

Abstract
Pancreatic ductal adenocarcinoma (PDAC) is a highly aggressive cancer with poor patient survival. Towards understanding the underlying molecular alterations that drive PDAC oncogenesis, we conducted comprehensive proteogenomic analysis of 140 pancreatic cancers, 67 normal adjacent tissues, and 9 normal pancreatic ductal tissues. Proteomic, phosphoproteomic, and glycoproteomic analyses were used to characterize proteins and their modifications. In addition, whole genome sequencing, whole exome sequencing, methylation, RNA-seq, and miRNA-seq were performed on the same tissues to facilitate an integrated proteogenomic analysis and determine the impact of genomic alterations on protein expression, signaling pathways, and post-translational modifications. To ensure robust, downstream analyses, tumor neoplastic cellularity was assessed via multiple, orthogonal strategies using molecular features, and verified via pathological estimation of tumor cellularity based on histological review. This integrated proteogenomic characterization of PDAC will serve as a valuable resource for the community, paving the way for early detection and identification of novel therapeutic targets.
Keywords: Pancreatic ductal adenocarcinoma, proteogenomics, KRAS, neoplastic cellularity, glycoproteins, kinase inhibitors, immune-cold tumors, endothelial cell, tumor subtyping, CPTAC
Introduction
With a five-year survival rate below 10%, pancreatic ductal adenocarcinoma (PDAC) is one of the deadliest solid malignancies, and is projected to become the second leading cause of cancer death in the US and in Europe by the year 2030 (Quante et al., 2016; Rahib et al., 2014). It has been estimated that 48,220 Americans died of PDAC in 2020 (American Cancer Society, 2021). Due to the lack of early signs and symptoms, as well as the dearth of reliable and effective methods for screening and early detection, the majority of patients (80%-85%) present with locally advanced or distant metastatic disease and are unresectable (Hruban et al., 2019; Motoi and Unno, 2020; Pereira et al., 2020; Singhi et al., 2019; Springfeld et al., 2019). Combination cytotoxic chemotherapy serves as first-line treatment of metastatic PDAC and is responsible for the modest survival improvement seen in this setting (Roth et al., 2020). Nevertheless, the median overall survival achieved in patients with metastatic disease is still less than 12 months (Vaccaro et al., 2011; Von Hoff et al., 2013).
Over the last decade, a number of drugs, including targeted therapies, have been developed for the treatment of PDAC. However, results have been disappointing, and new strategies are desperately needed. Comprehensive characterization of well-annotated tumor specimens has led to a better understanding of the key genomic and transcriptomic alterations in PDAC, including somatic mutations in KRAS, TP53, CDKN2A, SMAD4, and to molecular classifications of tumors based on gene expression patterns (Thompson et al., 2020). In addition, these studies have revealed tumor-specific therapeutic targets, such as poly(adenosine diphosphate–ribose) polymerase (PARP) inhibitors, for cancers with germline BRCA1 and BRCA2 mutations (Balachandran et al., 2019; Balsano et al., 2019; Elyada et al., 2019; Golan et al., 2019; Hessmann et al., 2020; Jones et al., 2008; Kaufman et al., 2015; Lowery et al., 2018). Unfortunately, these mutations only occur in a small number of pancreatic cancers, and resistance can emerge when single agents are given (Tao et al., 2020).
Somatic KRAS activating point mutations are the predominant genetic alteration in PDAC. However, KRAS is considered an undruggable target except for a specific mutant form, G12C (Janes et al., 2018). In addition to downstream intracellular changes, it has been increasingly appreciated that KRAS mutations also have a broad impact on the tumor microenvironment, contributing to promotion and maintenance of cancer malignancy, responses to immunotherapy, and drug delivery. Since most pancreatic cancers are notoriously immunologically “cold”, molecular classifiers are needed to identify the small fraction of patients with relative “hot” cancers that may benefit from immunotherapy and reveal mechanisms driving immune exclusion in the majority of PDACs (Ho et al., 2020). Although genomic and transcriptomic features are needed to identify critical signaling pathways active in PDAC that can be targeted and have started to unravel the importance of stroma and the immune environment, they alone are insufficient to fully understand this cancer and support advancements in precision oncology. A proteogenomic approach that integrates proteomics and post-translational modification (PTM) analysis provides a more comprehensive view of pancreatic cancer biology and supports discovery of targets for early detection and treatment.
Here, we performed a comprehensive proteogenomic characterization of treatment naive PDACs, paired normal adjacent tissues (NATs), and macro-dissected normal pancreatic duct tissues. We addressed the characteristic low neoplastic cellularity of pancreatic cancer by focusing on tumors with sufficient neoplastic cellularity as defined by several cross-validated methods, and explored the proteogenomic features specific to neoplastic ductal epithelial cells by applying molecular and histological deconvolution. The use of tissue isolated from normal pancreatic ducts allowed us to overcome the high acinar cell content of normal pancreatic parenchyma, and to compare directly neoplastic ductal epithelium to non-neoplastic ductal epithelium, confirming clinically relevant protein markers identified using PDACs and NATs. Moreover, integrated proteogenomic characterization revealed the phenotypic effects of genomic and epigenetic perturbations on proteins and protein modifications, and delineated PDAC molecular subtypes and cell microenvironment compositions. This dataset constitutes a rich resource for future studies focused on early detection and tumor classification-based patient stratification to guide treatment selection.
Results
Proteogenomic Landscape of the PDAC Cohort
For proteogenomic characterization of PDAC, 140 treatment-naive pancreatic tumors (135 PDACs and 5 pancreatic adenosquamous carcinomas), 67 paired NATs, and 9 normal pancreatic duct tissues were collected and homogenized via cryopulverization for genomic, epigenomic, transcriptomic, and proteomic analyses within the same portion of tissue. Clinical data, including age, sex, race, tumor site, and tumor stage, are summarized in Table S1. Whole-exome sequencing (WES), whole-genome sequencing (WGS), RNA sequencing (RNA-Seq), microRNA sequencing (miRNA-Seq), DNA methylation analysis, isobaric tandem mass tag (TMT) labeling based proteomics, phosphoproteomics, and glycoproteomics produced 8 sets of omics data (Figure 1A). RNA-Seq, miRNA-Seq, and methylation analysis identified 28,057 genes, 2,416 miRNAs, and 850,000 CpG sites, respectively. Proteomics, phosphoproteomics, and glycoproteomics analyses identified and quantified in total 11,662 proteins (8,781 proteins per sample on average), 51,469 phosphosites (25,764 phosphosites per sample on average), and 34,024 glycopeptides (30,660 N-linked glycopeptides and 3,364 O-linked glycopeptides; 8,706 N-linked glycopeptides per sample on average and 866 O-linked glycopeptides per sample on average) (Table S1). We found high measurement reproducibility of the quality control samples across the TMT plexes and no observable TMT-plex effect (Figure S1A). In this study, the median correlation between RNA and protein is 0.35 (Figure S1B), indicating the consistent disparity between RNA and protein expression, which was also observed in other cancer types such as colon cancer (mean=0.48, Vasaikar et al., 2019), ovarian cancer (median=0.45, Zhang et al., 2016), clear cell renal cell carcinoma (tumor median=0.43, NAT median=0.34, Clark et al., 2019), endometrial cancer (median 0.48, Dou et al., 2020), lung adenocarcinoma (tumor median=0.53, NAT median=0.15, Gillette et al., 2020), and head and neck cancer (median=0.52, Huang et al., 2021). To determine whether gene-wise correlations of tumors and NAT were different, we performed gene-wise correlation separately for tumors and NATs (Figure S1B), observing a decreased median correlation within the NAT-only group relative to the tumor-only group (tumor median=0.36, NAT median=0.26). This trend was noted in other tumor types (Clark et al., 2019; Gillette et al., 2020) potentially due to cell-type-specific translational regulation (Gonzalez et al., 2014).
Figure 1. Proteogenomic landscape of the PDAC cohort.
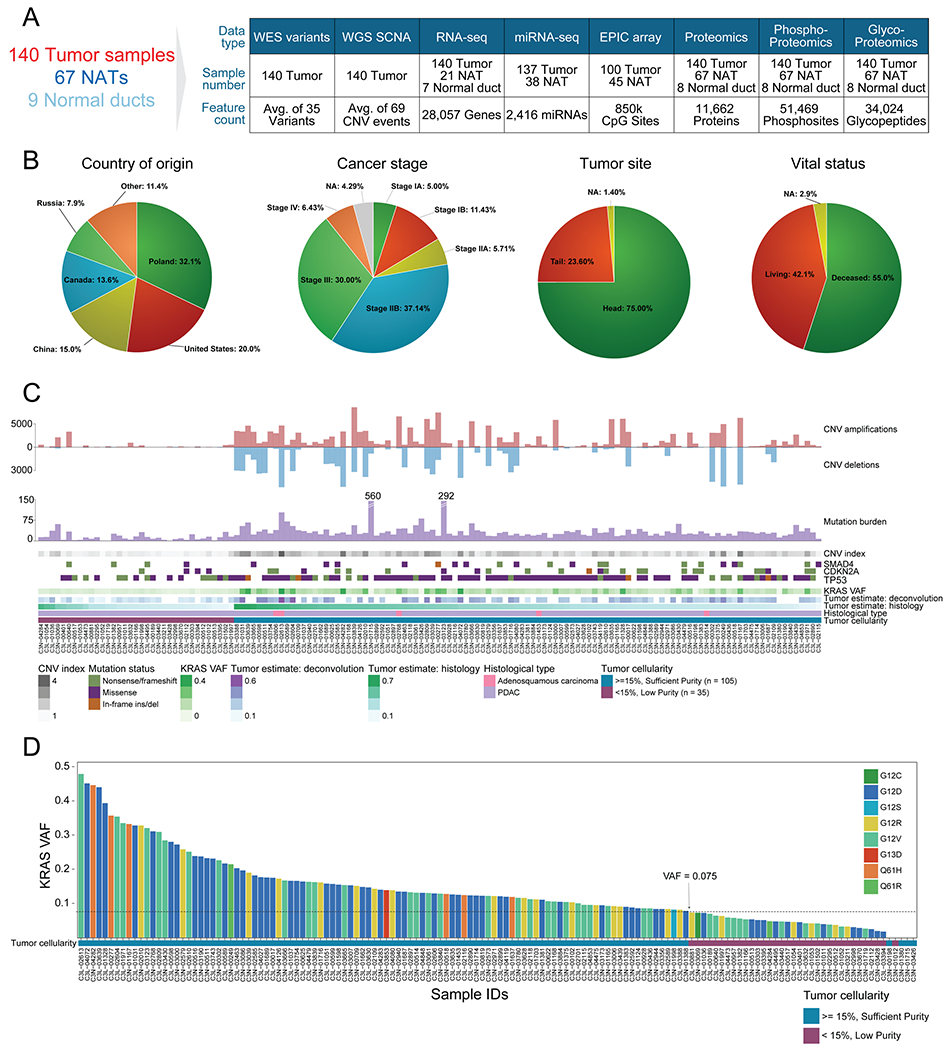
A) Sample numbers and omics data types of the cohort. B) Country of origin, cancer stage, tumor site, and vital status proportions in the cohort. C) Molecular and histology-based tumor estimates are used to classify samples into “sufficient” and “low” purity groups. D) KRAS VAF distribution in the cohort colored by KRAS hotspot amino acid change. The sufficient neoplastic purity KRAS VAF cutoff, denoted by a dashed line, is 0.075 (15% neoplastic cellularity). The 4 samples with no KRAS mutations detected were also included in the sufficient tumor cellularity group since they had high mutation burden (n > 25), high CNV (index > 1), and/or additional driver events in TP53, CDKN2A, and SMAD4.
A unique feature of this study is that samples were collected prospectively from multiple source sites worldwide, controlling for ischemia time to ensure high quality post-translational modification (PTM) analysis of proteins (Figure 1B). Our patient cohort reflected demographics of previous large-scale reports, with 75% of the resected tumors derived from the head of the pancreas (Becker et al., 2014; Kim et al., 2017; van Erning et al., 2018). As these were surgically resected cancers, the vast majority of patients were stage I-III with only nine stage IV patients, including 59% of patients with low stage tumors (Stage I or II), and 42% of patients that were alive at the time of the data freeze for the analysis reported in this study (Figure 1B). Several risk factors related to PDAC, including smoking history, chronic pancreatitis, obesity, and type II diabetes, were present in our cohort with rates of 37%, 22%, 11%, and 28%, respectively (Table S1). Importantly, KRAS alterations were detected in 97% of tumors (96% hotspot driver KRAS mutations and one KRAS amplification event), consistent with previous large-scale analyses (Bailey et al., 2016; The Cancer Genome Atlas Research Network, 2017b).
A major challenge in characterizing PDAC is that neoplastic ductal cells often comprise a minority of the cells in pancreatic tumors, with non-neoplastic cell components, such as acinar cells and stroma, making up a large proportion of the resected tissue. To address this limitation, we identified 105 samples with sufficient neoplastic purity based on several criteria: a minimum KRAS variant allele fraction (VAF) greater than or equal to a cutoff of 0.075 (equivalent to 15% neoplastic cellularity), or significant mutation burden and copy number alterations (Figures 1C, 1D). Among these, we included 4 tumors that do not carry detectable KRAS mutations, but harbor additional genetic features associated with PDAC, including mutations in other significantly mutated genes (SMGs), including TP53, CDKN2A, and SMAD4, and relatively high numbers of somatic mutations and copy number alterations. We did not detect any purity-related patterns with specific KRAS hotspot amino acid changes (Figures 1C, 1D). In addition to these KRAS VAF-based metrics, we used histology-based as well as DNA methylation- and RNA-based molecular deconvolution methods (Onuchic et al., 2016) to estimate neoplastic cellularity using different data modalities. These estimates of neoplastic cellularity significantly correlated with KRAS VAF estimates (Figure S1C). In particular, KRAS VAF was highly correlated with DNA methylation-based deconvolution (Spearman r = 0.81). We herein denote the remaining 35 tumors as “low purity” but emphasize that they do indeed contain neoplastic cells, evidenced by presence of other SMG alterations, low KRAS VAF, and pathology review (Figure 1C). When projected into principal component analysis (PCA) at RNA, protein, phosphorylation, and glycosylation levels, we observed that high purity tumors and NAT samples were separated but low purity samples were spatially localized -between the high purity tumors and NAT samples, which supported our purity classification (Figure S1D). Low purity samples were retained for selected analyses aimed at dissecting the tumor microenvironment and for tumor subtyping.
Previous molecular studies of PDAC used NATs in tumor-normal comparisons, despite the fact that NATs are mostly composed of non-neoplastic acinar cells, thus introducing cell-type-specific signatures that confound the analysis. In order to address this limitation, we included 9 normal macrodissected ductal tissues to serve as a true ductal/epithelial normal, in addition to NATs.
In summary, we leveraged genomic, histological, and computational approaches to address low tumor cellularity and high acinar content of NATs, and annotated tumor samples with sufficient tumor purity to delineate unique molecular features of PDAC tumors from NATs.
Impact of Genomic Alterations on Transcriptome, Proteome, and Phosphorylation
Previous genomic characterizations have delineated the most frequent genetic alterations associated with PDAC, with subsequent transcriptomic analyses resulting in the development of transcriptomic-based subtypes of PDAC (Bailey et al., 2016; Collisson et al., 2011; Moffitt et al., 2015; Singh et al., 2009). Despite substantial efforts, linking genomic alterations to the functional modules that drive the pathological phenotypes remains a challenge. Among 105 tissue samples with sufficient tumor cellularity, genomic alterations were detected in known pancreatic cancer driver genes, KRAS, TP53, CDKN2A, and SMAD4, at rates of 97%, 83%, 48%, and 29%, respectively (Figure 2A). These frequencies are comparable to previous reports, with somewhat higher percentages of CDKN2A and SMAD4 alterations due to the inclusion of copy number variations (CNVs) and fusions (Thompson et al., 2020). We found assessment and integration of CNVs to be critical, evidenced by the presence of more CDKN2A focal deletions than intragenic mutations in our cohort (Caldas et al., 1994). Hotspot KRAS mutations were largely G12D, G12V, and G12R (Figure 2A). Aside from these four major SMGs, we also detected ARID1A, RNF43, GNAS, KMT2C, KMT2D, TGFBR2, and RBM10 alterations in at least 5% of the tumors (Figure 2A).
Figure 2. Impact of genomic alterations on the transcriptome, proteome, and phosphoproteome.
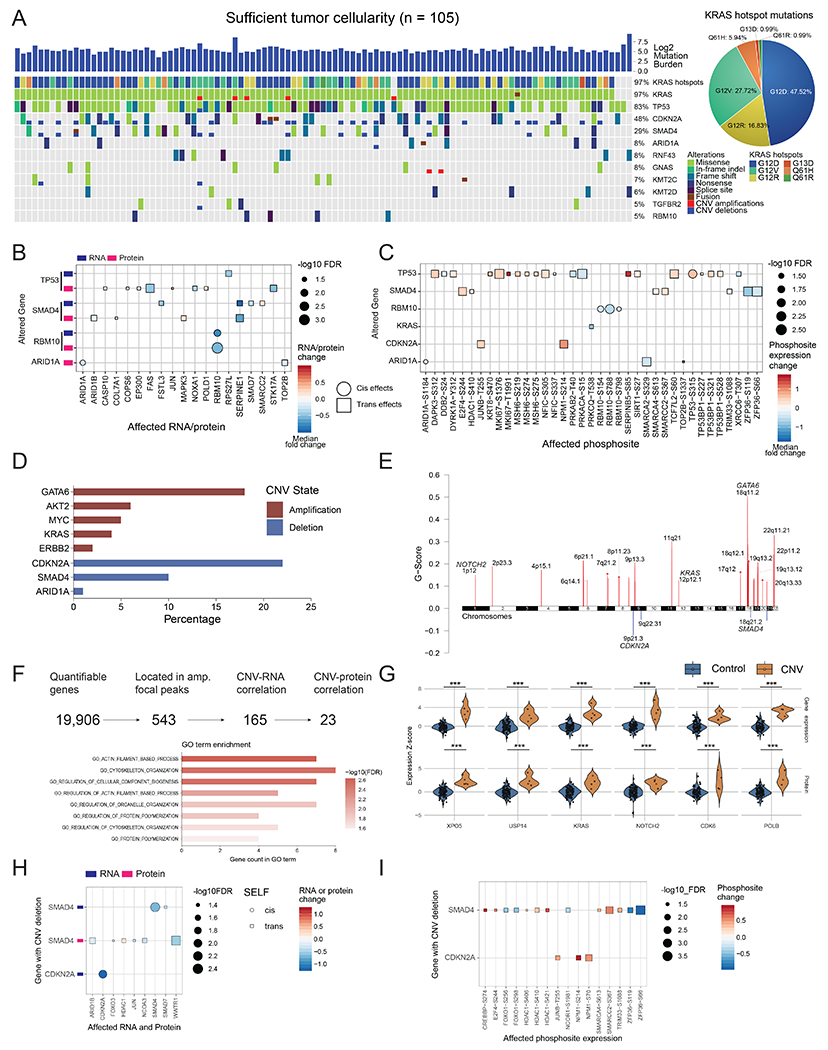
A) Genomic landscape of the cohort with sufficient tumor cellularity (n = 105) showing mutated genes with a frequency ≥0.05. All mutation types are considered, including missense, frameshift, splice-site, copy number alterations, and fusion events. G12D, G12R, and G12V are the most common KRAS driver mutations present in the cohort. B) Cis- and trans-effects of genomic alterations on RNA and protein levels. C) Cis- and trans-effects of genomic alterations on phosphosites. Protein levels are used as a covariate to remove protein abundance-related effects. In B and C, cis-effects are denoted by circles while trans-effects are denoted with squares. D) Major gene copy number amplification and deletion rates in the cohort. The log ratio cutoffs used are [−0.4, 0.4] (See STAR Methods). E) Significant arm level focal peaks detected using GISTIC. Several of these peaks contain known driver genes in PDAC such as GATA6, CDKN2A, and SMAD4. F) CNV driver approach schematic. From all genes with copy number events, 543 are located in the GISTIC focal peaks, of which 165 have RNA effects and 23 also have protein level effects. These 23 genes have roles in actin filament and cytoskeleton organization pathways. G) Violin plots showing the impact of copy number in a select number of proteins from these 23 putative CNV drivers. *** Denote p < 0.001. The control group in each comparison includes all samples without the copy number event for each gene or protein. The alterations of H) mRNA and proteins, and I) phosphosites associated with CDKN2A and SMAD4 deletions. Samples with wild-type CDKN2A and SMAD4 serve as controls.
We comprehensively characterized the impact of genetic alterations on RNA, protein, and phosphosite levels of the corresponding gene product (cis) or other interacting genes (trans) (Figures 2B and 2C) (STAR Methods). TP53 alterations had the most trans-effects at protein and phosphosite levels, with different targets identified at the RNA/protein levels and phosphosite levels, likely due to extensive post-translational regulation. Interestingly, mutations in TP53 were associated with an increase in phosphorylation of proteins involved in DNA damage repair pathways (e.g. MSH6, TP53, and TP53BP1), which suggests that these alterations play a role in maintaining genome integrity and preventing apoptosis (Figure 2C). In TP53 mutant tumors we also observed higher phosphorylation of MKI67, a marker for cellular proliferation, which implies that these mutations may lead to increased cell growth rates (Herr et al., 2020). We further explored the effects of TP53 missense mutations compared to truncating mutations. Samples carrying a frame-shift insertion or deletion, splice-site mutation, nonsense mutation, or CNV deletion in TP53 were included in the truncation group. The missense group was composed of samples with missense mutations in TP53. As expected, we observed a significantly greater cis-effect with higher TP53 protein expression and TP53-S315 phosphosite expression in the TP53 missense group compared to the wild-type group (Figure S2A), while there were no significant TP53 protein changes in cis-effects between the truncation and wild-type groups (Figure S2A). Evaluation of the trans-effects of disparate TP53 genomic alteration revealed impacts on three phosphosites, MKI67-S1376, MSH6-S219, and TP53BP1-S321. Interestingly, we observed similar trans-effects by both missense and truncation groups which were associated with to higher phosphorylation levels of these proteins (Figure S2A). SMAD4 mutations were associated with downregulation of SERPINE1, a known TGFβ pathway target (Dennler et al., 1998), at both the RNA and protein levels, as well as the up-regulation of MAPK3 protein expression and downstream MAPK signaling (E2F4 phosphorylation). These associations have been reported in vitro (Chen et al., 2002) and in other cancer types (Gomis et al., 2006). (Figure 2B). Additionally, we found that RBM10 mutations displayed a significant cis-effect leading to high expression of the cognate mRNA, protein and phosphoproteins (Figure 2B). RBM10 is a tumor suppressor that acts upstream of p53 and plays a role in RNA splicing (Hernandez et al., 2016). In a prior WES study of PDAC, RBM10 mutations were associated with better survival in patients with aggressive disease (Witkiewicz et al., 2015).
We also identified several arm level and focal level copy number variations (CNVs), including amplifications in 9p, 11q, 18q, and 22q arms, GATA6 focal amplifications, and CDKN2A deletions (Figures 2D and 2E) (Caldas et al., 1994; Fu et al., 2008; Iacobuzio-Donahue et al., 2004) that lead to significant expression changes in genes, proteins, and phosphoproteins (Figure S2B). Since we observed a much larger number of amplifications, we focused on identifying putative new CNV drivers within the amplified foci (Figure 2E, 2F). Of 543 genes within amplification peaks, 165 showed significant correlation of copy number with corresponding RNA levels, including 23 that displayed concordant protein expression (Figure 2F). Proteins identified by this approach, are representative of potentially novel cis-effects of CNV events, and associated with actin filament process and cytoskeleton organization (Figures 2F and 2G), with reorganization of actin fibers having been previously implicated in tumorigenesis and metastasis (Manoli et al., 2019; Stevenson et al., 2012).
To further investigate the impact of CNV, we analyzed the expression changes in gene, protein, and phosphorylation associated with CNV across the entire genome, visualized as correlation heatmaps depicting global cis- and trans-effects of CNVs (Figure S2C, Table S2). Most of the proteins regulated in trans by CNVs were located within chromosomes 7, 9, 17, and 18, while trans-effect of CNVs at the phospho-level was sporadic (Figure S2C).
We further examined any alterations associated with SMAD4 and CDKN2A loss, and observed lower SMAD4 and CDNK2A mRNA levels in samples with SMAD4 deletion and CNDK2A deletion, respectively, compared to corresponding wild-type group (Figure 2H). Regarding trans-effects of these deletions, higher phosphorylation of HDAC1 at S406, S410, and S421, and of SMARCA4 at S613 were associated with SMAD4 deletions, while CDKN2A deletions were associated with higher phosphorylation of NPM1 at S70 and S214 (Figure 2I). In addition, CNV loss of SMAD4 was associated with a higher phosphorylation level of CREBBP-S274 and lower phosphorylation of ZFP36-S66 and -S119 (Figure 2I). Although the functional role of ZFP36 phosphorylation is not fully characterized, this protein is down-regulated in several tumor types including pancreatic cancer, suggesting a potential role as a tumor suppressor in PDAC (Fallahi et al., 2014; Sun et al., 2015; Wei et al., 2016).
To identify proteins possibly regulated by DNA methylation in tumors, we correlated RNA, protein, and phosphoprotein levels with promoter DNA methylation (Figure S2D). Among others, GSTM1 methylation resulted in downregulation of the corresponding RNA and protein, in agreement with reports that implicate GSTM1 in multiple cancers (Wang et al., 2016; Zhang et al., 2017). The extent of promoter DNA methylation was lower in NATs than in tumors (Figure S2E–F). Tumors with KRAS G12D mutations also showed relatively higher DNA methylation compared to tumors with other KRAS mutations (Figure S2F). We applied the method for “Identification of epigenetically-silenced genes” as described by TCGA (The Cancer Genome Atlas Research Network, 2017b) on our methylation dataset and identified 86 epigenetically silenced genes, of which 22 were previously reported by TCGA (Table S2). Two genes (ZNF544 and THNSL2) that were epigenetically-silenced in more than 10% of tumors were significantly associated with patient survival (Figure S2G–I), and the methylation of both of these genes was confirmed in the TCGA data set (The Cancer Genome Atlas Research Network, 2017b). Two clusters (cluster M1 and M2) were identified by methylation-based subtyping (Table S2). Cluster M2 showed more extensive DNA hypermethylation relative to cluster M1 (Table S2). We also observed a positive association between tumor cellularity and methylation status in these tumors (Table S2), in line with the TCGA study (The Cancer Genome Atlas Research Network, 2017b).
In summary, we verified commonly mutated genes, CNVs, as well as DNA methylation events in PDAC, and linked these genomic and epigenetic perturbations to the functional modules that drive disease phenotype.
Discovery of Specific Molecular Features of Early Stage PDAC for Tumor Diagnosis and Prognosis
Approximately 80% of PDAC tumors are unresectable as patients are diagnosed at an advanced stage (Hruban et al., 2019). Thus, a panel of highly robust biomarkers for early detection may improve survival as treatment modalities for these patients emerge. Proteins, phosphorylation sites, and glycosylation sites that are dysregulated in tumors relative to NATs represent putative candidates for early detection/prognosis and may serve as novel drug targets. To identify tumor-associated proteins, we performed differential abundance analysis using high tumor cellularity samples (Table S3). Relative to NATs, 2,218 and 2,244 proteins were significantly down-regulated and up-regulated, respectively, in PDACs (Figure 3A, Wilcoxon signed rank test). As expected, proteins with high abundance in NATs (> 2 fold) were related to normal pancreatic functions, such as organic anion transport and digestion, while many of those upregulated in tumors (> 2 fold) were enriched for proteins involved in epidermal and endodermal development.
Figure 3. Identification of tumor-associated proteins and modification sites by comparison of tumor and normal tissues.
