

ABSTRACT
Paracetamol (acetaminophen) is undoubtedly one of the most widely used drugs worldwide. As an over-the-counter medication, paracetamol is the standard and first-line treatment for fever and acute pain and is believed to remain so for many years to come. Despite being in clinical use for over a century, the precise mechanism of action of this familiar drug remains a mystery. The oldest and most prevailing theory on the mechanism of analgesic and antipyretic actions of paracetamol relates to the inhibition of CNS cyclooxygenase (COX) enzyme activities, with conflicting views on the COX isoenzyme/variant targeted by paracetamol and on the nature of the molecular interactions with these enzymes. Paracetamol has been proposed to selectively inhibit COX-2 by working as a reducing agent, despite the fact that in vitro screens demonstrate low potency on the inhibition of COX-1 and COX-2. In vivo data from COX-1 transgenic mice suggest that paracetamol works through inhibition of a COX-1 variant enzyme to mediate its analgesic and particularly thermoregulatory actions (antipyresis and hypothermia). A separate line of research provides evidence on potentiation of the descending inhibitory serotonergic pathway to mediate the analgesic action of paracetamol, but with no evidence of binding to serotonergic molecules. AM404 as a metabolite for paracetamol has been proposed to activate the endocannabinoid and the transient receptor potential vanilloid-1 (TRPV1) systems. The current review gives an update and in some cases challenges the different theories on the pharmacology of paracetamol and raises questions on some of the inadequately explored actions of paracetamol.
List of Abbreviations: AM404, N-(4-hydroxyphenyl)-arachidonamide; CB1R, Cannabinoid receptor-1; Cmax, Maximum concentration; CNS, Central nervous system; COX, Cyclooxygenase; CSF, Cerebrospinal fluid; ED50, 50% of maximal effective dose; FAAH, Fatty acid amidohydrolase; IC50, 50% of the maximal inhibitor concentration; LPS, Lipopolysaccharide; NSAIDs, Non-steroidal anti-inflammatory drugs; PGE2, Prostaglandin E2; TRPV1, Transient receptor potential vanilloid-1
KEYWORDS: Paracetamol, acetaminophen, cyclooxygenase, pain, thermoregulation
Brief history of paracetamol
Paracetamol (acetaminophen, N-acetyl-p-aminophenol) is one of the most widely used over-the-counter analgesic antipyretic drugs. It was first synthesized by Joseph von Mering in (1893) by reacting p-nitrophenol with tin and glacial acetic acid. In the 1880s paracetamol and phenacetin (Figure 1) were found to possess antipyretic and later analgesic activity. Initially, phenacetin gained more popularity than paracetamol and was marketed in 1887; however, because of the serious side effects associated with phenacetin such as hemolytic anemia and methemoglobin formation, its clinical use declined, and attention focused on paracetamol, which was marketed in 1893 [1]. Additionally, more studies on phenacetin in the 1940s showed that paracetamol is one of its major metabolites and thus its pharmacological effects are attributed to paracetamol [2]. As a result, paracetamol became freely available from the 1950s and has become the most widely used over-the-counter non-narcotic analgesic agent for the treatment of mild to moderate pain and fever.
Figure 1.
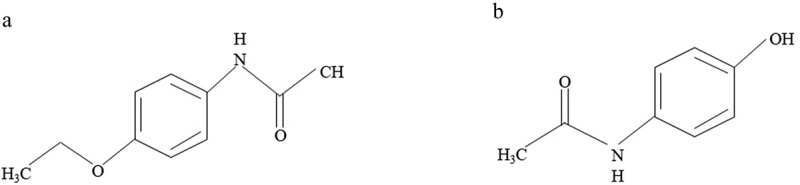
Chemical structures of A) paracetamol and B) phenacetin
Paracetamol now dominates the market of over-the-counter non-narcotic analgesic drugs following the demonstration of its safety profile at therapeutic doses and particularly after aspirin usage began to decline since the 1960s due to its gastrointestinal toxicity and association with Reye’s Syndrome in children [3]. Today paracetamol is the standard and first-line treatment for fever and acute pain and is believed to remain so for many years to come [4]. This is mainly due to its outstanding safety record at therapeutic doses when compared to the non-steroidal anti-inflammatory drugs (NSAIDs). Sales of paracetamol, most widely consumed over-the-counter analgesic drug, have been on the increase for the past few years – a trend that it is predicted to continue [5]
Pharmacological actions of paracetamol
Analgesic action of paracetamol
Paracetamol in adults is used for the management of various types of acute painful conditions that include headache [6], musculoskeletal pain [7], period pain [8], osteoarthritic pain [9], back pain [10], dental pain [11,12] also for the management of postoperative pain [13].
The standard therapeutic dose of paracetamol for adults is 2 tablets of 500 mg each taken orally every 4 hours up to a maximum of 8 tablets for any 24-hour period. In children, paracetamol is marketed in dosages depending on age and range from 60 mg (2–3 months) to 480–750 mg (12–16 year olds). Paracetamol is sold as a single pharmacologically active chemical entity or in formulations in combination with other analgesic drugs that include aspirin, caffeine or some opioid analgesic drugs [14,15].
Mechanistic research on the analgesic action of paracetamol
2.1.1.1. Does the inhibition of a centrally expressed cyclooxygenase enzyme explain the analgesic action of paracetamol?
One of the first published mechanistic research on paracetamol is the work by Flower and Vane (1972) who demonstrated potent inhibition of brain prostaglandin E2 (PGE2) synthesis (IC50 = 12.5 µg/ml) compared to PGE2 in the spleen (IC50 = 100 µg/ml) indicating 8x more potent inhibition of PGE2 synthesis in the brain than spleen, whereas several NSAIDs showed equipotent inhibition of brain and spleen PGE2 synthesis [16]. This work followed on from the Nobel Prize winning research by Sir John Vane who showed that the mechanism of action of aspirin and other NSAIDs is mediated through inhibition of the cyclooxygenase (COX) enzyme resulting in reduction of PGE2 synthesis [17].
The notion that the pharmacological actions of paracetamol are mediated through the selective inhibition of a centrally expressed COX enzyme was supported by several in vivo investigations in experimental animals [18–23] as well as in human subjects [24–26]. In addition, the analgesic action of paracetamol was shown not to be linked to inhibition of prostaglandin synthesis in the periphery [21,23,27,28]. The centrally mediated mechanism of action is also supported by the pharmacokinetic profile of paracetamol. Paracetamol is a moderately lipid-soluble weak organic acid with a pka value of 9.5 and is largely un-ionized over the physiological range of pH [29]. Its lipid solubility enables it to rapidly penetrate cellular membranes and to also readily cross the blood-brain barrier. Paracetamol is rapidly absorbed by passive diffusion in the small intestine [30]. A standard therapeutic dose of paracetamol produces 80% bioavailability and reaches peak plasma level (Cmax) of 18 mg/L (120 µM) after approximately 120 minutes and a cerebrospinal fluid (CSF) Cmax of 8.8 mg/ml at around 240 minutes [31]. Using functional magnetic resonance imaging in humans, paracetamol was shown to reduce firing within the spinothalamic tract in response to thermal noxious stimulation [26].
Which cyclooxygenase enzyme does paracetamol inhibit?
Products of the COX-1 and COX-2 enzymes, particularly PGE2, have been shown to have important roles in the transmission of nociceptive pain at the sites of pain initiation and also at the spinal and supraspinal nociceptive pathways, inhibition of which has been demonstrated to mediated the peripheral and in some cases central analgesic actions of NSAIDs [32]. Despite the potent inhibition by paracetamol of brain and spinal cord-derived prostaglandins in in vivo experiments, in vitro screening experiments demonstrated weak inhibitory activities on COX-1 and COX-2 enzymes by paracetamol [33]. In this study, IC50 values were not achievable for concentrations up to 1 mg/ml; hence, IC30 values of 2.7 µg/ml and 20 µg/ml were obtained against inhibition of COX-1 and COX-2, respectively. In this report, the authors conclude that the reduction of central nervous system (CNS) PGE2 synthesis by paracetamol may be mediated through inhibition of a COX enzyme yet to be discovered [33], a notion that has also been predicted by others [34,35].
Indeed, in 2002 a catalytically functional third COX enzyme was identified and was originally referred to as COX-3 by Professor Daniel Simmons’s laboratory [36] and was referred to COX-1b in some publications [37–40]. COX-3 was originally identified in canine brain tissues as a splice variant of COX-1. It was shown that COX-3 expression occurs when the intron-1 sequence of the COX-1 gene is retained in the mRNA and subsequently protein sequences resulting in the insertion of additional 33 amino acids that encode the hydrophobic signal peptide domain of the COX-3 protein. Paracetamol showed selectivity for inhibition of COX-3 (IC50 = 460 µM) over COX-1 and COX-2, which was dependent on the concentration of the substrate arachidonic acid suggesting competitive blockage at the active site [36]. Several reports disputed the expression of a fully functional COX-3 protein in human and rodent tissues as retention of intron-1 in the mRNA sequence introduced a stop codon early in the translational process resulting in a truncated protein with no catalytic activity [37–41]. The issue of an out-of-reading frame sequence for COX-3 in human and rodent cells was actually acknowledged by Professor Simmons original report on COX-3 [36]. In fact, mechanisms that might be important in correcting this out-of-reading frame sequence have been reported and include ribrosomal frame shifting. Indeed, Qin et al. (2005) have reported on fully functional COX-3 proteins in human cells in which the removal of one nucleotide from the 94 nucleotides long sequence of human COX-1 gene intron-1 brings the sequence back in frame leading to synthesis of a full length and catalytically active COX enzyme [42] (Figure 2). Using an antibody that recognizes the intron-1 sequence, we have also been able to detect an intron-1 retaining protein in rodent CNS tissues [23,43,44] (Figure 3).
Figure 2.
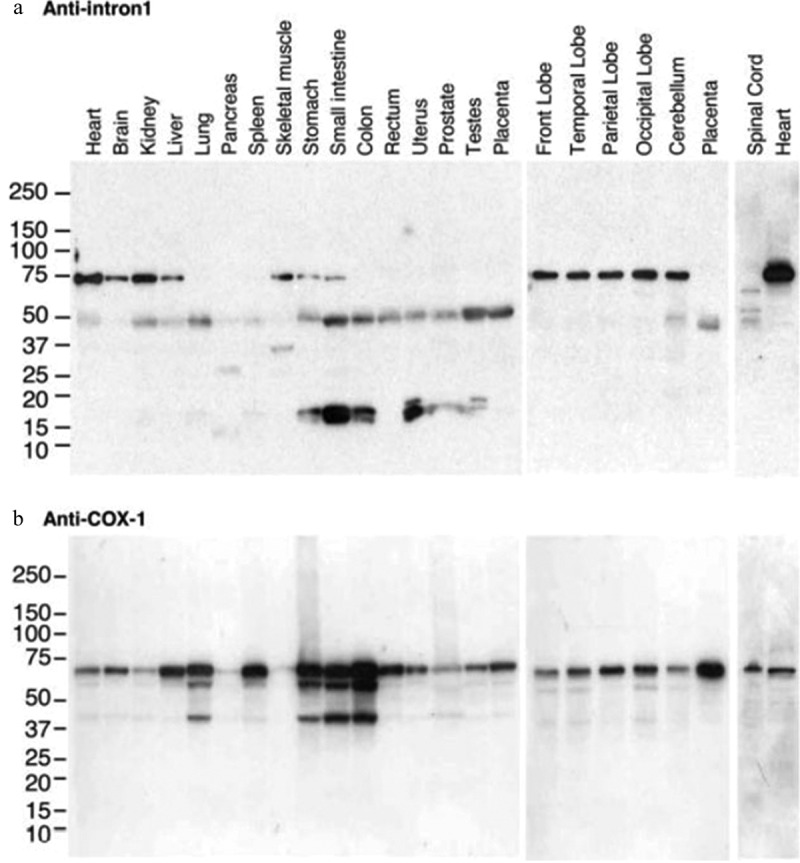
Tissue distribution of intron-1 retaining COX-1 proteins with 55 and 75KDa molecular weights in human tissues. Human multiple tissue total protein blots were probed with antihuman COX-1 intron 1 antibody (a) and re-probed with anti-human COX-1 antibody (b) using Western blotting analysis. Reproduced from Qin et al. 2005 [43]
Figure 3.
