

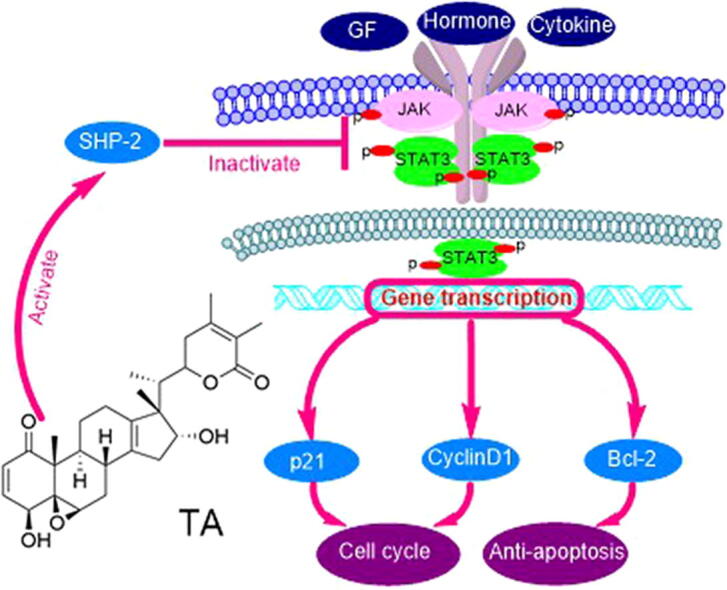
Graphical abstract
Keywords: Tubocapsenolide A, Osteosarcoma, SHP-2, JAK/STAT3
Abstract
Introduction
Previously, we have reported a withanolide-type steroid, named tubocapsenolide A (TA), which shows potent anti-proliferative activity in several cancer cell lines. However, its inhibitory effect on the Janus kinase/signal transducer and activator of transcription 3 (JAK/STAT3) pathway and therapeutic potential on osteosarcoma have not been reported.
Objectives
In the present study, we aimed to investigate the effect and molecular mechanism of TA in osteosarcoma.
Methods
The biological functions of TA in U2OS cells were investigated using colony formation, 5-ethynyl-20-deoxyuridine (EDU) staining, and cell cycle/apoptosis assays. The interaction between TA and Src homology 2 phosphatase 2 (SHP-2) was detected by enzyme activity and validated by target-identification methods such as drug affinity responsive target stability (DARTS), cellular thermal shift assay (CETSA), and biolayer interferometry (BLI). The in vivo anti-tumor efficacy of TA was analyzed in the xenograft tumor model. Western blotting analysis was performed to detect the protein expression levels.
Results
TA exhibited antitumor activity against osteosarcoma both in vitro and in vivo by regulating the JAK/STAT3 signaling pathway. Mechanically, TA interacted with SHP-2 directly and activated its phosphatase activity. Importantly, protein tyrosine phosphatase (PTP) inhibitor, SHP-2 inhibitor, and SHP-2 siRNA could reverse the inhibitory effect of TA on the JAK/STAT3 signaling pathway and restored the TA-induced cell death.
Conclusion
TA activated the phosphatase activity of SHP-2, which resulted in the inhibition of the JAK/STAT3 pathway and contributed to the antitumor efficacy of TA. Collectively, these findings suggested that TA could serve as a novel therapeutic agent for the treatment of osteosarcoma.
Introduction
As the most frequently diagnosed primary tumor in bone, osteosarcoma has a high incidence in pediatric patients [1]. The major cause of osteosarcoma progression is attributed to the lack of response to the standard chemotherapy regimen. To date, some chemotherapeutic drugs including high-dose methotrexate, doxorubicin, cisplatin, and ifosfamide, are used to eradicate osteosarcoma [2]. These drugs are widely used in various cancers with severe adverse effects. Therefore, it is urgently necessary to develop new drug candidates for the treatment of osteosarcoma.
Constitutively activated STAT3 plays important role in tumor cell growth, apoptosis, and metastasis [3], [4], [5]. Therefore, STAT3 is considered an attractive target for antitumor drug development. Compelling evidence from previous studies has revealed that JAK/STAT3 pathway plays a key role in the development of osteosarcoma, and STAT3 may become an attractive potential drug target for drug discovery of human osteosarcoma [6], [7], [8]. Therefore, agents that can suppress JAK/STAT3 activation have the potential as osteosarcoma therapeutics. Interestingly, tyrosine phosphatase family Src homology 2 phosphatase 1 (SHP-1), SHP-2, and protein tyrosine phosphatase 1B (PTP-1B) are the most important negative regulatory factors involved in JAK/STAT3 signaling through direct dephosphorylation of receptors or JAK tyrosine residues [9]. Additionally, several natural agents such as capillarisin [10], geranylnaringenin [11], and cryptotanshinone [12], have shown anticancer potentials in various cancers via SHP-1/SHP-2-mediated inhibition of the JAK/STAT3 signaling pathway. Nevertheless, such suppressive effect remains elusive in the progression of osteosarcoma, and there is a dearth of small-molecule that inhibits the JAK/STAT3 signaling pathway mediated by tyrosine phosphatases.
A great deal of attention has been paid to withanolides, a class of steroid compounds, due to their multiple bioactivities, such as immunosuppressive, anti-inflammation, antimicrobial, and antitumor properties [13], [14], [15]. Extensive studies have been carried out on the anticancer activity of the above-mentioned compounds. Especially, previous studies have shown that some withanolides can inhibit JAK/STAT3 signaling to exhibit the antitumor effect [16], [17]. However, the specific mechanism underlying the suppressive effects of these agents on STAT3 remains largely unexplored. TA, a withanolide isolated from Tubocapsicum anomalum, has been shown to possess potent anti-proliferative activity in several cancer cell lines [18], [19]. However, its in vitro and in vitro effects on osteosarcoma remain largely unknown. Particularly, whether it has effects on the JAK/STAT3 signaling pathway has not yet been documented. In the present study, we aimed to investigate the antitumor effect of TA on osteosarcoma and explore the underlying mechanisms. We found that TA exhibited potent cytotoxicity in osteosarcoma cells in vitro and in vivo. TA bound to SHP-2 directly and activated its phosphatase activity, leading to the inhibitory effect on the JAK/STAT3 signaling pathway, which contributed to the anti-proliferative effect in osteosarcoma.