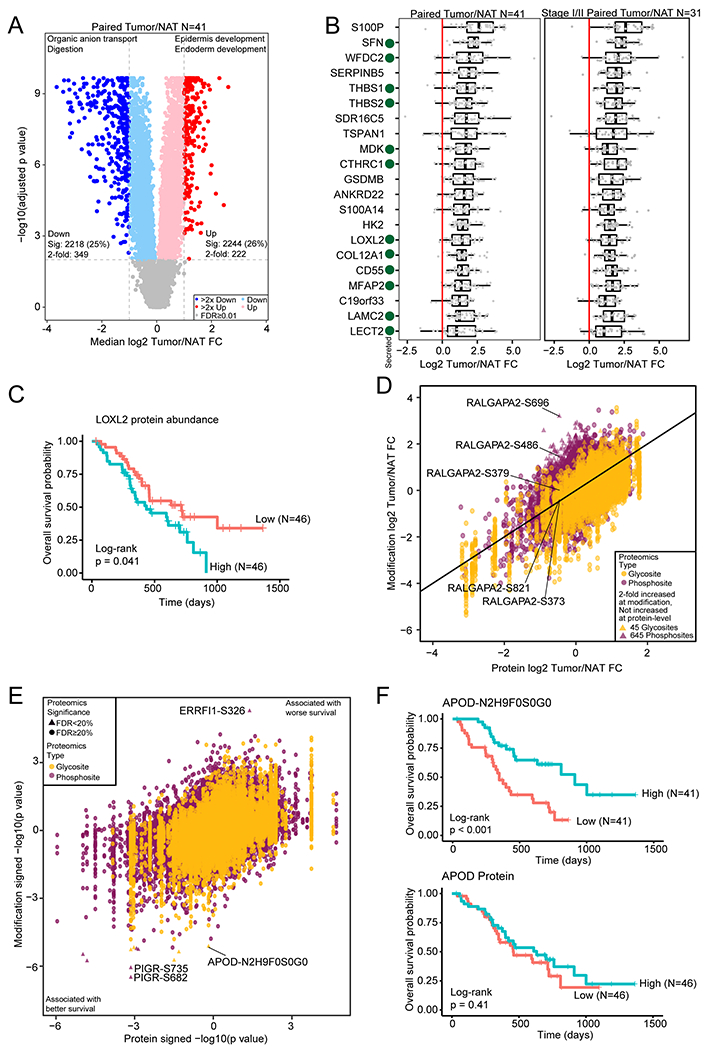
A) Differential protein abundance between tumors and paired NATs. Selected GO biological process terms for significantly increased and significantly decreased proteins are shown above the volcano plot. B) Proteins with a median fold change > 2 compared to matched NAT and with significantly increased abundance both compared to normal ductal tissues and after adjusting for epithelial content for all samples and the subset of stage I/II samples. Secreted proteins are indicated with a green dot. C) Kaplan-Meier curve for LOXL2 protein abundance association with overall survival. The two groups were separated by median LOXL2 abundance. D) Median phosphosite and N-linked glycosylation site fold change compared to the protein fold change in tumor compared to matched NAT. E) Cox regression signed p value for phosphosite and N-linked glycosylation site abundance association with survival compared to the protein association to survival. F) Kaplan-Meier survival curves for an N-linked glycosylation site on APOD and APOD protein abundance.
To identify proteins associated with PDACs, we focused on 222 proteins with more than two-fold increase in abundance in tumors relative to NATs (Figure 3A). To account for the inherent heterogeneity of pancreatic tumors, we adjusted for stromal and immune content using a linear mixed model. Twenty seven proteins remained significantly upregulated by more than two-fold in PDACs relative to NATs. We additionally found that the differential expression between PDACs and NATs was similar to that between PDACs and normal ductal tissues (Figure S3A), and 21 of 27 proteins were also up-regulated more than two-fold in PDACs compared to normal ductal tissues (Figure 3B, Table S3). Importantly, these proteins were similarly upregulated in early stage tumors (Figure 3B). In particular, 12 of these are secreted proteins and could serve as early detection markers in serum or pancreatic juice (Gonzalez-Borja et al., 2019). Among these putative biomarkers, two proteins, THBS2 and LAMC2, were reportedly elevated in sera from patients with PDAC (Kim et al., 2017; Kosanam et al., 2013). Expression of most of these proteins in pancreatic cancer is supported by immunohistochemistry (IHC) evidence in the Human Protein Atlas (Figure S3B). Eleven proteins are reported as elevated in the Pancreatic Cancer Database, with six proteins (HK2, LOXL2, COL12A1, C19orf33, TSPAN1, MDK) previously supported only by RNA or cell line proteomic evidence in the Pancreatic Cancer Database (Harsha et al., 2009; Thomas et al., 2014), with LOXL2 protein abundance associating with shorter overall survival (Figure 3C). Two were described elsewhere as elevated in PDAC (SDR16C5 and ANKRD22) (Caba et al., 2014; Chang et al., 2020), while we are the first to report elevated levels of GSDMB and LECT2 in PDAC. Fourteen out of twenty one tumor-associated proteins highlighted in Figure 3B as potential protein targets for early detection or prognostic markers were validated by the orthogonal method of data-independent acquisition (DIA) mass spectrometry analysis, indicating the reliability of our reported tumor-associated proteins (Table S3).
Tumor-specific changes in PTMs, including phosphorylation and glycosylation, could provide additional options for PDAC diagnosis and prognosis. Compared to NATs, 4,908 phosphorylation sites (30% of the quantified sites) and 1,727 N-linked glycosites showed significantly increased abundance in PDACs (Table S3, adjusted p < 0.01). The proteins containing these modifications were related to GTPase activity regulation, cytoskeleton organization, extracellular structure organization, and integrin-mediated signaling (Table S3) (Jones et al., 2008). In general, the differential abundance of PTMs was similar to the differential abundance at the protein level while 45 N-linked glycosylation sites and 645 phosphosites were upregulated more than 2-fold without a corresponding increase in protein abundance (Figure 3D). Interestingly, some phosphosites showed highly specific regulation at the phosphorylation level. For example, while the protein abundance of RALGAPA2 was decreased in PDACs (Figure S3C), two phosphosites were increased more than 2-fold at S486 and S696 while three others were decreased or similar to NAT (Figure S3D). RALGAPA2 is related to KRAS signaling in pancreatic cancer (Beel et al., 2020) and exploring the function of these specific sites in future studies may be warranted.
Finally, many of these PTMs, in addition to protein abundance, were associated with patient prognosis. Overall, the prognostic value of PTMs was similar to that of the protein (Figure 3E, Table S3). However, a particular N-linked glycosylation on APOD was associated with better overall survival, while total protein abundance did not (Figure 3F). Although decreased expression of APOD is associated with better prognosis in other cancer types, little is known about the role of this glycosylation site and its effect on APOD function (Ren et al., 2019). Additionally, two phosphosites on PIGR, which is involved in the epithelial-mesenchymal transition, are associated with better prognosis, while a site on ERRFI1, an ERBB signaling regulator, is associated with worse survival (Figure 3E).
Together, these proteins and PTMs, including phosphorylation and N-linked glycosylation, provide focused targets for future investigation as possible PDAC diagnostic and prognostic markers.
Targeting Glycoprotein Biosynthesis for Early Detection and Therapeutic Intervention
Cell surface, membrane, and secreted proteins are more likely to be glycosylated than proteins derived from other cellular compartments (Zhang et al., 2003). Aberrations in glycoprotein expression and their glycosylation play a critical role in cancer progression (Clarke et al., 2005; Engle et al., 2019; Hart and Copeland, 2010; Varki, 2017). Most importantly, extracellularly exposed proteins are easily accessible as potential immunotherapy targets and can be used to detect disease (Li et al., 2005); thus, glycoproteins not only make up the majority of tumor markers currently approved by US Food and Drug Administration (FDA), but also constitute the major biochemical class of therapeutic targets (Sokoll et al., 2008). Glycoproteomic analysis of PDACs and NATs identified 75 N-linked glycoproteins upregulated more than 2-fold in tumors (Figure 4A, S4A–B, and Table S4). Of these, 57 were reported in the Pancreatic Cancer Database (Harsha et al., 2009; Thomas et al., 2014), and 18 were newly identified in this study. Most of up-regulated N-linked glycoproteins were secreted or membrane proteins (Figure S4A). Forty-eight out of seventy-five tumor-associated glycoproteins were further validated by DIA analysis (Table S4). Gene Set Enrichment Analysis (GSEA) focused on altered N-linked glycoproteins showed that epithelial mesenchymal transition (EMT), collagen formation, and complement and coagulation cascades are the top three enriched pathways among up-regulated N-linked glycoproteins, while protein processing in ER, translation, N-glycan biosynthesis are the top three enriched pathways among down-regulated N-linked glycoproteins (Figure S4B). In addition, mucin-type O-linked glycoproteins, including MUC1, MUC3A, MUC5AC, MUC5B, MUC 13, and MUC16 associated with CA19-9 antigen (Akagi et al., 2001; Hollingsworth and Swanson, 2004; Yue et al., 2011) were significantly up-regulated in tumors, as well as early stage tumors, relative to NATs and/or normal duct tissues (Figure S4C). Of these, MUC1, MUC5AC, MUC5B, and MUC13 were further validated by DIA analysis (Table S4). We further discriminated tumor vs normal ductal tissue N-linked glycoprotein expression based on disparate hotspot KRAS mutations (G12D, G12V, G12R, Q61H) (Figure 4B). Interestingly, CEACAM5 and CEACAM6 were significantly upregulated in tumors with KRAS G12D, G12V, and Q61H, but not G12R, mutations (Figure 4B, Figure S4D). CEACAM5 and CEACAM6 are members of the carcinoembryonic antigen (CEA) family and are highly abundant cell surface glycoproteins serving as adhesion molecules in the extracellular matrix (ECM). CEACAM6 is a poor prognostic marker for patients with PDAC, and CEACAM6 overexpression has been associated with low cytolytic T-cell activity in PDAC (Pandey et al., 2019). A focused evaluation of N-linked glycoprotein expression in low stage tumors revealed several candidates for early detection or treatment (Figure 4B, Table S4) including galectin binding protein 3 (LGALS3BP) (Figure S4E). In addition to N-linked glycoprotein expression quantified from proteomic data, their glycosylated forms quantified from glycoproteomic data have provided unique expression patterns in tumors. Hemopexin (HPX) and collagen type VI alpha 1 chain (COL6A1) displayed similar expression across cancers, early stage cancers, NATs, and normal ductal tissues in total protein levels, while abundance differences of a sialylated glycan (N3H4S1) and a high mannose glycan (N2H8) on SWPAVGNCSSALR (HPX) and NFTAADWGQSR (COL6A1), respectively, was observed in PDAC tumors (Figure S4E).
Figure 4. Glycoproteomic characterization identified N-linked glycoproteins and glycosylation enzymes for the early detection or therapeutic intervention.
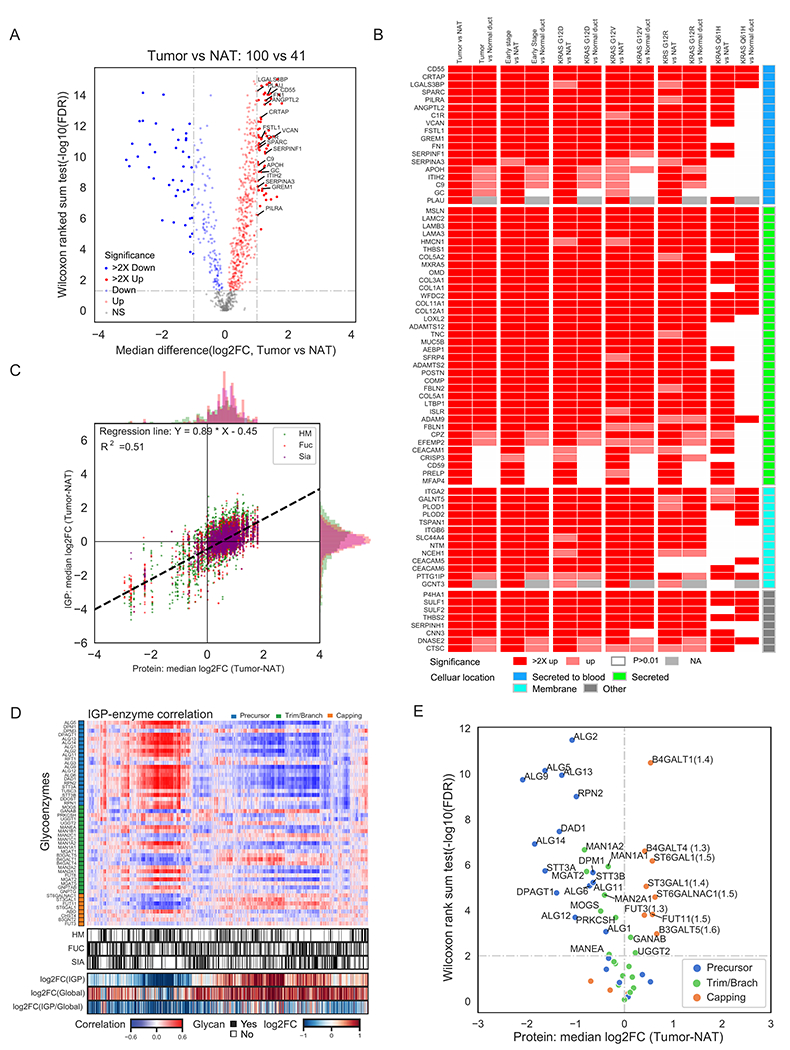
A) Differential expression analysis of N-linked glycoproteins in tumors to identify the most significant secreted (highlighted) and membrane N-linked glycoproteins elevated in tumors compared to NATs. B) Up-regulation of N-linked glycoproteins in all tumors, early stage tumors or tumors with different hotspot KRAS mutations relative to NATs and normal ductal tissues (Normal duct) at N-linked glycoprotein expression levels. C) Comparative analysis of the expression of global proteomics and glycoproteomics. IGP: intact glycopeptides; HM: high mannose type glycopeptides; Fuc: fucosylated glycopeptides; Sia: sialylated glycopeptides. D) Association of intact glycopeptide abundance and protein levels of glycosylation enzymes in tumors and NATs. E) Differential protein expression of N-linked glycosylation enzymes between tumors and NATs.
Further examination of our glycoproteomic dataset revealed variable glycan modifications on individual N-linked glycoproteins across individual tumors. This observation presents an opportunity to target glycoproteins harboring specific glycan forms as complementary to the current mRNA and protein alterations for diagnosis or therapeutic intervention. The biosynthesis of N-linked glycoproteins is regulated mainly by two factors, the glycoprotein substrates and glycosylation enzymes for glycan synthesis and conjugation to N-linked glycoproteins. We investigated the role of N-linked glycoprotein substrate abundance on the regulation of glycosylation. We first looked at the changes in intact glycopeptide (IGP) expression derived from glycoproteomic data and total N-linked glycoprotein expression derived from global proteomic data (Figure 4C). Although the alteration patterns of IGPs were mainly positively correlated to the protein abundances of N-linked glycoprotein substrates modified by different glycans (Figure 4C), IGP and protein features were not consistent, as we delineated heterogeneity of IGP abundances from the same protein displaying distinct glycan branching patterns across the pathological tissue types. Overall, N-linked glycoproteins upregulated in the tumors were most modified by complex glycans with sialic acids and/or fucoses, and N-linked glycoproteins downregulated in tumors were mainly modified by high mannose glycans (Figure 4C). These data indicate that focusing on sialylated and/or fucosylated glycans of the N-linked glycoproteins upregulated in PDAC may increase the specificity of markers for cancer. We next examined the intrinsic mechanism of these glycosylation alterations based on the abundance levels of glycosylation biosynthesis enzymes, wherein we correlated the abundance of IGPs from each tumor and non-tumor sample with the protein abundance of the glycosylation enzymes that were identified and quantified from the same sample using global proteomics (Figure 4D). We found that the intact glycopeptides with glycosylation of sialylated or fucosylated glycans were positively correlated with the expression of glycosylation enzymes involved in glycan trimming/branching and capping such as FUT3, B4GALT1, ST3GAL1, MGAT5, GANAB, B4GALT4, FUT11, and MAN2C1 (Figure 4D). We then compared the glycosylation enzyme expression levels between tumors and NATs, revealing up-regulation of glycosylation enzymes including ST6GAL1, ST3GAL1, FUT3, FUT11, B4GALT1, B4GALT4, B3GALT5, and ST6GALNAC1 in tumors (Figure 4E). Of these, FUT3, FUT11, B4GALT4, and B4GALT1 were further validated by DIA analysis (Table S4). Some of these glycosylation enzyme changes, such as elevated ST6GAL1, ST3GAL1, and B4GALT1, in tumors, were not observed at the transcriptomic level (Figure S4F), highlighting the added value of proteomics and glycoproteomics in our multi-omic analysis. ST6GAL1 and ST3GAL1 regulate sialylation, while FUT3 and FUT11 are responsible for fucosylation, in line with our observation that PDAC up-regulated proteins are mainly modified by sialylated and/or fucosylated glycans. ST3GAL1 is upregulated in several cancer types including thyroid cancer, lung cancer, liver cancer, pancreatic cancer, breast cancer, and ovarian cancer based on the transcriptomic data from TCGA studies (The Cancer Genome Atlas Research Network, 2011, 2012a, b, 2014a, b, 2017a, b) and has been reported to be associated with resistance to chemotherapy (Wu et al., 2018). Inhibition of these enzymes will likely attenuate increased sialylation and fucosylation glycan branching that was found on most tumor up-regulated glycoproteins, and serves as a potential therapeutic strategy for PDAC.
In summary, integrating global proteomic and glycoproteomic measurements identified proteins and glycoproteins overexpressed in PDACs, and these proteins and glycoproteins may find clinical utility as candidates for early detection and/or therapeutic intervention.
Kinase and Substrate Co-regulation Reveals Potential Therapeutic Targets
Since tumors with KRAS driver mutations are difficult to treat via targeted therapy, effective therapeutic intervention for PDAC, known to have high frequency for KRAS mutations, has remained elusive (Uprety and Adjei, 2020). Protein phosphorylation is heavily involved in various signaling pathways during pancreatic carcinogenesis (Furuse and Nagashima, 2017; Ruckert et al., 2019). To investigate signal transduction pathways downstream of activated KRAS in search of alternative therapeutic targets, we analyzed protein phosphorylation events regulated by kinases on their respective phosphorylation substrates. By analyzing differential abundance of phosphopeptides between 41 tumor/NAT paired tissues, we stratified five phospho-substrates (MCM2, FLNA, BAD, MAPK6, and STAT3) corresponding to five kinases (CDK7, AKT1, PAK1, PAK2, and SRC), for which inhibitors are either FDA-approved or under investigation (Wishart et al., 2018; Yeo et al., 2016) (Figure 5A). Previous studies have shown that elevated phosphorylated substrates are related to S-phase entry/progression (CDK7-MCM2), and inhibition of CDK7 can result in cell-cycle arrest and suppress tumor progression (Clark et al., 2019; Montagnoli et al., 2006; Sava et al., 2020). AKT1 is a kinase downstream of KRAS (Figure 5B). The elevation of AKT1 expression in almost all tumors is a consequence of nearly universal KRAS mutations, which in turn stimulate the progression of G1/S transition, with consequent stimulation of proliferative activity (Cai et al., 2018; Jones et al., 2008; Pelosi et al., 2017). A class I p21-activated kinase (PAK), PAK1, showed higher expression in more than 70% of tumors, with its subsequent activity in PDAC tumors supported by elevated phosphorylation of its substrate (BAD-S134) (Figures 5A). Apoptosis induced by BAD is inhibited upon phosphorylation of BAD-S134 by PAK1, thus promoting cell proliferation and survival (Polzien et al., 2011; Ye and Field, 2012). PAK1 can be activated by direct interaction with RAC1 (Fan, 2020), and RAC1 was up-regulated in most tumors (Figure 5B and 5C). PAK1 is an important effector of several receptor tyrosine kinases, such as MET (Rane and Minden, 2019; Zhou et al., 2014). We observed concordant up-regulation of MET and PAK1 (Figure 5B), and we found that MET was concordantly up-regulated with KRAS, RAC1, PAK1, PAK2 at both the transcription and protein levels in tumors as well (Figure 5C). The MET/PAK1 signaling axis drives pancreatic carcinogenesis via regulation of cell proliferation, motility, and regulation of cytoskeletal remodeling (Zhou et al., 2014). Furthermore, constitutive activation of the SRC/STAT3 signaling axis enhances hepatocyte growth factor (HGF) promoter activity, which in turn activates PAK1 via HGF/MET signaling (Aznar et al., 2001; Lee et al., 2019; Wojcik et al., 2006; Yuan et al., 2015). Another member of the class I PAKs, PAK2, was also up-regulated in almost 90% of tumors and likely responsible for elevated phosphorylation of MAPK6-S189 (Figures 5A–C). The phosphorylation process is critical for the formation of the MAPK6-Prak complex for MAPK6 signaling, suggesting an important role for PAK2 activity in regulating atypical MAPK signaling associated with cell motility (De la Mota-Peynado et al., 2011). Expanding the phosphoproteomic analysis to include the normal ductal tissues showed that the expression profiles of the class I PAKs and the other kinases as well as their substrates in PDAC tumors were substantially different from NATs and/or normal ductal tissues, suggesting that these proteins were PDAC-associated kinases (Figures 5D, S5A–B). Furthermore, the differential expression patterns of four of these kinases (PAK1, SRC, AKT1, and CDK7) were confirmed by DIA analysis (Table S5).
Figure 5. Kinase and substrate co-regulation.
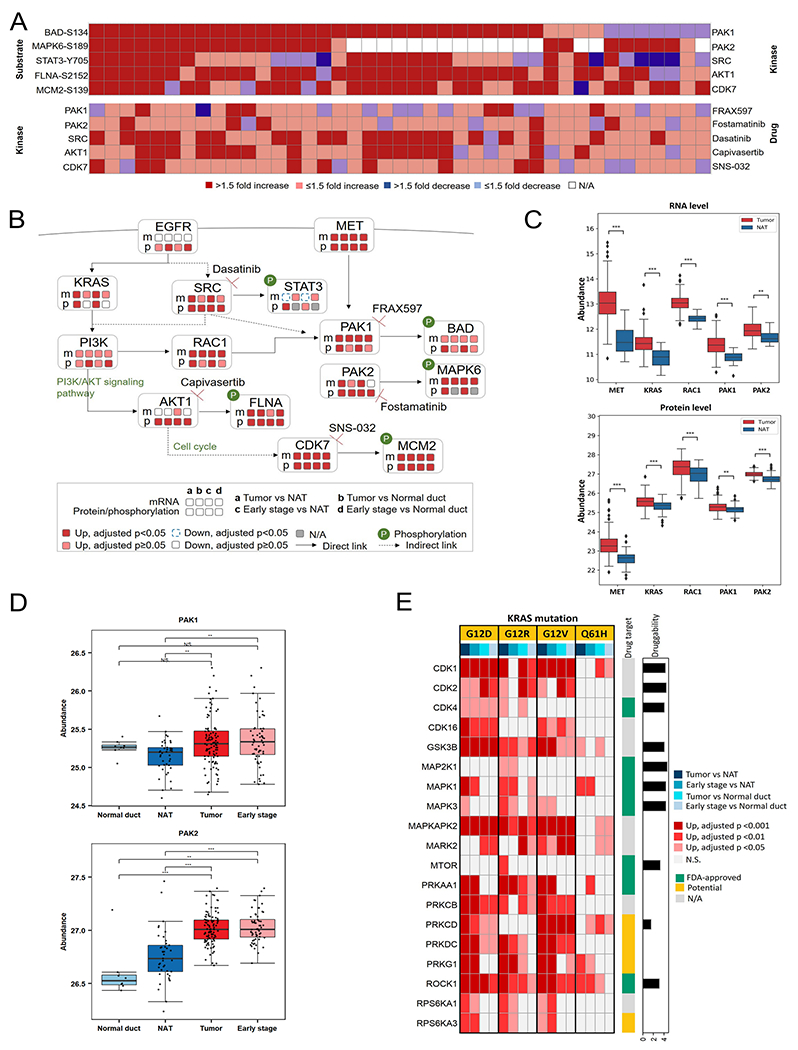
A) Differential abundances between 41 tumor/NAT paired tissues of stratified phospho-substrates (top) and their associated kinases (bottom). B) Pathways based on the selected phospho-substrates and kinases, with relevant drugs. Expression changes on mRNA and/or protein/phosphosites between PDAC tumors and NATs/Normal ductal tissues are labeled. C) Expression profiles of PAK1- and PAK2-associated proteins at transcriptomics and proteomics levels. D) Expression profiles of the class I p21-activated kinases (PAKs) in Normal duct, NAT, Tumor, and Early stage. E) Heatmap showing kinases elevated in different KRAS hotspot mutations. The kinases were identified based on their up-regulated phospho-substrates. The drug target annotation is from Human Protein Atlas (https://www.proteinatlas.org/) alongside with the log-transformed druggability score based on the drug sensitivity evaluated in PDAC cell lines from Genomics of Drug Sensitivity in Cancer (https://www.cancerrxgene.org/). Normal duct: normal ductal tissues; NAT: normal adjacent tissues; Tumor: all PDAC tumors; Early stage: Stage I and II PDAC tumors. Asterisks represent significant differences between two groups (Benjamini-Hochberg adjusted p): *p < 0.05; **p < 0.01; ***p<0.001; N.S., not significant. The list of kinase inhibitors/drugs is not exhaustive.
By evaluating phosphosite expression changes in tumors with different KRAS hotspot mutations relative to NATs (>2 fold increase with adjusted p<0.05, Figure S5C), we further stratified 19 kinases (Figure 5E, Table S5), including seven FDA-approved drug targets (Hobbs et al., 2020, Uhlen et al., 2015). These different patterns of kinase expression suggest alternative therapeutic targets associated with specific KRAS mutations. Given the importance of the class I PAKs to PDAC, combined inhibition of PAK1/2 and KRAS downstream pathways, such as MAPK/ERK and PI3K/AKT/mTOR, may increase therapeutic benefit by maximizing inhibition of tumor cell proliferation, motility, and signaling to the cytoskeleton (De la Mota-Peynado et al., 2011; Folkes et al., 2008; Ozkan-Dagliyan et al., 2020; Zhou et al., 2014).
In summary, we identified over-expressed substrates and their corresponding kinases, uncovering multiple potential targets that can be further explored with therapeutic intent.
Immune-Cold PDACs Associated with Endothelial Cell Remodeling, Glycolysis, and Cell Junction Dysregulation
One limitation of molecular analyses of tumor and normal tissues is that they do not fully dissect the interaction between tumor-intrinsic biology and microenvironment dynamics. This knowledge gap is particularly consequential for PDAC, which is heavily driven by tumor microenvironmental features (Collisson et al., 2019). Here, we classified tumors based on microenvironmental cell signatures, with an emphasis on delineating the degree of immune infiltration, as targeting immune modulators has shown promise in the treatment of a variety of cancer types (Yang, 2015). Unlike other tumors, such as melanoma, PDACs are resistant to immune checkpoint inhibitors in general (Hilmi et al., 2018), and leveraging a comprehensive proteogenomic approach may provide insight into this phenomenon.
We used a transcriptomics-based deconvolution method (Aran et al., 2017) to delineate the cellular composition of all 140 PDAC tumors in this study (i.e., including tumors with low neoplastic cellularity as these more fully represent stromal components of the tumor), which was further validated by DNA methylation-based tumor deconvolution (Figures 6A and S6A–C). Samples were classified into four clusters based on tumor/stromal/immune cell composition (Figure 6A). Of particular interest was a small portion (Cluster D) of tumors with higher CD8+ T-cell infiltration accompanied by increased expression of cytotoxic enzymes and immune checkpoint molecules. We annotated samples in this cluster as “immune hot” tumors. Histologic review of these cases confirmed prominent inflammatory infiltrates associated with the tumor component (Figures S6D). Nevertheless, in one case (C3N-00303), the immune signature was likely a result of the inclusion of a lymph node in the tissue harvested, highlighting the critical importance of histologic review of biosamples used in the study of any cancer type (Figure S6E). Samples in clusters A, B and C showed little immune infiltration. Because cluster A was enriched with non-neoplastic acinar and islet cells, as shown by the deconvolution and RNA subtyping, we considered only clusters B and C as true “immune cold” tumor groups (Figure S6F). Noticeably, immune hot tumors were also enriched with endothelial cells, and the enrichment was supported by an independent deconvolution tool (Becht et al., 2016) (Figure 6B). In addition, cell type association network analysis confirmed strong associations between endothelial cells and cytotoxic immune cells (Figure S6G).
Figure 6. Delineation of the cellular composition of PDAC tumors and identification of biological events accounting for the immune-cold phenotype.
