

Abstract
Zinc-finger nucleases (ZFNs), transcription activator-like endonucleases (TALENs), and CRISPR-associated Cas9 endonucleases are three major generations of genome editing tools. However, no parallel comparison about the efficiencies and off-target activity of the three nucleases has been reported, which is critical for the final clinical decision. We for the first time developed the genome-wide unbiased identification of double-stranded breaks enabled by sequencing (GUIDE-seq) method in ZFNs and TALENs with novel bioinformatics algorithms to evaluate the off-targets. By targeting human papillomavirus 16 (HPV16), we compared the performance of ZFNs, TALENs, and SpCas9 in vivo. Our data showed that ZFNs with similar targets could generate distinct massive off-targets (287–1,856), and the specificity could be reversely correlated with the counts of middle “G” in zinc finger proteins (ZFPs). We also compared the TALENs with different N-terminal domains (wild-type [WT]/αN/βN) and G recognition modules (NN/NH) and found the design (αN or NN) to improve the efficiency of TALEN inevitably increased off-targets. Finally, our results showed that SpCas9 was more efficient and specific than ZFNs and TALENs. Specifically, SpCas9 had fewer off-target counts in URR (SpCas9, n = 0; TALEN, n = 1; ZFN, n = 287), E6 (SpCas9, n = 0; TALEN, n = 7), and E7 (SpCas9, n = 4; TALEN, n = 36). Taken together, we suggest that for HPV gene therapies, SpCas9 is a more efficient and safer genome editing tool. Our off-target data could be used to improve the design of ZFNs and TALENs, and the universal in vivo off-target detection pipeline for three generations of artificial nucleases provided useful tools for genome engineering-based gene therapy.
Keywords: ZFN, TALEN, CRISPR, GUIDE-seq, HPV gene therapy
Graphical abstract
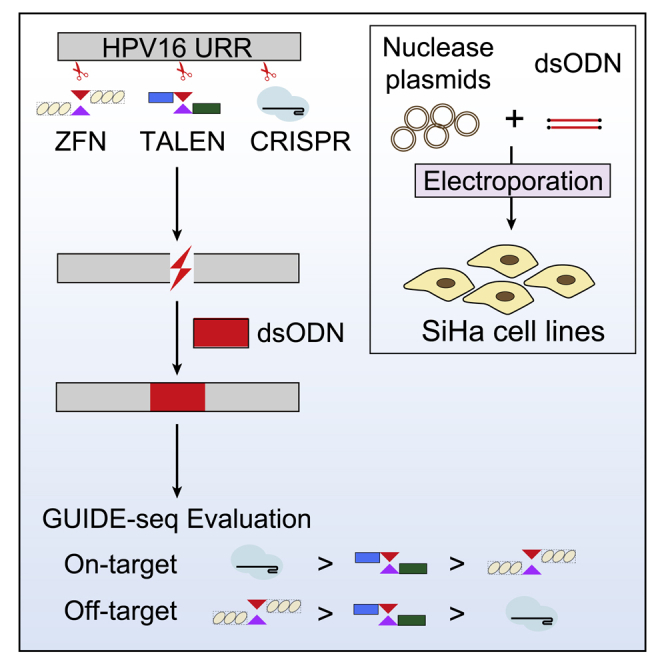
We compared the efficiency and specificity of ZFNs, TALENs, and SpCas9 by GUIDE-seq. SpCas9 outperformed ZFNs and TALENs with higher efficiency and specificity. We provided a universal pipeline to evaluate three generations of programmed nucleases to aid clinical decisions. Our data could help improve the designs of ZFNs and TALENs.
Introduction
Recent advances in genome editing nucleases has shown promising results in biotechnology, agriculture, and, in particular, biomedicine.1 Programmable nucleases, including zinc-finger nucleases (ZFNs), transcription activator-like endonucleases (TALENs), and CRISPR-associated Cas9 endonucleases, have been applied in the treatment of human cancer,2, 3, 4 infectious disease,5, 6, 7 and genetic disorders.8 By October 2020, there were 13, 6, and 42 registered clinical trials related to ZFNs, TALENs, and CRISPRs, respectively, in ClinicalTrials.gov. For example, ZFN-mediated CCR5 disruption of CD4+ T cells to resist HIV infection has entered seven phase I clinical trials (ClinicalTrials.gov: NCT00842634, NCT01044654, NCT02225665, NCT02388594, NCT03666871, NCT01252641, and NCT04201782), and one completed study demonstrated the safety and efficacy of ZFNs to efficiently reduce HIV DNA copy number in most patients.9 In addition, universal chimeric antigen receptors (UCART19) modified by two pairs of TALENs induced molecular remission in an 11-month-old infant with B-cell acute lymphoblastic leukemia (B-ALL), who was incapable of generating donor-derived CAR19 T cells.10 Currently, three clinical trials related to CAR T cells using TALEN technology are ongoing (ClinicalTrials.gov: NCT04142619, NCT03190278, and NCT04150497). Alternatively, the first registered clinical trial of CRISPR-Cas systems11,12 was launched in 2016. Since then, clinical trials of CRISPR increased rapidly and involved widespread diseases, including cancer (ClinicalTrials.gov: NCT03545815, NCT04037566, and NCT04244656), viral infection (ClincialTrials.gov: NCT04178382), hereditary diseases (ClinicalTrials.gov: NCT03855631 and NCT04122742), and hematological disorders (ClinicalTrials.gov: NCT03655678 and NCT03745287). The data from the above clinical investigations will generate valuable insights and will benefit patients in genome editing-based gene therapy.
In clinical applications, one major concern is the on-target efficiencies and the accompanied off-target activities of the programmed nucleases. To date, evolving technologies have been developed to achieve unbiased off-target detection of ZFNs,13, 14, 15 TALENs,16,17 and CRISPRs,18 but with inconsistent results. For instance, it has been reported that CRISPR-Cas9 showed higher editing efficacy than did TALENs,19, 20, 21 while the off-target activities of CRISPRs seemed more frequent than TALENs.22 In contrast, other studies found that CRISPR-Cas9 and TALENs could achieve comparable editing efficiency and high specificity in human induced pluripotent stem cell (iPSCs)23 and HEK293FT cells.23 Generally, the different conclusions of the above studies may be due to different off-target detections and their thresholds. Also, the data of direct comparison between ZFN, TALEN, and CRISPR are still lacking for a more comprehensive understanding toward the three generation of the artificial nucleases. Such evaluations will be important for further optimization of genome editing technologies in clinical trials.
In this study, we set out to systematically evaluate the on-target cleavage efficiencies and genome-wide off-target activities of ZFNs, TALENs, and CRISPRs in the setting of human papillomavirus 16 (HPV16) gene therapy. Of note, all three programmed nucleases have ongoing clinical trials in eliminating high-risk HPV infection (ClinicalTrials.gov: NCT02800369, NCT03226470, and NCT03057912). The pre-clinical studies of ZFNs,24 TALENs,7 and CRISPR-Cas925 all exhibited prospective applicability in the treatment of HPV infection. For the first time, we developed a new GUIDE-seq (genome-wide unbiased identification of double-stranded breaks enabled by sequencing) method to detect genome-wide off-targets of ZFNs and TALENs and established a feasible and universal experimental approach for the direct comparison of ZFNs, TALENs, and CRISPRs. Our aim is to help researchers in this field to design and select the best ZFNs, TALENs, and CRISPRs with maximum on-target effects and minimum off-target effects. Our data provide new insights into the clinical decision for genome editing strategies of eliminating HPV infections, as well as many other genome engineering-based gene therapies.
