

Abstract
Diffuse invasion is the primary cause of treatment failure of glioblastoma (GBM). Previous studies on GBM invasion have long been forced to use the resected tumor mass cells. Here, a strategy to reliably isolate matching pairs of invasive (GBM INV ) and tumor core (GBM TC ) cells from the brains of 6 highly invasive patient‐derived orthotopic models is described. Direct comparison of these GBM INV and GBM TC cells reveals a significantly elevated invasion capacity in GBM INV cells, detects 23/768 miRNAs over‐expressed in the GBM INV cells (miRNA INV ) and 22/768 in the GBM TC cells (miRNA TC ), respectively. Silencing the top 3 miRNAs INV (miR‐126, miR‐369‐5p, miR‐487b) successfully blocks invasion of GBM INV cells in vitro and in mouse brains. Integrated analysis with mRNA expression identifies miRNA INV target genes and discovers KCNA1 as the sole common computational target gene of which 3 inhibitors significantly suppress invasion in vitro. Furthermore, in vivo treatment with 4‐aminopyridine (4‐AP) effectively eliminates GBM invasion and significantly prolongs animal survival times (P = 0.035). The results highlight the power of spatial dissection of functionally accurate GBM INV and GBM TC cells in identifying novel drivers of GBM invasion and provide strong rationale to support the use of biologically accurate starting materials in understanding cancer invasion and metastasis.
Keywords: 4‐aminopyridine, glioblastoma, KCNA1, miRNA, patient derived orthotopic xenograft
Using 6 highly invasive glioblastoma (GBM) PDOX models, this study isolates matching pairs of invasive (GBM INV ) and tumor core (GBM TC ) cells, demonstrates an elevated invasion capacity in GBM INV cells, identifies a novel miRNA signature (miRNA INV ) of GBM invasion, and discovers a commonly shared gene, KCNA1, from miRNA INV regulated genes that can be pharmacologically inhibited to block GBM invasion in vivo.

1. Introduction
Glioblastoma multiforme (GBM) is the most malignant brain tumor in children and adults. Despite multimodal therapies and significant advances in the understanding of tumor biology[ 1 ] and molecular subgroups,[ 2 , 3 , 4 ] the prognosis of patients with GBM remains extremely poor,[ 5 ] with 5‐year survival rates between 5% and 15% in children[ 6 ] and a 1‐year survival rate of ≈10% in adults.[ 7 ] Diffuse infiltration of tumor cells into surrounding normal brain tissue, a hallmark of GBM growth, is the primary cause of tumor recurrence and treatment failure.[ 8 ] While major advances have been made in understanding the biology of GBM by studying cells from the tumor core, little is known about the invasive GBM cells (GBM INV ) that migrate deep into surrounding normal brain tissues despite the findings of “go or grow” mechanism[ 9 , 10 ] and epithelial to mesenchymal transition.[ 11 , 12 ] This is because these GBM INV cells are not amenable to surgical removal for study, as aggressive surgical resection of normal tissue carries the risk of serious and permanent neurological deficits.[ 6 , 13 ] Additionally, although it has been suggested that the blood‐brain barrier (BBB) could be compromised and consequently “leaky” in the GBM tumor core (GBM TC ), GBM INV cells are frequently protected by an intact and functional BBB, making them even less vulnerable to chemotherapeutic agents than GBM TC cells.
Most past and current studies on GBM invasion were and are still forced to utilize tumor tissues resected from the primary tumor mass for biologic analyses. Although these GBM TC cells may be capable of invasive growth, they are not actively invading at the time of harvest. Since tumor invasion is a complex biological process involving functional modifications and dynamic interactions between tumor cells and the microenvironment, there remains an urgent need for biologically‐ and functionally‐accurate GBM INV cells to identify key genetic drivers of GBM invasion.
To overcome this barrier, we developed a panel of patient‐derived orthotopic xenograft (PDOX or orthotopic PDX) mouse models through direct implantation of pediatric GBM (pGBM) surgical specimens into matched locations in the brains of SCID mice. Detailed characterization showed that these PDOX tumors replicate the histopathological features of pGBM, maintained key genetic abnormalities of the original patient tumors, and importantly, remained highly invasive.[ 14 , 15 , 16 ] These transplantable PDOX models thus provide a reliable resource for isolating paired and functionally accurate GBM INV cells from normal mouse brain tissues/parenchyma and GBM TC cells from the tumor core for biological studies of pGBM invasion. The inclusion of different molecular subtypes of pGBMs should further facilitate the discovery of commonly shared or subtype‐specific biological changes.
Accumulating data suggest that microRNAs (miRNA), noncoding RNAs of ≈20–23 bps,[ 17 , 18 ] play a key role in GBM invasion.[ 19 ] miRNAs often bind to target mRNAs through partial complementary pairing and either suppress mRNA translation or reduce mRNA stability. They are shown to regulate multiple cellular processes including cell division, differentiation, and death.[ 20 ] While the study of miRNAs in pGBM is still in its infancy, over‐expression of miRNAs, including miR‐34a, ‐124, ‐128, ‐137, and ‐145, has been detected in adult GBM and shown to suppress self‐renewal, inhibit tumorigenesis,[ 21 ] trigger cell cycle arrest,[ 22 ] or apoptosis,[ 23 , 24 ] and promote invasion in vitro in cultured GBM cells and in vivo in subcutaneous xenografts.[ 19 , 25 ]
In this study, we utilized 6 PDOX mouse models of pGBM of different subtypes to harvest functionally accurate, paired GBM INV cells (that have invaded into the surrounding normal mouse brain tissue) and GBM TC cells (from the primary tumor mass) to examine their functional differences in migrating into normal brains, followed by the analysis of differentially expressed miRNAs via global microRNA profiling, validation of the functional roles of the candidate driver miRNAs both in vitro and in vivo in mouse brains using purified GBM INV cells. We subsequently examined genes that mediate the driver miRNA‐induced pGBM invasion, and finally identified a novel candidate driver gene KCNA1 and demonstrated the therapeutic efficacy of pharmacological targeting of this invasion‐driver gene in blocking pGBM invasion and prolonging PDOX survival times.
2. Results
2.1. Patient‐Derived Orthotopic Xenograft Tumors Replicate the Highly Invasive Phenotype of Pediatric Glioblastoma In Vivo
Diffuse infiltration of tumor cells into surrounding normal brain tissue is one of the hallmarks of GBM. To confirm that our pGBM PDOX models replicated this important biological feature, we performed a systematic analysis of the invasive capacity and mode of tumor cell migration of pGBM cells in six PDOX models that had been sub‐transplanted in vivo in mouse brains for 6–8 generations as described previously (Figure 1a,b).[ 15 , 26 ] These models were subgrouped as proliferative, proneural, and mesenchymal through gene expression analysis (Figure 1a),[ 2 ] and MYCN, MID, G34, and pedRTKIII subtypes with DNA methylation profiling(Figure 1a).[ 27 , 28 ] When the tumor‐bearing mice became moribund, whole mouse brains were harvested, paraffin embedded, and serially sectioned (>160 sections/mouse brain). Standard H&E staining consistently revealed a large tumor mass surrounded by a characteristic “ragged edge” (Figure 1c and Figure S1, Supporting Information). To positively identify single and/or small clusters of invasive pGBM cells, we performed immunohistochemical (IHC) staining using human‐specific antibodies against vimentin (VIM)[ 15 , 29 , 30 ] and used a straight line reticle (eyepiece micrometer) to measure the distances of migration under a Nikon 3 head teaching microscope from which 2–3 investigators can examine the same fields of the same slides at the same time. In tumors with short invasion that can be captured in one image (from 4× to 20×), ImageJ was applied to digitally measure the distances. The border line between tumor core and invasion (or the edge of tumor core) was defined as the front of tumor core where tumor cells were lined up facing normal brain tissues; while the leading front of invasion was determined by the tumor cells that migrated the farthest (or deepest) into the normal brain. A line drawing from the leading front invasive cells perpendicular to the tumor core edge was used to measure the distances (Figure 1c). For each PDOX models, at least three mouse brains each with >3 slides of the largest cross sections of tumor mass were included. Despite the different molecular subtypes, diffuse invasion was detected in all six models (Figure 1c and Figure S1, Supporting Information). Most of the leading invasive edge was composed of single tumor cells followed by increasingly larger micro‐tumors closer to the tumor core, covering a distance ranging from 525 to 2083 µM (mean 1109 ± 375.2 µm). Migration along blood vessels, that is, perivascular invasion,[ 31 ] was detected in all 6 models, ranging from 289.2 to 2058.3 µm (914.4 ± 466.7 µm); while invasion along neural fibers ranged from 170 to 2666.7 µm (1158 ± 524 um). Seeding into the cerebral spinal fluid (CSF) was observed in some sections; the distances varied from 525 to 2750 µm (1881.3 ± 541.1 µm) (Figure 1c). Together, these data demonstrated maintenance of the invasive pGBM phenotype and all 3 routes of GBM tumor migration in vivo in our PDOX models, with single cell migration as the predominating mode.
Figure 1.
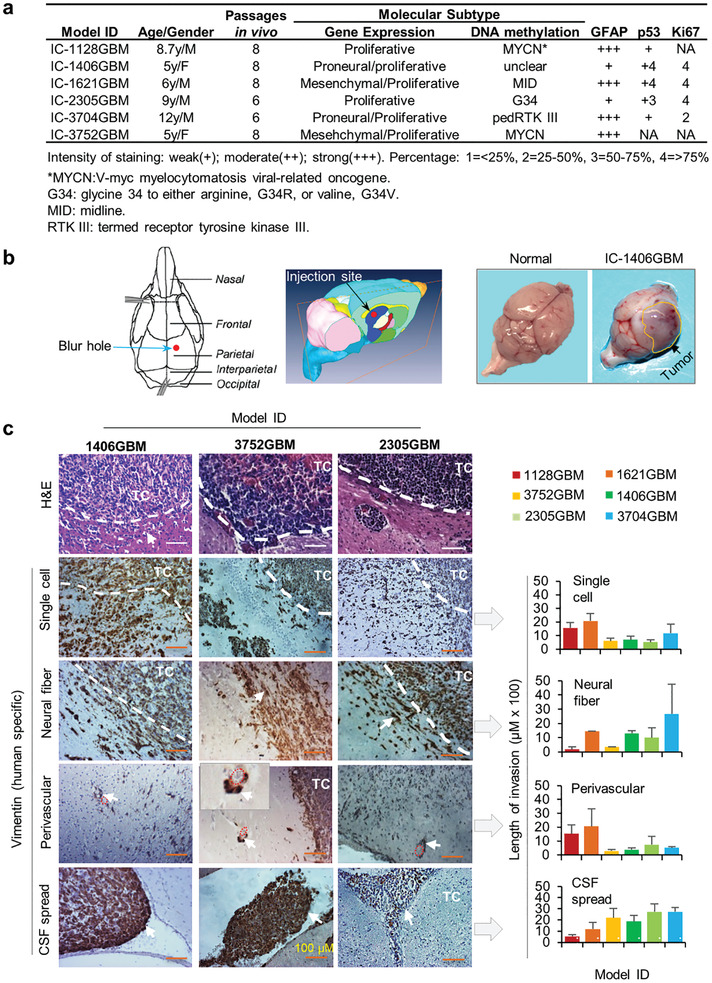
PDOX pGBM tumors were highly invasive in vivo. a) Clinical, pathological, and molecular subtype information of the six pGBM tumors. b) Orthotopic pGBM tumor implantation strategy and representative gross appearance of pGBM PDOX models. Tumor cells (1 × 105 in 2 µL) from six pGBMs were directly implanted into right cerebra of NOD/SCID mm mice (1.5 mm anterior and 3 mm deep) (left panel). The animals were monitored daily until they developed neurological deficits or became moribund, at which time they were euthanized. Formation of PDOX tumors can frequently be observed (arrow, right panel). c) Representative images showing the modes of intra‐cerebral invasion in PDOX models (left panel) and the quantitative analysis of migration distances of all six models (right panel). In additional to H&E staining, human pGBM xenograft cells were positively identified through IHC staining using human‐specific antibodies against vimentin (arrow). In all six models, invasion through single cells, along blood vessels (perivascular invasion), along neural fibers, and spread through cerebral spinal fluid (CSF) were observed. The distances between the invasive front and the border line of tumor core (white dotted line) were measured and graphed. Scale bar = 100 µM. n = 3 in each condition. Data are shown as mean ± SD.