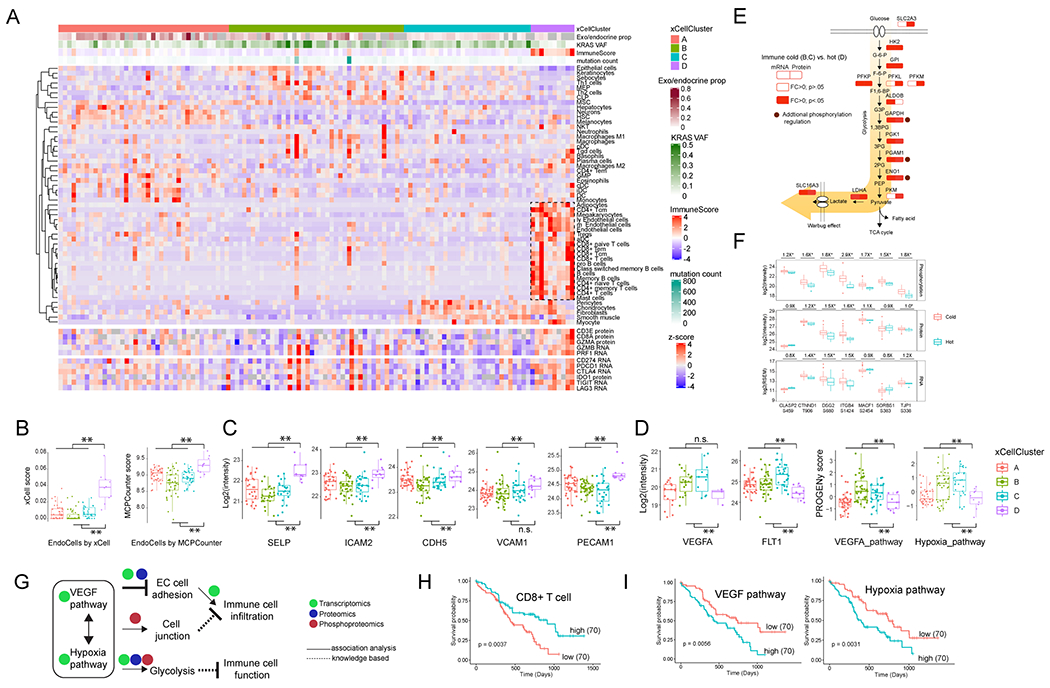
A) The 140 tumors were classified into four clusters based on tumor composition (upper heatmap). The cytotoxic T cells, together with endothelial cells enriched in the cluster D are highlighted by a rectangle. The expression of immune cytotoxic factor and checkpoint genes is shown in the sample order (lower heatmap). B) The comparison of endothelial cells between immune hot and cold samples based on the in silico deconvolution using either xCell or MCPCounter. C) Immune cold tumors have reduced endothelial adhesion proteins. D) Immune cold tumors have upregulated VEGF and hypoxia pathways. B-D: **p<0.01, n.s. not significant, Student’s t-test. E) The immune cold tumors had higher levels of glycolytic pathway components. Shown are the comparison of these components between immune cold vs hot at both the RNA and protein level. Some of the pathway components are identified with known functional phosphosites and are highlighted by brown circles. F) Phosphorylation pathway enrichment showed that the immune cold samples have higher phosphorylation levels of cell junction proteins. Shown are immune cold vs. hot fold changes for protein phosphorylation, protein expression and RNA expression. *p<0.05, Student’s t-test. G) The possible working model. VEGF and hypoxia pathways are associated with aberrant tumor vasculature and a hypoxic tumor microenvironment, and downregulated endothelial cell adhesion proteins, increased glycolysis and cell junction further inhibit the cytotoxic immune infiltration and function. H) The clinical outcome associated with CD8+ T cells. I) The clinical outcome associated with VEGF and hypoxia pathway activities. H-I): The p values were derived from logrank test and numbers in parentheses represent sample sizes for each group.
Endothelial cells represent a physical connection between the circulatory system and tumor cells (Klein, 2018), and endothelial cell adhesion proteins are essential for immune cell recruitment and frequently downregulated in tumor-associated vasculature (Schaaf et al., 2018). Accordingly, immune cold tumors in our cohort had reduced expression of endothelial adhesion proteins (Figure 6C). Meanwhile, these tumors also showed elevated activity of VEGF and hypoxia pathways, as indicated by expression of VEGF and its receptor, as well as the inferred pathway activities (Schubert et al., 2018) (Figure 6D). Both VEGF and hypoxia pathways are integral to the remodeling of endothelial cells during tumorigenesis (Petrova et al., 2018). Together, these results support an association of endothelial cell remodeling and suppressed immune infiltration in immune cold PDACs.
To further characterize the mechanisms underlying the immune cold phenotype, we performed pathway analysis using RNA, protein, and phosphorylation data. Immune cold samples had higher levels of glycolysis (Figures 6E and S6H), including enrichment of enzymes responsible for the generation and secretion of lactate, a known immune suppressor in tumor microenvironment (de la Cruz-Lopez et al., 2019). Phosphoproteomics data also showed increased phosphorylation of glycolysis pathway components, such as GAPDH, PGAM1 and ENO1. In addition, phosphorylation-specific pathway enrichment analysis showed that the immune cold samples had higher phosphorylation levels of cell junction proteins (Figure S6I); this feature was not as robustly detected at the transcriptomic or proteomic levels (Figure 6F). Cell junction proteins play an important role in regulating endothelial cell permeability for small molecules and immune cell infiltration (Daneman and Prat, 2015, Radeva and Waschke, 2018). Here, our results suggest that the dysregulation of protein phosphorylation in the cell junction components might represent an additional mechanism of immune exclusion in PDAC tumors.
Together, these data suggest that endothelial cell remodeling, accompanied by elevated VEGF and hypoxia pathways, increased glycolysis, and cell junction dysregulation might collectively inhibit immune cell infiltration and function (Figure 6G). Inhibiting these biological processes, especially glycolysis and endothelial cell remodeling, both of which have been actively targeted in multiple cancer types (Annan et al., 2020; Pelicano et al., 2006), may be therapeutically exploited to boost antitumor immunity in immune cold PDACs. This is supported by associating clinical outcomes with these processes. While CD8+ T cell infiltration was a favorable prognostic signature, elevated VEGF and hypoxia pathway signaling both were associated with decreased survival (Figure 6H–I).
In summary, multi-omics integration revealed immune-hot subtype tumors that may benefit from immunotherapy, as well as the underlying mechanisms associated with immune-cold subtypes, including endothelial cell remodeling, glycolysis, and cell junction dysregulation.
Proteogenomic Subtypes with Strong Prognostic Relevance
There are three main transcriptomics-based subtyping strategies for PDAC: Collisson (Collisson et al., 2011), Bailey (Bailey et al., 2016), and Moffitt (Moffitt et al., 2015). We applied them to the entire set of tumors to explore inter-sample heterogeneity (Figure S7A). Consistent with a previous report (The Cancer Genome Atlas Research Network, 2017b), some of these molecular classifications overlapped significantly, such as “ADEX” (Bailey) and “exocrine-like” (Collisson), “Classical (Collisson)” and “pancreatic progenitor” (Bailey), and “squamous” (Bailey), “quasimesenchymal” (Collisson) and “basal-like” (Moffitt) (p<0.0001, Fisher’s exact test). Notably, five adenosquamous carcinoma samples in our cohort were classified into “squamous” (Bailey), “quasimesenchymal” (Collisson), or “basal-like” (Moffit) groups, in line with the understanding for this histological pancreatic cancer subtype (Boecker et al., 2020; Lenkiewicz et al., 2020; Moffitt et al., 2015). Non-negative matrix factorization (NMF)-based proteogenomics subtyping using CNV, mRNA and protein expression, and phosphosite and glycosylation site abundance data from all 140 PDAC tumors (i.e., including tumors with low neoplastic cellularity) revealed four clusters with significant overlap with RNA-subtypes (Figures S7B–C, Table S7A). Both RNA-based and multi-omics-based subtyping results for the whole cohort were heavily confounded by tumor purity and cell type composition (Figures S7B–G).
In order to partially mitigate the impact of tumor purity on subtyping, we further limited the NMF-based proteogenomics subtyping to the 105 PDAC tumors with sufficient tumor neoplastic cellularity. This analysis revealed two clusters (C1 and C2, Figure 7A). The two clusters showed significant overlap with Moffitt classical and Moffitt basal-like RNA subtypes, respectively, and hereafter referred to as proteogenomic classical and proteogenomic basal-like subtypes. Since the Moffitt subtypes were derived by using a tumor-intrinsic gene signature (Moffitt et al., 2015), the difference between the two proteogenomic subtypes is more likely to reflect tumor-intrinsic biological signals. Pathway level analysis of cluster-specific features showed that the proteogenomic classical subtype was enriched with features associated with pancreas beta cells, bile acid metabolism, fatty acid metabolism, and KRAS signaling suppression, whereas the proteogenomic basal-like subtype was enriched with features associated with epithelial mesenchymal transition, DNA repair, glycolysis, hypoxia, apoptosis, reactive oxygen species pathway, and multiple proliferation and signaling pathways (Figure 7B).
Figure 7. Proteogenomic subtyping of 105 high-purity tumors using gene copy number, mRNA, protein, phosphosite, and glycosite abundances largely separated tumors to two subtypes.
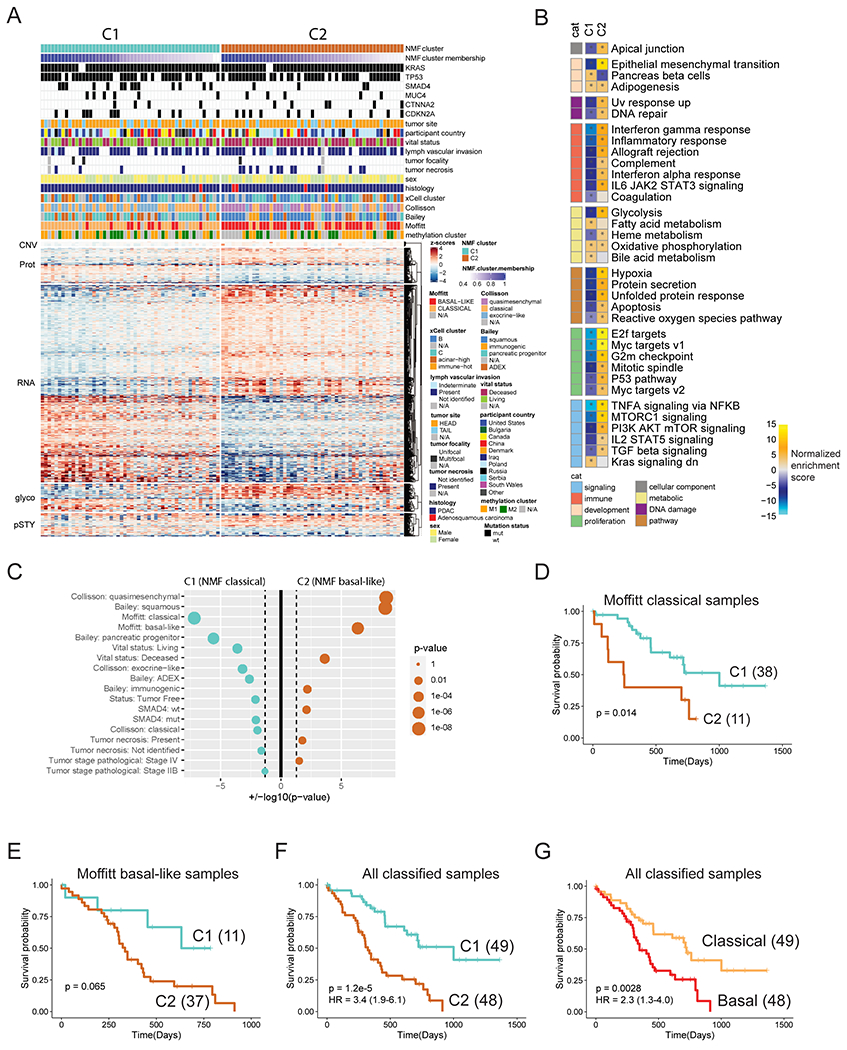
A) Heatmap depicting the z-scored abundances of proteogenomic features separating the two clusters as determined by NMF. Cluster membership scores indicating the strength of association of each sample with a given cluster were calculated as proportional weights. The columns of the matrix are ordered by proteogenomic subtype and decreasing cluster membership score. B) Pathway-level analysis on proteogenomic subtypes. Shown are pathway activity scores of cancer hallmark gene sets derived from single sample Gene Set Enrichment Analysis (ssGSEA) applied to the vector of feature weights characterizing each cluster. Asterisks indicate gene sets with FDR < 0.01. cat: category. C) Overrepresentation analysis of clinical variables, RNA-subtypes and somatic mutations in each proteogenomic subtype (Fisher’s exact test). Size of the dots scale with the significance of association. Cyan dots indicate association with the C1 (NMF classical), orange dots with the C2 (NMF basal-like) subtype. Vertical dashed lines correspond to nominal p-value of 0.05. D-G) Kaplan-Meier Plots comparing the survival outcomes between (D) Moffitt classical samples assigned into proteogenomic classical cluster (C1) and proteogenomic basal-like cluster (C2), (E) Moffitt basal-like samples assigned to the two proteogenomic clusters, (F) the two proteogenomic clusters, and (G) the two Moffitt subtypes. The p values were derived from logrank test and numbers in parentheses represent sample sizes for each group. The hazard ratios (HRs) were derived from Cox PH regression and shown as “HR (95% confidence interval)”.
Despite the overall concordance between proteogenomic subtypes derived from multi-omics data and the Moffitt subtypes derived from RNA-seq data alone, the classification of 22 tumors was inconsistent (Figures 7A–C). Eleven Moffitt basal-like tumors were classified as proteogenomic classical, and eleven Moffitt classical were classified as proteogenomic basal-like. Interestingly, splitting the Moffitt classical or basal tumors according to the proteogenomic clusters revealed a trend of distinct prognostic outcomes (Figures 7D–E). Concordantly, we observed that for the 97 PDAC samples with both proteogenomic and Moffitt assignments, the proteogenomics-dichotomized subtypes showed stronger prognostic separation than the Moffitt-dichotomized subtypes (CoxPH HR 3.4 vs 2.3 comparing favorable to adverse survival) (Figures 7F–G). We further interrogated the 392 proteins and 258 phosphosites with significant prognostic values (adjusted p-value <0.15, CoxPH regression, Tables S7E–F) and found that they were more likely to show differential abundance between the two proteogenomic subtypes (69% of the proteins and 78% of the phosphosites) than between the two Moffitt subtypes (39% of the proteins and 38% of the phosphosites).
Furthermore, we identified 1361/484 RNAs, 84/101 proteins, 364/217 phosphorylation sites, and 397/531 glycosylated peptides associated with proteogenomic subtype C1/C2 (Tables S7G–N). In order to examine alterations associated with C1 and C2, we focused on proteins, phosphorylation sites, and glycosylated peptides associated with the two subtypes (Table S7I–N). Among these, 37 and 47 proteins associated with C1 and C2 subtypes, respectively, have also been reported in the Pancreatic Cancer Database (Table S7I–J, Harsha et al., 2009; Thomas et al., 2014).
To perform in-depth analysis of the two subtypes, we correlated molecular markers, phosphorylation patterns of kinases, glycosylation enzymes, and therapeutic targets revealed by comprehensive proteogenomics (Figures 3–6) with C1 and C2 subtypes. The C2 subtype was associated with higher expression (adjusted p-value <0.05) of most of kinases highlighted in Figure S5 (e.g., AKT1 and CDK7) as well as those involved in MAPK/ERK and PI3K/AKT/mTOR signaling pathways, such as MTOR, MAP3K2, MAP4K4, and MET (Table S7O). We also performed GSEA correlating curated gene sets of targets of approved drugs and kinase inhibitors (downloaded from DSigDB, Yoo et al., 2015) with C1 and C2 subtypes (Table S7P). The analysis revealed an association of chemotherapeutic drugs with C1 (e.g. Docetaxel, Vinblastine, Cabazitaxel) and kinase inhibitors with C2 (e.g. PP-242 inhibits mTOR, CP466722 targets ATM/ATR, Sunitinib inhibits PDGFR/VEGFR), which were further supported by elevated expression of inferred corresponding drug targets in C1 or C2 subtypes (Figure S7H, Table S7P). For instance, elevated mTOR, AKT and ERK kinase expression and the enrichment of the PP-242 signature in C2 suggests that mTOR could be a potential therapeutic target in these patients. The glycan processing enzymes involving capping of elongated branches, e.g. FUT3, was upregulated in C1 subtype relative to C2 subtype (Table S7), suggesting potential therapeutic effects of systemic blockade of these capping processing of glycan synthesis in the treatment of PDAC patients belonging to C1 subtype. In addition, a comparative analysis of impact of genomic alterations on transcriptome, proteome, and phosphorylation between C1 and C2 subtypes revealed differences in the impact of genomic alterations (Table S7).
To investigate the association of clinical parameters with C1 and C2 subtypes, we used an expanded list of clinical features (Table S1). The C1 subtype correlated with longer patient survival and tumor free status, while C2 correlated with the presence of tumor necrosis and stage IV status (Figure 7C). Association of C2 with worse prognosis and tumor necrosis remained significant after excluding stage IV tumors.
Our binary proteogenomics subtyping focused only on the 105 samples with sufficient tumor purity to better understand tumor-intrinsic biology. On the other hand, the immune/microenvironment characterization was done using all 140 samples, as immune hot samples generally have low neoplastic purity (Figure S7I). When 140 samples were used for proteogenomic subtyping, four subtypes were observed (Figure S7B). Interestingly, eight out of nine immune hot samples comprised a subset of the C4 subtype (Figure S7I), which significantly overlapped with the Moffitt classical subtype (Figure S7B and C). By further comparing our microenvironment/immune profiling results to the studies focusing on the similar aspect of pancreatic cancer (Chan-Seng-Yue et al., 2020; Elyada et al., 2019; Maurer et al., 2019; Moffitt et al., 2015; Puleo et al., 2018), we identified that the microenvironmental features of immune-hot samples were more favorable for immune cell infiltration (Figure S6D).
Together, these results support the association of integrated proteogenomic subtyping with patient outcome. Further experimental investigation of the over activated proliferative and signaling pathways in the poor-prognosis proteogenomic basal-like subtype (Figure 7B) may facilitate the development of subtype-specific therapeutic strategies.
Discussion
In this report, we describe a comprehensive proteogenomic investigation of PDAC, that integrates multi-omic profiles to provide insights into the impact of genomic and epigenomic perturbations on gene and protein expression, as well as PTMs, including phosphorylation and, for the first time, glycosylation. To ensure a robust comparison of PDACs with pair-matched NATs and normal ductal tissues, we leveraged molecular, histological, and computational approaches to annotate neoplastic cellularity of the tumors in our cohort and the high acinar content of normal tissues, with a major aim of including only high quality samples in our analyses (Figure 1 and 2). Identification of high tumor cellularity samples based on KRAS VAF, mutation burden, methylation, and copy number alterations addressed the low neoplastic cellularity of this cancer type, allowing us to compare tumor-intrinsic molecular features among these tumors with disparate neoplastic cell content (Figure 1). The inclusion of 9 macrodissected normal ductal tissues facilitated appropriate cell type comparisons, enabling accurate comparisons of gene/protein expression patterns between ductal cancers and normal ducts, rather than normal acinar cells, yielded robust identification of potential targets for early detection, diagnosis, or therapeutic intervention (Figures 3, 4, 5, and 6). In addition, we matched these identified molecular changes and/or therapeutic options to our proteogenomic subtypes, suggesting potential targeted therapies that can be combined with first-line chemotherapies for subtype-specific therapeutic intervention (Figure 7).
We verified KRAS as the major driver gene in PDAC (Figure 1 and 2), in line with previous studies (Eser et al., 2014; The Cancer Genome Atlas Research Network, 2017b; Thompson et al., 2020). However, targeting the KRAS protein itself has failed due to its smooth surface topology and lack of a hydrophobic pocket for secure drug binding, leading to a dearth of approved KRAS-specific drugs, except for the compound, MRTX849, which has been approved for mutant KRAS G12C that is only present in <1% of PDACs (Berndt et al., 2011; Christensen et al., 2020; Vatansever et al., 2020). Interestingly, comparison of glycoprotein expression among tumors with different hotspot KRAS mutations (G12D, G12V, G12R, Q61H) revealed upregulation of CEACAM5 and CEACAM6 in PDACs with G12D, G12V, and Q61H mutations but not with mutant G12R (Figure 4B). CEACAM5/6 belong to the immunoglobulin superfamily, mediate cell migration, cell invasion, and cell adhesion via homophilic as well as heterophilic binding to other proteins, and protect neoplastic cells from undergoing anoikis (Beauchemin and Arabzadeh, 2013; Blumenthal et al., 2005b). Overexpression of CEACAM6 in pancreatic cancer has been associated with gemcitabine resistance as well as low cytolytic T-cell activity (Beauchemin and Arabzadeh, 2013; Pandey et al., 2019). Although the effect of anti-CEACAM5/6 monoclonal antibodies (mAbs) on normal tissues remains to be determined, mAbs MN-15 and MN-3 can impede metastasis in preclinical studies by reducing adhesion of tumor cells to endothelial cells and extracellular matrix (Blumenthal et al., 2005a; Blumenthal et al., 2005b; Govindan et al., 2009; Strickland et al., 2009). Thus, anti-CEACAM5/6 mAb coupled to first-line chemotherapies may benefit patients with PDACs harboring KRAS G12D, G12V, and/or Q61H mutations. Alternatively, inhibition of critical downstream targets and nodes orchestrated by constitutively activated KRAS is an attractive strategy for PDAC treatment. The MAPK/ERK and PI3K/AKT/mTOR pathways represent major targets for therapeutic intervention of PDAC, and multiple inhibitors of each of the pathways are clinically available (Eser et al., 2014). While drugs that block these pathways are being tested in the clinic, new efforts are underway to exploit previously unrecognized vulnerabilities, such as altered signaling networks, for novel targeted therapies (Ducreux et al., 2019; Sapalidis et al., 2019). Our integration of proteomic and phosphoproteomic measurements revealed that PAK1/PAK2 kinases were upregulated in most PDACs in our cohort (Figure 5) and these kinases have been reported to be critical effectors/regulators of vital signaling pathways that mediate cellular cytoskeletal motility, proliferation, and survival (Zhou et al., 2014). Positioned downstream of oncogene KRAS, inhibitors of PAK1/PAK2 have potential as new ways of targeting KRAS and could be coupled with inhibitors that target the canonical KRAS downstream MAPK/ERK and PI3K/AKT/mTOR pathways (Najahi-Missaoui et al., 2019; Semenova and Chernoff, 2017). Validating the therapeutic hypotheses through mechanistic experimentation (e.g. validating the importance of up-regulated kinases in PDAC patient-derived xenograft (PDX)-models using the inhibitors of these kinases) would be beyond the scope of this study since the tumors subjected to proteogenomic analyses from this study were not used to generate PDX-models. Nevertheless, the importance of some kinases and phosphorylation substrates identified in our study can be explored in the future or using publicly available datasets (Hobbs et al., 2020; Mer et al., 2019; Ozkan-Dagliyan et al., 2020).
PDAC is characterized by a highly suppressive tumor microenvironment, and intratumoral infiltration by cytotoxic T cells is low for most patients (Elyada et al., 2019; Hessmann et al., 2020). Although immunotherapies that target cytotoxic T lymphocyte antigen-4 (CTLA-4), programmed cell death protein-1 (PD-1), and programmed death-ligand 1 (PD-L1) significantly benefit patients with several solid malignancies such as melanomas, they are ineffective in patients with PDACs except for microsatellite instability-high (MSI-H) tumors that account for < 2% of PDACs (Goggins et al., 1998). The determinants of immune activation in PDAC are poorly understood, providing little therapeutic guidance (Balli et al., 2017). To dissect tumor microenvironment, we leveraged our multi-omics data and revealed that absence of endothelial cells associated with upregulation of VEGF and hypoxia pathway activities was, in turn, associated with immune cell exclusion in immune cold tumors (Figure 6). Modifying tumor endothelial cells into a normal endothelial cell phenotype could possibly be achieved by antiangiogenic therapy, such as sorafenib and NGR-TNF, with upregulation of leukocyte-endothelial cell adhesion molecules, and could possibly promote intratumoral immune cell infiltration (Allen et al., 2017; Elia et al., 2018; Ferrara et al., 2004). Hypoxia inducible factor-1 (HIF-1) is the main effector of the hypoxic microenvironment in pancreatic tumors and induces cell metabolism into glycolytic mode (Yuen and Diaz, 2014). Thus, therapies targeting HIF-1 activity, such as small molecules preventing the interactions of the HIF1-α and HIF1-β subunits, might also be beneficial for pancreatic immune cold tumors (Petrova et al., 2018).
We compared our microenvironment/immune profiling results to other studies. Among them, the study by Puleo et al., 2018 highlighted a subtype (named ‘desmoplastic’) with high expression of endothelial cell and immune cell marker genes. This is consistent with our overall characterization that the level of normal endothelial cells correlate with immune cell infiltration. We applied the Puleo subtyping scheme to our cohort and found that the immune hot samples were exclusively distributed in ‘desmoplastic’ and ‘immune classical’ subtypes, suggesting that our immune hot characterization was robust across different gene signatures.
The study by Moffitt et al., 2015 used NMF-based data dissection to extract tumor-intrinsic and stromal gene expression signatures. In our study, we have identified the Moffitt tumor-intrinsic subtypes (i.e. classical and basal-like) and found that they are consistent with our proteogenomics binary subtypes. To compare our microenvironment profiling to the Moffitt’s study, we further applied the Moffitt ‘normal’ and ‘activated’ stromal gene signature and found that the immune cold samples (i.e. xCell cluster B and C) showed reduced ‘normal’ stromal gene expression.
The study by Maurer et al., 2019 paired laser capture microdissection (LCM) and data deconvolution to understand tumor epithelial and non-epithelial events separately. We applied the tissue-specific gene signatures and the deconvolution method from Maurer’s study to our cohort. For the non-epithelial genes, we found that the immune hot samples (i.e. xCell cluster D) showed significantly higher expression of immune-related genes but not for extracellular matrix-related genes. The vast majority of PDAC tumors in our cohort display low CD8+ T-cell infiltration accompanied by decreased expression of cytotoxic enzymes and immune checkpoint molecules, in line with a predominantly immune-suppressive environment of PDAC revealed by single-cell RNA sequencing (Elyada et al., 2019).
These results corroborate our finding that the microenvironment of immune-hot samples (i.e. xCell cluster D) is more favorable for immune cell infiltration. Based on our multi-omics data, we further provide evidence to suggest that the lack of normal endothelial adhesion proteins accompanied by the activation of tumor-associated endothelial signals (e.g. VEGF signaling pathway) partially account for the compromised immune cell infiltration and function in the immune-cold samples.
N-linked glycosylation occurs in the endoplasmic reticulum (ER) and Golgi apparatus and is mediated by the activity of a series of glycosidases and glycosyltransferases (Bieberich, 2014; Cao et al., 2018). Abnormal expressions of sialylated glycoproteins have been uncovered in various solid malignancies including PDAC and have been associated with invasiveness and metastatic potential (Hsieh et al., 2017; Suzuki, 2019; Vajaria et al., 2016). Here we have shown that most up-regulated N-linked glycoproteins in PDAC are modified by sialylated glycans, consistent with up-regulation of ST6GAL1 and ST3GAL1 in PDACs relative to NATs (Figure 4). These tumor up-regulated N-linked glycoproteins were associated with vital signaling pathways involved in PDAC progression and metastasis, including EMT, TNFα, focal adhesion, and collagen formation (Figure S4B). Thus, inhibition of these sialyltransferases may attenuate PDAC cell growth, survival, and metastasis via abrogation of the functions of these N-linked glycoproteins (Garnham et al., 2019; Vajaria et al., 2016). Although administration of a sialic acid analog (3F-NeuAc) induced systemic blockade of sialylation in a mouse model, a deleterious “on target” effect was observed on liver and kidney function, suggesting the need to develop more selective sialyltransferase inhibitors for therapeutic use (Macauley et al., 2014).
Our proteogenomic subtyping focused on PDAC tumors with sufficient tumor neoplastic cellularity, revealing proteogenomic C1 and C2 subtypes that resemble Moffitt classical and basal-like subtypes, respectively (Figure 7). In addition, SMAD4 was more frequently mutated in C1 subtype and the TGF-beta signaling pathway was accordingly higher in C2 subtype (Figures 7B and 7C), both of which were consistent with the tumor-intrinsic distinctions of SMAD4 alterations and TGF-beta signaling pathway between classical and basal samples in previous study (Chan-Seng-Yue et al., 2020). We also correlated clinical parameters to our proteogenomic subtypes, and the C1 subtype correlated with longer survival and tumor free status relative to C2 subtype (Figure 7C), consistent with the observations in Moffitt classical subtype (Moffitt et al., 2015). Furthermore, the Moffitt classical tumors assigned to C2 had worse survival compared to those assigned C1, suggesting our proteogenomic subtyping may help to further stratify the Moffitt classification (Figure 7D and 7E). We linked our proteogenomic subtypes to molecular markers and/or treatment targets identified by our comprehensive proteogenomic characterization. Higher levels of key kinases and drug treatment features of kinase inhibitors were identified in C2 subtype, suggesting potential treatment benefit of combination of chemotherapy and kinase inhibitors for patients belonging to this subtype (Figure S7H, Table S4).