Materials and methods
Reagents
TA (Fig. 1A) was isolated from Tubocapsicum anomalum in our laboratory [18]. The compound was identified by MS, 1H NMR, and 13C NMR spectroscopy. TA was dissolved in dimethyl sulfoxide (DMSO) to obtain a stock solution of 50 mM and diluted in cell culture medium for use. RPMI 1640, DMEMA, and fetal bovine serum were obtained from Thermo Fischer Scientific (Waltham, MA, USA). 3-(4, 5-dimethyl-2-thiazolyl)-2, 5-diphenyl-2-H-tetrazolium bromide (MTT), and Doxorubicin hydrochloride (DOX) were purchased from Sigma–Aldrich (St. Louis, MO, USA). Rabbit IgG HRP, Mouse IgG HRP, Cell Cycle Regulation Antibody Sampler Kit 9932 T (CDK2, p27, Cyclin D1, CDK6, Cyclin D3, p21, CDK4), Cleaved Caspase Antibody Sampler Kit 9929 T (cleaved caspase-3, 7, 9 and cleaved PARP), Procaspase Antibody Sampler Kit 12742 T (Caspase-3, 7, 9; PARP), Bcl-2 (3498 T), Bax (2772 T), STAT3(9139 T), Phospho-STAT3 (Tyr705)(9145 T), Phospho-Src (Tyr416)(6943 T), Src (2109 T), Phospho-JAK Family Antibody Sampler Kit 97999 T [JAK1/2/3, Phospho-JAK1(Tyr1034/1035), Phospho-JAK2 (Tyr1008), Phospho-JAK3 (Tyr980/981)], SHP-1(3759S), SHP-2 (3397 T), PTP1B (5311S), PTEN(9188 T), SOCS-1(3950 T), and SOCS-3(52113S) were purchased from Cell Signaling Technology (Beverly, MA, USA).
Fig. 1.

TA suppresses constitutive tyrosine phosphorylation of STAT3 and the proliferation of osteosarcoma cells (A) Chemical structure of TA. (B) 143B, U2OS, MG63, and HOS cells were treated with the indicated concentrations of TA for 12, 24, and 36 h. Cell viability was determined by MTT assay. (C) U2OS cells were incubated with TA at indicated concentrations for 24 h, replaced with fresh medium, and grow into colonies for 10 days. The cells were fixed, stained with crystal violet, and photographed. (D) U2OS cells were incubated with TA and then EdU staining was observed using confocal microscopy. (E) Osteosarcoma cell lines were treated with 2 μM TA for 24 h. Whole-cell extracts were prepared and immunoblotted with p-STAT3/STAT3 antibodies. (F) STAT3 inhibition activity by TA was measured by Western blotting. (G) U2OS cells were incubated with or without 2 μM TA for 24 h and then analyzed for the distribution of p-STAT3 by immunocytochemistry, Bars, 20 µm. (H) Effect of TA (IL6 or EGF as positive control) on the DNA-binding activity of STAT3. Nuclear extracts were incubated with a 32P-labeled STAT3 consensus oligonucleotide and subjected to 6% PAGE.
Cell culture
The osteosarcoma cancer cell lines of MG63, U2OS, HOS, and 143B, were purchased from the Cell Bank of Shanghai Institute of Biochemistry and Cell Biology, Chinese Academy of Sciences (Shanghai, China). All cells had been authenticated and tested for Mycoplasma. MG63, U2OS, and 143B cells were cultured in PRMI-1640, and HOS cells were cultured in DMEM, supplemented with 10% fetal bovine serum, 100U/ml penicillin, and 100 mg/ml streptomycin at 37 °C with 5% CO2.
Cell viability
The cell viability was measured by MTT assay. Cells (5 × 103/well) were seeded into a 96-well culture plate for 24 h. Then cells were incubated with or without serial dilutions of TA for 12, 24, and 36 h. After treatment, 20 µL of MTT (5 mg/mL) was added and incubated for 4 h. After the medium was removed, 150 µL DMSO was added to dissolve the formazan crystals. Shaking for 5 min, the absorbance at 570 nm was measured by a microplate reader.
Colony-forming and EdU assays
Colony formation and EdU assays were performed to analyze cell proliferation as described in suppl. (1.1).
Flow cytometry
The apoptosis and cell cycle distribution were detected by flow cytometry as described previously [20], which is conducted as described suppl. (1.2).
Immunocytochemistry for STAT3 localization
Immunocytochemistry was performed as reported previously [21], which is described in the suppl. (1.3).
EMSA for STAT3-DNA binding
Electrophoretic mobility shift assay (EMSA) for STAT3-DNA binding was performed as described previously [22]. The detailed procedure is shown in the suppl. (1.4).
Detection of SHP-2 phosphatase activity
After treatment of TA, U2OS or xenograft tumor tissue protein extract were incubated with anti-SHP-2 antibody (Ab) in immunoprecipitation (IP) buffer (20 mM of Tris-HCl, 150 mM of NaCl, 1 mM of ethylenediaminetetraacetic acid, 1 mM DTT, 1% beta-mercaptoethanol, 1% TritonX-100, 1 mM MgCl2 and 0.1% BSA, 1% Protease inhibitor, pH 7.5) overnight. Protein A/G-Agarose Resin (Yeasen, Shanghai, China) was added to each group, followed by incubation for 3 h at 4 °C with rotation. After centrifugation, SHP-2 PTP activity was measured using the Tyrosine Phosphatase Assay Kit (Promega, Madison, WI, USA) as specified by its manufacturer. The purified recombinant SHP-2 proteins (Novus Biologicals, Littleton, CO, USA) were preincubated with different concentrations of TA in the reaction mixture. The detection method of purified SHP-2 PTP activity was as same as the IP samples.
Western blotting
Western or IP cell lysate was used to lyse the collected cells or RIPA buffer was used for tumor tissues and then centrifuged to obtain supernate. The lysates were analyzed by Western blotting according to the method described previously [11]. The detailed procedure is shown suppl. (1.5).
Coomassie blue staining
After electrophoresis, the gels were stained for 1 h at room temperature with gentle shaking. After protein bands become blue, discard the stain, the gels were left in the water and detected using the ChemiDOC system (Bio-Rad, Hercules, CA).
Transfection with siRNA
The sequence of the negative control (NC), SHP-1, SHP-2, PTEN, and PTP1B siRNA duplexes (General biosystems, Nanjing, Jiangsu, China) were as shown in the Supplementary Table S1. For RNA interference, cells were seeded in 6-well plates, siRNA duplexes designed against conserved targeting sequences were transfected into the cells using Lipofectamine 3000 (Invitrogen, Carlsbad, CA) according to the manufacturer’s instructions.