2.2. Spatial Dissection to Isolate Matching Pairs of Invasive (GBM INV ) and Tumor Core (GBM TC ) Cells
To isolate GBM INV and GBM TC cells, freshly harvested whole mouse brains were sectioned into 1 mm slices to enable gross identification of the primary tumor mass (Figure 2a) and to facilitate microscopic dissection of the tumor core (from which GBM TC cells were collected) from “normal” mouse brain tissue (from which GBM INV cells were collected) (Figure 2b).[ 32 , 33 , 34 ] To purify human GBM INV and GBM TC cells, we utilized FITC‐conjugated human HLA‐ABC antibodies and a cocktail of APC‐conjugated antibodies specific to mouse CD24, CD90, CD117, CD133 and performed florescence activated cell sorting (FACS). In the invasive front (from “normal” mouse brain tissues), human HLA‐ABC+ cells (GBM INV ) ranged from 10.6% to 54.4% (33.0 ± 0.68%) of the viable cells; in the tumor core, human GBM TC cell proportions ranged from 85.9% to 97.2% (93.1 ± 0.58%) (Figure 2c and Figure S2, Supporting Information). These data provided a quantitative estimate of the diffusive invasion of pGBM in mouse brains, using a novel strategy to harvest matched pairs of functionally distinct GBM INV and GBM TC cells for biological studies.
Figure 2.
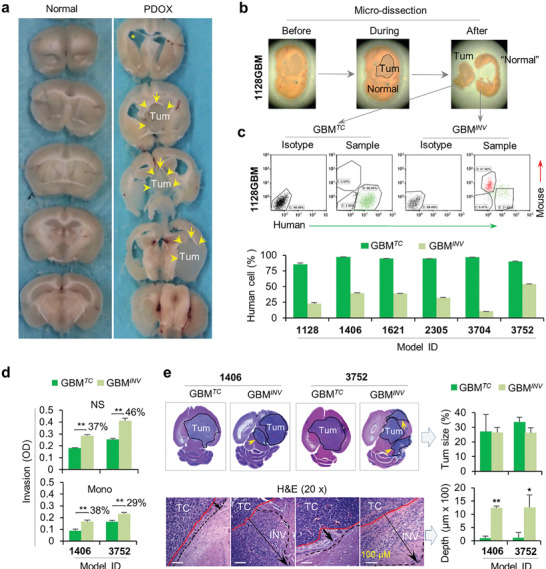
GBM INV cells possess stronger invasive capacity than their matching GBM TC cells. a) Slicing of fresh whole mouse brain to facilitate easy identification of tumor core. Whole mouse brains were placed on a mouse brain matrix and sliced at 1 mm thickness into 10–12 slices. b) Separation of tumor core (tumor) from “normal” mouse brains under stereotactic microscope. The border between tumor and normal (circle) was identified following general guidelines of human brain tumor resection during surgery.[ 32 , 33 , 34 ] “Normal” mouse brain tissues (containing GBM INV cells) and tumor mass (containing GBM TC cells) were placed in cold (4 °C) growth medium in separate Petri dishes and dissociated into single cell suspension using Gentle Dissociator (Miltenyi). c) Purification of GBM INV and GBM TC cells through FACS. Cell suspensions were incubated with FITC‐conjugated monoclonal antibodies against human HLA‐ABC and APC‐conjugated monoclonal antibodies against mouse major histocompatibility antigen by FACS. The mouse cells (APC‐positive and FITC‐negative) were gated out together with the dead cells (propidium iodine high). Data are shown as mean ± SD. d) In vitro assay showing significantly increased invasive capacity of GBM INV cells than the matching GBM TC cells in two models under two different growth conditions. The purified GBM INV and GBM TC cells from IC‐1406GBM (1406) and IC‐3752GBM (3752) were cultured as neurosphere (NS) in serum‐free media supplemented with EGF and bFGF and monolayer (Mono) cells in traditional FBS‐based medium. The invasive capacity of GBM TC and GBM INV was examined in triplicates by CytoSelect 96‐Well Cell invasion assay (*P < 0.05) (Data are shown as mean ± SD). e) In vivo validation of higher invasive capacity of GBM INV cells than that of the matching GBM TC cells. Purified GBM INV and GBM TC cells from IC‐1406GBM (1406) and IC‐3752GBM (3752) models were implanted separately into the brains of SCID mice. The animals were euthanized when they develop signs of neurologic deficits or become moribund. Paraffin sections were stained with H&E and the distances from the “border” of tumor core (red line) to the far front of the invasive edge (black line) were measured (arrow). Note the formation of invasive satellite tumors in mouse brains implanted with GBM INV cells (arrow in the upper panel). Tumor sizes and depths of pGBM invasion were quantitated by ImageJ (**P < 0.01, *P < 0.05) (n = 3 per group. Data are shown as mean ± SD).
2.3. GBM INV Cells Possess Significantly Stronger Invasive Capacity both In Vitro and In Vivo
To test our hypothesis that GBM INV cells possess stronger migratory capacity than those in the GBM TC cells, we first compared their invasive capability in vitro with a standard invasion assay. Since monolayer tumor cells maintained in traditional fetal bovine serum (FBS)‐based media do not share biological features with neurospheres propagated in serum‐free media (supplemented with EGF and βFGF), which favors the growth of cancer stem cells,[ 15 , 30 , 35 , 36 ] we incubated pGBM cells in both types of growth media to have a better coverage of cell subpopulations and to understand the differences between the monolayer and the neurosphere cells. GBM INV cells from two pGBM models (IC‐1406GBM and IC‐3752GBM) exhibited a 29–46% increase in invasion compared to the matching GBM TC cells (P <0.05), and 3D neurosphere cells were significantly more invasive (37–46% higher) than the monolayer cells (Figure 2d). Since neurospheres grew in suspension, the scratch assay that measures the cell motility was not performed.
To further validate these findings in vivo, we directly implanted freshly purified GBM INV and GBM TC cells from these two pGBM models into the brains of SCID mice and examined their invasive capacity on paraffin sections via H&E and IHC staining. As anticipated, invasion into surrounding normal brain was observed in the xenografts derived from GBM TC cells, confirming the maintenance of invasive capacity of a fraction of GBM TC cells even though they were not “invading” at the time of harvesting. In xenografts derived from GBM INV cells, however, the depth of invasion was significantly longer compared to GBM TC cells (1246 vs 105.1 µm in IC‐1406GBM and 1266.3 vs 114 µm in IC‐3752GBM) (Figure 2e). While GBM TC cells formed large intra‐cerebral tumor masses, it was the GBM INV cells that developed invasive satellite tumors (Figure 2e). These in vitro and in vivo data demonstrated the functional differences between GBM INV and GBM TC cells, thereby highlighting the importance of using functionally accurate GBM INV cells in understanding GBM invasion.
2.4. Novel miRNA Drivers of Pediatric Glioblastoma Invasion
MiRNAs have been implicated in driving biological processes of human cancers[ 37 ] including tumor invasion and metastasis.[ 38 , 39 ] To test our hypothesis that GBM INV cells depend on a unique set of miRNAs for invading into normal brain tissue, we compared the miRNA expression profiles of matched GBM INV and GBM TC cells derived from the 6 PDOX pGBM models using TaqMan MicroRNA array. In addition to four control assays provided by the vendor, we included normal human cerebral tissue obtained from warm autopsy (≈4 h postmortem) as a reference. Differences in miRNA expression between GBM INV and GBM TC cells were directly compared and the fold changes calculated by 2−ΔCT. Of the 768 miRNAs detected, 23 were significantly upregulated (>twofold) in the GBM INV cells (hereafter designated as miRNA INV ), and 22 miRNAs upregulated in GBM TC cells (hereafter referred as miRNA TC ) in at least 4 of the 6 pGBM models (Figure 3a and Table S1, Supporting Information).
Figure 3.
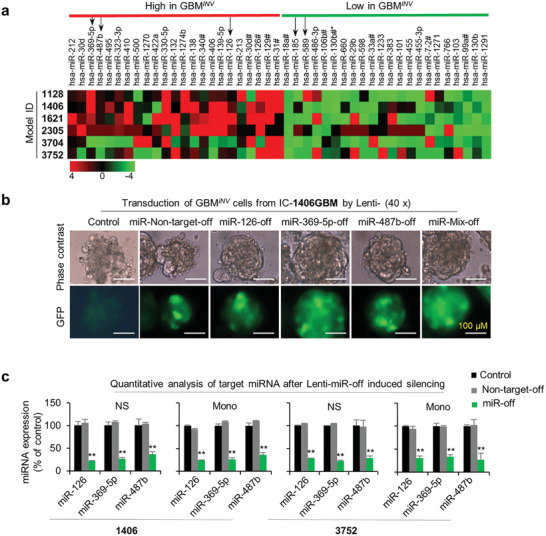
Differentially expressed miRNAs between GBM INV and GBM TC cells. a) Hierarchical clustering of over‐expressed and downregulated miRNA in GBM INV cells. miR‐126, miR‐487b, and miR‐369‐5p were selected for functional study from the 23 upregulated miRNAs, and miR‐185 and miR‐589 were selected from the 22 downregulated miRNAs in least 4 of the 6 pGBM models (P < 0.05). b) Representative images showing successful infection of pGBM cells by Lentivirus‐miR‐GFP. The purified GBM INV cells from IC‐1406GBM were grown as neurospheres and infected with lentivirus‐miRNA‐off (miR487b‐off, miR‐126‐off, miR‐369‐5p‐off, and mix‐off) for 72 h (MOI 1:1) and examined for the expression of GFP. Non‐infected cells were included control. c) Confirmation of lentivirus‐mediated miRNA knock‐down of miRNA INV using RT‐qPCR. Both neurosphere (NS) and monolayer (Mono) cells derived from GBM INV cells of IC‐1406GBM (1406) and IC‐3752GBM (3752) were tested. (**P < 0.05) (Data are shown as mean ± SD).
2.5. Silencing miRNA INV in GBM INV Cells Suppresses Invasion In Vitro
To examine the functional role of the upregulated miRNAs in the GBM INV cells (i.e., miRNA INV ) we selected two newly discovered miRNAs that exhibited high fold changes and high frequency, that is, miR‐369‐5p with >3.11‐folds in 5/6 models and miR‐487‐5p with > 3.3‐fold in 4/6 models, as well as, one miRNA (miR‐126, >2.7‐fold in 5/6 models) that was reported to be involved in tumor invasion.[ 40 , 41 ] We examined the effects of loss‐of‐function and gain‐of‐function of these 3 microRNAs through lentivirus‐mediated transduction assays. Successful transduction was confirmed with Lenti‐GFP (Figure 3b) and the efficient knock‐down of target miRNA INV (>70%) with RT‐qPCR (Figure 3c).
For the loss‐of‐function analysis of miRNA INV , the invasive capacity of puromycin‐selected and GFP+ GBM INV cells from two highly invasive PDOX models, IC‐1406GBM and IC‐3752GBM, was examined using CytoSelect 96‐Well Cell Invasion Assay in quadruplicates. As shown in Figure 4a, silencing miR‐126, ‐369‐5p, and ‐487b with Lenti‐miRNA‐126‐off, ‐369‐5p‐off, and ‐487b‐off, (MOI = 1:1 for 72 h) did not affect cell proliferation in either the GBM INV monolayer or neurosphere cultures but induced significant suppression of invasion in GBM INV cells grown as neurospheres (P < 0.05 compared to the untreated and the GBM INV cells transduced with Lenti‐non‐target‐off, n = 3) from both pGBM models. In the monolayer cells, only invasion of IC‐1406GBM INV cells was inhibited (Figure 4a and Figure S3a, Supporting Information). These data indicated the selectivity of the miRNA INV function in cell invasion, particularly in the 3D neurospheres that exhibited increased invasive capacity (in Section 2.3), and support their role as candidate miRNA drivers of pGBM invasion.
Figure 4.