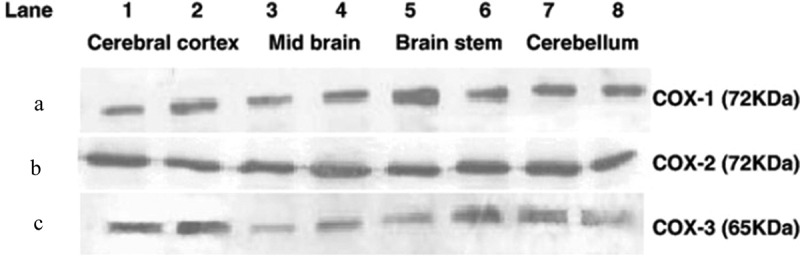
Western blotting analysis of COX isoenzyme proteins expression in different regions of mouse brains. Western blots for COX-1 (panel A), COX-2 (panel B) and COX-3 (panel C) from different brain regions of C57Bl/6 mice. Lanes 1 and 2 = cerebral cortex; lanes 3 and 4 = midbrain; lanes 5 and 6 = brain stem; lanes 7 and 8 = cerebellum. Reproduced from Ayoub et al. 2006 [23]
Using the acetic acid-induced abdominal constriction model of acute nociceptive pain, we showed that the analgesic action of paracetamol was accompanied by reduction of prostaglandin synthesis in the brain and spinal cord, but not in the peritoneum; effects that were abrogated in COX-1 homozygote knockout mice, but not in COX-2 homozygote knockout mice [23]. In addition, paracetamol blocked the centrally mediated abdominal constriction induced by the intraperitoneal administration of iloprost [23,45], more potently than constriction induced by acetic acid (ED50 values for paracetamol in the iloprost and acetic acid-induced constrictions were 149 and 172 mg/kg, respectively). In contrast, diclofenac, a peripherally active NSAID [46,47], inhibited the acetic acid-induced constriction with an ED50 value of 16 mg/kg, but had no effects on the ilorpost-induced constriction [23]. Given the loss of analgesia and inhibition of PGE2 synthesis by paracetamol in COX-1 knockout mice compared to wild-type littermate control mice (Figure 4) and the weak inhibition of COX-1 by paracetamol as reported by Mitchell et al. (1994) [33], the conclusion from this study is that the analgesic action of paracetamol in this model is mediated through inhibition of a COX-1 variant protein expressed in the brain and spinal cord. Other COX-3 selective inhibitors such as aminopyrine and antipyrine [36] showed similar pharmacological profiles to paracetamol [23]. The acetic acid-induced constriction model represents a human model of acute pain that shares similar pathologies to human pain. Injections of prostaglandin E1 and PGE2 were shown to induce the abdominal constriction response [48]. Within the peritoneum prostaglandin I2 (measured as its stable metabolite 6-keto-prostaglandin F1α) was shown to be released more than PGE2. In IP-receptor knockout mice, it was demonstrated that no constriction responses were induced in response to acetic acid [49]. Within the CNS COX enzymes products are also involved in mediating the nociceptive response in this model as both PGE2 and prostaglandin D2 administrated intracisternally to mice injected with acetic acid produced a biphasic effect, thus at low doses (5 ng and 15 ng, respectively) produced a hyperalgesic response and at high doses (5 µg and 50 ng, respectively) a hypoalgesic effect [50].
Figure 4.
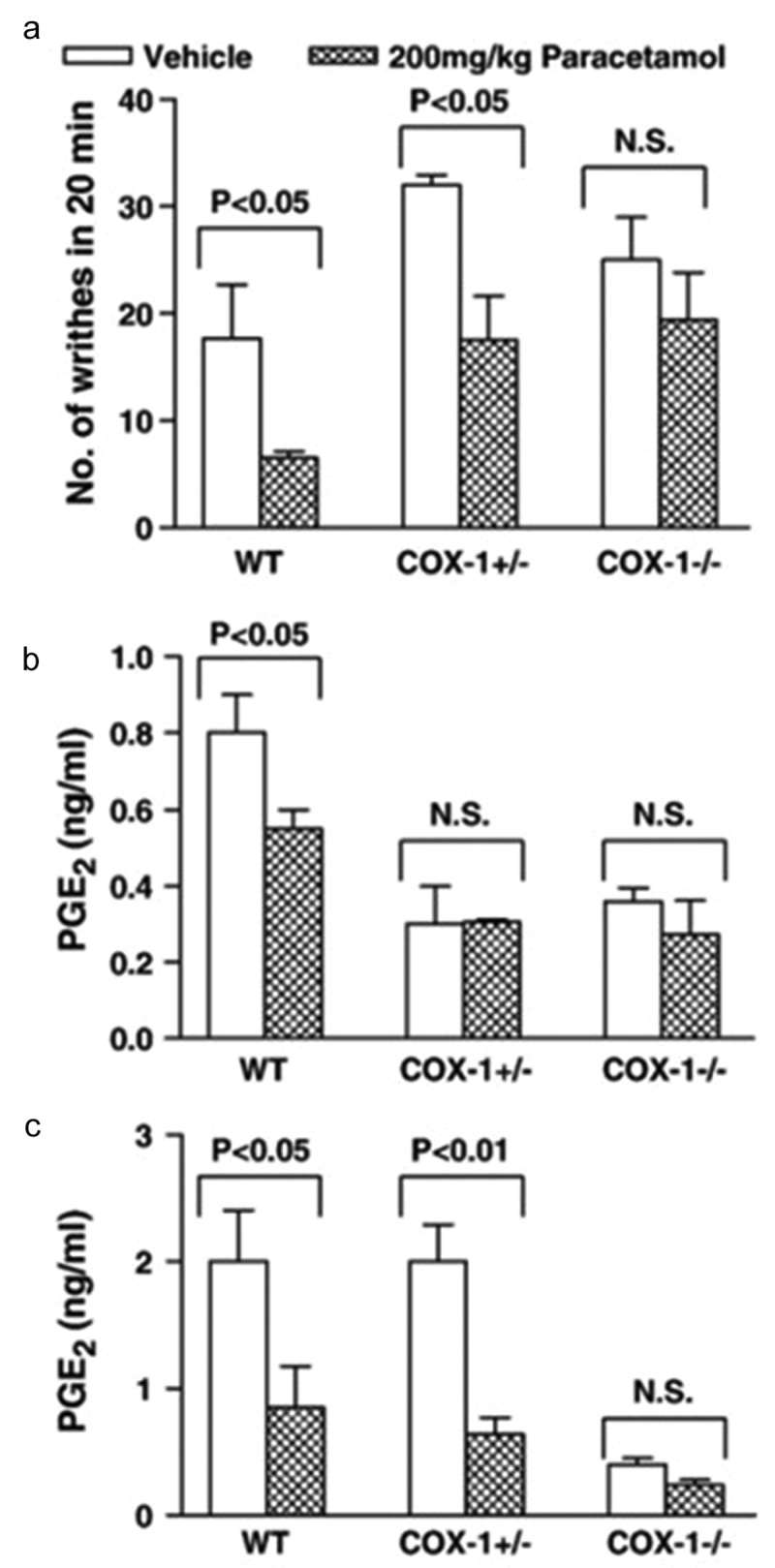
The analgesic action of 200 mg/kg paracetamol in the acetic acid-induced writhing test was abrogated in COX-1 knockout mice in comparison to wild-type littermate control mice (a). Similarly, the inhibition of PGE2 synthesis in brain (b) and spinal cord (c) tissues by paracetamol was abrogated in COX-1 knockout mice in comparison to wild-type littermate control mice. Reproduced from Ayoub et al. 2006 [23]
It has also been argued that paracetamol may produce its pharmacological actions through the selective inhibition of the COX-2 enzyme [51,52]. Hinz et al. (2007) demonstrated 4.4x more selectivity toward the inhibition of COX-2 (IC50 = 25.8 µmol/L) over COX-1 (IC50 = 113.7 µmol/L) from in vitro assays using whole human blood [51]. In this study, paracetamol demonstrated over 80% blockade of COX-2 activity comparable to NSAIDs and COX-2 selective inhibitors. It is not clear why there are significant discrepancies in relation to inhibition of COX-1 and COX-2 activities by paracetamol as reported by Hinz et al. [51] and Mitchell et al. [33]. The differences in the results are notwithstanding the similarities in the experimental procedures. The concentrations of paracetamol used in the two studies are within a similar range (Hinz et al. used 100 µM and Mitchell et al. used 50–600 µM). Other similarities between the two studies include the cell types used; whole human blood stimulated with 10 µg/ml Lipopolysaccharide (LPS, for the induction of COX-2) by Hinz et al. [51] and J774.2 macrophage cell line stimulated with 1 µg/ml LPS by Mitchell et al. [33].
Unlike the COX-2 selective inhibitors, which have been shown to produce severe cardiovascular toxicities [53–55], the induction of such cardiovascular toxicity by paracetamol is a matter of debate [56,57]. Paracetamol has also been claimed to have similar anti-inflammatory actions to COX-2 selective inhibitors [51] in reference to the work by Skjelbred and Lokken [58] and Bjornsson et al. [59] in which paracetamol has been shown to reduce dental edema. However, generally speaking paracetamol is regarded to have weak anti-inflammatory activities from clinical and preclinical studies [60–63]. The notion that paracetamol produces anti-inflammatory actions in such dental inflammatory reactions is based on the assumption that such inflammatory reactions present low-grade inflammation, an idea that has not been tested thoroughly.
As stated above, paracetamol showed potent analgesia and concomitant with inhibition of CNS PGE2 synthesis, but not peripheral prostaglandin synthesis in the abdominal constriction model [23]; a model of nociceptive pain that has been shown to be mediated by prostaglandin mediators derived from the COX-1 and not COX-2 gene products [64].
2.1.1.3. Does the lipid hydroproxide theory explain the weak anti-inflammatory actions of paracetamol?
One of the hypothesis that has been put forward to explain the weak anti-inflammatory action by paracetamol is related to its ability to work as a reducing agent [65] as opposed to blockade of the cyclooxygenase active site of the COX-1 and COX-2 enzymes. Structurally paracetamol is a phenolic compound and phenols are known to be good reducing agents. To be catalytically active, COX-1 and COX-2 enzymes need to be in the oxidized active state, which is ensured through the continuous oxidation of the tyrosine-385 residue at the COX active site to a tyrosyl radical through electron transfer. Generation of the tyrosyl radical is initiated at the peroxidase active site of the COX-1 and COX-2 enzymes by the reduction of an available hydroperoxide substrate [66]. Thus, supply of lipid hydroperoxides ensures that the enzyme remains in the oxidized active state. Paracetamol as a reducing agent has been proposed to work by lowering the intracellular lipid hydroperoxide tone driving COX-1 and COX-2 enzymes into the inactive reduced state ultimately reducing prostaglandin synthesis [65]. In an inflammatory milieu where the peroxide tone is believed to be high would render paracetamol inactive as a reducing agent, thus possibly explaining its weak anti-inflammatory action. It is noteworthy that this hypothesis, which has been claimed to explain the weak anti-inflammatory action of paracetamol and to also explain the mechanism through which paracetamol inhibits COX-1 and COX-2 activities [67–70], has to date not been tested in vivo. We found that by increasing the intracellular lipid peroxide tone using T-butyl hydroperoxide in J774.2 macrophage cells, paracetamol was still able to inhibit the catalytic activity of COX-2 induced by diclofenac (Figure 5). In the same cell line, paracetamol did not inhibit the catalytic activity induced by the pro-inflammatory stimulus LPS with the intracellular lipid hydroperoxide tone remaining unchanged after the addition of LPS [71].
Figure 5.
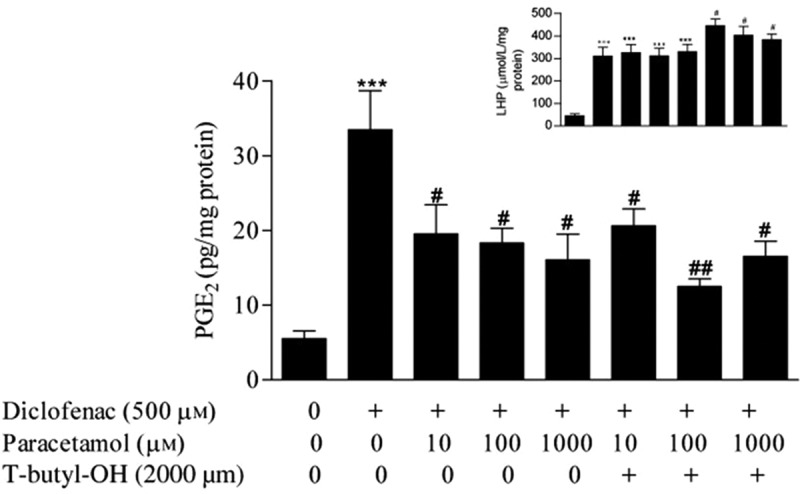
Elevation of the intracellular lipid hydroperoxide tone with T-butyl-OH does not antagonize the inhibitory effect of paracetamol on the diclofenac-induced cyclooxygenase-2 activity at 48 h in J774.2 macrophages. For the experimental protocol, refer to the methods section. ***P < 0.001 diclofenac-stimulated cells vs. unstimulated cells; #P < 0.05 and ##P < 0.01 diclofenac + paracetamol treatment ± T-butyl-OH vs. diclofenac-stimulated cells (ANOVA and Dunnett’s post hoc test). Inset: ***P < 0.001 cells stimulated with diclofenac ± 10, 100, 1000 µM paracetamol vs. unstimulated cells, # P < 0.05 cells stimulated with diclofenac and treated with T-butyl-OH and 10, 100 or 1000 µM paracetamol vs. cells stimulated with diclofenac. Reproduced from Ayoub et al. 2011 [72]
All the studies that provide evidence on the reducing property of paracetamol to explain the mechanism through which it inhibits COX-1 and COX-2 activities are entirely in vitro experiments in which the concentrations of reaction components are not necessarily representative of the concentrations found inside the cells in vivo. The cell-based assays reported by Boutaud et al. (2002) were performed to compare the inhibitory effect of paracetamol on COX-1 and COX-2 enzymes from different cell types [67]. This creates problems when considering the fact that the concentration of endogenous substrate, arachidonic acid, and perhaps other substances, apart from peroxides could be different in the different cell types. Assays based on purified COX-1 and COX-2 enzymes bear little resemblance to the in vivo conditions that the enzymes naturally exist in as the concentrations of co-factors required for the enzymatic activity are irrelevant [68]. The advantage that our experimental conditions offer is that we compared the interactions between paracetamol and COX-2 induced by two different stimuli in the same cell line under the same experimental conditions [71].
