

Abstract
Peptide identification by liquid chromatography-mass spectrometry (LC-MS) requires retention and elution of peptides from the LC column. Although medium and hydrophobic peptides are readily retained by the C18 columns that are commonly used in proteomics, short and hydrophilic peptides are not retained nor measured by MS due to their elution in the void volume after sample injection. These non-retained peptides can possess important post-translational modifications, such as glycosylation or phosphorylation. We describe a total retention LC-MS method that employs a reverse phase C18 column and porous graphitic carbon (PGC) column to retain both hydrophobic and hydrophilic peptides for LC-MS analysis. Our setup uses a single valve with a trapping column and two LC pumps run at low microliter/minute flow rates to deliver separate gradients to parallel capillary C18 and PGC columns. Our capillary LC system balances the need for high sensitivity with ease of implementation compared to other 2D LC systems that use nanocolumns with multiple trapping valves. We demonstrate the utility of the method identifying hydrophilic peptides that went undetected when only a C18 nanocolumn was used. These missed hydrophilic peptides include tripeptides and N-glycosylated species.
INTRODUCTION
Liquid chromatography-mass spectrometry (LC-MS) is widely used to identify proteins and characterize their post-translational modifications (PTMs).1–3 Typically, a protein or a mixture of proteins is digested with an enzyme like trypsin, and the resulting mixture of peptides is analyzed by LC-MS using a reverse phase (RP) C18 column. The peptides eluting from the LC are detected and fragmented by the MS, creating peptide mass fingerprint data that can be readily searched by several commonly used algorithms.4 Ideally, the amino acid-sequence coverage from the LC-MS analysis would be 100% for each protein. However, this is a challenging goal for samples of limited quantities run on nano or capillary RP columns because short peptides and peptides that contain several hydrophilic sites and are not well retained, elute with the sample matrix or void volume of the injection, and are not detected.5 These missed peptides are not required for protein identification because large numbers of other peptides are retained on the RP column, measured, and used for protein identification. However, these short and hydrophilic peptides that are not retained by an RP column often contain serine and threonine residues where phosphorylation or O-glycosylation may occur that only increases hydrophilicity.6 Serine and threonine also accompany asparagine as the site for N-glycosylation (e.g. the NXS/T motif).7 These short, unretained peptides will go undetected, and so will any phosphorylation or glycosylation PTMs associated with these peptides.
Early attempts to increase the retention of hydrophilic peptides on an RP column focused on the use of ion-pairing reagents such as trifluoroacetic acid (TFA) or heptafluorobutyric acid (HFBA).8–10 However, TFA and HFBA greatly reduce the ESI ionization efficiency and, therefore, the sensitivity of the MS analysis.11–12 Other approaches include chemically modifying the peptides with a non-polar group13 or use of a different enzyme from trypsin to make the hydrophilic amino acid region created by trypsin into a larger hydrophobic region.14 These approaches increase the amount of sample treatment and are not always sufficient to account for all the hydrophilic peptides.
Hydrophilic interaction chromatography (HILIC) has proven to be a good alternative to RP for analyzing short and hydrophilic peptides.15–16 HILIC columns have been shown to be particularly effective at analyzing glycosylated peptides.17 Although the HILIC mobile phase is ESI-MS compatible, the loading conditions require a high concentration of organic solvent for which many hydrophobic peptides have limited solubility. Therefore, to analyze the full complement of peptides, samples are often injected on separate RP and HILIC columns.18
A porous graphitic carbon (PGC) column uses similar mobile phases and gradients that are used for RP and also has the ability to retain hydrophilic peptides and glycosylated peptides, making the combination of RP-PGC easier to pair in a 2D LC system for complete proteome analysis of both hydrophobic and hydrophilic peptides.19–21 Lewandrowski and Sickmann described a 2D LC-MS system that simultaneously uses a C18 column, a PGC column, two trapping columns (C18 and PGC), two nano-gradient LC pumps, a loading pump, two ten-port valves, and a six-port valve.22 This setup has the benefit of retaining and analyzing both hydrophilic and hydrophobic species in a single analysis and was demonstrated for retention and identification of short and glycosylated peptides. Since then, several 2D and 3D LC-MS systems have been described incorporating a PGC column for proteomics analysis that includes measuring hydrophilic peptides. However, the systems used are also complex. For example, Lam et al. reported a 2D RP-RP-PGC setup for analyzing complex samples, but the system includes multiple switching valves to manage the high pH-RP, low pH-RP and PGC gradients.23 Zhao et al. described both 2D RP-PGC24 and 3D RP-PGC-HILIC25 systems for shotgun proteomics analysis that includes glycopeptide analysis. Although these systems demonstrate the ability to identify glycopeptides, they are complex to set up and operate. Stavenhagen et al. used pronase instead of trypsin to deliberately produce short peptides for glycopeptide analysis and designed an RP-PGC LC-MS system that would analyze the resulting peptides.26 Although their LC-MS system was simpler than other reported systems, it still requires multiple trapping valves, making it a challenge to configure and employ.