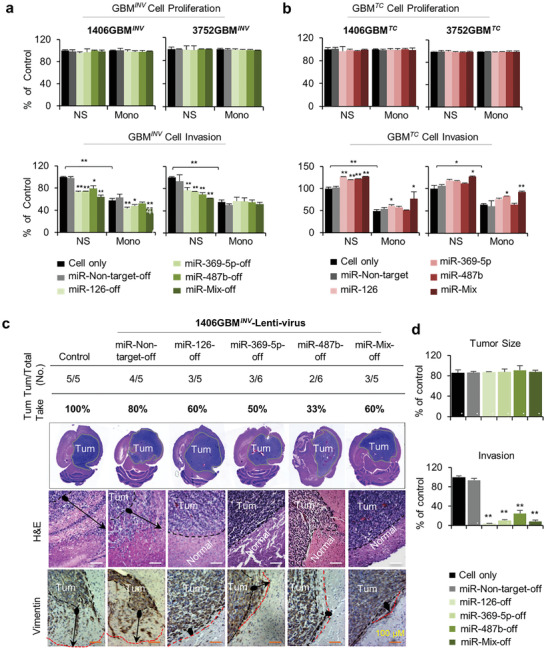
Functional validation of miRNA INV in GBM INV cell invasion both in vitro and vivo. a) In vitro loss‐of‐function assay showing the suppression of GBM INV cell invasion by lentivirus mediated silencing of miRNAs INV . The puromycin‐selected and FACS‐purified GFP+ GBM INV cells from IC‐1406GBM (1406GBM IN V ) and IC‐3752GBM (3752GBM INV ) were examined both as neurospheres (NS) and monolayer cells (Mono). Data were normalized to the cell only group of neurospheres and presented as % of control. While cell proliferation was not affected (P > 0.05) (top panel), silencing of miR‐126, ‐369‐5p, ‐487b alone, and in combination (miR‐mix‐off) with Lentivirus‐miRNA‐off led to significant suppression of cell invasion (lower panel, P < 0.05) as examined by CytoSelect 96‐Well Cell Invasion Assay. (**P < 0.01, *P < 0.05 compared to the control group, n = 3. Data are shown as mean ± SD). b) In vitro gain‐of‐function assay showing the activation of GBM TC cells following lentivirus mediated transduction of miRNA INV . GBM TC cells (that were not actively invading) were transduced with Lentivirus‐miRNA to increase the expression of the miRNA INV . Cell proliferation was not affected (upper panel), but cell invasion was increased, particularly in neurosphere (NS) cells from IC‐1406GBM (1406GBM TC ) and IC‐3752GBM (3752GBM TC ) (**P < 0.01, *P < 0.05 compared to the control group, n = 3. Data are shown as mean ± SD). c) In vivo confirmation of suppressed GBM INV cell invasion following the silencing of miRNA INV s. GBM INV cells from IC‐1406GBM (1406GBM IN V ) were transduced with Lentivirus‐miRNA‐off followed by puromycin‐selection. The FACS‐purified GFP+ GBM INV cells were implanted into the brains of NOD/SCID mice (1 × 105 cells per mouse brain) and monitored for signs of neurological deficits or moribund when the animals were euthanized. 1406GBM INV cells were identified through H&E staining of whole mouse brains and IHC staining of paraffin‐embedded sections using human‐specific antibodies against VIM. d) The slides with the largest cross section of intra‐cerebral xenografts were examined to quantitatively evaluate the tumor size and the distances (arrow)between the invasive front (red line) and tumor core “board line” (black dotted line) were measured by ImageJ (*P < 0.05). Scale bars represent 100 µM. Data are shown as mean ± SD.
To determine if gain‐of‐function of miRNA INV promotes invasion in GBM TC cells, which have lower levels of these 3 miRNAs, we transduced the non‐invading tumor core cells IC‐1406GBM TC and IC‐3752GBM TC cells with Lenti‐miRNA‐126, ‐487b, and ‐369‐5p in quadruplicates. The increased expression of these miRNA INV (alone or in combination) did not alter cell proliferation, similar to GBM INV cells (Figure 4b and Figure S3b, Supporting Information) but resulted in significantly elevated invasion in 3D neurospheres of IC‐1406GBM TC and in monolayers of IC‐3752GBM TC . In monolayers of IC‐1406GBM TC and neurospheres of IC‐3752GBM TC , overexpression of a single miRNA INV failed to promote invasion; however, simultaneous overexpression of all 3 miRNA INV caused a significant increase in invasion (Figure 4b and Figure S3b, Supporting Information). These data indicated collective/cooperative activities of these miRNA INV in promoting pGBM invasion and suggested a complex nature of the underlying biology of GBM invasion.
2.6. Silencing miRNA INV Significantly Suppresses Pediatric Glioblastoma Invasion in Mouse Brains
GBM invasion is an active process involving dynamic interactions between tumor cells and their microenvironment. To validate the functional roles of miRNA INV in vivo in a microenvironment similar to human brain tissue, FACS‐purified IC‐1406GBM INV cells were transduced with Lenti‐mir‐126‐off, ‐369‐5p‐off, and ‐487b‐off (MOI 1:1) to silence the 3 miRNA INV and subsequently implanted into the brains of NOD/SCID mice (1 × 105 cells/mouse brain, n = 5 per group). Compared with the 100% (5/5) tumor uptake rate seen with untreated IC‐1406GBM INV cells and 80% (4/5) in the non‐target lentivirus‐transduced group, the tumor take rates were reduced to 60% (3/5) after implantation of cells with Lenti‐miRNA‐126‐off, 50% (3/6) with Lenti‐miRNA‐369‐5p‐off, 33% (2 of 6) with Lenti‐miRNA‐487b‐off, and 60% (3/5) with a combination of all 3 Lenti‐miRNA‐off (Figure 4c). Animal survival times were not significantly different among the tumor‐bearing mice (Figure S5, Supporting Information).
We next examined whether silencing the 3 miRNA INV blocked GBM INV invasion in vivo in mouse brains. Except for the miR‐487b‐off group, in which 2 mouse brains were analyzed, there were 3 mouse brains in all the remaining groups (control, non‐target, miR126‐off, miR‐369‐5p‐off, and miR‐combination‐off). To quantitatively evaluate the invasive potential, slides with the largest cross‐section of intra‐cerebral xenografts were examined (Figure 4c). The non‐target control (miR‐non‐target‐off) and untreated IC‐1406GBM INV exhibited similar invasive capacity. Lentiviral‐mediated silencing of miR‐126, ‐369‐5p, and ‐487b caused significant reduction of invasion depth, ranging from >75% by miR‐487b‐off to ≈90% by miR‐369‐5p‐off and >95% by miR‐126‐off (Figure 4d). When the tumor sizes were compared, the differences among the six groups were not significantly different (Figure 4d) and IHC examination of stem cell (Nestin), neural (MAP2), glial (GFAP), cell proliferation (Ki67), and mitochondrial markers failed to reveal major differences as well (Figure S6, Supporting Information). Altogether, silencing the miRNA INV (miRNA‐487b, ‐369‐5p, and ‐126) in GBM INV cells blocked pGBM invasion and reduced tumorigenicity in vivo, supporting a critical role of these miRNA INV in maintaining the invasive phenotype of pGBM cells.
2.7. miRNAs INV Targeted a Set of Shared Genes and Signaling Pathways
miRNA‐mediated gene regulation is very complex.[ 42 ] The activity of a given miRNA on a transcript may result in target mRNA degradation, blockage of translation, or increased mRNA expression.[ 18 ] Further, a single miRNA can target multiple mRNAs to coordinately regulate their expression; in contrast, multiple miRNAs can target a single mRNA. To identify the mRNA targets of the 3 miRNA INV , we 1) searched TargetScan for an updated list of target genes of the 3 miRNA INV (miR‐126, ‐487b, and ‐369‐5p), 2) performed global gene expression profiling in the same six pairs of GBM INV and GBM TC cells using normal childhood cerebral RNA as control, and 3) generated a list of differentially expressed genes of the 3 miRNA INV between GBM INV and GBM TC , (fold difference >1.5 or <0.5, PINV/TC < 0.05). The P‐values of differentially expressed genes between GBM INV and normal human cerebral tissues (PINV/Normal ) and between GBM TC and normal tissues (PTC/Normal ) were also calculated.
For miR‐126, a total of 231 target genes were differentially expressed, including 126 downregulated (<0.5‐fold) and 105 upregulated genes (>1.5‐fold) in GBM INV compared with GBM TC cells (Figure 5a and Table S2, Supporting Information). For miRNA‐487b, there were 37 target genes (23 downregulated and 14 upregulated) (Figure 5a and Table S3, Supporting Information), 22 (62.8%) of which were shared targets of miR‐126 (Figure 5A). For miR‐369‐5p, which has not been associated with any human disease, only seven target genes were found (Figure 5a and Table S4, Supporting Information). The levels of most of the genes identified in the tumor core (GBM TC ) were not significantly different from those in normal human cerebral tissues, including 203/247 (81.8%) genes targeted by miR‐126, 33/35 (94.2%) by miR‐487b, and 6/7 (85.7%) by miR‐369‐5p (Tables S2–S4, Supporting Information). Therefore, these genes could have been missed if only GBM TC cells were utilized to compare with normal tissues.
Figure 5.
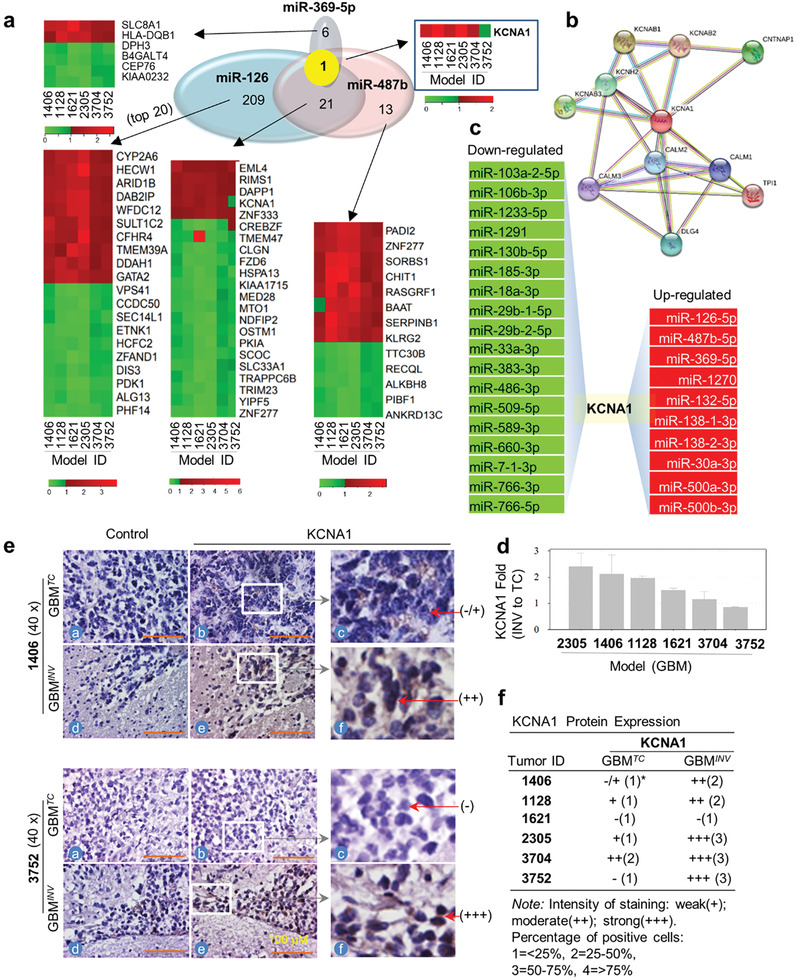
KCNA1 is a common target gene of miRNA INV . a) Integrated analysis of mRNA profiling with the 3 miRNA INV via TargetScan identified the private and shared target genes of miR‐126, ‐487b, and ‐369‐5p. KCNA1 is the only gene commonly targeted by all 3 miRNA INV .b) Protein‐protein interaction network analysis with STRING identified major binding partners of KCNA1. c) List of down‐ or upregulated miRNAs in the GBM INV cells that also target KCNA1. d) Over‐expression of KCNA1 mRNA in the GBM INV cells compared with the matching GBM TC cells in 4/6 pGBM models examined. Data are shown as mean ± SD. e,f) Elevated expression of KCNA1 protein in vivo in the invasive front (GBM INV ) as compared with the tumor core (GBM TC ) cells as analyzed with IHC in the six pGBM PDOX models. Slides incubated without the primary antibodies were uses as control.
