

Abstract
Metal-catalyzed mono-acylmethylation of pyridylcarboranes has been realized using α-carbonyl sulfoxonium ylides as a coupling partner. The reaction features high efficiency, excellent site-selectivity and good functional group tolerance. In the presence of pyridyl and enolizable acylmethyl groups, a post-coordination mode has been proposed and validated by in situ high resolution mass spectroscopy (HRMS) to rationalize the unique mono-substitution. Post-functionalization at the newly incorporated alkyl site provides additional utility of this method, including the construction of carborane-fused indoliziniums and quinoliziniums. We believe that these mono-alkylated carboranes, together with their post-functionalized derivatives, may find applications in luminescent materials and drug discovery in the near future.
Metal-catalyzed selective mono-acylmethylation of pyridylcarboranes has been realized, which provides further utility to construct carborane-fused indoliziniums and quinoliziniums.
Introduction
Icosahedral carboranes are molecular clusters1 that have triggered abundant research interest for decades as building blocks to construct versatile ligands,2 functional materials3 and tunable pharmacophores.4 The development of new methodologies to incorporate organic functionalities into carboranes holds great promise at the interface between cluster chemistry and organic chemistry. Central to the derivatization of carboranes has been metal-catalyzed B–H functionalization to generate B–C bonds.5 This strategy has emerged as a powerful strategy to provide otherwise inaccessible cluster molecules featuring mild reaction conditions, good functional group tolerance and improved step economy. Widely reported reactions include arylation, alkenylation, and alkynylation, while alkylation reactions that enable the formation of B–C(sp3) bonds have been less developed (Scheme 1a).5,6 Pioneering examples of the development of cage boron alkylated carboranes include electrophilic total-methylation of o-carborane, p-carborane or the mono-carborane anion [CB11H12]− with methyl triflate6a,b and transition metal-mediated hydroboration of alkenes with carboranes,6c followed by recent reports of transition-metal catalysis6d and nucleophilic substitutions.6e,f A fundamental issue in this direction is how to control the mono-substitution when two or more chemically similar B–H bonds exist in the carborane cluster. Even now, it is still challenging to suppress the double functionalization and drive the reaction in a mono-functionalized direction, presumably owing to uncontrolled metal reactivity including the “cage walking” behavior.7 Generally, stoichiometric control has been utilized to obtain one-fold functionalized products, although more-than-one-fold functionalized products can be usually generated.5 In particular, carboranes with mono-alkylated substituents4d–f,8 are highly desirable considering their utility for precise post-functionalization (such as cross-coupling reaction, annulation, bio-conjugation, etc.) at the alkyl site. In this work, readily accessible α-carbonyl sulfoxonium ylides9 have been utilized for the development of a general Rh(iii)Cp*-catalyzed selective alkylation of cage B–H bonds of 1-(2-pyridyl)-carboranes. It is noteworthy that metal–carbene intermediates in which sulfoxonium ylides function as precursors are known to undergo X–H (X = N, O, S, C) bond insertion reactions,9 while insertion into the B–H bonds of carboranes is yet to be established. A post-coordinating mode has been proposed to control the mono-substitution in metal-catalyzed B–H activation.
Scheme 1. Metal-catalyzed B–H functionalization of carboranes via B–C bond formation.

With this strategy, a vast number of mono-acylmethylated carboranes have been synthesized in high yields with excellent B(4)-vertex selectivity with the aid of the pyridyl directing group (Scheme 1b). The synthetic utility of these mono-acylmethylated pyridylcarboranes allows for the construction of carborane-fused heteroarenes including indoliziniums and quinoliziniums with carboranyl fusion. It is known that cationic heteroarenes are well-established DNA binders that can affect the biological activities of DNA.10 In particular, cationic quinolizinium alkaloids berberine and its derivatives (Scheme 2) exhibit antimalarial properties and other biological activities.11 Furthermore, drug design based on annulated indolizinium and quinolizinium derivatives (e.g. rhodacyanine derivatives) has attracted considerable attention.12 In extending the synthetic utility of these mono-alkylated products, we have realized the intramolecular annulation between the remaining pyridyl and newly attached alkyl groups to produce indoliziniums or quinoliziniums containing a five- or six-membered heterocycle. These carborane-fused indoliziniums or quinoliziniums represent a new type of boron-containing heteroarene.
Scheme 2. Illustration of indoliziniums, quinoliziniums and carborane-fuse analogues.