We believe the combination of C18 and PGC columns has great merit as demonstrated by the examples described above, but we feel a more simplified setup is warranted for the characterization of a protein PTMs from single sample of limited quantity. We report here a simplified, 2D C18-PGC column system that uses standard LC equipment to achieve the retention of both hydrophilic and hydrophobic peptides. We call our system “total retention LC-MS.” We demonstrate the utility of our system using two different human blood coagulation proteins. The first is tissue factor (TF), a small protein (approximately 30 kDa) that we have reported on previously regarding the identification and importance of its N-glycosylation.27–28 The second is factor Va light chain (FVaLC), a much larger protein (approximately 75 kDa) whose PTMs have not been fully characterized.
EXPERIMENTAL SECTION
Materials.
Water, acetonitrile, and trifluoroacetic acid were purchased from Fisher Scientific (Pittsburgh, PA), formic acid from EMD chemicals (Gibbstown, NJ), and modified trypsin and ProteaseMAX from Promega (Madison, WI). A Peptide CapTrap (0.5-mm ID × 2-mm that according to the manufacture produces C8-like retention and referred to hereafter as a C8 trap) and the 0.2-mm ID × 50-mm C18 AQ (3 μm) column were purchased from Bruker-Michrom (Auburn, CA). The PGC column was a Hypercarb® 0.18-mm ID × 100 mm (5 μm) from ThermoFisher (West Palm Beach, FL). LC-MS instrumentation included a Shimadzu Prominence HPLC (Columbia, MD), a ThermoFisher Surveyor HPLC pump and a ThermoFisher LTQ MS.
Sample preparation.
Human placenta-derived tissue factor (pTF) was purified from Thromborel S®, a commercial thromboplastin, by a procedure previously described.29 Vials of Thromborel S were resuspended in tris-buffered saline (pH = 7.4) containing 0.2% Triton X-100 and affinity purified using a sepharose-coupled anti-TF-5 mAb. Proteins were quantified by fluorescence-linked immunoassay by capture on anti-TF-5 mAb-coupled beads, probed with biotinylated anti-TF-48 mAb and detected using R-PE streptavidin.
Human FVa was isolated from platelet-poor plasma (10 mL) that was re-calcified to 50-mM CaCl2 and treated with thrombin (75 nM) for 10 min at 37 °C. The resulting fibrin clot was removed from the plasma by centrifugation (1,300× g, 25 °C, 10 min) and/or adherence to a 14.5-cm wooden applicator stick. Hirudin (112 nM), benzamidine (20 mM), soybean trypsin inhibitor (20 μg/mL), and AEBSF (2 μM) were added, followed by 1 mL of a sepharose-coupled anti-factor V monoclonal antibody resin slurry. The sample was rocked gently for two hours at 4 °C, at which point the resin was pelleted by centrifugation (180× g, 25 °C, 5 min), the supernatant was removed, and the resin was batch washed with repeated rounds of centrifugation and re-suspension in 1-mL low salt wash buffer (20 mM Tris pH 7.4, 150 mM NaCl, 10 mM CaCl2, 1 mM benzamidine) until the A280 of the wash solution was <0.1. The washing procedure was repeated with a mid-salt wash buffer (20 mM Tris pH 7.4, 350 mM NaCl, 10 mM CaCl2, 1-mM benzamidine). To elute the sample, 200 μL 10% SDS was added to the resin, the resulting slurry was incubated for 3 min at 25 °C, pelleted by centrifugation, and the supernatant recovered.30
The purified pTF and FVaLC were run separately on a 10% polyacrylamide SDS-PAGE gel and stained with Coomassie blue. The bands were excised, reduced, alkylated with iodoacetamide, and digested with trypsin in the presence of ProteaseMAX, according to the manufacturer’s instructions. The samples had final volumes of about 40 μL each, and 10 μL of each were injected onto the LC system.