Results
The capture of double-stranded oligodeoxynucleotides (dsODNs) for SpCas9, ZFNs, and TALENs
Programmed nucleases used in this study were designed based on three critical genes of HPV16, including the non-coding upstream regulatory region (URR), E6, and E7 (see Materials and methods). Using T7 endonuclease 1 (T7E1) and dsODN breakpoint PCR approaches,26 we were able to screen out efficient targets for each programmed nuclease (Table 1). Specifically, 3 ZFNs targeting the URR gene (out of 14 ZFN pairs) showed high gene editing efficiencies toward HPV16 DNA (Figure 1A). One TALEN targeting URR, two TALENs targeting E6, and one TALEN targeting the E7 gene were observed with cleavage activities (Figure 1A; Figure S1). For SpCas9, all three designed single guide RNAs (sgRNAs) (URR:1, E6:1, E7:1) displayed genome editing effects (Figure 1A; Figure S1). After cleavage of the programmed nuclease, double-stranded breaks (DSBs) created in situ could be integrated by dsODNs (Figure 1B), which acted as anchors in GUIDE-seq detection methods.27
Table 1.
ZFNs, TALENs, and SpCas9 sgRNAs involved in this study
| Nucleases | Target sites in HPV16 genes |
||
|---|---|---|---|
| URR | E6 | E7 | |
| ZFNs | 3 | – | – |
| TALENs | 1 | 2 | 1 |
| SpCas9 sgRNAs | 1 | 1 | 1 |
Figure 1.

The off-target detection basis of ZFNs, TALENs, and SpCas9
(A) Target design of ZFNs, TALENs, and SpCas9 in the HPV16 URR gene. (B) Schematic overview of dsODN integration after cleavage of ZFNs, TALENs, and SpCas9. (C) Parallel comparison of dsODN integration of ZFNs, TALENs, and SpCas9 targeting the HPV16 URR gene. The gel image showed products of dsODN-PCR under the same PCR cycles, and the dsODN incorporation was confirmed using Sanger sequencing.
Before GUIDE-seq library construction, dsODN breakpoint PCR could determine the activity of target-specific engineered nucleases and serve as a quality control. In the URR gene, for example, all sites of each programmed nuclease exhibited products of expected sizes (Figure 1C; ZFN Wolfe-161/162/165 ≈250 bp; TALEN T93≈350 bp; SpCas9 ≈330 bp). Sanger sequencing results showed the right integrations of dsODN tags, indicating that GUIDE-seq had the potential to detect off-targets for ZFNs, TALENs, and SpCas9.
Start positions of GUIDE-seq reads mapping revealed distinct DSB patterns induced by the three nucleases
First, we examined the distribution of start positions of GUIDE-seq reads on targets, which represented the dsODN tag integration sites. Generally, the variability levels of ZFNs (Figure 2A) and TALENs (Figure 2B) were higher than those of SpCas9 (Figure 2C), which may be due to unfixed cutting sites and the overhang DSBs generated by ZFNs and TALENs. Specifically, the dsODN integration sites of ZFNs located mainly around the spacer, and the most frequent locations of 72 and 96 h in spacers were different (at 72 h, ZFN Wolfe-161 = the 1st base of the right zinc finger, ZFN Wolfe-162 = the 5th base, and ZFN Wolfe-165 = the 2nd base; at 96 h, ZFN Wolfe-161 = the 2nd base, ZFN Wolfe-162 = the 5th base, and ZFN Wolfe-165 = the 6th base) (Figure 2A). The spacer length of TALENs was 14 bp and was longer than the 5–6 bp of ZFNs. Thus, the dsODN integration locations spanned in a wider range surrounding the spacers with scattered distant ones (Figure 2B), indicating relatively larger deletions in the TALENs repair process than that in the ZFN repair process.28 Still, the most frequent spacer integration sites of 72 and 96 h were different (at 72 h, T49-αN-NN = the 9th base, T49-βN-NN = the 10th base, and T49-wild-type (WT)-NN = the 9th base; at 96 h: T49-αN-NN = the 9th base, T49-βN-NN = the 9th base, and T49-WT-NN = the 10th base) (Figure 2B). In the contrast to ZFNs and TALENs, the intensely concentrated dsODN integration sites of SpCas9 exhibited consistent mapping peaks between the 72- and 96-h experiments (Figure 2C).
Figure 2.

Distribution of GUIDE-seq reads start positions for on-target sites of ZFNs, TALENs, and CRISPRs
Red shows experiments at 72 h; blue shows experiments at 96 h. (A) Distribution of GUIDE-seq read counts for ZFN Wolfe-161, ZFN Wolfe-162, and ZFN Wolfe-165’s on-target sites. Bars above and below the x axis refer to reads aligned to the plus strand and minus strand, respectively. The displaying window covers both the spacer (underlined in blue) and flanking sequences. The numeral positions were reranked from 1 in the start of the spacer, left and right ZFP. The most frequent dsODN integration positions are marked. (B) Distribution of GUIDE-seq reads start for T49-αN-NN, T49-βN-NN, and T49-WT-NN on-target sites. The displaying window covers both the spacer (underlined in blue) and 20-bp flanking sequences. The numeral positions were reranked from 1 in the start of the spacer, left and right TALE. The most frequent dsODN integration positions are marked. (C) Distribution of GUIDE-seq read counts for CRISPR E6, CRISPR E7, and CRISPR URR on-target sites. The displaying window covers both the 23-bp target and the 5-bp flanking sequences. The most frequent dsODN integration positions are marked.
GUIDE-seq detected massive off-target sites for ZFNs
The ZFN off-target identification was calculated based on three criteria: (1) the mismatches of each half-site were no more than four; (2) the distance between the dsODN integration site and the right zinc finger protein (ZFP) start site should be as short as possible, and (3) the ZFP pairs should be heterodimeric (Figure 3A; see Materials and methods).
Figure 3.

Massive off-target activities of three URR-targeted ZFNs
(A) Flowcharts of ZFN off-target identification algorithm. (B) Summary of log10-transformed normalized on-target reads, off-target reads, and off-target counts for three ZFNs at 72 and 96 h. (C) Schematic of on-targets and top 10 off-targets of three ZFNs at 72 and 96 h. The on-target was at the top line followed by off-targets with mismatches highlighted in color. The specific cleavage positions and normalized GUIDE-seq read counts are shown at the right of each site. (D) Correlation of normalized GUIDE-seq reads between 72 and 96 h in common off-target sites for three ZFNs. Spearman correlation coefficients are shown. (E) Summary of off-target counts of original and extended modes for three ZFNs. (F) Correlation between MIP indel rates and normalized GUIDE-seq reads for Wolfe-165 common off-targets at 72 and 96 h. The Spearman correlation coefficients for original and extended mode are shown separately. (G) WebLogo of ZFN Wolfe-161 off-targets. The contents form left to right of top panel are (1) the weblogs from off-targets sequences corresponding to left ZFP, (2) the distribution of spacer lengths from off-targets, and (3) the weblogs from off-target sequences corresponding to the right ZFP. The contents from left to right of bottom panel are (1) the distribution of off-target mismatches to intended sequences of the left ZFP and each zinc finger, (2) the distribution of off-target mismatches to intended sequences of Wolfe-161 ZFN, and (3) the distribution of off-target mismatches to intended sequences of right ZFP and each zinc finger.
To assess the specificity and efficiency of zinc fingers at different reaction times, we performed two sets of experiments, reacting for 72 and 96 h. In total, at 72 h, there were 966, 280, and 65 off-targets detected for Wolfe-161, Wolfe-162, and Wolfe-165, respectively, while at 96 h, there were 1,856, 1,365, and 287 off-target sites, respectively (Figure 3B; Table S1), indicating that the longer reaction time accumulated more off-targets. Still, off-target sites of Wolfe-161, Wolfe-162, and Wolfe-165 at 72 h shared 699, 259, and 61 common events with those at 96 h, respectively (Figure 3B; Table S1), accounting for a large proportion of off-target sites at 72 h (Wolfe-161, 72.4%; Wolfe-162, 92.5%; Wolfe-165. 93.9%). The on-target and top 10 common off-target diagrams of Wolfe-161, Wolfe-162, and Wolfe-165 showed that the number of mismatches varied from 1 to 3 bp (Figure 3C). For those common off-target sites, the normalized GUIDE-seq read count exhibited a high correlation between 72 and 96 h for three ZFNs (Figure 3D, Wolfe-161, R = 0.80; Wolfe-162, R = 0.81; Wolfe-165 R = 0.81).