Results and discussion
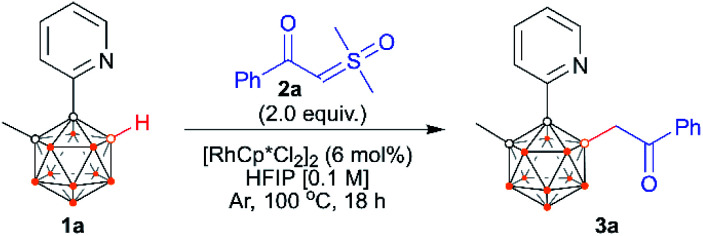
The reaction development is summarized in Table 1. At the outset of our studies, we chose 1-(2-pyridyl)-2-Me-o-carborane2l1a and α-carbonyl sulfoxonium ylide 2a as the model substrates. In the presence of 6 mol% of [RhCp*Cl2]2, the treatment of 1a with 2 equiv. of 2a in the absence of any additives in HFIP at 100 °C for 18 h afforded the B(4)/B(5)-mono-acylmethylated product 3a in an isolated yield of 72% (entry 1), and no di-acylmethylated product was observed. Attempts to replace the rhodium catalyst with either CoCp*(CO)I2, [IrCp*Cl2]2 or Cu(CH3CN)4PF6 were unsuccessful, while RhCp*(OAc)2·H2O and [Ru(p-cymene)Cl2]2 led to lower yields (entries 2–4). Introduction of Lewis bases or acids such as NaOAc or PivOH as an additive proved to be detrimental (entries 5 and 6). A prolonged or shortened period has little effect on yield (entries 7 and 8). At lower or higher temperature, the results were found to be slightly inferior to those observed under the standard conditions (entries 9 and 10). When other solvents such as toluene, 1,4-dioxane, DCE, THF, and CH3CN were used, no desired product was detected, even though TFE led to trace 3a (Table S1†). Reduction of 2a to 1.5 equiv. led to 3a in a slightly lower yield, even with a prolonged time period (entry 11). On the other hand, a lower yield was obtained when the catalyst loading was reduced (entry 12). Removal of the catalyst completely stopped the reaction (entry 13). It is noteworthy that the reaction still afforded a good yield when set up in an air atmosphere, which indicates that the method is rather robust (entry 14). In view of the yield, entry 1 was chosen as the optimal reaction conditions.
Reaction developmenta.
| ||
|---|---|---|
| Entry | Variation from the standard conditions | Yieldb [%] |
| 1 | None | 77 (72)c |
| 2 | CoCp*(CO)I2, [IrCp*Cl2]2, Cu(CH3CN)4PF6 instead of [RhCp*Cl2]2 | 0 |
| 3 | RhCp*(OAc)2·H2O instead of [RhCp*Cl2]2 | 59 |
| 4 | [Ru(p-cymene)Cl2]2 instead of [RhCp*Cl2]2 | 63 |
| 5 | With 1.0 equiv. of NaOAC | 58 |
| 6 | With 1.0 equiv. of PivOH | 40 |
| 7 | 36 h instead of 18 h | 76 |
| 8 | 12 h instead of 18 h | 74 |
| 9 | 120 °C instead of 100 °C | 74 |
| 10 | 80 °C instead of 100 °C | 65 |
| 11 | 1.5 equiv. of 2a, 36 h | 73 |
| 12 | 3 mol% [RhCp*Cl2]2 | 60 |
| 13 | Without [RhCp*Cl2]2 | 0 |
| 14 | Under air | 63 |
Reaction conditions: 1a (0.1 mmol), 2a (0.2 mmol), [RhCp*Cl2]2 (6 mol%), HFIP (1.0 mL), 100 °C, Ar, 18 h.
Determined by 1H NMR analysis of the crude reaction mixture.
Isolated yield. HFIP: hexafluoroisopropanol.
We then evaluated the scope of (hetero)aryl-substituted sulfoxonium ylides 2a–2z with 1a as a model substrate (Scheme 3). Substrates bearing electronically diverse substituents at different positions of the phenyl group were compatible, giving the corresponding products in moderate to excellent yields. Aryl sulfoxonium ylides having electron-donating groups such as –Me, –OMe, –OCF3 and –CONHPh provided products (3b–3d and 3r) in excellent yields (79–91%), despite the presence of reactive hydrogen. The lower yield for the substrate bearing –SCF3 may be attributed to its toxicity to the catalyst (3e). Notably, valuable halogen functional groups and ethers were well tolerated to afford the corresponding products in moderate to good yields which enables the late-stage manipulation of the initial products (3c, 3d, 3f–3i, 3n, 3p and 3s). Furthermore, aryl sulfoxonium ylides bearing electron-withdrawing –CF3, –Ph and –CO2Me furnished 3j–3l in moderate to good yields. Ylides having substituents at the ortho- or meta-position were all well tolerated (3m–3p). Substrates bearing multiple substituents were also examined, and the yields were comparable to those of the monosubstituted substrates (3q and 3s). Naphthalen-1-yl- and naphthalen-2-yl-substituted sulfoxonium ylides were applicable to the present method, affording 78% and 70% yields, respectively (3t and 3u). Substrates bearing furan scaffolds were effectively transformed into their corresponding products 3v (>97%) and 3x (60%). Similarly, thiophene and benzothiophene were compatible with the cross-coupling reaction (3w and 3y). Furthermore, reaction of 1a with adapalene13 derived sulfoxonium ylide afforded the adapalene analogue 3z, suggesting the potential of late-stage incorporation of the carboranyl motif into drug molecules using this reaction.
Scheme 3. Scope of (hetero)aryl-substituted sulfoxonium ylides. a Reaction conditions: 1a (0.1 mmol), 2a–2z (2.0 equiv.), [RhCp*Cl2]2 (6 mol%), HFIP (1.0 mL), 100 °C, 18 h, Ar atmosphere. b Isolated yield. c [Ru(p-cymene)Cl2]2 (6 mol%) as a catalyst. d With achiral coupling partners, the products are obtained as a racemic mixture because reaction at the B(4) and B(5) positions occurs with equal probability, and no attempts were made to separate any of the B(4)/B(5) isomers of the entire product series.

The scope of the alkyl-substituted sulfoxonium ylide coupling partners was also investigated (Scheme 4). With 1a as the model substrate, the coupling reactions with aliphatic sulfoxonium ylides smoothly led to the desired products in moderate to excellent yields. Both primary and secondary aliphatic acid derived ylides exhibited good to excellent reactivity (5a–5n). In addition, the sterically hindered tertiary alkyl-substituted sulfoxonium ylide could also give the desired product (5o). Piperidine units, key structural motifs in numerous natural products and pharmaceuticals, were well tolerated (5m and 5n). Tetrahydropyran was also a supportive motif (5l). Interestingly, cyclic secondary alkyl group containing substrates were converted into the respective products in nearly quantitative yields within 30 min (5g–5j). To showcase the practicality of the strategy, a scale-up reaction of 1a and 4g at a 1 mmol scale gave 5g with a comparable yield.
Scheme 4. Scope of alkyl-substituted sulfoxonium ylides. a Reaction conditions: 1a (0.1 mmol), 4a–4q (2.0 equiv.), RhCp*(OAc)2·H2O (6 mol%), HFIP (1.0 mL), 100 °C, 30 min, Ar atmosphere. b Isolated yield. c 12 h. d Sulfoxonium ylide was used as a racemic mixture. e no d.r. was determined. f 1 mmol scale. g 48 h. h 3 h. i Enantiopure 4p was used.