LC-MS setup and operation.
Our setup requires two separate gradient pumps (Pump 1 and Pump 2), an autosampler, a six-port valve, a C8 trap, a C18 column, a PGC column, and the mass spectrometer. Pump 1 is operated with solvents A (97.5% water, 2.5% acetonitrile, 0.05% TFA, 0.1% formic acid, v/v) and B (5% water, 95% acetonitrile, 0.05% TFA, 0.1% formic acid). Pump 2 solvents are A (water with 0.1% formic acid) and B (acetonitrile with 0.1% formic acid). Both pump 1 and pump 2 are connected to flow-splitting tees that allowed them to run at 90 μL/min but deliver 4 μL/min to the autosampler, C8 trap, and PGC and C18 columns. The PGC column was kept at 60 °C.
The sample is first injected via the autosampler (pump 1 at 100% A) onto the C8 trap connected across the 6-port valve (Figure 1A, blue-colored flow path). Hydrophobic peptides are retained on the C8 trap, but unretained hydrophilic peptides pass through to the PGC column. After 10 min, the 6-port valve is switched to connect the C8 trap to the C18 column (Figure 1B, red-colored flow path) where the flow to the C8 trap now comes from pump 2 (set at 5% acetonitrile). A gradient is started to the C18 column (pump 2) that reaches 40% acetonitrile at 30 min. During this time, hydrophobic peptides elute from the C18 column. Pump 1 continues to provide flow to the PGC column and remains at 100% A until 25 min when a gradient increases pump 1 to 40% B at 45 min. This gradient causes the hydrophilic peptides to elute from the PGC column. At this point all peptides have eluted from both the C18 and the PGC columns. The columns are cleaned by increasing pump 1 to 100 % B at 45 min and ramping pump 2 to 80% B at 51 min. The pumps are returned to their starting conditions at 55 min, and the 6-port valve returned to the initial setting. Allowing for column equilibration, the total time for an analysis is 70 min. Approximately 50 sample injections can be carried out on this setup prior to extensive washing of the PGC column as previously described.31
Figure 1.
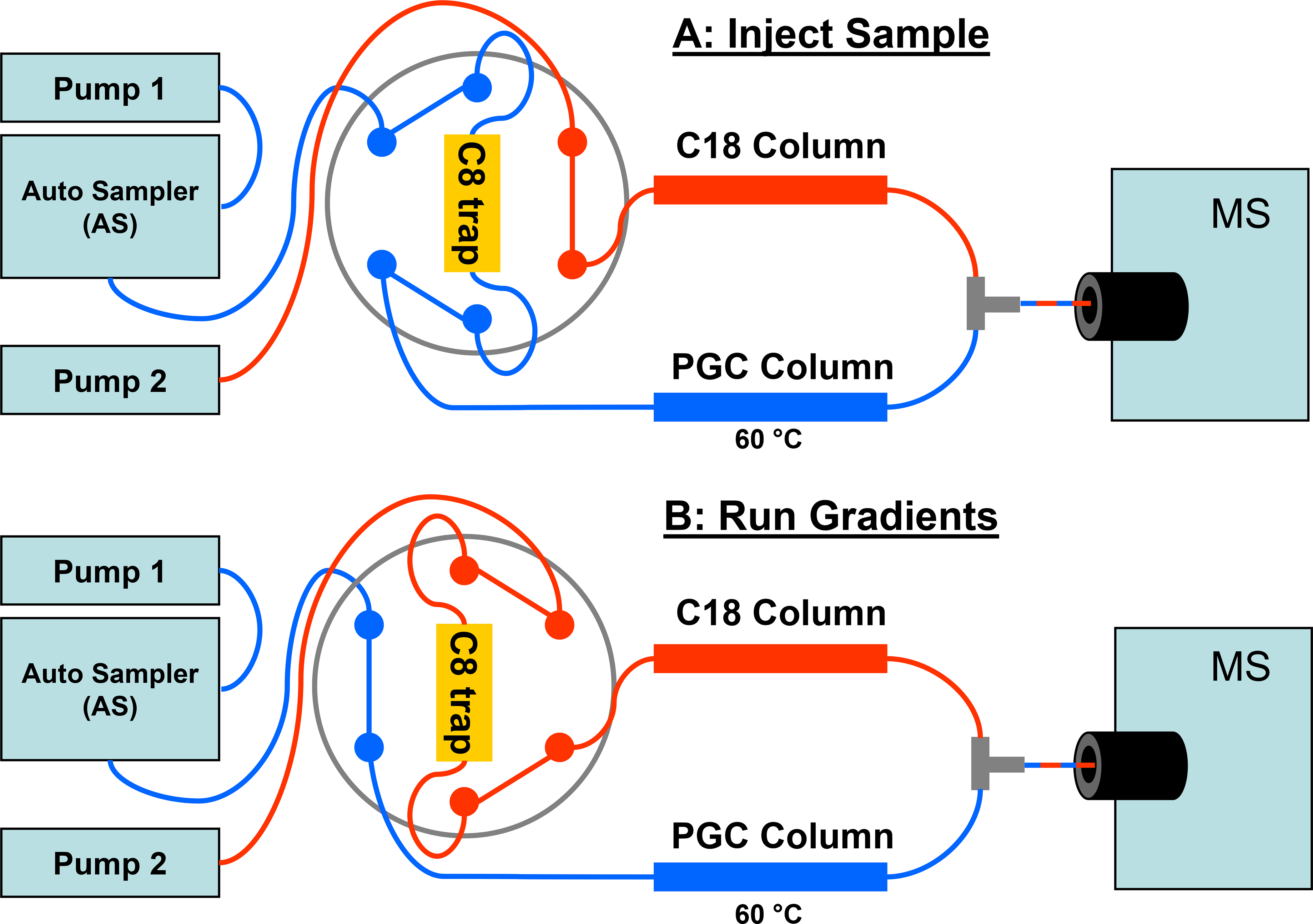