In addition to the original detection method,2, 3, 4 we also developed an extended mode by additional potential targeted sequences derived from the engineered endonuclease database EENdb (see Materials and methods). Using the extended mode, we were able to detect more off-targets of Wolfe-165, Wolfe-161, and Wolfe-162 (Figure 3E). Specifically, the off-target sites of Wolfe-161, Wolfe-162, and Wolfe-165 at the 72-h experiment increased by 1,518, 354, and 1, respectively, and the off-target sites of Wolfe-161, Wolfe-162, and Wolfe-165 at the 96-h experiment increased by 3,210, 2,128, and 6, respectively (Figure 3E; Table S2). To verify the accuracy of the two detection modes, we validated common off-target sites in original and 96-h off-targets in extended modes of Wolfe-165 by the molecular inversion probe (MIP) sequencing method (Table S3). The results showed that the verification rate of the original mode (ratio of 46/61, 75.4%) was higher than that of the extended mode (ratio of 2/6, 33.3%) (Table S3). We also observed a positive correlation between the indel ratios of MIP indel rates and the normalized GUIDE-seq read count in either the original mode (R = 0.65) or extended mode (R = 0.91) (Figure 3F).
To better understand the features of the off-target site, we constructed a web-logo plot for each ZFN (Figure 3G; Figures S2A and S2B), which involved distributions of spacer lengths and mismatches of zinc fingers. We noted the guanines were more conserved than other nucleotides, and the number of mismatches was mostly 0 when the intended target nucleotide triplets of the zinc fingers contained two guanine nucleotides (Figure 3G). Consistently, in top 10 common off-targets sites of three ZFNs, when the intended target nucleotide triplet had a higher G content (the C in display of the left ZFN target), there were fewer mismatches in those sites (Figure 3C).
The spacer length of ZFN was mainly 5 and 6, but the spacer lengths of a small part of Wolfe-161 off-target sites were 15 and 16 (common off-targets of 72 and 96 h in original mode; n = 34; Figure 3G). Mismatches of each zinc finger protein were mainly 0 and 1 bp (Wolfe-161, 86.3%; Wolfe-162, 86.9%; Wolfe-165, 91.3%). The total number of mismatches for the entire zinc finger was mostly distributed in 3–5 bp (Wolfe-161, 78.5%; Wolfe-162, 75.7%; Wolfe-165, 77.1%). Furthermore, there were few off-target sites when more than two mismatches were on both the left and right half-site simultaneously (Wolfe-161, n = 110; Wolfe-162, n = 22; Wolfe-165, n = 1). As the number of mismatches increased, there were significant declines in normalized GUIDE-seq reads at off-target sites (Wolfe-161, p = 1e−09; Wolfe-162, p = 0.0027; Wolfe-165, p = 0.00062; Figure S3), indicating that sites with more mismatches had weaker cleavage activities.
Genome-wide profiling of different TALEN backbones and modules by tailored GUIDE-seq pipeline
In this study, the TALEN off-target algorithm is described in Materials and methods (Figure 4A). In applications, many efforts have been made to evolve TALEN architectures to accommodate any N0 bases, such as the N-terminal domain modification from WT to αN/βN. This provided unconstrained DNA targeting capacity.29 Meanwhile the degeneracy of single-nucleotide repeat variable diresidue (RVD) codes of TALEN binding nature resulted in weighing between on-target effectiveness and off-target side effect. For example, a previous study showed that the NN module was permissive for both G and A while the NH module was more specific in binding G with a certain trade-off of efficiency.30 To investigate TALEN cleavage comprehensively, we applied the GUIDE-seq method in one URR-targeting, one E7-targeting (Figure S4), and seven E6-targeting TALENs (T46/T49 and derived variants with NN/NH modules or αN/βN N-terminal domains) (Figure 4B; Figure S1).
Figure 4.

TALEN off-target spectrum in HPV E6-targeted sites
(A) Flowcharts of TALEN off-target identification algorithm. (B) All TALEN variants targeted the HPV16 E6 gene. The engineered N-terminal domains, containing NN/NH RVDs and log10-transformed normalized GUIDE-seq on-target reads, are summarized for all variants. (C) Schematic of on-targets and top 10 off-targets of three T49 TALENs at 96 h. The on-target is at the top line followed by off-targets, with mismatches highlighted in color. The specific cleavage positions, TALEN-NT 2.0 scores of left and right TALEs, as well as log10-transformed normalized GUIDE-seq reads are displayed at the right of each site. (D) Bar plots of the log10-transformed normalized GUIDE-seq on-target reads, off-target reads, and off-target counts for each T49 TALEN. Both the 72- and 96-h data are shown. The double y axis represents log10-transformed normalized GUIDE-seq read counts (left) and off-target numbers (right). (E) Bar plots of the log10-transformed normalized GUIDE-seq on-target reads, off-target reads, and off-target counts for T46-αN-NN/NH and T46-βN-NN/NH TALENs. Both the 72- and 96-h data are shown. The double y axis represents log10-transformed normalized GUIDE-seq read counts (left) and off-target numbers (right). (F) Venn diagrams illustrating overlapped off-target of three T49 TALENs. Both the 72- and 96-h data were used. (G) Recognized nucleotide frequencies of each RVD derived from T49-WT-NN off-targets.
The detected off-targets (Figure 4C) showed that TALEN could tolerate numerous mismatches from 16 to 24 bp (Figure S5). We further compared the on-target of different backbones and modules for T49/T46. Briefly, for three different N-terminal TALENs, the on-target efficiency of αN and WT were comparable (T49-αN-NN, N_reads_72h = 4.57; N_reads_96h = 4.22; T49-WT-NN: N_reads_72h = 4.58; N_reads_96h = 4.30) and far exceeded βN N-terminal TALEN architecture (T49-βN-NN, N_reads_72h = 3.25; N_reads_96h = 3.98) (Figure 4D). Generally, the on-target performance of the NN module was better than that of the NH module with either αN N-terminal architecture (T46-αN-NN, N_reads_72h = 4.80; N_reads_96h = 3.45 versus T46-αN-NH, N_reads_72h = 2.98; N_reads_96h = 1.23) or βN N-terminal architecture (T46-βN-NN, N_reads_72h = 3.86; N_reads_96h = 3.18 versus T46-βN-NH, N_reads_72h = 0.94; N_reads_96h = 1.61) (Figure 4E). The above data were also in line with that the efficiency performance of αN outperformed that of βN with either the NN or NH module.
As for the off-target sites, in the 72-h experiment groups, we were able to detect four, three, and two off-targets of αN, βN, and WT N-terminal TALENs, respectively, in T49 (Figure 4D; Table S4). The T49 off-target numbers increased when editing time extended to 96 h (αN, n = 68; βN, n = 34; WT, n = 28; Figure 4D; Table S4). T46 with NH modules had no off-target activities with either αN or βN backbones in any experiments (Figure 4E). Meanwhile, T46 with NN modules showed increased off-target numbers (T46-αN-NN, from three to seven; T46-βN-NN, from five to six) when editing time extended from 72 to 96 h. The above data showed that although the αN backbone and NN module enhanced on-target activities of TALENs, they also induced more off-targets.