We also examined the scope of the reaction by varying the carbon substituents in 1-(2-pyridyl)-2-R1-o-carboranes using 4g as the model substrate (Scheme 5). Gratifyingly, the carborane substrates bearing different substituents at the cage C(2) position afforded the B(4)/B(5)-mono-substituted products (6b–6f) in nearly quantitative yields, indicating that the carbon substituent effect did not influence efficiency and selectivity in this reaction. In addition, substitution on the 2-pyridyl ring (–F, –Me and –OMe) did not affect the outcomes, giving 6g–6i in excellent yields. However, replacing the 2-pyridyl group with a phenyl or 3-pyridyl group totally shut down the reaction, which sheds light on the directing function of the 2-pyridyl moiety on the metal catalyst to activate the B–H bonds (ESI Schemes S8 and S9†). 1-(2-Pyridyl)-o-carborane and 1-(2-pyridyl)-p-carborane with free CH groups were also tolerated in this reaction, giving B(4)/B(5)-mono-acylmethylated products 7a–7d and corresponding para-carboranyl derivatives as the major products (Schemes 6a and 7). In these reactions, the post-coordination effect also plays a significant role in controlling the mono-substitution since the selectivity is usually low for the metal-catalyzed B–H functionalization reactions of o-carborane substrates with a free CH group.5a,6a,6i Interestingly, imine can also act as an effective directing group in the mono-acylmethylation reaction when using a ruthenium catalyst, giving corresponding products 7e and 7f in good yields (Scheme 6b). Furthermore, the imine group in 7e can be readily removed after treatment with HOAc in HFIP, affording aldehyde 7g in 81% yield (Scheme 6c). Subsequent removal of the aldehyde group in the presence of KMnO4 (1.0 equiv.) and Na2HPO4 (1.0 equiv.) led to 7h with a recovered CH site in 94% yield. In addition, competition experiments between ylides were examined as well. The B(4)–H alkylation between electron-rich (4-methoxy)- and electron-deficient (4-methoxycarbonyl) phenylsulfoxonium ylides (2c and 2l) provided 3c and 3l in 90% and 10% yields, respectively, indicating that the electron donating effect on the phenyl group of phenylsulfoxonium ylide is dominant (Scheme 8a). Competition experiments between phenylsulfoxonium ylide (2a) and cyclopropylsulfoxonium ylide (4g) gave 5g as the sole product in quantitative yield, demonstrating that alkylated sulfoxonium ylides 4g is more reactive than 2a (Scheme 8b).
Scheme 5. Scope of o-carboranyl with various carbon-substituents. a Reaction conditions: 1b–1i (0.1 mmol), 4g (2.0 equiv.), RhCp*(OAc)2·H2O (6 mol%), HFIP (1.0 mL), 100 °C, 30 min, Ar atmosphere. b Isolated yield.

Scheme 6. Reactions of 1-(2-pyridyl)-o-carborane and o-carboranyl N-arylimine with α-carbonyl sulfoxonium ylides (a and b). Directing group removal (c). a [Ru(p-cymene)Cl2]2 (6 mol%) as a catalyst.

Scheme 7. Reactions of 1-(2-pyridyl)-p-carborane with α-carbonyl sulfoxonium ylides.

Scheme 8. Intermolecular competition experiments (a and b) and deuteration reaction (c).

To gain insights into the reaction mechanism, control experiments were performed. The reactions in the presence of a radical quencher, TEMPO or 1,1-diphenylethylene, still provided product 5g in high yields (ESI Scheme S7†), implying that radical intermediates are not involved. The reaction of 1a and 4g under standard conditions (100 °C) in a mixed solution of D2O and HFIP led to the incorporation of deuterium at the α-carbon with a deuterium level of 75% (Scheme 8c). This result is in line with an in situ monitoring of the reaction in deuterated solvent HFIP-D2 at 25 °C, which led to increased deuterium incorporation (>95%), albeit with slightly decreased conversion (ESI Fig. S30†). These results indicate a facile enolization process at the newly incorporated acylmethyl group attached to boron.14 In sharp contrast, stirring 5g in D2O and HFIP at 25 °C in the absence of any other reagent or catalyst is not sufficient to enable deuterium incorporation at the enolizable positions. All of this demonstrates a reversible enolization and coordination process of the product with the rhodium catalyst.
On the basis of the above results as well as literature reports,9,15 a plausible reaction mechanism is depicted by using the reaction of 1a and 4g as an example (Scheme 9a). Firstly, the rhodium species RhCp*Cl2 coordinates with the pyridyl group of 1a to form intermediate A which subsequently induces B–H cleavage to generate a rhodacyclic complex B containing a Rh–B bond. Then B undergoes migratory insertion of α-carbonyl sulfoxonium ylide 4gvia a metal carbene intermediate C with loss of DMSO to give rise to D. This intermediate further delivers the final product 5gvia protodemetallation with concomitant regeneration of the RhCp*Cl2 catalyst. It is noteworthy that the conversion between D and the product should be reversible as indicated by the deuteration experiment. This post-coordination between the mono-alkylated product and the rhodium catalyst can prohibit the iterative B–H activation pathway, and thus control the selective mono-substitution (Scheme 9b). In situ NMR monitoring of the reaction suggests a clean catalytic conversion with the intermediacy of two new species, which may be related to B and D (ESI Fig. S23†). Trials to isolate these intermediates were unsuccessful. To our delight, HRMS measurements successfully detected the intermediates B and D across the different stages of the reaction (ESI Fig. S7–S22†). Furthermore, the α-deuterated analogue of D can also be detected in the deuteration reaction (ESI Fig. S27†). In addition, free DMSO liberated from sulfoxonium ylide was readily detected by 1H NMR in the catalytic reaction mixture (ESI Fig. S31†). All these results support the proposed mechanism.
Scheme 9. Proposed reaction cycle for Rh(iii)Cp*-catalyzed B(4)–H alkylation of o-carboranyl pyridine (a) and illustration of the post-coordination step (b).