Pathway enrichment analysis of the differentially expressed target genes was performed through Ingenuity with Fisher's exact test. When examined individually, each of the 3 miRNA INV affected a distinct collection of pathways. For example, the top canonical pathways affected by miR‐126 were dTMP novo biosynthesis, cardiomyocyte differentiation, cardiac‐adrenergic, cAMP‐mediated signaling, and ERK/MAPK signaling. The top miR‐487b‐affected pathways were protein citrullination, neutral pathway, pyrimidine ribonucleotide interconversion, and the Wnt/Ca+ pathway. Although miR‐369‐5p only targeted nine genes, the top affected pathways were B‐cell development, antigen presentation, and the autoimmune thyroid disease pathway (Figure S7a, Supporting Information). When analyzed for the shared pathways, all 3 miRNA INV regulated cell‐to‐cell signaling and cell interaction (Figure S7b, Supporting Information), while organismal injury and abnormalities, and nervous system development and function were affected by 2 of the 3 miRNA INV (Figure S7b, Supporting Information). Collectively, the 3 miRNA INV modulated ERK/MAPK and cAMP‐mediated signaling (in canonical pathways) (Figures S7a, S8b–S11b, Supporting Information), cancer, organismal injury (in disease) (Figure S7b, Supporting Information), protein synthesis, cellular development, cell death and survival, cell‐to‐cell signaling, and interaction (in molecular functions) (Figures S7b, S8b–S11b, Supporting Information), and embryonic, organ, organismal, reproductive system, and tissue development (in physiological systems involved in cancer invasion and metastasis) (Figures S8b–S11b, Supporting Information).
To further deduce the protein networks, we used STRING,[ 43 ] a database tool for predicting protein‐protein interactions directly and indirectly for analyzing miRNA INV targeted networks. In miR‐126 targeted genes, 2 major networks were observed centering on DNAJC10 (DnaJ heatshock protein family member C10)[ 44 ] and RAB33B (member RAS oncogene family) (Figure S8a, Supporting Information).[ 45 ] In miR‐487 target genes, MED28 (mediator complex subunit 28) was linked to the Mediator complex, a coactivator involved in the regulated transcription of nearly all RNA polymerase II‐dependent genes (Figure S9a, Supporting Information).[ 46 ] In miR‐369‐5p, a network of potassium voltage‐gated channel family surrounding KCNA1 was identified (Figure S10a, Supporting Information). For each of the miRNA INV target gene groups, there were genes that remain isolated. By combining all the target genes of the 3 miRNA INV s, we were able to expand the protein networks to link the nodes of DNAJC10, RAB33B, FLT1 (Fms Related Tyrosine Kinase 1)[ 47 ] and EXOC5 (Exocyst Complex Component 5) (Figure S11a, Supporting Information).[ 48 ] These sets of STRING identified novel protein networks critical to GBM invasion.
2.8. KCNA1 is a Common Computational Target Gene of the miRNAs INV
Since all 3 miRNA INV actively suppressed pGBM invasion, we examined if they shared common target gene(s) by comparing the target genes from each of the three miRNA INV s that were identified (detailed in the previous section). KCNA1 was found to be the sole computational target gene shared by miR‐369‐5p, miR‐126, and miR‐487b (Figure 5a–c). KCNA1 is potassium voltage‐gated channel subfamily A member 1, known to be involved in diverse physiological processes from repolarization of neuronal or cardiac action potentials to regulating calcium signaling and cell volume.[ 49 , 50 ] To functionally validate KCNA1 as a molecular target of miRNA INV , we examined its expression after the loss‐of‐function and gain‐of‐function of the 3 miRNA INV . In GBM INV cells, which express high levels of KCNA1 compared to GBM TC cells and normal brain tissue, silencing miR‐126, ‐369‐5p, and ‐487b significantly downregulated KCNA1 (25–45% compared with non‐target control) in both neurosphere and monolayer cultures of IC‐1406GBM INV and IC‐3752GBM INV cells (Figure 6a, top panel). Conversely, enhancing miRNA INV expression in GBM TC cells (in which KCNA1 expression was originally low) of IC‐1406GBM TC and IC‐3752GBM TC increased KCNA1 mRNA expression in neurospheres by miR‐369‐5p and ‐487b; in monolayer cells by miR‐487b and mild elevation by miR‐126 (Figure 6a, lower panel).
Figure 6.
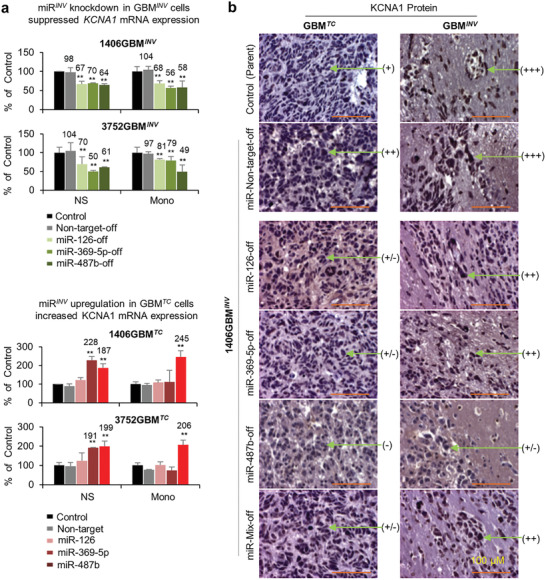
KCNA1 is a molecular target of miRNA INV . a) Silencing miRNA INV led to decreased KCNA1 mRNA expression in purified GBM INV cells from IC‐1406GBM (1406GBM INV ) and IC‐3752GBM (3752GBM INV ) grown as neurosphere (NS) and monolayer (Mono) (upper panel). Conversely, over‐expressing miRNA INV in GBM TC cells of these two models, resulted in elevated KCNA1 mRNA expression in both culture conditions (lower panel). KCNA1 expression was quantitated via qRT‐PCR (*P < 0.05). Data are shown as mean ± SD. b) KCNA1 protein expression was suppressed in vivo in the tumor core (GBM TC ) and invasive front (GBM INV ). GBM INV cells from IC‐1406GBM (1406GBM INV ) were transduced with Lentivirus‐miRNA off to silence miR‐126, ‐369‐5p, ‐487b alone (miR‐126‐off, miR‐369‐5p‐off, miR‐487‐off) and in combination (miR‐mix‐off), followed by implantation into the brains of SCID mice. KCNA1 protein expression was examined through IHC. Tumor cells receiving no (Control) or lentivirus‐non‐target‐off (miR‐Non‐target‐off) were included as references.
To validate KCNA1 as a molecular target of miRNAs in vivo, KCNA1 expression in the invasive pGBM cells were examined in IC‐1406GBM and IC‐3752GBM by IHC staining. Compared with strong (+++) KCNA1 positivity in GBM INV cells that migrated into the normal brains, silencing miRNA INV (miR‐126, ‐369‐5p, and ‐487b) significantly reduced KCNA1 expression to low (+/− to ++) levels (Figure 6b) in the invasive front, although the depth of invasion was significantly reduced after the silencing of miRNA INV .
Since a gene can be regulated by multiple miRNAs, we hypothesized that KCNA1 would be further regulated (fine‐tuned) by additional miRNAs if it is critical for pGBM invasion. A reverse scan of the miRNAs differentially expressed by GBM INV and GBM TC cells in the current study revealed that KCNA1 is the target (or putative target) gene redundantly regulated by 10/23 (47.8%) miRNA INV and 18/22 (81.8%) miRNA TC (Figure 5c), further supports a role of KCNA1 in pGBM invasion.
2.9. KCNA1 is Overexpressed in GBM INV Cells In Vivo in Patient‐Derived Orthotopic Xenograft Models and in Patient Glioblastoma Tumors
Although highly desired, examination of KCNA1 expression in the invasive front in human GBM patients is challenged by the difficulties of obtained normal brain tissues from patients. To further confirm KCNA1 over‐expression in the GBM INV cells, we compared its mRNA and protein levels between GBM INV and GBM TC cells in 6 PDOX models. Elevated (>1.5‐fold) KCNA1 mRNA expression in GBM INV cells was detected in 4/6 models (Figure 5d). KCNA1 protein expression was subsequently analyzed in whole mouse brain sections through IHC. Compared with low (− to ++) levels of KCNA1 in GBM TC cells, strong (++ to +++) KCNA1 positivity (KCNA1 +++) was detected in GBM INV cells in 5/6 models (Figure 5e,f). Altogether, over‐expression of KCNA1 mRNA and protein were detected in 4/6 models, while elevated expression of mRNA only was found in IC‐1621GBM and protein only in IC‐3752GBM TC (Figure 5e,f and Figure S12, Supporting Information). These data indicated that KCNA1 over‐expression, particularly at protein level, is frequent in the GBM INV cells.
To validate our discoveries in patient tumors, we extracted the RNAseq data from the IVY Atlas, where anatomic structures RNAseq was completed (in 120 samples from 10 tumors) in the leading edge as well as infiltrating tumor, cellular tumor, microvascular proliferation, and pseudopalisading cells around necrosis identified by H&E staining (https://glioblastoma.alleninstitute.org/static/home). Expression levels of the 239 unique target genes of miRNA INV were compared with the genes in the leading edges of patient GBMs (Table S5, Supporting Information). Among the 112 significantly (P < 0.05) over‐expressed miRNA INV target genes, 41 (36.6%) genes were discovered to be significantly elevated (P < 0.05) in the leading edges of patient GBM tumors found in the IVY project, including the KCNA1, which was 1.54‐fold (GBM INV /GBM TC ) in the PDOX and 4.79‐fold in the leading edges of patient tumors (group 1 in Table S5, Supporting Information). Of the 127 significantly downregulated miRNA INV target genes, 42 genes (33%) were also significantly different between the leading edges and tumor mass (group 2, Table S5, Supporting Information). Some discrepancies between the current study and the patient tumor findings were also noted, included 13 (11.6%) genes that were over‐expressed in the PDOX invasive cells but significantly downregulated in the patient tumors, and 30 (23.2%) downregulated in PDOX invasive cells but upregulated in the patient GBM leading edges (group 3 and 4, Table S5, Supporting Information). Despite the differences of invasive GBM cell isolation approaches, it is very encouraging to see the validation of > 60% of the miRNA INV target genes in patient GBM leading edges.
2.10. Pharmacological Targeting of KCNA1 Suppresses Pediatric Glioblastoma Invasion In Vitro and In Vivo
To explore the potential of KCNA1 inhibition in blocking GBM invasion, we treated 3 pairs of 3D neurosphere cultures of GBM TC and GBM INV cells (shown to have the strongest invasion capacity in vitro) with 3 KCNA1 inhibitors. Our aim was to confirm that the effects were not restricted to a single tumor model and could be reproduced by multiple inhibitors (i.e., not caused by off‐target activities). 3 KCNA1 inhibitors were employed, including ADWX‐1 and Agitoxin‐2 at 0.01–100 nM and 4‐aminopyridine (4‐AP) at 0.06–4 µM. For ADWX‐1 and Agitoxin‐2, only high doses (50 and 100 nM) caused minor suppression of cell proliferation (<20%) after 14 days’ exposure (Figure 7a and Figure S13a,b, Supporting Information). 4‐AP did not affect cell proliferation even at the highest dose (4 µM) after 7‐ and 10‐days’ exposure (Figure 7a and left of Figure S13c, Supporting Information).
Figure 7.