Activation of the serotonergic descending inhibitory pathway by paracetamol
Activation of the descending inhibitory serotonergic pathway by paracetamol has been proposed as a possible mechanism for the analgesic action of paracetamol in humans [72,73] and experimental animals [74–77]. This serotonergic pathway, which originates from the brain stem and terminates at the spinal cord dorsal horn, is important in the modulation of nociceptive signals. Decades of research have provided evidence on the significance of this pathway to mediate the analgesic action of paracetamol as it has been shown that selective blockade of particular serotonergic receptors to abolish the analgesic actions of paracetamol in acute models of pain. The serotonergic receptors that have been mostly implicated in these investigations include 5-HT1A [76–78] 5-HT3 [74,75,79,80] and 5-HT7 [81,82]. In addition, lesioning of the serotonergic bulbospinal pathways or depletion of serotonin levels has been shown to attenuate the analgesic action of paracetamol [83]. Activation of this pathway cannot fully explain the mechanism of analgesic action of paracetamol as it was shown that paracetamol did not have affinity for any of the serotonergic receptor types or subtypes or to any of the enzymes involved in the synthesis or degradation of serotonin [84]; hence, this interaction between paracetamol and the descending inhibitory serotonergic system is an indirect one and is perhaps a “by-product” of inhibition of a centrally expressed COX variant enzyme.
AM404 as a metabolite of paracetamol: A new introduction to the paracetamol dilemma
The metabolic pathways for paracetamol have been elucidated several decades back, nonetheless in 2005 Högestätt identified a new metabolite for paracetamol [85]. It was shown that when paracetamol-derived metabolite para-aminophenol, formed in the liver, enters the brain it conjugates with arachidonic acid through the action of fatty acid amidohydrolase (FAAH) to form N-(4-hydroxyphenyl)-arachidonamide (AM404). AM404 as a novel metabolite for paracetamol was shown to activate the endocannabinoid system and to also activate the transient receptor potential vanilloid-1 (TRPV-1) channel, both of which are involved in the modulation of pain signaling and proposed to mediate the analgesic action of paracetamol [86–90]. Prior to the work by Högestätt et al. (2005) AM404 was shown to have analgesic properties mediated through the inhibition of endocannabinoid reuptake, thereby by preventing the reuptake of anandamide from the synaptic cleft [91]. Experiments in which the conversion of p-aminophenol into AM404 has been interrupted using FAAH knockout mice or selective FAAH inhibitors, the analgesic actions of paracetamol were shown to be blocked [89,90,92,93]. The serotonergic system has also been shown to be targeted by the paracetamol-derived AM404 [94,95].
The potential interactions between paracetamol and TRP channels, in particular, the TRPV1 channel, have been reported by several groups. Mallet et al. (2010) showed attenuation of the paracetamol-induced analgesia in the formalin, tail immersion and von Frey tests of nociception in mice lacking FAAH and TRPV1 and that intracerebroventricular administration of the TRPV1 channel antagonist capsazepine to abolish the analgesic action of paracetamol [88]. In addition, the analgesic action of paracetamol was shown to be attenuated in FAAH knockout mice and in animals treated with the FAAH inhibitor URB937 in models of thermal hyperalgesia, chemical hyperalgesia, and mechanical allodynia [92]. Using patch clamp and calcium imaging techniques, Stueber et al. (2018) demonstrated TRPV1 activation by AM404 at concentrations below 1 µM in HEK 293 cells expressing human TRPV1 and in dorsal root ganglia neurons [96]. From a clinical perspective, paracetamol was shown to produce antinociception in a model of chemical nociception in individuals expressing a particular TRPV1 genetic polymorphism [97]. In support of a non-TRPV1 mediated action for AM404, in cultured hippocampal slices and microglial cells AM404 was shown to inhibit the LPS-mediated PGE2 production in a TRPV1 independent manner and was also able to decrease the expression of COX-2 protein [98]. Paracetamol-induced analgesia was not altered in mice lacking the TRPM8 channel in several animal models of pain [99].
Using similar experimental approaches to those reported above, we showed that the hypothermic action of paracetamol in mice is not mediated through AM404 and is not dependent on activation of the endocannabinoid or TRPV-1 pathways [100]. In addition, we showed that the hypothermic action of cannabinoid receptor-1 and TRPV-1 agonists is not mediated through the inhibition of prostaglandin synthesis demonstrating parallel rather than interdependent pathways [100].
We were the first to report on the presence of AM404 in human CSF and plasma samples following intravenous administration of 1 g paracetamol (in CSF samples of 14 of 26 patients at concentrations of 5.1–57.4 nmol.L−1) [101]. The clinical relevance of AM404 to the pharmacology of paracetamol in humans remain unclear, as the evidence to date on AM404 comes largely from animal studies. AM404 has been demonstrated in in vitro studies to inhibit reuptake of anandamide, block TRPV1 and inhibit COX-1 and COX-2 activities [86,98,102–105] in the high nanomolar to low micromolar concentration range, well above the concentrations were able to detect in human CSF [101]. Ex vivo animal studies have detected levels of AM404 in whole brain tissue equating to the low nanomolar range following systemic administration of standard analgesic doses of paracetamol in rats (300 mg.kg−1) [85]. Muramatsu et al. reported on the metabolism of paracetamol to AM404 in rats at doses of paracetamol similar to therapeutic doses in humans [16]. However, the authors report on a conversion rate of plasma paracetamol to AM404 of only 0.0013%, which would result in low concentrations expected to have negligible pharmacological activities. Our study reported on a relatively similar conversion rate of plasma paracetamol to AM404 CSF (0.009%) [106].
Research focussed on the role of other ion channels to mediate the pharmacological actions of paracetamol includes the work by Kerckhove et al. (2014) who demonstrated that the analgesic action of paracetamol was attenuated in mice in which the supraspinal calcium Cav3.2 channels are inhibited. Centrally administered AM404 resulted in antinociception which was lost in Ca(v)3.2(-/-) mice [107]. In addition, the reactive paracetamol metabolite N-acetyl-p-benzoquinone imine, which has been detected in the CNS, has recently been shown to reduce membrane excitability in rat dorsal root ganglion and spinal dorsal horn neurons accompanied by hyperpolarization resulting from increased currents through potassium KV7 channels [108].
Thermoregulatory action of paracetamol
Mechanistic research on the antipyretic and hypothermic actions of paracetamol
Paracetamol is widely used for its antipyretic action, particularly in children [109–111]. Similar to its analgesic action, the mechanism of antipyretic action of this drug remains poorly understood. Decades of research demonstrated that fever is generated when signaling molecules that include interleukin-6, interleukin-1β or tumor necrosis factor-α are released systemically following on from an inflammatory reaction in response to viral or bacterial pathogenic infections [112–114]. These molecules were shown to initiate a febrile response through activation of the vascular endothelial cells that line the pre-optic region of the hypothalamus, an important brain region in the regulation of body temperature [115–117]. These activated vascular endothelial cells respond through the induction of COX-2 (and not COX-1) that produces PGE2, which in turn resits the thermostatic control to a higher temperature [112,116,118]. LPS has been shown to work in a similar manner [118,119]. The resultant effector efferent signals drive the body toward temperature conservation, reduced heat loss, and increased heat generation [120]. Mice lacking the PGE2 receptor EP3 [112] or microsomal prostaglandin E synthase-1 enzyme [121,122] were shown to have impaired febrile responses. To demonstrate significance of central PGE2 in the development of fever, it was shown that selective deletion of the EP3 receptor in the median preoptic nucleus to abrogate the LPS-induced fever in mice [123].
Blockade of brain PGE2 synthesis by NSAIDs has been associated with the antipyretic action of these drugs, which has been suggested to be due to inhibition of inducible COX-2 produced by hypothalamic vascular endothelial cells [124,125]. It is therefore not surprising that SC560, a selective COX-1 inhibitor, does not exhibit antipyretic actions [125].
Inhibition of COX-2 by paracetamol as discussed earlier does not satisfactorily explain the mechanism of pharmacological actions of paracetamol, including its antipyretic action [33]. It is worth noting that in one of the earliest observations in which prostaglandin synthesis was shown to mediate fever, it was shown that paracetamol administered intraperitoneally was able to reduce body temperature and cerebrospinal fluid (CSF) prostaglandin E1 synthesis (collected from the third ventricle) within a short and insufficient timeframe to allow for the induction of COX-2 [126].
As NSAIDs are believed to induce antipyresis through inhibition of the inducible COX-2 enzyme expressed in hypothalamic vascular endothelial cells [112,127], Li et al. (2008) showed that the antipyretic (and hypothermic) actions of paracetamol were not attenuated in COX-1−/− mice in comparison to wild-type mice [128]. However, the antipyretic activity of paracetamol reported by these authors cannot be attributed to inhibition of inducible COX-2 protein as it was observed 1 h following on from LPS administration, which is insufficient time for the induction of COX-2 [129]. Engström et al. (2013) used COX-2+/− mice to study the mechanism of antipyretic action of paracetamol as COX-2−/− mice failed to develop fever to LPS. At a 50 mg/kg non-hypothermic dose, the antipyretic action of paracetamol was attenuated in COX-2± mice in comparison to COX-2+/+ mice [130]. The authors claim that by lowering the levels of COX-2 enzyme as is the case in COX-2± mice the sensitivity of inhibition of COX-2 by paracetamol increases. It is not clear how by losing one allele of the COX-2 gene would render paracetamol more effective at inhibition of the COX-2 enzyme.
Paracetamol is regarded generally speaking as a centrally acting temperature lowering drug [18,22,131] with the ability to temporally reduce brain PGE2 synthesis [22,132–134].
We recently showed that in COX-2-mediated endotoxin-induced fever model, paracetamol administered prophylactically and therapeutically was able to reduce fever with a concomitant reduction in hypothalamic PGE2 synthesis; effects that were both significantly attenuated in COX-1-/- mice when compared to littermate wild-type control mice [135]. Previously, we showed that the paracetamol-induced hypothermia in normothermic mice and concomitant reduction in brain PGE2 synthesis (Figure 6) were significantly attenuated in COX-1-/- mice in comparison to wild-type mice but not in COX-2-/- mice [22]. We concluded that the paracetamol-induced hypothermia and antipyresis are both mediated through inhibition of a COX-1 gene-derived enzyme a notion that is supported by the finding that therapeutically administered paracetamol-induced potent hypothermic and anti-pyretic actions in COX-1+/+ mice (with established pyrexia), which were partly lost in COX-1−/− mice. Similarly, the reduction of hypothalamic PGE2 synthesis by therapeutically administered paracetamol was significantly attenuated in COX-1-/- mice in comparison to littermate COX-1+/+ control mice (Figure 7) [135]. We, therefore, make the assumption that paracetamol reduces body temperature through the induction of hypothermia through inhibition of a constitutively expressed COX-1 variant enzyme [22,135]. The inhibition of PGE2 by hypothermic and antipyretic paracetamol is not attributed to inhibition of COX-1 since the selective COX-1 inhibitor SC560 and the dual COX-1/COX-2 inhibitor indomethacin at pharmacologically active doses [136–138], failed to induce hypothermia [135]. Selective inhibition of COX-2 by celecoxib does not result in hypothermia [135]. We therefore conclude that the target for the paracetamol-induced hypothermia is not COX-1 or COX-2 and is likely to be a variant of COX-1. A schematic representation of the action of paracetamol and NSAIDs on body temperature and the proposed mechanisms of action has been included in Figure 8.
Figure 6.