The utility of our method was developed. Compound 3h can be further modified at its C–Br site (Scheme 10). Sonogashira coupling of 3h with Me3SiC CH afforded 8 in 98% isolated yield. The coupling reaction of 3h with phenoxazine afforded product 9 in 90% isolated yield, which offers a new route to carborane B–H functionalized materials for potential application in light-emitting diodes. Note that C–H substituted carborane-based luminescent materials have been extensively studied,3d–f,i,16 but there are few examples of studies involving B–H substituted carborane derivatives.16b Product 9 exhibits a significant aggregation-induced emission (AIE) effect as indicated by the photoluminescence (PL) spectra in THF/H2O mixed solutions (Fig. 1, left). When the volumetric ratio of water (fw vol%) reaches 80%, a light-yellow emission starts to appear at 525 nm, indicative of the AIE-behavior. A solvatochromic phenomenon was observed as the emission peak exhibits a red shift from 457 nm (n-hexane) to 552 nm (CHCl3) (Fig. 1, right).
Scheme 10. Post-functionalization of 3hvia Pd-catalyzed cross-coupling reactions of C–Br bonds to form C–C and C–N bonds.

Fig. 1. Photophysical properties of 9. (Left) Luminescence photographs taken under UV light (365 nm) and fluorescence spectra with different water volume fractions (vH2O/vTHF%) of 9 (1.0 × 10−5 M, λex = 323 nm) under ambient conditions; (right) fluorescence spectra of 9 observed in different solvents (1.0 × 10−5 M, λex = 323 nm) under ambient conditions.

The carbonyl functionality in the alkylated products provides an additional opportunity for post-functionalization at either the carbonyl site or its α-carbon position (Scheme 11). The carbonyl group in 5j can be transformed into methylene by a borane reducing reagent, cleanly affording mono-alkylated product 10 (Scheme 11a). In addition, 3a could be reduced to o-carboranyl aliphatic alcohol 11 in the presence of NaBH4 which could be subsequently brominated and annulated with the 2-pyridyl group to provide carborane-fused quinolizinium. Deboronation at the B(6) position is observed in the isolated product, indicating the zwitterionic character of 12 (Scheme 11b). Besides the construction of quinoliziniums, carborane-fused indoliziniums can also be accessed by reacting the mono-acylmethylated products with Py·HBr3 at 60 °C. This process involves one-pot bromination at the α-carbon (next to boron), and subsequent annulation with the 2-pyridyl group. A series of aryl substituted analogues are tolerated in this reaction, giving carborane-fused indoliziniums 13a–13e in high yields (Scheme 11c). The alkyl group is not tolerated owing to competitive bromination at the other α-carbon position, as confirmed by the isolation of product 14 from the reaction with 5j. It is also worth noting that the carbonyl functionality in the acylmethylated pyridylcarboranes is essential for the construction of carborane-fused indoliziniums as the carbonyl could activate the adjacent C–H unit.
Scheme 11. Post-functionalization. (a) Reduction of the carbonyl group to afford mono-alkylated carborane. (b) Synthesis of a carborane-fused quinolizinium. (c) Synthesis of carborane-fused indoliziniums.

Basically, the carborane-fused indoliziniums and quinoliziniums contain two diastereomers as indicated by their NMR spectra. The molecular structures of 12 and 13a have been characterized by X-ray single crystal analysis (Fig. 2). The carboranyl group in the products is deboronated at the B(3)/B(6) position and the annulated compounds are zwitterionic. When comparing the crystal structures, it is obvious that the formation of compound 13a proceeds through deboronation at the B(3) position, which is different from the formation of compound 12. In the first case, it leads to B(9)-substituted nido-carborane, while in the second case it leads to B(4)-substituted nido-carborane. The 11B NMR spectra also confirm this. Moreover, as far as we know, this is the first example of such deboronation. Interestingly, the carborane-fused quinoliziniums and indoliziniums display luminescence under UV irradiation. 12 and 13a were selected as examples to investigate the photophysical properties (Fig. 3). Unexpectedly, the two compounds show efficient emission in the crystalline state and the emission wavelengths of the crystals are located in the yellowish green region (533 nm for 12 and 524 nm for 13a, respectively). The absolute quantum yields of 12 and 13a can reach 27% and 16%, respectively. In sharp contrast, those compounds prior to carborane fusion do not show emission either in solution or in the solid state, including the crystalline state. Obviously, the carborane-fused luminogens enable the aggregation caused quenching effect to be overcome. Recently, the synthesis of carborane-fused (hetero)arenes3i,17 has provided a new opportunity for these boron-containing compounds as optoelectronic materials or drug candidates. The incorporation of the three-dimensional, aromatic and high boron-content carboranyl unit into these heteroarenes may offer promising value for drug design as well as other theranostic related biotechnologies.
Fig. 2. X-ray structures of 12 (left) and 13a (right).

Fig. 3. Photophysical properties of 12 and 13a. (Left) PL spectra (λex = 365 nm) in the crystalline state under ambient conditions; (middle) luminescence photograph of the single crystal 12 taken under UV light (365 nm); (right) luminescence photograph of the single crystal 13a taken under UV light (365 nm).