DARTS and CETSA assay
DARTS was performed according to the previously described protocol [23] and CETSA was conducted using cell lysates as previously described [24]. The detailed procedure is conducted as described in the suppl. (1.6).
BLI assay
Binding kinetics measurements were performed by BLI assay using ForteBio Octet Red System (ForteBio, Inc., Menlo Park, USA). The purified recombinant his-tagged SHP-2 was immobilized onto NTA-Ni Biosensors, and the association and dissociation of TA were monitored in parallel to save time. The light shift distance (nm) corresponding to association/dissociation time. The constant Kd is calculated from the association and dissociation curve of TA binding with SHP-2.
Animal study
Ethical committee number for the study: CCPU 2019–094. All animal experimental procedures followed the National Institutes of Health guide for the care and use of laboratory animals and were performed under protocols approved by the Institutional Animal Care and Use Committee (IACUC) of China Pharmaceutical University Experimental Animal Center.
Xenograft tumor model Five-week-old male BALB/c-nu/nu mice were purchased from the Model Animal Research Center of Nanjing University (Nanjing, China). For the tumor xenograft assay, 5 × 106 U2OS cells were suspended in 200 μL PBS and subcutaneously injected into the right flank of the mice. When the tumors reached approximately 100 mm3, the tumor-bearing mice were randomly divided into four groups. Then, TA (5 or 10 mg/kg) and DOX (5 mg/kg) were intraperitoneally administered every two days for 20 days. When the tumor volume of the control group reached about 1000–2000 mm3, all of the mice were euthanized by intraperitoneal injection of pentobarbital sodium. Then, the tumors and visceral organs of each group were collected and fixed in 4% paraformaldehyde.
Immunohistochemistry
Solid tumors from control and various treatment groups were fixed with 4% formaldehyde solution, processed as described in the suppl. (1.7).
Statistical analysis
Statistical analysis was performed with ANOVA or Student’s t-test by using GraphPad Prism version 5.0 (GraphPad Software, San Diego, CA). For all the tests, P < 0.05 was considered statistically significant, the level of significance was *P < 0.05, **P < 0.01, ***P < 0.001.
Results
TA suppresses constitutive tyrosine phosphorylation of STAT3 and the proliferation of osteosarcoma cells
Due to the important roles of the JAK/STAT3 signaling pathway in the development and progression of osteosarcoma, we aimed to screen the inhibitors of STAT3 signaling, which might be developed as a novel strategy for the treatment of osteosarcoma. TA was selected in our study as it effectively decreased both the viability and STAT3 activity of osteosarcoma cells. For 143B, U2OS, MG63, and HOS cells, the IC50 value was 5.26 ± 1.04, 0.61 ± 0.13, 2.18 ± 0.31, and 1.14 ± 0.24 μM, respectively after TA treatment for 24 h (Fig. 1B). Since colony formation mimics tumor physiology and growth in vivo, we studied the effect of TA on colony formation. The colony formation of U2OS cells was greatly impeded by TA exposure, indicating that TA-treated cells lost their proliferative capacity (Fig. 1C). Correspondingly, EdU labeling assays also revealed that the cell viability of U2OS cells was suppressed by TA (Fig. 1D). To further investigate whether the STAT3 signaling was involved in the TA-induced anti-proliferative effect, STAT3 phosphorylation in different osteosarcoma cells was assessed. Fig. 1E shows that TA significantly blocked p-STAT3 activation in U2OS cells with hyperactive STAT3, which might explain different cytotoxicity in osteosarcoma cell lines. Because it possesses hyperactive STAT3, we further investigated the inhibitory effect of TA on STAT3 phosphorylation in U2OS cells. Western blotting analysis showed that TA decreased STAT3 phosphorylation in a time- and dose-dependent manner, while the protein level of total STAT3 remained unchanged (Fig. 1F). To regulate the expressions of its target genes, STAT3 must translocate from the cytoplasm to the nucleus. U2OS cells were exposed to TA for 24 h, followed by detection using immunofluorescence, and the nuclear translocation of STAT3 was largely inhibited when exposed to TA (Fig. 1G). Besides, EMSA assay demonstrated that TA treatment significantly inhibited the STAT3 DNA-binding activity in U2OS cells (Fig. 1H). Taken together, these results suggested that TA inhibited cell viability and STAT3 signaling in U2OS cells.
TA prevents IL-6/EGF-induced tyrosine phosphorylation of STAT3
STAT3 is phosphorylated by upstream kinases in response to cytokines and growth factors, including interleukin 6 (IL-6), and epidermal growth factor (EGF) [25], [26]. Therefore, we examined whether TA could inhibit IL-6/EGF-stimulated STAT3 phosphorylation. Fig. 2A illustrates that IL-6 treatment resulted in STAT3 activation, while pretreatment with TA significantly inhibited IL-6-induced STAT3 activation (Fig. 2B). Similar to IL-6, EGF stimulation increased the phosphorylated STAT3 level (Fig. 2C), and such increase was dampened by TA (Fig. 2D). Consistent with these data, the IL-6/EGF-increased density of the STAT3-probe-DNA band was significantly decreased when the cells were treated with TA (Fig. 2E). Therefore, TA could inhibit IL-6/EGF-induced activation of STAT3 in U2OS cells.
Fig. 2.

TA prevents IL-6/EGF-induced tyrosine phosphorylation of STAT3. (A, C) Cells were stimulated with different concentrations of IL-6 (A) or EGF (C) for 30 min, p-STAT3/STAT3 were detected. (B, D) Cells were pretreated with TA for 12 h and stimulated with IL-6 (B) or EGF (D) for 30 min, cell lysates were subjected to Western blotting to determine the p-STAT3/STAT3 level. (E) The DNA-binding activity of STAT3 was detected by EMSA. (F) U2OS cells were treated with indicated concentrations of TA, p-JAK1/2/3, and total JAK1/2/3 were examined by Western blotting. (G) U2OS cells were treated with the indicated concentration of pervanadate for 30 min, followed by 2 μM TA for 24 h, and the cell lysates were subjected to Western blotting. (H) The cells were treated with or without TA for 24 h and cell lysates were subjected to IP assay to determine the PTP activity of SHP-1, SHP-2, PTEN, and PTP1B, *P < 0.05, ***P < 0.001 versus control. (I) U2OS cells were transfected with negative control siRNA or SHP-1, SHP-2, PTEN, and PTP1B specific siRNA (100 nM). After 24 h, cells were treated with 2 μM TA for 24 h. Whole-cell extracts were prepared and immunoblotted with p-STAT3/STAT3 antibodies. (J) U2OS cells were pretreated with 25 μM NSC-87877 for 30 min, followed by 2 μM TA treatment for 24 h. Cell lysates were prepared and subjected to western blotting analysis.