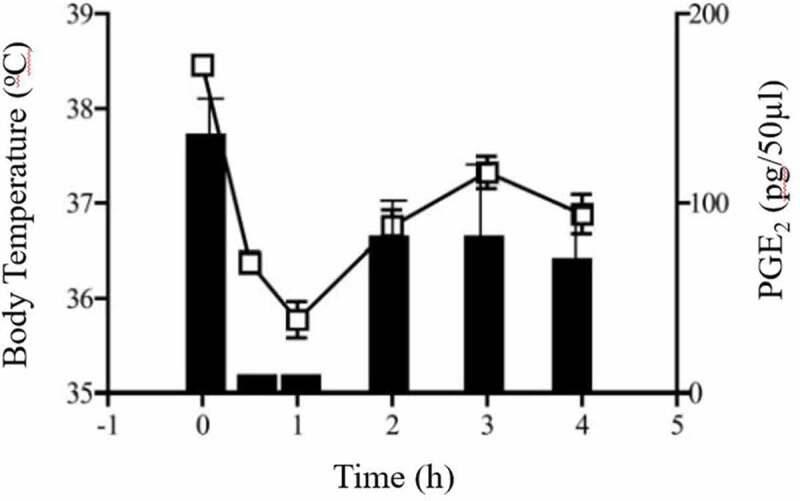
The reduction of basal body temperature (left y-axis) with 300 mg/kg paracetamol correlates with reduction of brain PGE2 levels (right y-axis) in male C57/BL6 mice. Reproduced from Ayoub et al. 2006 [23]
Figure 7.
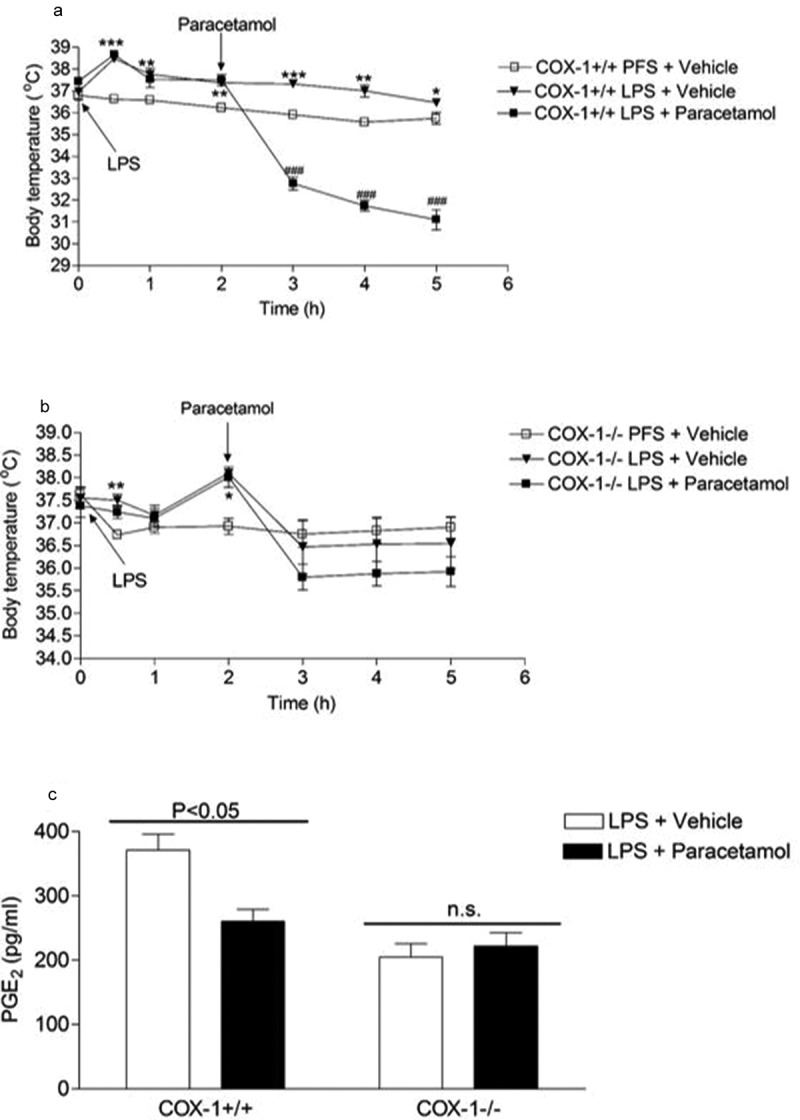
The antipyretic and inhibitory effect of therapeutically administered paracetamol on hypothalamic PGE2 synthesis was abolished in COX-1 knockout mice. The antipyretic effect of 200 mg/kg paracetamol administered subcutaneously 2 h after 10 µg/kg LPS was examined in COX-1+/+ (a) and COX-1−/- (b) mice. Panel C shows comparisons of the effect of therapeutically administered 200 mg/kg paracetamol on hypothalamic PGE2 levels 1 h after paracetamol administration. A: *P < 0.05, **P < 0.01 and ***P < 0.001 PFS and vehicle versus LPS and vehicle; ##P < 0.01 and ###P < 0.001 LPS and vehicle versus LPS and paracetamol. B: *P < 0.05 and **P < 0.01 PFS and vehicle versus LPS and vehicle; n = 4–5. Reproduced from Ayoub and Flower 2019 [130]
Figure 8.
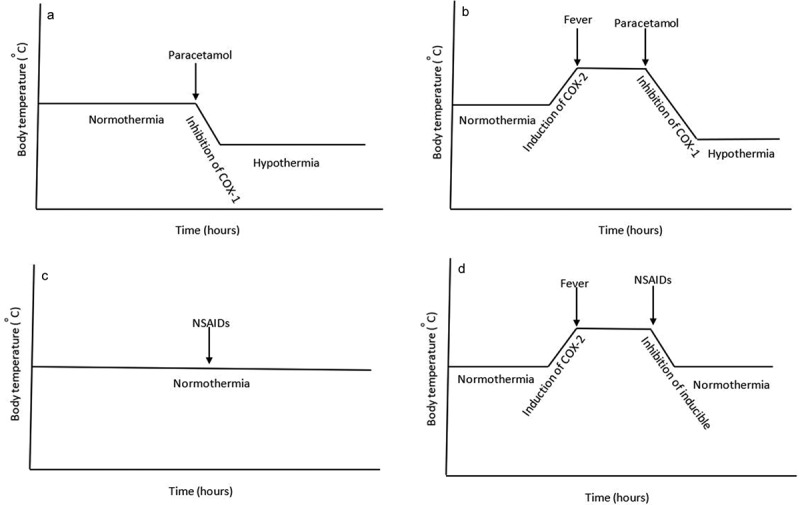
Proposed schematic representation of the effect of paracetamol and NSAIDs on body temperature. The paracetamol-induced hypothermia under normothermia (Panel A) and febrile conditions (Panel B) is proposed to be mediated through the inhibition of a hypothalamic COX-1 variant enzyme. Most NSAIDs are non-hypothermic (Panel C), but are able to reduce febrile temperature through inhibition of inducible hypothalamic COX-2 enzyme
Prophylactically administered paracetamol produced a similar magnitude of decrease in body temperature as therapeutically administered paracetamol in children in randomized controlled trials [110,139] supporting the notion that the antipyretic action of paracetamol is mediated through inhibition of a constitutively expressed enzyme. Additionally, given the fact that paracetamol is able to induce hypothermia and reduce brain PGE2 synthesis in the absence and presence of fever within 30 minutes after administration (insufficient time for the induction of COX-2) negates the notion that the thermoregulatory actions of paracetamol are mediated through inhibition of COX-2. Clinically, the paracetamol-induced hypothermia in humans has also been reported by several other research groups [140–145].
Our findings allude to a functional role for PGE2 in the maintenance of normothermia, despite the lack of substantial evidence to support this notion. Oka et al. (2003) showed that administration of EP1, EP3 and EP4 receptor agonists in the absence of LPS fever to induce an increase in body temperature and have suggested a counterregulatory role for the EP4 receptor [146], suggesting a significant role for PGE2 in the maintenance of normothermia mediated through the activation of different EP receptors.
The reported paracetamol-induced hypothermia in normothermic animals [22,38,135] and humans [140–145] is a reversable and nontoxic action that temporally correlates with the pharmacokinetic profile of paracetamol [22,135], which has been exploited therapeutically for the acute management of stroke in human adults alas with limited efficacy [144,145]. In mice paracetamol induces profound hypothermia (3°C); however, in humans paracetamol has been reported to induce very mild hypothermia (0.4°C), which is mostly related to the difference in body mass to surface area ration between mice and humans. In some reports, paracetamol has been demonstrated to induce hypothermia in children to temperatures below 35.6°C [109].
Identification of AM404 as a new metabolite for paracetamol [85] has prompted us to investigate the role of AM404 for the paracetamol-induced hypothermia. As AM404 has been proposed to work by activation of the endocannabinoid and TRPV1 systems, both of which when activated by cognate agonists result in hypothermia [147,148], we therefore measured hypothermia induced by paracetamol in mice lacking the cannabinoid receptor-1 (CB1R) receptor or TRPV1 channel. We found that paracetamol was able to produce a similar hypothermic response in the two transgenic lines when compared to their wild-type littermates [100]. The findings from these experiments were confirmed using selective pharmacological blockers of the CB1R and TRPV1 channel, which failed to prevent the development of hypothermia induced by paracetamol. We also showed that development of hypothermia by selective CB1R and TRPV1 agonists is not dependent on the inhibition of COX-1 or COX-2 enzymes. Using mice deficient of the enzyme FAAH, which has been shown to mediate the final step in the conversion of paracetamol into AM404, we demonstrated a similar hypothermic response by paracetamol in these mice in comparison to their littermate controls. Selective inhibition of FAAH with URB597 failed to prevent the development of hypothermia produced by paracetamol. Unlike reports by other researchers [149], in our study, exogenously administered AM404 failed to induce hypothermia. These findings demonstrate that the paracetamol-induced hypothermia is not mediated by AM404 and does not involve activation of the endocannabinoid or TRPV1 systems. Further support to this notion is provided by the work of Massey et al., (1982) who showed that direct intracerebroventricular administration of paracetamol to result in hypothermia within 20 minutes of administration [131]. Whilst supporting our conclusion on the absence of a functional role for TRPV1 in mediation of the hypothermic action of paracetamol, Gentry and colleagues (2015) provide evidence on a functional role for TRPA1, another member of the family of TRP channels [150].
Despite the fact that TRPV1 agonists are known to induce hypothermia as discussed earlier, not all TRPV1 antagonists were shown to induce hyperthermia [151]. In the report by Gavva et al. (2007) therapeutically administered 300 mg/kg paracetamol reduced the body temperature in rats in the absence and presence of the TRPV1 antagonist AMG8163 [152]. These data do not necessarily corroborate direct interactions with the TRPV1 channel to mediate the reduction in by temperature by paracetamol.
Corley (2009) showed that the cannabinoid and opioid systems are not involved in mediating the hypothermic action of paracetamol [153]. However, contrary to our conclusions on the association of PGE2 and the induction of hypothermia by cannabinoid drugs, Coupar and Taylor (1982) showed that administration of Δ9-tetrahydrocannabinol to reduce hypothalamic PGE2 synthesis in rats and that it correlated with the hypothermic action of this CB1R agonist [154].
Recent in vitro research suggested that the paracetamol-induced hypothermia is peripherally mediated and is depended on the inhibition of lipolysis in cultured adipocytes from brown adipose tissue, a process associated with thermogenesis [155]. Bashir et al. (2020) attempted to explain the mechanism of hypothermic action of paracetamol using adipocytes grown in culture and report on decrease of lipolysis with 1 and 10 mM of paracetamol.
It is noteworthy that the concentrations of paracetamol used by Bashir et al. (2020) are well above the plasma concentrations reported for paracetamol from pharmacological experiments in rodents. The 1 and 10 mM concentrations of paracetamol in these experiments [155] are well above the 700 µM (after 30 minutes administration) and 550 µM (after 1 hour of administration) plasma concentrations for the 200 mg/kg dose of paracetamol as reported by us [135]. Thus, despite being nontoxic to the cells, the concentrations of paracetamol used by these authors are pharmacologically irrelevant and do not represent plasma paracetamol concentrations in humans or rodents [31,135]. Such concentrations of paracetamol, thus represent a misinterpretation of the 200 mg/kg dose use by us [22,135,156] and others [83]. The authors also go on reference Fischer et al. (1981) in which the dose of paracetamol used to study the pharmacokinetics of a toxic dose of paracetamol in mice was 500 mg/kg [157], well above the 200 mg/kg and 300 mg/kg doses used by us [22,100,135]. Additionally, the authors provide justifications for the concentrations of paracetamol used on the basis of research by Orbach (2017), which is an entirely in vitro research [158].
Bashir et al. (2020) attempt to the challenge the notion that the paracetamol-induced hypothermia is mediated through the inhibition of a COX-1 variant (and other COX enzymes including COX-1 and COX-2), but present no data to support this argument [155]. In actual fact, the authors state that COX-3 has never been detected in human tissues, contrary to the work by Qin et al. (2005) [42]. The authors go on to extend their conclusion on the inhibition of lipolysis to explain the mechanism of action of NSAIDs and reference research that was conducted entirely in vitro, one of which is work done on renal tissues that represents toxicological as opposed to pharmacological actions of NSAIDs [159,160].