Schematic and operation of the total retention LC-MS system. (A) Shows the flow path when a sample is injected. Flow from pump 1 (in blue) carries the sample aliquot from the autosampler, to the C8 trap and onto the PGC column. During this period, flow from pump 2 (in red) goes only to the C18 column. (B) Shows the flow path during the analytical gradients for the C18 and PGC columns where the 6-port valve has been actuated to the alternate position. Flow from pump 2 (in red) now passes through the C8 trap and onto the C18 column. Flow from pump 1 (in blue) goes directly to the PGC column. The PGC and C18 columns are connected to a tee prior to the MS.
The flows from both columns are joined together by a tee that is connected to a Michrom Captive Spray source that is attached directly to a Thermo Scientific LTQ MS. The Captive Spray source was operated according to the manufacturer’s recommendations: 1.6kV for the capillary voltage, 170 °C for the capillary temperature, and an auxiliary gas flow of 8 units. The Thermo Scientific LTQ MS was operated in a data-dependent acquisition mode as previously described.27 In addition to these MS parameters, an additional scan segment was added where an MS3 scan was triggered based on the presence of an oxonium (sugar) loss detected in the MS/MS scan. Table S-1 shows the neutral loss mass list that we used to take into account the loss of a sugar unit for doubly and triply charges glycopeptides. The use of a sugar neutral loss to trigger MS3 spectra to identify glycopeptides has been described previously.32
Data analysis.
The LC-MS data were viewed in Qualbrowser and also searched using the SEQUEST algorithm in Proteome Discoverer and a reduced database containing only the amino acid sequences for TF and FVaLC. The Xcorr versus charge state (z) were set to 1.00 for z = 1, 2.00 for z = 2, and 3.00 for z = 3. The identified peptide MS/MS spectra were also manually inspected to eliminate the possibility of false positive identification. We also manually extracted the peptide precursor ions (for z = 1, 2, and 3) for peptide sequences that were not identified by SEQUEST and manually identified the b-type and y-type fragment ions. We identified potential glycopeptides by reviewing in Qualbrowser the MS/MS that had triggered an MS3 scan and identifying the glycan composition based on specific mass losses of different sugars as described previously.33–34 By subtracting the glycan molecular weight from the molecular weight of the glycopeptide, we determined the peptide molecular weight and matched this weight to the predicted peptide sequences from the tryptic peptides, taking into account possible miscleavages by trypsin.