TA inhibits phosphorylation of JAKs
Since JAKs, Src, and extracellular-signal-regulated kinase (ERK) are major upstream tyrosine families that regulate STAT3 activation [9], we examined the upstream signaling involved in TA-mediated STAT3 inactivation. The results showed that TA suppressed the constitutive activation of JAK1, JAK2, and JAK3 in a dose- and time-dependent manner (Fig. 2F). However, under the same condition, both the phosphorylated and total protein levels of Src were not affected by TA treatment (Fig. S1). Interestingly, the total protein levels of JAK1 and JAK2 were also altered by TA treatment, while the level of JAK3 was not changed, indicating that there might be other molecular mechanisms regulating this signaling pathway. Furthermore, we detected ERK to determine whether it was involved in the inhibitory effect of STAT3. Fig. S2 shows that pretreatment of 50 µM U0126 could not rescue TA-induced inactivation of STAT3, indicating that the inhibited STAT3 activation was not mediated by ERK. In conclusion, our results demonstrated that the inhibition of STAT3 phosphorylation was likely due to the inhibition of JAK activity.
Tyrosine phosphatase SHP-2 is involved in the TA-induced inactivation of STAT3
Because suppressors of cytokine signaling (SOCS) and PTPs have been reported to be negative regulators of the JAK/STAT3 signaling pathway [9], we assessed the potential role of upstream SOCS and PTPs in TA-induced inhibition of STAT3 in U2OS cells. TA did not significantly affect SOCS protein levels (Fig. S3), similar results were obtained in all known PTPs (SHP-1, SHP-2, PTEN, and PTP-1B) mediating STAT3 activity (Fig. S4). However, treatment with pervanadate (Na3VO4), a broad-acting tyrosine phosphatase inhibitor, prevented TA-induced suppression of STAT3 activation (Fig. 2G). These results suggested that PTPs played important roles in STAT3 inactivation by TA. To identify which PTP was affected by TA, the PTP activity was analyzed in U2OS cells. The dephosphorylation capacity of SHP-1 and PTEN remained unchanged, the PTP-1B activity was slightly elevated, while the SHP-2 activity was significantly enhanced by TA treatment under the same experiment conditions (Fig. 2H). Next, the effects of PTP gene silencing were explored. The U2OS cells were transfected with siRNAs of SHP-1, SHP-2, PTEN, and PTP-1B, or negative control, respectively, followed by treatment with 2 μM TA for 24 h. Fig. 2I shows that depletion of SHP-2 dramatically reversed the inhibitory effect of TA on STAT3, while the siRNAs of SHP-1, PTP-1B, and PTEN had no obvious effect on the reduction of dephosphorylated STAT3 by TA. Subsequently, we employed the SHP-1/2 inhibitor to confirm whether SHP-2 activation was essential for TA-mediated inhibition of STAT3. Indeed, treatment with NSC 87,877 significantly increased the levels of phosphorylated STAT3 (Fig. 2J). Furthermore, we found that Na3VO4, SHP-1/2 inhibitor, and SHP-2 depletion by siRNA efficiently abrogated TA-induced dephosphorylation of JAKs (Fig. S5-7). These results confirmed that TA directly interacted with SHP-2, leading to the inactivation of the JAK/STAT3 signaling pathway.
TA activates SHP-2 PTP activity by interacting with SHP-2 directly
DARTS and CETSA assay have been widely used to identify target proteins of drugs [27], [28]. Thereby, they were applied to determine the interaction between TA and SHP-2. Compared with the control lysates, TA-treated lanes showed a band with enhanced intensity between 60 and 75 kD (Fig. 3A). Immunoblotting was carried out to confirm whether the present band was SHP-2. Results showed that TA partially prevented the pronase-mediated digestion of SHP-2 in a dose-dependent manner (Fig. 3B). In contrast, the stability of other potential signaling proteins involved in the JAK/STAT3 signaling pathway was not affected in TA-treated cell lysate (Fig. S8). Next, CETSA was performed in U2OS cell lysate. Compared with DMSO-treated cell lysate, the thermal stability of SHP-2 was increased in TA-treated cells between 52 °C and 64 °C (Fig. 3C). We also tested whether SHP-2 stability was dose-dependent. Fig. 3D reveals that in contrast with the stability of GAPDH, SHP-2 accumulation was markedly increased as the TA concentration was increased. These results confirmed that TA directly interacted with SHP-2 in cells and enhanced the SHP-2 affinity target stability.
Fig. 3.

TA interacts with SHP-2 directly. (A, B) U2OS cell lysates were incubated in the presence or absence of TA for 1 h at room temperature, followed by proteolysis with indicated ratios of pronase for 20 min, the lysates were analyzed by Western blotting (A) The SDS-PAGE gels were stained by coomassie blue. (B) The content of SHP-2 was analyzed by SHP-2 antibody, *P < 0.05, **P < 0.01, ***P < 0.001 versus control. (C) The stabilizing effects of TA on SHP-2 at increasing temperature up to 64 °C were analyzed by Western blotting. (D) TA increased the thermal stability of SHP-2 compared with DMSO-treated U2OS cell lysates. U2OS lysates were mixed with the indicated doses of TA at 50 or 37 °C to evaluate the thermal stability of SHP-2.
The kinetic parameters of the interaction between TA and SHP-2 were further determined by biolayer interferometry. Fig. 4A shows that TA was bound to SHP-2 with Kd = 4.7 ± 0.69 μM. To further determine the action mode of TA on SHP-2, the SHP-2 activity in vitro and in vivo was detected. TA effectively up-regulated the SHP-2 activity both in SHP-2-containing cell extracts (Fig. 4B) and purified recombinant SHP-2 proteins (Fig. 4C), suggesting that TA activated SHP-2 through direct interaction with SHP-2. Consistent with this observation, the PTP inhibitors or the siRNAs effectively attenuated the SHP-2 phosphatase activity (Fig. 4D) and restored TA-induced cell death (Fig. 4E). Collectively, we demonstrated that TA activated SHP-2, which negatively regulated JAK/STAT3 phosphorylation, whereas inhibition of phosphatase SHP-2 rescued TA-induced cell death. Taken together, these results confirmed that TA directly interacted with SHP-2 and SHP-2 was a target protein of TA for modulation of JAK/STAT3 activation.