Conclusions
In summary, we have presented an efficient metal-catalyzed selective B(4)/B(5)–H mono-acylmethylation protocol of pyridylcarboranes by using sulfoxonium ylides as the alkyl sources. The reaction shows broad substrate scope and gives access to a series of carborane-substituted carbonyl compounds with a B–C(sp3) linkage at the α-carbon position. These products are difficult to synthesize by other means. The post-coordination of 2-pyridyl and enol moieties to the metal catalyst prohibits the iterative B–H activation event and leads to finely controlled mono-substitution. The products contain pyridyl, carbonyl and other newly incorporated organic functional groups, which provides new opportunities for post-functionalization. Notably, carborane-fused indoliziniums and quinoliziniums can be accessed, providing synthetic utility of this method in drug and luminescent molecule design.
Data availability
All experimental and crystallographic data are available in the ESI.†
Author contributions
H. Y. and H.-J. C. conceived and designed the present work. H.-J. C. and X. Z. wrote the manuscript. H. Y. and C. L. supervised the study. H.-J. C. performed and analyzed the experiments with assistance of X. W. and F. S. All authors contributed to the final manuscript.
Conflicts of interest
There are no conflicts to declare.
Supplementary Material
Acknowledgments
We are grateful for financial support from the National Natural Science Foundation of China (21820102004, 21531004 and 91961104). We thank Prof. Yong Liang and Prof. Zhuangzhi Shi for their helpful discussions and suggestions.
Electronic supplementary information (ESI) available: Experimental details, compound characterization and X-ray data in CIF format. CCDC 2004239–2004241, 2025251, 2110467, 2088268–2088269 and 2089805. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc05296a
Notes and references
- (a) Grimes R. N., Carboranes, Academic Press, Amsterdam, 3rd edn, 2016 [Google Scholar]; (b) Hosmane N. S. and Eagling R., Handbook of Boron Science, World Scientific, Singapore, 2019 [Google Scholar]
- For selected examples, see: ; (a) Hosmane N. S. and Maguire J. A., in Comprehensive Organometallic Chemistry III, ed. D. M. P. Mingos and R. H. Crabtree, Elsevier, Oxford, 2007, ch. 5, vol. 3 [Google Scholar]; (b) Cui P.-F. Liu X.-R. Guo S.-T. Lin Y.-J. Jin G.-X. J. Am. Chem. Soc. 2021;143:5099–5105. doi: 10.1021/jacs.1c00779. [DOI] [PubMed] [Google Scholar]; (c) Sivaev I. B. Bregadze V. I. Coord. Chem. Rev. 2019;392:146–176. doi: 10.1016/j.ccr.2019.04.011. [DOI] [Google Scholar]; (d) Coburger P. Demeshko S. Rödl C. Hey-Hawkins E. Wolf R. Angew. Chem., Int. Ed. 2017;129:16087–16091. doi: 10.1002/ange.201709140. [DOI] [PubMed] [Google Scholar]; (e) Eleazer B. J. Smith M. D. Popov A. A. Peryshkov D. V. J. Am. Chem. Soc. 2016;138:10531–10538. doi: 10.1021/jacs.6b05172. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Spokoyny A. M. Machan C. W. Clingerman D. J. Rosen M. S. Wiester M. J. Kennedy R. D. Stern C. L. Sarjeant A. A. Mirkin C. A. Nat. Chem. 2011;3:590–596. doi: 10.1038/nchem.1088. [DOI] [PubMed] [Google Scholar]; (g) Zhang X. Dai H. Yan H. Zou W. Cremer D. J. Am. Chem. Soc. 2016;138:4334–4337. doi: 10.1021/jacs.6b01249. [DOI] [PubMed] [Google Scholar]; (h) Jude H. Disteldorf H. Fischer S. Wedge T. Hawkridge A. M. Arif A. M. Hawthorne M. F. Muddiman D. C. Stang P. J. J. Am. Chem. Soc. 2005;127:12131–12139. doi: 10.1021/ja053050i. [DOI] [PubMed] [Google Scholar]; (i) Jaiswal K. Malik N. Tumanskii B. Ménard G. Dobrovetsky R. J. Am. Chem. Soc. 2021;143:9842–9848. doi: 10.1021/jacs.1c03568. [DOI] [PMC free article] [PubMed] [Google Scholar]; (j) Coburger P. Leitl J. Scott D. J. Hierlmeier G. Shenderovich I. G. Hey-Hawkins E. Wolf R. Chem. Sci. 2021:11225–11235. doi: 10.1039/D1SC02948G. [DOI] [PMC free article] [PubMed] [Google Scholar]; (k) Jayaweera H. D. A. C. Rahman M. M. Pellechia P. J. Smith M. D. Peryshkov D. V. Chem. Sci. 2021:10441–10447. doi: 10.1039/D1SC02252K. [DOI] [PMC free article] [PubMed] [Google Scholar]; (l) Coult R. Fox M. A. Gill W. R. Herbertson P. L. MacBride J. A. H. Wade K. J. Organomet. Chem. 1993;462:19–29. doi: 10.1016/0022-328X(93)83337-U. [DOI] [Google Scholar]