RESULTS AND DISCUSSION
Human placenta-derived tissue factor (pTF).
Our goal was to identify all possible PTMs on pTF, an integral membrane protein that plays a key role in initiation of blood coagulation in vivo.35 We have previously reported achieving 71% sequence coverage of pTF using a combination of trypsin and GluC digestions by conventional nano-ESI LC-MS/MS using a C18 nanocolumn.27 Most of the missing peptides contained a serine, threonine, or both. Because this protein has been well characterized,27–28 it was chosen to test our system and potentially increase the amino acid sequence coverage by LC-MS.
Figure 2 shows the base peak plot of 2 pmol of pTF sample digested with trypsin and analyzed by our setup where full MS spectra were collected from 260–2000 m/z. Three groups of peaks elute and enter the MS between 1 and 45 min prior to the column wash and re-equilibration. Region 1 represents hydrophilic peptides that were not retained on the C8 trap and only weakly retained on the PGC column. These peptides eluted in the first 10 min under the initial starting conditions and continued for an additional 3 min. Region 2 shows peptides that were retained on the C8 trap, then eluted to the C18 column and separated on this column during the C18 gradient by/before 30 min (end of pump 2 gradient, Figure 2 red trace). Region 3 shows peptides that were not retained on the C8 trap, but were retained by the PGC column and separated by this column during the pump 1 gradient (from 25 to 45 min, Figure 2 blue trace). At 45 min, pump 1 to the PGC column went to 100% B, and pump 2 went to 80% B at 51 min to wash both columns before equilibrating for the next analysis. No identifiable peptides eluted off either column during this high acetonitrile wash period.
Figure 2.
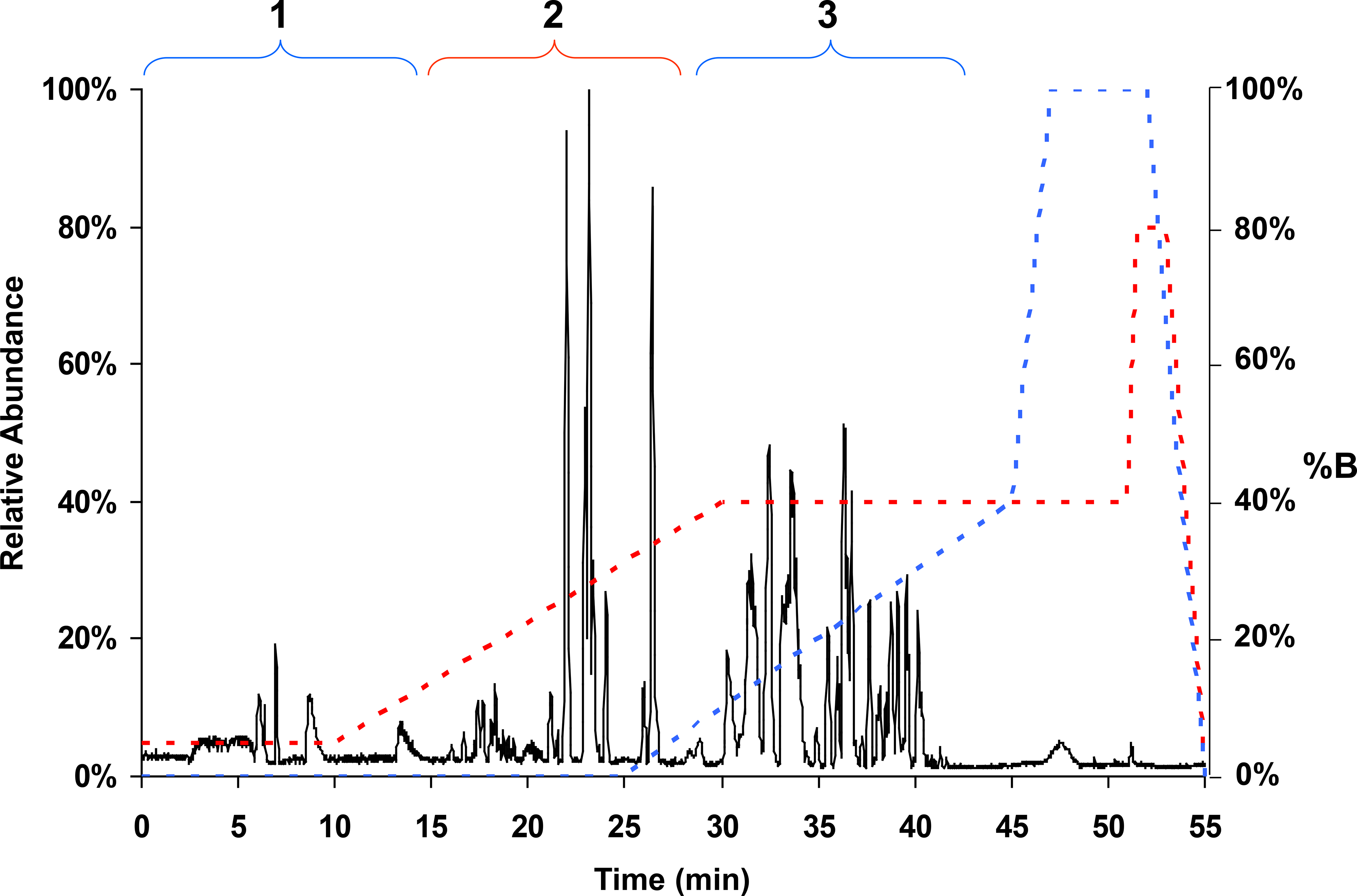
