

Abstract
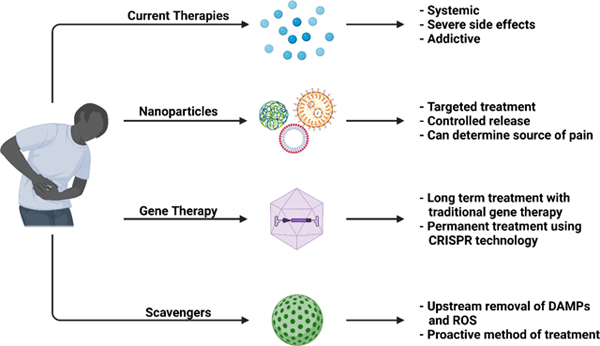
Pain is one of the most common medical conditions and affects more Americans than diabetes, heart disease, and cancer combined. Current pain treatments mainly rely on opioid analgesics and remain unsatisfactory. The life-threatening side effects and addictive properties of opioids demand new therapeutic approaches. Nanomedicine may be able to address these challenges as it allows for sensitive and targeted treatments without some of the burdens associated with current clinical pain therapies. This review discusses the physiology of pain, the current landscape of pain treatment, novel targets for pain treatment, and recent and ongoing efforts to effectively treat pain using nanotechnology-based approaches. We highl ight advances in nanoparticle-based drug delivery to reduce side effects, gene therapy to tackle the source of pain, and nanomaterials-based scavenging to proactively mediate pain signaling.
Keywords: pain, nanomedicine, drug delivery, gene therapy, CRISPR, ROS scavenging
Graphical Abstract
1. Introduction
Pain is among the most common reasons for medical care visits [1]. Globally, an estimated 20% of all patients experience pain, and 10% are diagnosed with chronic pain [2]. Over 40% of patients treated for primary pain report inadequate pain relief [3], and many pain relievers have debilitating side effects such as hepatotoxicity, depression, respiratory depression and addiction. The recent opioid epidemic—the leading cause of medication-induced overdose—highlights the urgent need for better treatment options for chronic pain. Chronic pain affects over 20% of the adult population in the United States [4, 5], and is associated with diseases such as cancer, diabetes, cystic fibrosis, and inflammatory diseases, and with trauma due to injury or surgery. Sufferers of chronic pain have the additional risk of anxiety and depressive disorders, sleep disorders, addiction, and disability [6]. The burden of pain for an individual includes not only physical and mental impairment but also medical costs, strained social relationships, and reduced work productivity. Chronic pain is also a financial burden for countries, costing the United States an estimated $635 billion annually [7, 8], due to the socioeconomic costs of healthcare expenses and lost productivity. Chronic pain is more prevalent as the aging population grows. Ultimately, pain negatively impacts the quality of life and is one of the leading causes of long-term disability. Despite this clear need, chronic pain remains difficult to treat effectively and without undesirable side effects.
Nanomedicine is a rapidly growing field, but its application to pain management has been limited by the complexity of pain physiology and the intractable nature of chronic pain. Nevertheless, nanotechnology is playing a major role in the next generation of pain treatments. New nanomaterials serve as drug carriers that target specific tissues, cell types and organelles with stimuli-sensitive release, and as nanodevices that detect the molecular source of pain. Nanoparticle drug carriers exhibit improved efficacy with smaller analgesic doses and longer-term relief of pain symptoms. Gene therapy delivery using nanoparticles is improving the long-term treatment of chronic pain, and both viral and non-viral vectors for gene therapy have proven effective in clinical trials. CRISPR is being used to modulate gene expression to reduce pain without eliminating sensitization. Scavengers of proinflammatory reactive oxygen species and free nucleic acids represent a proactive approach to pain management: instead of treating the symptoms of pain, scavengers remove molecules that trigger nociceptors and that cause sensitization. The application of nanotechnology to pain management represents a frontier for nanomedicine and is the subject of this review.
2. The physiology of pain
A better understanding of the physiology of pain is needed to develop new therapies that act on specific targets to reduce dosage and toxicity. Pain is an unpleasant, multifaceted sensory and emotional experience associated with actual or potential tissue damage [9], and involves physical, emotional, and psychosocial elements. Pain is difficult to treat and study in part because it is subjective; the perception of pain and its severity varies between individuals. Multimodal pain care regimens are often used to address the complex nature of pain. Pharmaceutical treatments are mechanism-based and considers both pain physiology and psychological factors. To better assess pain and provide personalized pain treatment, and to develop more effective nanotherapeutics, the physiological mechanisms underlying different types of pain must be better understood.
2.1. Acute and chronic pain
Pain is categorized as acute or chronic. Acute pain is temporary and resolves once the primary cause is removed (e.g., by wound healing), and functions as a signal to prevent further harm. Treatments for acute pain typically address the underlying cause, which is often injury or disease. Chronic pain is long-lasting, often arises without injury or disease, and does not always resolve once the primary cause is removed. The biological purpose of chronic pain is unclear, and often there is no recognizable endpoint. The mechanisms underlying chronic pain and the transition from acute pain to chronic pain remain poorly understood.
2.2. Pain pathways
Pain pathways involve both the peripheral and central nervous systems (Figure 1). Pain sensation occurs when mechanical, chemical, or thermal stimuli activate receptors called nociceptors, which are located on sensory neurons called A- or C-ty pe primary afferent fibers. Aδ-type fibers are large, myelinated fibers that rapidly conduct sharp, well-localized pain; in contrast, C-type fibers are small, unmyelinated fibers that transmit slow, dull, poorly-localized pain. Noxious stimuli (stimuli that have the potential to damage tissue) cause epithelial cells and cells in the immune and circulatory systems to release molecules that stimulate G-protein coupled receptors (GPCRs), ionotropic receptors, and tyrosine kinase receptors on the peripheral terminals of primary spinal afferent neurons; these released stimulatory molecules includes lipids (e.g., prostaglandins), proteases, neurotrophins (e.g., nerve growth factor), and peptides. Neurogenic inflammation occurs when the terminals release neuropeptides such as substance P and calcitonin gene-related proteins (CGRP) that activate receptors within the vasculature, on epithelial cells and immune cells [10]. Activation of receptors and channels of primary sensory neurons evokes central transmission of action potentials and subsequent release of glutamate, substance P, and CGRP within the dorsal horn of the spinal cord. These transmitters activate receptors on second-order neurons in the dorsal horn of the spinal cord. Pain perception occurs when these signals are transmitted through the spinothalamic tract to the cortex.
Figure 1. Pain pathways and current pain treatments.

The ascending pathway transmits pain and sensory information from the periphery to the brain. Painful stimuli activate primary afferent nociceptors of mechanosensitive Aδ and mechanothermal C fibers, which send signals to second-order neurons in the spinal cord. This information is transmitted up the spinothalamic tract to tertiary neurons in the thalamus, and pain is perceived in the somatosensory cortex. The descending pathway inhibits pain via noradrenergic/serotonergic neurons and Aβ fibers. Upon activation, interneurons in the substantia gelatinosa (central box) release enkephalin (ENK) or endogenous opioids that inhibit ascending impulses. Conventional pain treatments (blue text on the left) and their locations of action (circled numbers) are shown. Abbrev: NSAIDs, nonsteroidal anti-inflammatory drugs; α2 agonists, α2 adrenergic receptor agonists; TCA, tricyclic antidepressants; SSRI, selective serotonin reuptake inhibitor; SP, substance P; +, stimulation; -, inhibition.
2.3. Central and peripheral sensitization
Structural and functional changes in pain pathways such as increases in long-term potentiation at synapses and neuronal hypersensitivity prevent further harm following injury or damage. Elevated sensitivity to noxious stimuli causes hyperalgesia (enhanced sensitivity to pain), and can occur following surgery or opioid use; non-noxious stimuli such as light touch or warmth can also elicit pain (allodynia or pain from stimuli that are not normally painful), which can occur due to other medical disorders or following injury. Hypersensitivity via increased intracellular Ca2+ can occur by activation of N-methyl-D-aspartate (NMDA) receptors following injury. Influx of calcium ions causes upregulation of a-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors. Increase of AMPA receptors enhances postsynaptic excitation and activates protein kinases such as calmodulin dependent protein kinase II (a kinase that plays a role in synaptic plasticity, learning, and memory). Calcium influx also upregulates calcium-dependent kinases including cyclooxygenases (COXs) and nitric oxide synthases. This results in production of prostaglandin E2 and nitric oxide that causes neurotransmitter release and activation of downstream second messenger signaling via the cyclic adenosine monophosphate (cAMP)/protein kinase A (PKA) pathways.
Peripheral and central sensitization (heightened sensitivity to stimuli) play critical roles in chronic pain. Central sensitization occurs when nociceptive neurons in the central nervous system fire at subthresholds, resulting in neuronal hyperexcitability. Activated neurons in the dorsal horn of the spinal cord release glutamate and neuropeptides that bind receptors and generate action potential firing. Microglia and astrocytes in the spinal cord release cytokines and chemokines that stimulate neuronal firing [11]. Peripheral sensitization is hyperexcitability at primary afferent neurons. Activation of peripheral receptors is regulated by ion channels that include transient receptor potential ion channels (TRPs) such as transient receptor potential ankyrin 1 (TRPA1) and transient receptor potential vanilloid type 1 (TRPV1), and sodium channels such as Nav1.7, Nav1.8, and Nav1.9 [12, 13].
2.4. Pain and inflammation
Pain and inflammation are tightly connected. Damage to vascularized tissue triggers inflammatory responses, causing T cells, neutrophils, mast cells, and macrophages to release inflammatory mediators such as hydrogen ions, adenosine triphosphate (ATP), serotonin, and substance P, which in turn induce vasodilation, increased vascular permeability, and plasma extravasation. These inflammatory molecules also activate pain receptors, increasing an inflow of calcium and sodium ions into neurons and inducing action potential firing. Proinflammatory mediators promote the release of injury byproducts such as prostaglandins, bradykinin, and histamines that stimulate pain neurons to release additional inflammatory neuropeptides and cytokines that exacerbate inflammation. Proinflammatory chemokines (CCL2, CXCL5) and cytokines such as tumor necrosis factor-α (TNF-α) and interleukin 1β (IL-1β) bind receptors and ion channels to sustain the inflammatory response [14]. Damaged cells release phospholipids that are converted to prostaglandin H2 (PGH2) via COXs. Prostaglandin synthases convert PGH2 to PGE2, prostacyclin (PGI2), and PGF2, which mediate fever, enhanced pain, and inflammation, or to thromboxane A2 (TXA2) which mediates platelet aggregation. Inflammation usually subsides when damaged tissues have recovered, but can become chronic inflammation, which continues past the healing period and persists for months or years.
Following inflammatory response, phospholipase A2 is released, which is then converted into arachidonic acid. The COX enzymatic pathway, which includes COX-1 and COX-2 is responsible for converting arachidonic acid into prostaglandins (PGs). Normally, COX-1 produces thromboxane and PGs in platelets, GI mucosal cells, and renal tubule cells. COX-2 is upregulated at sites of inflammation and produces PGs that cause inflammation and pain. Inhibition of COX-2 reduces production of PGs to result in anti-inflammatory and analgesic effects.
2.5. Nociceptive, neuropathic, and nociplastic pain
Identifying the pathophysiological origin of pain is important for determining an appropriate treatment. Pain is classified into neuropathic, nociceptive, and nociplastic pain. Nociceptive pain arises through nociceptor activation from noxious stimuli (mechanical, chemical, or thermal stimuli that have the potential to damage tissue). Nerve cells are responsible for propagation of pain signals from peripheral nerve fibers to the spinal cord and the brain. Nociceptive pain typically results from physical injury and presents as somatic pain, a well-defined, precisely-located pain from injury to skin, joints, and muscles, or visceral pain, a type of pain due to injury to internal organs or viscera that is often diffuse and difficult to localize [15].
Neuropathic pain originates from injury or dysfunction of the somatosensory system and is categorized into central and peripheral neuropathic pain. Central neuropathic pain stems from injury lesions to the spinal cord or brain and can be caused by diseases such as Parkinson’s disease. Peripheral neuropathic pain results from nerve damage, which often occurs in the hands and feet and manifests as a chronic stabbing or burning sensation. Roughly 20% of patients who experience chronic pain suffer from neuropathic pain [16].
Nociplastic pain is a new mechanistic descriptor that encompasses pain with an unknown origin or altered nociception. The mechanisms underlying nociplastic pain include changes in nociceptive signaling that result in peripheral and central sensitization. While traditionally pain has been considered a symptom of injury or damage to the nervous system, nociplastic pain considers forms of chronic pain without a clear origin to be disease states themselves. Common examples of nociplastic pain include chronic musculoskeletal and visceral pain including fibromyalgia and lower back pain [17].
3. Current pain treatments and new targets
3.1. Current pain treatments
3.1.1. Non-opioid pain medications
Treatment for chronic pain typically begins with a low-risk, non-opioid analgesic, such as acetaminophen (Tylenol), non-steroidal anti-inflammatory drugs (NSAIDs), and adjuvant medications (e.g., antidepressants, anticonvulsants, and corticosteroids). Acetaminophen is a first line treatment for mild musculoskeletal pain (e.g., osteoarthritis, lower back pain). Acetaminophen block proinflammatory prostaglandin synthesis by oxidized cyclooxygenases (COX), with analgesic and antipyretic (fever-reducing) effects [18, 19]. Acetaminophen is effective in low doses for short duration, but long-term use or high doses can cause hepatotoxicity [20].
NSAIDs such as aspirin, ibuprofen (Advil, Motrin), and naproxen (Aleve) are the most common first line treatments for inflammation-associated pain. Unlike acetaminophen, NSAIDs relieve both pain and inflammation. Many NSAIDs are COX inhibitors that reduce prostaglandin production to relieve inflammation; these include COX-1 inhibitors (low-dose aspirin), COX-2 inhibitors (celecoxib), and non-selective COX inhibitors (ibuprofen, naproxen). However, since cyclooxygenases mediate multiple physiological functions, prolonged use of NSAIDs at high dosage can have negative effects such as gastric bleeding, peptic ulcers, kidney damage, myocardial infarction, or stroke.
Adjuvant analgesics such as antidepressants and anticonvulsants are increasingly being used to treat neuropathic and nociplastic pain. Antidepressants do not act as acute analgesics but can be used to treat chronic pain. The requirement of a longer treatment duration when using antidepressants suggests that long-term neuronal plasticity is involved in chronic pain. Antidepressants used to treat neuropathic pain include tricyclic antidepressants (TCAs) such as amitriptyline, serotonin-norepinephrine inhibitors (SNRIs) such as duloxetine, and selective serotonin reuptake inhibitors (SSRIs) such as paroxetine. TCAs inhibit the presynaptic reuptake of norepinephrine and serotonin, and block α2 adrenergic, H1-histaminergic, and muscarinic cholinergic receptors, and are effective in 33–50% of patients with chronic pain [21]. SNRIs are balanced noradrenaline and serotonergic inhibitors that rely on drug dosage and concentration and are effective in 20–25% of patients. Duloxetine has a high affinity for norepinephrine and serotonin reuptake transporters and is effective for treatment of diabetic peripheral neuropathic pain [22]. Only ~14% of patients are relieved of pain with SSRIs, which block serotonin reuptake [23]. The differing efficacy of antidepressants with different mechanisms of action suggests that noradrenaline plays a more important role in relieving pain than serotonin. The anticonvulsants gabapentin and pregabalin are currently used to treat neuropathic pain, especially postherpetic neuralgia and peripheral diabetic neuropathy. Gabapentin is a gamma-amino-butyric acid (GABA) analog that binds the α2δ subunit of the voltage-gated calcium channel complex to block the presynaptic neurotransmitter release. Like gabapentin, pregabalin binds the calcium channel α2δ subunit, but with six times the potency. These anticonvulsants address the increased sensitivity associated with chronic pain and work by reducing action potential firing at nerve terminals.