Meanwhile, we found only three common off-target sites shared by three different N-terminal backbones, which reflected that the N-terminal structure affected TALEN off-targets largely through different binding affinities (Figure 4F). Furthermore, we validated the 96 h-generated off-target sites of T49-WT and T49-βN by using MIP sequencing methods. There were 11 (11/28, 39.3%) and 9 (9/34, 26.5%) off-target sites for T49-WT-NN and T49-αN-NN with ≥0.1% indel rates (Table S5). Similar to ZFNs, the validated indel rates of TALEN off-targets exhibited positive correlation with normalized GUIDE-seq reads (R = 0.43; Figure S6). For the specificity comparison of two TALENs targeting HPV-E6, we observed more off-targets in the T49 (96 h: T49-αN-NN, n = 68; T49-βN-NN, n = 34) than T46 (96 h: T46-αN-NN, n = 7; T46-βN-NN, n = 6), which was due to the shorter DNA-binding sequence of T49 (L19_S14_R16) than that of T46 (L20_S14_R20).16
To explore the relationship between modules and nucleotides, we calculated the frequency of 4 nt in each target position of three T49 backbones using 96-h off-target data (Figure 4G; Figures S7A and S7B; Table S6). We found that any module had non-exclusive binding activities yet with the dominant nucleotide following the RVD codes. For instance, the most frequent binding nucleotides of NN modules were G (n = 16), A (n = 11), C (n = 2), and T (n = 1) for three TALENs (Table S6), consistent with the conclusion that the frequency of nucleotides recognized by NN is sorted as follows: G>A>C>T (3). Of note, we observed that the most frequent nucleotide was not T or C in the N0 base among off-targets of three-TALEN architecture (WT, left N0 G = 34%, right N0 T = 31%; αN, left N0 T = C = G = 28%, right N0 T = 28%; βN, left N0 G = 31%, right N0 C = 34%; Table S6), contrasting to the reported stringent affinity of the N0 domain binding to T or C.31
Parallel comparison of cleavage efficiencies and genome-wide off-targets among SpCas9, ZFNs, and TALENs
GUIDE-seq results of three SpCas9 sgRNAs showed high specificity and efficiency. No off-targets were identified for URR sgRNA while E6 (72 h, n = 4; 96 h, n = 2; common = 2) and E7 sgRNA (96 h, n = 4) had several detectable off-targets (Figure 5A). All of the above off-targets were validated by MIP sequencing (threshold, ≥0.1% indel rate) (Figure 5A; Table S7), and the indel rates showed a positive correlation with normalized GUIDE-seq reads (R = 0.88; Figure S8). Log10-transformed normalized GUIDE-seq reads revealed that the cleavage efficiencies of SpCas9 were better at 96 h (URR on-target, 4.84; E6 on-target, 4.88; total off-target, 4.53; E7 on-target, 4.87; total off-target, 3.06) than at 72 h (URR on-target, 3.24; E6 on-target, 3.26; total off-target, 3.19; E7 on-target, 3.59) (Figures 5B–5D). As the off-target activity of SpCas9 was related to cell line diversity, we also transfected the same three sgRNAs into pB-HPV16 stable cell lines (HEK293T containing the whole HPV16 genome; see Materials and methods) and harvested them in 96 h (Figure S9A). Interestingly, we were able to detect the same off-target sites in pB-HPV16 cell lines as in SiHa cell lines (Figure S9).
Figure 5.

SpCas9 off-targets and comparison among three nucleases
(A) Schematic of GUIDE-seq on-target and off-target sites for three HPV-targeted sgRNAs. The on-target was at the top line followed by off-targets with mismatches highlighted in color. The specific cleavage positions, log10-transformed normalized GUIDE-seq reads, and MIP validated indel rates are at the right of each site. (B–D) Bar plot of log10-transformed normalized GUIDE-seq on-target reads, off-target reads, and off-target counts for each Spcas9 sgRNA-targeted URR (B), E6 (C), and E7 (D) genes. Both the 72- and 96-h data are shown. The double y axis represents log10-transformed normalized GUIDE-seq read count (left) and off-target number (right). (E) Bar plot of log10-transformed normalized GUIDE-seq on-target reads, off-target reads, and off-target counts for the URR-targeted SpCas9, TALENs and ZFNs. The 96-h data were used. The double y axis represents log10-transformed normalized GUIDE-seq read counts (left) and off-target numbers (right). (F) Bar plot of log10-transformed normalized GUIDE-seq on-target reads, off-target reads, and off-target counts for E6-targeted SpCas9 and TALENs. The 96-h data were used. The double y axis represents log10-transformed normalized GUIDE-seq read counts (left) and off-target numbers (right). (G) Bar plot of log10-transformed GUIDE-seq normalized on-target reads, off-target reads, and off-target counts among E7-targeted SpCas9 and TALENs. The 96-h data were used. The double y axis represents log10-transformed normalized GUIDE-seq read counts (left) and off-target numbers (right). (H) Plots of log10-transformed normalized GUIDE-seq reads of dsODN integration sites for all used nucleases in this study. Both the 72- and 96-h data are involved and distinguished by different shapes. The corresponding on-target and off-targets are highlighted in blue and red, respectively.
Eventually, parallel comparisons of the on/off-target activities of ZFNs, TALENs, and SpCas9 targeting URR, E6, and E7 of HPV16 were performed. Based on URR editing data at 96 h (Figure 5E), the cutting efficiency of the three gene editing tools ranked as following: CRISPR (4.84) > TALEN (3.89) > ZFN (3.38), while off-target damage was opposite (CRISPR, no off-target; TALEN, 1.91/1 site; ZFN, 5.11/287 sites). We further compared the specificity and efficiencies of SpCas9 and TALEN at the E6 and E7 sites (Figures 5E–5G). SpCas9 outperformed TALENs in E6 targets (SpCas9 on-target reads: 4.88, off-target count of 2; T46-αN-NN on-target reads: 3.45, off-target count of 7; T49-αN-NN on-target reads: 4.22, off-target count of 68) and E7 targets (SpCas9 on-target reads: 4.87, off-target count of 4; T164 on-target reads: 2.85, off-target count of 36). Therefore, as for HPV genome editing, SpCas9 had enhanced cutting efficiency and fewer undesired off-targets than did TALENs and ZFNs.
Furthermore, we compared the dsODN toxicity of three programmed nucleases through measurement of ODN insertion sites. We extracted qualified dsODN integration sites for all nucleases used in this study (Figure 5H; Tables S8 and S9). Compared with the control group (n = 16), tremendous dsODN integration sites were observed in ZFNs (Wolfe-161, 19,274; Wolfe-162, 10,404; Wolfe-165, 1,296), followed by TALENs (average = 1,169.5) and SpCas9 (average = 573), indicating that SpCas9 also triggered less genome stress than did TALENs and ZFNs.
Discussion
In this study, we established a universal approach to evaluate the efficiency and specificity of ZFNs, TALENs, and SpCas9 in vivo. For the first time, we employed GUIDE-seq methods in detecting off-targets of ZFNs and TALENs and developed novel bioinformatics pipelines correspondingly. Our data provide new insights into the intrinsic cleavage features of three programmed nucleases.