- For selected examples, see: ; (a) Gan L. Chidambaram A. Fonquernie P. G. Light M. E. Choquesillo-Lazarte D. Huang H. Solano E. Fraile J. Viñas C. Teixidor F. Navarro J. A. R. Stylianou K. C. Planas J. G. J. Am. Chem. Soc. 2020;142:8299–8311. doi: 10.1021/jacs.0c01008. [DOI] [PubMed] [Google Scholar]; (b) Saha A. Oleshkevich E. Vinas C. Teixidor F. Adv. Mater. 2017;29:1704238. doi: 10.1002/adma.201704238. [DOI] [PubMed] [Google Scholar]; (c) Nunez R. Romero I. Teixidor F. Vinas C. Chem. Soc. Rev. 2016;45:5147–5173. doi: 10.1039/C6CS00159A. [DOI] [PubMed] [Google Scholar]; (d) Wei X. Zhu M.-J. Cheng Z. Lee M. Yan H. Lu C. Xu J.-J. Angew. Chem., Int. Ed. 2019;58:3162–3166. doi: 10.1002/anie.201900283. [DOI] [PubMed] [Google Scholar]; (e) Wee K.-R. Cho Y.-J. Song J. K. Kang S. O. Angew. Chem., Int. Ed. 2013;52:9682–9685. doi: 10.1002/anie.201304321. [DOI] [PubMed] [Google Scholar]; (f) Ochi J. Tanaka K. Chujo Y. Angew. Chem., Int. Ed. 2020;59:9841–9855. doi: 10.1002/anie.201916666. [DOI] [PubMed] [Google Scholar]; (g) Dash B. P. Satapathy R. Gaillard E. R. Maguire J. A. Hosmane N. S. J. Am. Chem. Soc. 2010;132:6578–6587. doi: 10.1021/ja101845m. [DOI] [PubMed] [Google Scholar]; (h) Zhang Y. Yang L. Wang L. Duttwyler S. Xing H. Angew. Chem., Int. Ed. 2019;58:8145–8150. doi: 10.1002/anie.201903600. [DOI] [PubMed] [Google Scholar]; (i) Nunez R. Tarres M. Ferrer-Ugalde A. de Biani F. F. Teixidor F. Chem. Rev. 2016;116:14307–14378. doi: 10.1021/acs.chemrev.6b00198. [DOI] [PubMed] [Google Scholar]; (j) Hablot D. Ziessel R. Alamiry M. A. H. Bahraidah E. Harriman A. Chem. Sci. 2013;4:444–453. doi: 10.1039/C2SC21505E. [DOI] [Google Scholar]; (k) Kirlikovali K. O. Axtell J. C. Gonzalez A. Phung A. C. Khan S. I. Spokoyny A. M. Chem. Sci. 2016;7:5132–5138. doi: 10.1039/C6SC01146B. [DOI] [PMC free article] [PubMed] [Google Scholar]; (l) Li X. Tong X. Yin Y. Yan H. Lu C. Huang W. Zhao Q. Chem. Sci. 2017;8:5930–5940. doi: 10.1039/C7SC00160F. [DOI] [PMC free article] [PubMed] [Google Scholar]; (m) You D. K. So H. Ryu C. H. Kim M. Lee K. M. Chem. Sci. 2021;12:8411–8423. doi: 10.1039/D1SC00791B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For selected examples, see: ; (a) Iijima T. Endo Y. Tsuji M. Kawachi E. Kagechika H. Shudo K. Chem. Pharm. Bull. 1999;47:398–404. doi: 10.1248/cpb.47.398. [DOI] [PubMed] [Google Scholar]; (b) Leśnikowski Z. J. J. Med. Chem. 2016;59:7738–7758. doi: 10.1021/acs.jmedchem.5b01932. [DOI] [PubMed] [Google Scholar]; (c) Barry N. P. E. Sadler P. J. Chem. Soc. Rev. 2012;41:3264–3279. doi: 10.1039/C2CS15300A. [DOI] [PubMed] [Google Scholar]; (d) Issa F. Kassiou M. Rendina L. M. Chem. Rev. 2011;111:5701–5722. doi: 10.1021/cr2000866. [DOI] [PubMed] [Google Scholar]; (e) Yin Y. Ochi N. Craven T. W. Baker D. Takigawa N. Suga H. J. Am. Chem. Soc. 2019;141:19193–19197. doi: 10.1021/jacs.9b09106. [DOI] [PubMed] [Google Scholar]; (f) Stockmann P. Gozzi M. Kuhnert R. Sárosi M. B. Hey-Hawkins E. Chem. Soc. Rev. 2019;48:3497–3512. doi: 10.1039/C9CS00197B. [DOI] [PubMed] [Google Scholar]; (g) Scholz M. Hey-Hawkins E. Chem. Rev. 2011;111:7035–7062. doi: 10.1021/cr200038x. [DOI] [PubMed] [Google Scholar]; (h) Hey-Hawkins E. and Teixidor C. V., Boron-Based Compounds: Potential and Emerging Applications in Medicine, John Wiley & Sons, 2018 [Google Scholar]; (i) Otero R. Seoane S. Sigüeiro R. Belorusova A. Y. Maestro M. A. Pérez-Fernández R. Rochel N. Mouriño A. Chem. Sci. 2016;7:1033–1037. doi: 10.1039/C5SC03084F. [DOI] [PMC free article] [PubMed] [Google Scholar]; (j) Fujiki K. Kanayama Y. Yano S. Sato N. Yokokita T. Ahmadi P. Watanabe Y. Haba H. Tanaka K. Chem. Sci. 2019;10:1936–1944. doi: 10.1039/C8SC04747B. [DOI] [PMC free article] [PubMed] [Google Scholar]; (k) Couto M. Alamón C. Nievas S. Perona M. Dagrosa M. A. Teixidor F. Cabral P. Viñas C. Cerecetto H. Chem.–Eur. J. 2020;26:14335–14340. doi: 10.1002/chem.202002963. [DOI] [PubMed] [Google Scholar]; (l) Couto M. Alamón C. García M. F. Kovacs M. Trias E. Nievas S. Pozzi E. Curotto P. Thorp S. Dagrosa M. A. Teixidor F. Viñas C. Cerecetto H. Cells. 2020;9:1408. doi: 10.3390/cells9061408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For related reviews, see: ; (a) Quan Y. Xie Z. Chem. Soc. Rev. 2019;48:3660–3673. doi: 10.1039/C9CS00169G. [DOI] [PubMed] [Google Scholar]; (b) Duttwyler S. Pure Appl. Chem. 2018;90:733–744. doi: 10.1515/pac-2017-1202. [DOI] [Google Scholar]; (c) Zhang X. Yan H. Coord. Chem. Rev. 2019;378:466–482. doi: 10.1016/j.ccr.2017.11.006. [DOI] [Google Scholar]; (d) Yu W.-B. Cui P.-F. Gao W.-X. Jin G.-X. Coord. Chem. Rev. 2017;350:300–319. doi: 10.1016/j.ccr.2017.07.006. [DOI] [Google Scholar]; (e) Au Y. K. Xie Z. Bull. Chem. Soc. Jpn. 2021;94:879–899. doi: 10.1246/bcsj.20200366. [DOI] [Google Scholar]