LC-MS base-peak chromatogram from a trypsin digestion of human placenta-derived tissue factor. The y-axis left side is relative ion intensity or abundance, and the y-axis right side shows %B for the gradient profiles for pump 1 (blue) and pump 2 (red). Region 1 (1 to 13 min) represents elution of hydrophilic peptides that were not retained on the C8 trap and are weakly retained on the PGC column. This region extends into the beginning of the pump 2 gradient. Region 2 shows peptides that were retained on the C8 trap and sequentially eluted onto and separated by the C18 column during the C18 gradient. Region 3 (25 to 45 min) shows hydrophilic peptides that were retained by the PGC column and eluted during the pump 1 gradient.
We show in Figure 3 the extracted ion chromatograms for some of the hydrophilic peptides from the trypsin digest of pTF. The peptides DVK, SSSSGK, and SSSSGKK were not detected when injected onto a C18 nanocolumn alone.27 They were not retained by the C8 trap, but were weakly retained by the PGC column and eluted between 6–7 min (Figures 3A–3C region 1). We also did not previously detect the hydrophilic peptides TLVR, STNFK, and DVFGK.27 On our setup, these peptides were also not retained on the C8 trap, but were well retained on the PGC column, eluting between 33 and 36 min (Figures 3D–3G region 3). Peptide QTYLAR appeared as two peaks at 14.4 min and at 39.6 min (Figure 3G). This peptide had some retention on the C8 trap, thereby splitting the peptide between the PGC and C18 columns. Having two LC-MS peaks for one peptide is not a disadvantage for a discovery analysis as long as one of the peaks has sufficient intensity to be chosen for MS/MS and yields good fragment ion data. If it became desirable to have this peptide completely retained on the PGC column, we would increase the amount of acetonitrile during the sample injection, thereby decreasing this peptide’s interaction with the C8 trap and moving 100% of QTYLAR onto the PGC column.
Figure 3.