In total, this report exemplifies the unique and useful insights that can be gained when characterizing the disease state at multiple “omics” levels, enabling a deeper understanding of the functional consequences of genomic aberrations associated with PDAC. Integrating measurements of the transcriptome, proteome, phosphoproteome, and glycoproteome, and comparative profiling of PDACs, NATs, and normal ductal tissues enabled our detection of proteoforms associated with early stage PDAC, as well as identification of potential therapeutic targets that may find utility in the clinical setting. Overall, our study delineates the molecular features that drive the PDAC phenotypes, and provides a rich bioinformatic resource for future hypothesis-driven translational research.
Limitations of the Study
The objectives of this study were to comprehensively characterize PDAC tumors and NATs using genomics, epigenomics, transcriptomics, proteomics, phosphoproteomics, and glycoproteomics as well as to provide proteogenomic resources to decipher the impacts of genomic alterations in gene expression, protein abundances, and protein modifications. For these purposes, tissues collected by the CPTAC program are treatment-naïve and surgically resected. Consequently, there are inherent limitations to this study. First, although data on adjuvant patient treatment and outcome was sought, the present cohort comprises treatment-naïve samples, which limits extrapolation to metastatic disease treated with systemic therapy. In addition, adjuvant treatment regimens were non-standardized across multiple institutions participating in tissue collection, resulting in heterogeneous adjuvant therapies utilized for patient treatment. While treatment data from therapeutic drug clinical trials are needed to investigate treatment outcomes related to the observed proteogenomic subtypes, currently such data are limited in that clinical trials only generate transcriptomic data (O’Kane et al., 2020). Second, proteogenomic data provide rich resources for correlating different molecular alterations that are essential for hypothesis generation to decipher molecular functions or prediction of treatment options. However, causal effects of the correlations can not be determined from this study. The biological hypothesis or treatment prediction would need further validation using cell lines, PDX models, publicly available datasets, or clinical trials. Third, proteogenomic measurements of this study are deployed using bulk tumor and NAT tissues, where the impact of heterogeneity in cellularity and tumor microenvironment cannot be fully accounted for. Here, we addressed this limitation by selecting a subset of tissue samples with sufficient tumor cellularity for focused analyses. However, enrichment of tumor cellularity using laser capture microdissection or characterization of tissues through single cell analyses would be beneficial (Elyada et al., 2019; Maurer et al., 2019).
STAR Methods
LEAD CONTACT AND MATERIALS AVAILABILITY
This study did not generate new unique reagents. Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Hui Zhang (huizhang@jhu.edu).
DATA AND CODE AVAILABILITY
The raw proteomic data files generated during this study are available at the Proteomic Data Commons (PDC, https://pdc.cancer.gov/pdc/). Genomic, epigenomic, and transcriptomic data generated for this publication are available at the Genomic Data Commons (GDC, https://gdc.cancer.gov/). All processed data tables are available at PDC (https://pdc.cancer.gov/pdc/publications) and LinkedOmics (http://www.linkedomics.org/data_download/CPTAC-PDAC/).
The workflow described under ‘Multi-omics clustering’ has been implemented as a module for PANOPLY (https://github.com/broadinstitute/PANOPLY/) running on Broad’s cloud platform Terra (https://app.terra.bio/). The docker containers encapsulating the source code and required R-packages for NMF clustering and ssGSEA are available on Dockerhub (broadcptacdev/pgdac_mo_nmf:15, broadcptac/pgdac_ssgsea:5). The data evaluation tool has been implanted as a R package available in OmicsEV (https://github.com/bzhanglab/OmicsEV/). The codes for genomics data processing pipelines are available in https://github.com/ding-lab/.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Human Subjects
A total of 140 cases of patients with pancreatic cancer (135 PDACs and 5 pancreatic adenosquamous carcinoma) were carefully evaluated histologically and included in this study. Institutional review boards at tissue source sites, reviewed protocols and consent documentation adhering to the Clinical Proteomic Tumor Analysis Consortium (CPTAC) guidelines for study participation.
Clinical Data Annotation
A total of 140 participants (74 males, 66 females between the age group of 31-85) were collected for this study by 11 different tissue source sites (TSS) from 7 different countries. Clinical data were obtained from tissue source sites and aggregated by an internal database called the CDR (Comprehensive Data Resource) that synchronizes with the CPTAC DCC (Data Coordinating Center). Clinical data can be accessed and downloaded from the DCC. Demographics, histopathologic information, treatment and patient outcome information were collected and reviewed for consistency before deposition into the Proteomic Data Commons (PDC) and Genomic Data Commons (GDC). All histologic and radiologic details can be accessed from The Cancer Imaging Archive (TCIA) Public Access portal. The genotypic, clinical, geographical and other associated metadata is summarized in Table S1. The cohort consists of 53% male (n=74) and 47% female (n=66), in line with the previous observation of a slightly higher incident rate in men than in women (Kim et al., 2017; The Cancer Genome Atlas Research Network, 2017). Age distributions [31-40 (2.9%), 41-50 (9.3%), 51-60 (16.4%), 61-70 (42.9%), 71-80 (25.7%), and 81-90 (2.9%)] and stage distributions [I (16.4%), II (42.9%), III (30.0%), and IV (6.4%)] of the patients reflect the general incidence of surgically resected PDAC (The Cancer Genome Atlas Research Network, 2017).
METHOD DETAILS
Specimen Acquisition
The tumor, normal adjacent tissue (NAT), and whole blood samples used in this manuscript were prospectively collected for the CPTAC project. Treatment naïve patients scheduled for surgical treatment for a pancreatic mass suspected to be pancreatic ductal adenocarcinoma were considered. Patients who underwent cancer treatment more than ten years prior were included if the cancer was at a site other than the pancreas. Only histopathologically-defined adult pancreatic ductal adenocarcinoma (135) and adenosquamous carcinoma (5) were considered for analysis. 67 out of the 140 had matched normal tissue from non-neoplastic pancreatic tissue as an acceptable normal. To supplement the normal cohort, 9 additional normal macrodissected main pancreatic ductal tissues were collected from unmatched patients who underwent surgery for benign neoplasms. The tumor specimen weights ranged from 150 to 1000 milligrams. The average tissue mass was 258 mg. For most cases, three to four tumor specimens were collected. Each tissue specimen endured cold ischemia for less than 30 minutes prior to freezing in liquid nitrogen; the average ischemic time was 20 minutes from resection/collection to freezing. Specimens were flash frozen in liquid nitrogen. Histologic sections obtained from top and bottom portions from each case were reviewed by a board-certified pathologist and a disease specific expert pathologist to confirm the assigned pathology. Although there was no tumor nuclei cutoff, for samples to be deemed acceptable, the top and bottom sections had to contain an average of less than 20% necrosis as assessed histologically. Specimens were shipped overnight from the tissue source sites to the biospecimen core resource (BCR) located at Van Andel Research Institute, Grand Rapids, MI using a cryoport that maintained an average temperature of less than −140 °C. At the biospecimen core resource, specimens were confirmed for pathology qualification and prepared for genomic, transcriptomic, and proteomic analyses. Selected specimens were cryopulverized using a Covaris CryoPREP instrument and material aliquoted for subsequent molecular characterization. Genomic DNA and total RNA were extracted and sent to the genome sequencing centers. The whole exome and whole genome DNA sequencing and methylation EPIC array analyses were performed at the Broad Institute, Cambridge. Total RNA and miRNA sequencing were performed at the University of North Carolina, Chapel Hill, NC. Material for proteomic analyses were sent to the Proteomic Characterization Center (PCC) at the Johns Hopkins University, Baltimore, MD.
Genomic, Epigenomic, and Transcriptomic Sample Preparation and Data Acquisition
Sample Processing for Genomic DNA and Total RNA Extraction
Each primary tumor was obtained from a single sample from surgical resections, with a requirement of a minimum of 125 mg of tumor tissue and 50 mg of adjacent normal tissue. DNA and RNA were extracted from the tumor and normal specimens using a co-isolation protocol (Qiagen’s QIAsymphony DNA Mini Kit and QIAsymphony RNA Kit). Genomic DNA was also isolated from peripheral blood (3-5 mL) to serve as matched normal reference material. The Qubit™ dsDNA BR Assay Kit was used with the Qubit® 2.0 Fluorometer to determine the concentration of dsDNA in an aqueous solution. Only samples with enough DNA yield that passed quality control were sent for genomic characterization. RNA quality was quantified using the NanoDrop 8000 and had its quality assessed using Agilent Bioanalyzer. Only samples that passed RNA quality control and had a minimum RIN (RNA integrity number) score of 7 underwent RNA sequencing. Identity matching for germline, normal adjacent tissue, and tumor tissue was assayed at the BCR using the Illumina Infinium QC array.
Whole Exome Sequencing
Library Construction.
Library construction was performed as described in (Fisher et al., 2011), with the following modifications: the initial genomic DNA input shearing was reduced from 3 μg to 20-250 ng in 50 μL of solution. For adapter ligation, Illumina paired-end adapters were replaced with palindromic forked adapters, purchased from IDT, with unique dual-indexed molecular barcode sequences to facilitate downstream pooling. Kapa HyperPrep reagents in 96-reaction kit format were used for end repair/A-tailing, adapter ligation, and library enrichment PCR. During post-enrichment SPRI cleanup, elution volume was reduced to 30 μL to maximize library concentration and a vortexing step was added to maximize the amount of template eluted.
In-solution Hybrid Selection.
Libraries were pooled into groups of up to 96 samples. Hybridization and capture were performed using the relevant components of Illumina’s Nextera Exome Kit, following the manufacturer’s suggested protocol, except for a few modifications. The following modifications were made: all libraries within a library construction plate were pooled prior to hybridization, the Midi plate from Illumina’s Nextera Exome Kit was replaced with a skirted PCR plate to facilitate automation, and all hybridization and capture steps were automated on the Agilent Bravo liquid handling system.
Preparation of Libraries for Cluster Amplification and Sequencing.
After post-capture enrichment, library pools were quantified using qPCR (automated assay on the Agilent Bravo) using a kit purchased from KAPA Biosystems with probes specific to the ends of the adapters. Based on qPCR quantification, libraries were normalized to 2 nM.
Cluster Amplification and Sequencing.
Cluster amplification of DNA libraries was performed according to the manufacturer’s protocol (Illumina) using exclusion amplification chemistry and flowcells. Flowcells were sequenced utilizing sequencing-by-synthesis chemistry. Flowcells were then analyzed using RTA v.2.7.3 or later. Each pool of whole exome libraries was sequenced on paired 76 cycle runs with two 8 cycle index reads across the number of lanes needed to meet coverage for all libraries in the pool. Pooled libraries were then run on HiSeq 4000 paired-end runs targeting a depth of coverage of 300x for 122 tumor sample libraries and 150x for the remaining 18 tumor sample libraries and the NAT and blood normal sample libraries. The raw Illumina sequence data were demultiplexed and converted to fastq files with adapter and low-quality sequences trimmed out.
PCR-Free Whole Genome Sequencing
Preparation of Libraries for Cluster Amplification and Sequencing.
An aliquot of genomic DNA (350 ng in 50 μL) was used as the input into DNA fragmentation. Shearing was performed acoustically using a Covaris focused-ultrasonicator, targeting 385bp fragments. Following fragmentation, additional size selection was performed using a SPRI cleanup. Library preparation was performed using akit from KAPA Biosystems (KAPA Hyper Prep without the amplification module) and with palindromic forked adapters with unique 8-base index sequences embedded within the adapter (purchased from IDT). Following sample preparation, libraries were quantified using qPCR (KAPA Biosystems) with probes specific to adapter ends. This assay was automated using Agilent’s Bravo liquid handling platform. Based on qPCR quantification, libraries were normalized to 1.7 nM and pooled into 24-plexes.
Cluster Amplification and Sequencing (HiSeq X).
Sample pools were combined with HiSeq X Cluster Amp Reagents EPX1, EPX2, and EPX3 into single wells on a strip tube using the Hamilton Starlet Liquid Handling system. Cluster amplification of the templates was performed according to the manufacturer’s protocol (Illumina) with the Illumina cBot. Flowcells were sequenced with a target of 15x to 30x depth on HiSeq X utilizing sequencing-by-synthesis kits to produce 151bp paired-end reads.
Illumina Infinium Methylation EPIC Bead Chip Array
The MethylationEPIC array uses an 8-sample version of the Illumina Beadchip capturing > 850,000 DNA methylation sites per sample. 250 ng of DNA was used for the bisulfite conversation using the Infinium MethylationEPIC BeadChip Kit. The EPIC array includes sample plating, bisulfite conversion, and methylation array processing. After scanning, the data was processed through an automated genotype calling pipeline.
RNA Sequencing
Quality Assurance and Quality Control of RNA Analytes.
All RNA analytes were assayed for RNA integrity, concentration, and fragment size. Samples for total RNA-seq were quantified on a TapeStation system (Agilent, Inc. Santa Clara, CA). Samples with RINs > 8.0 were considered high quality.
Total RNA-seq Library Construction.
Total RNA-seq library construction was performed from the RNA samples using the TruSeq Stranded RNA Sample Preparation Kit and bar-coded with individual tags following the manufacturer’s instructions (Illumina). Libraries were prepared on an Agilent Bravo Automated Liquid Handling System. Quality control was performed at every step and the libraries were quantified using the TapeStation system.
Total RNA Sequencing.
Indexed libraries were prepared and run on HiSeq 4000 paired end 75 base pairs to generate a minimum of 120 million reads per sample library with a target of greater than 90% mapped reads, typically with four-sample pools. The raw Illumina sequence data were then demultiplexed and converted to FASTQ files, and adapter and low-quality sequences were quantified. Reads were mapped to the hg38 human genome reference and underwent significant QC steps: estimating the total number of reads that mapped, amount of RNA mapping to coding regions, amount of rRNA in sample, number of genes expressed, and relative expression of housekeeping genes. Samples passing this QA/QC were then clustered with other expression data from similar and distinct tumor types to confirm expected expression patterns. Atypical samples were then SNP typed from the RNA data to confirm source analyte.
miRNA-seq Library Construction.
miRNA-seq library construction was performed from the RNA samples using the NEXTflex Small RNA-Seq Kit (v3, PerkinElmer) and bar-coded with individual tags following the manufacturer’s instructions. Libraries were prepared on the Sciclone Liquid Handling Workstation and quality control checks were performed at every step. Libraries were quantified using a TapeStation system and an Agilent Bioanalyzer using the Small RNA analysis kit. Pooled libraries were then size selected according to NEXTflex Kit specifications using a Pippin Prep system (Sage Science).
miRNA Sequencing.
Indexed libraries were loaded on the Hiseq 4000 to generate a minimum of 10 million reads per library with a minimum of 90% reads mapped. The raw Illumina sequence data were demultiplexed and converted to FASTQ files for downstream analysis. Samples were assessed for the number of miRNAs called, species diversity, and total abundance.
Genomic, Epigenomic, and Transcriptomic Data Processing
Genome alignment
WGS, WES, and RNA-Seq data were harmonized by NCI Genomic Data Commons (GDC) https://gdc.cancer.gov/about-data/gdc-data-harmonization, to the hg38 human reference genome, version GRCh38.d1.vd1.
Whole-genome copy number variation
Copy number variation (CNV) was detected using BIC-Seq2 (module versions NBICseq-seg v0.7.2 and NBICseq-norm v0.2.4) (Xi et al., 2016) from WGS tumor and normal paired BAMs. A bin size of 100bp and a lambda of 3 for segmentation smoothing was used. We used the mean of overlapping segment data to further summarize the CNV data into gene-level copy number changes. We also used GISTIC2 v2.0.22 (Mermel et al., 2011) to integrate results from individual patients and identify focal genomic regions recurrently amplified or deleted in our samples. The threshold for gene or arm-level CNV status was 0.4 for copy number gains and −0.4 for losses.
Somatic Variant Calling
Somatic variants were called from WES tumor and normal paired BAMs using SomaticWrapper v1.6. SomaticWrapper merges and filters variant calls from four callers: Strelka v2.9.2 (Kim et al., 2018), VarScan v2.3.8 (Koboldt et al., 2012), Pindel v0.2.5 (Ye et al., 2009), and MuTect v1.1.7 (Cibulskis et al., 2013). SNV calls were obtained from Strelka, Varscan, and Mutect. Indel calls were obtained from Stralka2, Varscan, and Pindel. The following filters were applied to get variant calls of high confidence:
Normal VAF ≤ 0.02 and tumor VAF ≥ 0.01. The tumor VAF cutoff is set lower to account for the unique low neoplastic cellularity in PDAC.
Read depth in tumor ≥ 14 and normal ≥ 8
Indel length < 100 bp
All variants must be called by 2 or more callers
All variants must be exonic
Exclude variants in dbSNP but not in COSMIC
Germline Variant Calling
Germline variant calling was performed using the GermlineWrapper v1.1 pipeline, which implements multiple tools for the detection of germline INDELs and SNVs. Germline SNVs were identified using VarScan v2.3.8 (with parameters: --min-var-freq 0.10 --p-value 0.10, --min-coverage 3 --strand-filter 1) operating on a mpileup stream produced by samtools v1.2 (with parameters: -q 1 -Q 13) and GATK v4.0.0.0 (McKenna et al., 2010) using its haplotype caller in single-sample mode with duplicate and unmapped reads removed and retaining calls with a minimum quality threshold of 10. All resulting variants were limited to the coding region of the full-length transcripts obtained from Ensembl release 95 plus additional two base pairs flanking each exon to cover splice donor/acceptor sites. We required variants to have allelic depth ≥ 5 reads for the alternative allele in both tumor and normal samples. We used bam-readcount v0.8 for reference and alternative alleles quantification (with parameters: -q 10 -b 15) in both normal and tumor samples. Additionally, we filtered all variants with ≥ 0.05% frequency in gnomAD v2.1 (Karczewski et al., 2020) and The 1000 Genomes Project (Auton et al., 2015).
Pathogenic Germline Variant Classification
To predict the pathogenicity of germline variants, we annotate each variant with Variant Effect Predictor (VEP) and process them using the CharGer pipeline with the parameters from a previous pan-cancer TCGA study (Huang et al., 2018; Scott et al., 2019). Briefly, the CharGer pipeline considers pathogenic peptide changes from ClinVar, hotspot variants, minor allele frequency from ExAC, and several in silico analyses (such as Sift and Polyphen). Each predicted pathogenic variant was then manually reviewed.
DNA Methylation Microarray Processing
Raw methylation idat files were downloaded from CPTAC DCC and GDC. Beta values of CpG loci were reported after functional normalization, quality check, common SNP filtering, and probe annotation using Li Ding Lab’s methylation pipeline v1.1 (https://github.com/ding-lab/cptac_methylation). To derive the gene-level methylation, we focused on the probes located in the promoter region and simultaneously located in annotated CpG island and aggregate their levels by median (Clark et al., 2019). Resulting beta values of methylation were used for downstream analysis.
mRNA and Circular RNA Quantification
The hg38 reference genome and RefSeq annotations were used for the RNAseq data analysis and were downloaded from the UCSC table browser. First, CIRI (v2.0.6) was used to call circular RNA with default parameters and BWA (version 0.7.17-r1188) was used as the mapping tool. The cutoff of supporting reads for circRNAs was set to 10. Then we used a pseudo-linear transcript strategy to quantify gene and circular RNA expression (Li et al., 2017). In brief, for each sample, linear transcripts of circular RNAs were extracted and 75bp (read length) from the 3’ end was copied to the 5’ end. The modified transcripts were called pseudo-linear transcripts. Transcripts of linear genes were also extracted and mixed with pseudo-linear transcripts. RSEM (version 1.3.1) with Bowtie2 (version 2.3.3) as the mapping tool was used to quantify gene and circular RNA expression based on the mixed transcripts. After quantification, the upper quantile method was applied for normalization. The normalized matrix was log2-transformed and separated into gene and circular RNA expression matrices.
Gene Fusion Detection
We used three callers, STAR-Fusion v1.5.0 (Haas et al., 2019), INTEGRATE v0.2.6 (Zhang et al., 2016), and EricScript v0.5.5 (Benelli et al., 2012), to call consensus fusion/chimeric events in our samples. Calls by each tool using tumor and normal RNA-Seq data were then merged into a single file and extensive filtering is done. As STAR-Fusion has higher sensitivity, calls made by this tool with higher supporting evidence (defined by fusion fragments per million total reads, or FFPM > 0.1) were required, or a given fusion must be reported by at least 2 callers. We then removed fusions present in our panel of blacklisted or normal fusions, which included uncharacterized genes, immunoglobulin genes, mitochondrial genes, and others, as well as fusions from the same gene or paralog genes and fusions reported in TCGA normal samples (Gao et al., 2018), GTEx tissues (reported in STAR-Fusion output), and non-cancer cell studies (Babiceanu et al., 2016).
miRNA Quantification
miRNA-Seq FASTQ files were downloaded from GDC. We reported the mature miRNA and precursor miRNA expression in TPM (Transcripts Per Million) after adapter trimming, quality check, alignment, annotation, reads counting using Li Ding Lab’s miRNA pipeline https://github.com/ding-lab/CPTAC_miRNA. The mature miRNA expression was calculated irrespective of its gene of origin by summing the expression from its precursor miRNAs.
Proteomic, Phosphoproteomic, and Glycoproteomic Sample Preparation and Data Acquisition
Sample Processing for Protein Extraction and Tryptic Digestion
All samples for the current study were prospectively collected for the CPTAC PDAC project as described above and processed for mass spectrometry (MS) analysis at Johns Hopkins University. Tissue lysis and downstream sample preparation for global proteomic, phosphoproteomic, and glycoproteomic analysis were carried out as previously described (Mertins et al., 2018; Yang et al., 2018a). Each of cryo-pulverized pancreatic cancer tissues, normal adjacent tissues (NAT), and normal ductal tissues was lysed in lysis buffer (8 M urea, 75 mM NaCl, 50 mM Tris (pH 8.0), 1 mM EDTA, 2 μg/mL aprotinin, 10 μg/mL leupeptin, 1 mM PMSF, 10 mM NaF, Phosphatase Inhibitor Cocktail 2 and Phosphatase Inhibitor Cocktail 3 [1:100 dilution], and 20 μM PUGNAc) by vortexing for 20 sec. The lysed tissue was placed on ice for 15 min. This process was repeated one time. Cell debris was removed by centrifugation at 20,000 x g for 10 min at 4°C. The protein-containing supernatant was collected and measured by BCA assay (Pierce). The sample concentration was adjusted to 8 mg/ml with the lysis buffer, and an appropriate volume of the protein solution was used for the downstream reduction, alkylation and digestion. Proteins were reduced and alkylated with dithiothreitol (DTT, ThermoFisher, 5 mM, 37°C, 1h) and iodoacetamide (IAM, Sigma, 10 mM, room temperature (RT) in the dark, 45 min), respectively. The reduced proteins were diluted 1:4 with 50 mM Tris-HCl (pH 8.0) to reduce urea concentration and digested with LysC (Wako Chemicals, an enzyme-to-substrate ratio of 1 mAU:50 mg, RT, 2h) followed by trypsin (Promega, an enzyme-to-substrate ratio of 1:50, RT, 16h). The proteolytic reaction was quenched by adjusting pH to <3 with 50% of formic acid (FA, Fisher Chemicals). The peptides were desalted on reversed phase C18 SPE columns (Waters) and dried using Speed-Vac (Thermo Scientific).
Tandem Mass Tag (TMT) Labeling of Peptides
Dried peptides from each sample were labeled with 11-plex TMT reagents (Thermo Fisher Scientific). Peptides (300 μg) were dissolved in 60 μL of 100 ml HEPES (pH 8.5). A reference sample was created by pooling an aliquot from 129 pancreatic cancer tissues and 52 NAT tissues (representing ~85% of the sample cohort), and was included in all TMT 11-plex sets as a reference channel. An internal quality control (QC) sample that was an NCI-7 Cell Line Panel sample (Clark et al., 2018) was interspersed among TMT 11-plex sets. 140 pancreatic cancer tissues and 67 NATs were co-randomized to 24 TMT 11-plex sets, while 8 normal ductal tissues were assigned to the 25th TMT 11-plex set. TMT reagents were dissolved in 250 μL of anhydrous acetonitrile (Sigma), and 20 μL of each TMT reagent was added to the corresponding aliquot of peptides. The reaction was incubated at RT for 1h with shaking and quenched with 5% hydroxylamine at RT for 15 min. The labelled peptides were desalted on reversed phase C18 SPE columns (Waters) and dried using Speed-Vac (Thermo Scientific).
Peptide Fractionation by Basic Reversed-phase Liquid Chromatography (bRPLC)
The desalted peptides from each TMT set were dissolved in 900 μL of 5 mM ammonium formate (pH 10) and 2% acetonitrile (ACN) and fractionated with a 4.6 mmx 250 mm Zorbax Extend-C18 analytical column (3.5 μm beads, Agilent) lined up with an Agilent 1220 Series HPLC. Buffer A and B were 5 mM ammonium formate in 2% ACN (pH 10) and 5 mM ammonium formate in 90% ACN, respectively. Peptides were separated by using a non-linear gradient: 0% buffer B (7 min), 0% to 16% buffer B (6 min), 16% to 40% buffer B (60 min), 40% to 44% buffer B (4 min), 44% to 60% buffer B (5 min) and then held at 60% buffer B for 14 min. The flow rate was set at 1 mL/min. Collected fractions were concatenated into 24 fractions as described previously. (Mertins et al., 2018) Eight percent of each of the 24 fractions was aliquoted, cleaned up with strong cation exchange (SCX) stage tip, and dried in a Speed-Vac. Samples were resuspended in 3% ACN, 0.1% FA prior to ESI-LC-MS/MS analysis. The remaining sample was utilized for phosphopeptide enrichment.
Enrichment of Phosphopeptides by Fe-IMAC
The remaining 92% of the sample was further concatenated into 12 fractions before being subjected to phosphopeptide enrichment using immobilized metal affinity chromatography (IMAC) as previously described (Mertins et al., 2018). Ni-NTA agarose beads (QIAGEN) were conditioned and incubated with a 10mM FeCl3 aqueous solution at RT for 1h to prepare Fe3+-NTA agarose beads. Peptides from each fraction were reconstituted in 80% ACN, 0.1% trifluoroacetic acid (TFA) and incubated with 10 μL of the Fe3+-IMAC beads at RT for 30 min. The bead solution was spun down at 1,000 g for 1 min. The supernatant containing unbound peptides was separated from beads and collected for subsequent glycopeptide enrichment. The beads were resuspended in 80% ACN (0.1% TFA) and loaded onto conditioned C18 stage tip. The tip was washed twice with 80% ACN (0.1% TFA) followed by 1% FA. The flowthroughs were collected and combined with the unbound peptides for subsequent glycopeptide enrichment. Peptides were eluted from beads to C18 stage tip with 70 μL of 500 mM dibasic potassium phosphate, pH 7.0 three times. The tip was then washed twice with 1% FA to remove salts. Peptides were eluted twice with 80% ACN (0.1% FA), dried down, and redissolved in 3% ACN, 0.1% FA prior to ESI-LC-MS/MS analysis.
Enrichment of Glycopeptides
All unbound peptides from phosphopeptide enrichment were desalted on reversed phase C18 SPE column (Waters). The glycopeptides were enriched with OASIS MAX solid-phase extraction (Waters). The MAX cartridge was conditioned with 3 x 1 mL ACN, then 3 x 1 mL of 100 mM triethylammonium acetate buffer, followed by 3 x 1 mL of water, and finally 3 x 1 mL of 95%ACN (1%TFA). The peptides were loaded twice. The cartridge was washed with 4 x 1 mL of 95% ACN (1% TFA) to remove non-glycosylated peptides. The glycopeptide fraction was eluted with 50% ACN (0.1% TFA), dried down, and reconstituted in 3% ACN, 0.1% FA prior to ESI-LC-MS/MS analysis.