Other non-opioid pain treatments include local anesthetics and steroids. Local anesthetics such as lidocaine are commonly used for short-acting pain relief. Lidocaine reduces sharp burning pain such as postherpetic neuralgia in shingles by blocking voltage-dependent sodium channels to mediate pain transmission. Lidocaine can be applied topically as a local anesthetic to relieve pain or carefully injected as a nerve block to lessen pain and discomfort from medical procedures. Capsaicin is a topical cream that targets nociceptors and is a highly selective agonist of noxious heat-sensing TRPV1 in nociceptors. Persistent activation of TRPV1 by capsaicin reduces receptor function and pain sensitivity for an extended period of time [24]. Steroids are also used for chronic pain management. Glucocorticoids relieve pain by targeting proinflammatory responses associated with pain, for example by blocking prostaglandin synthesis and reducing vascular permeability to treat inflammation and tissue edema [25]. Dexamethasone, a synthetic corticosteroid, is the most frequently used steroid for pain relief due to its high potency, long half-life, and low mineralocorticoid activity which results in less fluid retention. However, the side effects of dexamethasone include gastric bleeding and muscle myopathy. Prednisolone, another steroid used for pain relief, has fewer side effects than dexamethasone and acts by stimulating glucocorticoid receptors to address the inflammatory component of pain. Recently, α2-adrenergic agonists have been used for anesthetic management alone or in combination with local anesthetics. Clonidine, an α2-adrenergic agonist in combination with local anesthetics extends the length of peripheral nerve blocks. Dexmedetomidine, a more selective α2-adrenergic agonists, has also been used in combination with local anesthetics to prolong the anesthetic effects with both central and peripheral nerve blockers [26–29].
3.1.2. Opioids
Opioids are used when nociceptive symptoms become more severe and when non-opioid analgesic regimens are inadequate. Opioids are potent analgesics and have been considered the most effective pain medications for non-neuropathic pain. Opioid medications act like endogenous opioids, which bind opioid receptors throughout the peripheral and central nervous systems. Opioid receptors are GPCRs that, when activated on the presynaptic terminal, cause the beta-gamma subunit to inhibit voltage-gated calcium channels, preventing release of the neurotransmitter glutamate and the neuropeptides substance P and CGRP [30]. When opioid receptors are activated on postsynaptic terminals, G protein-coupled inwardly rectifying potassium channels (GIRK) are opened to allow outflow of potassium, preventing depolarization of the neuron. The Gα subunit also binds phospholipase C and adenylyl cyclase to cause downstream signaling such as cAMP production to modulate neurotransmitter release [31]. Overall, activation of opioid receptors is antinociceptive by reducing action potential firing and neuronal sensitivity. Activation of opioid receptors at the brainstem and spinal cord removes inhibition of GABAergic neurons, causing GABA release and hyperpolarization to prevent pain transmission.
Many opioid drugs activate the μ and κ opioid receptors for pain relief. Morphine is a natural opiate used to treat moderate to severe pain. Synthetic opioids, including fentanyl, hydrocodone, methadone, and oxycodone, mimic endogenous opioid peptides but with higher potency [32]. Methadone is used to relieve both nociceptive and neuropathic pain since it antagonizes NMDA receptors and acts as a serotonin-norepinephrine inhibitor.
Although opioids effectively relieve acute pain, prolonged use causes serious side effects. Constipation is a common on-target effect due to the presence of opioid receptors in the small intestine that control gut motility. Nausea occurs with opioid use due to chemoreceptor binding in the medulla [33]. Dose-dependent respiratory depression is a dangerous side effect of opioid drug use. A high dosage of opioids can lead to activation of opioid receptors of interneurons in the pons and the Pre-botzinger complex of the medulla, leading to suppression of respiratory activity. Other dangerous side effects of opioids are related to addiction, dependence, and tolerance. Opioid drugs activate opioid receptors in the brainstem and in the ventral tegmental area of the brain, which inhibits GABA release at presynaptic terminals, promoting dopaminergic activity in the reward system [34]. Chronic opioid usage causes receptor desensitization and tolerance. When opioid use is reduced or stopped, withdrawal symptoms include diarrhea, anxiety, and dysphoria. The recent opioid epidemic was driven by increased opioid prescriptions and overuse, which led to addiction, overdoses and deaths [35]. Opioid abuse is now thought to be responsible for more deaths than motor vehicle accidents and suicide combined. The devastation of the recent opioid epidemic highlights the urgent need for better treatment options to address chronic pain.
3.1.3. Other pain treatments
Other methods of pain treatment include nerve blockers and electrical stimulation. Nerve blockers are used to treat chronic pain when other drugs do not provide relief or to avoid side effects, and include epidural steroid injections and peripheral nerve blockers [36]. Local anesthetics and neurotoxins are two common forms of nerve block agents. Epidural steroid injections are commonly administered for spine-related pain. Continuous peripheral nerve blockers, which have been traditionally used for perioperative or postoperative periods, are now also used for chronic pain. Continuous administration of peripheral nerve blockers uses a lower initial bolus, resulting in reduced systemic toxicity and reduced supplemental opioid usage and side effects [37].
Transcutaneous electrical nerve stimulation (TENS) is a non-pharmacological method of pain relief. TENS uses a small battery-powered device to apply a mild electrical current to activate endogenous inhibitory mechanisms in the central nervous system. TENS activates opioid receptors in the descending inhibitory pathway of the rostral ventromedial medulla, spinal cord, and periaqueductal gray [38]. TENS also activates muscarinic receptors and GABA-A receptors in the spinal cord to reduce hyperalgesia. A spinal cord stimulator (SCS) is an implanted device that is inserted into the dorsal epidural space that sends low currents of electricity into the spinal cord for chronic neuropathic pain relief. The specific mechanism of action of SCS is unclear, but has been shown to increase the release of GABA to suppress dorsal horn neuronal hyperexcitability [39].
3.2. New targets
Current pain medications are inadequate due to lack of specificity and serious side effects. Recent studies have investigated novel pain targets and novel methods for pain treatment. Advances in pain therapy include specific targeting of ion channels, pain receptors, and mediators of inflammation, described below.
3.2.1. Voltage-gated sodium channels
Voltage-gated sodium channels are an attractive target for pain treatment. An influx of sodium through the channel shifts a neuron’s membrane potential towards action potential depolarization and neuronal firing. Sodium channel Nav1.7, which is expressed in peripheral sensory neurons, dorsal horn neurons, and sympathetic ganglion neurons, is associated with pain transmission [40]. Loss-of-function mutations in the gene encoding Nav1.7, SCN9A, leads to congenital insensitivity to pain, and gain-of-function mutations are associated with familial pain disorders such as paroxysmal extreme pain disorder and inherited primary erythromelalgia [41]. Recent studies have targeted the Nav1.7 channel with a monoclonal antibody specific to voltage-sensor regions that allosterically control channel gating [16].
3.2.2. Nerve growth factor and TrkA
Another target for pain treatment is nerve growth factor (NGF) and its receptor, tropomyosin-related kinase A (TrkA). NGF is a neurotrophin that is released from all innervated peripheral tissues, immune cells, CNS, and PNS, and promotes the growth and survival of sensory and sympathetic neurons and ganglia. NGF levels increase in response to noxious stimuli from injury, neuroinflammation, and chronic pain. The binding of NGF to TrkA receptors in Aδ- and C-type fibers and mast cells releases proinflammatory mediators such as histamine and protons, and exacerbates inflammation. Tanezumab is a humanized monoclonal IgG2 antibody that blocks NGF-TrkA binding, and was fast-tracked by the FDA for patients with osteoarthritis and chronic lower back pain [42]. The cost of Tanezumab is high, but it can be administered only once every eight weeks and does not have the adverse side effects seen with opioids and some NSAIDs. Tanezumab can also be administered at home with a single subcutaneous injection, avoiding medical visit costs [43]. Another promising antibody for treating osteoarthritis is fasinumab, by Regeneron, a recombinant fully-human anti-NGF antibody that is currently in clinical trials.
3.2.3. Endosomal targets
Endosomes are commonly described as conduits for biomolecule degradation or recycling, but are also the site of persistent signals from GPCRs that control pain transmission and thus are a promising target for treating chronic pain. GPCRs in pain pathways were once thought to signal solely at the plasma membrane, and drug discovery was focused on targeting receptors at the cell surface. However, many of these drugs were found to be unsuccessful in clinical trials. Although such drugs might fail for multiple reasons, one possibility could be related to their inability to antagonize GPCRs within the acidic microenvironment of endosomes. Thus, the targeted delivery of GPCR agonists and antagonists to endosomes may result in more effective mediation of GPCR pain signaling.
Endosomal signaling from GPCRs such as the neurokinin 1 receptor (NK1R), calcitonin-like receptor (CLR), and protease-activated receptor 2 (PAR2) might regulate the expression of genes in the nucleus and the activity of ion channels at the plasma membrane that control neuronal excitation and chronic pain [44–46]. For example, substance P, a ligand of the neurokinin 1 receptor, causes increased activation of extracellular signal-regulated kinase (ERK) in the nucleus and protein kinase C (PKC) and cAMP in the cytosol [44]. These signals mediate sustained excitation of spinal neurons and pain transmission in the spinal cord. Inhibitors of clathrin and dynamin suppress substance P-induced signaling by ERK, PKC, and cAMP as well as abolishing persistent neuronal firing, suggesting that endosomal signaling mediates neuronal excitability. Studies are now examining GPCRs in endosomes as a therapeutic target for chronic pain treatment. Conjugation of transmembrane lipid cholestenol with an NK1R antagonist promotes drug delivery to endosomes, allowing antagonism of endosomal NK1R signaling. Nanoparticle technology (described in section 4) is being used to deliver antagonists of pronociceptive receptors to endosomes—which have an acidic and reducing environment that can be exploited for targeted delivery of these GPCR inhibitors.
3.2.4. Other targets
Other targets for pain treatment include purinergic P2X receptor channels and the angiotensin II receptor. P2X receptors are ligand-gated cation channels found on peripheral afferents (the axons of sensory neurons). Damaged and inflamed tissues release ATP which binds and activates P2X receptors, leading to influx of Ca2+ and Na+ into the cytoplasm for membrane potential depolarization. Animals with a P2X3 knockdown or siRNA-silenced P2X3 expression exhibit decreased pain behavior [47]. P2X3 antagonists are a potential therapy for neuropathic pain [48]. Abbott Laboratories developed the P2X3 antagonist A-317491, which reduced pain in chronic and inflammatory pain models. Afferent Pharmaceuticals’ potent P2X3 antagonist, AF-219, is currently in Phase 2 trials for cystitis/bladder pain syndrome. Additional, second-generation P2X3 antagonists that have a reduced risk of hyperbilirubinemia are being developed [49].
The angiotensin II-receptor (AT2R) is another target for treating chronic pain. Angiotensin II is a mediator of the renin-angiotensin system and has been implicated in pain modulation. Gαs-coupled AT2R signaling modulates sensory neuron firing, and Gαi-coupled AT2R signaling leads to analgesia in mice [50]. Activation of AT2R on macrophages causes mechanical and cold pain hypersensitivity in mouse models of neuropathic pain and chronic inflammatory pain [51]. Other targets of chronic pain drugs currently in development include CGRP pathways, TNF-α, epidermal growth factor receptor, and TRP channels.
4. Nanoparticles for pain management
Nanomedicine aims to apply nanotechnology to enhancing the efficacy and safety of drugs, for example by encapsulating naked drugs in biocompatible nanocarriers such as nanoparticles, liposomes, micelles, and dendrimers. Nanoparticulate drug delivery systems (NDDSs, Figure 2) have design parameters such as size, shape, surface charge, and cargo dose that can be optimized to prolong drug circulation and to target specific tissues or subcellular organelles [52, 53]. NDDS surfaces can be functionalized with cell-penetrating peptides or ligands to deliver therapeutics across the blood-brain barrier and to the central nervous system. NDDSs can achieve enhanced therapeutic efficacy by regulating spatial localization and reducing dosage and side effects. Therapeutic potency can be enhanced by using a nanocarrier containing multiple analgesics or by using small molecules that target pain signaling receptors. Such approaches might overcome the redundancy that is inherent in essential processes, such as pain transmission. NDDSs are being developed to treat systemic, neuropathic, localized, and disease-associated pain with reduced risk of addiction. Theragnostic nanoparticles are also being developed to detect the source of pain.
Figure 2. Nanoparticles for pain relief.

Design considerations for analgesic nanoparticulate drug delivery systems include the type and location of pain (top left), what drugs are clinically available (top right), nanocarrier composition (middle), route of administration (lower left), and accessible external stimuli (lower right).
4.1. Analgesic nanoparticulate drug delivery systems
Analgesic nanoparticulate drug delivery systems (Figure 2) can be used for relief of systemic, neuropathic, and inflammation-related pain by serving as nanocarriers of drug cargo and targeting molecules. For example, targeting opioid receptors to create safer drugs is an active area of research, and pain medicine is moving towards more effective delivery of non-opioid analgesics and less addictive opioids. Intraoral, intranasal, and transdermal administration are preferred routes of administration for patient compliance, while local and systemic administration via injection in clinics is useful for treatments that require longer time periods between doses. Localized administration of local anesthetic-loaded NDDSs can block pathways related to perioperative pain. Neurotoxins traditionally considered too dangerous can benefit from NDDSs to become new local anesthetic candidates.
4.1.1. Systemic pain: opioids and new approaches
Conventional pain treatments with naked drugs provide uncontrolled drug release; often, several doses are taken daily to achieve and maintain sufficient plasma concentrations. However, such intermittent administration causes fluctuations in plasma drug levels, which can fall below the effective concentration or exceed the toxic concentration threshold [54]. Liposomes and polymeric nanoparticles have been used since the 1990s to encapsulate opioids for extended-release (ER) and reduced systemic toxicity [55–58]. These efforts led to FDA approval and commercialization of two ER morphine NDDSs, Depodur and Avinza. Depodur uses proprietary DepoFoam, a multivesicular liposomal delivery system that encompasses numerous non-concentric aqueous chambers containing a drug [59]. Single epidural injection of Depodur achieves 48 h of analgesia [60]. Orally delivered Avinza contains ER morphine capsules in proprietary beads consisting of ammonium-methacrylate copolymers that are solubilized by gastrointestinal fluids [61]. The drug solution then diffuses out of the capsule, providing therapeutic plasma levels for up to 24 h [59].
Other formulations of opioids with ER profiles have been studied extensively and are commercially available [59]. Liposomes and polymeric nanoparticles used in these ER formulations are generally considered as safe carriers at therapeutic concentrations. Modifications such as liposome PEGylation and cationic coating can potentially improve safety only when the inherent toxicity of the functionalization is accounted for. ER opioids offer advantages such as stabilized plasma drug levels, but suffer from misuse and abuse, and drug tolerance further complicates their safety and analgesic efficacy. A growing number of investigations are focused on therapeutics with lower abuse potential [62, 63].
Enkephalin (ENK) is an attractive neuropeptide analgesic; this endogenous neuropeptide preferentially binds δ-opioid receptors, which are less correlated with abuse and tolerance than μ-opioid receptors [64]. Leu-enkephalin (LENK) has been conjugated with lipid squalene to target proinflammatory mediators [65]. LENK-squalene bioconjugate nanoformulated in dextrose allowed a higher drug payload than ENK-loaded liposomes or poly(lactic-co-glycolic acid) (PLGA) nanoparticles. Animal studies showed that an intravenous injection of LENK-squalene nanoparticles achieves a greater anti-hyperalgesic effect than morphine, without causing tolerance. Further, using a microparticulate formulation of clustered nanoparticles, intranasal administration can be used to deliver LENK-squalene specifically to the brain [66].
As an alternative to opioids, new pain medications in development target GPCRs including adrenergic, cannabinoid, and serotonin receptors [67]. PLGA-PEG nanoparticles containing the synthetic cannabinoid CB13 have achieved an analgesic effect for up to 11 days after one oral dose in a murine neuropathic pain model [68]. Mesoporous silica nanoparticles (MSNs) are well-suited for systemic and local delivery due to their dual surfaces (internal cylindrical pores and exterior particle surface), which enable a multistage delivery. MSNs loaded with the cannabinoid Δ9-THC and the erythropoietin-derived polypeptide ARA290 provide sustained systemic and neuropathic pain relief. THC-MSN-ARA290 nanocomplexes represent a combinatorial delivery system in which THC diffuses into the circulation while ARA290 is released upon the cleavage of a disulfide bond triggered by glutathione. With two intraperitoneal (IP) injections, an analgesic effect was seen for four weeks in mouse models of thermal hyperalgesia and mechanical allodynia [69].
pH-responsive MSNs functionalized with a PEGylated liposome coating (lipoMSN) and loaded with a δ-opioid receptor agonist DADLE ([D-Ala2, D-Leu5]-Enkephalin) can target endosomal δ-opioid receptors and provide sustained inflammatory pain relief. The pH-responsiveness of the lipoMSN allows for preferential delivery to the acidified endosome while the DADLE-functionalized liposomal coating helps to cloak the MSN core and selectively target δ-opioid receptor-expressing neurons. One intrathecal injection of the lipoMSN can provide an analgesic effect lasting for 6 hours in a mouse model of inflammatory nociception [70]. This study suggests that endosomal signaling of DOPr may provide relief from inflammatory pain, which presents a unique opportunity for NDDSs because of the natural and efficient trafficking of nanoparticles to endosomes.