As the first generation of the genome-editing tool, the three URR-targeted ZFNs in our studies showed distinct massive off-target counts (96 h: Wolfe-161, n = 1,856; Wolfe-162, n = 1,365; Wolfe-165, n = 287; Figures 1A and 3B). In contrast, a previous in vivo genome-wide off-target detection method for ZFN only identified four off-targets for CCR5-targeted ZFN,13 which may indeed possess 15,882 potential off-targets identified by another in vitro machine-learning method.15 In this study, the massive off-targets of Wolfe-161 and Wolfe-162 indicated that our detection method for ZFN had much higher sensitivity compared to the previous in vivo method.13 Additionally, the verification rate of Wolfe-165 achieved 75.4%, further confirming the reliability of our methods. Taken together, we think that our data can bring two aspects of value to researchers in this field. First, the massive off-targets generated by our method could provide precious materials for machine learnings to predict ZFN off-targets and improve ZFN designs.15,32 For instance, that three similar ZFNs differed largely in specificity was due to the counts of middle G in six ZFPs (Wolfe-161, n = 0; Wolfe-162, n = 2; Wolfe-165, n = 4), which was reversely correlated with off-target counts. Second, our ZFN off-target detection approach could be applied widely in genome editing-based gene therapy, especially for clinical studies to evaluate ZFN genome editing performance.3,5,24
Compared to ZFNs, the specificity of TALENs was considered better than that of ZFNs. Still, different TALEN target sites exhibited distinct off-target profiles. URR-targeted TALEN T93 had only one off-target detected in 96 h (Figure 5E), while the E6-targeted TALEN T49-αN-NN had 68 off-targets in 96 h (Figure 5F). Meanwhile, we for the first time described and compared the off-targets of TALENs with different N-terminal architectures and NN/NH modules (for G recognition). Our data showed that the modification of both N-terminal architectures (96 h on-target, T46-αN-NN = 3.45 versus T46-βN-NN = 3.18) and RVD modules (96 h on-target, T46-αN-NN = 3.45 versus T46-αN-NH = 1.23) increased on-target efficiency, but induced more off-targets (96 h off-target, T46-αN-NN = 7 versus T46-βN-NN = 6, T46-αN-NN = 7 versus T46-αN-NH = 0; Figure 4E). Therefore, the actual application of TALENs required screenings of specific and effective targets and architectures, and our method provides an easy-to-perform option for this purpose. Furthermore, our study profiled massive off-targets of TALENs, which could help to explore the mismatch patterns of TALEN binding requirements and furthermore improve the design of TALENs. For example, the non-T/C of the N0 observation in our off-target data suggested that we should consider all kinds of upstream nucleotides in predicting off-targets of TALENs.
The specificity of SpCas9 is the biggest concern in its clinical application. Unlike the common concept that SpCas9 was less specific than TALENs,22 our data showed that SpCas9 outperformed TALENs in both on-target efficacy and off-target side effects in all three HPV16 critical target genes (Figures 5E–5G). This is probably due to the improved CRISPR designing algorithm to pick the best sgRNA with maximized on-targets and minimized off-targets.33,34 We think that the same improvement in the designing software for ZFNs and TALENs could be made by learning the patterns of massive off-targets of ZFNs and TALENs that were produced in this study. For the undergoing clinical trials using ZFNs, TALENs, and CRISPRs to eliminate HPV16 infections, we suggested that SpCas9 was the most effective and safest programmed nuclease.35 Additionally, we also noticed that the on-target activity of SpCas9 was higher at 96 h than at 72 h, while the on-target activities of ZFNs and TALENs were higher at 72 h than at 96 h. Meanwhile, from 72 to 96 h, the off-targets of ZFNs and TALENs increased significantly in the cells. The results indicated that the clinical application of ZFNs and TALENs should be more cautious with respect to duration and clearance.
In summary, our study for the first time compared the on-target/off-target effects of ZFNs, TALENs, and SpCas9 in parallel. Based on our data, SpCas9 was the most effective and safest programmed nuclease in treating HPV infection. Furthermore, we provided a new universal pipeline for evaluating the three generations of programmed nucleases. Our data could help to improve the designs of ZFNs, TALENs, and CRISPRs and guide clinical decisions in many gene therapies.
Materials and methods
Cas9 sgRNA design and plasmid construction
For SpCas9, sgRNAs targeting HPV16 genes were designed using Cas-designer36 (http://www.rgenome.net/cas-designer), and we chose sgRNAs with the highest on-target efficacy and low off-numbers in human GRCh38 references. SpCas9 plasmid was purchased from Addgene (plasmid #42229). Then, SpCas9 and sgRNA sequences were synthesized and purified by GENEWIZ Biotech (Suzhou, China) and cloned into the PX330 and PXZ vectors, respectively. All sgRNA sequences targeting the HPV16 genome are listed in Table S10.
ZFNs design and plasmid construction
For ZFNs targeting the critical genes of HPV16, we designed six and eight paired ZFNs using online tools (https://mccb.umassmed.edu/ZFPmodularsearch.html) and ZiFiT software32 of the CoDA module (http://zifit.partners.org/ZiFiT/Disclaimer.aspx) with default parameters, respectively. The customized pcDNA3.1 ZFN plasmids with heterodimeric EL/KK ForkI were constructed in a previous study.24 Then, the Zinc Finger sequences were synthesized and cloned into pcDNA3.1 ZFN plasmids (GENEWIZ Biotech, Suzhou, China). All 14 plasmids were confirmed using Sanger DNA sequencing. Protein and target sequences of three effective ZFNs are listed in Table S11.
Design and plasmid construction of TALENs
TALENs targeting HPV16 URR, E6, and E7 genes were designed using TALEN Targeter 2.0 online software (https://tale-nt.cac.cornell.edu).31 The customized design parameters are as follows: (1) fixed spacer length of 14 bp, which had the highest cleavage effect in NΔ152/C+18 TALEN architectures;37,38 (2) the upstream bases were T or C; (3) module NN and NH were both attempted to bind G nucleotides; and (4) the genome of Homo sapiens was used to predict off-target sites. To maximize the successful rate by obtaining dispersed target sites of each gene, for predicted results, we further selected sites with high scores of on-target activities, including one, two, and one sites targeting URR, E6, and E7 genes, respectively. The protocol for the assembly of the TALENs targeting HPV16 was described previously.13 The module and array plasmids of each TALEN were ultimately constructed into pcDNA3.1(+) backbone plasmid (V790-20, Invitrogen) with heterodimeric EL/KK ForkI and an NΔ152/C+18 backbone. RVD and target sequences of each TALEN are provided in Table S12.
Plasmid extraction
The two-in-one SpCas9-sgRNA plasmids, ZFN plasmids, and TALEN plasmids were extracted using E.Z.N.A. Plasmid Mini kit II (Omega Bio-tek).
pB-HPV16-transfected stable cell line
The pB-HPV16 plasmid was constructed by amplifying the HPV16 whole genome from pBR322-HPV16 plasmid39 and cloning it into the pB-CMV-MCS-EF1α-GFP-Puro plasmid (purchased from Miaoling, Wuhan City, China). Then, the HEK293T cell line (purchased from the American Type Culture Collection [ATCC]) was transfected with 2 μg of pB-HPV16 plasmid using X-tremeGENE HP (Roche) and maintained in Dulbecco’s modified Eagle’s medium (C11995500BT, Gibco, USA) containing 3 μg/mL puromycin for 5 days. The HPV16 DNA was validated by PCR and Sanger sequencing before nuclease transfection (Figure S10).
SiHa cell line culture and transfection
The human cervical cancer cell line SiHa (HPV16-positive) was purchased from ATCC and cultured in Dulbecco’s modified Eagle’s medium (C11995500BT, Gibco, USA) supplemented with 10% fetal bovine serum (10270-106, Gibco, USA) at 37°C in a humidified 5% CO2 chamber.
SiHa cells (program DN-100) were transfected on a Lonza 4D-Nucleofector using the SE cell line 4D-Nucleofector X kit (V4XC-1024, Lonza, Germany) according to the manufacturer’s protocol. The same dsODN as used in a previous study40 was synthesized by Sangon Biotech (Shanghai, China). For SpCas9, 2 μg of Cas9, 2 μg of sgRNA plasmid, and 100 pmol of dsODN were transfected into six-well plates. Cells were harvested 96 h after transfection. For TALENs and ZFNs, 2 μg of each pair of plasmids and 100 pmol of dsODN were transfected into six-well plates. The DNA was harvested after 72 and 96 h to fully exposure the off-target sites.
DNA extraction and dsODN-tag breakpoint PCR
DNA was extracted with a kit (69,506, QIAGEN, Germany). The PCR reaction was conducted using Q5 hot start high-fidelity 2× master mix (M0494S, New England Biolabs, MA, USA). The primers used are listed in Table S13. The dsODN-PCR process is displayed in Figure S11.