ESI-LC-MS/MS for Global Proteome, Phosphoproteome, and Glycoproteome Analysis
The TMT-labeled global proteome, phosphoproteome, and glycoproteome fractions were analyzed using Orbitrap Fusion Lumos mass spectrometer (Thermo Scientific). Approximately 0.8 μg of peptides were separated on an in-house packed 28 cm x 75 mm diameter C18 column (1.9 mm Reprosil-Pur C18-AQ beads (Dr. Maisch GmbH); Picofrit 10 mm opening (New Objective)) lined up with an Easy nLC 1200 UHPLC system (Thermo Scientific). The column was heated to 50°C using a column heater (Phoenix-ST). The flow rate was set at 200 nl/min. Buffer A and B were 3% ACN (0.1% FA) and 90% ACN (0.1% FA), respectively. The peptides were separated with a 6–30% B gradient in 84 min. Peptides were eluted from the column and nanosprayed directly into the mass spectrometer. The mass spectrometer was operated in a data-dependent mode. Parameters for global proteomic samples were set as follows: MS1 resolution – 60,000, mass range – 350 to 1800 m/z, RF Lens – 30%, AGC Target – 4.0e5, Max injection time – 50 ms, charge state include – 2-6, dynamic exclusion – 45 s. The cycle time was set to 2 s, and within this 2 s the most abundant ions per scan were selected for MS/MS in the orbitrap. MS2 resolution – 50,000, high-energy collision dissociation activation energy (HCD) – 37, isolation width (m/z) – 0.7, AGC Target – 2.0e5, Max injection time – 105 ms. Parameters for phosphoproteomic samples were set as follows: MS1 resolution – 60,000, mass range – 350 to 1800 m/z, RF Lens – 30%, AGC Target – 4.0e5, Max injection time – 50 ms, charge state include – 2-6, dynamic exclusion – 45 s. The cycle time was set to 2 s, and within this 2 s the most abundant ions per scan were selected for MS/MS in the orbitrap. MS2 resolution – 50,000, high-energy collision dissociation activation energy (HCD) – 34, isolation width (m/z) – 0.7, AGC Target – 2.0e5, Max injection time – 100 ms. Parameters for glycoproteomic samples were set as follows: MS1 resolution – 60,000, mass range – 500 to 2000 m/z, RF Lens – 30%, AGC Target – 5.0e5, Max injection time – 50 ms, charge state include – 2-6, dynamic exclusion – 45 s. The cycle time was set to 2 s, and within this 2 s the most abundant ions per scan were selected for MS/MS in the orbitrap. MS2 resolution – 50,000, high-energy collision dissociation activation energy (HCD) – 35, isolation width (m/z) – 0.7, AGC Target – 1.0e5, Max injection time – 100 ms.
ESI-LC-MS/MS for Global Proteome Data-Independent Acquisition (DIA) Analysis
Unlabeled, digested peptide material from individual tissue samples (PDAC and NAT) was spiked with index Retention Time (iRT) peptides (Biognosys) and subjected to DIA analysis. Approximately 1 μg of peptides were separated on an in-house packed 28 cm x 75 mm diameter C18 column (1.9 mm Reprosil-Pur C18-AQ beads (Dr. Maisch GmbH); Picofrit 10 mm opening (New Objective)) lined up with an Easy nLC 1200 UHPLC system (Thermo Scientific). The column was heated to 50°C using a column heater (Phoenix-ST). The flow rate was set at 200 nl/min. Buffer A and B were 3% ACN (0.1% FA) and 90% ACN (0.1% FA), respectively. The peptides were separated with a 7–30% B gradient in 118 min. Peptides were eluted from the column and nanosprayed directly into Orbitrap Fusion Lumos mass spectrometer (Thermo Scientific). The mass spectrometer was operated in a data-independent mode. The DIA segment consisted of one MS1 scan (350-1650 m/z range, 120K resolution) followed by 30 MS2 scans (variable m/z range, 30K resolution). Additional parameters were as follows: MS1: RF Lens – 30%, AGC Target 3.0e6, Max IT – 60 ms, charge state include – 2-6; MS2: isolation width (m/z) – 0.7, AGC Target – 3.0e6, Max IT – 120 ms.
Spectral Library generation for Data-Independent Acquisition Analysis
For spectral library generation, an aliquot (2 mg) of unlabeled, digested peptide material from individual tissue samples (PDAC and NAT) was pooled and subjected to bRPLC. Ninety-six fractions were collected into a 96-well plate. These fractions were pooled every eight fraction (e. g., combining fractions #1, #9, #17, #25, #33, #41, #49, #57, #65, #73, #81, and #89; #2, #18, #26, #34, #42, #58, #66, #74, #82, and #90; and so on). The resulting 8 fractions were dried in a speed vacuum centrifuge, resuspended in 3% ACN, 0.1% formic acid, and spiked with iRT peptides prior to ESI-LC-MS/MS analysis. Parameters were the same as previously described for ESI-LC-MS/MS for TMT-labeled global proteome analysis with a high-energy collision dissociation activation energy (HCD) – 34.
Proteomic, Phosphoproteomic, and Glycoproteomic Data Processing
Proteomic and Phosphoproteomic Data Processing
MS/MS spectra were searched using the MSFragger version 3.0 (Kong et al., 2017) against a CPTAC3 RefSeq human protein sequence database appended with an equal number of decoy sequences. For the analysis of whole proteome data, MS/MS spectra were searched using a precursor-ion mass tolerance of 10 ppm, and allowing C12/C13 isotope errors (−1/0/1/2/3). MS and MS/MS mass calibration, MS/MS spectral deisotoping, and parameter optimization were enabled (Yu et al., 2020). Cysteine carbamidomethylation (+57.0215), lysine TMT labeling (+229.1629), and peptide N-terminal TMT labeling were specified as fixed modifications. Methionine oxidation (+15.9949) and serine TMT labeling (+229.1629) were specified as variable modifications. The search was restricted to tryptic and semi-tryptic peptides, allowing up to two missed cleavage sites. For phosphopeptide enriched data, the set of variable modifications also included phosphorylation (+79.9663) of serine, threonine, and tyrosine residues, but excluded the serine TMT labeling, and with C12/C13 isotope errors parameter set to (0/1/2).
The post-processing of the search results was done using the Philosopher toolkit version v3.2.8 (da Veiga Leprevost et al., 2020). MSFragger output files (in pepXML format) were processed using PeptideProphet (Keller et al., 2002) (with the high–mass accuracy binning and semi-parametric mixture modeling options) to compute the posterior probability of correct identification for each peptide to spectrum match (PSM). In the phosphopeptide-enriched dataset, PeptideProphet files were additionally processed using PTMProphet (Shteynberg et al., 2019) to localize the phosphorylation sites. The resulting pepXML files from PeptideProphet (or PTMProphet) from all 25 TMT 11-plex experiments were then processed together to assemble peptides into proteins (protein inference) and to create a combined file (in protXML format) of high confidence protein groups.
The combined protXML file and the individual PSM lists for each TMT 11-plex were further processed using the Philosopher filter command as follows. Each peptide was assigned either as a unique peptide to a particular protein group or set as a razor peptide to a single protein group with the most peptide evidence. The protein groups assembled by ProteinProphet (Nesvizhskii et al., 2003) were filtered to 1% protein-level False Discovery Rate (FDR) using the best peptide approach (allowing both unique and razor peptides) and applying the picked FDR target-decoy strategy. In each TMT 11-plex, the PSM lists were filtered using a sequential FDR strategy, retaining only those PSMs with PeptideProphet probability of 0.9 or higher (which in these data corresponded to less than 1% PSM-level FDR) and mapped to proteins that also passed the global 1% protein-level FDR filter. For each PSM that passed these filters, the corresponding precursor ion MS1 intensity was extracted using the Philosopher label-free quantification module, using 10 p.p.m mass tolerance and 0.4 min retention time window for extracted ion chromatogram peak tracing.
Also, for all PSMs corresponding to a TMT-labeled peptide, eleven TMT reporter ion intensities were extracted from the MS/MS scans (using 0.002 Da window). The precursor ion purity scores were calculated using the intensity of the sequenced precursor ion and that of other interfering ions observed in MS1 data (within a 0.7 Da isolation window). All supporting information for each PSM, including the accession numbers and names of the protein/gene selected based on the protein inference approach with razor peptide assignment and quantification information (MS1 precursor-ion intensity and the TMT reporter ion intensities), was summarized in the output PSM.tsv files, one file for each TMT 11-plex experiment.
To generate summary reports on different levels (gene, peptide, and protein for global and phosphopeptide enriched data; additional modification site report for phosphopeptide data), all PSM.tsv files were processed together using TMT-Integrator (Djomehri et al., 2020). Each PSM in a PSM.tsv file that passed the following criteria were kept for creating integrated reports, including (1) having a TMT label at peptide N-terminus, (2) having non-zero intensity in the reference channel, (2) precursor-ion purity above 50%, (3) summed reported ion intensity (across all channels) not in the lower 5% of all PSMs (2.5% for phosphopeptide enriched data), (4) fully tryptic peptides, (5) peptide with phosphorylation (for phosphopeptide enriched data). For a peptide with redundant PSMs in the same MS run, only the PSM with the highest summed TMT intensity was kept for later analysis. PSMs mapping to common external contaminant proteins was excluded, and both unique and razor peptides were used for quantification. Next, the reporter ion intensities of each PSM were log2 transformed and normalized by the reference channel intensity (i.e., subtracted log2 reference intensity from those log2 report ion intensities), therefore the intensities were converted into a log2-based ratio (denoted as ‘ratios’ in the following paragraphs). After converting the intensities to ratios, the PSMs were grouped based on the predefined level (i.e., gene, protein, peptide, and site-level). The interquartile range (IQR) algorithm was then applied to remove the outliers in each PSM group, and the remaining ratios were median centered. The ratios were converted back to abundances using the weighted sum of the MS1 intensities of the top three most intense peptide ions, with the weighting factor (computed for each PSM) taken as the ratio of the reference channel intensity to the summed reporter ion intensity (across all channels). In generating the site-level reports (phosphopeptide-enriched data), sites with PTMProphet computed localization probability equal or greater than 0.75 were considered as confidently localized. Additional details regarding these steps can be found in (Clark et al., 2019).
Glycoproteomic Data Processing
Glycoproteomic and phosphoproteomic raw data files were converted to universal format mzML files using the msconvert tool from ProteoWizard, and searched with the GPQuest search engine (version 2.1) with the following modifications: dynamic oxidation (+15.9949 Da) on Met, and static carbamidomethylation (+57.021464 Da) on Cys residues. GPQuest was applied to identify intact N-linked glycopeptides to MS/MS spectra using two approaches: searching spectra containing oxonium ions (‘oxo-spectra’) and identifying intact N-linked glycopeptides. The oxonium ions were used as the signature features of the glycopeptides from the MS/MS spectra, which were caused by the fragmentation of glycans attached to intact glycopeptides in the mass spectrometer. In this study, the MS/MS spectra containing the oxonium ions (m/z 204.0966) in the top 10 abundant peaks (N-linked glycopeptide search) and top 1000 abundant peaks (O-linked glycopeptide search) after removing TMT reporter ions were considered as the potential glycopeptide candidates. The intact N-linked glycopeptides were identified by using GPQuest to search against the glycopeptide database of glycositeatlas (Sun et al., 2019) and a glycan database collected from the public database of GlycomeDB (Ranzinger et al., 2011). Each tandem mass spectrum was first processed in a series of preprocessing procedures, including removing reporter ions, spectrum de-noising, intensity square root transformation (Liu et al., 2007), oxonium ions evaluation and glycan type prediction (Toghi Eshghi et al., 2016). The top 100 peaks in each preprocessed spectrum were matched to the fragment ion index generated from a peptide sequence database to identify all the candidate peptides. All the qualified (>= 6 fragment ions matchings) candidate peptides were compared with the spectrum again to calculate the Morpheus scores (Wenger and Coon, 2013) by considering all the peptide fragments, glycopeptide fragments, and their isotope peaks. The peptide having the highest Morpheus score was then assigned to the spectrum. The mass gap between the assigned peptide and the precursor mass was searched in the glycan database to find the associated glycan. The best hits of all ‘oxo-spectra’ were filtered by precursor isotopes distribution fitting score and then ranked by the Morpheus score in descending order, in which those with FDR <1% and covering >10% total intensity of each tandem spectrum were reserved as qualified identifications. The precursor mass tolerance was set as 10ppm, and the fragment mass tolerance was 20 ppm.
For the identification of O-linked glycopeptides, the LC-MS/MS data were searched against a peptide database generated from 2,225 O-linked glycoproteins and 84 Functional Glycomics Gateway (CFG) O-linked glycan database. The 2,225 O-linked glycoproteins were collected from glycoproteins identified using EXoO method (Yang et al., 2018b; Yang et al., 2020) and O-glycoprotein database (www.oglyp.org). An IR score of over 0.2 and Morpheus score of at least 7 were used to filter the data and the decoy identification was used to calculate the FDR for the identification of O-linked glycopeptides. The identified O-linked glycopeptides were compared to the list of N-linked glycopeptides identified in this study, and the overlapped glycopeptides were removed from the final list of O-linked glycopeptides.
Peptide-spectrum matches (PSMs) were quantified using the MS-PyCloud proteomics pipeline (https://bitbucket.org/mschnau/ms-pycloud/downloads/). TMT correction factors were applied in order to correct the MS2 intensity of each PSM. Only fully-tryptic peptides with up to two missed cleavages were retained. Glycopeptide (peptide + glycan) false discovery rate (FDR) was restricted to less than or equal to 1 percent by applying a PSM-level FDR filter of less than or equal to 0.25 percent, requiring a minimum of two PSMs per peptide, and a minimum of one peptide per protein. Sample log2 ratios were calculated for each PSM relative to the pooled reference for that sample’s TMTplex after median normalizing each sample in the TMTplex to the pooled reference. PSM log2 ratios were then rolled up to glycopeptide-level by taking the median of PSMs that map to the same glycopeptide. The glycopeptide log2 ratio matrix was then median normalized across all samples. The glycopeptide abundance matrix was derived from the log2 ratio matrix by adding the median log2 value of all TMTplex pooled reference summed MS2 intensities to each sample log2 ratio for a given glycopeptide.
Protein Database Searching and Quantification of Global DIA Data
Raw mass spectrometry files from DIA and data dependent acquisition (DDA) platforms were processed using the DIA-Umpire (Tsou et al., 2015) based pipeline to generate a combined spectral library that integrated DDA and DIA search results (Cho et al., 2020; Clark et al., 2019). The combined library was then converted to Spectronaut (Biognosys) format and loaded into Spectronaut. The DIA data was searched using default settings of Spectronaut (Cho et al., 2020; Clark et al., 2019), and the results were exported without normalization. The protein abundances were further grouped by unique gene names using sum of all the protein abundances belonging to the identical gene name. The protein abundances in the protein expression matrix were log2-transformed. The missing value excluded median abundance Mi = median (Aij, j = 1,…p) of all p proteins in each sample i were calculated. The median abundance of the first sample (C3L-01124-T) was selected as the reference M0. The abundances in each sample were median centered to M0 (normalized Aij = Aij-Mi+M0).
Data Quality Control
Different normalization methods for global proteomics, phosphoproteomics and glycoproteomics data were evaluated using OmicsEV (https://github.com/bzhanglab/OmicsEV/) and an optimal normalization method was then selected for each data type. After the data were normalized, batch effect was also evaluated using OmicsEV both visually by correlation heatmaps ordered by TMT-plex and by PCA. For each PC, the Pearson correlation coefficient to the batch covariate was calculated and significance was assessed by using one-way ANOVA. None of the first 3 PCs were significantly correlated to the TMT-plex, indicating the lack of a batch effect. Pairwise comparisons between replicate samples and samples within TMT plexes were conducted using the square of the Pearson correlation coefficient (R2) based on the data generated using a virtual reference-based method. In the correlation analysis, only features without any missing value were used. The virtual reference of proteomic, phosphoproteomic, N-linked glycoproteomic, and O-linked glycoproteomic data was calculated as the median PSM intensity from all channels in the TMTplex, with zero value intensities being omitted.
In addition to extensive QC during data acquisition, RNA sequencing data quality was assessed using FastQC. To detect potential sample swaps or mislabeling across data types, genome-wide correlations at all omics levels (e.g. RNA-Protein) were used to determine sample identity concordance. Of note, the gender of one case (C3N-02295) was predicted to be male based on mRNA data, which was inconsistent with the clinical data provided. We decided to include this case in our cohort since we did not perform any gender-related analysis.
Integrated Analysis
Mutation Impact on the RNA, Proteome, and Phosphoproteome
We examined the cis- and trans-effects of 11 genes with somatic mutations that were significantly mutated in previous large scale PDAC studies (Bailey et al., 2018; Weinstein et al., 2013) on the RNA, proteome, and phosphoproteome. We collected a set of interacting proteins partners from OmniPath (downloaded on 2018-03-29) (Turei et al., 2016), DEPOD (downloaded on 2018-03-29) (Duan et al., 2015), CORUM (downloaded on 2018-06-29) (Ruepp et al., 2010), Signor2 (downloaded on 2018-10-29) (Perfetto et al., 2016), and Reactome (downloaded on 2018-11-01) (Fabregat et al., 2018). We used this interaction set to assess the trans-effects of these genes. After excluding silent mutations, samples were separated into mutated and WT groups for each gene of interest, removing samples with missing values. We used the Wilcoxon rank-sum test to report differentially expressed features (RNA, proteins, or phosphosites) between the two groups, requiring at least 3 samples in each comparison group. Differentially enriched features passing an FDR <0.05 cut-off were separated into two categories based on cis- and trans-effects.
Copy Number Impacts on Gene and Protein Levels
To infer focal-level significant somatic copy number alterations (SCNA) we used GISTIC2 (Mermel et al., 2011) with the default parameters except for increased thresholds for amplifications and deletions (i.e., -ta and -td parameters of GISTIC2), that were set to 0.4, and confidence level set to 0.95. This analysis was performed on the segment-level SCNA data for the autosomes.
We first filtered all the genes to those with quantifiable copy number, gene expression, and proteomics (N=11,623). Next, we also filtered genes for those occurring in the focal amplified regions identified by GISTIC2 with Q value < 0.25 (N = 543). Finally, we filtered the genes by their CN-mRNA correlation and CN-protein correlation to keep the genes with significant CN cis-effect (FDR < 0.05, Spearman’s correlation). The resulting set of genes (N=23) was used for the gene set enrichment analysis to identify significantly enriched GO-biological processes (Subramanian et al., 2005).
DNA Methylation Associations with RNA, Protein, and Phosphorylation
To investigate the association between methylation and proteomics expression, for each gene, we first calculated Z scores for its mRNA expression, protein, and phosphorylation levels and beta values for DNA methylation. We then calculated Pearson correlation scores with its associated significance between methylation and gene expression, protein, and phosphorylation levels for all pairs of genes, respectively.
The 69 tumors and 9 normal adjacent tissues (NATs) covered in both RNA and DNA methylation data sets were involved in this identification of epigenetically-silenced genes. Probes that were located in CpG Islands (CpGIs) and transcript start sites (TSS) were selected except those located on X and Y chromosomes. Hierarchical clustering analysis (Scipy 1.5.2, Python package) showed that the tumor and NAT tissues can be separated based on the beta value of DNA methylation except one NAT sample (Figure S2E). The approach of identification of epigenetically-silenced genes is similar to the TCGA project (The Cancer Genome Atlas Research Network, 2017). Level 3 RNA-seq RSEM data were log2-transformed [log2 (RSEM+1)] and used to assess the expression levels associated with DNA methylation changes. DNA methylation and gene expression data were merged by Entrez Gene IDs. We removed the CpG sites that were methylated in the NATs (mean β-value > 0.2). We then dichotomized the DNA methylation data using a β-value of > 0.3 to definite positive DNA methylation, and further eliminated CpG sites methylated in fewer than 3% of the tumor samples. For each probe/gene pair, we applied the following algorithm: 1) classify the tumors as either methylated (β > 0.3) or unmethylated (β ≤0.3); 2) compute the mean expression in the methylated and unmethylated groups; 3) compute the standard deviation of the expression in the unmethylated group. We then selected probes for which the mean expression in the methylated group was lower than 1.64 (10% of one-sided Z distribution) standard deviations of the mean expression in the unmethylated group and the NAT tissues. We labeled each individual tumor sample as epigenetically silenced for a specific probe/gene pair selected from above if: a) it belonged to the methylated group and b) the expression of the corresponding gene was lower than the mean of the unmethylated group of samples. If there were multiple probes associated with the same gene, a sample identified as epigenetically silenced at more than or equal to half the probes for the corresponding gene was also labeled as epigenetically silenced at the gene level. The methylation status of ZNF544 was also found significantly correlated with survival time by using the Python package lifelines (version 0.25.4, DOI: 10.5281/zenodo.4002777).
Differential Abundance Analysis
Paired differential abundance analysis between tumor and NATs was performed using the Wilcoxon signed-rank test. At least 50% of the paired samples were required to have non-missing values. Significance was determined to be Benjamini-Hochberg corrected p value < 0.01 and fold change was calculated as the median log2 fold change. Unpaired differential abundance analysis was performed using the Wilcoxon rank sum test. At least 4 samples in both groups were required to have non-missing values. Adjusted p values and fold changes were calculated as above. Immunohistochemistry data were collected from the Human Protein Atlas (Uhlen et al., 2015) for pancreatic cancer samples. The list of secretable proteins was also collected from the Human Protein Atlas.
Glycoproteomics Analysis
Tumor related glycoproteins identification.
The Wilcoxon rank sum test was used to compare the global protein expression difference of each protein containing at least one glycopeptide identified in the glycoproteomics data (termed glycoprotein) in tumors and NATs. At least 50% of all samples were required to have non-missing values. At least 4 samples in each group were required to have non-missing values. The p values were corrected to FDR values using the Benjamini-Hochberg method (statsmodels.stats.multitest.multipletests, version 0.12.0, Python 3.7). The statistical significant up-/down-regulations were determined by using FDR <0.01, while the median log2 fold changes (log2FC) = 1 or −1 were applied to further dichotomize the significant changes to ‘2x up/down’ and ‘up/down’ respectively (Figure 4A). The secreted-to-blood glycoproteins were annotated with their gene names. The protein subcellular location information (Table S4) was collected from two resources: The Human Protein Atlas (www.proteinatlas.org) (Thul et al., 2017) and UniprotKB (www.uniprot.org) (The UniProt Consortium, 2017). The sunburst plot (Plotly, Python package) of the up-/down-regulated glycoproteins and their corresponding cellular locations were shown in Figure S4A. The gene set enrichment analysis of altered glycoproteins was achieved by Webgestalt (http://www.webgestalt.org/).
Impact of early Stage and KRAS hotspot mutations on N-linked glycoprotein expression.
We compared the glycoprotein expression measured in tumors (including subsets of early stage: stage I and II, and four most common KRAS hotspot mutations: G12D, G12R, G12V, and Q61H) and normal tissues (including NATs and normal duct tissues). The Wilcoxon rank sum test was applied on each pair of comparison (stats, R package) to investigate the secreted glycoproteins significantly up-regulated in tumors (Figure 4B, Table S4). The P-values were adjusted by Benjamini-Hochberg procedures. If the p value was < 0.01, the result was annotated as “>2x up” when the fold change > 2, otherwise it was annotated as “up”. The Wilcoxon tests were also applied in the investigation of tumor (all tumors, early stage tumors, and four KRAS mutant subsets: G12D, G12V, G12R, and Q61H) and normal (NATs or normal duct tissues) comparison for other proteins, including MUC family proteins (Figure S4C), CEACAM5 and CEACAM6 (Figure S4D), and LGALS3BP, HPX, COL6A1 and their corresponding glycopeptides (Figure S4E).
Protein glycosylation comparison on protein level and intact glycopeptide level.
The log2 fold change (FC) of the intact glycopeptides and the corresponding global protein expression were shown in Figure 4C. The associated glycans on the intact glycopeptides were classified to three groups of oligomannose (HM), fucose (Fuc), and sialic acid (Sia) based on the composition of the glycans. The projection of the distribution of log2 FC values were shown in the top and right side for protein and intact glycopeptides, respectively.
Correlation between the glycosylation enzymes and intact glycopeptide expression and investigation of glycosylation biosynthetic pathways.
The intact glycopeptide expression was hypothesized to be influenced at least by the expression of substrate glycoproteins and glycosylation enzymes. The Spearman’s rank correlation coefficient was used to measure the correlation between the abundance (log2 ratio values) of intact glycopeptides and the abundance of glycosylation enzymes identified from the global proteomic data in this study. The correlation matrix was further arranged by the order of enzymes in the glycosylation synthetic pathways and visualized in Figure 4D. The glycan compositions were linked to the intact glycopeptides in the middle panel of Figure 4D. The result of Wilcoxon ranked sum tests on the tumor/NAT abundance comparison of the glycosylation enzymes was shown in Figure 4E to illustrate the overall trend of down-regulated precursor pathway and up-regulated capping pathways of glycosylation in tumors on protein level. The same approach was applied on mRNA data and shown in Figure S4F.
Kinase and Substrate Co-regulation
To discover the phosphorylation events that were relevant to PDAC, we utilized phosphosite abundance data to examine the relationship between phospho-substrates and their associated kinases. The kinase-substrate association was first extracted from PhosphoSitePlus (Hornbeck et al., 2015) to eliminate phosphosites that were not reported as well as those without associated kinases identified in our global proteome data. Next, we inspected any substantial differences among 41 tumor/NAT pairs, especially those showing higher changes in tumors, by calculating the fold change (log2 scale) between each paired sample as well as groups (median log2 fold change). Finally, we ranked each tumor (> 1.5 fold increase) among different kinase-substrate pairs to obtain the high ranked phospho-substrate events in the majority of tumors. We identified five phospho-substrate events of five kinases with inhibitors that are either FDA-approved or under investigation. Data was analyzed using Omic-Sig (https://github.com/hzhangjhu/Omic-Sig) (Lih et al., 2019). Kinases enriched in different hotspot KRAS mutations were stratified from the phospho-substrates (at least 2 substrates) showing elevated expression profiles (>2 fold increase with adjusted p<0.05) in the differential analysis between KRAS mutant tumors and NATs. The differential analysis was conducted using Wilcoxon rank-sum test (unpaired samples) and Wilcoxon signed-rank test (paired samples) in transcriptomics, global proteomics, or phosphoproteomics data (at least 50% of all samples were required to have non-missing values) between PDAC tumors and NATs/normal ductal tissues as well as between early stage PDAC tumors and NATs/normal ductal tissues (proteins/phosphosites quantified in at least 4 samples in both groups). The p-value was adjusted using the Benjamini Hochberg method. The druggability score was calculated by summing the number of PDAC cell lines with positive drug response from Genomics of Drug Sensitivity in Cancer (https://www.cancerrxgene.org/) and then log transformed.
RNA Subtyping
The RNA subtyping was performed similarly to the TCGA PDAC study (The Cancer Genome Atlas Research Network, 2017). Specifically, for the three RNA subtyping schemes reported previously (i.e. Collisson, Bailey, and Moffitt), the gene signatures were obtained from the original publications (Bailey et al., 2016; Collisson et al., 2011; Moffitt et al., 2015). The harmonized RNA expression matrices (with zero counts less than 50% for all the genes) for these gene signatures (Collisson N=61; Bailey N=488 and Moffitt N=49) were normalized by z-scoring in the gene-wise manner. Next, we applied consensus clustering (Wilkerson and Hayes, 2010) to these subsetted RNA matrices to identify sample groups with distinct expression patterns for these signature genes. We chose the K value (i.e. cluster number) equal to the reported subtype numbers of each subtyping scheme, after we checked the Consensus Cumulative Distribution Function (CDF) plot and the Delta Area plot to ensure that these Ks indeed represented the best cohort partition (Wilkerson and Hayes, 2010). These clusters were further labelled by interrogating their expression of the signature genes used at the first place.
Multi-omics Clustering
Non-negative matrix factorization (NMF)-based multi-omics clustering was performed similar to as previously described (Gillette et al., 2020; Huang et al., 2021; Krug et al., 2020; Wang et al., 2021). Briefly, NMF was used to perform unsupervised clustering of tumor samples using gene copy number aberrations, mRNA and protein expression, and phosphorylation and glycosylation sites abundances. To enable integrative multi-omics clustering, we required all data types (and converted if necessary) to represent ratios to either a common reference measured in each TMT plex (proteome, phosphorylation and glycosylation sites) or an in-silico common reference calculated as the median abundance across all samples. All data tables were then concatenated and only features quantified in all tumors were used for subsequent analysis (no missing values were allowed). Features with the lowest standard deviation (bottom 5th percentile) across all samples were deemed uninformative and were removed from the dataset. Each column in the data matrix was further scaled and standardized such that all features from different data types were represented as z-scores. Since NMF requires a non-negative input matrix, the data matrix of z-scores was further converted into a non-negative matrix as follows:
Create one data matrix with all negative numbers zeroed.