4.1.2. Neuropathic pain: local anesthetics
NDDSs can enhance the therapeutic potential of local anesthetics to for perioperative pain management. Local anesthetics such as lidocaine and prilocaine are widely used for perioperative pain management, and act by blocking specific nerve pathways [71]. ER local anesthetics have been developed to prolong their analgesic effect while preventing adverse events.
Traditional local anesthetic formulations for postsurgical analgesia have a short duration of effect, lasting no longer than 24 h with a single injection [72, 73]. Several approaches have been used to encapsulate local anesthetics in polymeric nanoparticles (e.g., PLA, PLGA, PCL, alginate, chitosan, and copolymers), resulting in long-term stability, sustained release, and enhanced anesthetic efficacy in vivo [74–77]. The only FDA-approved liposomal bupivacaine, Exaparel, which also uses the DepoFoam platform, can reduce postoperative pain for up to 3 days after a single infiltration [78].
The Nav1.4 inhibitor lamotrigine has demonstrated efficacy for neuropathic pain treatment in multiple randomized controlled trials [79, 80]. However, its clinical applications in neuropathic pain are limited by the risk of severe rash, and it has a poor pharmacokinetic profile due to nonselective distribution to organs other than the brain. Lamotrigine-carrying PLGA nanoparticles were functionalized with transferrin or lactoferrin to enhance blood-brain barrier permeability [79]. Preferential distribution of these nanoparticles to the brain and reduced accumulation in non-target organs were observed in a partial sciatic nerve injury mouse model, with lactoferrin being superior to transferrin as the targeting ligand.
In labor pain, epidural local anesthetics are injected into the lower spinal nerves. Epidurals have a short-lasting effect and can have side effects such as infection and nerve damage. Solid lipid nanoparticles (SLNs) can be used as drug carriers for epidurals, and can double their longevity via controlled release and reduce side effects [81]. Lidocaine-loaded SLNs allow longer-lasting effects than free lidocaine with more effective sensory and motor blocks [82]. However, the toxicity of SLNs is not well characterized; ongoing research on nanoparticles for delivering epidurals aims to reduce motor weakness and systemic absorption, optimize controlled release, and reduce the dosage required for an analgesic effect.
4.1.3. Neuropathic pain: neurotoxins
NDDSs can enable the safe use of otherwise toxic analgesic molecules. For example, conventional local anesthetics are nonspecific Nav channel blockers, and their use can result in rare but life-threatening systemic toxicity upon leakage into the cardiovascular system or central nervous system [83–85]. Neurotoxins are also potent and specific Nav blockers with slightly less serious complications (e.g., muscle paralysis) [86]. Guanidinium toxins, tetrodotoxin (TTX) and saxitoxin (STX), are Nav blockers that synergistically prolong anesthesia when combined with other local anesthetics [87, 88]. Clinical use of these neurotoxins has been limited due to their systemic toxicity. One way to circumvent this toxicity is to slowly release a therapeutic amount. Conjugating TTX with poly(triol dicarboxylic acid)-co-PEG (TDP) has achieved nerve blocks in rat sciatic nerves from several hours to 3 days, depending on the dose. Minimal systemic or local toxicity was induced, and TTX release could be adjusted by tuning the hydrophilicity of the TDP polymer [89]. Local administration is another method to circumvent toxicity while simultaneously increase efficacy. Local injection of hollow silica nanoparticles loaded with TTX to the sciatic nerve increased the duration of nerve block while decreasing toxicity. The nanoparticles could penetrate the sciatic nerve in a size dependent manner, enhancing efficacy while improving safety [90].STX and dexamethasone have also been encapsulated in liposomes for treatment of neuropathic pain [91]; a single percutaneous injection of STX-dexamethasone nanoparticles provided a nerve block that lasts for about a week in a rat spared nerve injury model [92]. Crotoxin, a rattlesnake venom-derived neurotoxin with prolonged anti-inflammatory and antinociceptive activity, was encapsulated in inert SBA-15 MSNs to treat neuropathic pain, resulting in reduced toxicity of crotoxin and enhanced analgesic effect after subcutaneous and oral delivery in a mouse neuropathic pain model [93].
4.1.4. Chronic pain
NSAIDs and acetaminophen are generally safe in low doses, but prolonged use can cause side effects in the stomach and liver, respectively. NDDSs are effective chronic pain treatment options due to their controlled release kinetics and versatility of nanoformulation.
Drug-induced acute liver failure has a high morbidity and mortality rate, with the leading cause being acetaminophen overdose [94]. Milk thistle-extracted silymarin has shown hepatoprotective properties due to its antioxidant, anti-inflammatory, and antifibrotic effects [95]. Silymarin nanoparticles entrap acetaminophen via nanoprecipitation, and upon intraperitoneal injection, glutathione is generated to counter hepatic damage [96]. In an animal model of acetaminophen-induced hepatotoxicity, no death occurred even when the drug was administered after established hepatic necrosis. Similar NDDS-based approaches can reduce the side effects of long-term NSAID use for chronic pain.
Osteoarthritis is a disease of the cartilage and bone and is marked by chronic pain. Most osteoarthritis drugs are aimed at mediating this pain. Osteoarthritis is typically treated with NSAIDs, cyclooxygenase-2 inhibitors, or experimental therapeutics such as MAPK-inhibiting drugs. Targeting these drugs to the cartilage matrix and subchondral bone can be achieved by using nanocarriers (<40 nm diameter) with positive surface charges, such as micelles and dendrimers. Targeting the cartilage surface, synovial membrane, intra-articular space, or infrapatellar fat pad require larger nanoparticles (>60 nm) to avoid penetration into cartilage, making liposomes, high-generation dendrimer micelles, and other larger nanoparticles more suitable nanocarriers for these applications. The combination of osteoarthritis drugs with appropriate nanocarriers for targeting will lead to more effective treatments of osteoarthritis-associated pain with fewer side effects [97].
Other sources of chronic pain include receptor signaling from subcellular compartments, such as the GPCR cascade. Endocytosed neurokinin 1 receptor (NK1R), a GPCR in the central and peripheral nervous systems, mediates pain and offers a new target for treating chronic pain [44]. pH-responsive nanoparticles loaded with the NK1R antagonist aprepitant deliver the drug to acidic endosomes environment to block NK1R signaling [98]. These nanoparticles exhibit greater and more sustained pain relief than standard therapy with free drug in animal models of nociceptive, neuropathic, and inflammatory pain (Figure 3).
Figure 3. pH-responsive nanoparticles target NK1R in the endosome to target chronic pain.

A) Structure of pH-responsive DIPMA and pH-non-responsive BMA nanoparticles. The nanoparticles share the same hydrophilic shell, P(PEGMA-co-DMAEMA), but have different hydrophobic cores. B) Accumulation of nanoparticles in spinal neurons, the target of the encapsulated Aprepitant. C) pH-responsive nanoparticles target NK1R in endosomes. D) DIPMA-Aprepitant (AP) nanoparticles are more effective than morphine in mouse models of inflammatory pain [98].
4.1.4. Localized pain
Localized pain in joints, burns, surgical sites, and in many diseases is commonly treated with NSAIDs and pain receptor inhibitors, but opioids are often used when the pain becomes severe. NDDSs can target specific pain receptors and treat the underlying source of localized pain.
Functionalization of liposomes with monoclonal antibodies or antibody fragments (immunoliposomes) is a popular targeted drug delivery strategy that reduces doses and thus side effects [99]. For example, the antidiarrheal loperamide was converted to the first peripherally-selective analgesic by intravenous use of anti-intracellular adhesion molecule 1 (ICAM-1) immunoliposomes [100]. This NDDS showed antinociceptive and anti-inflammatory effects exclusively in peripheral inflamed tissue in a rat local inflammation model. In a follow-up study, conjugation of the NDDS with anti-oxytocin receptor increased immunoliposome localization at the uterus of pregnant mice by 7-fold; localization was not detected in the maternal brain or fetus, preventing inflammation-induced preterm labor [101].
For migraine treatment, Girotra et al. encapsulated the GPCR agonists sumatriptan and zolmitriptan in various nanoparticles (chitosan solid lipid, ApoE-bovine serum albumin, and PLGA-poloxamer) to enhance brain targeting [102–104]. This group applied in silico models to virtually screen ligands from Drugbank, and identified nystatin as the lead ligand against four receptors that are responsible for migraine pathogenesis, including CGRP (PDB ID: 3N7R). Mice studies using nystatin-chitosan nanoparticles revealed an analgesic effect via IP injection and greater accumulation of nanoparticles in the brain than in other organs such as the liver and spleen [104].
Metastatic cancer can be excruciatingly painful, and the success rate of treatment is low. Between 30–50% of patients with tumors receiving active treatment and 70–90% with advanced-stage disease experience chronic pain [105]. Prostate cancer tends to metastasize to the bone, where it often becomes untreatable and causes intractable pain. Gdowski et al. developed alendronate-conjugated PLGA-cabazitaxel nanoparticles to target bone metastases to treat bone pain. In mice orthoptic bone tumor models, the targeted nanoparticle-treated group showed lower pain as well as reduced tumor burden and improved maintenance of bone structure than the free drug-treated group, alleviating long-term pain and other complications [106].
4.2. Enhancing drug targeting
Conventional pain treatment relies on drugs with continuous release profiles to sustain the pharmacological effect until the payload is exhausted. Most NDDSs aim to prolong the therapeutic effect; however, an alternative approach is to use external stimuli-responsive NDDSs that allow drug release on demand.
Current treatment of perioperative and other acute pain rely on opioids and local anesthetics. By using stimuli such as light, heat, ultrasound, magnetic field, and electric field, the location and timing of drug release can be controlled to maximize efficacy and reduce opioid use to minimize side effects. For example, emerging evidence suggests that chronotherapy of NSAIDs can be effective, and on-demand drug release may improve pain relief by limiting treatment to the active phase of the circadian rhythm [107]. In addition, theragnostic nanoparticles can be designed to accumulate in targets of interest to both detect pain and deliver a drug on demand, for precision pain management [108].
4.2.1. Light-responsive NDDSs
Light used as a non-invasive exogenous trigger can enable multiple drug administrations with precise spatiotemporal control. Light-activated NDDSs include photosensitive molecules with labile bonds that are photochemically cleaved upon ultraviolet (UV), visible, or near-infrared (NIR) light irradiation [109]. Short-wavelength light (UV) is potent enough to disrupt chemical structures but can damage DNA and proteins [109, 110]. NIR-triggered NDDSs have been developed since NIR can achieve deeper tissue penetration than UV or visible light [110]. The mechanisms of NIR-triggered NDDS include photodynamic reactions via photosensitizer-loaded liposomes and the photothermal effect via plasmonic nanoparticles [111].
Rwei et al. developed NIR-light-triggered liposomes loaded with TTX and photosensitizer, allowing peroxidation of liposomal lipids and drug release upon irradiation at 730 nm. This NDDS exhibited adjustable on-demand local anesthesia lasting 14 h following injection in a rat sciatic nerve [112]. The photosensitivity and repeatability of this system was enhanced by an additional tethering of gold nanorods excitable at the same NIR wavelength as the photosensitizer [113].
By combining the photothermal effect of copper sulfide (CuS) nanoparticles upon NIR excitation and the thermoresponsive behavior of amine-terminated copolymer P(MEO2MA-co-OEGMA), de Solorzano et al. achieved repeated on-demand release of bupivacaine after NIR excitation [114]. This copolymer can be functionalized with disulfides for gold nanoparticle binding [115]. These studies showed a successful drug release of ~50%, demonstrating the potential for POEGMA-based light-activated systems for pain management.
NIR-triggered NDDSs have also been applied to patient-controlled transdermal analgesia systems. Microneedles composed of PCL, plasmonic lanthanum hexaboride nanoparticles, and lidocaine can release drug in a pulsatile and programmed manner by varying the duration of irradiation and turning a laser on and off. Lidocaine delivered via implanted microneedle is rapidly absorbed into the blood circulation within 10 min and has a bioavailability of at least 95% relative to subcutaneous injection(Figure 4A-B) [116].
Figure 4. Local on-demand delivery of analgesia using external stimuli.

A) Schematic of NIR-triggered NDDS and implanted polymeric microneedles for on-demand transdermal delivery of lidocaine. The plot shows an in vitro drug release profile after intermittent laser irradiation. B) Histological sections of rat skin with microneedles after NIR exposure for 0 and 3 min [116]. C) Ultrasound (US)-triggered release of liposome-PPIX-red dye. Insonation is indicated by arrows. D) Combined use of liposome-PPIX-TTX and liposome-DMED shows initial nerve block of 35 h, followed by repeated US-triggered analgesia [119]. E) Schematic of magnetic microgels containing iron oxide (magnetite) nanoparticles and ropivacaine. Magnetic nanoparticles in circulation are attracted to the ankle upon magnet application. F) Withdrawal latency trends of untreated left paw and treated right paw [130].
One limitation of NIR light as a trigger is that its tissue penetration is only 1–5 mm; cytotoxicity and burning are risks of deeper penetration [117–119]. Moreover light-responsive NDDSs are designed to be controlled by the intensity and localization of the light. However, there can be variability in the depth of light penetration from patient to patient due to factors including tissue thickness, tissue type, ratio of muscle vs fat, and amount of body hair in the effected region, all of which affect the translatability of such a platform.
4.2.2. Ultrasound-responsive NDDSs
Ultrasound, with its proven clinical utility and tissue penetration, which is an order of magnitude deeper than NIR, is well-suited as a non-invasive external trigger for on-demand local anesthesia. Ultrasound alone or combined with contrast agent microbubbles is widely used clinically to deliver drugs and to diagnose cancers, stroke, osteoarthritis, and chronic pain [120–122]. Sonoporation, cavitation, and hyperthermia are well-known biophysical effects of ultrasound that can be applied to enhance the efficacy of pain relievers [123]. Local anesthetics and hydrophilic molecules such as TTX are impeded by tissue barriers that restrict access to nerve cells. Using ultrasound alone, the peripheral nerve blockade capacity of TTX is enhanced, but the same effect is not seen with the more hydrophobic bupivacaine [124]. While ultrasound is a highly translatable method to control drug targeting due to its safety and deep tissue penetration, it does suffer from poor spatial resolution compared to other methods.
Rwei et al. have shown that the timing, intensity, and duration of nerve blocks can be controlled when using ultrasound-triggered delivery of anesthetic via liposomes by varying ultrasound parameters (Figure 4C-D). Upon insonation, the encapsulated sonosensitizer protoporphyrin IX (PPIX) produces ROS that react with the liposomal membrane, leading to TTX release. The liposome-PPIX-TTX induces an initial nerve block that lasts for over 8 h in rats; subsequent insonation can reproduce nerve blocks twice more for 0.7 and 0.2 h. Co-administration of liposome-DMED and liposome-PPIX-TTX significantly extends the initial nerve block to 35 h. As the duration of anesthesia depends on the extent and intensity of insonation, further development of similar NDDSs could achieve ultrasound-triggered local anesthesia with shorter or longer initial nerve blocks or a greater number of triggerable events. Such control will provide on-demand, personalized pain treatment [119].
Kim et al. have developed theragnostic PVAX nanoparticles that serve as ultrasonographic contrast agents and therapeutic agents by leveraging poly(vanillyl alcohol-co-oxalate) (PVAX) nanoparticles that generate CO2 bubbles through H2O2-triggered hydrolysis. The PVAX nanoparticles rapidly scavenge H2O2 and exert antioxidant and anti-inflammatory effects for musculoskeletal injuries associated with overproduction of H2O2 [125]. This group also loaded curcumin in PVAX (CUR-PVAX) nanoparticles to increase therapeutic capacity. Along with suppression of the proinflammatory cytokines TNFα and IL-1β, significantly enhanced VEGF and PECAM-1 levels led to blood perfusion into ischemic mice tissues [126].
4.2.3. Magnetic field-responsive NDDSs
Targeted delivery of chemotherapeutics with magnetic nanoparticles (MNPs) has been achieved in animals and humans. MNPs improve spatiotemporal localization of therapeutics by controlling hyperthermia (magnetite, maghemite, and ferrite MNPs), mechanical deformation, and magnetic guiding [84, 111]. In hybrid NDDS approaches, alginate-based ferrogels and chitosan-based nanoparticles have been used to induce pore formation and drug release upon magnetic stimulation [84].