T7E1 analysis
Genomic DNA was prepared with the DNeasy blood and tissue kit (69504, QIAGEN, Hilden, Germany). For the PCR amplification of sgRNA-targeting genome regions with the corresponding primers, we used the TransTaq DNA polymerase high-fidelity (HiFi) kit (K10222, TransGen Biotech, Beijing, China) according to the supplier’s protocols. Then, the purified PCR products were digested with 0.5 μL of T7 nuclease (M0302L, NEB, USA) in a 50-μL volume at 37°C for 20 min.
GUIDE-seq experiments
The dsODN anchor is a quantitative measurement for cleavage efficiency, which is adopted by GUIDE-seq technology.27 Specifically, after the genome cleavage induced by programmed nucleases, the end-protected dsODN was incorporated into DSBs through non-homologous end joining (NHEJ) pathways. The frequencies of dsODN integration actually reflected the DSB amounts cut by programmed nucleases. Meanwhile, the end-protected strategy improved the dsODN integration frequencies, which helped the detection of low-frequency off-targets. Therefore, both the on-targets and off-targets were able to be captured and quantified through measuring the efficiency of the ODN anchor integration into the dsDNA breaks. Furthermore, by adding unique molecular identifier (UMI) tags during the PCR process, GUIDE-seq library construction could avoid the PCR bias and truly detect the on-targets and off-targets quantitatively. In this study, GUIDE-seq experiments were performed as previously described.27,40 GUIDE-seq libraries were sequenced using Illumina NextSeq500 sequencer with customized settings for 16-bp UMIs.
General data analysis of GUIDE-seq
Data were first demultiplexed using Illumina’s bcl2fastq software and then analyzed using GUIDEseq v1.1 as described previously.41 The off-targets of CRISPRs were identified using the original standards with mismatches ≤7.27 For ZFNs and TALENs, we developed different algorithms to identify off-target sites. Finally, for parallel comparison, the sequencing reads of GUIDE-seq were normalized (referred to as N_reads) according to the formula: normalized reads = reads/consolidated reads × 1,000,000. Consolidated read numbers were GUIDE-seq reads after merging UMI molecules and removing duplication.
ZFN off-target identification algorithm
For ZFNs, to identify the binding half-site of ZFNs around the dsODN integration sites, we extracted the 60-bp window sequences flanking each dsODN integration site based on the characteristic that repair of ZFN-induced DSBs had deletions shorter than 60 bp.42 Meanwhile, most off-targets (57/58; 98.3%) of three-finger ZFNs had fewer than or equal to four mismatches at each half-site among validated off-targets of ZFN targeted to VEGFA in vivo (Table S14) in a previous study,15 and the heterodimeric ZFNs had no cleavage effect at homodimer off-target sites as a previous study reported.13 Therefore, we scanned only heterodimer off-target sites in the 60-bp flanking region around the dsODN integration site for the nearest and most homologous sequences (each half-site ≤ 4-bp mismatches), and results above were referred to as the “original mode.”
Additionally, 7-aa variable regions of each finger bind to a triplet component. The binding rule is still not fully understood. For example, the first variable region “RSDNLTK” had eight documented binding triplets in EENdb (CTG, GTA, GTC, GCG, GTG, GTC, GTG, and GCG).43 Therefore, we developed an “extended” mode for off-target identification of ZFNs. Specifically, we listed all possible ZFP-binding targets based on the EENdb resource and scanned the 60-bp flanking region for the nearest and most homologous sequences to each binding target (each half-site ≤ 4-bp mismatches).
TALEN off-target identification algorithm
For TALEN, in silico tools of TAL Effector-Nucleotide Targeter (TALE-NT) 2.0 can predict off-targets of TAL effectors to aid researchers in selecting a highly targeting TALEN design with the fewest off-target number.31 The original C-coded version restricted the N0 base (the 5′-most base of a TALE-bound sequence) of potential off-target sites to either T or C. To accommodate any N0 bases, we modified the C-code of the “pairedtalesf” program, and the modified program had an 88.9% (32/36) consistency rate (Figure S12; Table S15) using 36 validated off-target sites from a previous study.16 Specifically, among a total of 36 validated off-target sites, there were 32 predicted sites with scores lower than 4-fold the best possible score for the RVD sequence, including 29 sites (N0 base: T/C) and 3 sites (N0 base: not T or C) (Figure S12). Only four off-target sites had sores greater than 4-fold the best possible score for the RVD sequence. The above data confirmed the ability of the modified TALEN-NT 2.0 algorithm to predict off-targets in further GUIDE-seq downstream detection. Then, we implemented the modified algorithm after the general GUIDE-seq analysis. Specifically, after identification of the dsODN integration sites, we extracted the upstream/downstream 60-bp sequences as potential off-target windows. Then, we ran the modified script with parameters “-t 4 -d 1 -m 12 -x 30” to detect off-target sites according to the following criteria: (1) any N0 base, (2) heterodimeric model, (3) the potential spacer length is 12–30, and (4) the score of each monomer should be lower than 4-fold the best possible score for the RVD sequence (recommended by TALE-NT 2.0 software).31
MIP probe design and data analysis
To validate off-target sites, we designed MIP probes by MIPgen44 with the parameter “-min_capture_size 150” and “-max_capture_size 250,” and synthesized them through GENEWIZ Biotech (Suzhou, China). For data analysis, after quality control of the raw sequencing data by fastp with default parameters,45 pair-end reads were merged into one read by FLASH software.46 Then, all reads were aligned to designed MIP sequences by BWA-MEM.47 The Python package pysam and the rewritten python script from the CRISPRMatch package48 were used to extract and count sequences flanking 10 nt around the predicted targets, and indel rates were calculated, including sequences with either insertions or deletions. Control experiments were conducted without adding nucleases to exclude the indels caused by background mutation or sequence errors. The cutting efficiency of each site was represented by the mean indel rate of the experiment group subtracting that of the control group.
Statistical analysis
All tests were performed using R programming language 3.4.2 with a two-sided p value provided. p < 0.05 was considered statistically significant.
Acknowledgments
High-performance computing systems and services were provided by the Tianhe 2 supercomputer in Guangzhou city. This work was supported by the National Science and Technology Major Project of the Ministry of science and technology of China (grant no. 2018ZX10301402); The National Natural Science Foundation of China (grant no. 32171465, 82102392 and 82172584); General Program of Natural Science Foundation of Guangdong Province of China (grant no. 2021A1515012438); the National Postdoctoral Program for Innovative Talent (grant no. BX20200398); the China Postdoctoral Science Foundation (grant no. 2020M672995); Guangdong Basic and Applied Basic Research Foundation (grant no. 2020A1515110170); Characteristic Innovation Research Project of University Teachers (grant no. 2020SWYY07); the National Ten Thousand Plan-Young Top Talents of China; Characteristic Innovation Research Project of University Teachers (grant no. 2020SWYY07); the Research Team of Precise Diagnosis and Treatment for Gynecologic Malignant Tumors (grant no. SZSM201812041); the Foundation of Health Commission of Hubei Province of China (grant no. WJ2019Q008 to X.T.); the Foundation of Wuhan Municipal Health Commission (grant no. WX19M02 to X.T.); Hubei Provincial Science and Technology Plan Project Youth Fund Project (2019CFB292).
Author contributions
Conceptualization, Z.C., H.L., H.Z., and Zhaoyue H.; methodology, Z.C., W.F., J.L., and Y.C.; investigation, Z.J., L.L., L.C., S.Z., Zheying H., W.X., J.M., and H.X.; visualization, Z.C.; funding acquisition, Zheng H., X.T., B.L., and R.T.; project administration, B.C.D., K.S., I.I.H., P.R.D., and Zheng H.; supervision, Zheng H., X.T., and H.W.; writing – original draft, Z.C. and Zhaoyue H.; writing – review & editing, Zheng H. and X.T.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtn.2021.08.008.