The evidence that support the paracetamol-induced hypothermia and indeed the paracetamol-induced antipyresis and analgesia are centrally mediated is supported by a large volume of published literature that spans 6 decades [18–23,27,28,96,131–135], which comes from direct in vivo experiments in which the pharmacological actions of paracetamol are directly correlated with reduction in PGE2 synthesis in the CNS and in some papers with clear absence of peripheral actions. As an example, the work by Feldberg (1973) in which anti-pyresis was correlated by direct and real-time reduction in brain prostaglandin E1 synthesis [132]. One of the key observations that demonstrate a central mechanism of the hypothermic action of paracetamol is the induction of hypothermia with intracerebroventricularly administered paracetamol [131,161].
Our in vivo research demonstrates that induction of hypothermia is not dependent on the ambient temperature [22,135]. We found paracetamol to be able to induce comparable hypothermia in mice housed below their thermoneutral temperature (22°C), in which heat loss exceeds heat gain and at their thermoneutral temperature zone (30°C) in which heat gain and loss are equal. Bashir et al. (2020) argue state that freshly isolated brown adipocytes to have high level of basal lipolysis as a result of the cells being freshly isolated from mice and is attributed to the cells acclimatizing to ambient temperature, below the animals’ thermoneutral zone. It is noteworthy that such thermoregulatory changes are whole organism modifications and are centrally mediated [120].
Is paracetamol a sleep-inducing drug?
As a widely available over-the-counter drug, paracetamol is known to be used for purposes other than for its analgesic and antipyretic actions. This include the use of paracetamol for the induction of sleep, which is based on anecdotal personal experiences [162]. It is logical to reason that such sleep promoting action by paracetamol is a consequence of improvement of the patients’ pain experience or is merely a placebo effect. Pilot-controlled clinical trials failed to demonstrate a positive correlation between paracetamol administration and improvement of sleep [163–165]. Considering the thermoregulatory actions of paracetamol are believed to be mediated through inhibition of PGE2 within the hypothalamus, it is thought provoking to reason that paracetamol might have mild sleeping inducing properties, particularly when bearing in mind the fact that PGE2 is known to induce wakefulness [166–168] inhibition of which would promote sleepiness. It is feasible to believe that paracetamol affects common neuronal circuitry mechanisms within the hypothalamus that regulate sleep and body temperature, a paradigm that would be worth further investigation (Reference 169 provides an overview on the CNS neural circuitry and role of prostaglandins in the development of sickness syndrome/behavior that includes fever and increased sleepiness). Indeed, the suprachiasmatic nucleus within the anterior medial zone of the hypothalamus is known to be involved in circadian control of sleep-wake cycle and body temperature.
Final remarks and future considerations on paracetamol
Owing to the fatal hepatotoxicity associated with paracetamol over-dose [170,171], it has been debated whether paracetamol should be withdrawn from the market or to be re-classified. At therapeutic dose paracetamol is a safe drug, but with a narrow therapeutic window, it is easy to accidently or deliberately over-dose. Such debates between clinicians, scientists and drug regulators have been ongoing for some time with the general population rarely being involved in such dialogs. It is predicted that withdrawal of paracetamol from global markets or even its re-classification would not be well received by the general population. As on over-the-counter, most people self-medicate with paracetamol for the management of acute/mild pain and fever. Paracetamol became particularly important during the current global SARS-CoV-2 pandemic as ibuprofen, another over-the-counter analgesic antipyretic drug, was initially contraindicated for these patients [172], a notion that was later rejected [173,174]. Undoubtedly, paracetamol holds a unique place as a familiar and widely used analgesic antipyretic drug which for many decades has puzzled pharmacologists in regards to its mechanism of pharmacological actions (Prevailing theories on the mechanisms of pharmacological actions discussed in this review are summarized in Figure 9). From a clinical perspective, withdrawal of paracetamol from the market would leave a void for the management of mild pain and fever whether through physicians recommendation or patients’ own self-medication endeavors and the search for a drug to replace paracetamol may be the way ahead, but equally not necessarily provide a safer alternative. It is worth remembering that a wealth of knowledge on the paracetamol-induced toxicity has accumulated over the many decades of clinical use. Several measures have been put in place to help reduce the paracetamol-induced toxicity that include limits on package size, which has had limited impact [175,176]. Therefore, more steps to help prevent overdosing with paracetamol are needed. Such steps may include helping to provide general awareness on the risks linked to overdosing with paracetamol [177–179]. From a pharmacological perspective, the search for the molecular target for paracetamol continues, which may provide us with a new way to treat pain and fever in the future.
Figure 9.
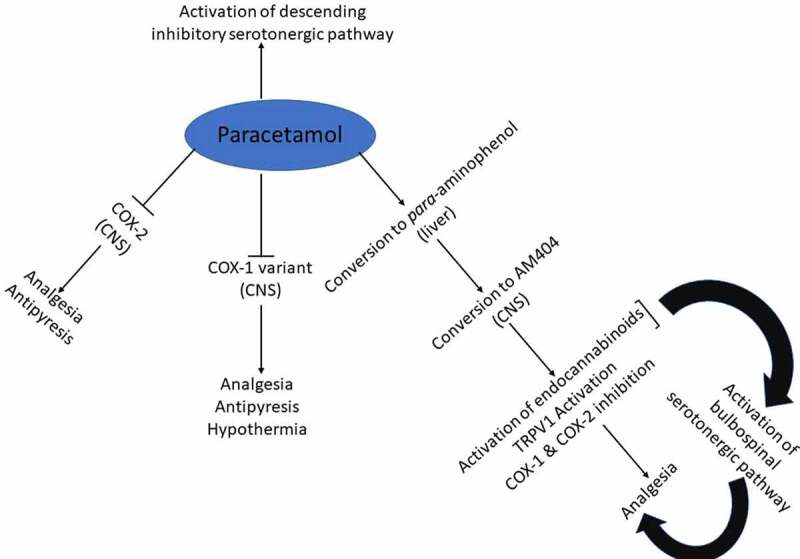
Schematic representation of the prevailing theories on the mechanisms of pharmacological actions of paracetamol discussed in this review
Biography
Dr Ayoub is a Senior Lecturer in Pharmacology at the University of East London. He acquired his PhD training at the William Harvey Research Institute, London, which was followed by a post-doctoral post funded by the Leverhulme Trust as an Early Career Fellowship; during which time he was also a visiting scientist at Professor Daniel Simmons’s laboratory at the Brigham Young University, USA. Before joining the University of East London, Dr Ayoub worked as a Research Associate at the Institute of Heart and Lung, Imperial College London.
Dr Ayoub’s research focus has been on the pharmacology of paracetamol and the non-steroidal anti-inflammatory drugs in inflammation, pain and thermoregulation and has been the first to provide substantial evidence on implicating cyclooxygenase variant enzymes in mediating the thermoregulatory and analgesic actions of paracetamol. Dr Ayoub continues to be active in this area of research as well as in elucidating the role of interleukin-4 and cycloogenase-2 in the resolution of inflammation and macrophage polarization. In recent years, Dr Ayoub has been collaborating with colleagues at the University of East London in research aimed at elucidation of mechanisms involved in the development of addiction behaviors to alcohol in Drosophila.
Disclosure statement
No potential conflict of interest was reported by the author.
References
- [1].Von Mering J. Beiräge zur Kenntniss der Antipyretica. Therap. Monatsch.1893;7:558–577. [Google Scholar]
- [2].Brodie BB, Axelrod J. The fate of acetophenetidin (phenacetin) in man and methods for the estimation of acetophenetidin and its metabolites in biological material. J Pharmacology Exp Ther.1949;97:58–67. [PubMed] [Google Scholar]
- [3].Schrör K. Aspirin and Reye syndrome: a review of the evidence. Paediatr Drugs. 2007;9(3):195–204. [DOI] [PubMed] [Google Scholar]
- [4].Prescott LF. Paracetamol: past, present and future. Am J Ther. 2000;7:143–147. [PubMed] [Google Scholar]
- [5].Hider-Mlynarz K, Cavalié P, Maison P. Trends in analgesic consumption in France over the last 10 years and comparison of patterns across Europe. Br J Clin Pharmacol. 2018;84(6):1324–1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Wober C, Wober-Bingol C. Clinical management of young patients presenting with headache. Funct Neurol. 2000;15(3): 89–105. Suppl. [PubMed] [Google Scholar]
- [7].Woo WW, Man SY, Lam PK, et al. Randomized double-blind trial comparing oral paracetamol and oral nonsteroidal anti-inflammatory drugs for treating pain after musculoskeletal injury. Ann Emerg Med. 2005;46(4):352–361. [DOI] [PubMed] [Google Scholar]
- [8].Pendergrass PB, Scott JN, Ream LJ, et al. Effect of small doses of aspirin and acetaminophen on total menstrual loss and pain of cramps and headache. Gynecol Obstet Invest. 1985;19(1):32–37. [DOI] [PubMed] [Google Scholar]
- [9].Hochberg MC, Dougados M. Pharmacological therapy of osteoarthritis. Best Pract Res Clin Rheumatol. 2001;15(4):583–593. [DOI] [PubMed] [Google Scholar]
- [10].Porter RW, Ralston SH. Pharmacological management of back pain syndromes. Drugs. 1994;48(2):189–198. [DOI] [PubMed] [Google Scholar]
- [11].Dionne RA, Campbell RA, Cooper SA, et al. Suppression of postoperative pain by preoperative administration of ibuprofen in comparison to placebo, acetaminophen, and acetaminophen plus codeine. J Clin Pharmacol. 1983;23:37–43. [DOI] [PubMed] [Google Scholar]
- [12].Haglund B, Von Bültzingslöwen I. Combining paracetamol with a selective cyclooxygenase‐2 inhibitor for acute pain relief after third molar surgery: a randomized, double‐blind, placebo‐controlled study. Eur J Oral Sci. 2006;114(4):293–301. [DOI] [PubMed] [Google Scholar]
- [13].Dahl V, Raeder JC. Non-opioid postoperative analgesia. Acta Anaesthesiol. Scand. 2000;44:1191–1203. [DOI] [PubMed] [Google Scholar]
- [14].HKH L, SM T, FL L. A randomised control trial comparing the efficacy of tramadol and paracetamol against ketorolac and paracetamol in the management of musculoskeletal pain in the emergency department.J Emerg Crit Care Med. 2008;15(1):5–11. [Google Scholar]
- [15].Moore RA, et al. Minimum efficacy criteria for comparisons between treatments using individual patient meta-analysis of acute pain trials: examples of etoricoxib, paracetamol, ibuprofen, and ibuprofen/paracetamol combinations after third molar extraction. Pain. 2011;152(5):982–989. [DOI] [PubMed] [Google Scholar]
- [16].Flower RJ, Vane JR. Inhibition of prostaglandin synthetase in brain explains the anti-pyretic activity of paracetamol (4-acetamidophenol). Nature. 1972;240:410–411. [DOI] [PubMed] [Google Scholar]
- [17].Vane JR. Inhibition of prostaglandin synthesis as a mechanism of action for aspirin-like drugs. Nature New Biol.1971;231:232–235. [DOI] [PubMed] [Google Scholar]
- [18].Feldberg W, Gupta KP, Milton AS, et al. Effect of bacterial pyrogen and antipyretics on prostaglandin activity in cerebrospinal fluid of unanaesthetised cats. BrJ Pharmacol. 1972;46:550P-551P. [PMC free article] [PubMed] [Google Scholar]
- [19].Yaksh TL, Malmberg AB. Spinal actions of NSAIDs in blocking spinally mediated hyperalgesia: the role of cyclooxygenase products. Agents Actions. 1993;41:89–100. [PubMed] [Google Scholar]
- [20].Malmberg AB, Yaksh TL. Capsaicin-evoked prostaglandin E2 release in spinal cord slices: relative effect of cyclooxygenase inhibitors. Eur J Pharmacol. 1994;271(2–3):293–299. [DOI] [PubMed] [Google Scholar]
- [21].Muth-Selbach US, Tegeder I, Brune K, et al. Acetaminophen inhibits spinal prostaglandin E2 release after peripheral noxious stimulation. Anesthesiology. 1999;91:231–239. [DOI] [PubMed] [Google Scholar]