Fig. 4.

TA activates SHP-2 PTP activity and mediates the cell death induced by TA. (A) The kinetic curve of the interaction between TA and SHP-2 was determined by BLI. (B) Cell lysates from U2OS cells treated with TA for 24 h were subjected to SHP-2 phosphatase activity assay, *P < 0.05, ***P < 0.001 versus 0 μM TA group. (C) Purified SHP-2 protein was incubated with TA for 2 h at 25 °C, and then its phosphatase activity was detected. (D) SHP-2 phosphatase activity assay was detected by IP, #P < 0.05 versus inhibitors treatment, ***P < 0.001 versus control (E) Cell viability was assessed by MTT. ***P < 0.001 versus TA treatment group.
TA induces cell cycle arrest and apoptosis
TA suppressed both the constitutive and inducible STAT3 activation, which regulates the expressions of various genes involved in the cell cycle and apoptosis [3], [17], [21]. Therefore, cell cycle analysis was used to test the effect of TA on cell cycle progression in U2OS cells. Fig. 5A and B show that 3 h and 6 h treatment resulted in a cell-cycle arrest at the G0-G1 phase, whereas the G2/M arrest was continuously increased in a time-dependent manner, suggesting that TA induced moderate sub-G1 accumulation and cell cycle arrest at the G2/M phase. Moreover, we examined cell cycle-related proteins by Western blotting analysis. Fig. 5C shows that TA induced the expressions of p21 and p27 and reduced the protein levels of CDK2/4/6 and cyclin D1/D3/E1. These results revealed that TA suppressed cell proliferation in human osteosarcoma cells by blocking cell cycle progress. Cell apoptosis plays a significant role in the anti-proliferative effect by external factors. Fig. 5D reveals that TA treatment time-dependently increased the proportion of apoptotic cells, suggesting that TA induced apoptosis in U2OS cells. Bcl-2 family proteins, caspase proteins, and PARP play important roles in the apoptosis pathway. To further explore the mechanism of TA in apoptosis, we examined the expressions of these proteins. TA up-regulated the ratio of Bax/Bcl-2, reduced the inactive enzymatic form of caspase-9/7/3/PARP, and increased the cleavage of caspase-9/7/3/PARP in a dose- and time-dependent manner (Fig. 5E). These results further confirmed that the antitumor effect of TA was mediated through activation of the apoptotic signaling pathway in human osteosarcoma cells.
Fig. 5.

TA induces cell cycle arrest and apoptosis. (A) U2OS cells were treated with TA (2 μM) for different periods and the cell cycle distribution was examined by flow cytometry. (B) The histogram represents the percentage of cells in each phase of the cell cycle. (C) Cells were treated with indicated concentrations and periods of TA and evaluated for the expression of CDK2/4/6, cyclin D1/D3/E1, p21, and p27 proteins. (D) The apoptotic rate of U2OS cells after treatment with indicated periods of TA (2 μM) was determined by Annexin V- FITC and PI staining. **P < 0.01, ***P < 0.001 versus 0 μM treatment group. (E) Cells were treated with indicated concentrations and times of TA, the proteolytic activation of procaspase-7, −9, −3, cleavage of PARP, Bcl-2, and Bax was detected.
TA inhibits the growth of xenograft tumors
To evaluate the antitumor capacity of TA in vivo, we established a xenograft model by subcutaneously injecting U2OS cells into nude mice. Fig. 6A shows that the growth of the xenograft tumor was significantly inhibited by TA. Tumor weight and tumor volume were decreased observably after treatment with TA (Fig. 6B and C). Furthermore, TA did not significantly change the body weight of mice (Fig. 6D), whereas the DOX group showed a bodyweight loss, suggesting that TA had few side effects on the mouse body at our therapeutic concentration. Pathologically, no obvious morphological changes were observed in the organs of the tumor-bearing mice treated with TA (Fig. 6E), while animals treated with DOX showed cardiac toxicity and hepatic steatosis, which is consistent with the previous study [29], [30]. All of these results revealed that TA exerted antitumor activity with less toxicity compared with the DOX group in vivo.
Fig. 6.

TA inhibits the growth of xenograft tumors. (A-E) U2OS tumor-bearing mice treated with TA (10 and 5 mg/kg) or DOX (5 mg/kg) for 21 days. **P < 0.01, ***P < 0.001 versus control group (A) Tumor volume (B) Morphology of tumor (C) Changes of tumor weight (D) body weight (E) Hearts, livers, spleen, lungs, and kidneys were harvested and sectioned for H&E staining. Scale bars = 50 μm.
To further confirm the tumor inhibitory mechanism of TA, we carried out H&E and IHC staining against Ki67 in the tumor tissue. The results demonstrated the density of osteosarcoma cells in xenografts was much lower compared with the control group when treated with TA. Consistently, the expression of Ki67 was also reduced after TA treatment (Fig. 7A), indicating that TA suppressed the proliferation of tumor cells. Moreover, the expression of p-STAT3 was down-regulated in the TA treatment groups (Fig. 7B), which was consistent with the Western blotting analysis. Accordingly, the STAT3 target proteins such as Cyclin D1, p21, and Bcl-2 were also affected by TA administration (Fig. 7C). To confirm the in vivo effect of TA on SHP-2, we measured its tyrosine phosphatase activity in tumor tissues. We found that after TA treatment, the tyrosine phosphatase activity of SHP-2 was significantly increased (Fig. 7D). Taken together, these results proved that TA significantly suppressed U2OS xenograft tumor growth via SHP-2-mediated regulation of STAT3 target proteins in vivo.
Fig. 7.

TA activates SHP-2 and regulates the expression of STAT3 target proteins in tumor tissue. (A) Tumor sections were subjected to H&E staining and IHC against Ki-67. Scale bars = 50 μm. ***P < 0.001 versus control group. (B) Representative images of p-STAT3 IHC staining. Scale bars = 50 μm. ***P < 0.001 versus control group. (C) Western blotting analysis of p-STAT3, STAT3, CyclinD1, p21, Bcl-2 in tumor tissues. (D) The samples from the tumor tissue on day 21 were used in detecting the tyrosine phosphatase activity of SHP-2. **P < 0.01, ***P < 0.001 versus control group.