Preemptive nerve blocking at the ankle is a common technique to provide analgesia before foot surgeries for reduced central sensitization, postoperative pain, and analgesic consumption [127]. The use of ultrasound-guided techniques has become the gold standard for regional anesthesia or peripheral nerve blocks, providing minimal complications [128]. However, rare but devastating complications such as nerve injury, catheter infection, bleeding, and LAST may arise, calling for finer spatiotemporal control of therapy [129].
In proof-of-concept studies, intravenous injections of MNP complexes with ropivacaine and bupivacaine followed by magnet application at the ankle significantly improved anesthesia [130, 131]. Using magnetic nanogels of PM(EO)2MA, magnetite, and ropivacaine, Mantha et al. showed increased thermal antinociceptive response and ankle ropivacaine concentration when an external magnet was applied for 30 min (Figure 4E-F) [130]. Similar results were obtained from nanogels containing NIPAAM-MAA and bupivacaine [131]. The plasma concentration of complexed ropivacaine was several-fold higher than for direct drug injection [130].
The lack of formal toxicity assessments in these studies means that further research is required before clinical translation. Several reports indicate that MNPs can have significant dose-dependent cytotoxicity as seen in both morphological changes and apoptosis in chicken embryos and human umbilical vein endothelial cells [108, 132–135]. In contrast, dose-dependent pain relief by ultrasmall (6–10 nm) magnetite (Fe3O4) nanoparticles even without drug cargo has been shown to reduce inflammatory cells, proinflammatory markers, and ROS production in rat paw lesions [136]. The ability of MNPs to scavenge free radicals provides a safer and more effective alternative to traditional pain management, and is discussed further in section 6.
Several iron oxide nanoparticles (IONPs) including Feridex, Gastromark, and Feraheme are FDA-approved for contrast enhancement in magnetic resonance imaging (MRI) [137]. With greater bioavailability and visibility with MRI, IONPs offer optimal pain treatment. Superparamagnetic iron oxide nanoparticles (SPIONs) also increase the blood circulation time of quercetin, a well-established anti-inflammatory, antioxidant, and analgesic agent [138, 139].
4.3. Nanoparticles to detect molecular sources of pain
Successful pain treatment relies on locating the source of pain, yet this process is currently imprecise and laborious. A point-of-care system that accurately and efficiently determines the origins of pain by using specific pain biomarkers has the potential to streamline the process, eliminating weeks-long testing and allowing rapid treatment of patients. Researchers are elucidating biomarkers for pain in disease states such as matrix metalloproteinases (MMPs) in neuropathic pain, IL-6 in osteoarthritis, various serum markers in lower back pain, and cytokine IL-6 and P neuropeptide in cerebrospinal fluid in fibromyalgia.
4.3.1. Multiplexed detection of pain markers
Multiplexed point-of-care detection of pain biomarkers can be achieved using nanotechnology, as demonstrated with cancer biomarkers [136, 140]. Quantum dot nanoparticles (Qdots) are particularly applicable, owing to their tunable optical properties [82, 141]. Bioconjugated Qdots with varying diameters, emission spectra, and antibody motifs can determine pain sources from patient samples [82]. This system allows pain-specific biomarkers to be quantified in a point-of-care modality based on the unique fluoroscopic signature of the Qdot, obviating the need for a physician to run multiple tests to check for individual biomarkers, for determination of the specific source of pain. This system is unique in that it tests for a variety of biomarkers and pain sources at once in a rapid manner, rather than by using multiple biomarker tests. Efficient determination of the pain source will facilitate localized treatment and reduce unnecessary systemic treatments that are commonplace today.
4.3.2. Localization of neuropathic pain
Neuropathic pain is a consequence of neural pathology such as nerve lesions that interrupt axonal continuity and cause peripheral sensitization, or diseases such as diabetes mellitus that are associated with nerve damage. Neuropathy is a common form of chronic pain and remains difficult to treat. Diagnosis and treatment of neuropathic pain require locating the lesion or pain source; however, current clinical determination of neuropathic pain relies on questionnaires and electrodiagnostic tests that are unable to locate the exact source of pain [142]. Nanoparticles are uniquely suited to determine sources of lesions as they can be modified to target regions with high levels of biomarkers and can be imaged. The largest obstacle to using nanoparticles for locating lesions is the lack of well-defined biomarkers.
Recently, Husain et al. illustrated the feasibility and efficacy of using nanoparticles to locate lesions responsible for neuropathic pain by targeting MMPs. MMPs are upregulated after nerve injury and have elevated levels for ~20 days as they maintain neuroinflammation. To test the hypothesis that MMP upregulation is a biomarker for peripheral and spinal lesions, the group used magnetic IONPs to target MMP-12 in spinal nerve ligations. MRI scans and histological studies showed significant uptake of the MMP-12-targeted probe at the lesion. Stable and non-toxic in vitro, the IONP probe appears promising as a tool for harvesting biomarkers for clinical determination of neuropathic pain sources. Other proteins which are over-expressed in injured nerves, such as aquaporin-4, interleukin 1 receptor-like 1, and periaxin, can be targeted using a similar approach [142].
4.4. Future use of nanoparticles in pain management
Successful pain treatment requires determining biomarkers to identify the location of pain and to target the source of pain. Using biomarkers to locate the source of pain will be a major breakthrough in the field as it will allow pain to be managed locally instead of through systemic treatments; this will lower dosages, side effects, and cytotoxicity while providing better pain therapies to patients. Another new and attractive area is treating pain by targeting intracellular signaling molecules to mitigate nociception and neuropathy at the source. Nanoparticles play a crucial role in this effort as they can target receptors and allow controlled release of drugs at the receptor location [82]. Nanoparticles are also being used to replace opioids via receptor targeting. Compounds such as MAPK inhibitors are being developed to treat a wide variety of chronic pain, but their delivery cannot be systemic. Nanoparticles represent a major step towards treating pain in a site-specific manner with minimal systemic uptake, which is vital to long-term chronic pain management without negative systemic side effects and addiction [97].
5. Gene therapy for pain
Gene therapy allows specific targeting of the pain source by tailoring three parameters, vector, transgene, and promoter, to a known pathophysiology. This level of control makes gene therapy powerful by enabling both specific targeting of a disease or gene causing the pain, and localized delivery to the source of the pain. Co-treatment with other approved drugs can enhance the palliative effect of gene therapy. For treatment of chronic pain, transgenes can reduce nociception by inducing overexpression of analgesic genes and anti-inflammatory cytokines or by inhibiting a pain-producing gene (Figure 5).
Figure 5. Methods of viral gene therapy for pain treatment.

Schematic showing various methods of gene therapy for pain treatment. 1) Repression of genes (Nav1.3, Nav1.7) or inflammatory cytokines (IL-1, TNF-α) to reduce pain signaling and inflammation in affected areas. 2) Expression of preproenkephalin (PENK) and enkephalin (ENK), which act as endogenous opioids by binding to opioid receptors and mediating pain. 3) Overexpression of anti-inflammatory cytokines (IL-2, IL-10, GAD65, BDNF) to reduce inflammation, immune response, and inflammatory pain. Adapted from Moreno et al.[195].
Recently, extensive research efforts have developed safe viral vectors that transfer therapeutic genetic materials. Herpes simplex virus type 1 (HSV-1), adeno-associated viruses (AAVs), adenoviruses (AVs), and lentiviruses (LVs) have become the four main viral vectors for pain gene therapy as they can target non-dividing cells such as neurons(Figure 6). Retroviruses cannot transfect non-dividing cells and thus have not been useful in targeting chronic pain. HSV-1 is an ideal viral carrier for pain treatment given its high packaging capacity and innate neurotropism, allowing delivery to be as simple as a dermal application or subdermal injection. AAVs are commonly used as carriers to produce opioids. AAVs are used to deliver genes via intrathecal injection, targeting, and triggering neuronal cells to secrete opiate-like proteins in low and sustained amounts. This novel treatment can potentially reduce pain without exposing patients to the risk of opiate abuse [143].
Figure 6. Comparison of common viral vector carriers used in gene therapy for pain:

herpes simplex virus 1 (HSV-1), adeno-associated virus (AAV), adenovirus (AV), and lentivirus (LV).
5.1. Vectors for delivery of gene therapy for pain
5.1.1. Herpes simplex virus type 1
HSV-1 is one of the most commonly used viral vectors for pain management in large part due to its high packaging capacity and neurotropism. HSV-1 has become the vector of choice in a number of disease models for pain management after its proven efficacy in the NP2 clinical trial escribed in section 5.2. A common use of HSV is to express ENK and PENK, naturally occurring endogenous opioids that, upon transfection, can improve the body’s ability to release endogenous opioids.
The anti-nociceptive, anti-neuropathic, and anti-inflammatory effects of HSV vectors expressing ENK and PENK have been demonstrated in a number of in vivo models, including pancreatic inflammation [144], rheumatoid arthritis using the adjuvant-induced polyarthritis model, [145], facial pain from the infraorbital nerve constriction [146], arthritis induced by injection of complete–Freund’s adjuvant [147], nerve injury [147], and bone cancer pain [148]. Induction of glycine receptor (GlyR) expression using HSV can function as an endogenous opioid that is not ordinarily present in sensory neurons, maximizing therapeutic selectivity and minimizing immunogenicity [149].
HSV vectors have also been used to express IL-10 in a model of type I diabetes to alleviate pain by reducing the Toll-like receptor 4 (TLR4) expression, which reduces macrophage activation and inhibits painful neuropathy [150]. Another application of HSV vectors is suppressing neuropathic pain induced by HIV by transfecting the gad1 gene that expresses GAD67, which synthesizes GABA for neuronal activity [151, 152]. The expression of TRPV1 using HSV vectors has been found effective in treating interstitial cystitis/bladder pain syndrome [153].
5.1.2. Adenoviruses
Adenoviruses can be used for gene transfer to both dividing and non-dividing cells and are commonly used in gene therapy due to their low host specificity and high immunogenicity, as most people have been exposed to AV serotypes 2 and 5. AVs have moderate packaging ability and short-term transgene expression, making them ideal for acute pain treatment. AVs have been used as a vector for GAD65 and IL-10. AVs expressing GAD65 and targeting glial cells were shown to be effective in a facial pain model, where GAD expression reduced pain by acting on GABA receptors on neurons [154]. AVs encoding IL-10 blocked both nerve pain and allodynia in three models of neuropathic pain nerve injury [155]. Researchers have used AVs to express IL-2 to mediate nociceptive pain. IL-2 has analgesic effects in both the PNS and CNS, mediated by opioid receptor binding. AVs expressing IL-2 delivered to nerve injury (CCI) models via intrathecal injection have a nearly week-long effect [156]. GLT-1, a glial glutamate transporter, has been expressed by AVs and delivered to the spinal cord to treat inflammatory and neuropathic pain. GLT-1 attenuates the induction of inflammatory and neuropathic pain but has little effect on mediating pre-existing pain, making it an excellent candidate to administer in clinical procedures that induce pain, such as chemotherapy [157].
5.1.3. Adeno-associated viruses
Adeno-associated viruses are similar to AVs but have deficiencies in their replication and pathogenicity, making them safer than AVs. AAVs have been used as a vector for pain management to knock down Nav1.3 in a diabetic model to alleviate tactile allodynia, and in a nerve injury neuropathic pain model. Nav1.3 is a voltage-gated sodium channel that is upregulated in both the PNS and CNS after nerve injury and in dorsal root ganglion neurons in diabetes. The increase in Nav1.3 contributes to chronic pain. Knocking down Nav1.3 via siRNA to reduce Nav1.3 levels via AAV is effective in alleviating diabetic allodynia (neuropathic pain) and nerve injury-induced neuropathic pain [158, 159].
Overexpression of GAD65 after peripheral nerve injury is effective in alleviating neuropathic pain by increasing GABA levels. However, the increased levels of GAD65 remain for less than a week from the time of injury. Recombinant AAVs expressing GAD65 have attenuated neuropathic pain for longer periods via administration to the sciatic nerve and dorsal root ganglion [160, 161].
The use of AAVs to express the analgesic prepro-β-endorphin and IL-10 through lumbar puncture reduced neuropathic pain in a L5 spinal ligation (SNL) chronic neuropathic pain model [143], as did overexpression of brain-derived neurotrophic factor (BDNF) via injection into the dorsal root ganglion after chronic constriction injury of the sciatic nerve (CCI model of neuropathic pain) [162].
5.1.4. Lentiviruses
Lentiviruses naturally integrate with non-diving cells and provide stable long-term expression of transgenes with low immunogenicity, making them uniquely suited for pain therapy. Knocking down the transcription factor NF-κB using siRNA has been a major focus of research, as NF-κB controls multiple genes that encode inflammatory and pain responses. Selectively knocking down NF-κB super-repressor IκBα results in inhibition of the NF-κB pathway in nerve injury models and attenuation of neuropathic pain [163]. Using lentiviral vectors to deliver short hairpin DNA targeting NF-κB65 to silence NF-κB inhibits proinflammatory TNF-α, IL-1β, and IL-6 and moderates neuropathic pain and allodynia for over four weeks [164].
Lentiviral vectors have also been used to knock down PKC to treat nerve injury-based neuropathic pain and reverse morphine tolerance in patients with chronic pain. PKCγ is an important second messenger as its activation is involved in chronic neuropathic pain. Lentiviral delivery of RNAi can silence the PKCγ gene and reduce pain and allodynia in rat nerve-injury models for over six weeks [165]. PKCγ is also thought to play a role in morphine tolerance. To combat increased tolerance, lentiviral vectors of PKCγ short hairpin RNA are delivered to morphine-tolerant rats via intrathecal injection. After injection, downregulation of expression of PKCγ was observed along with a reversal in morphine tolerance, which is useful for patients already taking opioids [166].
5.1.5. Non-viral vectors
While most gene therapy for pain is accomplished using viral vectors, many non-viral vectors are also to treat pain. Non-viral vectors are less immunogenic, more stable, and safer than their viral counterparts, but are much less efficient [167]. Non-viral vectors include cationic lipids and polymers, plasmids, naked DNA, and lipid-polymers. Non-viral vectors have been extensively used in gene therapy-based treatment of peripheral and coronary artery disease using VEGF165 and VEGF-2; however, clinical trials using plasmid DNA (phVEGF165 and phVEGF-2) have shown varying degrees of success [168–174].
IL-2 and IL-10 have become popular targets for non-viral gene therapy of neuropathic pain. IL-2 is unsuitable as an analgesic as it is short-lived in vivo and requires constant administration. However, IL-2 gene therapy may be suitable for short-term neuropathic pain therapy. Humanized IL-2-expressing plasmids administered via a spinal catheter in CCI rat models have shown dose-dependent pain reduction [175]. Long-term control of neuropathic pain has also been established using IL-10 to control glial inflammation, mediating neuropathic pain [176–179].
One form of non-viral treatment requires an intrathecal ‘priming’ injection of DNA to induce accumulation of immune cells and short-term pain reversal before a second intrathecal injection; one DNA used was a naked plasmid-encoded IL-10F129S transgene for long-term pain reduction. The injections achieved pain relief for over three months in peripheral nerve injuries. The priming shot, given from 5 h to 3 d before the second injection, potentiated long-term pain relief in a time- and dose-dependent manner [180].
Intrathecal IL-10 transgene expression induces an anti-inflammatory environment in the dorsal root ganglion and in the lumbar spinal cord. Co-injection of naked IL-10-encoded plasmids with D-mannose, an immune cell adjuvant, allows stable long-term neuropathic pain relief following a single intrathecal injection in CCI and IL-10 deficient rat models [167]. D-mannose is a mannose receptor-specific ligand that increases mannose receptor expression, which is associated with anti-inflammatory macrophage polarization, anti-inflammatory signaling, and transient pain relief. Treatment with D-mannose optimizes IL-10 transgene expression, and co-injection of mannose with a 25-fold lower transgene dose produces prolonged pain suppression in CCI rat models [178].
The μ-opioid receptor OPRM1 has been a target of non-viral gene delivery to attenuate cancer-associated pain. A non-viral hybrid vector, modified HIV-1 Tat, was used to transfect HSC-3 (human tongue squamous cell carcinoma) cells with OPRM1. These cells were then inoculated into athymic SCC (oral cancer) mouse models and were found to have an analgesic effect. This non-viral approach is superior to viral approaches as the vector has a much smaller size, allowing greater transfection efficiency and lower sufficient dosages [181].