Create another data matrix with all positive numbers zeroed and the signs of all negative numbers removed.
Concatenate both matrices resulting in a data matrix twice as large as the original, but with positive values only and zeros and hence appropriate for NMF.
The resulting matrix was then subjected to NMF analysis leveraging the NMF R-package (Gaujoux and Seoighe, 2010) and using the factorization method described in (Brunet et al., 2004). Given a factorization rank k (where k is the number of clusters), NMF decomposes a p x n data matrix V into two matrices W and H such that multiplication of W and H approximates V. Matrix H is a k x n matrix whose entries represent weights for each sample (1 to N) to contribute to each cluster (1 to k), whereas matrix W is a p x k matrix representing weights for each feature (1 to p) to contribute to each cluster (1 to k). Matrix H was used to assign samples to clusters by choosing the k with maximum score in each column of H. For each sample, we calculated a cluster membership score as the maximal fractional score of the corresponding column in matrix H.
To determine the optimal factorization rank k (number of clusters) for the multi-omic data matrix, a range of clusters between k=2 and 10 was tested. For each k we factorized matrix V using 50 iterations with random initializations of W and H. To determine the optimal factorization rank we calculated two metrics for each k: 1) cophenetic correlation coefficient measuring how well the intrinsic structure of the data was recapitulated after clustering and 2) the dispersion coefficient of the consensus matrix as defined in (Kim and Park, 2007) measuring the reproducibility of the clustering across 50 iterations. The optimal k was defined as the maximum of the product of both metrics for cluster numbers between k=2 and 10. Having determined the optimal factorization rank k, and in order to achieve robust factorization of the multi-omics data matrix, the NMF analysis was repeated using 500 iterations with random initializations of W and H.
Matrix W containing the weights of each feature in a certain cluster was used to derive a list of representative features separating the clusters using the method proposed in (Kim and Park, 2007). Cluster-specific features were further subjected to a 2-sample moderated t-test (Ritchie et al., 2015) comparing the feature abundance between the respective cluster and all other clusters. Derived p-values were adjusted for multiple hypothesis testing using the method proposed by Benjamini and Hochberg (Benjamini and Hochberg, 1995).
In order to functionally characterize the clustering results, normalized enrichment scores (NES) of cancer-relevant gene sets were calculated by projecting the matrix of signed multi-omic feature weights (Wsigned) onto Hallmark pathway gene sets (Liberzon et al., 2015) using ssGSEA (Barbie et al., 2009). To derive a single weight for each gene measured across multiple omics data types (protein, RNA, phosphorylation site, acetylation site) we retained the weight with maximal absolute amplitude. We used the ssGSEA implementation available on https://github.com/broadinstitute/ssGSEA2.0 using the following parameters:
gene.set.database=“h.all.v6.2.symbols.gmt”
sample.norm.type=“rank”
weight=1
statistic=”area.under.RES”
output.score.type=“NES”
nperm=1000
global.fdr=TRUE
min.overlap=5
correl.type=”z.score”
The association between the resulting clusters and inferred phenotypes (e.g. RNA subtypes) and clinical variables, either a Fisher’s exact test (R function fisher.test) for discrete variables or a Wilcoxon rank-sum test (ggpubr R-package) in case of continuous variables was used to assess overrepresentation in tumors assigned to each cluster.
Inference of subtype-specific drug signatures (DSigDB GSEA)
Gene Set Enrichment Analysis (GSEA) implemented in the WebGestaltR R-package (Liao et al., 2019) was used to infer signatures of approved drugs (D1, 1,202 gene sets) and kinase inhibitors (D2, 1,220 gene sets) available in the drug signatures database (DSigDB, Yoo et al., 2015, http://dsigdb.tanlab.org/DSigDBv1.0/). Based on the signed multi-omic feature weights (Wsigned) for the two proteogenomic clusters described above, a single weight for each protein was derived by retaining the weight with maximal amplitude. Negative weights indicated in cluster C1, positive weights proteins with specific expression in cluster C2, respectively. The resulting vector of protein weights (n=5,773) was used as ranking in WebGestaltR. Normalized enrichment scores and p-values were based on 1,000 permutations. Other relevant parameters in WebGestaltR were set as follows: sigMethod=”top”, topThr=10, minNum=5, fdrThr = 0.05, sigMethod = “fdr”, fdrMethod = “BH”.
Methylation-based Deconvolution
We used an established methylation-based deconvolution method, EDec (Onuchic et al., 2016) to dissect the composition of different cell types within the whole bulk tumor. In brief, EDec assumes that the methylation observed from the whole bulk tumor is a linear combination of the methylation from each constituent cell type, weighted by their proportion within the whole bulk tumor. Mathematically, EDec applies the NMF algorithm to the methylation matrix (i.e. a # methylation feature by # samples matrix, with entries being beta values) profiled from the whole bulk tumor and generates a # methylation feature by # cell-type matrix (with entries being beta vale) and a # cell type by # samples matrix (with entries being cell type proportions). To ensure that the second matrix reflects the cell type composition, EDec uses only the methylation features (i.e. probe-level methylation) that are known to have differential levels across the presumed cell types within the tumor. We selected such methylation features from cell lines or physically purified tissues that are available in the public database (Table S6, adapted from (Lurie et al., 2020)). Based on the robustness of NMF matrix decomposition, the methylation-based deconvolution resulted into four cell types: tumor epithelial cells, immune cells, stromal cells and mature exocrine and endocrine cells.
Tumor Microenvironment Inference
The RNA-based tumor microenvironment inference tool ESTIMATE (Yoshihara et al., 2013) was used to derive the overall immune score and stromal score for each sample. In addition, two established RNA-based immune cell inference tools, xCell (Aran et al., 2017) and MCPCounter (Becht et al., 2016), were used to dissect the relative level of different immune cell infiltration. For these tools, we used RNA expression quantified as upper-quantile normalized RSEM and kept only genes with zero counts less than 50% as the input. We found that the xCell results were sparse for some immune cells and further filtered out immune cell types with zero readout in >80% of samples. The remaining cell types were used to derive the microenvironment-based grouping using consensus clustering (Wilkerson and Hayes, 2010). To further explore the relationship between these tumor microenvironment components, a correlation network was built by performing the Pearson’s correlation for all pairs of xCell components and linking the pairs with adjusted p value < 0.05 with edges. The network module discovery was performed by edge betweenness implanted in the R package ‘igraph’.
To contextualize our immune profiling results with current understanding in the field, we extracted the gene signatures from relevant publications, including the “normal stromal” and “activated stromal” genes from Moffitt et al., 2015, “immune” and “ECM” genes from Maurer et al., 2019, and ‘classical’ (signature 1 and 6) and ‘basal-like’ (signature 2 and 10) genes from Chan-Seng-Yue et al., 2020 and used single sample GSEA (ssGSEA that was implemented in GSVA R package (Hanzelmann et al., 2013) to infer the corresponding microenvironment activities. For the ssGSEA scores derived from Maurer et al., 2019, we applied the data deconvolution method reported in the publication and used stroma-specific gene expression for the analysis. In addition, we generated the subtypes reported by Puleo et al, 2018 by centroid-based subtyping using the reported gene signatures from the publication. The comparisons of the xCell-based immune subtyping and the results using these published gene signatures and methods were discussed in the discussion section.
Adjustment for Epithelial Content
For the fifteen patients with an ESTIMATE score for both tumor and NAT samples, a linear mixed model was used to correct for non-epithelial content. The lmerTest package (doi = {10.18637/jss.v082.i13}) in R was used. Proteomics data had to be available for both the tumor and normal samples for at least 10 of the patients. The tumor type and z-scored ESTIMATE score were fixed effects and the patient was a random effect. P value for expression was adjusted using Benjamini-Hochberg, with 0.01 considered significant. A beta value > 1 for expression was used to filter tumor-associated proteins.
Over-representation Analysis
Over-representation analysis of Gene Ontology Biological Process terms was performed using WebGestaltR (Liao et al., 2019) with the > 2-fold increased or decreased proteins in tumors vs NAT compared to a background of all quantified proteins (proteins non-missing in at least 50% of the paired samples). For PTMs, proteins containing at least one PTM that was > 2-fold increased or decreased in tumors vs NAT were compared to a background of proteins containing at least one quantified site (non-missing in at least 50% of the paired samples). A Benjamini-Hochberg corrected p value of 0.01 was considered significant.
Survival Analysis
Cox proportional hazards regression (from the R package survival) on overall survival was performed to test the association between survival outcomes to continuous variables. Logrank test (from the R package survminer) was used to test the differential survival outcomes between categorical variables. Samples with a death event within 30 days of surgery were excluded. For the survival association analysis for the tumor proteomics data, the proteins were filtered to keep the ones with no-missing values for at least 10 patients.
Supplementary Material
Figure S3. Tumor-associated protein and phosphosites, related to Figure 3. A) Median log2 protein fold change between tumor and paired NAT or unpaired normal ductal tissue. B) Immunohistochemistry levels for tumor-associated proteins in pancreatic cancer samples in Human Protein Atlas. C) RALGAPA2 protein abundance in tumor and NAT. D) RALGAPA2 phosphosite abundance in tumor and NAT. ** p<0.001 Wilcoxon signed rank test, NS=not significant.
Figure S2. Impacts of genomic and epigenetic alterations, related to Figure 2. A) Cis- and trans-effects of the TP53 truncation and missense mutation groups. Samples with wild-type TP53 serve as the control. B) CNV impacts on gene, protein, and phosphoprotein of the most common proteins with CNV events. The control group in each comparison includes all samples without the copy number event for each gene or protein. C) Functional impacts of CNVs on gene/protein/phosphorylation expression. The data in the top panel show the correlations of CNVs to mRNA, protein, and phosphorylation levels with significant positive correlations in red and significant negative correlations in blue (adjusted p-value < 0.05). The data in the bottom panel show the summed number of significant positive and negative correlations at mRNA, protein, and phosphopeptide. D) Correlation and anti-correlation of gene, protein, and phosphoprotein levels with DNA methylation levels. E) The two dimensional hierarchical clustering of methylation probes (Y-axis) and tumor and NAT samples (X-axis) on PDAC DNA methylation data. F) DNA methylation status between tumors with different pathological stages and NATs. G) Scatter plot of epigenetically silenced genes on survival significance and silenced samples counting. H) Scatter plot of beta values of probe cg21747958 and RNA expression of ZNF544 in tumors and NATs. I) Kaplan-Meier Plot of ZNF544 methylation association with overall survival. The two groups were separated by whether ZNF544 is silenced in the sample.
Figure S4. Differential expression of glycoproteins and glycosylation enzymes between PDAC tumors, early stage PDAC tumors, NATs, and normal duct tissues, related to Figure 4. A) Cellular distribution of tumor up-regulated and down-regulated N-linked glycoproteins. B) Analysis of significantly differentially regulated pathways between tumors and NATs at the N-linked glycoprotein expression level. C) Expression pattern of mucin 1 (MUC1), mucin 3A (MUC3A), mucin 5AC (MUC5AC), mucin 5B (MUC5B), mucin 13 (MUC13), and mucin 16 (MUC16) in tumors, early stage tumors, NATs, and normal duct tissues. The significance is defined as: N.S. P > 0.05, * P ≤ 0.05, **: P ≤ 0.01, ***: P ≤ 0.001 D) Expression pattern of CEA cell adhesion molecule 5 (CEACAM5) and CEA cell adhesion molecule 6 (CEACAM6) in tumors, early stage tumors, tumors with different KRAS hotspot mutations, NATs, and normal duct tissues. E) Expression pattern of galectin binding protein 3 (LGALS3BP), hemopexin (HPX), and collagen type VI alpha 1 chain (COL6A1) in tumors, early stage tumors, NATs, and normal duct tissues. F) Differential gene expression of N-linked glycosylation enzymes between tumors and NATs.
Figure S5. Kinase-substrate pairs and phosphosites enriched in KRAS mutant tumors, related to Figure 5. A) Expression profiles of SRC, AKT1, and CDK7 in Normal duct, NAT, Tumor, and Early stage. B) Expression profiles of BAD-S134, MAPK6-S189, STAT3-Y705, FLNA-S2152, and MCM2-S139 in Normal duct, NAT, Tumor, and Early stage. C) Bar charts showing the phospho-substrates enriched (>2-fold increase with adjusted p-value<0.05) in the KRAS mutant tumors relative to NATs that were used for identifying kinases enriched in different KRAS hotspot mutant tumors. Normal duct: normal ductal tissues; NAT: normal adjacent tissues; Tumor: all PDAC tumors; Early stage: Stage I and II PDAC tumors. Asterisks represent significant differences between two groups (Benjamini-Hochberg adjusted p): *p < 0.05; **p < 0.01; ***p<0.001; N.S., not significant.
Figure S6. Identification of biological processes associated with immune-cold tumors, related to Figure 6. A-C) The relative levels of tissue components (A: immune; B: Epithelial; C: exo/endocrine) inferred by xCell correlate with the counterparts derived from methylation-based deconvolution. Note that xCell gene signatures do not explicitly contain exo/endocrine cells and the neurons were instead chosen as they are similar to exo/endocrine cells in terms of the regulatory factors utilized during development and differentiation (Arntfield and van der Kooy, 2011). Shown are Pearson’s correlation coefficients and p values. D) Representative histological images showing samples with immune cell infiltration (C3L-04848, ‘immune hot’) and samples with immune cell depletion (C3L-02809, ‘immune cold’). E) Representative histological image showing an example of contamination of lymph nodes (C3N-00303, to be excluded from ‘immune hot’). F) Boxplots comparing the abundance of exo/endocrine cells (from methylation-based deconvolution) and gene expression of amylase (AMY) and insulin (INS). G) Correlation network showing the relationship among all cell types inferred by xCell. Several network modules are identified to reflect the close connection for certain cell types and denoted by different colors. The endothelial cells are highlighted by dashed circles.H) Pathway enrichment using the differentially expressed proteins between immune hot (cluster D) and immune cold (cluster B and C) samples. Shown are pathways that are statistically significant (FDR<0.05) for either directions. I) Pathway enrichment of genes with phospho-specific regulation in the immune cold samples (FDR<0.05). Pathways highlighted by black borders are the ones discussed in the main text.
Figure S1. Characterization of PDAC cohort and proteogenomic data, related to Figure 1. A) Pairwise comparisons between replicate samples and samples within TMT plex. B) Gene-wise correlation of mRNA and protein expression in NATs, tumors, and all tumors and NATs combined. C) Estimation of neoplastic cellularity by histology, DNA methylation-based deconvolution, and transcript proportion-based methods and their correlation with KRAS VAF tumor fraction estimates. D) PCAs at the RNA, protein, phosphorylation, and N-linked glycosylation levels of sufficient purity, low purity, and NAT samples.
Figure S7. Unsupervised, non-negative matrix factorization (NMF)-based proteogenomic subtyping of 140 PDAC tumors using gene copy number, mRNA and protein expression, and phosphosite and glycosylation site abundances and clinical relevance of binary proteogenomic subtypes from the 105 tumors and the 4 proteogenomic subtypes from all 140 tumor samples, related to Figure 7. A) Heatmap showing the clustering of samples by the three published RNA subtyping schemes. Shown are also the histological diagnosis (PDAC vs. pancreatic adenosquamous carcinoma) and selected signature gene expression. B) Heatmap depicting the contribution of samples (x-axis) to each of the four clusters (C1-C4, y-axis). Sample weights have been normalized by the maximum weight in each column. Cluster membership scores indicating the strength of association of each sample with a given cluster were calculated as proportional weights. The columns of the matrix are ordered by NMF subtype and decreasing cluster membership score. C) Overrepresentation analysis of clinical variables and RNA-subtypes in each proteogenomic cluster (Fisher’s exact test) shown in panel A). All clusters showed significant overlap with RNA-subtypes (Bailey, Collisson, and Moffitt). Low purity tumors were enriched in C2 and C4. D-G) Distribution of inferred cell types (y-axis) across NMF-subtypes (x-axis). H) Cluster-specific drug signatures inferred by Gene Set Enrichment Analysis of DSigDB gene sets using NMF weights of protein features (derived from binary proteogenomic subtypes from the 105 tumors) as a ranking. NMF classical cluster (C1) showed enrichment of gene sets targeted by various chemotherapeutics, while the NMF basal-like (C2) cluster was enriched for gene sets of several kinase inhibitors targeting mTOR, ATM, PDGFR, VEGF or BTK (FDR < 0.05). I) Sankey diagram depicting the association of samples classified into xCell-based subtypes (center) with binary proteogenomic subtypes from 105 tumors (left) and the 4 proteogenomic subtypes from all 140 tumor samples (right). Nodes are colored according to the NMF or xCell subtype, links between nodes are colored by xCell subtype.
Table S1 Clinical information and molecular phenotypes of the samples analyzed in this study, related to Figure 1 and Figure S1.
Table S2 Copy number variation (CNV) and methylation analysis, related to Figure 2 and Figure S2.
Table S3 Tumor-associated proteins, phosphosites, and glycosites, related to Figure 3 and Figure S3.
Acknowledgments
This work was supported by grants U24CA210955, U24CA210985, U24CA210986, U24CA210954, U24CA210967, U24CA210972, U24CA210979, U24CA210993, U01CA214114, U01CA214116, and U01CA214125 from the National Cancer Institute (NCI) Clinical Proteomic Tumor Analysis Consortium (CPTAC), National Institutes of Health (NIH). A part of this research was funded through the NIH/NCI Cancer Center Support Grant P30CA008748 and by the Troper Wojcicki Foundation, the Rolfe Pancreatic Cancer Foundation, and the Evelyn Grollman Glick Scholar Award.
Consortia
Mitual Amin, Eunkyung An, Christina Ayad, Oliver F. Bathe, Thomas Bauer, Chet Birger, Michael J. Birrer, Simina M. Boca, William Bocik, Emily S. Boja, Melissa Borucki, Shuang Cai, Liwei Cao, Song Cao, Steven A. Carr, Sandra Cerda, Daniel W. Chan, Huan Chen, Lijun Chen, Steven Chen, David Chesla, Arul M. Chinnaiyan, David J. Clark, Antonio Colaprico, Sandra Cottingham, Daniel Cui Zhou, Felipe da Veiga Leprevost, Ludmila Danilova, Magdalena Derejska, Saravana M. Dhanasekaran, Li Ding, Marcin J. Domagalski, Yongchao Dou, Brian J. Druker, Elizabeth Duffy, Maureen A. Dyer, Hariharan Easwaran, Nathan J. Edwards, Matthew J. Ellis, Jennifer Eschbacher, David Fenyö, Alicia Francis, Jesse Francis, Stacey Gabriel, Nikolay Gabrovski, Johanna Gardner, Gad Getz, Michael A. Gillette, Charles A. Goldthwaite Jr, Pamela Grady, Shuai Guo, Benjamin Haibe-Kains, Pushpa Hariharan, Tara Hiltke, Barbara Hindenach, Katherine A. Hoadley, Galen Hostetter, Ralph H. Hruban, Yingwei Hu, Chen Huang, Jasmine Huang, Scott D. Jewell, Wen Jiang, Corbin D. Jones, Karen A. Ketchum, Christopher R. Kinsinger, Jennifer M. Koziak, Karsten Krug, Katarzyna Kusnierz, Qing Kay Li, Yize Li, Yuxing Liao, T. Mamie Lih, Ruiyang Liu, Tao Liu, Wenke Liu, Jiang Long, Rita Jui-Hsien Lu, David Mallery, D. R. Mani, Sailaja Mareedu, Ronald Matteotti, Nicollette Maunganidze, Peter B. McGarvey, Arvind Singh Mer, Mehdi Mesri, Parham Minoo, Alexey I. Nesvizhskii, Chelsea J. Newton, Gilbert S. Omenn, Oxana V. Paklina, Jianbo Pan, Amanda G. Paulovich, Samuel H. Payne, Olga Potapova, Barbara Pruetz, Liqun Qi, Ana I. Robles, Nancy Roche, Karin D. Rodland, Henry Rodriguez, Michael H. Roehrl, Daniel C. Rohrer, Peter Ronning, Sara R. Savage, Eric E. Schadt, Michael Schnaubelt, Alexey V. Shabunin, Troy Shelton, Zhiao Shi, Yvonne Shutack, Shilpi Singh, Michael Smith, Richard D. Smith, James Suh, Nadezhda V. Terekhanova, Ratna R. Thangudu, Mathangi Thiagarajan, Shirley X. Tsang, Ki Sung Um, Dana R. Valley, Negin Vatanian, Joshua M. Wang, Pei Wang, Wenyi Wang, Bo Wen, George D. Wilson, Maciej Wiznerowicz, Yige Wu, Matthew A. Wyczalkowski, Weiming Yang, Seungyeul Yoo, Bing Zhang, Hui Zhang, Zhen Zhang, Grace Zhao, Houxiang Zhu
Footnotes
Declaration of Interests
Ralph H. Hruban has the potential of receiving royalty payments from Thrive Earlier Diagnosis for the GNAS invention in a relationship overseen by Johns Hopkins University. All other authors have no conflicts of interest to declare.