Discussion
SHP-2 is an important negative regulator of the JAK/STAT3 pathway which is involved in many pro-oncogenic signals to promote tumor development [31]. Therefore, agents that can inhibit STAT3 signal can be used in the treatment and prevention of cancer. TA is identified as a withanolide exhibiting cytotoxicity against a variety of human tumor cell lines [19]. However, the molecular mechanism and inhibitory effect of TA on JAK/STAT3 signaling remain largely unknown. In the present study, we showed that TA had powerful antitumor activity against osteosarcoma cells by suppressing the JAK/STAT3 pathway. Further results demonstrated that such inhibitory effect was mediated by activating SHP-2 tyrosine phosphatase activity. TA bound to SHP-2 directly and resulted in the inactivation of JAK/STAT3-related proteins, contributing to the antitumor efficacy of TA in osteosarcoma.
Constitutive STAT3 activation has been shown to prevent apoptosis. Some natural products and derivatives have been found to possess inhibitory effects on STAT3 activation such as curcumin [32], resveratrol [33], and others. In our study, we found that TA could suppress both constitutive and inducible STAT3 phosphorylation in osteosarcoma cells and these effects were especially significant in U2OS cells with hyperactive STAT3. We also observed that TA suppressed nuclear translocation and DNA binding activity of STAT3. Furthermore, we showed that TA inhibited the proliferation of osteosarcoma cells and such inhibition was correlated with the reduced STAT3 activation. The downregulation of STAT3 activation led to the suppression of various proteins such as Cyclin D1, p21, Bcl-2, which are involved in the survival and proliferation of tumor cells. As a result, TA induced osteosarcoma and the accumulation of cells in the G2/M phase of the cell cycle.
Inhibitors of STAT3 can be split into direct and indirect categories. Direct inhibitors interact with the STAT3 protein itself. Indirect inhibitors interfere with its ligands or upstream kinases that phosphorylate STAT3 or activate its negative regulators such as SOCS, ERK kinases, and phosphatase (SHP-1, SHP-2, PTP1B, PTEN, and so on) [9], [34], [35], [36]. To investigate the mechanism of TA-induced STAT3 inhibition in osteosarcoma cells, we analyzed the upstream signals involved in the inactivation of STAT3. Results showed that TA inhibited the phosphorylation of JAK1/2/3 in a concentration- and time-dependent manner. However, the p-Src or SOCS levels remained unchanged after TA treatment, while the TA-induced inhibition of the STAT3 signaling pathway could not be rescued by U0126, indicating that all these known upstream signals did not contribute to the inhibition of STAT3 by TA.
PTPs, such as SHP-1, SHP-2, and PTEN, negatively regulate JAK/STAT3 activation. PTPs can directly dephosphorylate the receptor tyrosine kinases, thus ensuring STAT3 signaling termination [36]. Previous studies have documented that natural products, such as farnesol and diosgenin, regulate STAT3 activation through up-regulating SHP-1 and SHP-2 [21], [37]. Given the critical role of PTPs in STAT3 activation and function, it has been considered an attractive targetable site in STAT3 inhibition. In our present study, a suppressed p-STAT3 level was rescued by pervanadate treatment, suggesting that PTPs played important roles in the action of TA. However, TA-induced suppression of p-STAT3 was not linked to the induced expression of SHP-1 or SHP-2 (Fig. S4). This finding suggested that TA regulated STAT3 activation via different modes of action. Interestingly, we noted that the transfection with SHP-2 siRNA reversed the inhibitory effect of TA on STAT3 (Fig. 2I), indicating that TA should modulate the level of p-STAT3 in an SHP-2-dependent manner. Our results were consistent with previous reports that SHP-2 is a negative regulator of JAK/STAT3 and a tumor suppressor in hepatocellular carcinogenesis and metachondromatosis [38], [39].
Furthermore, we applied target identification approaches, such as DARTS, CETSA, and BLI, to validate the direct interactions between SHP-2 and TA. After incubation with TA, the hydrolysis of SHP-2 by pronase was prevented, and the thermal stability of SHP-2 was increased in a dose-dependent manner. Moreover, the kinetic parameters of the interaction between TA and SHP-2 showed a Kd = 4.7 ± 0.69 μM. These results provided evidence that TA directly interacted with SHP-2. To confirm the specific action of TA on SHP-2, the enzyme activity in vitro and in vivo was detected. TA dramatically activated SHP-2 PTP activity both in SHP-2-containing cell extracts and purified recombinant SHP-2 proteins.
One of the important findings in this study was that TA inhibited xenograft tumor cell growth and suppressed the expression of genes known to drive uncontrolled cellular growth of cancer, such as Cyclin D1, p21, and Bcl-2. Previous studies indicated STAT3 activation is rapid and transient in non-cancerous cells, but constitutively activated in the majority of cancers [4]. This implies constitutively activated STAT3 is essential for cancer development at different levels. In line with this, our work uncovered TA significantly inhibited osteosarcoma cell proliferation by inhibiting the target genes of STAT3. Notably, TA displayed a satisfactory therapeutic effect in an osteosarcoma xenograft model without clear organ toxicity at our therapeutic concentration and showed a much better therapeutic effect compared with the DOX group. These results provided strong evidence that TA possessed potential therapeutic effects on osteosarcoma. However, it is still plausible that TA may have other targets involved in tumor progression. For example, the total protein level of JAK2 and JAK1 also altered by TA treatment could not be explained in the current study. Further investigations are still required to explore its other targets in the anti-proliferative activity of cancer cells. Besides, combination with standard-of-care agents for osteosarcoma therapy will be interesting to study if they complement each other.
Conclusions
We confirmed the direct interactions between TA and SHP-2 using target identification methods and measurements of the enzymatic activity. TA significantly suppressed osteosarcoma cell proliferation in vitro and in vivo through regulating the expressions of various JAK/STAT3 target proteins, such as Cyclin D1, p21, and Bcl-2, resulting in cell cycle arrest and apoptosis. These results demonstrated that TA suppressed osteosarcoma cell proliferation via SHP-2-mediated inhibition of the JAK/STAT3 signaling pathway. Thereby, our findings highlighted the important potential of TA as a drug candidate for the treatment of osteosarcoma.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
Acknowledgments
This work was supported financially by the National Natural Science Foundation of China (81872983), the Natural Science Foundation of Jiangsu Province (BK20181329), and the Drug Innovation Major Project (2018ZX09711-001-007).
Authors Contributions
Dongrong and Xiaoqin performed the experiments and analyzed the data. Dongrong and Chen wrote the manuscript. Sibei and Jiangmin isolated and identified the compound of TA. Hao, Lingyi, and Jianguang corrected the manuscript and approved the final version.