- For selected examples, see: ; (a) Jiang W. Knobler C. B. Mortimer M. D. Hawthorne M. F. Angew. Chem., Int. Ed. 1995;34:1332–1334. doi: 10.1002/anie.199513321. [DOI] [Google Scholar]; (b) King B. T. Janoušek Z. Grüner B. Trammell M. Noll B. C. Michl J. J. Am. Chem. Soc. 1996;118:3313–3314. doi: 10.1021/ja9541443. [DOI] [Google Scholar]; (c) Hewes J. D. Kreimendahl C. W. Marder T. B. Hawthorne M. F. J. Am. Chem. Soc. 1984;106:5757–5759. doi: 10.1021/ja00331a072. [DOI] [Google Scholar]; (d) Wang Q. Tian S. Zhang C. Li J. Wang Z. Du Y. Zhou L. Lu J. Org. Lett. 2019;21:8018–8021. doi: 10.1021/acs.orglett.9b03009. [DOI] [PubMed] [Google Scholar]; (e) Tang C. Zhang J. Xie Z. Angew. Chem., Int. Ed. 2017;56:8642–8646. doi: 10.1002/anie.201702347. [DOI] [PubMed] [Google Scholar]; (f) Tang C. Zhang J. Zhang J. Xie Z. J. Am. Chem. Soc. 2018;140:16423–16427. doi: 10.1021/jacs.8b10270. [DOI] [PubMed] [Google Scholar]; (g) Zhang X. Zheng H. Li J. Xu F. Zhao J. Yan H. J. Am. Chem. Soc. 2017;139:14511–14517. doi: 10.1021/jacs.7b07160. [DOI] [PubMed] [Google Scholar]; (h) Zhang X. Yan H. Chem. Sci. 2018;9:3964–3969. doi: 10.1039/C8SC01154K. [DOI] [PMC free article] [PubMed] [Google Scholar]; (i) Zhang Z.-Y. Zhang X. Yuan J. Yue C.-D. Meng S. Chen J. Yu G.-A. Che C.-M. Chem.–Eur. J. 2020;26:5037–5050. doi: 10.1002/chem.201905647. [DOI] [PubMed] [Google Scholar]; (j) Liang Y.-F. Yang L. Jei B. B. Kuniyil R. Ackermann L. Chem. Sci. 2020:10764–10769. doi: 10.1039/D0SC01515F. [DOI] [PMC free article] [PubMed] [Google Scholar]; (k) Xu T.-T. Cao K. Zhang C.-Y. Wu J. Ding L.-F. Yang J. Org. Lett. 2019;21:9276–9279. doi: 10.1021/acs.orglett.9b03790. [DOI] [PubMed] [Google Scholar]; (l) Lin F. Yu J.-L. Shen Y. Zhang S.-Q. Spingler B. Liu J. Hong X. Duttwyler S. J. Am. Chem. Soc. 2018;140:13798–13807. doi: 10.1021/jacs.8b07872. [DOI] [PubMed] [Google Scholar]; (m) Mirabelli M. G. L. Sneddon L. G. J. Am. Chem. Soc. 1988;110:449–453. doi: 10.1021/ja00210a023. [DOI] [Google Scholar]; (n) Shen Y. Zhang K. Liang X. Dontha R. Duttwyler S. Chem. Sci. 2019;10:4177–4184. doi: 10.1039/C9SC00078J. [DOI] [PMC free article] [PubMed] [Google Scholar]; (o) Zhang Y. Sun Y. Lin F. Liu J. Duttwyler S. Angew. Chem., Int. Ed. 2016;55:15609–15614. doi: 10.1002/anie.201607867. [DOI] [PubMed] [Google Scholar]; (p) Teixidor F. Barberà G. Vaca A. Kivekäs R. Sillanpää R. Oliva J. Viñas C. J. Am. Chem. Soc. 2005;127:10158–10159. doi: 10.1021/ja052981r. [DOI] [PubMed] [Google Scholar]; (q) Puga A. V. Teixidor F. Sillanpää R. Kivekäs R. Viñas C. Chem. Commun. 2011;47:2252–2254. doi: 10.1039/C0CC05151A. [DOI] [PubMed] [Google Scholar]
- Guo C. Qiu Z. Xie Z. ACS Catal. 2021;11:2134–2140. doi: 10.1021/acscatal.0c05639. [DOI] [Google Scholar]
- For selected examples, see: ; (a) Lee, Jr. M. W. Sevryugina Y. V. Khan A. Ye S. Q. J. Med. Chem. 2012;55:7290–7294. doi: 10.1021/jm300740t. [DOI] [PubMed] [Google Scholar]; (b) Koshino M. Tanaka T. Solin N. Suenaga K. Isobe H. Nakamura E. Science. 2007;316:853. doi: 10.1126/science.1138690. [DOI] [PubMed] [Google Scholar]; (c) Kobr L. Zhao K. Shen Y. Comotti A. Bracco S. Shoemaker R. K. Sozzani P. Clark N. A. Price J. C. Rogers C. T. Michl J. J. Am. Chem. Soc. 2012;134:10122–10131. doi: 10.1021/ja302173y. [DOI] [PubMed] [Google Scholar]; (d) Wang J. Chen L. Ye J. Li Z. Jiang H. Yan H. Stogniy M. Y. Sivaev I. B. Bregadze V. I. Wang X. Biomacromolecules. 2017;18:1466–1472. doi: 10.1021/acs.biomac.6b01845. [DOI] [PubMed] [Google Scholar]