References
- Akagi J, Takai E, Tamori Y, Nakagawa K, and Ogawa M (2001). CA19-9 epitope a possible marker for MUC-1/Y protein. International journal of oncology 18, 1085–1091. [DOI] [PubMed] [Google Scholar]
- Allen E, Jabouille A, Rivera LB, Lodewijckx I, Missiaen R, Steri V, Feyen K, Tawney J, Hanahan D, Michael IP, et al. (2017). Combined antiangiogenic and anti-PD-L1 therapy stimulates tumor immunity through HEV formation. Science translational medicine 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- American Cancer Society (2021). Cancer Facts & Figures 2021. Atlanta: American Cancer Society. [Google Scholar]
- Annan DA, Kikuchi H, Maishi N, Hida Y, and Hida K (2020). Tumor Endothelial Cell-A Biological Tool for Translational Cancer Research. International journal of molecular sciences 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aran D, Hu Z, and Butte AJ (2017). xCell: digitally portraying the tissue cellular heterogeneity landscape. Genome Biol 18, 220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aznar S, Valeron PF, del Rincon SV, Perez LF, Perona R, and Lacal JC (2001). Simultaneous tyrosine and serine phosphorylation of STAT3 transcription factor is involved in Rho A GTPase oncogenic transformation. Mol Biol Cell 12, 3282–3294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey P, Chang DK, Nones K, Johns AL, Patch AM, Gingras MC, Miller DK, Christ AN, Bruxner TJ, Quinn MC, et al. (2016). Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 531, 47–52. [DOI] [PubMed] [Google Scholar]
- Balachandran VP, Beatty GL, and Dougan SK (2019). Broadening the Impact of Immunotherapy to Pancreatic Cancer: Challenges and Opportunities. Gastroenterology 156, 2056–2072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balli D, Rech AJ, Stanger BZ, and Vonderheide RH (2017). Immune Cytolytic Activity Stratifies Molecular Subsets of Human Pancreatic Cancer. Clinical cancer research: an official journal of the American Association for Cancer Research 23, 3129–3138. [DOI] [PubMed] [Google Scholar]
- Balsano R, Tommasi C, and Garajova I (2019). State of the Art for Metastatic Pancreatic Cancer Treatment: Where Are We Now? Anticancer research 39, 3405–3412. [DOI] [PubMed] [Google Scholar]
- Beauchemin N, and Arabzadeh A (2013). Carcinoembryonic antigen-related cell adhesion molecules (CEACAMs) in cancer progression and metastasis. Cancer metastasis reviews 32, 643–671. [DOI] [PubMed] [Google Scholar]
- Becht E, Giraldo NA, Lacroix L, Buttard B, Elarouci N, Petitprez F, Selves J, Laurent-Puig P, Sautes-Fridman C, Fridman WH, et al. (2016). Estimating the population abundance of tissue-infiltrating immune and stromal cell populations using gene expression. Genome Biol 17, 218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker AE, Hernandez YG, Frucht H, and Lucas AL (2014). Pancreatic ductal adenocarcinoma: risk factors, screening, and early detection. World journal of gastroenterology 20, 11182–11198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beel S, Kolloch L, Apken LH, Jurgens L, Bolle A, Sudhof N, Ghosh S, Wardelmann E, Meisterernst M, Steinestel K, et al. (2020). kappaB-Ras and Ral GTPases regulate acinar to ductal metaplasia during pancreatic adenocarcinoma development and pancreatitis. Nat Commun 11, 3409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berndt N, Hamilton AD, and Sebti SM (2011). Targeting protein prenylation for cancer therapy. Nat Rev Cancer 11, 775–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bieberich E (2014). Synthesis, Processing, and Function of N-glycans in N-glycoproteins. Advances in neurobiology 9, 47–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blumenthal RD, Hansen HJ, and Goldenberg DM (2005a). Inhibition of adhesion, invasion, and metastasis by antibodies targeting CEACAM6 (NCA-90) and CEACAM5 (Carcinoembryonic Antigen). Cancer research 65, 8809–8817. [DOI] [PubMed] [Google Scholar]
- Blumenthal RD, Osorio L, Hayes MK, Horak ID, Hansen HJ, and Goldenberg DM (2005b). Carcinoembryonic antigen antibody inhibits lung metastasis and augments chemotherapy in a human colonic carcinoma xenograft. Cancer immunology, immunotherapy: CII 54, 315–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boecker W, Tiemann K, Boecker J, Toma M, Muders MH, Loning T, Buchwalow I, Oldhafer KJ, Neumann U, Feyerabend B, et al. (2020). Cellular organization and histogenesis of adenosquamous carcinoma of the pancreas: evidence supporting the squamous metaplasia concept. Histochemistry and cell biology 154, 97–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caba O, Prados J, Ortiz R, Jimenez-Luna C, Melguizo C, Alvarez PJ, Delgado JR, Irigoyen A, Rojas I, Perez-Florido J, et al. (2014). Transcriptional profiling of peripheral blood in pancreatic adenocarcinoma patients identifies diagnostic biomarkers. Digestive diseases and sciences 59, 2714–2720. [DOI] [PubMed] [Google Scholar]
- Cai J, Lu W, Du S, Guo Z, Wang H, Wei W, and Shen X (2018). Tenascin-C Modulates Cell Cycle Progression to Enhance Tumour Cell Proliferation through AKT/FOXO1 Signalling in Pancreatic Cancer. Journal of Cancer 9, 4449–4462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caldas C, Hahn SA, da Costa LT, Redston MS, Schutte M, Seymour AB, Weinstein CL, Hruban RH, Yeo CJ, and Kern SE (1994). Frequent somatic mutations and homozygous deletions of the p16 (MTS1) gene in pancreatic adenocarcinoma. Nature genetics 8, 27–32. [DOI] [PubMed] [Google Scholar]
- Cao L, Diedrich JK, Ma Y, Wang N, Pauthner M, Park SR, Delahunty CM, McLellan JS, Burton DR, Yates JR, et al. (2018). Global site-specific analysis of glycoprotein N-glycan processing. Nat Protoc 13, 1196–1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan-Seng-Yue M, Kim JC, Wilson GW, Ng K, Figueroa EF, O’Kane GM, Connor AA, Denroche RE, Grant RC, McLeod J, et al. (2020). Transcription phenotypes of pancreatic cancer are driven by genomic events during tumor evolution. Nat Genet 52, 231–240. [DOI] [PubMed] [Google Scholar]
- Chang X, Yang MF, Fan W, Wang LS, Yao J, Li ZS, and Li DF (2020). Bioinformatic Analysis Suggests That Three Hub Genes May Be a Vital Prognostic Biomarker in Pancreatic Ductal Adenocarcinoma. Journal of computational biology: a journal of computational molecular cell biology. [DOI] [PubMed] [Google Scholar]
- Chen CR, Kang Y, Siegel PM, and Massague J (2002). E2F4/5 and p107 as Smad cofactors linking the TGFbeta receptor to c-myc repression. Cell 110, 19–32. [DOI] [PubMed] [Google Scholar]
- Christensen JG, Olson P, Briere T, Wiel C, and Bergo MO (2020). Targeting Kras(g12c) -mutant cancer with a mutation-specific inhibitor. Journal of internal medicine 288, 183–191. [DOI] [PubMed] [Google Scholar]
- Clark DJ, Dhanasekaran SM, Petralia F, Pan J, Song X, Hu Y, da Veiga Leprevost F, Reva B, Lih TM, Chang HY, et al. (2019). Integrated Proteogenomic Characterization of Clear Cell Renal Cell Carcinoma. Cell 179, 964–983 e931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke R, Leonessa F, and Trock B (2005). Multidrug resistance/P-glycoprotein and breast cancer: review and meta-analysis. Seminars in oncology 32, S9–15. [DOI] [PubMed] [Google Scholar]
- Collisson EA, Bailey P, Chang DK, and Biankin AV (2019). Molecular subtypes of pancreatic cancer. Nature reviews Gastroenterology & hepatology 16, 207–220. [DOI] [PubMed] [Google Scholar]
- Collisson EA, Sadanandam A, Olson P, Gibb WJ, Truitt M, Gu S, Cooc J, Weinkle J, Kim GE, Jakkula L, et al. (2011). Subtypes of pancreatic ductal adenocarcinoma and their differing responses to therapy. Nature medicine 17, 500–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daneman R, and Prat A (2015). The blood-brain barrier. Cold Spring Harb Perspect Biol 7, a020412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Cruz-Lopez KG, Castro-Munoz LJ, Reyes-Hernandez DO, Garcia-Carranca A, and Manzo-Merino J (2019). Lactate in the Regulation of Tumor Microenvironment and Therapeutic Approaches. Front Oncol 9, 1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De la Mota-Peynado A, Chernoff J, and Beeser A (2011). Identification of the atypical MAPK Erk3 as a novel substrate for p21-activated kinase (Pak) activity. J Biol Chem 286, 13603–13611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dennler S, Itoh S, Vivien D, ten Dijke P, Huet S, and Gauthier JM (1998). Direct binding of Smad3 and Smad4 to critical TGF beta-inducible elements in the promoter of human plasminogen activator inhibitor-type 1 gene. The EMBO journal 17, 3091–3100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dou Y, Kawaler EA, Cui Zhou D, Gritsenko MA, Huang C, Blumenberg L, Karpova A, Petyuk VA, Savage SR, Satpathy S, et al. (2020). Proteogenomic Characterization of Endometrial Carcinoma. Cell 180, 729–748 e726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ducreux M, Seufferlein T, Van Laethem JL, Laurent-Puig P, Smolenschi C, Malka D, Boige V, Hollebecque A, and Conroy T (2019). Systemic treatment of pancreatic cancer revisited. Seminars in oncology 46, 28–38. [DOI] [PubMed] [Google Scholar]
- Elia AR, Grioni M, Basso V, Curnis F, Freschi M, Corti A, Mondino A, and Bellone M (2018). Targeting Tumor Vasculature with TNF Leads Effector T Cells to the Tumor and Enhances Therapeutic Efficacy of Immune Checkpoint Blockers in Combination with Adoptive Cell Therapy. Clinical cancer research: an official journal of the American Association for Cancer Research 24, 2171–2181. [DOI] [PubMed] [Google Scholar]
- Elyada E, Bolisetty M, Laise P, Flynn WF, Courtois ET, Burkhart RA, Teinor JA, Belleau P, Biffi G, Lucito MS, et al. (2019). Cross-Species Single-Cell Analysis of Pancreatic Ductal Adenocarcinoma Reveals Antigen-Presenting Cancer-Associated Fibroblasts. Cancer discovery 9, 1102–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engle DD, Tiriac H, Rivera KD, Pommier A, Whalen S, Oni TE, Alagesan B, Lee EJ, Yao MA, Lucito MS, et al. (2019). The glycan CA19-9 promotes pancreatitis and pancreatic cancer in mice. Science 364, 1156–1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eser S, Schnieke A, Schneider G, and Saur D (2014). Oncogenic KRAS signalling in pancreatic cancer. Br J Cancer 111, 817–822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fallahi M, Amelio AL, Cleveland JL, and Rounbehler RJ (2014). CREB targets define the gene expression signature of malignancies having reduced levels of the tumor suppressor tristetraprolin. PLoS One 9, e115517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan G (2020). FER mediated HGF-independent regulation of HGFR/MET activates RAC1-PAK1 pathway to potentiate metastasis in ovarian cancer. Small GTPases 11, 155–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrara N, Hillan KJ, Gerber HP, and Novotny W (2004). Discovery and development of bevacizumab, an anti-VEGF antibody for treating cancer. Nature reviews Drug discovery 3, 391–400. [DOI] [PubMed] [Google Scholar]
- Folkes AJ, Ahmadi K, Alderton WK, Alix S, Baker SJ, Box G, Chuckowree IS, Clarke PA, Depledge P, Eccles SA, et al. (2008). The identification of 2-(1H-indazol-4-yl)-6-(4-methanesulfonyl-piperazin-1-ylmethyl)-4-morpholin-4-yl-t hieno[3,2-d]pyrimidine (GDC-0941) as a potent, selective, orally bioavailable inhibitor of class I PI3 kinase for the treatment of cancer. Journal of medicinal chemistry 51, 5522–5532. [DOI] [PubMed] [Google Scholar]
- Fu B, Luo M, Lakkur S, Lucito R, and Iacobuzio-Donahue CA (2008). Frequent genomic copy number gain and overexpression of GATA-6 in pancreatic carcinoma. Cancer biology & therapy 7, 1593–1601. [DOI] [PubMed] [Google Scholar]
- Furuse J, and Nagashima F (2017). Emerging protein kinase inhibitors for treating pancreatic cancer. Expert opinion on emerging drugs 22, 77–86. [DOI] [PubMed] [Google Scholar]
- Garnham R, Scott E, Livermore KE, and Munkley J (2019). ST6GAL1: A key player in cancer. Oncology letters 18, 983–989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillette MA, Satpathy S, Cao S, Dhanasekaran SM, Vasaikar SV, Krug K, Petralia F, Li Y, Liang WW, Reva B, et al. (2020). Proteogenomic Characterization Reveals Therapeutic Vulnerabilities in Lung Adenocarcinoma. Cell 182, 200–225 e235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goggins M, Offerhaus GJ, Hilgers W, Griffin CA, Shekher M, Tang D, Sohn TA, Yeo CJ, Kern SE, and Hruban RH (1998). Pancreatic adenocarcinomas with DNA replication errors (RER+) are associated with wild-type K-ras and characteristic histopathology. Poor differentiation, a syncytial growth pattern, and pushing borders suggest RER+. The American journal of pathology 152, 1501–1507. [PMC free article] [PubMed] [Google Scholar]
- Golan T, Hammel P, Reni M, Van Cutsem E, Macarulla T, Hall MJ, Park JO, Hochhauser D, Arnold D, Oh DY, et al. (2019). Maintenance Olaparib for Germline BRCA-Mutated Metastatic Pancreatic Cancer. The New England journal of medicine 381, 317–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomis RR, Alarcon C, Nadal C, Van Poznak C, and Massague J (2006). C/EBPbeta at the core of the TGFbeta cytostatic response and its evasion in metastatic breast cancer cells. Cancer cell 10, 203–214. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Borja I, Viudez A, Goni S, Santamaria E, Carrasco-Garcia E, Perez-Sanz J, Hernandez-Garcia I, Sala-Elarre P, Arrazubi V, Oyaga-Iriarte E, et al. (2019). Omics Approaches in Pancreatic Adenocarcinoma. Cancers 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez C, Sims JS, Hornstein N, Mela A, Garcia F, Lei L, Gass DA, Amendolara B, Bruce JN, Canoll P, et al. (2014). Ribosome profiling reveals a cell-type-specific translational landscape in brain tumors. J Neurosci 34, 10924–10936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Govindan SV, Cardillo TM, Moon SJ, Hansen HJ, and Goldenberg DM (2009). CEACAM5-targeted therapy of human colonic and pancreatic cancer xenografts with potent labetuzumab-SN-38 immunoconjugates. Clinical cancer research: an official journal of the American Association for Cancer Research 15, 6052–6061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harsha HC, Kandasamy K, Ranganathan P, Rani S, Ramabadran S, Gollapudi S, Balakrishnan L, Dwivedi SB, Telikicherla D, Selvan LD, et al. (2009). A compendium of potential biomarkers of pancreatic cancer. PLoS medicine 6, e1000046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hart GW, and Copeland RJ (2010). Glycomics hits the big time. Cell 143, 672–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez J, Bechara E, Schlesinger D, Delgado J, Serrano L, and Valcarcel J (2016). Tumor suppressor properties of the splicing regulatory factor RBM10. RNA biology 13, 466–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herr P, Bostrom J, Rullman E, Rudd SG, Vesterlund M, Lehtio J, Helleday T, Maddalo G, and Altun M (2020). Cell Cycle Profiling Reveals Protein Oscillation, Phosphorylation, and Localization Dynamics. Mol Cell Proteomics 19, 608–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hessmann E, Buchholz SM, Demir IE, Singh SK, Gress TM, Ellenrieder V, and Neesse A (2020). Microenvironmental Determinants of Pancreatic Cancer. Physiological reviews 100, 1707–1751. [DOI] [PubMed] [Google Scholar]
- Hilmi M, Bartholin L, and Neuzillet C (2018). Immune therapies in pancreatic ductal adenocarcinoma: Where are we now? World journal of gastroenterology 24, 2137–2151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho WJ, Jaffee EM, and Zheng L (2020). The tumour microenvironment in pancreatic cancer - clinical challenges and opportunities. Nature reviews Clinical oncology 17, 527–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hobbs GA, Baker NM, Miermont AM, Thurman RD, Pierobon M, Tran TH, Anderson AO, Waters AM, Diehl JN, Papke B, et al. (2020). Atypical KRAS(G12R) Mutant Is Impaired in PI3K Signaling and Macropinocytosis in Pancreatic Cancer. Cancer Discov 10, 104–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollingsworth MA, and Swanson BJ (2004). Mucins in cancer: protection and control of the cell surface. Nat Rev Cancer 4, 45–60. [DOI] [PubMed] [Google Scholar]
- Hruban RH, Gaida MM, Thompson E, Hong SM, Noe M, Brosens LA, Jongepier M, Offerhaus GJA, and Wood LD (2019). Why is pancreatic cancer so deadly? The pathologist’s view. J Pathol 248, 131–141. [DOI] [PubMed] [Google Scholar]
- Hsieh CC, Shyr YM, Liao WY, Chen TH, Wang SE, Lu PC, Lin PY, Chen YB, Mao WY, Han HY, et al. (2017). Elevation of beta-galactoside alpha2,6-sialyltransferase 1 in a fructoseresponsive manner promotes pancreatic cancer metastasis. Oncotarget 8, 7691–7709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang C, Chen L, Savage SR, Eguez RV, Dou Y, Li Y, da Veiga Leprevost F, Jaehnig EJ, Lei JT, Wen B, et al. (2021). Proteogenomic insights into the biology and treatment of HPV-negative head and neck squamous cell carcinoma. Cancer Cell 39, 361–379 e316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iacobuzio-Donahue CA, van der Heijden MS, Baumgartner MR, Troup WJ, Romm JM, Doheny K, Pugh E, Yeo CJ, Goggins MG, Hruban RH, et al. (2004). Large-scale allelotype of pancreaticobiliary carcinoma provides quantitative estimates of genome-wide allelic loss. Cancer research 64, 871–875. [DOI] [PubMed] [Google Scholar]
- Janes MR, Zhang J, Li LS, Hansen R, Peters U, Guo X, Chen Y, Babbar A, Firdaus SJ, Darjania L, et al. (2018). Targeting KRAS Mutant Cancers with a Covalent G12C-Specific Inhibitor. Cell 172, 578–589 e517. [DOI] [PubMed] [Google Scholar]
- Jones S, Zhang X, Parsons DW, Lin JC, Leary RJ, Angenendt P, Mankoo P, Carter H, Kamiyama H, Jimeno A, et al. (2008). Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science 321, 1801–1806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufman B, Shapira-Frommer R, Schmutzler RK, Audeh MW, Friedlander M, Balmana J, Mitchell G, Fried G, Stemmer SM, Hubert A, et al. (2015). Olaparib monotherapy in patients with advanced cancer and a germline BRCA1/2 mutation. Journal of clinical oncology: official journal of the American Society of Clinical Oncology 33, 244–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Bamlet WR, Oberg AL, Chaffee KG, Donahue G, Cao XJ, Chari S, Garcia BA, Petersen GM, and Zaret KS (2017). Detection of early pancreatic ductal adenocarcinoma with thrombospondin-2 and CA19-9 blood markers. Science translational medicine 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein D (2018). The Tumor Vascular Endothelium as Decision Maker in Cancer Therapy. Front Oncol 8, 367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosanam H, Prassas I, Chrystoja CC, Soleas I, Chan A, Dimitromanolakis A, Blasutig IM, Ruckert F, Gruetzmann R, Pilarsky C, et al. (2013). Laminin, gamma 2 (LAMC2): a promising new putative pancreatic cancer biomarker identified by proteomic analysis of pancreatic adenocarcinoma tissues. Mol Cell Proteomics 12, 2820–2832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H, Jeong AJ, and Ye SK (2019). Highlighted STAT3 as a potential drug target for cancer therapy. BMB reports 52, 415–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenkiewicz E, Malasi S, Hogenson TL, Flores LF, Barham W, Phillips WJ, Roesler AS, Chambers KR, Rajbhandari N, Hayashi A, et al. (2020). Genomic and Epigenomic Landscaping Defines New Therapeutic Targets for Adenosquamous Carcinoma of the Pancreas. Cancer research. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Orlandi R, White CN, Rosenzweig J, Zhao J, Seregni E, Morelli D, Yu Y, Meng XY, Zhang Z, et al. (2005). Independent validation of candidate breast cancer serum biomarkers identified by mass spectrometry. Clinical chemistry 51, 2229–2235. [DOI] [PubMed] [Google Scholar]
- Lowery MA, Kelsen DP, Capanu M, Smith SC, Lee JW, Stadler ZK, Moore MJ, Kindler HL, Golan T, Segal A, et al. (2018). Phase II trial of veliparib in patients with previously treated BRCA-mutated pancreas ductal adenocarcinoma. Eur J Cancer 89, 19–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macauley MS, Arlian BM, Rillahan CD, Pang PC, Bortell N, Marcondes MC, Haslam SM, Dell A, and Paulson JC (2014). Systemic blockade of sialylation in mice with a global inhibitor of sialyltransferases. J Biol Chem 289, 35149–35158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manoli S, Coppola S, Duranti C, Lulli M, Magni L, Kuppalu N, Nielsen N, Schmidt T, Schwab A, Becchetti A, et al. (2019). The Activity of Kv 11.1 Potassium Channel Modulates F-Actin Organization During Cell Migration of Pancreatic Ductal Adenocarcinoma Cells. Cancers 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurer C, Holmstrom SR, He J, Laise P, Su T, Ahmed A, Hibshoosh H, Chabot JA, Oberstein PE, Sepulveda AR, et al. (2019). Experimental microdissection enables functional harmonisation of pancreatic cancer subtypes. Gut 68, 1034–1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mer AS, Ba-Alawi W, Smirnov P, Wang YX, Brew B, Ortmann J, Tsao MS, Cescon DW, Goldenberg A, and Haibe-Kains B (2019). Integrative Pharmacogenomics Analysis of Patient-Derived Xenografts. Cancer Res 79, 4539–4550. [DOI] [PubMed] [Google Scholar]
- Moffitt RA, Marayati R, Flate EL, Volmar KE, Loeza SG, Hoadley KA, Rashid NU, Williams LA, Eaton SC, Chung AH, et al. (2015). Virtual microdissection identifies distinct tumor- and stroma-specific subtypes of pancreatic ductal adenocarcinoma. Nature genetics 47, 1168–1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montagnoli A, Valsasina B, Brotherton D, Troiani S, Rainoldi S, Tenca P, Molinari A, and Santocanale C (2006). Identification of Mcm2 phosphorylation sites by S-phase-regulating kinases. J Biol Chem 281, 10281–10290. [DOI] [PubMed] [Google Scholar]
- Motoi F, and Unno M (2020). Neoadjuvant treatment for resectable pancreatic adenocarcinoma: What is the best protocol? Annals of gastroenterological surgery 4, 100–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Najahi-Missaoui W, Quach ND, Jenkins A, Dabke I, Somanath PR, and Cummings BS (2019). Effect of P21-activated kinase 1 (PAK-1) inhibition on cancer cell growth, migration, and invasion. Pharmacology research & perspectives 7, e00518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Kane GM, Grunwald BT, Jang GH, Masoomian M, Picardo S, Grant RC, Denroche RE, Zhang A, Wang Y, Lam B, et al. (2020). GATA6 Expression Distinguishes Classical and Basal-like Subtypes in Advanced Pancreatic Cancer. Clin Cancer Res 26, 4901–4910. [DOI] [PubMed] [Google Scholar]
- Onuchic V, Hartmaier RJ, Boone DN, Samuels ML, Patel RY, White WM, Garovic VD, Oesterreich S, Roth ME, Lee AV, et al. (2016). Epigenomic Deconvolution of Breast Tumors Reveals Metabolic Coupling between Constituent Cell Types. Cell Rep 17, 2075–2086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozkan-Dagliyan I, Diehl JN, George SD, Schaefer A, Papke B, Klotz-Noack K, Waters AM, Goodwin CM, Gautam P, Pierobon M, et al. (2020). Low-Dose Vertical Inhibition of the RAF-MEK-ERK Cascade Causes Apoptotic Death of KRAS Mutant Cancers. Cell Rep 31, 107764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandey R, Zhou M, Islam S, Chen B, Barker NK, Langlais P, Srivastava A, Luo M, Cooke LS, Weterings E, et al. (2019). Carcinoembryonic antigen cell adhesion molecule 6 (CEACAM6) in Pancreatic Ductal Adenocarcinoma (PDA): An integrative analysis of a novel therapeutic target. Sci Rep 9, 18347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelicano H, Martin DS, Xu RH, and Huang P (2006). Glycolysis inhibition for anticancer treatment. Oncogene 25, 4633–4646. [DOI] [PubMed] [Google Scholar]
- Pelosi E, Castelli G, and Testa U (2017). Pancreatic Cancer: Molecular Characterization, Clonal Evolution and Cancer Stem Cells. Biomedicines 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereira SP, Oldfield L, Ney A, Hart PA, Keane MG, Pandol SJ, Li D, Greenhalf W, Jeon CY, Koay EJ, et al. (2020). Early detection of pancreatic cancer. The lancet Gastroenterology & hepatology 5, 698–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrova V, Annicchiarico-Petruzzelli M, Melino G, and Amelio I (2018). The hypoxic tumour microenvironment. Oncogenesis 7, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polzien L, Baljuls A, Albrecht M, Hekman M, and Rapp UR (2011). BAD contributes to RAF-mediated proliferation and cooperates with B-RAF-V600E in cancer signaling. J Biol Chem 286, 17934–17944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puleo F, Nicolle R, Blum Y, Cros J, Marisa L, Demetter P, Quertinmont E, Svrcek M, Elarouci N, Iovanna J, et al. (2018). Stratification of Pancreatic Ductal Adenocarcinomas Based on Tumor and Microenvironment Features. Gastroenterology 155, 1999–2013 e1993. [DOI] [PubMed] [Google Scholar]
- Quante AS, Ming C, Rottmann M, Engel J, Boeck S, Heinemann V, Westphalen CB, and Strauch K (2016). Projections of cancer incidence and cancer-related deaths in Germany by 2020 and 2030. Cancer medicine 5, 2649–2656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radeva MY, and Waschke J (2018). Mind the gap: mechanisms regulating the endothelial barrier. Acta Physiol (Oxf) 222. [DOI] [PubMed] [Google Scholar]
- Rahib L, Smith BD, Aizenberg R, Rosenzweig AB, Fleshman JM, and Matrisian LM (2014). Projecting cancer incidence and deaths to 2030: the unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer research 74, 2913–2921. [DOI] [PubMed] [Google Scholar]
- Rane CK, and Minden A (2019). P21 activated kinase signaling in cancer. Semin Cancer Biol 54, 40–49. [DOI] [PubMed] [Google Scholar]
- Ren L, Yi J, Li W, Zheng X, Liu J, Wang J, and Du G (2019). Apolipoproteins and cancer. Cancer medicine 8, 7032–7043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth MT, Cardin DB, and Berlin JD (2020). Recent advances in the treatment of pancreatic cancer. F1000Research 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruckert MT, de Andrade PV, Santos VS, and Silveira VS (2019). Protein tyrosine phosphatases: promising targets in pancreatic ductal adenocarcinoma. Cellular and molecular life sciences: CMLS 76, 2571–2592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sapalidis K, Kosmidis C, Funtanidou V, Katsaounis A, Barmpas A, Koimtzis G, Mantalobas S, Alexandrou V, Aidoni Z, Koulouris C, et al. (2019). Update on current pancreatic treatments: from molecular pathways to treatment. Journal of Cancer 10, 5162–5172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sava GP, Fan H, Coombes RC, Buluwela L, and Ali S (2020). CDK7 inhibitors as anticancer drugs. Cancer metastasis reviews 39, 805–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaaf MB, Garg AD, and Agostinis P (2018). Defining the role of the tumor vasculature in antitumor immunity and immunotherapy. Cell death & disease 9, 115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schubert M, Klinger B, Klunemann M, Sieber A, Uhlitz F, Sauer S, Garnett MJ, Bluthgen N, and Saez-Rodriguez J (2018). Perturbation-response genes reveal signaling footprints in cancer gene expression. Nat Commun 9, 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semenova G, and Chernoff J (2017). Targeting PAK1. Biochem Soc Trans 45, 79–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh A, Greninger P, Rhodes D, Koopman L, Violette S, Bardeesy N, and Settleman J (2009). A gene expression signature associated with “K-Ras addiction” reveals regulators of EMT and tumor cell survival. Cancer Cell 15, 489–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singhi AD, Koay EJ, Chari ST, and Maitra A (2019). Early Detection of Pancreatic Cancer: Opportunities and Challenges. Gastroenterology 156, 2024–2040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sokoll LJ, Wang Y, Feng Z, Kagan J, Partin AW, Sanda MG, Thompson IM, and Chan DW (2008). [−2]proenzyme prostate specific antigen for prostate cancer detection: a national cancer institute early detection research network validation study. The Journal of urology 180, 539–543; discussion 543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Springfeld C, Jager D, Buchler MW, Strobel O, Hackert T, Palmer DH, and Neoptolemos JP (2019). Chemotherapy for pancreatic cancer. Presse Med 48, e159–e174. [DOI] [PubMed] [Google Scholar]
- Stevenson RP, Veltman D, and Machesky LM (2012). Actin-bundling proteins in cancer progression at a glance. Journal of cell science 125, 1073–1079. [DOI] [PubMed] [Google Scholar]
- Strickland LA, Ross J, Williams S, Ross S, Romero M, Spencer S, Erickson R, Sutcliffe J, Verbeke C, Polakis P, et al. (2009). Preclinical evaluation of carcinoembryonic cell adhesion molecule (CEACAM) 6 as potential therapy target for pancreatic adenocarcinoma. J Pathol 218, 380–390. [DOI] [PubMed] [Google Scholar]
- Sun XJ, Liu BY, Yan S, Jiang TH, Cheng HQ, Jiang HS, Cao Y, and Mao AW (2015). MicroRNA-29a Promotes Pancreatic Cancer Growth by Inhibiting Tristetraprolin. Cell Physiol Biochem 37, 707–718. [DOI] [PubMed] [Google Scholar]
- Suzuki O (2019). Glycosylation in lymphoma: Biology and glycotherapy. Pathology international 69, 441–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao H, Liu S, Huang D, Han X, Wu X, Shao YW, and Hu Y (2020). Acquired multiple secondary BRCA2 mutations upon PARPi resistance in a metastatic pancreatic cancer patient harboring a BRCA2 germline mutation. American journal of translational research 12, 612–617. [PMC free article] [PubMed] [Google Scholar]
- The Cancer Genome Atlas Research Network (2011). Integrated genomic analyses of ovarian carcinoma. Nature 474, 609–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Cancer Genome Atlas Research Network (2012a). Comprehensive genomic characterization of squamous cell lung cancers. Nature 489, 519–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Cancer Genome Atlas Research Network (2012b). Comprehensive molecular portraits of human breast tumours. Nature 490, 61–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Cancer Genome Atlas Research Network (2014a). Comprehensive molecular profiling of lung adenocarcinoma. Nature 511, 543–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Cancer Genome Atlas Research Network (2014b). Integrated genomic characterization of papillary thyroid carcinoma. Cell 159, 676–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Cancer Genome Atlas Research Network (2017a). Comprehensive and Integrative Genomic Characterization of Hepatocellular Carcinoma. Cell 169, 1327–1341 e1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Cancer Genome Atlas Research Network (2017b). Integrated Genomic Characterization of Pancreatic Ductal Adenocarcinoma. Cancer cell 32, 185–203 e113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas JK, Kim MS, Balakrishnan L, Nanjappa V, Raju R, Marimuthu A, Radhakrishnan A, Muthusamy B, Khan AA, Sakamuri S, et al. (2014). Pancreatic Cancer Database: an integrative resource for pancreatic cancer. Cancer biology & therapy 15, 963–967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson ED, Roberts NJ, Wood LD, Eshleman JR, Goggins MG, Kern SE, Klein AP, and Hruban RH (2020). The genetics of ductal adenocarcinoma of the pancreas in the year 2020: dramatic progress, but far to go. Modern pathology: an official journal of the United States and Canadian Academy of Pathology, Inc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhlen M, Fagerberg L, Hallstrom BM, Lindskog C, Oksvold P, Mardinoglu A, Sivertsson A, Kampf C, Sjostedt E, Asplund A, et al. (2015). Proteomics. Tissue-based map of the human proteome. Science 347, 1260419. [DOI] [PubMed] [Google Scholar]
- Uprety D, and Adjei AA (2020). KRAS: From undruggable to a druggable Cancer Target. Cancer treatment reviews 89, 102070. [DOI] [PubMed] [Google Scholar]
- Vaccaro V, Sperduti I, and Milella M (2011). FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. The New England journal of medicine 365, 768–769; author reply 769. [DOI] [PubMed] [Google Scholar]
- Vajaria BN, Patel KR, Begum R, and Patel PS (2016). Sialylation: an Avenue to Target Cancer Cells. Pathology oncology research: POR 22, 443–447. [DOI] [PubMed] [Google Scholar]