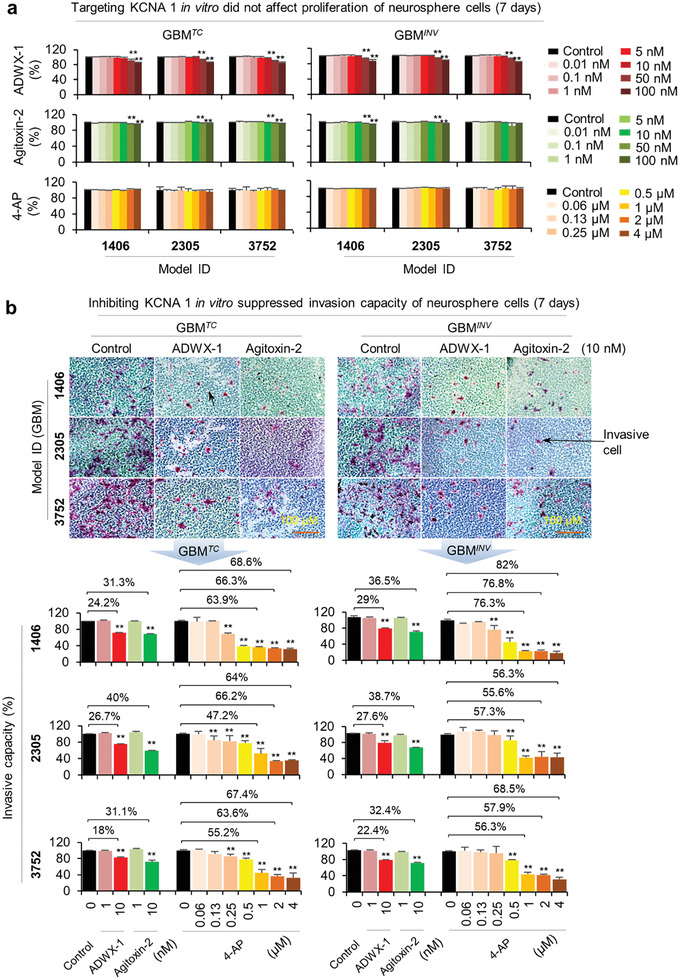
Pharmacological inhibition of KCNA1 suppresses pGBM invasion in vitro. a) Impact of 3 KCNA1 inhibitors on cell proliferation were examined in 3 pairs of neurosphere cultures of GBM TC and GBM INV cells derived from 3 PDOX models, IC‐1406GBM (1406), IC‐2305GBM (2305), and IC‐3752GBM (3752). Tumor cells were exposed to ADWX‐1 and Agitoxin‐2 at 0.01–100 nM for 1–14 days or 4‐AP (ranging from 0.06 to 4 µM) for 7, 10 days, respectively. Data on day 7 were shown (See left of Figure S13a–c, Supporting Information, for results of additional time points). Changes of cell proliferation were examined with CCK‐8 kit assay (*P < 0.05). b) KCNA1 inhibitors suppressed in vitro invasion of both GBM TC and GBM INV cells derived from the 3 pGBM mouse models. Cells were treated with 1 and 10 nM of ADWX‐1 and Agitoxin‐2 for 3, 7, and 14 days or with 0.06, 0.13, 0.25, 0.5, 1, 2, and 4 µM of 4‐AP for 7, 10 days. KCNA1 inhibitors also examined for changes of cell invasion using the CytoSelect 24‐Well Cell Invasion Assay (top panel). Data from the treated groups were normalized to the untreated (Control). Results from 7‐day treatment of 3 KCNA1 inhibitors were presented (lower panel) (*P < 0.05), and those from other time points summarized in supplemental right of Figure S13a,b, Supporting Information. Data are shown as mean ± SD.
The effects on cell invasion were thus investigated with doses that did not affect cell proliferation by focused on the neurosphere cultures that have exhibited a stronger invasive capacity than the monolayer cells in vitro. While no significant changes were noted in the 1 nM groups, treatment with ADWX‐1 and Agitoxin‐2 at 10 nM resulted in significant inhibition of cell invasion in GBM TC and GBM INV cells starting from day 3 through 14 (Figure 7b and right of Figure S13a,b, Supporting Information). Agitoxin‐2 was more potent than ADWX‐1, exhibiting stronger inhibition in GBM INV cells 1.5 ± 0.3‐fold higher than that in GBM TC cells (35% ±1.7%, vs 24.1% ± 6.7%). 7 days of treatment led to peak inhibition in both GBM INV (35.9% ± 4.5%) and GBM TC (34.1% ± 4%) cells, while prolonged exposure to14 days did not further inhibit cell invasion. Since 4‐AP did not affect cell proliferation, we examined its activities in a more detailed time‐ (7–10 days) and dose‐setting (0.06, 0.13, 0.25, 0.5, 1, 2, and 4 µM). Significant dose‐dependent inhibition of cell invasion up to 82% at 4 µM, the highest among 3 inhibitors, was observed starting from day 7 and lasted to day 10 (Figure 7b and Figure S13c, Supporting Information). The overall levels of suppression in GBM INV neurosphere cells were higher than that in the matching GBM TC cells. In summary, invasion was suppressed by all 3 KCNA1 inhibitors in 3 pairs (n = 6) of pGBM neurosphere cultures.
Compared with high molecular weight of ADWX‐1 (M W = 4071.86) and Agitoxin‐2 (M W = 4090.87), which are >ninefold higher than the theoretical molecular weight threshold of 450 Dalton for molecules able to pass through the BBB,[ 51 ] 4‐AP is very small (M W = 94.11) and shown to be able to penetrate the BBB.[ 52 , 53 ] Additionally, 4‐AP is a FDA approved drug for Lambert‐Eaton myasthenic syndrome (a rare autoimmune disorder characterized by muscle weakness of the limbs) and multiple sclerosis.[ 54 ] We therefore prioritized 4‐AP to assess its anti‐invasion and therapeutic efficacy. Mice bearing IC‐3752 GBM INV 3 were treated with 4‐AP (5 mg kg−1) daily after tumor implantation. Although 4‐AP is potent convulsant and can generate seizures in animals,[ 55 ] we only observed signs of excitement (increased activities) without significant (>15%) loss of body weight. Median animal survival times in IC‐3752GBM models were prolonged from 42 days in the vehicle group to 45 days in the treatment group (P = 0.035; Figure 8a). This extension was equivalent to the efficacy of fractionated radiation (2 Gy/day × 5 days). Since reducing implanted tumor cells from 10 000 to 1000 per mouse did not change animal survival times, which highlighted the highly malignant and progressive nature of IC‐3752GBM, the significantly extended animal survival times indicated the efficacy of 4‐AP as a single agent.
Figure 8.
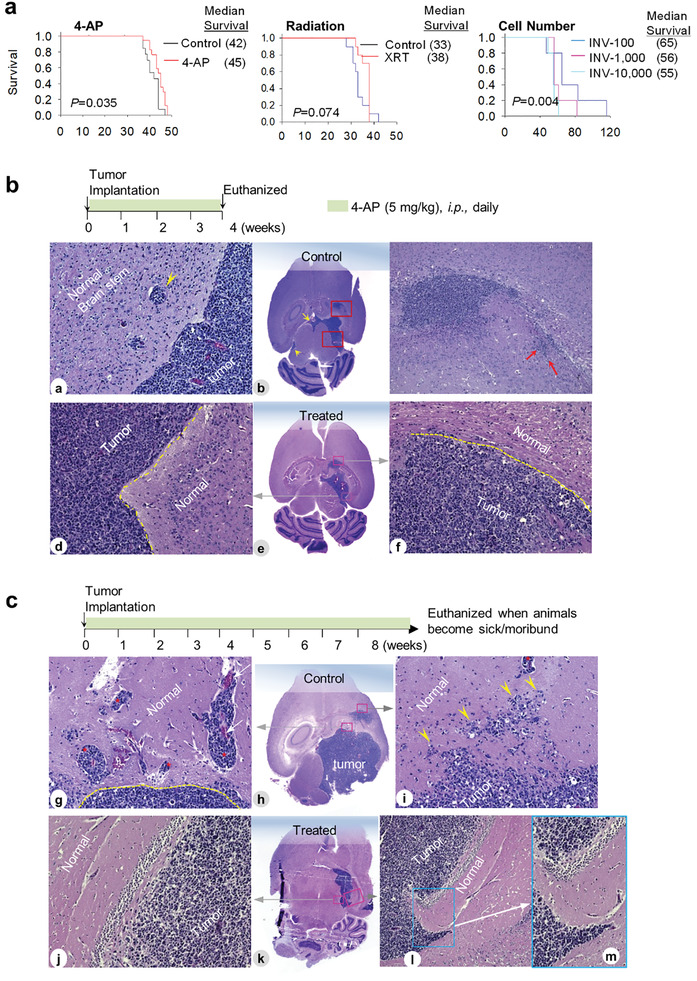
Suppression of pGBM invasion by 4‐AP in vivo in IC‐3752GBM. a) Log‐rank analysis of animal survival times. Mice (n = 10–15/group) bearing IC‐3752GBM were treated with 4‐AP daily (left panel) or fractionated radiation (2 Gy/day × 5 days) or injected with different cell numbers and compared with those administered with vehicle (PBS). b) Changes of pGBM invasion during 4‐AP treatment. Mouse brains were harvested on 4 weeks post drug treatment (top panel) followed by H&E staining of paraffin sections. In the untreated tumors (a–c), para‐vascular invasion (a, arrow), CSF spread (b, arrow), and deep invasion (c, arrow) were prominent. Tumors treated with 4‐AP treatment (d–f) for four weeks exhibited sharp margins between tumor and normal tissues (e and f, dotted yellow lines). c) Reduced invasion after long‐term in vivo treatment with KCNA1 inhibitor 4‐AP. The animals were continuously treated daily until they were euthanized due to sickness or moribund (top panel). Representative images from the untreated control (g–i) showing large foci of para‐vascular migration (g, *) from tumor into normal brains, and diffuse single cell invasion (i, arrowhead) as compared with well‐defined tumor and normal border with very limited or no tumor invasion (j–m). Magnification: 10× (c and i), 20× (all others).
To examine if the reduction of pGBM invasion mediated the extended animal survival times (since 4‐AP did not affect pGBM proliferation in vitro), we euthanized 2 mice at the end of 4‐week drug administration (Figure 8b). Compared with the frequent invasion (single cell, perivascular) and CSF spread in the untreated mice (Figure 8a–c), the treated tumors exhibited a sharp margin between the normal tissue and the tumor with low to no local invasion (Figure 8b,d–f). Similar reduction of tumor invasion was confirmed in the remnant tumors of mice in the survival group in which the treated GBM tumors were clearly circumscribed with few invasive cells and dramatically reduced CSF spread (Figure 8c), although the tumor sizes were similar between the treated and the control groups. These data demonstrated the in vivo inhibition of pGBM invasion by a single agent 4‐AP, which in turn has contributed to the significantly extended animal survival times.
3. Discussion
In this study, we utilized a novel set of highly invasive PDOX pGBM models to harvest and functionally validate matching pairs of GBM INV and GBM TC cells. Through global miRNA profiling, we identified a novel set of miRNAs that were differentially expressed between GBM INV (miRNA INV ) and GBM TC (miRNA TC ) cells of different molecular subtypes. Silencing the top 3 miRNA INV (miR‐126, ‐369‐5p, and ‐487b) led to significant suppression of GBM INV cell invasion (without affecting cell proliferation) in vitro and blocked pGBM invasion in vivo in the brains of SCID mice, thereby establishing these miRNA INV as novel drivers of pGBM invasion. Mechanistically, we confirmed KCNA1 as the sole common computational target gene of the 3 miRNAs INV . Using a set of KCNA1 inhibitors, we demonstrated that pharmacological targeting of KCNA1 significantly suppressed GBM INV cell invasion in vitro, blocked GBM invasion in vivo and prolonged animal survival times in a highly invasive pGBM model.
Pediatric GBM invasion is a complex biological process involving a dynamic interplay between tumor cells and surrounding normal brain tissues. Due to the risk of severe neurological deficits, extended surgical resection of normal brain tissues is rarely attempted, making it nearly impossible to obtain GBM INV cells for biological studies of GBM invasion. Our PDOX models that replicated the diffuse invasion of GBM have thus provided an opportunity to overcome this barrier by allowing for the isolation of purified matching pairs of GBM INV and GBM TC cells. Our finding of elevated invasive capacity of GBM INV cells in vitro and in vivo showed that the GBM invasion is not a random event but a capacity endowed with a selected subpopulations of tumor cells. This discovery demonstrated the power and highlighted the importance of using functionally‐ and biologically‐ accurate source cells (i.e., GBM INV ) for future biological studies and pre‐clinical drug testing for targeting pGBM invasion.
Identifying molecular drivers of pGBM invasion is an important step in developing new anti‐invasion therapies. Given the critical roles of miRNA in regulating critical and complex biological activities, we performed global miRNA profiling of matching pairs of GBM INV and GBM TC cells and identified a novel miRNA signature overexpressed in the invasive front (miRNA INV ) and in the tumor core (miRNA TC ), respectively. In addition to miRNAs that have been previously associated with tumor invasion/metastasis, such as miR‐126,[ 40 , 41 ] we also discovered novel miRNAs that may contribute to GBM invasion. Of note, many of these differentially expressed miRNA INV and miRNA TC would have been missed if only normal brain tissues were utilized as references, which again underscores the importance of using functionally‐accurate cell sub‐populations to address critical biological questions.