Non-viral gene transfer has also been used to prevent drug-induced neuropathy. Cisplatin is a powerful chemotherapeutic but causes dose-dependent neuropathy with slow and often partial recovery. Neurotrophin-3 (NT-3) is a promising agent for preventing and treating cisplatin-induced neuropathy as it readily reaches the dorsal root ganglion, the main target of cisplatin toxicity. However, the administration of NT-3 is complicated as its plasma half-life is ~1 min. Non-viral gene transfer of NT-3 using a recombinant plasmid followed by electroporation can protect against cisplatin-induced neuropathy. NT-3-encoded plasmids were intramuscularly injected followed by four square-wave pulses of 100 V and 20 ms duration delivered at a frequency of 1 Hz in a cisplatin-treated mouse model. This treatment caused only slight muscle toxicity and no general side effects while reducing neuropathic pain, making it a robust platform to treat chemo-induced neuropathy and peripheral neuropathies [182].
5.1.6. Future use of gene therapy for treating chronic pain
Future opportunities for applications of gene editing to pain are expansive. Current gene therapy can be enhanced, for example, by designing a specific transgene to allow better targeting of cells of interest and longer-lasting expression of the genetic modification. With improved knowledge of patient profiles and how they correspond to transgene selection, treatments can be made more effective. AAV-mediated transfer of Kv1.2 sense RNA for reduction of dorsal root ganglion neuronal excitability [183], and viral vector-mediated overexpression of anti-inflammatory cytokines to counter over-inflammation are promising methods to treat pain using gene therapy. Other long-term goals for gene therapy include specific delivery to the brain to target pain control centers, which is currently difficult due to the complexity of the neural circuits of the brain in comparison to the spinal cord.
5.2. Clinical trials
Gene therapy was proven effective for treating pain in humans in 2011, in the first clinical trial of gene transfer as a treatment for pain. In the phase 1 trial, cancer patients were treated with NP2, a replication-defective HSV-based vector expressing human preproenkephalin (PENK). PENK induces the release of enkephalin peptides which activate opioid receptors, inhibiting the transmission of pain signals to neurons. NP2 was transdermally injected into the pain location as perceived by the patients. NP2 was well tolerated and caused no adverse effects, and patients given moderate to high doses of NP2 saw pain relief over the course of treatment [184, 185]. A phase 2 clinical trial of NP2 was conducted with 33 participants with intractable pain due to malignant cancer in 2013 [185].
A phase 1 trial to treat osteopathic pain using XT-150 was conducted by Xalud Pharmaceuticals. Instead of blocking pain signaling, XT-150 treats the inflammation responsible for chronic pain through the expression of a variant of IL-10, a naturally occurring anti-inflammatory protein that suppresses TNF-α, IL-1β, and IL-6, down-regulates cytokine receptors, and upregulates cytokine antagonists. Prior to clinical trials, upregulation of IL-10 to mediate pain was conducted in CCI rat models of neuropathic pain with positive results [176]. XT-150 is similar to XT-101, a predecessor that was shown to successfully treat pain in models of multiple sclerosis (MS) and enhanced pain states in rats [177, 179, 180]. In this trial, XT-150 was administered via injection into the knee synovial capsule. The study followed patients for six months, monitoring their pain levels and blood levels of the IL-10 variant. While phase 1 results are yet to be published, phase 2 trials of XT-150 for elderly patients with musculoskeletal pain are currently underway [186, 187].
An ongoing FDA fast-tracked Phase 1/2 trial to treat refractory angina using XC001 is being conducted by XyloCor Therapeutics. Refractory angina is chronic chest pain in coronary artery disease that cannot be treated otherwise. Angina in these patients is severe and debilitating, affecting daily activities and quality of life. XC001, also known as AdVEGF-All6A+, is a novel gene therapy consisting of a replication-deficient adenovirus vector that expresses a hybrid variant of VEGF. XC001 is being used to treat angina by promoting angiogenesis (revascularization), which would increase myocardial blood flow. Angiogenesis can relieve myocardial ischemia and improve ventricular performance [188, 189].
There is also an ongoing phase 1 trial to treat refractory angina using Ad5FGF-4 (AFFIRM). Ad5FGF-4 is a replication-deficient serotype 5 adenovirus expressing the gene for human fibroblast growth factor-4 (FGF-4) driven by a cytomegalovirus promoter [190]. Ad5FGF-4 was previously tested in clinical trials AGENT-3 and AGENT-4 in 2008 to treat chronic angina. After preclinical successes, the trials were cut short after it became clear that 12 weeks would not be long enough to reach significance. The AGENT trials enrolled over 500 participants and found that while not effective in men, Ad5FGF-4 was effective in women. This was the first clinical report to show a gender difference in the treatment of angina [191, 192]. The purpose of this ongoing study is to determine whether Ad5FGF-4 is effective in reducing debilitation from angina, including increasing the duration of exercise, reducing the frequency of angina attacks, and improving overall quality of life [193].
5.3. CRISPR-Cas for pain
CRISPR-Cas offers a new mechanism to combat chronic pain. CRISPR-Cas is a gene-editing system that allows genes to be added, deleted, or altered at particular locations in the genome. CRISPR-Cas9 is one form of CRISPR-Cas, and is adapted from a naturally occurring genome editing system in bacteria. CRISPR-Cas9 is faster, cheaper, more accurate, and more efficient than other gene-editing tools. One obstacle when using CRISPR is that the target must be specific to the cells being modified—this is particularly important in the context of pain. The goal of CRISPR in the context of pain therapy is to edit cells to make them more resistant to pain. Off-target editing or over-editing could lead to cells that are completely resistant to pain, which would have serious negative repercussions. Since CRISPR permanently edits cells, CRISPR-based therapies must be extensively tested to ensure that they are not too potent and that they can be delivered in a strictly targeted manner. Pain is biologically important to alert and protect the body from harm; permanently removing pain sensation via CRISPR would be detrimental, while limiting the amount of pain in specific cells could bring relief to those suffering from debilitating chronic pain.
5.3.1. Repressing Nav1.7 via SCN9A
One way to make CRISPR safe and controlled for pain management is to use inactivated or ‘dead’ Cas9 (dCas9). dCas9 does not cleave DNA but maintains other functions—binding to guide RNA and the DNA strand being targeted—and can modify and employ transcriptional activators to modulate gene expression. This dCas9 mechanism is being studied in the context of the rare SCN9A gene mutation. SCN9A is responsible for production of Nav1.7, which plays a role in transmitting pain from nerves to the brain. Loss-of-function mutations in Nav1.7 cause congenital insensitivity to pain (CIP), a phenomenon that can lead to lack of pain perception to noxious stimuli [194].
Some mutations of the SCN9A gene cause people to feel more or less pain, or, in the extreme case of CIP where SCN9A has been disabled completely, no pain at all. While this discovery has led to advances in pain treatment research, it also shows why researchers need to be cognizant of the level of pain attenuation. Pain is essential for survival, as can be seen from those who suffer from CIP: individuals with CIP are often mistakenly injured as evidenced by limping or missing pieces of their tongue that they unknowingly bit off because their bodies lack a mechanism to indicate damage. The goal of using CRISPR should not be to eliminate pain but to attenuate it, such that people do not suffer from debilitating chronic pain, while retaining the ability to feel pain [16, 40, 41]. The Mali group has been studying this mutation and how to pair it with CRISPR to mediate pain in people with chronic pain conditions. CRISPR is advantageous for blocking NaV1.7 as small molecules and antibodies targeting Nav1.7 have overwhelming off-target effects in the Nav family. CRISPR was used to block Nav1.7 in mice, in the first use of CRISPR for pain management. AAV was the vector for CRISPR-dCas9 (inactivated Cas9) and zinc finger protein (ZFP), which was injected into the spine to infiltrate neuron cells in inflammatory, neuropathic, and BzATP-induced pain models. CRISPR and ZFP reduced neuropathic (lesion and chemotherapy-induced) and nociceptive pain. Knockdown of Nav1.7 did not affect inflammation. These CRISPR-based systems are a successful proof of concept, but must be further tested to see how long Nav1.7 stays knocked out; researchers expect the Nav1.7 knockout period to be six months to one year [195]. dCAS9 can activate or repress a gene of interest without creating permanent changes. This behavior is ideal because gene expression can be modulated to suit the patient’s needs and can be reversed. This study serves as a platform for gene therapy that would last for months at a time, ideal for shorter-lived chronic pain such as that of chemotherapy patients.
5.3.2. Blocking proinflammatory signaling
The use of CRISPR to treat pain has been studied by blocking proinflammatory signaling in vitro. CRISPR can prevent tissue damage and chronic pain by modulating gene expression to reduce proinflammatory signaling. Inhibition of TNF-α and IL-1, which upregulate NF-κB, can reduce inflammation. Researchers built lentiviral vectors encoding TNF-α and IL-1 receptors, TNFR1 and IL1R1, and targeted CRISPR-based transcriptional repressors (Figure 5). These vectors inhibit NF-κB activation while promoting cell survival, demonstrating that CRISPR-based epigenome editing can be used to modulate inflammation [196].
5.3.3. Alleviating osteoarthritic pain
Osteoarthritis is marked by chronic pain and inflammation in joints, affects over 10% of adults, and has no cure. CRISPR-Cas9 provides a new platform for osteoarthritis therapy. Osteoarthritis is marked by upregulation of NGF, IL-1β, and MMP13. AAVs expressing CRISPR-Cas9 have been used to target NGF, IL-1β, and/or MMP13 via injection into arthritic sites in a surgical mouse model. Shutting off NGF resulted in reduction of pain, but joint damage increased. Shutting off NGF, IL-1β, and MMP13 together reduced pain and inhibited disease progression [197], suggesting that CRISPR-based gene editing can be useful in treating osteoarthritis.
5.3.4. Future use of CRISPR-Cas9 for treating chronic pain
From these studies, the potential for CRISPR-based gene editing and replacement in pain therapy is clear. CRISPR can treat chronic pain by editing the genes that are responsible for pain in a specific manner, reducing the use of pain medication, and can be done in a way that relieves pain but preserves some healthy sensation of pain. CRISPR can also be used to modulate gene expression, for example to upregulate expression of anti-inflammatory cytokines, an exciting new direction for chronic pain management.
A new CRISPR approach uses nanoparticles rather than viral vectors to deliver CRISPR-Cas machinery. These nanoparticle delivery systems, such as CRISPR-Gold, have been administered successfully and with high specificity [198]. Nanoparticle-mediated delivery minimizes the immunogenicity, risk of genomic damage [198], barriers to large-scale production, and limited insertion size [199] associated with viral delivery. Nanoparticle systems can be developed to tag specific cell types and overcome physiological barriers to aid in localized delivery. Examples of specialized nanoparticle carriers are CRISPR-Gold, which can target neurons and muscle cells [198]; selective organ targeting lipid nanoparticles, which selectively target the lung, spleen, and liver [200]; biomimetic mineralized ribonucleoprotein nanoparticles [201]; and magnetic nanoparticles; some of these systems have unique properties such as the ability to pass through cellular barriers or magnetic field-responsiveness for magnetic field-triggered genome editing [202].
These advances in using CRISPR will allow the development of platforms for monitoring patients’ chronic pain and inflammation and modulating their gene expression to healthy levels as needed.
6. Scavengers
Acute pain can cause and reinforce the accumulation of molecules that cause unwanted immune activation and central sensitization, which in turn can increase pain and cause chronic pain. Scavengers of such molecules can improve therapeutic outcomes without off-target effects and loss of biological activity of immune agents. Scavengers are therapeutic immunomodulatory nanomaterials that are uniquely designed to proactively remove overproduced molecules to reduce chronic pain. Scavengers are a promising agent for treating chronic pain and inflammatory pain due to their structure and mechanism of action. Two of the most promising types of scavengers are nucleic acid-binding scavengers (NABS) and ROS scavengers.
NABS are highly charged polymers and nanoparticles that recognize danger- and pathogen-associated molecular patterns (DAMPs and PAMPs) that stimulate TLRs and activate an innate immune response. DAMPs and PAMPs effectively regulate immune response in healthy cells, but in chronic disease, they overstimulate TLRs leading to chronic pain and inflammation. NABS can reduce TLR overactivation, relieving inflammation and pain.
ROS scavengers remove excess ROS that are yet to be metabolized by cellular enzymes. Increased levels of ROS cause central sensitization and promote chronic pain. Scavenging excess ROS reverses central sensitization and reduces pain by increasing the threshold for pain.
6.1. Nucleic acid-binding scavengers
In chronic pain, TLRs are over-activated and cause undesirable chronic immune responses. Nucleic acid-binding scavengers (NABS) that remove the DAMPs and PAMPs that cause chronic inflammation and pain reduce both inflammation and pain. These scavengers function proactively (Figure 7A) [203]. Instead of treating the symptoms of pain, scavengers eliminate the cause of pain by removing the agonists that cause TLR overexpression. Scavengers are unique in that the immune response is reduced in a dose-dependent manner, which can eliminate overactivation without eliminating baseline healthy activation.
Figure 7. Role of scavengers in pain mediation.

A) The role of scavengers in mediating PAMP-, DAMP-, bacteria-, and ROS-associated pain pathways. B) Nucleic acid binding PLGA-b-PDMA nanoparticles in a rheumatoid arthritis model [203]. C) Water soluble Gd@C82-(ethylenediamine)8 nanoparticles act as efficient and biocompatible ROS scavengers as can be seen through decreased electron paramagnetic resonance (EPR) signal. These nanoparticles also exhibit a cytoprotective effect [219].
DAMPs and PAMPs are molecular signaling molecules that activate an immune response. DAMPs are released by damaged cells and injured tissue into the blood and tissue fluid; PAMPs result from infection, bacteria, and viruses. Both are recognized by pattern recognition receptors (PRRs) and trigger intracellular signaling cascades, leading to upregulation of inflammatory cytokines and type 1 interferons (Figure 7A).TLRs are a type of PRR that recognize specific molecular patterns associated with pathogens and damaged tissue, which allow them to act as a ‘guard’ of the innate immune system. When TLRs recognize a PAMP or DAMP, they immediately activate an innate immune response, which leads to expression of inflammatory cytokines, immune-stimulatory cytokines, and chemokines that destroy invading pathogens and promote tissue regeneration [204]. However, inappropriate activation of TLRs contributes to the development of diseases such as autoimmune disease [205], inflammatory disease [203, 206, 207], sepsis [208], arthritis [203], and cancer [209] (Figure 7B), making TLRs an attractive therapeutic target for disease-associated pain, tissue damage-associated neuropathic pain [210], and inflammatory pain [211].
NABSs are highly cationic polymers and nanoparticles that act as molecular scavengers and counteract the activity of nucleic acid aptamers, as well as inhibiting RNA- and DNA-mediated activation of TLRs and inflammation. Their positive charge allows them to bind nucleic acids and other free negatively-charged molecules, including DAMPs and PAMPs. When NABSs capture nucleic acids, the ability of those DAMPs and PAMPs to activate TLRs is neutralized. NABSs block TLR activation by nucleic acids in a controlled and localized manner without interfering with the normal course of an immune response or compromising TLR responses to non-nucleic acid, pathogenic stimulators. NABSs cannot neutralize the ability of non-nucleic acid DAMPS to induce cell death.
6.2. ROS-scavenging molecules
Reactive oxygen species are byproducts of cellular functions such as oxidative phosphorylation, an act as secondary messengers in cell-to-cell signaling and pathogen defense. In healthy cells, ROS levels are maintained by specialized enzymes, but in pathological conditions, excess ROS cause inflammation, cell and tissue damage, and pain [212, 213]. Excess ROS has long been known to have a role in persistent inflammatory and neuropathic pain [214]. Elevated ROS phosphorylate NMDA receptors in the spinal cord which leads to central sensitization, a persistent state of high reactivity where the threshold of pain is reduced, creating a state of chronic pain. Reducing ROS in neuropathic pain models has dramatic analgesic effects by rapidly and effectively reversing central sensitization [214]. One way to reduce ROS levels to treat chronic pain is to use ROS-scavenging molecules.
ROS scavengers include alpha-phenyl N-tertiary-butyl nitrone (PBN), 5,5-dimethyl-pyrroline N-oxide (DMPO), 2,2,6,6-tetramethylpiperidine-N-oxyl (TEMPO), 4-hydroxy-2,2,6,6-tetramethylpiperidine-1-oxyl (TEMPOL), and vitamin E [213–216]. ‘Spin trap’ reagents (e.g. PBN and DMPO) are the most potent ROS scavengers as they covalently react with radicals to create stable adducts. However, these ROS scavengers are nonspecific, lack self-propulsion, and can be cytotoxic, limiting clinical translation [217]. The next generation of ROS scavengers is addressing these issues for greater efficiency and biocompatibility.