- van Erning FN, Mackay TM, van der Geest LGM, Groot Koerkamp B, van Laarhoven HWM, Bonsing BA, Wilmink JW, van Santvoort HC, de Vos-Geelen J, van Eijck CHJ, et al. (2018). Association of the location of pancreatic ductal adenocarcinoma (head, body, tail) with tumor stage, treatment, and survival: a population-based analysis. Acta Oncol 57, 1655–1662. [DOI] [PubMed] [Google Scholar]
- Varki A (2017). Biological roles of glycans. Glycobiology 27, 3–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasaikar S, Huang C, Wang X, Petyuk VA, Savage SR, Wen B, Dou Y, Zhang Y, Shi Z, Arshad OA, et al. (2019). Proteogenomic Analysis of Human Colon Cancer Reveals New Therapeutic Opportunities. Cell 177, 1035–1049 e1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vatansever S, Erman B, and Gumus ZH (2020). Comparative effects of oncogenic mutations G12C, G12V, G13D, and Q61H on local conformations and dynamics of K-Ras. Computational and structural biotechnology journal 18, 1000–1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Von Hoff DD, Ervin T, Arena FP, Chiorean EG, Infante J, Moore M, Seay T, Tjulandin SA, Ma WW, Saleh MN, et al. (2013). Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. The New England journal of medicine 369, 1691–1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang SC, Huang CC, Shen CH, Lin LC, Zhao PW, Chen SY, Deng YC, and Liu YW (2016). Gene Expression and DNA Methylation Status of Glutathione S-Transferase Mu1 and Mu5 in Urothelial Carcinoma. PloS one 11, e0159102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei ZR, Liang C, Feng D, Cheng YJ, Wang WM, Yang DJ, Wang YX, and Cai QP (2016). Low tristetraprolin expression promotes cell proliferation and predicts poor patients outcome in pancreatic cancer. Oncotarget 7, 17737–17750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wishart DS, Feunang YD, Guo AC, Lo EJ, Marcu A, Grant JR, Sajed T, Johnson D, Li C, Sayeeda Z, et al. (2018). DrugBank 5.0: a major update to the DrugBank database for 2018. Nucleic acids research 46, D1074–D1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witkiewicz AK, McMillan EA, Balaji U, Baek G, Lin WC, Mansour J, Mollaee M, Wagner KU, Koduru P, Yopp A, et al. (2015). Whole-exome sequencing of pancreatic cancer defines genetic diversity and therapeutic targets. Nat Commun 6, 6744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wojcik EJ, Sharifpoor S, Miller NA, Wright TG, Watering R, Tremblay EA, Swan K, Mueller CR, and Elliott BE (2006). A novel activating function of c-Src and Stat3 on HGF transcription in mammary carcinoma cells. Oncogene 25, 2773–2784. [DOI] [PubMed] [Google Scholar]
- Wu X, Zhao J, Ruan Y, Sun L, Xu C, and Jiang H (2018). Sialyltransferase ST3GAL1 promotes cell migration, invasion, and TGF-beta1-induced EMT and confers paclitaxel resistance in ovarian cancer. Cell death & disease 9, 1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y (2015). Cancer immunotherapy: harnessing the immune system to battle cancer. The Journal of clinical investigation 125, 3335–3337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye DZ, and Field J (2012). PAK signaling in cancer. Cellular logistics 2, 105–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeo D, He H, Patel O, Lowy AM, Baldwin GS, and Nikfarjam M (2016). FRAX597, a PAK1 inhibitor, synergistically reduces pancreatic cancer growth when combined with gemcitabine. BMC cancer 16, 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo M, Shin J, Kim J, Ryall KA, Lee K, Lee S, Jeon M, Kang J, and Tan AC (2015). DSigDB: drug signatures database for gene set analysis. Bioinformatics 31, 3069–3071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan J, Zhang F, and Niu R (2015). Multiple regulation pathways and pivotal biological functions of STAT3 in cancer. Sci Rep 5, 17663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue T, Partyka K, Maupin KA, Hurley M, Andrews P, Kaul K, Moser AJ, Zeh H, Brand RE, and Haab BB (2011). Identification of blood-protein carriers of the CA 19-9 antigen and characterization of prevalence in pancreatic diseases. Proteomics 11, 3665–3674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuen A, and Diaz B (2014). The impact of hypoxia in pancreatic cancer invasion and metastasis. Hypoxia (Auckl) 2, 91–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Li XJ, Martin DB, and Aebersold R (2003). Identification and quantification of N-linked glycoproteins using hydrazide chemistry, stable isotope labeling and mass spectrometry. Nat Biotechnol 21, 660–666. [DOI] [PubMed] [Google Scholar]
- Zhang H, Liu T, Zhang Z, Payne SH, Zhang B, McDermott JE, Zhou JY, Petyuk VA, Chen L, Ray D, et al. (2016). Integrated Proteogenomic Characterization of Human High-Grade Serous Ovarian Cancer. Cell 166, 755–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Wu Y, Hu X, Wang B, Wang L, Zhang S, Cao J, and Wang Z (2017). GSTT1, GSTP1, and GSTM1 genetic variants are associated with survival in previously untreated metastatic breast cancer. Oncotarget 8, 105905–105914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou W, Jubb AM, Lyle K, Xiao Q, Ong CC, Desai R, Fu L, Gnad F, Song Q, Haverty PM, et al. (2014). PAK1 mediates pancreatic cancer cell migration and resistance to MET inhibition. J Pathol 234, 502–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auton A, Brooks LD, Durbin RM, Garrison EP, Kang HM, Korbel JO, Marchini JL, McCarthy S, McVean GA, and Abecasis GR (2015). A global reference for human genetic variation. Nature 526, 68–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babiceanu M, Qin F, Xie Z, Jia Y, Lopez K, Janus N, Facemire L, Kumar S, Pang Y, Qi Y, et al. (2016). Recurrent chimeric fusion RNAs in non-cancer tissues and cells. Nucleic acids research 44, 2859–2872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey MH, Tokheim C, Porta-Pardo E, Sengupta S, Bertrand D, Weerasinghe A, Colaprico A, Wendl MC, Kim J, Reardon B, et al. (2018). Comprehensive Characterization of Cancer Driver Genes and Mutations. Cell 173, 371–385 e318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbie DA, Tamayo P, Boehm JS, Kim SY, Moody SE, Dunn IF, Schinzel AC, Sandy P, Meylan E, Scholl C, et al. (2009). Systematic RNA interference reveals that oncogenic KRAS-driven cancers require TBK1. Nature 462, 108–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benelli M, Pescucci C, Marseglia G, Severgnini M, Torricelli F, and Magi A (2012). Discovering chimeric transcripts in paired-end RNA-seq data by using EricScript. Bioinformatics 28, 3232–3239. [DOI] [PubMed] [Google Scholar]
- Benjamini Y, and Hochberg Y (1995). Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. Journal of the Royal Statistical Society Series B (Methodological) 57, 289–300. [Google Scholar]
- Brunet JP, Tamayo P, Golub TR, and Mesirov JP (2004). Metagenes and molecular pattern discovery using matrix factorization. Proc Natl Acad Sci U S A 101, 4164–4169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho KC, Clark DJ, Schnaubelt M, Teo GC, Leprevost FDV, Bocik W, Boja ES, Hiltke T, Nesvizhskii AI, and Zhang H (2020). Deep Proteomics Using Two Dimensional Data Independent Acquisition Mass Spectrometry. Anal Chem 92, 4217–4225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cibulskis K, Lawrence MS, Carter SL, Sivachenko A, Jaffe D, Sougnez C, Gabriel S, Meyerson M, Lander ES, and Getz G (2013). Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat Biotechnol 31, 213–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark DJ, Hu Y, Bocik W, Chen L, Schnaubelt M, Roberts R, Shah P, Whiteley G, and Zhang H (2018). Evaluation of NCI-7 Cell Line Panel as a Reference Material for Clinical Proteomics. J Proteome Res 17, 2205–2215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- da Veiga Leprevost F, Haynes SE, Avtonomov DM, Chang HY, Shanmugam AK, Mellacheruvu D, Kong AT, and Nesvizhskii AI (2020). Philosopher: a versatile toolkit for shotgun proteomics data analysis. Nat Methods 17, 869–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Djomehri SI, Gonzalez ME, da Veiga Leprevost F, Tekula SR, Chang HY, White MJ, Cimino-Mathews A, Burman B, Basrur V, Argani P, et al. (2020). Quantitative proteomic landscape of metaplastic breast carcinoma pathological subtypes and their relationship to triple-negative tumors. Nat Commun 11, 1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan G, Li X, and Kohn M (2015). The human DEPhOsphorylation database DEPOD: a 2015 update. Nucleic acids research 43, D531–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabregat A, Jupe S, Matthews L, Sidiropoulos K, Gillespie M, Garapati P, Haw R, Jassal B, Korninger F, May B, et al. (2018). The Reactome Pathway Knowledgebase. Nucleic acids research 46, D649–D655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher S, Barry A, Abreu J, Minie B, Nolan J, Delorey TM, Young G, Fennell TJ, Allen A, Ambrogio L, et al. (2011). A scalable, fully automated process for construction of sequence-ready human exome targeted capture libraries. Genome Biol 12, R1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Q, Liang WW, Foltz SM, Mutharasu G, Jayasinghe RG, Cao S, Liao WW, Reynolds SM, Wyczalkowski MA, Yao L, et al. (2018). Driver Fusions and Their Implications in the Development and Treatment of Human Cancers. Cell Rep 23, 227–238 e223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaujoux R, and Seoighe C (2010). A flexible R package for nonnegative matrix factorization. BMC bioinformatics 11, 367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas BJ, Dobin A, Li B, Stransky N, Pochet N, and Regev A (2019). Accuracy assessment of fusion transcript detection via read-mapping and de novo fusion transcript assembly-based methods. Genome Biol 20, 213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanzelmann S, Castelo R, and Guinney J (2013). GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinformatics 14, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hornbeck PV, Zhang B, Murray B, Kornhauser JM, Latham V, and Skrzypek E (2015). PhosphoSitePlus, 2014: mutations, PTMs and recalibrations. Nucleic acids research 43, D512–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Y, Shah P, Clark DJ, Ao M, and Zhang H (2018). Reanalysis of Global Proteomic and Phosphoproteomic Data Identified a Large Number of Glycopeptides. Anal Chem 90, 8065–8071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang C, Chen L, Savage SR, Eguez RV, Dou Y, Li Y, da Veiga Leprevost F, Jaehnig EJ, Lei JT, Wen B, et al. (2021). Proteogenomic insights into the biology and treatment of HPV-negative head and neck squamous cell carcinoma. Cancer Cell. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang KL, Mashl RJ, Wu Y, Ritter DI, Wang J, Oh C, Paczkowska M, Reynolds S, Wyczalkowski MA, Oak N, et al. (2018). Pathogenic Germline Variants in 10,389 Adult Cancers. Cell 173, 355–370 e314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber W, Carey VJ, Gentleman R, Anders S, Carlson M, Carvalho BS, Bravo HC, Davis S, Gatto L, Girke T, et al. (2015). Orchestrating high-throughput genomic analysis with Bioconductor. Nat Methods 12, 115–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iorio F, Knijnenburg TA, Vis DJ, Bignell GR, Menden MP, Schubert M, Aben N, Goncalves E, Barthorpe S, Lightfoot H, et al. (2016). A Landscape of Pharmacogenomic Interactions in Cancer. Cell 166, 740–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alfoldi J, Wang Q, Collins RL, Laricchia KM, Ganna A, Birnbaum DP, et al. (2020). The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 581, 434–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller A, Nesvizhskii AI, Kolker E, and Aebersold R (2002). Empirical statistical model to estimate the accuracy of peptide identifications made by MS/MS and database search. Anal Chem 74, 5383–5392. [DOI] [PubMed] [Google Scholar]
- Kim H, and Park H (2007). Sparse non-negative matrix factorizations via alternating non-negativity-constrained least squares for microarray data analysis. Bioinformatics 23, 1495–1502. [DOI] [PubMed] [Google Scholar]
- Kim S, Scheffler K, Halpern AL, Bekritsky MA, Noh E, Kallberg M, Chen X, Kim Y, Beyter D, Krusche P, et al. (2018). Strelka2: fast and accurate calling of germline and somatic variants. Nat Methods 15, 591–594. [DOI] [PubMed] [Google Scholar]
- Koboldt DC, Zhang Q, Larson DE, Shen D, McLellan MD, Lin L, Miller CA, Mardis ER, Ding L, and Wilson RK (2012). VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome research 22, 568–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong AT, Leprevost FV, Avtonomov DM, Mellacheruvu D, and Nesvizhskii AI (2017). MSFragger: ultrafast and comprehensive peptide identification in mass spectrometry-based proteomics. Nat Methods 14, 513–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krug K, Jaehnig EJ, Satpathy S, Blumenberg L, Karpova A, Anurag M, Miles G, Mertins P, Geffen Y, Tang LC, et al. (2020). Proteogenomic Landscape of Breast Cancer Tumorigenesis and Targeted Therapy. Cell 183, 1436–1456 e1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krug K, Mertins P, Zhang B, Hornbeck P, Raju R, Ahmad R, Szucs M, Mundt F, Forestier D, Jane-Valbuena J, et al. (2019). A Curated Resource for Phosphosite-specific Signature Analysis. Mol Cell Proteomics 18, 576–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, and Durbin R (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Xie X, Zhou J, Sheng M, Yin X, Ko EA, Zhou T, and Gu W (2017). Quantifying circular RNA expression from RNA-seq data using model-based framework. Bioinformatics 33, 2131–2139. [DOI] [PubMed] [Google Scholar]
- Liao Y, Wang J, Jaehnig EJ, Shi Z, and Zhang B (2019). WebGestalt 2019: gene set analysis toolkit with revamped UIs and APIs. Nucleic acids research 47, W199–W205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liberzon A, Birger C, Thorvaldsdottir H, Ghandi M, Mesirov JP, and Tamayo P (2015). The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell systems 1, 417–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lih TM, Clark DJ, and Zhang H (2019). Omic-Sig: Utilizing Omics Data to Explore and Visualize Kinase-Substrate Interactions. BioRxiv, 10.1101/746123. [DOI] [Google Scholar]
- Liu J, Bell AW, Bergeron JJ, Yanofsky CM, Carrillo B, Beaudrie CE, and Kearney RE (2007). Methods for peptide identification by spectral comparison. Proteome science 5, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lurie E, Liu D, LaPlante EL, Thistlethwaite LR, Yao Q, and Milosavljevic A (2020). Histoepigenetic analysis of the mesothelin network within pancreatic ductal adenocarcinoma cells reveals regulation of retinoic acid receptor gamma and AKT by mesothelin. Oncogenesis 9, 62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, et al. (2010). The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome research 20, 1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mermel CH, Schumacher SE, Hill B, Meyerson ML, Beroukhim R, and Getz G (2011). GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. Genome Biol 12, R41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mertins P, Tang LC, Krug K, Clark DJ, Gritsenko MA, Chen L, Clauser KR, Clauss TR, Shah P, Gillette MA, et al. (2018). Reproducible workflow for multiplexed deep-scale proteome and phosphoproteome analysis of tumor tissues by liquid chromatography-mass spectrometry. Nat Protoc 13, 1632–1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nesvizhskii AI, Keller A, Kolker E, and Aebersold R (2003). A statistical model for identifying proteins by tandem mass spectrometry. Anal Chem 75, 4646–4658. [DOI] [PubMed] [Google Scholar]
- Perfetto L, Briganti L, Calderone A, Cerquone Perpetuini A, Iannuccelli M, Langone F, Licata L, Marinkovic M, Mattioni A, Pavlidou T, et al. (2016). SIGNOR: a database of causal relationships between biological entities. Nucleic acids research 44, D548–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranzinger R, Herget S, von der Lieth CW, and Frank M (2011). GlycomeDB--a unified database for carbohydrate structures. Nucleic acids research 39, D373–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, and Smyth GK (2015). limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic acids research 43, e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruepp A, Waegele B, Lechner M, Brauner B, Dunger-Kaltenbach I, Fobo G, Frishman G, Montrone C, and Mewes HW (2010). CORUM: the comprehensive resource of mammalian protein complexes−-2009. Nucleic acids research 38, D497–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott AD, Huang KL, Weerasinghe A, Mashl RJ, Gao Q, Martins Rodrigues F, Wyczalkowski MA, and Ding L (2019). CharGer: clinical Characterization of Germline variants. Bioinformatics 35, 865–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shteynberg DD, Deutsch EW, Campbell DS, Hoopmann MR, Kusebauch U, Lee D, Mendoza L, Midha MK, Sun Z, Whetton AD, et al. (2019). PTMProphet: Fast and Accurate Mass Modification Localization for the Trans-Proteomic Pipeline. J Proteome Res 18, 4262–4272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun S, Hu Y, Ao M, Shah P, Chen J, Yang W, Jia X, Tian Y, Thomas S, and Zhang H (2019). N-GlycositeAtlas: a database resource for mass spectrometry-based human N-linked glycoprotein and glycosylation site mapping. Clinical proteomics 16, 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Cancer Genome Atlas Research Network (2017). Integrated Genomic Characterization of Pancreatic Ductal Adenocarcinoma. Cancer cell 32, 185–203 e113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The UniProt Consortium (2017). UniProt: the universal protein knowledgebase. Nucleic acids research 45, D158–D169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thul PJ, Akesson L, Wiking M, Mahdessian D, Geladaki A, Ait Blal H, Alm T, Asplund A, Bjork L, Breckels LM, et al. (2017). A subcellular map of the human proteome. Science 356. [DOI] [PubMed] [Google Scholar]
- Toghi Eshghi S, Yang W, Hu Y, Shah P, Sun S, Li X, and Zhang H (2016). Classification of Tandem Mass Spectra for Identification of N- and O-linked Glycopeptides. Sci Rep 6, 37189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsou CC, Avtonomov D, Larsen B, Tucholska M, Choi H, Gingras AC, and Nesvizhskii AI (2015). DIA-Umpire: comprehensive computational framework for data-independent acquisition proteomics. Nat Methods 12, 258–264, 257–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turei D, Korcsmaros T, and Saez-Rodriguez J (2016). OmniPath: guidelines and gateway for literature-curated signaling pathway resources. Nat Methods 13, 966–967. [DOI] [PubMed] [Google Scholar]
- Wang LB, Karpova A, Gritsenko MA, Kyle JE, Cao S, Li Y, Rykunov D, Colaprico A, Rothstein JH, Hong R, et al. (2021). Proteogenomic and metabolomic characterization of human glioblastoma. Cancer Cell. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinstein JN, Collisson EA, Mills GB, Shaw KR, Ozenberger BA, Ellrott K, Shmulevich I, Sander C, and Stuart JM (2013). The Cancer Genome Atlas Pan-Cancer analysis project. Nature genetics 45, 1113–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenger CD, and Coon JJ (2013). A proteomics search algorithm specifically designed for high-resolution tandem mass spectra. J Proteome Res 12, 1377–1386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkerson MD, and Hayes DN (2010). ConsensusClusterPlus: a class discovery tool with confidence assessments and item tracking. Bioinformatics 26, 1572–1573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xi R, Lee S, Xia Y, Kim TM, and Park PJ (2016). Copy number analysis of whole-genome data using BIC-seq2 and its application to detection of cancer susceptibility variants. Nucleic acids research 44, 6274–6286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang G, Hu Y, Sun S, Ouyang C, Yang W, Wang Q, Betenbaugh M, and Zhang H (2018a). Comprehensive Glycoproteomic Analysis of Chinese Hamster Ovary Cells. Anal Chem 90, 14294–14302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang W, Ao M, Hu Y, Li QK, and Zhang H (2018b). Mapping the O-glycoproteome using site-specific extraction of O-linked glycopeptides (EXoO). Molecular systems biology 14, e8486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang W, Song A, Ao M, Xu Y, and Zhang H (2020). Large-scale site-specific mapping of the O-GalNAc glycoproteome. Nat Protoc 15, 2589–2610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye K, Schulz MH, Long Q, Apweiler R, and Ning Z (2009). Pindel: a pattern growth approach to detect break points of large deletions and medium sized insertions from paired-end short reads. Bioinformatics 25, 2865–2871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo M, Shin J, Kim J, Ryall KA, Lee K, Lee S, Jeon M, Kang J, and Tan AC (2015). DSigDB: drug signatures database for gene set analysis. Bioinformatics 31, 3069–3071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshihara K, Shahmoradgoli M, Martinez E, Vegesna R, Kim H, Torres-Garcia W, Trevino V, Shen H, Laird PW, Levine DA, et al. (2013). Inferring tumour purity and stromal and immune cell admixture from expression data. Nat Commun 4, 2612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu F, Teo GC, Kong AT, Haynes SE, Avtonomov DM, Geiszler DJ, and Nesvizhskii AI (2020). Identification of modified peptides using localization-aware open search. Nat Commun 11, 4065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, White NM, Schmidt HK, Fulton RS, Tomlinson C, Warren WC, Wilson RK, and Maher CA (2016). INTEGRATE: gene fusion discovery using whole genome and transcriptome data. Genome research 26, 108–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
SI Reference
- Arntfield ME, and van der Kooy D (2011). beta-Cell evolution: How the pancreas borrowed from the brain: The shared toolbox of genes expressed by neural and pancreatic endocrine cells may reflect their evolutionary relationship. Bioessays 33, 582–587. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S3. Tumor-associated protein and phosphosites, related to Figure 3. A) Median log2 protein fold change between tumor and paired NAT or unpaired normal ductal tissue. B) Immunohistochemistry levels for tumor-associated proteins in pancreatic cancer samples in Human Protein Atlas. C) RALGAPA2 protein abundance in tumor and NAT. D) RALGAPA2 phosphosite abundance in tumor and NAT. ** p<0.001 Wilcoxon signed rank test, NS=not significant.
Figure S2. Impacts of genomic and epigenetic alterations, related to Figure 2. A) Cis- and trans-effects of the TP53 truncation and missense mutation groups. Samples with wild-type TP53 serve as the control. B) CNV impacts on gene, protein, and phosphoprotein of the most common proteins with CNV events. The control group in each comparison includes all samples without the copy number event for each gene or protein. C) Functional impacts of CNVs on gene/protein/phosphorylation expression. The data in the top panel show the correlations of CNVs to mRNA, protein, and phosphorylation levels with significant positive correlations in red and significant negative correlations in blue (adjusted p-value < 0.05). The data in the bottom panel show the summed number of significant positive and negative correlations at mRNA, protein, and phosphopeptide. D) Correlation and anti-correlation of gene, protein, and phosphoprotein levels with DNA methylation levels. E) The two dimensional hierarchical clustering of methylation probes (Y-axis) and tumor and NAT samples (X-axis) on PDAC DNA methylation data. F) DNA methylation status between tumors with different pathological stages and NATs. G) Scatter plot of epigenetically silenced genes on survival significance and silenced samples counting. H) Scatter plot of beta values of probe cg21747958 and RNA expression of ZNF544 in tumors and NATs. I) Kaplan-Meier Plot of ZNF544 methylation association with overall survival. The two groups were separated by whether ZNF544 is silenced in the sample.
Figure S4. Differential expression of glycoproteins and glycosylation enzymes between PDAC tumors, early stage PDAC tumors, NATs, and normal duct tissues, related to Figure 4. A) Cellular distribution of tumor up-regulated and down-regulated N-linked glycoproteins. B) Analysis of significantly differentially regulated pathways between tumors and NATs at the N-linked glycoprotein expression level. C) Expression pattern of mucin 1 (MUC1), mucin 3A (MUC3A), mucin 5AC (MUC5AC), mucin 5B (MUC5B), mucin 13 (MUC13), and mucin 16 (MUC16) in tumors, early stage tumors, NATs, and normal duct tissues. The significance is defined as: N.S. P > 0.05, * P ≤ 0.05, **: P ≤ 0.01, ***: P ≤ 0.001 D) Expression pattern of CEA cell adhesion molecule 5 (CEACAM5) and CEA cell adhesion molecule 6 (CEACAM6) in tumors, early stage tumors, tumors with different KRAS hotspot mutations, NATs, and normal duct tissues. E) Expression pattern of galectin binding protein 3 (LGALS3BP), hemopexin (HPX), and collagen type VI alpha 1 chain (COL6A1) in tumors, early stage tumors, NATs, and normal duct tissues. F) Differential gene expression of N-linked glycosylation enzymes between tumors and NATs.
Figure S5. Kinase-substrate pairs and phosphosites enriched in KRAS mutant tumors, related to Figure 5. A) Expression profiles of SRC, AKT1, and CDK7 in Normal duct, NAT, Tumor, and Early stage. B) Expression profiles of BAD-S134, MAPK6-S189, STAT3-Y705, FLNA-S2152, and MCM2-S139 in Normal duct, NAT, Tumor, and Early stage. C) Bar charts showing the phospho-substrates enriched (>2-fold increase with adjusted p-value<0.05) in the KRAS mutant tumors relative to NATs that were used for identifying kinases enriched in different KRAS hotspot mutant tumors. Normal duct: normal ductal tissues; NAT: normal adjacent tissues; Tumor: all PDAC tumors; Early stage: Stage I and II PDAC tumors. Asterisks represent significant differences between two groups (Benjamini-Hochberg adjusted p): *p < 0.05; **p < 0.01; ***p<0.001; N.S., not significant.
Figure S6. Identification of biological processes associated with immune-cold tumors, related to Figure 6. A-C) The relative levels of tissue components (A: immune; B: Epithelial; C: exo/endocrine) inferred by xCell correlate with the counterparts derived from methylation-based deconvolution. Note that xCell gene signatures do not explicitly contain exo/endocrine cells and the neurons were instead chosen as they are similar to exo/endocrine cells in terms of the regulatory factors utilized during development and differentiation (Arntfield and van der Kooy, 2011). Shown are Pearson’s correlation coefficients and p values. D) Representative histological images showing samples with immune cell infiltration (C3L-04848, ‘immune hot’) and samples with immune cell depletion (C3L-02809, ‘immune cold’). E) Representative histological image showing an example of contamination of lymph nodes (C3N-00303, to be excluded from ‘immune hot’). F) Boxplots comparing the abundance of exo/endocrine cells (from methylation-based deconvolution) and gene expression of amylase (AMY) and insulin (INS). G) Correlation network showing the relationship among all cell types inferred by xCell. Several network modules are identified to reflect the close connection for certain cell types and denoted by different colors. The endothelial cells are highlighted by dashed circles.H) Pathway enrichment using the differentially expressed proteins between immune hot (cluster D) and immune cold (cluster B and C) samples. Shown are pathways that are statistically significant (FDR<0.05) for either directions. I) Pathway enrichment of genes with phospho-specific regulation in the immune cold samples (FDR<0.05). Pathways highlighted by black borders are the ones discussed in the main text.
Figure S1. Characterization of PDAC cohort and proteogenomic data, related to Figure 1. A) Pairwise comparisons between replicate samples and samples within TMT plex. B) Gene-wise correlation of mRNA and protein expression in NATs, tumors, and all tumors and NATs combined. C) Estimation of neoplastic cellularity by histology, DNA methylation-based deconvolution, and transcript proportion-based methods and their correlation with KRAS VAF tumor fraction estimates. D) PCAs at the RNA, protein, phosphorylation, and N-linked glycosylation levels of sufficient purity, low purity, and NAT samples.
Figure S7. Unsupervised, non-negative matrix factorization (NMF)-based proteogenomic subtyping of 140 PDAC tumors using gene copy number, mRNA and protein expression, and phosphosite and glycosylation site abundances and clinical relevance of binary proteogenomic subtypes from the 105 tumors and the 4 proteogenomic subtypes from all 140 tumor samples, related to Figure 7. A) Heatmap showing the clustering of samples by the three published RNA subtyping schemes. Shown are also the histological diagnosis (PDAC vs. pancreatic adenosquamous carcinoma) and selected signature gene expression. B) Heatmap depicting the contribution of samples (x-axis) to each of the four clusters (C1-C4, y-axis). Sample weights have been normalized by the maximum weight in each column. Cluster membership scores indicating the strength of association of each sample with a given cluster were calculated as proportional weights. The columns of the matrix are ordered by NMF subtype and decreasing cluster membership score. C) Overrepresentation analysis of clinical variables and RNA-subtypes in each proteogenomic cluster (Fisher’s exact test) shown in panel A). All clusters showed significant overlap with RNA-subtypes (Bailey, Collisson, and Moffitt). Low purity tumors were enriched in C2 and C4. D-G) Distribution of inferred cell types (y-axis) across NMF-subtypes (x-axis). H) Cluster-specific drug signatures inferred by Gene Set Enrichment Analysis of DSigDB gene sets using NMF weights of protein features (derived from binary proteogenomic subtypes from the 105 tumors) as a ranking. NMF classical cluster (C1) showed enrichment of gene sets targeted by various chemotherapeutics, while the NMF basal-like (C2) cluster was enriched for gene sets of several kinase inhibitors targeting mTOR, ATM, PDGFR, VEGF or BTK (FDR < 0.05). I) Sankey diagram depicting the association of samples classified into xCell-based subtypes (center) with binary proteogenomic subtypes from 105 tumors (left) and the 4 proteogenomic subtypes from all 140 tumor samples (right). Nodes are colored according to the NMF or xCell subtype, links between nodes are colored by xCell subtype.
Table S1 Clinical information and molecular phenotypes of the samples analyzed in this study, related to Figure 1 and Figure S1.
Table S2 Copy number variation (CNV) and methylation analysis, related to Figure 2 and Figure S2.
Table S3 Tumor-associated proteins, phosphosites, and glycosites, related to Figure 3 and Figure S3.
Data Availability Statement
The raw proteomic data files generated during this study are available at the Proteomic Data Commons (PDC, https://pdc.cancer.gov/pdc/). Genomic, epigenomic, and transcriptomic data generated for this publication are available at the Genomic Data Commons (GDC, https://gdc.cancer.gov/). All processed data tables are available at PDC (https://pdc.cancer.gov/pdc/publications) and LinkedOmics (http://www.linkedomics.org/data_download/CPTAC-PDAC/).
The workflow described under ‘Multi-omics clustering’ has been implemented as a module for PANOPLY (https://github.com/broadinstitute/PANOPLY/) running on Broad’s cloud platform Terra (https://app.terra.bio/). The docker containers encapsulating the source code and required R-packages for NMF clustering and ssGSEA are available on Dockerhub (broadcptacdev/pgdac_mo_nmf:15, broadcptac/pgdac_ssgsea:5). The data evaluation tool has been implanted as a R package available in OmicsEV (https://github.com/bzhanglab/OmicsEV/). The codes for genomics data processing pipelines are available in https://github.com/ding-lab/.