To validate the role of miRNA INV candidates in driving pGBM invasion, we selected 3 miRNA INV for in vitro and in vivo functional studies, including 2 newly identified miRNA INV (miR‐369‐5p and ‐487b) and miR‐126, whose role in invasion/metastasis of multiple human cancers has been previously reported.[ 40 , 41 ] Silencing these miRNAs led to significant inhibition of GBM INV cell invasion without affecting cell proliferation in vitro, particularly in the 3D neurospheres, in 2 distinct pGBM cell cultures, indicating the functional specificity of these miRNA INV in pGBM invasion. Subsequent in vivo confirmation of blocked invasion in mouse brains after miRNA silencing provided strong functional evidence to support the role of these miRNA INV in driving pGBM invasion.
Since silencing all 3 candidate miRNA INV did not completely eliminate pGBM invasion, additional drivers may exist. Indeed, we have discovered a total of 23 miRNA INV and 20 miRNA TC that may regulate pGBM invasion, which highlights the need for additional functional studies. However, even converting a diffusely invasive pGBM into a tumor with “limited invasive capacity,” for example, infiltrating only several mm into surrounding normal brain parenchyma, may still improve the likelihood of complete surgical resection and overall survival in children with GBM.
Although the function of many miRNAs is still unknown, our integrated analysis of miRNA INV and global gene expression of the matching GBM INV and GBM TC cells linked differentially expressed miRNA with their target genes and pathways. While each of the 3 miRNA INV affected distinct canonical pathways, they shared some overlapping functions in disease and biology as well as molecular/cellular and physiological system development, thereby supporting the notion that they collectively promoted pGBM invasion. Our discovery of KCNA1 as a commonly shared computational target gene by the 3 miRNA INV not only suggested a new biological node of miRNA INV regulation of pGBM invasion but also provided a therapeutic target. Validation of KCNA1 over‐expression (together with an additional 40 upregulated miRNA INV target genes) in the leading edges of a precious set of carefully prepared patient GBM tumors provided much‐needed data to support the role KCNA1 in GBM invasion and our strategy in understanding GBM invasion. Although miRNAs usually downregulate target genes, there are reports showing that miRNAs can also positively regulate gene expression.[ 56 , 57 ] KCNA1 is a potassium voltage‐gated channel subfamily A member 1 involved in diverse physiological processes from repolarization of neuronal or cardiac action potentials to regulating calcium signaling and cell volume.[ 49 , 50 ] It is a newly identified molecular marker for Group 4 medulloblastoma[ 58 ] and is involved in breast cancer proliferation and regulation of oncogene‐induced senescence and transformation.[ 59 ] Since channel proteins can be accessed from the extracellular milieu,[ 60 ] KCNA1 as a therapeutic target potentially allows for the use of lower drug doses thus decreased toxicities. Indeed, a series of KCNA1 (also known as Kv) inhibitors have been developed.[ 50 ] In addition to show anti‐invasion activities of 3 chemical inhibitors of KCNA1 in 3 sets of GBM INV cells, we demonstrated that 4‐AP, the smallest KCNA1 inhibitor that can penetrate the BBB,[ 52 , 53 ] effectively blocked pGBM invasion in vivo and significantly prolonged animal survival times in a highly invasive pGBM PDOX model. It is particularly encouraging that 4‐AP as single agent was able to extend animal survival times similar to that produced by fractionated radiation therapy. Although the overall level of survival time extension was not high (as 4‐AP did not suppress cell proliferation), inhibition of GBM invasion can improve the chances of completed tumor resection, thereby preventing tumor recurrence. Given the mild to moderate toxicity profiles, the recently identified neuroprotective properties, and the commercially available tablets for sustained release and long‐term administration in patients,[ 52 , 53 , 54 , 61 ] 4‐AP represents an ideal candidate drug for repurposing to target GBM invasions.
In summary, we demonstrated that not all pGBM cells have equal capacity of invasion (GBM TC cells are far less invasive than GBM INV cells), identified a novel miRNA INV signature through direct analysis of six matching pairs of biologically accurate GBM INV and GBM TC cells, completed functional validation of 3 miRNA INV (miR‐126, ‐369‐5p, and ‐487b) in driving pGBM invasion, discovered KCNA1 as a druggable molecular target of the miRNA INV , and confirmed the pre‐clinical therapeutic efficacy of KCNA1 inhibitors as novel anti‐invasion agents both in vitro and in vivo. Our strategy of isolating matching pairs of biologically accurate invasive and tumor core cells is applicable to most of human cancer studies and can potentially cause a paradigm shift in the study of cancer invasion and metastasis.
4. Experimental Section
Patient‐Derived Orthotopic Xenograft Mouse Models
Freshly resected brain tumor specimens were collected from six children undergoing surgery at Texas Children's Hospital in Houston, TX (Figure 1a). Signed informed consent was obtained from the patient or legal guardian prior to sample acquisition in accordance with our local Institutional Review Board (IRB) approved protocol. NOD/SCID mice were bred and housed in a specific pathogen‐free animal facility at Texas Children's Hospital. All animal experiments were conducted using an Institutional Animal Care and Use Committee (IACUC)—approved protocol as described previously.[ 14 , 15 , 62 ] Tumor tissues were mechanically dissociated within 60 min of surgical removal. After the cell suspensions were passed through 40 and 100 µm cell strainers, viable tumor cells were dissociated into single cells, and small clumps (≈5–10 cells) were counted with Trypan blue staining. Tumor cells (1 × 105) were then suspended in 2 µL of culture medium and injected into mice brains 1 mm to the right of the midline, 1.5 mm anterior (for intra‐cerebral tumors) to the lambdoid suture, and 3 mm deep via a 10 µL 26‐gauge Hamilton Gastight 1701 syringe needle (Figure 1b). The animals were monitored daily until they developed signs of neurological deficits or became moribund, at which time they were euthanized.
Immunohistochemistry
IHC was performed using a Vectastain Elite kit (Vector Laboratories, Burlingame, CA) as described previously.[ 15 , 30 ] Primary antibodies included the human‐specific mitochondria monoclonal antibody (1:50) (MAB1273MI, fisher scientific) and mouse anti‐glial fibrillary acidic protein (GFAP) (1:200) (M0761, AGILENT TECHNOLOGIES INC), VIM (1:200) (M0725, Dako North America,), MAP‐2 (1:200) (AB7756, Abcam Inc), Ki67 (1:20) (ab833‐500, Abcam Inc), and rabbit anti‐Nestin (NES) (1:500) (ABD69, EMD Milipore). After slides were incubated with primary antibodies for 90 min at room temperature, the appropriate biotinylated secondary antibodies (1:200) were applied and incubated for 30 min. The final signal was developed using the 3,3’‐diaminobenzidine substrate kit for peroxidase. IHC staining was assessed by combining the intensity and extent of immunopositivity.[ 15 , 30 ]
In Vivo Quantitative Analysis of Pediatric Glioblastoma Invasion
Quantitative analysis of pGBM invasion was performed using whole mouse brains that were harvested when the tumor‐bearing mice became moribund and then fixed and paraffin embedded. Serial paraffin sections were then subjected to standard H&E and IHC staining with human‐specific antibodies against MT and VIM to positively identify human pGBM cells in the mouse brain and antibodies recognizing both human and mouse vWF to identify micro‐blood vessels. The modes of GBM invasion, that is, single cell migration, along neural fibers, infiltrating to CSF and following micro‐blood vessels, were identified under the microscope (10–40×). To achieve a comprehensive and quantitative evaluation of GBM invasion, the digitally captured gross and microscopic images (4–40×) on the largest cross sections (mean ± SD) from at least 3 (3.5 ± 1.2) whole mouse brains were analyzed used a straight line reticle (eyepiece micrometer) to measure the distances (in µM) of migration.[ 29 ] Representative images were also taken and analyzed with ImageJ software to quantify the longest distance between the leading edge of invasive tumor cells and the edge of the primary tumor mass.
Isolation of Functionally Validated Invasive (GBM INV ) and Tumor Core (GBM TC ) Cells
To isolate matched pairs of GBM INV cells and non‐invading GBM TC cells, freshly harvested whole mouse brains were placed on a mouse brain matrix and sliced at 1 mm thickness into 10–12 slices (Figure 2a). These brain slices were then submerged in cold (4 °C) growth medium and the tumor core/mass (GBM TC cells) was subsequently dissected from the “normal” mouse brain tissues (containing GBM INV cells) with a surgical scalpel under a surgical microscope (Figure 2b).[ 32 , 33 , 34 ] The 2 compartments of tumor tissues, the “normal” brain (containing GBM INV cells) and the tumor mass (with GBM TC cells) from the same mouse brain were pooled and mechanically dissociated into single cell suspensions using Gentle Dissociator (Miltenyi). After passing through a stack of cell strainers (100 and 40 µm), viable tumor cells were counted after being stained with trypan blue.
Florescence Activated Cell Sorting
To isolate pure human GBM INV and GBM TC tumor cells without mouse cell contamination, particularly from the “normal” mouse brain fractions, cell suspensions were incubated with FITC‐conjugated monoclonal antibodies against human HLA‐ABC (BD Biosciences, 555552, Franklin Lakes, NJ), and an APC‐conjugated cocktail of monoclonal antibodies against mouse major histocompatibility antigen for 15 min. The mouse antibody cocktail included APC‐conjugated monoclonal antibodies against mouse CD24, CD90, CD117, CD133 (BD Biosciences, Franklin Lakes, NJ), or mouse major histocompatibility antigen (MHC) H2 haplotype (H‐2 Db‐APC). The stained cells were then washed and subjected to FACS (Figure 2c and Figure S2, Supporting Information) to gate out mouse cells and dead cells (propidium iodine high).
Whole Genome miRNA Profiling
Whole genome miRNA profiling was completed using a two‐card set of TaqMan Array MicroRNA Cards (Cards A and B) for a total of 754 unique assays specific to human miRNAs (Invitrogen) following the manufacturer's instructions. The presence of the target was detected in real time through cleavage of the TaqMan probe by polymerase 5′–3′ exonuclease activity. Briefly, total RNAs were extracted using the mirVana miRNA isolation kit. To synthesize single‐stranded cDNA, 500 ng of total RNA were reverse transcribed using the Taqman MicroRNA reverse transcription kit and the Megaplex RT Primers on a 7900HT real‐time PCR system. 6 µL of Megaplex RT product was then mixed with 450 µL of TaqMan Universal PCR Master Mix and 444 µL of nuclease‐free water and before being dispensed in 100 µL aliquots into each port of the array card. The card was briefly centrifuged, sealed, and loaded on to the Applied Biosystems 7900HT fast real‐time PCR system for amplification. Relative quantitation of target miRNA expression levels was performed through comparative CT analysis using the same threshold setting for all arrays.
Global Gene Expression Profiling
Global gene expression profiling was performed using Affymetrix U133 Plus 2.0 chips (Affymetrix, Santa Clara, CA). For RNA purification and cDNA amplification, total RNA was extracted with RNeasy (Qiagen), processed, and cDNA amplified using the Affymetrix RNA amplification and biotinylation kit. Fragmented and biotinylated cDNA was then hybridized to Affymetrix U133 Plus 2.0 chips following the manufacturer's instructions. The stained chips were then scanned using the Affymetrix GeneChip scanner 3000. The data file was imported to Bioconductor and Partek for data analyses and deposited at the Gene Expression Omnibus (GEO) Web site.
Microarray data analysis and quality control of the Affymetrix U133 Plus 2.0 gene‐expression chips were performed using the BioConductor package affyQCReport. β‐Actin and GAPDH ratios, as well as, signal distribution, were assessed to determine the outlier cases. Normalization and probe set summarization was done in BRB‐Arraytools (http://linus.nci.nih.gov/BRB‐ArrayTools. html) using the Robust Multichip Average algorithm. Hierarchical clustering was performed using centered correlation and average linkage. The significance analysis of microarrays algorithm was used for analysis of differential expression, with a false discovery rate of 0.01. Heatmaps and other graphics were created using Multi‐Experiment Viewer, part of the TM4 Microarray Software Suite. The log intensity values were exported and analyzed in Bioconductor.[ 63 ] To perform clustering, all samples were included using the hierarchical clustering algorithm provided in the “stats” R library. To determine the correlations, all elements were used to calculate the correlation coefficient (R 2 values) of all groups of samples. Pair‐wise comparison was done between pGBM cells from the invasive front, invasive rim, and the central core to identify the deregulated genes.