Nanoparticles can also be used to scavenge ROS. Like other nanoscavengers, mesoporous silica nanoparticles (MSN) have low motility and difficulty reaching some intracellular locations. Hollow MSN loaded with hemin has harnessed the chemical free energy of catalytic reactions and achieved 3.5-fold higher average speed than solid nanoparticles [217]. The motility can be controlled by modulating the thickness of the nanoparticle shell and presents as a new model to scavenge ROS in a more controlled manner [217].
MSN have also been decorated with ultrasmall ceria nanocrystals to create a ROS-scavenging nanocomposite that scavenges ROS in a localized manner and facilitates wound repair [136]. MSN-ceria nanocomposites can be useful in inflammatory pain especially in cases of chronic inflammation that causes tissue damage. The MSN-ceria scavenge ROS while facilitating tissue repair, reducing the likelihood of future neuropathic pain.
Another way to improve ROS scavenging is to render the nanoparticulate surface more biocompatible. Endohedral metallofullerenol nanoparticles are ROS-scavengers that inhibit lipid peroxidation, protect cells from further oxidative stress, and can potentially reverse central sensitization long-term [218]. These nanoparticles could be useful as their ability to protect cells from further stress would be beneficial when reversing central sensitization, as they could reduce oxidative stress for extended periods. Metallofullerene Gd@C82 nanoparticles have been modified with ethylenediamine (EDA) to create a positive zeta potential and a water-soluble surface (Figure 7C) [218]. Even at low concentrations, Gd@C82-(EDA)8 nanoparticles exhibit excellent hydroxyl radical scavenging and cytoprotective effects suitable for antioxidants. Moreover, the naked amino groups on the surface can be sites of further surface functionalization, making Gd@C82-(EDA)8 attractive for a host of applications, including biomaterials and dietary supplements [219].
pH-responsive scavengers have been developed for targeted ROS scavenging. pH-responsive nitroxide radical-containing nanoparticles were developed to disintegrate in acidic lesions and release nitroxide radicals locally, neutralizing ROS [215]. This scavenging approach is attractive for localized injury as it can remove excess ROS in a lesion, relieving neuropathic pain without affecting the rest of the body.
NABSs may also be useful in mediating ROS-induced pain as NABSs can remove DAMPs and PAMPs before ROS generation, thereby proactively preventing ROS-related pain sensitization (Figure 7A).
7. Conclusions
Nanomedicine has become an important field in therapeutic research, but nanotherapeutics have only begun to be explored in the context of pain management, in part due to the complex nature of pain. Chronic pain is associated with many diseases and with postoperative care, is difficult to treat, and costs the U.S. healthcare system over $635 billion annually. Current therapeutics for chronic pain do not provide adequate relief and many debilitating side effects. Advances in nanomaterials and nanoparticles are improving the targeting and detection of the molecular sources of pain to reduce dosage and improve long-term efficacy and safety. Gene therapy is also enabling more effective and longer-term treatment of chronic pain, with both viral and non-viral vectors for gene therapy showing effectiveness in clinical trials. CRISPR allows modulating the gene expression of newly identified targets to mediate pain without eliminating sensitization. Scavengers represent a proactive approach to treating pain by removing molecules that cause pain and pain sensitization (such as free nucleic acids and reactive oxygen species) rather than merely treating the symptoms of pain. Applying nanotechnology to new molecular pain targets and to detecting the molecular sources of pain is a frontier in nanomedicine and pain management.
Highlights.
Nanoparticle based drug delivery devices allow for targeted pain treatment with enhanced therapeutic efficacy
Gene therapy allows for long term pain treatment using both viral and nonviral vectors
Pro-active scavenging of overproduced molecules using immunomodulatory nanomaterials can reduce pain
Acknowledgments:
Support by National Institutes of Health (NS102722, DE026806, DK118971, DE029951, NWB, BLS) and Department of Defense (W81XWH1810431, NWB, BLS) is acknowledged. Images were created with the assistance of Biorender.com.
Biographies

Divya Bhansali, MS
Divya Bhansali obtained her Bachelors of Science in Biomedical Engineering from the University of Miami. She is currently a Ph.D. student in the Department of Biomedical Engineering at Columbia University. Her research interests include investigating nanoparticles and their application in therapeutic platforms for cancer treatment, pain management, and inflammation modulation. She is a recipient the NSF Graduate Research Fellowship and Columbia University’s Blavatnik Presidential Fellowship.

Shavonne L. Teng, MS
Shavonne L. Teng obtained her Bachelors of Science in Neuroscience and Behavioral Biology from Emory University. She is currently a Ph.D. graduate student in the Department of Physiology and Cellular Biophysics at Columbia University Medical Center. She is mentored by Dr. Kam Leong at Columbia and Dr. Nigel Bunnett at NYU. Her research interests include utilizing nanoparticles to probe the mechanisms of endosomal signaling of GPCRs and to understand the role of GPCR signaling in chronic pain.

Caleb S. Lee, MS
Caleb Sangchul Lee received his B.S. in biological engineering and materials science and engineering at University of California, Berkeley. He is a Biomedical Engineering Ph.D. student at Columbia University jointly advised by Dr. Kam Leong and Dr. Raju Tomer to investigate brain disorders and nanomedicine.

Brian L. Schmidt, DDS, MD, PhD
Dr. Schmidt is a surgeon and scientist. He earned his advanced degrees at the University of California, San Francisco. He is a Professor in the Departments of Oral and Maxillofacial Surgery and Neuroscience and Physiology. He directs Bluestone Center for Clinical Research at New York University as well as the NYU Oral Cancer Center. Dr. Schmidt’s clinical efforts are dedicated to comprehensive surgical management of oral cancer; his scientific work explores the neurobiology of oral cancer pain. His clinical and basic science research have been funded by the NIH since 2005.

Nigel Bunnett, PhD
Nigel Bunnett obtained a Ph.D. from the University of Cambridge. Nigel serves as Professor and Chair of the Department of Molecular Pathobiology, Professor of Neuroscience and Physiology, and Member of Neuroscience Institute at New York University College. Nigel’s laboratory investigates the mechanisms by which G protein-coupled receptors signal chronic pain and neurogenic inflammation. His laboratory has developed novel therapies that target receptors in endosomes and provide more effective and selective relief from chronic pain and inflammation. Nigel has ~350 peer-reviewed publications, which have received >36,000 citations. Nigel is Editor-in-Chief of the American Journal of Physiology, Gastrointestinal and Liver Physiology.

Kam W. Leong, PhD
Kam W. Leong is the Samuel Y. Sheng Professor of Biomedical Engineering at Columbia University. He received his PhD in Chemical Engineering from the University of Pennsylvania. He has published ~450 peer-reviewed and holds more than 60 issued patents. The lab focuses on three major research directions: 1) Design of biomaterials for inflammation modulation; 2) Construction of patient-specific iPSC-derived brain organoids for disease modeling and drug discovery; 3) Development of nonviral gene editing systems in vivo. He is the Editor-in-Chief of Biomaterials, a member of the USA National Academy of Inventors, National Academy of Engineering, and National Academy of Medicine.
Footnotes
Competing interests. Nigel W. Bunnett is a founding scientist of Endosome Therapeutics Inc.
CRediT author statement
Divya Bhansali: Conceptualization; Writing - Original Draft; Writing - Review & Editing; Visualization
Shavonne Teng: Writing - Original Draft; Writing - Review & Editing
Caleb Lee: Writing - Original Draft; Writing - Review & Editing; Visualization
Brian Schmidt: Writing - Review & Editing
Nigel Bunnett: Writing - Review & Editing
Kam Leong: Conceptualization; Writing - Review & Editing
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Schappert SM, Burt CW, Vital Health Stat 13, (2006) 1–66. [PubMed] [Google Scholar]
- [2].Enright A, Goucke R, Anesth Analg, 123 (2016) 529–530. [DOI] [PubMed] [Google Scholar]
- [3].Whitten CE, Donovan M, Cristobal K, Perm J, 9 (2005) 9–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Tsang A, Von Korff M, Lee S, Alonso J, Karam E, Angermeyer MC, Borges GL, Bromet EJ, Demytteneare K, de Girolamo G, de Graaf R, Gureje O, Lepine JP, Haro JM, Levinson D, Oakley Browne MA, Posada-Villa J, Seedat S, Watanabe M, J Pain, 9 (2008) 883–891. [DOI] [PubMed] [Google Scholar]
- [5].Dahlhamer J, Lucas J, Zelaya C, Nahin R, Mackey S, DeBar L, Kerns R, Von Korff M, Porter L, Helmick C, MMWR Morb Mortal Wkly Rep, 67 (2018) 1001–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Davis JA, Robinson RL, Le TK, Xie J, J Pain Res, 4 (2011) 331–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Gaskin DJ, Richard P, J Pain, 13 (2012) 715–724. [DOI] [PubMed] [Google Scholar]
- [8].Smith TJ, Hillner BE, JAMA Network Open, 2 (2019) e191532-e191532. [DOI] [PubMed] [Google Scholar]
- [9].Suppl Pain, 3 (1986) S1–226. [PubMed] [Google Scholar]
- [10].Geppetti P, Veldhuis NA, Lieu T, Bunnett NW, Neuron, 88 (2015) 635–649. [DOI] [PubMed] [Google Scholar]
- [11].Gosselin RD, Suter MR, Ji RR, Decosterd I, Neuroscientist, 16 (2010) 519–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Premkumar LS, Sikand P, Curr Neuropharmacol, 6 (2008) 151–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Habib AM, Wood JN, Cox JJ, Handb Exp Pharmacol, 227 (2015) 39–56. [DOI] [PubMed] [Google Scholar]
- [14].Zhou YQ, Liu Z, Liu ZH, Chen SP, Li M, Shahveranov A, Ye DW, Tian YK, J Neuroinflammation, 13 (2016) 141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Cervero F, Dig Dis, 27 Suppl 1 (2009) 3–10. [DOI] [PubMed] [Google Scholar]
- [16].Cohen SP, Mao J, BMJ : British Medical Journal, 348 (2014) f7656. [DOI] [PubMed] [Google Scholar]
- [17].Trouvin AP, Perrot S, Best Pract Res Clin Rheumatol, 33 (2019) 101415. [DOI] [PubMed] [Google Scholar]
- [18].Ghanem CI, Pérez MJ, Manautou JE, Mottino AD, Pharmacological Research, 109 (2016) 119–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Karandikar YS, Belsare P, Panditrao A, Indian J Pharmacol, 48 (2016) 281–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Yoon E, Babar A, Choudhary M, Kutner M, Pyrsopoulos N, J Clin Transl Hepatol, 4 (2016) 131–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Sindrup SH, Otto M, Finnerup NB, Jensen TS, Basic & Clinical Pharmacology & Toxicology, 96 (2005) 399–409. [DOI] [PubMed] [Google Scholar]
- [22].Lunn MP, Hughes RA, Wiffen PJ, Cochrane Database Syst Rev, (2014) Cd007115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Obata H, Int J Mol Sci, 18 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Neill J, Brock C, Olesen AE, Andresen T, Nilsson M, Dickenson AH, Pharmacological Reviews, 64 (2012) 939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Vyvey M, Can Fam Physician, 56 (2010) 1295–e1415. [PMC free article] [PubMed] [Google Scholar]
- [26].Memiş D, Turan A, Karamanlioğlu B, Pamukçu Z, Kurt I, Anesth Analg, 98 (2004) 835–840, table of contents. [DOI] [PubMed] [Google Scholar]
- [27].Calasans-Maia JA, Zapata-Sudo G, Sudo RT, J Pharm Pharmacol, 57 (2005) 1415–1420. [DOI] [PubMed] [Google Scholar]
- [28].Kanazi GE, Aouad MT, Jabbour-Khoury SI, Al Jazzar MD, Alameddine MM, Al-Yaman R, Bulbul M, Baraka AS, Acta Anaesthesiol Scand, 50 (2006) 222–227. [DOI] [PubMed] [Google Scholar]
- [29].Yoshitomi T, Kohjitani A, Maeda S, Higuchi H, Shimada M, Miyawaki T, Anesth Analg, 107 (2008) 96–101. [DOI] [PubMed] [Google Scholar]
- [30].Ashish KR, Amteshwar SJ, Nirmal S, CNS & Neurological Disorders - Drug Targets, 12 (2013) 112–125. [DOI] [PubMed] [Google Scholar]
- [31].Corder G, Castro DC, Bruchas MR, Scherrer G, Annual Review of Neuroscience, 41 (2018) 453–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Kiyatkin EA, Neuropharmacology, 151 (2019) 219–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Khademi H, Kamangar F, Brennan P, Malekzadeh R, Arch Iran Med, 19 (2016) 870–876. [DOI] [PubMed] [Google Scholar]
- [34].Del Vecchio G, Spahn V, Stein C, ACS Chem Neurosci, 8 (2017) 1638–1640. [DOI] [PubMed] [Google Scholar]
- [35].Skolnick P, Annu Rev Pharmacol Toxicol, 58 (2018) 143–159. [DOI] [PubMed] [Google Scholar]
- [36].Hayek SM, Shah A, Neurosurg Clin N Am, 25 (2014) 809–817. [DOI] [PubMed] [Google Scholar]
- [37].Aguirre J, Del Moral A, Cobo I, Borgeat A, Blumenthal S, Anesthesiology Research and Practice, 2012 (2012) 560879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Leonard G, Goffaux P, Marchand S, Pain, 151 (2010) 215–219. [DOI] [PubMed] [Google Scholar]
- [39].Wolter T, J Pain Res, 7 (2014) 651–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Dib-Hajj SD, Yang Y, Black JA, Waxman SG, Nat Rev Neurosci, 14 (2013) 49–62. [DOI] [PubMed] [Google Scholar]
- [41].Faber CG, Hoeijmakers JG, Ahn HS, Cheng X, Han C, Choi JS, Estacion M, Lauria G, Vanhoutte EK, Gerrits MM, Dib-Hajj S, Drenth JP, Waxman SG, Merkies IS, Ann Neurol, 71 (2012) 26–39. [DOI] [PubMed] [Google Scholar]
- [42].Bannwarth B, Kostine M, Drugs, 77 (2017) 1377–1387. [DOI] [PubMed] [Google Scholar]