- [22].Ayoub SS, Botting RM, Goorha S, et al. Acetaminophen-induced hypothermia in mice is mediated by a prostaglandin endoperoxide synthase 1 gene-derived protein. Proc Natl Acad Sci U S A. 2004;101:11165–11169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Ayoub SS, Colville-Nash PR, Willoughby DA, et al. The involvement of a cyclooxygenase 1 gene-derived protein in the antinociceptive action of paracetamol in mice. Eur J Pharmacol. 2006;538(1–3):57–65. [DOI] [PubMed] [Google Scholar]
- [24].Derendorf H, Drehsen G, Rohdewald P. Cortical-evoked potentials and saliva levels as basis for the comparison of pure analgesics to analgesic combinations. Pharmacology. 1982;25(4):227–236. [DOI] [PubMed] [Google Scholar]
- [25].Bromm B, Forth W, Richter E, et al. Effects of acetaminophen and antipyrine on non-inflammatory pain and EEG activity. Pain. 1992;50(2):213–221. [DOI] [PubMed] [Google Scholar]
- [26].Pickering G, Kastler A, Macian N, et al. The brain signature of paracetamol in healthy volunteers: a double-blind randomized trial. Drug Des Devel Ther. 2015;9:3853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Shetty N1, AK P, SV G, et al. Comparison of the effects of ibuprofen and acetaminophen on PGE2 levels in the GCF during orthodontic tooth movement: a human study. Prog Orthod. 2013;14:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Seppälä E, Laitinen O, Vapaatalo H. Comparative study on the effects of acetylsalicylic acid, indomethacin and paracetamol on metabolites of arachidonic acid in plasma, serum and urine in man. Int J Clin Pharmacol Res. 1983;3(4):265–269. [PubMed] [Google Scholar]
- [29].ClissoldS P. Paracetamol and phenacetin. Drugs. 1986;32(4): 46–59. Suppl. [DOI] [PubMed] [Google Scholar]
- [30].Gwilt JR, Robertson A, Goldman L, et al. The absorption characteristics of paracetamol in man. J. Pharm Pharmacol.1963;15:445. [DOI] [PubMed] [Google Scholar]
- [31].Langford RA, Hogg M, Bjorksten AR, et al. Comparative plasma and cerebrospinal fluid pharmacokinetics of paracetamol after intravenous and oral administration. Anesth Analg. 2016;123(3):610–615. [DOI] [PubMed] [Google Scholar]
- [32].Appleton I. Non-steroidal anti-inflammatory drugs and pain. In: The pharmacology of pain. Berlin, Heidelberg: Springer; 1997. p. . 43–60. [Google Scholar]
- [33].Mitchell JA, Akarasereenont P, Thiemermann C, et al. Selectivity of non-steroidal anti-inflammatory drugs as inhibitors of constitutive and inducible cyclooxygenase. proc. Natl. Acad. Sci. U S A.1993;90:11693–11697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Botting RM. Mechanism of action of acetaminophen: is there a cyclooxygenase 3? Clin Infect Dis. 2000;31(5): S202‐S210. Suppl. [DOI] [PubMed] [Google Scholar]
- [35].Willoughby DA, Moore AR, Colville-Nash PRCOX-1. COX-2, and COX-3 and the future treatment of chronic inflammatory disease. Lancet. 2000;355(9204):646‐648. [DOI] [PubMed] [Google Scholar]
- [36].Chandrasekharan NV, Dai H, Roos KL, et al. a cyclooxygenase-1 variant inhibited by acetaminophen and other analgesic/antipyretic drugs: cloning, structure and expression. Proc Natl Acad Sci U S A. 2002;99:13926–13931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Reinauer C, Censarek P, Kaber G, et al. Expression and translation of the COX-1b gene in human cells–no evidence of generation of COX-1b protein. Biol Chem. 2013;394(6):753–760. [DOI] [PubMed] [Google Scholar]
- [38].Li S, Dou W, Tang Y, et al. Acetaminophen: antipyretic or hypothermic in mice? In either case, PGHS-1b (COX-3) is irrelevant. Prostaglandins Other Lipid Mediat. 2008;85(3–4):89–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Kis B, Snipes JA, Gaspar T, et al. Cloning of cyclooxygenase-1b (putative COX-3) in mouse. Inflamm Res. 2006;55(7):274–278. [DOI] [PubMed] [Google Scholar]
- [40].Snipes JA, Kis B, Shelness GS, et al. Cloning and characterization of cyclooxygenase-1b (putative cyclooxygenase-3) in rat. J Pharmacol Exp Ther.2005;313(2):668–676. [DOI] [PubMed] [Google Scholar]
- [41].Schwab JM, Beiter T, Linder JU, et al. COX-3-a virtual pain target in humans. Faseb J. 2003;17:2174–2175. [DOI] [PubMed] [Google Scholar]
- [42].Qin N, Zhang SP, Reitz TL, et al. Cloning, expression, and functional characterization of human cyclooxygenase-1 splicing variants: evidence for intron 1 retention. J Pharmacol Exp Ther.2005;315(3):1298–1305. [DOI] [PubMed] [Google Scholar]
- [43].Ayoub SS, Yazid S, Flower RJ. Increased susceptibility of annexin‐A1 null mice to nociceptive pain is indicative of a spinal antinociceptive action of annexin‐A1. Br J Pharmacol. 2008;154(5):1135–1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Ayoub SS, Wood EG, Hassan SU, et al. Cyclooxygenase expression and prostaglandin levels in central nervous system tissues during the course of chronic relapsing experimental autoimmune encephalomyelitis (EAE). Inflamm Res. 2011;60(10):919. [DOI] [PubMed] [Google Scholar]
- [45].Ayoub SS, Botting RM. Iloprost-induced nociception: determination of the site of anti-nociceptive action of cyclooxygenase inhibitors and the involvement of cyclooxygenase products in central mechanisms of nociception. In: Ayoub SS, Flower RJ, Seed M, editors. Cyclooxygenases. US: Humana Press; 2010. p. 207–221. [DOI] [PubMed] [Google Scholar]
- [46].Ku EC, Wasvary JM, Cash WD. Diclofenac sodium (GP 45840, Voltaren), a potent inhibitor of prostaglandin synthetase. Biochem Pharmacol. 1975;24(5):641–643. [DOI] [PubMed] [Google Scholar]
- [47].Tonussi CR, Ferreira SH. Mechanism of diclofenac analgesia: direct blockade of inflammatory sensitisation. Eur J Pharmacol. 1994;14(2–3):73–179. [DOI] [PubMed] [Google Scholar]
- [48].Gyires K, Knoll J. Inflammation and writhing syndrome inducing effect of PGE1, PGE2 and the inhibition of these actions. Pol J Pharmacol Pharm. 1975;27:257–264. [PubMed] [Google Scholar]
- [49].Murata T, Ushikubi F, Matsuoka T, et al. Altered pain perception and inflammatory response in mice lacking prostacyclin receptor. Nature. 1997;388:678–682. [DOI] [PubMed] [Google Scholar]
- [50].Horiguchi S, Ueno R, Hyodo M, et al. Alterations in nociception after intracisternal administration of prostaglandin D2, E2, or F2α to conscious mice. Eur J Pharmacol. 1986;122:173–179. [DOI] [PubMed] [Google Scholar]
- [51].Hinz B, Cheremina O, Brune K. Acetaminophen (paracetamol) is a selective cyclooxygenase-2 inhibitor in man. Faseb J. 2007;22:383–390. [DOI] [PubMed] [Google Scholar]
- [52].Sciulli MG, Seta F, Tacconelli S, et al. Effects of acetaminophen on constitutive and inducible prostanoid biosynthesis in human blood cells. Br J Pharmacol. 2003;138(4):634–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Solomon DH. Selective cyclooxygenase 2 inhibitors and cardiovascular events. Arthritis Rheum. 2005;52(7):1968–1978. [DOI] [PubMed] [Google Scholar]
- [54].Ott E, Nussmeier NA, Duke PC, et al. The multicenter study of perioperative ischemia (McSPI) research group, and the ischemia research and education foundation (IREF) Investigators. efficacy and safety of the cyclooxygenase 2 inhibitors parecoxib and valdecoxib in patients undergoing coronary artery bypass surgery. J Thorac Cardiovasc Surg. 2003;125:1481–1492. [DOI] [PubMed] [Google Scholar]
- [55].Bombardier C, Laine L, Reicin A, et al. The VIGOR study group. VIGOR study group: comparison of upper gastrointestinal toxicity of rofecoxiband naproxen in patients with rheumatoid arthritis. N Engl J Med. 2000;343:1520–1528. [DOI] [PubMed] [Google Scholar]
- [56].Roberts E, Nunes VD, Buckner S, et al. Paracetamol: not as safe as we thought? A systematic literature review of observational studies. Ann Rheum Dis. 2016;75(3):552–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Graham GG, IG R, Day RO. Comparative analgesia, cardiovascular and renal effects of celecoxib, rofecoxib and acetaminophen (paracetamol). Curr Pharm Des. 2002;8(12):1063–1075. [DOI] [PubMed] [Google Scholar]
- [58].Skjelbred P, Løkken P. Paracetamol versus placebo: effects on post-operative course. Eur J Clin Pharmacol. 1979;15(1):27–33. [DOI] [PubMed] [Google Scholar]
- [59].Bjørnsson GA, Haanaes HR, Skoglund LA. A randomized, double‐blind crossover trial of paracetamol 1000 mg four times daily vs ibuprofen 600 mg: effect on swelling and other postoperative events after third molar surgery. Br J Clin Pharmacol. 2003;55(4):405–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Boardman PL, Hart FD. Clinical measurement of the anti-inflammatory effects of salicylates in rheumatoid arthritis. BMJ. 1967;4(5574):264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Ring EF, Collins AJ, Bacon PA, et al. 1974. Quantitation of thermography in arthritis using multi-isothermal analysis. II. Effect of nonsteroidal anti-inflammatory therapy on the thermographic index. Ann Rheum Dis. 1974;33(4):353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Seppälä E, Nissilä M, Isomäki H, et al. Comparison of the effects of different anti-inflammatory drugs on synovial fluid prostanoid concentrations in patients with rheumatoid arthritis. Clin Rheumatol. 1985;4(3):315. [DOI] [PubMed] [Google Scholar]
- [63].Moore PK, Marshall M. Nitric oxide releasing acetaminophen (introacetaminophen). Dig Liver Dis. 2003;35(2): S49–S60. Suppl. [DOI] [PubMed] [Google Scholar]
- [64].Ballou LR, Botting RM, Goorha S, et al. Nociception in cyclooxygenase isoenzyme-deficient mice. Proc Natl Acad Sci U S A. 2000;97:10272–10276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Hanel AM, Lands WE. Modification of anti-inflammatory drug effectiveness by ambient lipid peroxides. Biochem Pharmacol. 1982;31:3307–3311. [DOI] [PubMed] [Google Scholar]
- [66].Marnett LJ. Cyclooxygenase mechanisms. Curr Opin Chem Biol. 2000;4(5):545–552. [DOI] [PubMed] [Google Scholar]
- [67].Boutaud O, Aronoff DM, Richardson JH, et al. Determinants of the cellular specificity of acetaminophen as an inhibitor of prostaglandin H(2) synthases. Proc Natl Acad Sci U S A. 2002;99:7130–7135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Ouellet M, Percival MD. Mechanism of acetaminophen inhibition of cyclooxygenase isoforms. Arch Biochem Biophys. 2001;387:273–280. [DOI] [PubMed] [Google Scholar]
- [69].Lucas R, Warner TD, Vojnovic I, et al. Cellular mechanisms of acetaminophen: role of cyclo-oxygenase. Faseb J. 2005;19:635–637. [DOI] [PubMed] [Google Scholar]
- [70].Schildknecht S, Daiber A, Ghisla S, et al. Acetaminophen inhibits prostanoid synthesis by scavenging the PGHS-activator peroxynitrite. Faseb J. 2008;22:215–224. [DOI] [PubMed] [Google Scholar]
- [71].Ayoub SS, Joshi A, Chol M, et al. Inhibition of the diclofenac‐induced cyclooxygenase‐2 activity by paracetamol in cultured macrophages is not related to the intracellular lipid hydroperoxide tone. Fundam Clin Pharmacol. 2011;25(2):186–190. [DOI] [PubMed] [Google Scholar]
- [72].Pickering G, Loriot MA, Libert F, et al. Analgesic effect of paracetamol in humans: first evidence of a central serotonergic mechanism. Clin Pharmacol Ther. 2006;79:371–378. [DOI] [PubMed] [Google Scholar]
- [73].Pickering G, Estève V, Loriot MA, et al. Paracetamol reinforces descending inhibitory pain pathways. Clin Pharmacol Ther. 2008;84:47–51. [DOI] [PubMed] [Google Scholar]
- [74].Alloui A, Pelissier T, Dubray C, et al. Tropisetron inhibits the antinociceptive effect of intrathecally administered paracetamol and serotonin. Fundam Clin Pharmacol. 1996;10:406–407. [DOI] [PubMed] [Google Scholar]
- [75].Girard P, Pansart Y, M-C C, et al. Modulation of paracetamol and nefopam antinociception by serotonin 5-HT3 receptor antagonists in mice. Pharmacology. 2009;83:243–246. [DOI] [PubMed] [Google Scholar]