DNA Methylation Profiling
DNA methylation profiling of the six PDOX models was performed using Illumina Infinium Human Methylation450 arrays as previously described.[ 27 ] Tumors were classified based on their DNA methylation profiles using the DKFZ brain tumor methylation classifier (http://www.molec ularneuropathology.org), which is based on a reference cohort of 2801 CNS tumors and normal tissues representing 82 distinct tumor and 9 distinct non‐neoplastic brain methylation classes.[ 27 ]
Infection of Pediatric Glioblastoma Cells with Lentiviral miRNAs
To silence the expression of miRNA, five miRNA‐specific lentiviral transduction particles were purchased from Applied Biological Materials Inc. (ABM, MC, Canada), including LentimiRa‐Off‐has‐miR‐126 (hereafter referred as Lenti‐miR‐126‐off), LentimiRa‐Off‐has‐miR‐487b (Lenti‐miR‐487b‐off), LentimiRa‐Off‐has‐miR‐369‐5p (Lenti‐miR‐369‐5p‐off), LentimiRa‐Off‐has‐miR‐589 (Lenti‐miR‐589‐off), and LentimiRa‐Off‐has‐miR‐185 (Lenti‐miR‐185‐off). To increase miRNA expression, an additional five lentiviruses were obtained, including LentimiRa‐GFP‐has‐miR‐126 (Lenti‐miR‐126), LentimiRa‐GFP‐has‐miR‐487b (Lenti‐miR‐487b), Lenti‐miRa‐GFP‐has‐miR‐369‐5p (Lenti‐miR‐369‐5p), LentimiRa‐GFP‐has‐miR‐589 (Lenti‐miR‐589), and LentimiRa‐GFP‐has‐miR‐185 (Lenti‐miR‐185). Lentiviruses expressing GFP (Lenti‐off‐GFP and Lenti‐GFP) were used to monitor transduction efficiency, and those containing no miRNA or non‐target miRNA of Lenti‐III‐miR‐Off Control (Lenti‐off‐non‐target‐GFP) and Lenti‐III‐miR‐GFP Control (Lenti‐nontarget‐GFP) were included as non‐target controls. To silence or activate target miRNAs, purified GBM INV and GBM TC cells were seeded in traditional FBS‐containing DMEM and serum‐free media supplemented with EGF (50 ng mL−1), bFGF (50 ng mL−1), B2 (1×), and N27 (1×). The culture media was removed 24 h later, replaced with 100 µL of Polybrene (2 µg mL−1) media mixture, and exposed to lentiviral‐miRNAs for 72 h (MOI 1:1). The infection efficiency was monitored by flow cytometry, florescent microscopy examination of GFP expression, and quantitative RT‐PCR (RT‐qPCR). GFP−positive cells were selected by puromycin (1 µg mL−1) for 4 days before being counted and seeded to 96‐well plates.
Quantitative RT‐PCR
Total RNAs enriched with miRNAs were extracted using miRNeasy Mini Kit (Qiagen) following the manufacturer's instructions and reverse transcribed into cDNA using miRNA cDNA Synthesis Kit (G270, Applied Biological Materials Inc, Richmond, BC, Canada). RT‐qPCR was performed with a miScript SYBR PCR kit (Qiagen) using specific primers for miR‐126, miR‐369‐5p, miR‐487b, miR‐185, and miR‐589 (Applied Biological Materials, Inc., Richmond, BC, Canada). The relative levels of miRNA transcripts were normalized to SNORD44, a control miRNA recommended by the manufacturer (Applied Biological Materials, Inc.); KCNA1 mRNA expression was quantitated with qRT‐PCR (forward primer 5’ CTGAGCAGGAAGGAAACCAG3’ and reverse primer 5’ CCTTAGAGTGGCGGGAGAG3’)[ 16 ] and normalized to GAPDH (forward primer 5’AAGGTGAAGGTCGGAGTCAA3’ and reverse primer 5’ AATGAAGGGGTCATTGATGG3’) through standard ΔΔCt method as described previously.[ 14 ]
In Vitro Cell Invasion Assay
A CytoSelect 96‐well cell invasion assay (Cell Biolabs, San Diego, CA) was used following the manufacturer's instructions. Briefly, 1 × 105 cells in starvation medium were plated onto the upper chamber of an 8 µm pore polycarbonate membrane and exposed to a lower chamber filled with FBS growth or/and serum‐free media. After incubation at 37 °C for 24 h, the inserts were taken out, and the cells that migrated through the membrane were stained with CyQuant GR dye solution for 20 min (CBA‐112, Cell Biolabs) for quantification. The intensity of fluorescence was measured using a Synergy 2 Microplate Reader (excitation at 485 nm and emission at 528 nm) (Synergy, BioTek).
In Vitro Treatment with ADWX‐1, Agitoxin‐2, and 4‐AP
To determine the time‐ and dose‐dependent effects of 3 KCNA1 inhibitors of ADWX‐1, Agitoxin‐2, and 4‐AP, paired GBM TC and GBM INV cells from IC‐1406GBM (1406), IC‐2305GBM (2305), and IC‐3752GBM (3752) were seeded into 96‐well plates in quadruplicates and exposed to 7 doses of ADWX‐1 and Agitoxin‐2 (ranging from 0.01 to 100 nM), of 4‐AP (ranging from 0.06 to 4 µM) or vehicle control, respectively. Cell viability was measured at day 1, 3, 7, 10, and 14 for ADWX‐1 and Agitoxin‐2 and at day 7 and 10 for 4‐AP using the cell counting kit‐8 (CCK8; Dojindo Molecular Technologies) as ref. [ 15 , 30 ] and cell invasion capacity was detected by invasion assay at the meantime as described in method above.
In Vivo Treatment of Patient‐Derived Orthotopic Xenograft Tumors with 4‐AP
4‐AP was dissolved in PBS to achieve a final concentration of 0.5 mg mL−1. The same day of intracerebral IC‐3752 GBM INV tumor cell implantation, mice were intraperitoneal administrated with 4‐AP (5 mg kg−1) once daily till the endpoint of mice. To determine any survival benefits from 4‐AP treatment, the mice were monitored daily until they developed signs of neurologic deficit or became moribund, at which time they were euthanized, and their brains were removed for analysis.
Analysis of the Protein Interaction Network
STRING (http://www.string‐db.org), a database and a tool for predicting direct and indirect protein–protein interactions was utilized. This database is derived from the following sources: previous knowledge, high‐throughput experiments, genomic context, and conserved co‐expression. In addition to the miRNA target genes, 20 necessary proteins were also inputted into STRING to generate an output network of the protein‒protein interactions between the targeted genes and those necessary added proteins.
Statistical Analysis
Values were presented as mean and standard deviation (mean ± SD) with potential differences analyzed with the Student's t‐test and two‐way ANOVA. P < 0.05 is considered significant. Changes of animal survival times were examined through log‐rank analysis using Sigmaplot 14.
Conflict of Interest
The authors declare no conflict of interest.
Author Contributions
Y.H. and L.Q. contributed equally to this work. X.‐N.L. and Y.H. conceived the project; X.‐N.L. led the experimental design; Y.H., L.Q., M.K., Y.D., S.X., H.Z., H.L., S.Z., P.B., and X.‐N.L. performed the in vivo studies; S.B., M.K., and S.M.P. led the molecular sub‐ classification of PDOX models; Y.H., L.Q., F.K.B., and X.‐N.L. carried out the miRNA profiling and in vitro studies; Y.H., L.F.H., L.Q., and X.‐N.L. performed the computational studies; J.M.S., C.K.M., J.Y., M.C., D.W.P., Z.W., and Y.Z. participated in the experiment design and data analysis; A.A. provided the histopathology evaluation of the in vivo models; L.P., Z.W., Y.Z., and W.‐Y.T. provided the technology guidance of in vivo study; S.B., M.K., and S.M.P. contributed the analysis of gene profiling; X.‐N.L. and L.Q. wrote the manuscript; and all authors reviewed the manuscript.
Supporting information
Supporting Information
Supplemental Table 1‐5
Acknowledgements
The authors wish to thank all the veterinarians and veterinary technicians of the Center of Comparative Medicine in Baylor College of Medicine and staff members of the Feigin Center animal facility at Texas Children's Hospital for their excellent support of our animal experiments.
Huang Y., Qi L., Kogiso M., Du Y., Braun F. K., Zhang H., Huang L. F., Xiao S., Teo W.‐Y., Lindsay H., Zhao S., Baxter P., Su J. M. F., Adesina A., Yang J., Brabetz S., Kool M., Pfister S. M., Chintagumpala M., Perlaky L., Wang Z., Zhou Y., Man T.‐K., Li X.‐N., Spatial Dissection of Invasive Front from Tumor Mass Enables Discovery of Novel microRNA Drivers of Glioblastoma Invasion. Adv. Sci. 2021, 8, 2101923. 10.1002/advs.202101923
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
- 1. Vinci M., Burford A., Molinari V., Kessler K., Popov S., Clarke M., Taylor K. R., Pemberton H. N., Lord C. J., Gutteridge A., Forshew T., Carvalho D., Marshall L. V., Qin E. Y., Ingram W. J., Moore A. S., Ng H. K., Trabelsi S., Brahim D. H.‐B., Entz‐Werle N., Zacharoulis S., Vaidya S., Mandeville H. C., Bridges L. R., Martin A. J., Al‐Sarraj S., Chandler C., Sunol M., Mora J., de Torres C., et al., Nat. Med. 2018, 24, 1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Korshunov A., Schrimpf D., Ryzhova M., Sturm D., Chavez L., Hovestadt V., Sharma T., Habel A., Burford A., Jones C., Zheludkova O., Kumirova E., Kramm C. M., Golanov A., Capper D., von Deimling A., Pfister S. M., Jones D. T. W., Acta Neuropathol. 2017, 134, 507. [DOI] [PubMed] [Google Scholar]
- 3. Puchalski R. B., Shah N., Miller J., Dalley R., Nomura S. R., Yoon J. G., Smith K. A., Lankerovich M., Bertagnolli D., Bickley K., Boe A. F., Brouner K., Butler S., Caldejon S., Chapin M., Datta S., Dee N., Desta T., Dolbeare T., Dotson N., Ebbert A., Feng D., Feng X., Fisher M., Gee G., Goldy J., Gourley L., Gregor B. W., Gu G., Hejazinia N., et al., Science 2018, 360, 660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Sturm D., Pfister S. M., Jones D. T. W., J. Clin. Oncol. 2017, 35, 2370. [DOI] [PubMed] [Google Scholar]
- 5. Aldape K., Brindle K. M., Chesler L., Chopra R., Gajjar A., Gilbert M. R., Gottardo N., Gutmann D. H., Hargrave D., Holland E. C., Jones D. T. W., Joyce J. A., Kearns P., Kieran M. W., Mellinghoff I. K., Merchant M., Pfister S. M., Pollard S. M., Ramaswamy V., Rich J. N., Robinson G. W., Rowitch D. H., Sampson J. H., Taylor M. D., Workman P., Gilbertson R. J., Nat. Rev. Clin. Oncol. 2019, 16, 509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wells E. M., Ullrich N. J., Seidel K., Leisenring W., Sklar C. A., Armstrong G. T., Diller L., King A., Krull K. R., Neglia J. P., Stovall M., Whelan K., Oeffinger K. C., Robison L. L., Packer R. J., Neuro‐Oncology 2018, 20, 132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Stupp R., Mason W. P., van den Bent M. J., Weller M., Fisher B., Taphoorn M. J., Belanger K., Brandes A. A., Marosi C., Bogdahn U., Curschmann J., Janzer R. C., Ludwin S. K., Gorlia T., Allgeier A., Lacombe D., Cairncross J. G., Eisenhauer E., Mirimanoff R. O., N. Engl. J. Med. 2005, 352, 987. [DOI] [PubMed] [Google Scholar]