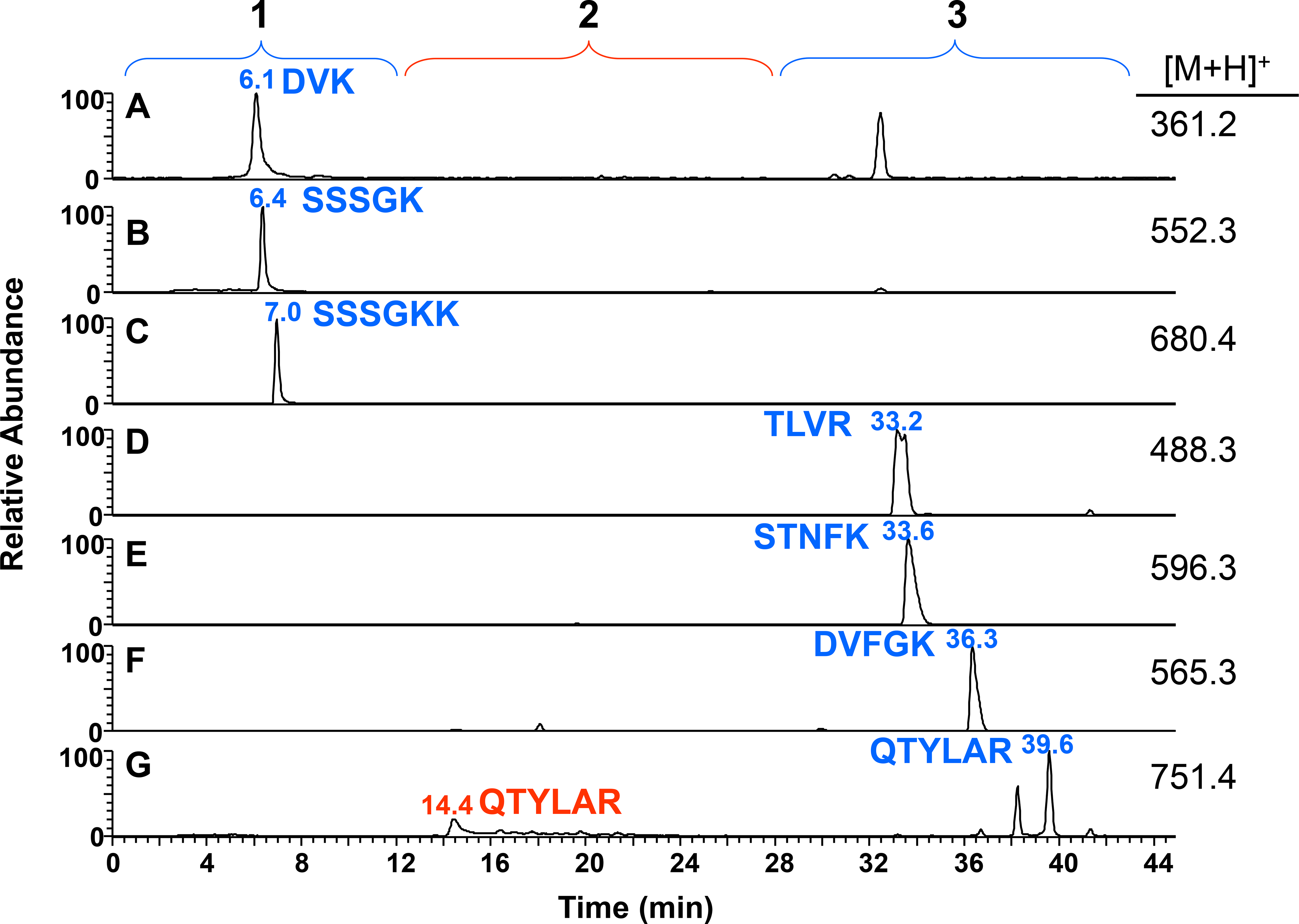
Extracted ion chromatograms of hydrophilic peptides from a trypsin digestion of 2 pmol of tissue factor. Peptides that were not retained on the C8 trap and only weakly retained on the PGC column are region 1 (blue): (A) DVK, (B) SSSSGK, and (C) SSSSGKK. Peptides that were well retained by the PGC column and eluted from the column during the PGC gradient are region 3 (blue): (D) TLVR, (E) STNFK, and (F) DVFGK. One peptide was split between the PGC and C18 columns due to some retention on the C8 trap, region 2 (red): (G) QTYLAR.
Table S-2 provides the theoretical hydrophobicity index of each peptide calculated using SSRCalc (http://hs2.proteome.ca/SSRCalc/SSRCalcQ.html).36 A negative or low positive index value indicates low hydrophobicity and limited retention on a C18 column. As expected, the hydrophobicity indices are very low (−5.5 to −2.4) for region 1 peptides that were not retained by the C8 trap, but weakly retained on the PGC column. The indices were higher (1.1 to 5.2) for region 3 peptides that were also not retained on the C8 trap, but were retained and eluted from the PGC column. Peptides identified as retained on the C8 trap and eluting from the C18 column (region 2) had intermediate to higher (5 to 20) indices.