Contributor Information
Haiyan Weng, Email: Whaiyan1166@163.com.
Xun Tian, Email: tianxun@zxhospital.com.
Zheng Hu, Email: huzheng1998@163.com.
Supplemental information
References
- 1.Gupta R.M., Musunuru K. Expanding the genetic editing tool kit: ZFNs, TALENs, and CRISPR-Cas9. J. Clin. Invest. 2014;124:4154–4161. doi: 10.1172/JCI72992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhan T., Rindtorff N., Betge J., Ebert M.P., Boutros M. CRISPR/Cas9 for cancer research and therapy. Semin. Cancer Biol. 2019;55:106–119. doi: 10.1016/j.semcancer.2018.04.001. [DOI] [PubMed] [Google Scholar]
- 3.Provasi E., Genovese P., Lombardo A., Magnani Z., Liu P.Q., Reik A., Chu V., Paschon D.E., Zhang L., Kuball J., et al. Editing T cell specificity towards leukemia by zinc finger nucleases and lentiviral gene transfer. Nat. Med. 2012;18:807–815. doi: 10.1038/nm.2700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nyquist M.D., Li Y., Hwang T.H., Manlove L.S., Vessella R.L., Silverstein K.A., Voytas D.F., Dehm S.M. TALEN-engineered AR gene rearrangements reveal endocrine uncoupling of androgen receptor in prostate cancer. Proc. Natl. Acad. Sci. USA. 2013;110:17492–17497. doi: 10.1073/pnas.1308587110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Perez E.E., Wang J., Miller J.C., Jouvenot Y., Kim K.A., Liu O., Wang N., Lee G., Bartsevich V.V., Lee Y.L., et al. Establishment of HIV-1 resistance in CD4+ T cells by genome editing using zinc-finger nucleases. Nat. Biotechnol. 2008;26:808–816. doi: 10.1038/nbt1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang J., Quake S.R. RNA-guided endonuclease provides a therapeutic strategy to cure latent herpesviridae infection. Proc. Natl. Acad. Sci. USA. 2014;111:13157–13162. doi: 10.1073/pnas.1410785111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hu Z., Ding W., Zhu D., Yu L., Jiang X., Wang X., Zhang C., Wang L., Ji T., Liu D., et al. TALEN-mediated targeting of HPV oncogenes ameliorates HPV-related cervical malignancy. J. Clin. Invest. 2015;125:425–436. doi: 10.1172/JCI78206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dever D.P., Bak R.O., Reinisch A., Camarena J., Washington G., Nicolas C.E., Pavel-Dinu M., Saxena N., Wilkens A.B., Mantri S., et al. CRISPR/Cas9 β-globin gene targeting in human haematopoietic stem cells. Nature. 2016;539:384–389. doi: 10.1038/nature20134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tebas P., Stein D., Tang W.W., Frank I., Wang S.Q., Lee G., Spratt S.K., Surosky R.T., Giedlin M.A., Nichol G., et al. Gene editing of CCR5 in autologous CD4 T cells of persons infected with HIV. N. Engl. J. Med. 2014;370:901–910. doi: 10.1056/NEJMoa1300662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Qasim W., Amrolia P.J., Samarasinghe S., Ghorashian S., Zhan H., Stafford S., Butler K., Ahsan G., Gilmour K., Adams S., et al. First clinical application of TALEN engineered universal CAR19 T cells in B-ALL. Blood. 2015;126:2046. [Google Scholar]
- 11.Gasiunas G., Young J.K., Karvelis T., Kazlauskas D., Urbaitis T., Jasnauskaite M., Grusyte M.M., Paulraj S., Wang P.H., Hou Z., et al. A catalogue of biochemically diverse CRISPR-Cas9 orthologs. Nat. Commun. 2020;11:5512. doi: 10.1038/s41467-020-19344-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shmakov S., Abudayyeh O.O., Makarova K.S., Wolf Y.I., Gootenberg J.S., Semenova E., Minakhin L., Joung J., Konermann S., Severinov K., et al. Discovery and functional characterization of diverse class 2 CRISPR-Cas systems. Mol. Cell. 2015;60:385–397. doi: 10.1016/j.molcel.2015.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gabriel R., Lombardo A., Arens A., Miller J.C., Genovese P., Kaeppel C., Nowrouzi A., Bartholomae C.C., Wang J., Friedman G., et al. An unbiased genome-wide analysis of zinc-finger nuclease specificity. Nat. Biotechnol. 2011;29:816–823. doi: 10.1038/nbt.1948. [DOI] [PubMed] [Google Scholar]
- 14.Pattanayak V., Ramirez C.L., Joung J.K., Liu D.R. Revealing off-target cleavage specificities of zinc-finger nucleases by in vitro selection. Nat. Methods. 2011;8:765–770. doi: 10.1038/nmeth.1670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sander J.D., Ramirez C.L., Linder S.J., Pattanayak V., Shoresh N., Ku M., Foden J.A., Reyon D., Bernstein B.E., Liu D.R., Joung J.K. In silico abstraction of zinc finger nuclease cleavage profiles reveals an expanded landscape of off-target sites. Nucleic Acids Res. 2013;41:e181. doi: 10.1093/nar/gkt716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Guilinger J.P., Pattanayak V., Reyon D., Tsai S.Q., Sander J.D., Joung J.K., Liu D.R. Broad specificity profiling of TALENs results in engineered nucleases with improved DNA-cleavage specificity. Nat. Methods. 2014;11:429–435. doi: 10.1038/nmeth.2845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Juillerat A., Dubois G., Valton J., Thomas S., Stella S., Maréchal A., Langevin S., Benomari N., Bertonati C., Silva G.H., et al. Comprehensive analysis of the specificity of transcription activator-like effector nucleases. Nucleic Acids Res. 2014;42:5390–5402. doi: 10.1093/nar/gku155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pattanayak V., Lin S., Guilinger J.P., Ma E., Doudna J.A., Liu D.R. High-throughput profiling of off-target DNA cleavage reveals RNA-programmed Cas9 nuclease specificity. Nat. Biotechnol. 2013;31:839–843. doi: 10.1038/nbt.2673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Osborn M.J., Webber B.R., Knipping F., Lonetree C.L., Tennis N., DeFeo A.P., McElroy A.N., Starker C.G., Lee C., Merkel S., et al. Evaluation of TCR gene editing achieved by TALENs, CRISPR/Cas9, and megaTAL nucleases. Mol. Ther. 2016;24:570–581. doi: 10.1038/mt.2015.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nerys-Junior A., Braga-Dias L.P., Pezzuto P., Cotta-de-Almeida V., Tanuri A. Comparison of the editing patterns and editing efficiencies of TALEN and CRISPR-Cas9 when targeting the human CCR5 gene. Genet. Mol. Biol. 2018;41:167–179. doi: 10.1590/1678-4685-GMB-2017-0065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.He Z., Proudfoot C., Whitelaw C.B., Lillico S.G. Comparison of CRISPR/Cas9 and TALENs on editing an integrated EGFP gene in the genome of HEK293FT cells. Springerplus. 2016;5:814. doi: 10.1186/s40064-016-2536-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang J., Liu J., Yang W., Cui M., Dai B., Dong Y., Yang J., Zhang X., Liu D., Liang H., Cang M. Comparison of gene editing efficiencies of CRISPR/Cas9 and TALEN for generation of MSTN knock-out cashmere goats. Theriogenology. 2019;132:1–11. doi: 10.1016/j.theriogenology.2019.03.029. [DOI] [PubMed] [Google Scholar]