- [76].Yogita SK, Peeyush B, Aditi P. Effect of drugs modulating serotonergic system on the analgesic action of paracetamol in mice. Indian J Pharmacol. 2016;48(3):281–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Roca-Vinardell A, Berrocoso E, Llorca-Torralba M, et al. Involvement of 5-HT1A/1B receptors in the antinociceptive effect of paracetamol in the rat formalin test. Neurobiol Pain. 2018;3:15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Bonnefont J, Chapuy E, Clottes E, et al. Spinal 5-HT1A receptors differentially influence nociceptive processing according to the nature of the noxious stimulus in rats: effect of WAY-100635 on the antinociceptive activities of paracetamol, venlafaxine and 5-HT. Pain. 2005;114(3):482–490. [DOI] [PubMed] [Google Scholar]
- [79].Pelissier T, Alloui A, Caussade F, et al. Paracetamol exerts a spinal antinociceptive effect involving an indirect interaction with 5-hydroxytryptamine3-receptors: in vivo and in vitro evidence. J Pharmacol Exp Ther. 1996;278:8–14 [PubMed] [Google Scholar]
- [80].Alloui A, Chassaing C, Schmidt J, et al. Paracetamol exerts a spinal, tropisetron-reversible, antinociceptive effect in an inflammatory pain model in rats. Eur J Pharmacol. 2002;443(1–3):71–77. [DOI] [PubMed] [Google Scholar]
- [81].Dogrul A, Seyrek M, AkgulEO, et al. Systemic paracetamol-induced analgesic and antihyperalgesic effects through activation of descending serotonergic pathways involving spinal 5-HT7 receptors. Eur J Pharmacol. 2012;677(1–3):93–101. [DOI] [PubMed] [Google Scholar]
- [82].Liu J, ReidA R, Sawynok J. Antinociception by systemically-administered acetaminophen (paracetamol) involves spinal serotonin 5-HT7 and adenosine A1 receptors, as well as peripheral adenosine A1 receptors. Neurosci Lett. 2013;536:64–68. [DOI] [PubMed] [Google Scholar]
- [83].Pini LA, Sandrini M, Vitale G. The antinociceptive action of paracetamol is associated with changes in the serotonergic system in the rat brain. Eur J Pharmacol. 1996;308(1):31–40. [DOI] [PubMed] [Google Scholar]
- [84].Raffa RB, Codd EE. Lack of binding of acetaminophen to 5-HT receptor or uptake sites (or eleven other binding/uptake assays). Life Sci. 1996;59(2):PL37-PL40. [DOI] [PubMed] [Google Scholar]
- [85].Högestätt ED, Jönsson BA, Ermund A, et al. Conversion of acetaminophen to the bioactive N-acylphenolamine AM404 via fatty acid amide hydrolase-dependent arachidonic acid conjugation in the nervous system. J Biol Chem. 2005;280(36):31405‐31412. [DOI] [PubMed] [Google Scholar]
- [86].Costa B, Siniscalco D, Trovato A, et al. AM404, an inhibitor of anandamide uptake, prevents pain behaviour and modulates cytokine and apoptotic pathways in a rat model of neuropathic pain. Br J Pharmacol. 2006;148(7):1022–1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Barrière DA, Mallet C, Blomgren A, et al. Fatty acid amide hydrolase-dependent generation of antinociceptive drug metabolites acting on TRPV1 in the brain. PLoS One. 2013;8(8):e70690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Mallet C, Barrière DA, Ermund A, et al. TRPV1 in brain is involved in acetaminophen-induced antinociception. PLoS One. 2010;5(9):e12748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Soukupová M, Palazzo E, De Chiaro M, et al. Effects of URB597, an inhibitor of fatty acid amide hydrolase (FAAH), on analgesic activity of paracetamol. Neuro Endocrinol Lett. 2010;31(4):507‐511. [PubMed] [Google Scholar]
- [90].Ottani A, Leone S, Sandrini M, et al. The analgesic activity of paracetamol is prevented by the blockade of cannabinoid CB1 receptors. Eur J Pharmacol. 2006;531(1–3):280‐281. [DOI] [PubMed] [Google Scholar]
- [91].Kathuria FD, MercierR S, Li C, et al. Anandamide transport is independent of fatty-acid amide hydrolase activity and is blocked by the hydrolysis-resistant inhibitor AM1172. Proc Natl Acad Sci U S A. 2004;101(23):8756–8761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Dalmann R, Daulhac L, Antri M, et al. Supra-spinal FAAH is required for the analgesic action of paracetamol in an inflammatory context. Neuropharmacology. 2015;91:63‐70. [DOI] [PubMed] [Google Scholar]
- [93].Barrière DA, Boumezbeur F, Dalman R, et al. Acetaminophen, a centrally‐acting analgesic involving the periaqueductal grey. Br J Pharmacol. 2020;77(8):1773–1792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Mallet C, Daulhac L, Bonnefont J, et al. Endocannabinoid and serotonergic systems are needed for acetaminophen-induced analgesia. Pain. 2008;139(1):190‐200. [DOI] [PubMed] [Google Scholar]
- [95].Ruggieri V, Vitale G, PiniL A, et al. Differential involvement of opioidergic and serotonergic systems in the antinociceptive activity of N-arachidonoyl-phenolamine (AM404) in the rat: comparison with paracetamol. Naunyn Schmiedebergs Arch Pharmacol. 2008;377(3):219‐229. [DOI] [PubMed] [Google Scholar]
- [96].Stueber T, Meyer S, Jangra A, et al. Activation of the capsaicin-receptor TRPV1 by the acetaminophen metabolite N-arachidonoylaminophenol results in cytotoxicity. Life Sci. 2018;194:67–74. [DOI] [PubMed] [Google Scholar]
- [97].Pickering G, Creveaux I, Macian N, et al. Paracetamol and pain modulation by TRPV1, UGT2B15, SULT1A1 genotypes: a randomized clinical trial in healthy volunteers. Pain Med. 2020;21(4):661–669. [DOI] [PubMed] [Google Scholar]
- [98].Zygmunt PM, Chuang -H-H, Movahed P, et al. The anandamide transport inhibitor AM404 activates vanilloid receptors. Eur J Pharmacol. 2000;396(1):39–42. [DOI] [PubMed] [Google Scholar]
- [99].Liu B, Fan L, Balakrishna S, et al. TRPM8 is the principal mediator of menthol-induced analgesia of acute and inflammatory pain. Pain. 2013. Oct;154(10):2169–2177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Ayoub SS, Pryce G, Seed MP, et al. Paracetamol-induced hypothermia is independent of cannabinoids and transient receptor potential vanilloid-1 and is not mediated by AM404. Drug Metab Dispos. 2011;39(9):1689–1695. [DOI] [PubMed] [Google Scholar]
- [101].Sharma CV, Long JH, Shah S, et al. First evidence of the conversion of paracetamol to AM404 in human cerebrospinal fluid. J Pain Res. 2017;10:2703‐2709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Beltramo M, Stella N, Calignano A, et al. Functional role of high-affinity anandamide transport, as revealed by selective inhibition. Science. 1997;277(5329):1094–1097. [DOI] [PubMed] [Google Scholar]
- [103].Giuffrida A, Beltramo M, Piomelli D. Mechanisms of endocannabinoid inactivation: biochemistry and pharmacology. J Pharmacol Exp Ther. 2001;298(1):7–14. [PubMed] [Google Scholar]
- [104].Fu J, Bottegoni G, Sasso O, et al. A catalytically silent FAAH-1 variant drives anandamide transport in neurons. Nat Neurosci. 2012;15(1):64–69. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Saliba SW, Marcotegui AR, Fortwängler E, et al. AM404, paracetamol metabolite, prevents prostaglandin synthesis in activated microglia by inhibiting COX activity. J Neuroinflammation. 2017. Dec 13;14(1):246. Erratum in: J Neuroinflammation. 2018 Feb 6;15(1):34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Muramatsu S, Shiraishi S, Miyano K, et al. Metabolism of AM404 from acetaminophen at human therapeutic dosages in the rat brain. Anesth Pain Med. 2016;6(1):e32873. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Kerckhove N, Mallet C, François A, et al. Ca(v)3.2 calcium channels: the key protagonist in the supraspinal effect of paracetamol. Pain. 2014;155(4):764–772. [DOI] [PubMed] [Google Scholar]
- [108].Ray S, Salzer I, Kronschläger MT, et al. The paracetamol metabolite N-acetylp-benzoquinone imine reduces excitability in first- and second-order neurons of the pain pathway through actions on KV7 channels. Pain. 2019;160(4):954–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Walson PD, Galletta G, Chomilo F, et al. Comparison of multidose ibuprofen and acetaminophen therapy in febrile children. Am J Dis Child. 1992;146(5):626–632. [DOI] [PubMed] [Google Scholar]
- [110].Prymula R, Siegrist CA, Chlibek R, et al. Effect of prophylactic paracetamol administration at time of vaccination on febrile reactions and antibody responses in children: two open-label, randomised controlled trials. Lancet. 2009;374(9698):1339–1350. [DOI] [PubMed] [Google Scholar]
- [111].Karbasi SA, Modares-Mosadegh M, Golestan M. Comparison of antipyretic effectiveness of equal doses of rectal and oral acetaminophen in children. 2010;86(3):228–232. Journal De Pediatria. [DOI] [PubMed] [Google Scholar]
- [112].Li S, L R B, Morham SG, et al. Cyclooxygenase-2 mediates the febrile response of mice to interleukin-1β. Brain Res. 2001;910(1–2):163–173. [DOI] [PubMed] [Google Scholar]
- [113].Harden LM, Du Plessis I, Poole S, et al. Interleukin (IL)-6 and IL-1β act synergistically within the brain to induce sickness behavior and fever in rats. Brain Behav Immun. 2008;22(6):838–849. [DOI] [PubMed] [Google Scholar]
- [114].Klir JJ, Roth J, Szelenyi Z, et al. 1993. Role of hypothalamic interleukin-6 and tumor necrosis factor-alpha in LPS fever in rat. Am J Physiol Regul Integr Comp Physiol. 1993;265(3):R512–R517. [DOI] [PubMed] [Google Scholar]
- [115].Cao C, Matsumura K, Yamagata K, et al. Endothelial cells of the rat brain vasculature express cyclooxygenase-2 mRNA in response to systemic interleukin-1β: a possible site of prostaglandin synthesis responsible for fever. Brain Res. 1996;733(2):263–272. [DOI] [PubMed] [Google Scholar]
- [116].Matsumura K, Cao C, Ozaki M, et al. Brain endothelial cells express cyclooxygenase-2 during lipopolysaccharide-induced fever: light and electron microscopic immunocytochemical studies. 1998;18(16):6279–6289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Cao C, Matsumura K, Ozaki M, et al. Lipopolysaccharide injected into the cerebral ventricle evokes fever through induction of cyclooxygenase-2 in brain endothelial cells Journal of Neurosci. 1999;19(2):716–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Li S, Wang Y, Matsumura KRBL, et al. The febrile response to lipopolysaccharide is blocked in cyclooxygenase-2−/−, but not in cyclooxygenase-1−/− mice. Brain Res. 1999;825(1–2):86–94. [DOI] [PubMed] [Google Scholar]
- [119].Steiner AA, Rudaya AY, Robbins JR, et al. Expanding the febrigenic role of cyclooxygenase-2 to the previously overlooked responses. Am J Physiol Regul Integr Comp Physiol. 2005;289(5):R1253–R1257. [DOI] [PubMed] [Google Scholar]
- [120].Nakamura K. Central circuitries for body temperature regulation and fever. Am J Physiol Regul Integr Comp Physiol. 2011;301(5):R1207–R1228. [DOI] [PubMed] [Google Scholar]
- [121].Ushikubi F, Segi E, Sugimoto Y, et al. Impaired febrile response in mice lacking the prostaglandin E receptor subtype EP 3. Nature. 1998;395(6699):281–284. [DOI] [PubMed] [Google Scholar]
- [122].Saha S, Engstrom L, Mackerlova L, et al. Impaired febrile responses to immune challenge in mice deficient in microsomal prostaglandin E synthase-1. Am J Physiol Regul Integr Comp Physiol. 2005;288(5):R1100–R1107. [DOI] [PubMed] [Google Scholar]
- [123].Lazarus M, Yoshida K, Coppari R, et al. EP3 prostaglandin receptors in the median preoptic nucleus are critical for fever responses. Nat Neurosci. 200710(9):1131–1133. [DOI] [PubMed] [Google Scholar]
- [124].Taniguchi Y, Yokoyama K, Inui K, et al. Inhibition of brain cyclooxygenase-2 activity and the antipyretic action of nimesulide. Eur J Pharmacol. 1997;330(2–3):221–229. [DOI] [PubMed] [Google Scholar]
- [125].Steiner AA, Li S, Llanos QJ, et al. Differential inhibition by nimesulide of the early and late phases of intravenous- and intracerebroventricular-LPS-induced fever in guinea pigs. Neuroimmunomodulation. 2001;9(5):263–275. [DOI] [PubMed] [Google Scholar]
