD, Umetani M, Shaul PW, Mangelsdorf DJ & McDonnell DP (2008) 27‐hydroxycholesterol is an endogenous selective estrogen receptor modulator. Mol Endocrinol 22, 65–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Wu Q, Ishikawa T, Sirianni R, Tang H, McDonald JG, Yuhanna IS, Thompson B, Girard L, Mineo C, Brekken RA et al. (2013) 27‐Hydroxycholesterol promotes cell‐autonomous, ER‐positive breast cancer growth. Cell Rep 5, 637–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Nelson ER (2018) The significance of cholesterol and its metabolite, 27‐hydroxycholesterol in breast cancer. Mol Cell Endocrinol 466, 73–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Touvier M, Fassier P, His M, Norat T, Chan DS, Blacher J, Hercberg S, Galan P, Druesne‐Pecollo N & Latino‐Martel P (2015) Cholesterol and breast cancer risk: a systematic review and meta‐analysis of prospective studies. Br J Nutr 114, 347–357. [DOI] [PubMed] [Google Scholar]
- 119. Le Cornet C, Walter B, Sookthai D, Johnson TS, Kühn T, Herpel E, Kaaks R & Fortner RT (2020) Circulating 27‐hydroxycholesterol and breast cancer tissue expression of CYP27A1, CYP7B1, LXR‐β, and ERβ: results from the EPIC‐Heidelberg cohort. Breast Cancer Res 22, 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Lu DL, Le Cornet C, Sookthai D, Johnson TS, Kaaks R & Fortner RT (2019) Circulating 27‐hydroxycholesterol and breast cancer risk: results from the EPIC‐Heidelberg cohort. J Natl Cancer Inst 111, 365–371. [DOI] [PubMed] [Google Scholar]
- 121. Lipidomics Standards Initiative (2019) Lipidomics needs more standardization. Nat Metab 1, 745–747. [DOI] [PubMed] [Google Scholar]
- 122. Meaney S, Heverin M, Panzenboeck U, Ekstrom L, Axelsson M, Andersson U, Diczfalusy U, Pikuleva I, Wahren J, Sattler W et al. (2007) Novel route for elimination of brain oxysterols across the blood‐brain barrier: conversion into 7alpha‐hydroxy‐3‐oxo‐4‐cholestenoic acid. J Lipid Res 48, 944–951. [DOI] [PubMed] [Google Scholar]
- 123. Setchell KD, Schwarz M, O'Connell NC, Lund EG, Davis DL, Lathe R, Thompson HR, Weslie Tyson R, Sokol RJ & Russell DW (1998) Identification of a new inborn error in bile acid synthesis: mutation of the oxysterol 7alpha‐hydroxylase gene causes severe neonatal liver disease. J Clin Invest 102, 1690–1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Kakiyama G, Marques D, Martin R, Takei H, Rodriguez‐Agudo D, LaSalle SA, Hashiguchi T, Liu X, Green R, Erickson S et al. (2020) Insulin resistance dysregulates CYP7B1 leading to oxysterol accumulation: a pathway for NAFL to NASH transition. J Lipid Res 61, 1629–1644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. de Medina P, Paillasse MR, Segala G, Poirot M & Silvente‐Poirot S (2010) Identification and pharmacological characterization of cholesterol‐5,6‐epoxide hydrolase as a target for tamoxifen and AEBS ligands. Proc Natl Acad Sci USA 107, 13520–13525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Vanier MT, Gissen P, Bauer P, Coll MJ, Burlina A, Hendriksz CJ, Latour P, Goizet C, Welford RW, Marquardt T et al. (2016) Diagnostic tests for Niemann‐Pick disease type C (NP‐C): a critical review. Mol Genet Metab 118, 244–254. [DOI] [PubMed] [Google Scholar]
- 127. Voisin M, de Medina P, Mallinger A, Dalenc F, Huc‐Claustre E, Leignadier J, Serhan N, Soules R, Segala G, Mougel A et al. (2017) Identification of a tumor‐promoter cholesterol metabolite in human breast cancers acting through the glucocorticoid receptor. Proc Natl Acad Sci USA 114, E9346–E9355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Soulès R, Audouard‐Combe F, Huc‐Claustre E, de Medina P, Rives A, Chatelut E, Dalenc F, Franchet C, Silvente‐Poirot S, Poirot M et al. (2019) A fast UPLC‐HILIC method for an accurate quantification of dendrogenin A in human tissues. J Steroid Biochem Mol Biol 194, 105447. [DOI] [PubMed] [Google Scholar]
- 129. de Medina P, Paillasse MR, Segala G, Voisin M, Mhamdi L, Dalenc F, Lacroix‐Triki M, Filleron T, Pont F, Saati TA et al. (2013) Dendrogenin A arises from cholesterol and histamine metabolism and shows cell differentiation and anti‐tumour properties. Nat Commun 4, 1840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Segala G, David M, de Medina P, Poirot MC, Serhan N, Vergez F, Mougel A, Saland E, Carayon K, Leignadier J et al. (2017) Dendrogenin A drives LXR to trigger lethal autophagy in cancers. Nat Commun 8, 1903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Mast N, Annalora AJ, Lodowski DT, Palczewski K, Stout CD & Pikuleva IA (2011) Structural basis for three‐step sequential catalysis by the cholesterol side chain cleavage enzyme CYP11A1. J Biol Chem 286, 5607–5613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Strushkevich N, MacKenzie F, Cherkesova T, Grabovec I, Usanov S & Park H‐W (2011) Structural basis for pregnenolone biosynthesis by the mitochondrial monooxygenase system. Proc Natl Acad Sci USA 108, 10139–10143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Song BL, Javitt NB & DeBose‐Boyd RA (2005) Insig‐mediated degradation of HMG CoA reductase stimulated by lanosterol, an intermediate in the synthesis of cholesterol. Cell Metab 1, 179–189. [DOI] [PubMed] [Google Scholar]
- 134. Chen L, Ma MY, Sun M, Jiang LY, Zhao XT, Fang XX, Man Lam S, Shui GH, Luo J, Shi XJ et al. (2019) Endogenous sterol intermediates of the mevalonate pathway regulate HMGCR degradation and SREBP‐2 processing. J Lipid Res 60, 1765–1775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Yang C, McDonald JG, Patel A, Zhang Y, Umetani M, Xu F, Westover EJ, Covey DF, Mangelsdorf DJ, Cohen JC et al. (2006) Sterol intermediates from cholesterol biosynthetic pathway as liver X receptor ligands. J Biol Chem 281, 27816–27826. [DOI] [PubMed] [Google Scholar]
- 136. Sever N, Mann RK, Xu L, Snell WJ, Hernandez‐Lara CI, Porter NA & Beachy PA (2016) Endogenous B‐ring oxysterols inhibit the Hedgehog component Smoothened in a manner distinct from cyclopamine or side‐chain oxysterols. Proc Natl Acad Sci USA 113, 5904–5909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Soroosh P, Wu J, Xue X, Song J, Sutton SW, Sablad M, Yu J, Nelen MI, Liu X, Castro G et al. (2014) Oxysterols are agonist ligands of RORgammat and drive Th17 cell differentiation. Proc Natl Acad Sci USA 111, 12163–12168. [DOI] [PMC free article] [PubMed] [Google Scholar]