- 23.Smith C., Gore A., Yan W., Abalde-Atristain L., Li Z., He C., Wang Y., Brodsky R.A., Zhang K., Cheng L., Ye Z. Whole-genome sequencing analysis reveals high specificity of CRISPR/Cas9 and TALEN-based genome editing in human iPSCs. Cell Stem Cell. 2014;15:12–13. doi: 10.1016/j.stem.2014.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ding W., Hu Z., Zhu D., Jiang X., Yu L., Wang X., Zhang C., Wang L., Ji T., Li K., et al. Zinc finger nucleases targeting the human papillomavirus E7 oncogene induce E7 disruption and a transformed phenotype in HPV16/18-positive cervical cancer cells. Clin. Cancer Res. 2014;20:6495–6503. doi: 10.1158/1078-0432.CCR-14-0250. [DOI] [PubMed] [Google Scholar]
- 25.Gao X., Jin Z., Tan X., Zhang C., Zou C., Zhang W., Ding J., Das B.C., Severinov K., Hitzeroth I.I., et al. Hyperbranched poly(β-amino ester) based polyplex nanopaticles for delivery of CRISPR/Cas9 system and treatment of HPV infection associated cervical cancer. J. Control. Release. 2020;321:654–668. doi: 10.1016/j.jconrel.2020.02.045. [DOI] [PubMed] [Google Scholar]
- 26.Niu G., Jin Z., Zhang C., He D., Gao X., Zou C., Zhang W., Ding J., Das B.C., Severinov K., et al. An effective vaginal gel to deliver CRISPR/Cas9 system encapsulated in poly (β-amino ester) nanoparticles for vaginal gene therapy. EBioMedicine. 2020;58:102897. doi: 10.1016/j.ebiom.2020.102897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tsai S.Q., Zheng Z., Nguyen N.T., Liebers M., Topkar V.V., Thapar V., Wyvekens N., Khayter C., Iafrate A.J., Le L.P., et al. GUIDE-seq enables genome-wide profiling of off-target cleavage by CRISPR-Cas nucleases. Nat. Biotechnol. 2015;33:187–197. doi: 10.1038/nbt.3117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fine E.J., Cradick T.J., Zhao C.L., Lin Y., Bao G. An online bioinformatics tool predicts zinc finger and TALE nuclease off-target cleavage. Nucleic Acids Res. 2014;42:e42. doi: 10.1093/nar/gkt1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lamb B.M., Mercer A.C., Barbas C.F., 3rd Directed evolution of the TALE N-terminal domain for recognition of all 5′ bases. Nucleic Acids Res. 2013;41:9779–9785. doi: 10.1093/nar/gkt754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cong L., Zhou R., Kuo Y.C., Cunniff M., Zhang F. Comprehensive interrogation of natural TALE DNA-binding modules and transcriptional repressor domains. Nat. Commun. 2012;3:968. doi: 10.1038/ncomms1962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Doyle E.L., Booher N.J., Standage D.S., Voytas D.F., Brendel V.P., Vandyk J.K., Bogdanove A.J. TAL Effector-Nucleotide Targeter (TALE-NT) 2.0: tools for TAL effector design and target prediction. Nucleic Acids Res. 2012;40:W117–W122. doi: 10.1093/nar/gks608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sander J.D., Maeder M.L., Reyon D., Voytas D.F., Joung J.K., Dobbs D. ZiFiT (Zinc Finger Targeter): An updated zinc finger engineering tool. Nucleic Acids Res. 2010;38 doi: 10.1093/nar/gkq319. W462–S468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Doench J.G., Fusi N., Sullender M., Hegde M., Vaimberg E.W., Donovan K.F., Smith I., Tothova Z., Wilen C., Orchard R., et al. Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat. Biotechnol. 2016;34:184–191. doi: 10.1038/nbt.3437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lin J., Wong K.C. Off-target predictions in CRISPR-Cas9 gene editing using deep learning. Bioinformatics. 2018;34:i656–i663. doi: 10.1093/bioinformatics/bty554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Javed M.R., Sadaf M., Ahmed T., Jamil A., Nawaz M., Abbas H., Ijaz A. CRISPR-Cas system: History and prospects as a genome editing tool in microorganisms. Curr. Microbiol. 2018;75:1675–1683. doi: 10.1007/s00284-018-1547-4. [DOI] [PubMed] [Google Scholar]
- 36.Park J., Bae S., Kim J.S. Cas-Designer: A web-based tool for choice of CRISPR-Cas9 target sites. Bioinformatics. 2015;31:4014–4016. doi: 10.1093/bioinformatics/btv537. [DOI] [PubMed] [Google Scholar]
- 37.Mussolino C., Morbitzer R., Lütge F., Dannemann N., Lahaye T., Cathomen T. A novel TALE nuclease scaffold enables high genome editing activity in combination with low toxicity. Nucleic Acids Res. 2011;39:9283–9293. doi: 10.1093/nar/gkr597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Christian M.L., Demorest Z.L., Starker C.G., Osborn M.J., Nyquist M.D., Zhang Y., Carlson D.F., Bradley P., Bogdanove A.J., Voytas D.F. Targeting G with TAL effectors: A comparison of activities of TALENs constructed with NN and NK repeat variable di-residues. PLoS ONE. 2012;7:e45383. doi: 10.1371/journal.pone.0045383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Saunier M., Monnier-Benoit S., Mauny F., Dalstein V., Briolat J., Riethmuller D., Kantelip B., Schwarz E., Mougin C., Prétet J.L. Analysis of human papillomavirus type 16 (HPV16) DNA load and physical state for identification of HPV16-infected women with high-grade lesions or cervical carcinoma. J. Clin. Microbiol. 2008;46:3678–3685. doi: 10.1128/JCM.01212-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chen Y., Jiang H., Wang T., He D., Tian R., Cui Z., Tian X., Gao Q., Ma X., Yang J., et al. In vitro and in vivo growth inhibition of human cervical cancer cells via human papillomavirus E6/E7 mRNAs’ cleavage by CRISPR/Cas13a system. Antiviral Res. 2020;178:104794. doi: 10.1016/j.antiviral.2020.104794. [DOI] [PubMed] [Google Scholar]
- 41.Tsai S.Q., Topkar V.V., Joung J.K., Aryee M.J. Open-source guideseq software for analysis of GUIDE-seq data. Nat. Biotechnol. 2016;34:483. doi: 10.1038/nbt.3534. [DOI] [PubMed] [Google Scholar]
- 42.Honma M., Sakuraba M., Koizumi T., Takashima Y., Sakamoto H., Hayashi M. Non-homologous end-joining for repairing I-SceI-induced DNA double strand breaks in human cells. DNA Repair (Amst.) 2007;6:781–788. doi: 10.1016/j.dnarep.2007.01.004. [DOI] [PubMed] [Google Scholar]
- 43.Xiao A., Wu Y., Yang Z., Hu Y., Wang W., Zhang Y., Kong L., Gao G., Zhu Z., Lin S., Zhang B. EENdb: A database and knowledge base of ZFNs and TALENs for endonuclease engineering. Nucleic Acids Res. 2013;41:D415–D422. doi: 10.1093/nar/gks1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Boyle E.A., O’Roak B.J., Martin B.K., Kumar A., Shendure J. MIPgen: Optimized modeling and design of molecular inversion probes for targeted resequencing. Bioinformatics. 2014;30:2670–2672. doi: 10.1093/bioinformatics/btu353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chen S., Zhou Y., Chen Y., Gu J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics. 2018;34:i884–i890. doi: 10.1093/bioinformatics/bty560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Magoč T., Salzberg S.L. FLASH: Fast length adjustment of short reads to improve genome assemblies. Bioinformatics. 2011;27:2957–2963. doi: 10.1093/bioinformatics/btr507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li H., Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.You Q., Zhong Z., Ren Q., Hassan F., Zhang Y., Zhang T. CRISPRMatch: An automatic calculation and visualization tool for high-throughput CRISPR genome-editing data analysis. Int. J. Biol. Sci. 2018;14:858–862. doi: 10.7150/ijbs.24581. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.