Footnotes
Peer review under responsibility of Cairo University.
Supplementary data to this article can be found online at https://doi.org/10.1016/j.jare.2021.06.004.
Contributor Information
Lingyi Kong, Email: lykong@cpu.edu.cn.
Jianguang Luo, Email: luojg@cpu.edu.cn.
Appendix A. Supplementary material
The following are the Supplementary data to this article:
References
- 1.Yan G.N., Lv Y.F., Guo Q.N. Advances in osteosarcoma stem cell research and opportunities for novel therapeutic targets. Cancer Lett. 2016;370(2):268–274. doi: 10.1016/j.canlet.2015.11.003. [DOI] [PubMed] [Google Scholar]
- 2.Isakoff M.S., Bielack S.S., Meltzer P., Gorlick R. Osteosarcoma: Current Treatment and a Collaborative Pathway to Success. J Clin Oncol. 2015;33(27):3029–3035. doi: 10.1200/JCO.2014.59.4895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chun J., Li R.J., Cheng M.S., Kim Y.S. Alantolactone selectively suppresses STAT3 activation and exhibits potent anticancer activity in MDA-MB-231 cells. Cancer Lett. 2015;357(1):393–403. doi: 10.1016/j.canlet.2014.11.049. [DOI] [PubMed] [Google Scholar]
- 4.Devarajan E., Huang S. STAT3 as a central regulator of tumor metastases. Curr Mol Med. 2009;9(5):626–633. doi: 10.2174/156652409788488720. [DOI] [PubMed] [Google Scholar]
- 5.Zhang X., Sun Y., Pireddu R., Yang H., Urlam M.K., Lawrence H.R., et al. A novel inhibitor of STAT3 homodimerization selectively suppresses STAT3 activity and malignant transformation. Cancer Res. 2013;73(6):1922–1933. doi: 10.1158/0008-5472.CAN-12-3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Angulo P., Kaushik G., Subramaniam D., Dandawate P., Neville K., Chastain K., et al. Natural compounds targeting major cell signaling pathways: a novel paradigm for osteosarcoma therapy. J Hematol Oncol. 2017;10(1):1–10. doi: 10.1186/s13045-016-0373-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ryu K., Choy E., Yang C., Susa M., Hornicek F.J., Mankin H., et al. Activation of signal transducer and activator of transcription 3 (Stat3) pathway in osteosarcoma cells and overexpression of phosphorylated-Stat3 correlates with poor prognosis. J Orthop Res. 2010;28(7):971–978. doi: 10.1002/jor.21088. [DOI] [PubMed] [Google Scholar]
- 8.Wang W., Li J., Ding Z., Li Y., Wang J., Chen S., et al. Tanshinone I inhibits the growth and metastasis of osteosarcoma via suppressing JAK/STAT3 signalling pathway. J Cell Mol Med. 2019;23(9):6454–6465. doi: 10.1111/jcmm.14539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Du Z., Shen Y., Yang W., Mecklenbrauker I., Neel B.G., Ivashkiv L.B. Inhibition of IFN-α signaling by a PKC- and protein tyrosine phosphatase SHP-2-dependent pathway. Proc. Natl Acad Sci U S A. 2005;102(29):10267–10272. doi: 10.1073/pnas.0408854102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lee J.H., Chiang S.Y., Nam D., Chung W.S., Lee J., Na Y.S., et al. Capillarisin inhibits constitutive and inducible STAT3 activation through induction of SHP-1 and SHP-2 tyrosine phosphatases. Cancer Lett. 2014;345(1):140–148. doi: 10.1016/j.canlet.2013.12.008. [DOI] [PubMed] [Google Scholar]
- 11.Jin Y., Yoon Y.J., Jeon Y.J., Choi J., Lee Y.J., Lee J., et al. Geranylnaringenin (CG902) inhibits constitutive and inducible STAT3 activation through the activation of SHP-2 tyrosine phosphatase. Biochem Pharmacol. 2017;142:46–57. doi: 10.1016/j.bcp.2017.06.131. [DOI] [PubMed] [Google Scholar]
- 12.Lu L., Zhang S., Li C., Zhou C., Li D., Liu P., et al. Cryptotanshinone inhibits human glioma cell proliferation in vitro and in vivo through SHP-2-dependent inhibition of STAT3 activation. Cell Death Dis. 2017;8(5) doi: 10.1038/cddis.2017.174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen L.X., He H., Qiu F. Natural withanolides: an overview. Nat Prod Rep. 2011;28(4):705–740. doi: 10.1039/c0np00045k. [DOI] [PubMed] [Google Scholar]
- 14.Dar N., Hamid A., Ahmad M. Pharmacologic overview of Withania somnifera, the Indian Ginseng. Cell Mol Life Sci. 2015;72(23):4445–4460. doi: 10.1007/s00018-015-2012-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wei S.S., Gao C.Y., Li R.J., Kong L.Y., Luo J. Withaminimas A-F, six withanolides with potential anti-inflammatory activity from Physalis minima. Chin J Nat Med. 2019;17(6):469–474. doi: 10.1016/S1875-5364(19)30054-8. [DOI] [PubMed] [Google Scholar]
- 16.Um H.J., Min K.J., Kim D.E., Kwon T.K. Withaferin A inhibits JAK/STAT3 signaling and induces apoptosis of human renal carcinoma Caki cells. Biochem Biophys Res Commun. 2012;427(1):24–29. doi: 10.1016/j.bbrc.2012.08.133. [DOI] [PubMed] [Google Scholar]
- 17.Yang Y.W., Yang L., Zhang C., Gao C.Y., Ma T., Kong L.Y. Physagulide Q suppresses proliferation and induces apoptosis in human hepatocellular carcinoma cells by regulating the ROS-JAK2/Src-STAT3 signaling pathway. RSC Adv. 2017;7(21):12793–12804. [Google Scholar]
- 18.Chen W.Y., Chang F.R., Huang Z.Y., Chen J.H., Wu Y.C., Wu C.C. Tubocapsenolide A, a novel withanolide, inhibits proliferation and induces apoptosis in MDA-MB-231 cells by thiol oxidation of heat shock proteins. J Biol Chem. 2008;283(25):17184–17193. doi: 10.1074/jbc.M709447200. [DOI] [PubMed] [Google Scholar]