- [43].Nair AS, Indian J Palliat Care, 24 (2018) 384–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Jensen DD, Lieu T, Halls ML, Veldhuis NA, Imlach WL, Mai QN, Poole DP, Quach T, Aurelio L, Conner J, Herenbrink CK, Barlow N, Simpson JS, Scanlon MJ, Graham B, McCluskey A, Robinson PJ, Escriou V, Nassini R, Materazzi S, Geppetti P, Hicks GA, Christie MJ, Porter CJH, Canals M, Bunnett NW, Sci Transl Med, 9 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Yarwood RE, Imlach WL, Lieu T, Veldhuis NA, Jensen DD, Klein Herenbrink C, Aurelio L, Cai Z, Christie MJ, Poole DP, Porter CJH, McLean P, Hicks GA, Geppetti P, Halls ML, Canals M, Bunnett NW, Proc Natl Acad Sci U S A, 114 (2017) 12309–12314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Jimenez-Vargas NN, Pattison LA, Zhao P, Lieu T, Latorre R, Jensen DD, Castro J, Aurelio L, Le GT, Flynn B, Herenbrink CK, Yeatman HR, Edgington-Mitchell L, Porter CJH, Halls ML, Canals M, Veldhuis NA, Poole DP, McLean P, Hicks GA, Scheff N, Chen E, Bhattacharya A, Schmidt BL, Brierley SM, Vanner SJ, Bunnett NW, Proc Natl Acad Sci U S A, 115 (2018) E7438–e7447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Müller CE, Purinergic Signal, 6 (2010) 145–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Jung YH, Kim YO, Lin H, Cho JH, Park JH, Lee SD, Bae J, Kang KM, Kim YG, Pae AN, Ko H, Park CS, Yoon MH, Kim YC, ACS Chem Neurosci, 8 (2017) 1465–1478. [DOI] [PubMed] [Google Scholar]
- [49].Ginnetti AT, Paone DV, Stauffer SR, Potteiger CM, Shaw AW, Deng J, Mulhearn JJ, Nguyen DN, Segerdell C, Anquandah J, Calamari A, Cheng G, Leitl MD, Liang A, Moore E, Panigel J, Urban M, Wang J, Fillgrove K, Tang C, Cook S, Kane S, Salvatore CA, Graham SL, Burgey CS, Bioorg Med Chem Lett, 28 (2018) 1392–1396. [DOI] [PubMed] [Google Scholar]
- [50].Marion E, Song OR, Christophe T, Babonneau J, Fenistein D, Eyer J, Letournel F, Henrion D, Clere N, Paille V, Guerineau NC, Saint Andre JP, Gersbach P, Altmann KH, Stinear TP, Comoglio Y, Sandoz G, Preisser L, Delneste Y, Yeramian E, Marsollier L, Brodin P, Cell, 157 (2014) 1565–1576. [DOI] [PubMed] [Google Scholar]
- [51].Shepherd AJ, Mickle AD, Golden JP, Mack MR, Halabi CM, de Kloet AD, Samineni VK, Kim BS, Krause EG, Gereau R.W.t., Mohapatra DP, Proc Natl Acad Sci U S A, 115 (2018) E8057–E8066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Almeida JP, Chen AL, Foster A, Drezek R, Nanomedicine (Lond), 6 (2011) 815–835. [DOI] [PubMed] [Google Scholar]
- [53].Blanco E, Shen H, Ferrari M, Nat Biotechnol, 33 (2015) 941–951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Noordin MI, Advance Delivery System Dosage Form for Analgesic, Their Rationale, and Specialty, 2017. [Google Scholar]
- [55].Bernards CM, Luger TJ, Malmberg AB, Hill HF, Yaksh TL, Anesthesiology, 77 (1992) 529–535. [DOI] [PubMed] [Google Scholar]
- [56].Grant GJ, Vermeulen K, Zakowski MI, Stenner M, Turndorf H, Langerman L, Anesth Analg, 79 (1994) 706–709. [DOI] [PubMed] [Google Scholar]
- [57].Yaksh TL, Provencher JC, Rathbun ML, Kohn FR, Anesthesiology, 90 (1999) 1402–1412. [DOI] [PubMed] [Google Scholar]
- [58].Alyautdin RN, Petrov VE, Langer K, Berthold A, Kharkevich DA, Kreuter J, Pharm Res, 14 (1997) 325–328. [DOI] [PubMed] [Google Scholar]
- [59].Sao Pedro A, Fernandes R, Villarreal CF, Fialho R, Albuquerque EC, J Microencapsul, 33 (2016) 18–29. [DOI] [PubMed] [Google Scholar]
- [60].Alam M, Hartrick CT, Pain Pract, 5 (2005) 349–353. [DOI] [PubMed] [Google Scholar]
- [61].Balch RJ, Trescot A, J Pain Res, 3 (2010) 191–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Park K, Otte A, Annu Rev Biomed Eng, 21 (2019) 61–84. [DOI] [PubMed] [Google Scholar]
- [63].Simon K, Ther Adv Drug Saf, 6 (2015) 198–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Contet C, Kieffer BL, Befort K, Curr Opin Neurobiol, 14 (2004) 370–378. [DOI] [PubMed] [Google Scholar]
- [65].Cárdeno A, Aparicio-Soto M, Montserrat-de la Paz S, Bermudez B, Muriana FJG, Alarcónde-la-Lastra C, Journal of Functional Foods, 14 (2015) 779–790. [Google Scholar]
- [66].Feng J, Lepetre-Mouelhi S, Gautier A, Mura S, Cailleau C, Coudore F, Hamon M, Couvreur P, Sci Adv, 5 (2019) eaau5148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Oladosu FA, Maixner W, Nackley AG, Mayo Clin Proc, 90 (2015) 1135–1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Berrocoso E, Rey-Brea R, Fernandez-Arevalo M, Mico JA, Martin-Banderas L, Nanomedicine, 13 (2017) 2623–2632. [DOI] [PubMed] [Google Scholar]
- [69].Xie J, Xiao D, Zhao J, Hu N, Bao Q, Jiang L, Yu L, Adv Healthc Mater, 5 (2016) 1213–1221. [DOI] [PubMed] [Google Scholar]
- [70].Jimenez-Vargas NN, Gong J, Wisdom MJ, Jensen DD, Latorre R, Hegron A, Teng S, DiCello JJ, Rajasekhar P, Veldhuis NA, Carbone SE, Yu Y, Lopez-Lopez C, Jaramillo-Polanco J, Canals M, Reed DE, Lomax AE, Schmidt BL, Leong KW, Vanner SJ, Halls ML, Bunnett NW, Poole DP, Proceedings of the National Academy of Sciences, 117 (2020) 15281–15292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Muniz BV, Baratelli D, Di Carla S, Serpe L, da Silva CB, Guilherme VA, Ribeiro LNM, Cereda CMS, de Paula E, Volpato MC, Groppo FC, Fraceto LF, Franz-Montan M, Sci Rep, 8 (2018) 17972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Gadsden J, Long WJ, Open Orthop J, 10 (2016) 94–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Ilfeld BM, Anesth Analg, 113 (2011) 904–925. [DOI] [PubMed] [Google Scholar]
- [74].De Melo NF, De Araújo DR, Grillo R, Moraes CM, De Matos AP, de Paula E, Rosa AH, Fraceto LF, J Pharm Sci, 101 (2012) 1157–1165. [DOI] [PubMed] [Google Scholar]
- [75].Silva de Melo NF, Campos EV, Goncalves CM, de Paula E, Pasquoto T, de Lima R, Rosa AH, Fraceto LF, Colloids Surf B Biointerfaces, 121 (2014) 66–73. [DOI] [PubMed] [Google Scholar]
- [76].Melo NFS, Campos EVR, Franz-Montan M, Paula E, Silva C, Maruyama CR, Stigliani TP, Lima R, Araujo DR, Fraceto LF, J Nanosci Nanotechnol, 18 (2018) 4428–4438. [DOI] [PubMed] [Google Scholar]
- [77].Ramos Campos EV, Silva de Melo NF, Guilherme VA, de Paula E, Rosa AH, de Araújo DR, Fraceto LF, J Pharm Sci, 102 (2013) 215–226. [DOI] [PubMed] [Google Scholar]
- [78].Malik O, Kaye AD, Kaye A, Belani K, Urman RD, J Anaesthesiol Clin Pharmacol, 33 (2017) 151–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Lalani J, Patil S, Kolate A, Lalani R, Misra A, AAPS PharmSciTech, 16 (2015) 413–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Nakatani Y, Masuko H, Amano T, J Pharmacol Sci, 123 (2013) 203–206. [DOI] [PubMed] [Google Scholar]
- [81].Leng F, Wan J, Liu W, Tao B, Chen X, Reg Anesth Pain Med, 37 (2012) 159–165. [DOI] [PubMed] [Google Scholar]
- [82].Chakravarthy KV, Boehm FJ, Christo PJ, Pain Med, 19 (2018) 232–243. [DOI] [PubMed] [Google Scholar]
- [83].El-Boghdadly K, Pawa A, Chin KJ, Local Reg Anesth, 11 (2018) 35–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].He Y, Qin L, Huang Y, Ma C, Nanoscale Res Lett, 15 (2020) 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Sekimoto K, Tobe M, Saito S, Acute Med Surg, 4 (2017) 152–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Vadhanan P, Tripaty DK, Adinarayanan S, Journal of anaesthesiology, clinical pharmacology, 31 (2015) 384–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Kohane DS, Yieh J, Lu NT, Langer R, Strichartz GR, Berde CB, Anesthesiology, 89 (1998) 119–131. [DOI] [PubMed] [Google Scholar]
- [88].Barnet CS, Tse JY, Kohane DS, Pain, 110 (2004) 432–438. [DOI] [PubMed] [Google Scholar]
- [89].Zhao C, Liu A, Santamaria CM, Shomorony A, Ji T, Wei T, Gordon A, Elofsson H, Mehta M, Yang R, Kohane DS, Nat Commun, 10 (2019) 2566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Liu Q, Santamaria CM, Wei T, Zhao C, Ji T, Yang T, Shomorony A, Wang BY, Kohane DS, Nano Letters, 18 (2018) 32–37. [DOI] [PubMed] [Google Scholar]
- [91].Curley J, Castillo J, Hotz J, Uezono M, Hernandez S, Lim JO, Tigner J, Chasin M, Langer R, Berde C, Anesthesiology, 84 (1996) 1401–1410. [DOI] [PubMed] [Google Scholar]
- [92].Shankarappa SA, Tsui JH, Kim KN, Reznor G, Dohlman JC, Langer R, Kohane DS, Proc Natl Acad Sci U S A, 109 (2012) 17555–17560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Sant’Anna MB, Lopes FSR, Kimura LF, Giardini AC, Sant’Anna OA, Picolo G, Toxins (Basel), 11 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Woolbright BL, Jaeschke H, J Hepatol, 66 (2017) 836–848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Federico A, Dallio M, Loguercio C, Molecules, 22 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Das S, Roy P, Auddy RG, Mukherjee A, Int J Nanomedicine, 6 (2011) 1291–1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Chinnagounder Periyasamy P, Leijten JCH, Dijkstra PJ, Karperien M, Post JN, Journal of Nanomaterials, 2012 (2012) 673968. [Google Scholar]
- [98].Ramirez-Garcia PD, Retamal JS, Shenoy P, Imlach W, Sykes M, Truong N, Constandil L, Pelissier T, Nowell CJ, Khor SY, Layani LM, Lumb C, Poole DP, Lieu T, Stewart GD, Mai QN, Jensen DD, Latorre R, Scheff NN, Schmidt BL, Quinn JF, Whittaker MR, Veldhuis NA, Davis TP, Bunnett NW, Nat Nanotechnol, 14 (2019) 1150–1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Eloy JO, Petrilli R, Trevizan LNF, Chorilli M, Colloids Surf B Biointerfaces, 159 (2017) 454–467. [DOI] [PubMed] [Google Scholar]
- [100].Hua S, Cabot PJ, Pain Physician, 16 (2013) E199–216. [PubMed] [Google Scholar]
- [101].Paul JW, Hua S, Ilicic M, Tolosa JM, Butler T, Robertson S, Smith R, Am J Obstet Gynecol, 216 (2017) 283.e281–283.e214. [DOI] [PubMed] [Google Scholar]
- [102].Girotra P, Singh SK, Pharm Res, 33 (2016) 1682–1695. [DOI] [PubMed] [Google Scholar]
- [103].Girotra P, Singh SK, Kumar G, Int J Biol Macromol, 85 (2016) 92–101. [DOI] [PubMed] [Google Scholar]
- [104].Girotra P, Thakur A, Kumar A, Singh SK, Int J Biol Macromol, 96 (2017) 687–696. [DOI] [PubMed] [Google Scholar]
- [105].Portenoy RK, Lesage P, Lancet, 353 (1999) 1695–1700. [DOI] [PubMed] [Google Scholar]
- [106].Gdowski AS, Ranjan A, Sarker MR, Vishwanatha JK, Nanomedicine (Lond), 12 (2017) 2083–2095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Al-Waeli H, Nicolau B, Stone L, Abu Nada L, Gao Q, Abdallah MN, Abdulkader E, Suzuki M, Mansour A, Al Subaie A, Tamimi F, Scientific Reports, 10 (2020) 468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Liu G, Gao J, Ai H, Chen X, Small, 9 (2013) 1533–1545. [DOI] [PubMed] [Google Scholar]
- [109].Mendoza G, Arruebo M, Expert Opin Drug Deliv, 17 (2020) 627–633. [DOI] [PubMed] [Google Scholar]
- [110].Linsley CS, Wu BM, Ther Deliv, 8 (2017) 89–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Wang Y, Kohane DS, Nature Reviews Materials, 2 (2017) 17020. [Google Scholar]
- [112].Rwei AY, Lee JJ, Zhan C, Liu Q, Ok MT, Shankarappa SA, Langer R, Kohane DS, Proc Natl Acad Sci U S A, 112 (2015) 15719–15724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Rwei AY, Wang BY, Ji T, Zhan C, Kohane DS, Nano Lett, 17 (2017) 7138–7145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Ortiz de Solorzano I, Alejo T, Abad M, Bueno-Alejo C, Mendoza G, Andreu V, Irusta S, Sebastian V, Arruebo M, J Colloid Interface Sci, 533 (2019) 171–181. [DOI] [PubMed] [Google Scholar]
- [115].Alejo T, Andreu V, Mendoza G, Sebastian V, Arruebo M, J Colloid Interface Sci, 523 (2018) 234–244. [DOI] [PubMed] [Google Scholar]
- [116].Chen MC, Chan HA, Ling MH, Su LC, J Mater Chem B, 5 (2017) 496–503. [DOI] [PubMed] [Google Scholar]
- [117].Padalkar MV, Pleshko N, Analyst, 140 (2015) 2093–2100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Vedunova MV, Mishchenko TA, Mitroshina EV, Ponomareva NV, Yudintsev AV, Generalova AN, Deyev SM, Mukhina IV, Semyanov AV, Zvyagin AV, RSC Advances, 6 (2016) 33656–33665. [Google Scholar]
- [119].Rwei AY, Paris JL, Wang B, Wang W, Axon CD, Vallet-Regi M, Langer R, Kohane DS, Nat Biomed Eng, 1 (2017) 644–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Yang Q, Nanayakkara GK, Drummer C, Sun Y, Johnson C, Cueto R, Fu H, Shao Y, Wang L, Yang WY, Tang P, Liu LW, Ge S, Zhou XD, Khan M, Wang H, Yang X, Front Physiol, 8 (2017) 818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Miller DL, Smith NB, Bailey MR, Czarnota GJ, Hynynen K, Makin IR, Bioeffects M. Committee of the American Institute of Ultrasound in, J Ultrasound Med, 31 (2012) 623–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Aiyer R, Noori SA, Chang KV, Jung B, Rasheed A, Bansal N, Ottestad E, Gulati A, Pain Med, (2019). [DOI] [PubMed] [Google Scholar]
- [123].Tharkar P, Varanasi R, Wong WSF, Jin CT, Chrzanowski W, Front Bioeng Biotechnol, 7 (2019) 324–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Cullion K, Petishnok LC, Sun T, Santamaria CM, Pemberton GL, McDannold NJ, Kohane DS, Drug Deliv Transl Res, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Kim GW, Kang C, Oh YB, Ko MH, Seo JH, Lee D, Theranostics, 7 (2017) 2463–2476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Jung E, Noh J, Kang C, Yoo D, Song C, Lee D, Biomaterials, 179 (2018) 175–185. [DOI] [PubMed] [Google Scholar]
- [127].van Walsem A, Pandhi S, Nixon RM, Guyot P, Karabis A, Moore RA, Arthritis Res Ther, 17 (2015) 66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].Falyar CR, AANA J, 83 (2015) 357–364. [PubMed] [Google Scholar]
- [129].Jeng CL, Torrillo TM, Rosenblatt MA, Br J Anaesth, 105 Suppl 1 (2010) i97–107. [DOI] [PubMed] [Google Scholar]
- [130].Mantha VR, Nair HK, Venkataramanan R, Gao YY, Matyjaszewski K, Dong H, Li W, Landsittel D, Cohen E, Lariviere WR, Anesth Analg, 118 (2014) 1355–1362. [DOI] [PubMed] [Google Scholar]
- [131].Nadri S, Mahmoudvand H, Eatemadi A, J Microencapsul, 33 (2016) 656–662. [DOI] [PubMed] [Google Scholar]
- [132].Patel S, Jana S, Chetty R, Thakore S, Singh M, Devkar R, Drug Chem Toxicol, 42 (2019) 1–8. [DOI] [PubMed] [Google Scholar]
- [133].Agotegaray MA, Campelo AE, Zysler RD, Gumilar F, Bras C, Gandini A, Minetti A, Massheimer VL, Lassalle VL, Biomater Sci, 5 (2017) 772–783. [DOI] [PubMed] [Google Scholar]