- 8. Cuddapah V. A., Robel S., Watkins S., Sontheimer H., Nat. Rev. Neurosci. 2014, 15, 455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Oliveira A. I., Anjo S. I., de Castro J. V., Serra S. C., Salgado A. J., Manadas B., Costa B. M., Cell Commun. Signaling 2017, 15, 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Horing E., Harter P. N., Seznec J., Schittenhelm J., Buhring H. J., Bhattacharyya S., von Hattingen E., Zachskorn C., Mittelbronn M., Naumann U., Acta Neuropathol. 2012, 124, 83. [DOI] [PubMed] [Google Scholar]
- 11. Rajesh Y., Biswas A., Mandal M., Exp. Cell Res. 2017, 359, 299. [DOI] [PubMed] [Google Scholar]
- 12. Dongre A., Weinberg R. A., Nat. Rev. Mol. Cell Biol. 2019, 20, 69. [DOI] [PubMed] [Google Scholar]
- 13. Mangiola A., de Bonis P., Maira G., Balducci M., Sica G., Lama G., Lauriola L., Anile C., Cancer 2008, 113, 841. [DOI] [PubMed] [Google Scholar]
- 14. Kogiso M., Qi L., Braun F. K., Injac S. G., Zhang L., Du Y., Zhang H., Lin F. Y., Zhao S., Lindsay H., Su J. M., Baxter P. A., Adesina A. M., Liao D., Qian M. G., Berg S., Muscal J. A., Li X. N., Clin. Cancer Res. 2018, 24, 2159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Shu Q., Wong K. K., Su J. M., Adesina A. M., Yu L. T., Tsang Y. T., Antalffy B. C., Baxter P., Perlaky L., Yang J., Dauser R. C., Chintagumpala M., Blaney S. M., Lau C. C., Li X. N., Stem Cells 2008, 26, 1414. [DOI] [PubMed] [Google Scholar]
- 16. Zhao X., Liu Z., Yu L., Zhang Y., Baxter P., Voicu H., Gurusiddappa S., Luan J., Su J. M., Leung H. C., Li X. N., Neuro‐Oncology 2012, 14, 574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ambros V., Nature 2004, 431, 350. [DOI] [PubMed] [Google Scholar]
- 18. Bartel D. P., Cell 2009, 136, 215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zhang S., Han L., Wei J., Shi Z., Pu P., Zhang J., Yuan X., Kang C., Int. J. Oncol. 2015, 46, 1589. [DOI] [PubMed] [Google Scholar]
- 20. Fineberg S. K., Kosik K. S., Davidson B. L., Neuron 2009, 64, 303. [DOI] [PubMed] [Google Scholar]
- 21. Fabbri M., Garzon R., Cimmino A., Liu Z., Zanesi N., Callegari E., Liu S., Alder H., Costinean S., Fernandez‐Cymering C., Volinia S., Guler G., Morrison C. D., Chan K. K., Marcucci G., Calin G. A., Huebner K., Croce C. M., Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 15805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Truesdell S. S., Mortensen R. D., Seo M., Schroeder J. C., Lee J. H., LeTonqueze O., Vasudevan S., Sci. Rep. 2012, 2, 842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kofman A. V., Kim J., Park S. Y., Dupart E., Letson C., Bao Y., Ding K., Chen Q., Schiff D., Larner J., Abounader R., Cell Cycle 2013, 12, 3500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sharif S., Ghahremani M. H., Soleimani M., Stem Cell Rev. 2018, 14, 236. [DOI] [PubMed] [Google Scholar]
- 25. Gabriely G., Wurdinger T., Kesari S., Esau C. C., Burchard J., Linsley P. S., Krichevsky A. M., Mol. Cell. Biol. 2008, 28, 5369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Spijker S., Dissection of Rodent Brain Regions. In Neuroproteomics. Neuromethods (Eds: Li K.) Humana Press, Totowa, NJ: 2011, vol 57. 10.1007/978-1-61779-111-6_2. [DOI] [Google Scholar]
- 27. Capper D., Jones D. T. W., Sill M., Hovestadt V., Schrimpf D., Sturm D., Koelsche C., Sahm F., Chavez L., Reuss D. E., Kratz A., Wefers A. K., Huang K., Pajtler K. W., Schweizer L., Stichel D., Olar A., Engel N. W., Lindenberg K., Harter P. N., Braczynski A. K., Plate K. H., Dohmen H., Garvalov B. K., Coras R., Holsken A., Hewer E., Bewerunge‐Hudler M., Schick M., Fischer R., et al., Nature 2018, 555, 469.29539639 [Google Scholar]
- 28. Sturm D., Bender S., Jones D. T., Lichter P., Grill J., Becher O., Hawkins C., Majewski J., Jones C., Costello J. F., Iavarone A., Aldape K., Brennan C. W., Jabado N., Pfister S. M., Nat. Rev. Cancer 2014, 14, 92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kogiso M., Qi L., Lindsay H., Huang Y., Zhao X., Liu Z., Braun F. K., Du Y., Zhang H., Bae G., Zhao S., Injac S. G., Sobieski M., Brunell D., Mehta V., Tran D., Murray J., Baxter P. A., Yuan X. J., Su J. M., Adesina A., Perlaky L., Chintagumpala M., Parsons D. W., Lau C. C., Stephan C. C., Lu X., Li X. N., Oncotarget 2017, 8, 87455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Yu L., Baxter P. A., Voicu H., Gurusiddappa S., Zhao Y., Adesina A., Man T. K., Shu Q., Zhang Y. J., Zhao X. M., Su J. M., Perlaky L., Dauser R., Chintagumpala M., Lau C. C., Blaney S. M., Rao P. H., Leung H. C., Li X. N., Neuro‐Oncology 2010, 12, 580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Smith S. J., Diksin M., Chhaya S., Sairam S., Estevez‐Cebrero M. A., Rahman R., Int. J. Mol. Sci. 2017, 18, 2452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lim J., Park Y., Ahn J. W., Hwang S. J., Kwon H., Sung K. S., Cho K., PLoS One 2021, 16, e0244325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ahmadipour Y., Kaur M., Pierscianek D., Gembruch O., Oppong M. D., Mueller O., Jabbarli R., Glas M., Sure U., El Hindy N., J. Neurol. Surg. A 2019, 80, 262. [DOI] [PubMed] [Google Scholar]
- 34. Gupta S., Mallick S., Benson R., Haresh K. P., Julka P. K., Rath G. K., Child's Nerv. Syst. 2017, 33, 951. [DOI] [PubMed] [Google Scholar]
- 35. Zhao X., Zhao Y. J., Lin Q., Yu L., Liu Z., Lindsay H., Kogiso M., Rao P., Li X. N., Lu X., Neuro‐Oncology 2015, 17, 965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Laks D. R., Masterman‐Smith M., Visnyei K., Angenieux B., Orozco N. M., Foran I., Yong W. H., Vinters H. V., Liau L. M., Lazareff J. A., Mischel P. S., Cloughesy T. F., Horvath S., Kornblum H. I., Stem Cells 2009, 27, 980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lu J., Getz G., Miska E. A., Alvarez‐Saavedra E., Lamb J., Peck D., Sweet‐Cordero A., Ebert B. L., Mak R. H., Ferrando A. A., Downing J. R., Jacks T., Horvitz H. R., Golub T. R., Nature 2005, 435, 834. [DOI] [PubMed] [Google Scholar]
- 38. Moller H. G., Rasmussen A. P., Andersen H. H., Johnsen K. B., Henriksen M., Duroux M., Mol. Neurobiol. 2013, 47, 131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ma L., Teruya‐Feldstein J., Weinberg R. A., Nature 2007, 449, 682. [DOI] [PubMed] [Google Scholar]
- 40. Musiyenko A., Bitko V., Barik S., J. Mol. Med. 2008, 86, 313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Huang T. H., Chu T. Y., Oncogene 2014, 33, 3636. [DOI] [PubMed] [Google Scholar]
- 42. Wilczynska A., Bushell M., Cell Death Differ. 2015, 22, 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Szklarczyk D., Franceschini A., Wyder S., Forslund K., Heller D., Huerta‐Cepas J., Simonovic M., Roth A., Santos A., Tsafou K. P., Kuhn M., Bork P., Jensen L. J., von Mering C., Nucleic Acids Res. 2015, 43, D447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Cunnea P. M., Miranda‐Vizuete A., Bertoli G., Simmen T., Damdimopoulos A. E., Hermann S., Leinonen S., Huikko M. P., Gustafsson J. A., Sitia R., Spyrou G., J. Biol. Chem. 2003, 278, 1059. [DOI] [PubMed] [Google Scholar]
- 45. Starr T., Sun Y., Wilkins N., Storrie B., Traffic 2010, 11, 626. [DOI] [PubMed] [Google Scholar]
- 46. Gerhard D. S., Wagner L., Feingold E. A., Shenmen C. M., Grouse L. H., Schuler G., Klein S. L., Old S., Rasooly R., Good P., Guyer M., Peck A. M., Derge J. G., Lipman D., Collins F. S., Jang W., Sherry S., Feolo M., Misquitta L., Lee E., Rotmistrovsky K., Greenhut S. F., Schaefer C. F., Buetow K., Bonner T. I., Haussler D., Kent J., Kiekhaus M., Furey T., Brent M., et al., Genome Res. 2004, 14, 2121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Shibuya M., Yamaguchi S., Yamane A., Ikeda T., Tojo A., Matsushime H., Sato M., Oncogene 1990, 5, 519. [PubMed] [Google Scholar]
- 48. Hamada D., Takata Y., Osabe D., Nomura K., Shinohara S., Egawa H., Nakano S., Shinomiya F., Scafe C. R., Reeve V. M., Miyamoto T., Moritani M., Kunika K., Inoue H., Yasui N., Itakura M., Arthritis Rheum. 2005, 52, 1371. [DOI] [PubMed] [Google Scholar]
- 49. Glaudemans B., van der Wijst J., Scola R. H., Lorenzoni P. J., Heister A., van der Kemp A. W., Knoers N. V., Hoenderop J. G., Bindels R. J., J. Clin. Invest. 2009, 119, 936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Wulff H., Castle N. A., Pardo L. A., Nat. Rev. Drug Discovery 2009, 8, 982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Banks W. A., Nat. Rev. Drug Discovery 2016, 15, 275. [DOI] [PubMed] [Google Scholar]
- 52. Bever C. T., Judge S. I., Expert Opin. Invest. Drugs 2009, 18, 1013. [DOI] [PubMed] [Google Scholar]
- 53. Blight A. R., Henney H. R. III, Clin. Ther. 2009, 31, 328. [DOI] [PubMed] [Google Scholar]
- 54. Jensen H. B., Ravnborg M., Dalgas U., Stenager E., Ther. Adv. Neurol. Disord. 2014, 7, 97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Yamaguchi S., Rogawski M. A., Epilepsy Res. 1992, 11, 9. [DOI] [PubMed] [Google Scholar]
- 56. Orom U. A., Nielsen F. C., Lund A. H., Mol. Cell 2008, 30, 460. [DOI] [PubMed] [Google Scholar]
- 57. Dragomir M. P., Knutsen E., Calin G. A., Cell 2018, 174, 1038. [DOI] [PubMed] [Google Scholar]
- 58. Northcott P. A., Korshunov A., Witt H., Hielscher T., Eberhart C. G., Mack S., Bouffet E., Clifford S. C., Hawkins C. E., French P., Rutka J. T., Pfister S., Taylor M. D., J. Clin. Oncol. 2011, 29, 1408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Lallet‐Daher H., Wiel C., Gitenay D., Navaratnam N., Augert A., Le Calve B., Verbeke S., Carling D., Aubert S., Vindrieux D., Bernard D., Cancer Res. 2013, 73, 5253. [DOI] [PubMed] [Google Scholar]
- 60. Arcangeli A., Pillozzi S., Becchetti A., Curr. Med. Chem. 2012, 19, 683. [DOI] [PubMed] [Google Scholar]
- 61. Dietrich M., Hartung H. P., Albrecht P., Neurol. Neuroimmunol. Neuroinflammation 2021, 8, e976. [Google Scholar]
- 62. Yu L., Baxter P. A., Voicu H., Gurusiddappa S., Zhao Y., Adesina A., Man T. K., Shu Q., Zhang Y. J., Zhao X. M., Su J. M., Perlaky L., Dauser R., Chintagumpala M., Lau C. C., Blaney S. M., Rao P. H., Leung H. C., Li X. N., Neuro‐Oncology 2010, 12, 580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Gentleman R. C., Carey V. J., Bates D. M., Bolstad B., Dettling M., Dudoit S., Ellis B., Gautier L., Ge Y., Gentry J., Hornik K., Hothorn T., Huber W., Iacus S., Irizarry R., Leisch F., Li C., Maechler M., Rossini A. J., Sawitzki G., Smith C., Smyth G., Tierney L., Yang J. Y., Zhang J., Genome Biol. 2004, 5, R80. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Supplemental Table 1‐5
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.