- [126].Feldberg W, Gupta KP. Pyrogen fever and prostaglandin‐like activity in cerebrospinal fluid. J Physiol. 1973;228(1):41–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Abe M, Oka T, Hori T, et al. Prostanoids in the preoptic hypothalamus mediate systemic lipopolysaccharide-induced hyperalgesia in rats. Brain Res. 2001;1:1–9. [DOI] [PubMed] [Google Scholar]
- [128].Li S, Dou W, Tang Y, et al. Acetaminophen: antipyretic or hypothermic in mice? In either case, PGHS-1b (COX-3) is irrelevant. Prostaglandins Other Lipid Mediat. 2008;85(3–4):89–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Ryseck RP, Raynoschek C, Macdonald-Bravo H, et al. Identification of an immediate early gene, pghs-B, whose protein product has prostaglandin synthase/cyclooxygenase activity. Cell Growth Differ. 1992;3:443–450. [PubMed] [Google Scholar]
- [130].Engström RL, Wilhelms DB, Eskilsson A, et al. Acetaminophen reduces lipopolysaccharide-induced fever by inhibiting cyclooxygenase-2. Neuropharmacology. 2013;71:124–129. [DOI] [PubMed] [Google Scholar]
- [131].Massey TE, Walker RM, McElligott TF, et al. Acetaminophen-induced hypothermia in mice: evidence for a central action of the parent compound. Toxicology. 1982;25(2–3):187–200. [DOI] [PubMed] [Google Scholar]
- [132].Feldberg W, Gupta KP, Milton AS, et al. Effect of pyrogen and antipyretics on prostaglandin activity in cisternal c.s.f. of unanaesthetized cats. J Physiol. 1973;234:279–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].Kanashiro A, Pessini AC, Machado RR, et al. Characterization and pharmacological evaluation of febrile response on zymosan-induced arthritis in rats. Am J Physiol Regul Integr Comp Physiol. 2009;296:R1631–R1640. [DOI] [PubMed] [Google Scholar]
- [134].Mirrasekhian E, Nilsson JL, Shionoya K, et al. The antipyretic effect of paracetamol occurs independent of transient receptor potential ankyrin 1‐mediated hypothermia and is associated with prostaglandin inhibition in the brain. The FASEB J.2018;32(10):5751–5759. [DOI] [PubMed] [Google Scholar]
- [135].Ayoub SS, Flower RJ. Loss of hypothermic and anti-pyretic action of paracetamol in cyclooxygenase-1 knockout mice is indicative of inhibition of cyclooxygenase-1 variant enzymes. Eur J Pharmacol. 2019;861:172609. [DOI] [PubMed] [Google Scholar]
- [136].Crawford IL, Kennedy JI, Lipton JM, et al. Effects of central administration of probenecid on fevers produced by leukocytic pyrogen and PGE2 in the rabbit. J Physiol. 1979;287:519–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [137].Masferrer JL, Zweifel BS, Manning PT, et al. Selective inhibition of inducible cyclooxygenase 2 in vivo is antiinflammatory and nonulcerogenic. Proc Natl Acad Sci U S A. 1994;91:3228–3232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].Smith CJ, Zhang Y, Koboldt CM, et al. Pharmacological analysis of cyclooxygenase-1 in inflammation. Proc Natl Acad Sci U S A. 1998;95:13313–13318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [139].Yalçin SS, Gümüş A, Yurdakök K. Prophylactic use of acetaminophen in children vaccinated with diphtheria-tetanus-pertussis. World J Pediatr. 2008;4:112–127. [DOI] [PubMed] [Google Scholar]
- [140].Van Tittelboom T, Govaerts-Lepicard M. Hypothermia: an unusual side effect of paracetamol. Vet Hum Toxicol. 1989;31(1):57–59. [PubMed] [Google Scholar]
- [141].Nabulsi M, Tamim H, Sabra R, et al. Equal antipyretic effectiveness of oral and rectal acetaminophen: a randomized controlled trial [ISRCTN11886401]. BMC Pediatr. 2005;5:35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [142].Foster J, Mauger A, Thomasson K, et al. Effect of acetaminophen ingestion on thermoregulation of normothermic, non-febrile humans. Front Pharmacol. 2016;7:54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [143].Foster J, AR M, Govus A, et al. Acetaminophen (paracetamol) induces hypothermia during acute cold stress. Clin Drug Investig. 2017;37(11):1055–1065. [DOI] [PubMed] [Google Scholar]
- [144].Dippel DWJ, Van Breda EJ, HMA VG, et al. Effect of paracetamol (acetaminophen) on body temperature in acute ischemic stroke: a double-blind, randomized phase II clinical trial. Stroke. 2001;32(7):1607–1612. [DOI] [PubMed] [Google Scholar]
- [145].Dippel DWJ, Van Breda EJ, Van Der Worp HB, et al. Timing of the effect of acetaminophen on body temperature in patients with acute ischemic stroke. Neurology. 2003;61(5):677–679. [DOI] [PubMed] [Google Scholar]
- [146].Oka T, Oka K, Saper CB. Contrasting effects of E type prostaglandin (EP) receptor agonists on core body temperature in rats. Brain Res. 2003;968:256–262. [DOI] [PubMed] [Google Scholar]
- [147].Pryce G, Giovannoni G, Baker D. Mifepristone or inhibition of 11beta-hydroxylase activity potentiates the sedating effects of the cannabinoid receptor-1 agonist Delta(9)-tetrahydrocannabinol in mice. Neurosci Lett. 2003;341:164–166. [DOI] [PubMed] [Google Scholar]
- [148].Varga A, SzabÓ ŃJ, McDougall A, et al. Effects of the novel TRPV1 receptor antagonist SB366791 in vitro and in vivo in the rat. Neurosci Lett. 2005;385:137–142. [DOI] [PubMed] [Google Scholar]
- [149].Rawls SM, Ding Z, Cowan A. Role of TRPV1 and cannabinoid CB1 receptors in AM404-evoked hypothermia in rats. Pharmacol Biochem Behav. 2006;83:508–516. [DOI] [PubMed] [Google Scholar]
- [150].Gentry C, Andersson DA, Bevan S. TRPA1 mediates the hypothermic action of acetaminophen. Sci Rep. 2015;5(1):1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [151].Garami A, Shimansky YP, Rumbus Z, et al. Hyperthermia induced by transient receptor potential vanilloid-1 (TRPV1) antagonists in human clinical trials: insights from mathematical modeling and meta-analysis. Pharmacol Ther. 2020. Apr;208:107474. [DOI] [PubMed] [Google Scholar]
- [152].Gavva NR, Bannon AW, Hovland DNJ, et al. Repeated administration of vanilloid receptor TRPV1 antagonists attenuates hyperthermia elicited by TRPV1 blockade.J Pharmacol Exp Ther. 2007;323(1):128–137. [DOI] [PubMed] [Google Scholar]
- [153].Corley G, Rawls SM. Opioid, cannabinoid CB1 and NOP receptors do not mediate APAP-induced hypothermia in rats. Pharmacol Biochem Behav. 2009;92(3):503–507. [DOI] [PubMed] [Google Scholar]
- [154].Coupar IM, Taylor DA. Alteration in the level of endogenous hypothalamic prostaglandins induced by delta 9-tetrahydrocannabinol in the rat. Br J Pharmacol. 1982;76(1):115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [155].Bashir S, Elegunde B, Morgan WA. Inhibition of lipolysis: a novel explanation for the hypothermic actions of acetaminophen in non-febrile rodents. Biochem Pharmacol. 2020;172:113774. [DOI] [PubMed] [Google Scholar]
- [156].Satinoff E. Salicylate: action on normal body temperature in rats. Science. 1972;176:532–533. [DOI] [PubMed] [Google Scholar]
- [157].Fischer LJ, Green MD, Harman AW. Levels of acetaminophen and its metabolites in mouse tissues after a toxic dose. J Pharmacol Exp Ther.1981;219(2):281–286. [PubMed] [Google Scholar]
- [158].Orbach SM, Cassin ME, Ehrich MF, et al. Investigating acetaminophen hepatotoxicity in multi-cellular organotypic liver models. Toxicol in Vitro. 2017;42:10–20. [DOI] [PubMed] [Google Scholar]
- [159].Erman A, Schwartzman M, Raz A. Indomethacin but not aspirin inhibits basal and stimulated lipolysis in rabbit kidney. Prostaglandins. 1980;20(4):689–702. [DOI] [PubMed] [Google Scholar]
- [160].Vázquez-Meza H, De Piña MZ, Pardo JP, et al. Non-steroidal anti-inflammatory drugs activate NADPH oxidase in adipocytes and raise the H2O2 pool to prevent cAMP-stimulated protein kinase a activation and inhibit lipolysis. BMC Biochem. 2013;14(1):13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [161].Clark WG, Alderdice MT. Inhibition of leukocytic pyrogen-induced fever by intracerebroventricular administration of salicylate and acetaminophen in the cat. Proc Soc Exp Biol Med. 1972;140(2):399–403. [DOI] [PubMed] [Google Scholar]
- [162].Sproule BA, BustoU E, Buckle C, et al. The use of non‐prescription sleep products in the elderly. Int J Geriatr Psychiatry. 1999;14(10):851–857. [PubMed] [Google Scholar]
- [163].Murphy PJ, Badia P, Myers BL, et al. Nonsteroidal anti-inflammatory drugs affect normal sleep patterns in humans. Physiol Behav. 1994;55(6):1063–1066. [DOI] [PubMed] [Google Scholar]
- [164].Baker FC, Driver HS, Paiker J, et al. Acetaminophen does not affect 24-h body temperature or sleep in the luteal phase of the menstrual cycle. J Appl Physiol. 2002;92(4):1684–1691. [DOI] [PubMed] [Google Scholar]
- [165].Van De Glind EM, Hooft L, Tulner LR, et al. Acetaminophen for self-reported sleep problems in an elderly population (ASLEEP): a randomized double-blind placebo-controlled trial. Int J Geriatr Psychiatry. 2016;31(8):955–957. [DOI] [PubMed] [Google Scholar]
- [166].Matsumura H, Goh Y, Ueno R, et al. 1988. Awaking effect of PGE2 microinjected into the preoptic area of rats. Brain Res. 1988;444(2):265–272. [DOI] [PubMed] [Google Scholar]
- [167].Hayaishi O. Molecular mechanisms of sleep‐wake regulation: roles of prostaglandins D2 and E2. Faseb J. 1991;5(11):2575–2581. [PubMed] [Google Scholar]
- [168].Matsumura H, Honda K, Choi WS, et al. Evidence that brain prostaglandin E2 is involved in physiological sleep-wake regulation in rats. Proc Natl Acad Sci. 1989;86(14):5666–5669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [169].Saper CB, Romanovsky AA, Scammell TE. Neural circuitry engaged by prostaglandins during the sickness syndrome. Nat Neurosci. 2012. Jul 26;15(8):1088–1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [170].Tittarelli R, Pellegrini M, Scarpellini MG, et al. Hepatotoxicity of paracetamol and related fatalities. Eur Rev Med Pharmacol Sci. 2017;21(1): 95–101. Suppl. [PubMed] [Google Scholar]
- [171].Smilkstein MJ, Knapp GL, Kulig KW, et al. Efficacy of oral N-acetylcysteine in the treatment of acetaminophen overdose. Analysis of the national multicenter study (1976 to 1985). N Engl J Med. 1988;319(24):1557–1562. [DOI] [PubMed] [Google Scholar]
- [172].Day M. Covid-19: ibuprofen should not be used for managing symptoms, say doctors and scientists. Covid-19: ibuprofen should not be used for managing symptoms, say doctors and scientists. BMJ. 2020;368:m1086. [DOI] [PubMed] [Google Scholar]
- [173].Moore N, Carleton B, Blin P, et al. Does Ibuprofen Worsen COVID-19? Drug Saf. 2020;43:611–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [174].Sridharan GK, Kotagiri R, Chandiramani VH, et al. COVID-19 and avoiding ibuprofen. How good is the evidence? Am J Ther. 2020;27(4):e400–e402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [175].Hawkins LC, Edwards JN, Dargan PI. Impact of restricting paracetamol pack sizes on paracetamol poisoning in the United Kingdom. Drug Saf. 2007;30(6):465–479. [DOI] [PubMed] [Google Scholar]
- [176].Bateman DN. Pack size and paracetamol overdose: 16 years later. Clin Toxicol. 2014;52(8):821–823. [DOI] [PubMed] [Google Scholar]
- [177].Shah AD, Wood DM, Dargan PI. Internet survey of home storage of paracetamol by individuals in the UK. QJM. 2013;106(3):253–259. [DOI] [PubMed] [Google Scholar]
- [178].McNicholl BP. Toxicity awareness and unintended suicide in drug overdoses. Emerg Med J. 1992;9(2):214–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [179].Hawton K, Ware C, Mistry H, et al. Why patients choose paracetamol for self-poisoning and their knowledge of its dangers. BMJ. 1995;310(6973):164. [DOI] [PMC free article] [PubMed] [Google Scholar]