- [134].Toropova YG, Golovkin AS, Malashicheva AB, Korolev DV, Gorshkov AN, Gareev KG, Afonin MV, Galagudza MM, Int J Nanomedicine, 12 (2017) 593–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].Agotegaray M, Palma S, Lassalle V, J Nanosci Nanotechnol, 14 (2014) 3343–3347. [DOI] [PubMed] [Google Scholar]
- [136].Wu H, Li F, Wang S, Lu J, Li J, Du Y, Sun X, Chen X, Gao J, Ling D, Biomaterials, 151 (2018) 66–77. [DOI] [PubMed] [Google Scholar]
- [137].Zhu L, Zhou Z, Mao H, Yang L, Nanomedicine (Lond), 12 (2017) 73–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].Amanzadeh E, Esmaeili A, Abadi REN, Kazemipour N, Pahlevanneshan Z, Beheshti S, Sci Rep, 9 (2019) 6876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [139].Enteshari Najafabadi R, Kazemipour N, Esmaeili A, Beheshti S, Nazifi S, BMC Pharmacol Toxicol, 19 (2018) 59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [140].Bilan R, Nabiev I, Sukhanova A, ChemBioChem, 17 (2016) 2103–2114. [DOI] [PubMed] [Google Scholar]
- [141].Pisanic TR 2nd, Zhang Y, Wang TH, The Analyst, 139 (2014) 2968–2981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [142].Husain SF, Lam RWM, Hu T, Ng MWF, Liau ZQG, Nagata K, Khanna S, Lam Y, Bhakoo K, Ho RCM, Wong HK, Pain Res Manag, 2019 (2019) 9394715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [143].Storek B, Reinhardt M, Wang C, Janssen WGM, Harder NM, Banck MS, Morrison JH, Beutler AS, Proceedings of the National Academy of Sciences, 105 (2008) 1055–1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [144].Lu Y, McNearney TA, Lin W, Wilson SP, Yeomans DC, Westlund KN, Molecular therapy : the journal of the American Society of Gene Therapy, 15 (2007) 1812–1819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [145].Braz J, Beaufour C, Coutaux A, Epstein AL, Cesselin F, Hamon M, Pohl M, J Neurosci, 21 (2001) 7881–7888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [146].Meunier A, Latrémolière A, Mauborgne A, Bourgoin S, Kayser V, Cesselin F, Hamon M, Pohl M, Mol Ther, 11 (2005) 608–616. [DOI] [PubMed] [Google Scholar]
- [147].Hu C, Cai Z, Lu Y, Cheng X, Guo Q, Wu Z, Zhang Q, Neuroscience Letters, 634 (2016) 87–93. [DOI] [PubMed] [Google Scholar]
- [148].Goss JR, Harley CF, Mata M, O’Malley ME, Goins WF, Hu X, Glorioso JC, Fink DJ, Ann Neurol, 52 (2002) 662–665. [DOI] [PubMed] [Google Scholar]
- [149].Goss JR, Cascio M, Goins WF, Huang S, Krisky DM, Clarke RJ, Johnson JW, Yokoyama H, Yoshimura N, Gold MS, Glorioso JC, Molecular Therapy, 19 (2011) 500–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [150].Thakur V, Gonzalez M, Pennington K, Chattopadhyay M, Molecular and Cellular Neuroscience, 72 (2016) 46–53. [DOI] [PubMed] [Google Scholar]
- [151].Kanda H, Kanao M, Liu S, Yi H, Iida T, Levitt RC, Candiotti KA, Lubarsky DA, Hao S, Gene therapy, 23 (2016) 340–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [152].Kanao M, Kanda H, Huang W, Liu S, Yi H, Candiotti KA, Lubarsky DA, Levitt RC, Hao S, Anesth Analg, 120 (2015) 1394–1404. [DOI] [PubMed] [Google Scholar]
- [153].Majima T, Funahashi Y, Takai S, Goins WF, Gotoh M, Tyagi P, Glorioso JC, Yoshimura N, Human Gene Therapy, 26 (2015) 734–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [154].Vit J-P, Ohara PT, Sundberg C, Rubi B, Maechler P, Liu C, Puntel M, Lowenstein P, Castro M, Jasmin L, Molecular pain, 5 (2009) 42–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [155].Milligan ED, Soderquist RG, Malone SM, Mahoney JH, Hughes TS, Langer SJ, Sloane EM, Maier SF, Leinwand LA, Watkins LR, Mahoney MJ, Neuron Glia Biol, 2 (2006) 293–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [156].Yao MZ, Gu JF, Wang JH, Sun LY, Liu H, Liu XY, Gene Therapy, 10 (2003) 1392–1399. [DOI] [PubMed] [Google Scholar]
- [157].Maeda S, Kawamoto A, Yatani Y, Shirakawa H, Nakagawa T, Kaneko S, Molecular pain, 4 (2008) 65–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [158].Tan AM, Samad OA, Dib-Hajj SD, Waxman SG, Molecular Medicine, 21 (2015) 544–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [159]. Samad OA, Tan AM, Cheng X, Foster E, Dib-Hajj SD, Waxman SG, Molecular Therapy, 21 (2013) 49–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [160].Kim J, Kim SJ, Lee H, Chang JW, Biochem Biophys Res Commun, 388 (2009) 73–78. [DOI] [PubMed] [Google Scholar]
- [161].Lee B, Kim J, Kim SJ, Lee H, Chang JW, Biochemical and Biophysical Research Communications, 357 (2007) 971–976. [DOI] [PubMed] [Google Scholar]
- [162].Eaton MJ, Blits B, Ruitenberg MJ, Verhaagen J, Oudega M, Gene Therapy, 9 (2002) 1387–1395. [DOI] [PubMed] [Google Scholar]
- [163].Meunier A, Latrémolière A, Dominguez E, Mauborgne A, Philippe S, Hamon M, Mallet J, Benoliel J-J, Pohl M, Molecular Therapy, 15 (2007) 687–697. [DOI] [PubMed] [Google Scholar]
- [164].Sun T, Luo J, Jia M, Li H, Li K, Fu Z, European Journal of Pharmacology, 682 (2012) 79–85. [DOI] [PubMed] [Google Scholar]
- [165].Song Z, Zou W, Liu C, Guo Q, The Journal of Gene Medicine, 12 (2010) 873–880. [DOI] [PubMed] [Google Scholar]
- [166].Zou W, Song Z, Guo Q, Liu C, Zhang Z, Zhang Y, Hum Gene Ther, 22 (2011) 465–475. [DOI] [PubMed] [Google Scholar]
- [167].Vanderwall AG, Noor S, Sun MS, Sanchez JE, Yang XO, Jantzie LL, Mellios N, Milligan ED, Brain, Behavior, and Immunity, 69 (2018) 91–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [168].Isner JM, Walsh K, Symes J, Pieczek A, Takeshita S, Lowry J, Rossow S, Rosenfield K, Weir L, Brogi E, Schainfeld R, Circulation, 91 (1995) 2687–2692. [DOI] [PubMed] [Google Scholar]
- [169].Baumgartner I, Pieczek A, Manor O, Blair R, Kearney M, Walsh K, Isner JM, Circulation, 97 (1998) 1114–1123. [DOI] [PubMed] [Google Scholar]
- [170].Isner JM, Baumgartner I, Rauh G, Schainfeld R, Blair R, Manor O, Razvi S, Symes JF, Journal of Vascular Surgery, 28 (1998) 964–975. [DOI] [PubMed] [Google Scholar]
- [171].Losordo DW, Vale PR, Symes JF, Dunnington CH, Esakof DD, Maysky M, Ashare AB, Lathi K, Isner JM, Circulation, 98 (1998) 2800–2804. [DOI] [PubMed] [Google Scholar]
- [172].Vale PR, Losordo DW, Milliken CE, McDonald MC, Gravelin LM, Curry CM, Esakof DD, Maysky M, Symes JF, Isner JM, Circulation, 103 (2001) 2138–2143. [DOI] [PubMed] [Google Scholar]
- [173].Losordo DW, Vale PR, Hendel RC, Milliken CE, Fortuin FD, Cummings N, Schatz RA, Asahara T, Isner JM, Kuntz RE, Circulation, 105 (2002) 2012–2018. [DOI] [PubMed] [Google Scholar]
- [174].Kastrup J, Jørgensen E, Rück A, Tägil K, Glogar D, Ruzyllo W, Bøtker HE, Dudek D, Drvota V, Hesse B, Thuesen L, Blomberg P, Gyöngyösi M, Sylvén C, A randomized double-blind placebo-controlled study: The Euroinject One trial, 45 (2005) 982–988. [DOI] [PubMed] [Google Scholar]
- [175].Yao MZ, Gu JF, Wang JH, Sun LY, Lang MF, Liu J, Zhao ZQ, Liu XY, Neuroscience, 112 (2002) 409–416. [DOI] [PubMed] [Google Scholar]
- [176].Milligan ED, Langer SJ, Sloane EM, He L, Wieseler-Frank J, O’Connor K, Martin D, Forsayeth JR, Maier SF, Johnson K, Chavez RA, Leinwand LA, Watkins LR, European Journal of Neuroscience, 21 (2005) 2136–2148. [DOI] [PubMed] [Google Scholar]
- [177].Soderquist RG, Milligan ED, Harrison JA, Chavez RA, Johnson KW, Watkins LR, Mahoney MJ, J Biomed Mater Res A, 93 (2010) 1169–1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [178].Dengler EC, Alberti LA, Bowman BN, Kerwin AA, Wilkerson JL, Moezzi DR, Limanovich E, Wallace JA, Milligan ED, Journal of Neuroinflammation, 11 (2014) 92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [179].Grace PM, Loram LC, Christianson JP, Strand KA, Flyer-Adams JG, Penzkover KR, Forsayeth JR, van Dam A-M, Mahoney MJ, Maier SF, Chavez RA, Watkins LR, Brain, Behavior, and Immunity, 59 (2017) 49–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [180].Sloane E, Langer S, Jekich B, Mahoney J, Hughes T, Frank M, Seibert W, Huberty G, Coats B, Harrison J, Klinman D, Poole S, Maier S, Johnson K, Chavez R, Watkins LR, Leinwand L, Milligan E, Gene Therapy, 16 (2009) 1210–1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [181].Yamano S, Viet CT, Dang D, Dai J, Hanatani S, Takayama T, Kasai H, Imamura K, Campbell R, Ye Y, Dolan JC, Kwon WM, Schneider SD, Schmidt BL, Pain, 158 (2017) 240–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [182].Pradat PF, Finiels F, Kennel P, Naimi S, Orsini C, Delaere P, Revah F, Mallet J, Hum Gene Ther, 12 (2001) 367–375. [DOI] [PubMed] [Google Scholar]
- [183].Guedon J-MG, Wu S, Zheng X, Churchill CC, Glorioso JC, Liu C-H, Liu S, Vulchanova L, Bekker A, Tao Y-X, Kinchington PR, Goins WF, Fairbanks CA, Hao S, Molecular pain, 11 (2015) 27-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [184].Fink DJ, Wechuck J, Mata M, Glorioso JC, Goss J, Krisky D, Wolfe D, Ann Neurol, 70 (2011) 207–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [185]. https://ClinicalTrials.gov/show/NCT01291901.
- [186]. https://ClinicalTrials.gov/show/NCT03477487.
- [187]. https://ClinicalTrials.gov/show/NCT04124042.
- [188].Therapeutics X, http://xylocor.com/angina_trial.html, 2021.
- [189]. https://ClinicalTrials.gov/show/NCT04125732.
- [190].Grines C, Rubanyi GM, Kleiman NS, Marrott P, Watkins MW, The American Journal of Cardiology, 92 (2003) 24–31. [DOI] [PubMed] [Google Scholar]
- [191].Henry TD, Grines CL, Watkins MW, Dib N, Barbeau G, Moreadith R, Andrasfay T, Engler RL, Journal of the American College of Cardiology, 50 (2007) 1038–1046. [DOI] [PubMed] [Google Scholar]
- [192]. https://ClinicalTrials.gov/show/NCT00346437.
- [193]. https://ClinicalTrials.gov/show/NCT02928094.
- [194].McDermott LA, Weir GA, Themistocleous AC, Segerdahl AR, Blesneac I, Baskozos G, Clark AJ, Millar V, Peck LJ, Ebner D, Tracey I, Serra J, Bennett DL, Neuron, 101 (2019) 905–919 e908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [195].Moreno AM, Alemán F, Catroli GF, Hunt M, Hu M, Dailamy A, Pla A, Woller SA, Palmer N, Parekh U, McDonald D, Roberts AJ, Goodwill V, Dryden I, Hevner RF, Delay L, Gonçalves dos Santos G, Yaksh TL, Mali P, Science Translational Medicine, 13 (2021) eaay9056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [196].Farhang N, Brunger JM, Stover JD, Thakore PI, Lawrence B, Guilak F, Gersbach CA, Setton LA, Bowles RD, Tissue Engineering Part A, 23 (2017) 738–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [197].Zhao L, Huang J, Fan Y, Li J, You T, He S, Xiao G, Chen D, Annals of the Rheumatic Diseases, 78 (2019) 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [198].Lee B, Lee K, Panda S, Gonzales-Rojas R, Chong A, Bugay V, Park HM, Brenner R, Murthy N, Lee HY, Nature biomedical engineering, 2 (2018) 497–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [199].Hryhorowicz M, Grześkowiak B, Mazurkiewicz N, Śledziński P, Lipiński D, Słomski R, Mol Biotechnol, 61 (2019) 173–180. [DOI] [PubMed] [Google Scholar]
- [200].Cheng Q, Wei T, Farbiak L, Johnson LT, Dilliard SA, Siegwart DJ, Nature Nanotechnology, 15 (2020) 313–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [201].Li S, Song Z, Liu C, Chen X-L, Han H, ACS Applied Materials & Interfaces, 11 (2019) 47762–47770. [DOI] [PubMed] [Google Scholar]
- [202].Rohiwal SS, Dvorakova N, Klima J, Vaskovicova M, Senigl F, Slouf M, Pavlova E, Stepanek P, Babuka D, Benes H, Ellederova Z, Stieger K, Scientific Reports, 10 (2020) 4619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [203].Liang H, Peng B, Dong C, Liu L, Mao J, Wei S, Wang X, Xu H, Shen J, Mao H-Q, Gao X, Leong KW, Chen Y, Nature Communications, 9 (2018) 4291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [204].Holl EK, Shumansky KL, Pitoc G, Ramsburg E, Sullenger BA, PLoS One, 8 (2013) e69413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [205].Holl EK, Shumansky KL, Borst LB, Burnette AD, Sample CJ, Ramsburg EA, Sullenger BA, Proc Natl Acad Sci U S A, 113 (2016) 9728–9733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [206].Lee J, Jackman JG, Kwun J, Manook M, Moreno A, Elster EA, Kirk AD, Leong KW, Sullenger BA, Biomaterials, 120 (2017) 94–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [207].Lee J, Sohn JW, Zhang Y, Leong KW, Pisetsky D, Sullenger BA, Proc Natl Acad Sci U S A, 108 (2011) 14055–14060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [208].Liu F, Sheng S, Shao D, Xiao Y, Zhong Y, Zhou J, Quek CH, Wang Y, Hu Z, Liu H, Li Y, Tian H, Leong KW, Chen X, Nano Letters, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [209].Naqvi I, Gunaratne R, McDade JE, Moreno A, Rempel RE, Rouse DC, Herrera SG, Pisetsky DS, Lee J, White RR, Sullenger BA, Mol Ther, 26 (2018) 1020–1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [210].Holl EK, Bond JE, Selim MA, Ehanire T, Sullenger B, Levinson H, Plast Reconstr Surg, 134 (2014) 420e–433e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [211].Pisetsky DS, Lee J, Leong KW, Sullenger BA, Expert Rev Clin Immunol, 8 (2012) 1–3. [DOI] [PubMed] [Google Scholar]
- [212].Mittal M, Siddiqui MR, Tran K, Reddy SP, Malik AB, Antioxid Redox Signal, 20 (2014) 1126–1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [213].Kim HK, Park SK, Zhou JL, Taglialatela G, Chung K, Coggeshall RE, Chung JM, Pain, 111 (2004) 116–124. [DOI] [PubMed] [Google Scholar]
- [214].Gao X, Kim HK, Chung JM, Chung K, Pain, 131 (2007) 262–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [215].Yoshitomi T, Hirayama A, Nagasaki Y, Biomaterials, 32 (2011) 8021–8028. [DOI] [PubMed] [Google Scholar]
- [216].Fidanboylu M, Griffiths LA, Flatters SJ, PLoS One, 6 (2011) e25212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [217].Lian M, Xue Z, Qiao X, Liu C, Zhang S, Li X, Huang C, Song Q, Yang W, Chen X, Wang T, Chem, 5 (2019) 2378–2387. [Google Scholar]
- [218].Yin JJ, Lao F, Meng J, Fu PP, Zhao Y, Xing G, Gao X, Sun B, Wang PC, Chen C, Liang XJ, Mol Pharmacol, 74 (2008) 1132–1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [219].Li J, Guan M, Wang T, Zhen M, Zhao F, Shu C, Wang C, ACS Appl Mater Interfaces, 8 (2016) 25770–25776. [DOI] [PubMed] [Google Scholar]