- 19.Wang S.B., Zhu D.R., Nie B., Li J., Zhang Y.J., Kong L.Y., et al. Cytotoxic withanolides from the aerial parts of Tubocapsicum anomalum. Bioorg Chem. 2018;81:396–404. doi: 10.1016/j.bioorg.2018.08.034. [DOI] [PubMed] [Google Scholar]
- 20.Chen G., Hu K., Sun H., Zhou J., Song D., Xu Z., et al. A novel phosphoramide compound, DCZ0847, displays in vitro and in vivo anti-myeloma activity, alone or in combination with bortezomib. Cancer Lett. 2020;478:45–55. doi: 10.1016/j.canlet.2020.03.006. [DOI] [PubMed] [Google Scholar]
- 21.Lee J.H., Kim C., Kim S.H., Sethi G., Ahn K.S. Farnesol inhibits tumor growth and enhances the anticancer effects of bortezomib in multiple myeloma xenograft mouse model through the modulation of STAT3 signaling pathway. Cancer Lett. 2015;360(2):280–293. doi: 10.1016/j.canlet.2015.02.024. [DOI] [PubMed] [Google Scholar]
- 22.Ahn K.S., Sethi G., Sung B., Goel A., Ralhan R., Aggarwal B.B. Guggulsterone, a Farnesoid X Receptor Antagonist, Inhibits Constitutive and Inducible STAT3 Activation through Induction of a Protein Tyrosine Phosphatase SHP-1. Cancer Res. 2008;68(11):4406–4415. doi: 10.1158/0008-5472.CAN-07-6696. [DOI] [PubMed] [Google Scholar]
- 23.Pai M.Y., Lomenick B., Hwang H., Schiestl R., McBride W., Loo J.A., et al. Drug affinity responsive target stability (DARTS) for small-molecule target identification. Methods Mol Biol. 2015;1263:287–298. doi: 10.1007/978-1-4939-2269-7_22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Molina D.M., Jafari R., Ignatushchenko M., Seki T., Larsson E.A., Dan C., et al. Monitoring Drug Target Engagement in Cells and Tissues Using the Cellular Thermal Shift Assay. Science. 2013;341(6141):84–87. doi: 10.1126/science.1233606. [DOI] [PubMed] [Google Scholar]
- 25.Lakkaraju A.K., van der Goot F.G. Calnexin controls the STAT3-mediated transcriptional response to EGF. Mol Cell. 2013;51(3):386–396. doi: 10.1016/j.molcel.2013.07.009. [DOI] [PubMed] [Google Scholar]
- 26.Xu S., Grande F., Garofalo A., Neamati N. Discovery of a novel orally active small-molecule gp130 inhibitor for the treatment of ovarian cancer. Mol Cancer Ther. 2013;12(6):937–949. doi: 10.1158/1535-7163.MCT-12-1082. [DOI] [PubMed] [Google Scholar]
- 27.Chang Y., Schlebach J.P., VerHeul R.A., Park C. Simplified proteomics approach to discover protein–ligand interactions. Protein Sci. 2012;21(9):1280–1287. doi: 10.1002/pro.2112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Park C., Marqusee S. Pulse proteolysis: A simple method for quantitative determination of protein stability and ligand binding. Nat Methods. 2005;2(3):207–212. doi: 10.1038/nmeth740. [DOI] [PubMed] [Google Scholar]
- 29.Doroshow J.H., Locker G.Y., Ifrim I., Myers C.E. Prevention of doxorubicin cardiac toxicity in the mouse by N-acetylcysteine. J Clin Invest. 1981;68(4):1053–1064. doi: 10.1172/JCI110328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ganey P.E., Kauffman F.C., Thurman R.G. Oxygen-dependent hepatotoxicity due to doxorubicin: role of reducing equivalent supply in perfused rat liver. Mol Pharmacol. 1988;34(5):695–701. [PubMed] [Google Scholar]
- 31.Al Zaid Siddiquee K., Turkson J. STAT3 as a target for inducing apoptosis in solid and hematological tumors. Cell Res. 2008;18(2):254–267. doi: 10.1038/cr.2008.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bharti A.C., Donato N., Aggarwal B.B. Curcumin (diferuloylmethane) inhibits constitutive and IL-6-inducible STAT3 phosphorylation in human multiple myeloma cells. J Immunol. 2003;171(7):3863–3871. doi: 10.4049/jimmunol.171.7.3863. [DOI] [PubMed] [Google Scholar]
- 33.Kotha A., Sekharam M., Cilenti L., Siddiquee K., Khaled A., Zervos A.S., et al. Resveratrol inhibits Src and Stat3 signaling and induces the apoptosis of malignant cells containing activated Stat3 protein. Mol Cancer Ther. 2006;5(3):621–629. doi: 10.1158/1535-7163.MCT-05-0268. [DOI] [PubMed] [Google Scholar]
- 34.Darnell J.E., Jr, Kerr I.M., Stark G.R. Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science. 1994;264(5164):1415–1421. doi: 10.1126/science.8197455. [DOI] [PubMed] [Google Scholar]
- 35.Krebs D.L., Hilton D.J. SOCS Proteins: Negative Regulators of Cytokine Signaling. Stem Cells. 2001;19(5):378–387. doi: 10.1634/stemcells.19-5-378. [DOI] [PubMed] [Google Scholar]
- 36.Valentino L., Pierre J. JAK/STAT signal transduction: Regulators and implication in hematological malignancies. Biochem Pharmacol. 2006;71(6):713–721. doi: 10.1016/j.bcp.2005.12.017. [DOI] [PubMed] [Google Scholar]
- 37.Li F., Fernandez P.P., Rajendran P., Hui K.M., Sethi G. Diosgenin, a steroidal saponin, inhibits STAT3 signaling pathway leading to suppression of proliferation and chemosensitization of human hepatocellular carcinoma cells. Cancer Lett. 2010;292(2):197–207. doi: 10.1016/j.canlet.2009.12.003. [DOI] [PubMed] [Google Scholar]
- 38.Bard-Chapeau E.A., Li S., Ding J., Zhang S.S., Zhu H.H., Princen F., et al. Ptpn11/Shp2 acts as a tumor suppressor in hepatocellular carcinogenesis. Cancer Cell. 2011;19(5):629–639. doi: 10.1016/j.ccr.2011.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yang W., Neel B.G. From an orphan disease to a generalized molecular mechanism: PTPN11 loss-of-function mutations in the pathogenesis of metachondromatosis. Rare Dis. 2013;1 doi: 10.4161/rdis.26657. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.