- For selected examples, see: ; (a) Zhou T. Qian P.-F. Li J.-Y. Zhou Y.-B. Li H.-C. Chen H.-Y. Shi B.-F. J. Am. Chem. Soc. 2021;143:6810–6816. doi: 10.1021/jacs.1c03111. [DOI] [PubMed] [Google Scholar]; (b) Momo P. B. Leveille A. N. Farrar E. H. E. Grayson M. N. Mattson A. E. Burtoloso A. C. B. Angew. Chem., Int. Ed. 2020;59:15554–15559. doi: 10.1002/anie.202005563. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Barday M. Janot C. Halcovitch N. R. Muir J. Aissa C. Angew. Chem., Int. Ed. 2017;56:13117–13121. doi: 10.1002/anie.201706804. [DOI] [PubMed] [Google Scholar]; (d) Vaitla J. Bayer A. Hopmann K. H. Angew. Chem., Int. Ed. 2017;56:4277–4281. doi: 10.1002/anie.201610520. [DOI] [PubMed] [Google Scholar]; (e) Xu Y. W. Zhou X. K. Zheng G. F. Li X. W. Org. Lett. 2017;19:5256–5259. doi: 10.1021/acs.orglett.7b02531. [DOI] [PubMed] [Google Scholar]
- For selected examples, see: ; (a) Li J. J., Heterocyclic Chemistry in Drug Discovery, John Wiley & Sons, Hoboken, 2013 [Google Scholar]; (b) Banerjee S. Veale E. B. Phelan C. M. Murphy S. A. Tocci G. M. Gillespie L. J. Frimannsson D. O. Kelly J. M. Gunnlaugsson T. Chem. Soc. Rev. 2013;42:1601–1618. doi: 10.1039/C2CS35467E. [DOI] [PubMed] [Google Scholar]; (c) Basili S. Bergen A. Dall'Acqua F. Faccio A. Granzhan A. Ihmels H. Moro S. Viola G. Biochemistry. 2007;46:12721–12736. doi: 10.1021/bi701518v. [DOI] [PubMed] [Google Scholar]; (d) Rescifina A. Zagni C. Varrica M. G. Pistarà V. Corsaro A. Eur. J. Med. Chem. 2014;74:95–115. doi: 10.1016/j.ejmech.2013.11.029. [DOI] [PubMed] [Google Scholar]
- For selected examples, see: ; (a) Iwasa K. Kim H.-S. Wataya Y. Lee D.-U. Eur. J. Med. Chem. 1998;33:65–69. doi: 10.1016/S0223-5234(99)80077-4. [DOI] [Google Scholar]; (b) Iwasa K. Nishiyama Y. Ichimaru M. Moriyasu M. Kim H.-S. Wataya Y. Yamori T. Takashi T. Lee D.-U. Eur. J. Med. Chem. 1999;34:1077–1083. doi: 10.1016/S0223-5234(99)00127-0. [DOI] [Google Scholar]
- For selected examples, see: ; (a) Jortzik E. Zocher K. Isernhagen A. Mailu B. M. Rahlfs S. Viola G. Wittlin S. Hunt N. H. Ihmels H. Becker K. Antimicrob. Agents Chemother. 2016;60:115–125. doi: 10.1128/AAC.01066-15. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Morisaki D. Kim H.-S. Inoue H. Terauchi H. Kuge S. Naganuma A. Wataya Y. Tokuyama H. Ihara M. Takasu K. Chem. Sci. 2010;1:206–209. doi: 10.1039/C0SC00125B. [DOI] [Google Scholar]; (c) Takasu K. Morisaki D. Kaiser M. Brun R. Ihara M. Heterocycles. 2005;66:161–166. doi: 10.3987/COM-05-S(K)67. [DOI] [Google Scholar]
- Kolli S. S. Pecone D. Pona A. Cline A. Feldman S. R. Am. J. Clin. Dermatol. 2019;20:345–365. doi: 10.1007/s40257-019-00423-z. [DOI] [PubMed] [Google Scholar]
- (a) He Z. Zajdlik A. Yudin A. K. Dalton Trans. 2014;43:11434–11451. doi: 10.1039/C4DT00817K. [DOI] [PubMed] [Google Scholar]; (b) Diaz D. B. Scully C. C. G. Liew S. K. Adachi S. Trinchera P. St Denis J. D. Yudin A. K. Angew. Chem., Int. Ed. 2016;55:12659–12663. doi: 10.1002/anie.201605754. [DOI] [PubMed] [Google Scholar]; (c) Trinchera P. Corless V. B. Yudin A. K. Angew. Chem., Int. Ed. 2015;54:9038–9041. doi: 10.1002/anie.201504271. [DOI] [PubMed] [Google Scholar]; (d) He Z. Yudin A. K. J. Am. Chem. Soc. 2011;133:13770–13773. doi: 10.1021/ja205910d. [DOI] [PubMed] [Google Scholar]; (e) St. Denis J. D. He Z. Yudin A. K. ACS Catal. 2015;5:5373–5379. doi: 10.1021/acscatal.5b00790. [DOI] [Google Scholar]
- (a) Zhang Q. Shi B.-F. Acc. Chem. Res. 2021;54:2750–2763. doi: 10.1021/acs.accounts.1c00168. [DOI] [PubMed] [Google Scholar]; (b) Hu P. Kong L. Wang F. Zhu X. Li X. Angew. Chem., Int. Ed. 2021;60:20424–20429. doi: 10.1002/anie.202106871. [DOI] [PubMed] [Google Scholar]; (c) Sun L. Chen H. Liu B. Chang J. Kong L. Wang F. Lan Y. Li X. Angew. Chem., Int. Ed. 2021;60:8391–8395. doi: 10.1002/anie.202012932. [DOI] [PubMed] [Google Scholar]
- (a) Li X. Tong X. Yan H. Lu C. Zhao Q. Huang W. Chem.–Eur. J. 2016;22:17282–17290. doi: 10.1002/chem.201603340. [DOI] [PubMed] [Google Scholar]; (b) Dash B. P. Satapathy R. Gaillard E. R. Maguire J. A. Hosmane N. S. J. Am. Chem. Soc. 2010;132:6578–6587. doi: 10.1021/ja101845m. [DOI] [PubMed] [Google Scholar]
- For selected examples, see: ; (a) Yang Z. Zhao W. Liu W. Wei X. Chen M. Zhang X. Zhang X. Liang Y. Lu C. Yan H. Angew. Chem., Int. Ed. 2019;58:11886–11892. doi: 10.1002/anie.201904940. [DOI] [PubMed] [Google Scholar]; (b) Sun Y. Zhang J. Zhang Y. Liu J. van der Veen S. Duttwyler S. Chem.–Eur. J. 2018;24:10364–10371. doi: 10.1002/chem.201801602. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All experimental and crystallographic data are available in the ESI.†