We show in Figure 4 the amino acid sequence for pTF and use color to denote the peptides we identified. The amino acid sequence displayed in red represents peptides that were identified using the C18 column alone. Three of these peptides required manual identification due to N-glycosylation (sites Asn-11, Asn-124, and Asn-137), as previously described.27 Another very large peptide that consisted of 49 amino acids (Val-75 to Lys-122) was also identified manually. About 74% of the protein’s peptides were retained on the C8 trap and separated by the C18 column. We show in blue the hydrophilic peptides detected by our setup. These hydrophilic peptides accounted for a 14% increase in the pTF amino acid-sequence coverage (88% total coverage versus 74% from the C18 column alone). These peptides also have ten potential phosphorylation sites. If any of these sites were phosphorylated, the PTM would have gone unidentified using a conventional C18 column. We show in red and underline in blue the peptide sequence that was partially retained on the C8 trap and therefore identified on both C18 and PGC columns. The remaining unidentified amino acids from pTF are shown in black and represent the very hydrophobic trans-membrane domain that we did not expect to elute from the C18 column. Because peptides GEFR and CR are on either side of the trans-membrane domain, we believe their absence in our LCMS data represents them remaining bound to the trans-membrane domain and not cleaved by trypsin during the digestion step.
Figure 4.

Amino acid sequence coverage for tissue factor. Peptides that were identified and came from the C18 column are shown in red. Peptides identified from the PGC column are shown in blue. Peptides that came from both the C18 and PGC columns due to some retention on the C8 trap are shown in red with a blue underline. Peptides that were not identified are shown in black. The amino acids in red and blue represent 88% of tissue factor.
We found that heating the PGC column and using some TFA in the PGC gradient eliminated LC peak tailing that occurred when running the column at room temperature and using formic acid alone in the mobile phase. By teeing together the flows from the PGC column (0.05% TFA) and the C18 column (no TFA), we are able to decrease the TFA concentration by half at the ESI source as well as blend 40% acetonitrile coming from the C18 column during the elution of hydrophilic peptides from the PGC column during the PGC gradient. Diluting the TFA concentration and the additional acetonitrile added prior to MS analysis helped to decrease the negative impact TFA has on ESI.37 We observed that the signal intensities for peptides retained by the PGC column were higher when the PGC gradient was run after the C18 gradient, (compared to running the PGC gradient first when only 5% acetonitrile was coming from the C18 column). Hence, by teeing together the flows exiting both columns and running the PGC gradient after the C18 gradient, we were able to optimize the ionization of the hydrophilic peptides. Although no new PTMs were discovered for pTF using this system, the proof of concept demonstrated here gave us the confidence to apply this system to another protein derived from human factor Va.
Plasma-derived human factor Va light chain.
Although N-glycosylation has been identified on FVaLC after treatment with PNGase F,38 glycan compositions have not been defined, nor have all the potential N-glycosylation sites been identified based on the NXS/T motif using mass spectrometry until recently.39 One potential site that we have focused upon is Asn-1982 that would be contained in the short hydrophilic tryptic peptide GNSTR, a peptide that we had been unable to measure using a standard C18 column (data not shown).
Figure 5A shows the base peak plot of 2 pmol of trypsin-digested FVaLC injected onto our total retention LC-MS system where full MS spectra was collected between 400–2000 m/z. By reviewing the MS/MS and MS3, we found spectra that were typical of glycopeptides. We identified the glycan composition based on specific mass losses and were able to determine the peptide molecular weight for each group of glycopeptides. Figure 5B shows the extracted ion current for the full MS that represents the glycopeptide on Asn-2181 (m/z = 1356, where z = 2). The glycan attached to the asparagine consisted of the penta-saccharide core (GlcNAc2Man3),40 which we will refer to hereafter as the “core,” plus two N-acetyl hexosamines and two hexoses. The type of MS analysis we used is limited to defining glycan composition, and the glycan figure shown in Figure 5B is only meant to account for that composition. We did not carry out an analysis to determine the specific glycan structure. The Asn-2181 glycopeptide was retained by the C8 trap and eluted from the C18 column in region 2 of Figure 5B. The hydrophobicity index for the peptide sequence without the glycan from SSRCalc is 11.0.
Figure 5.
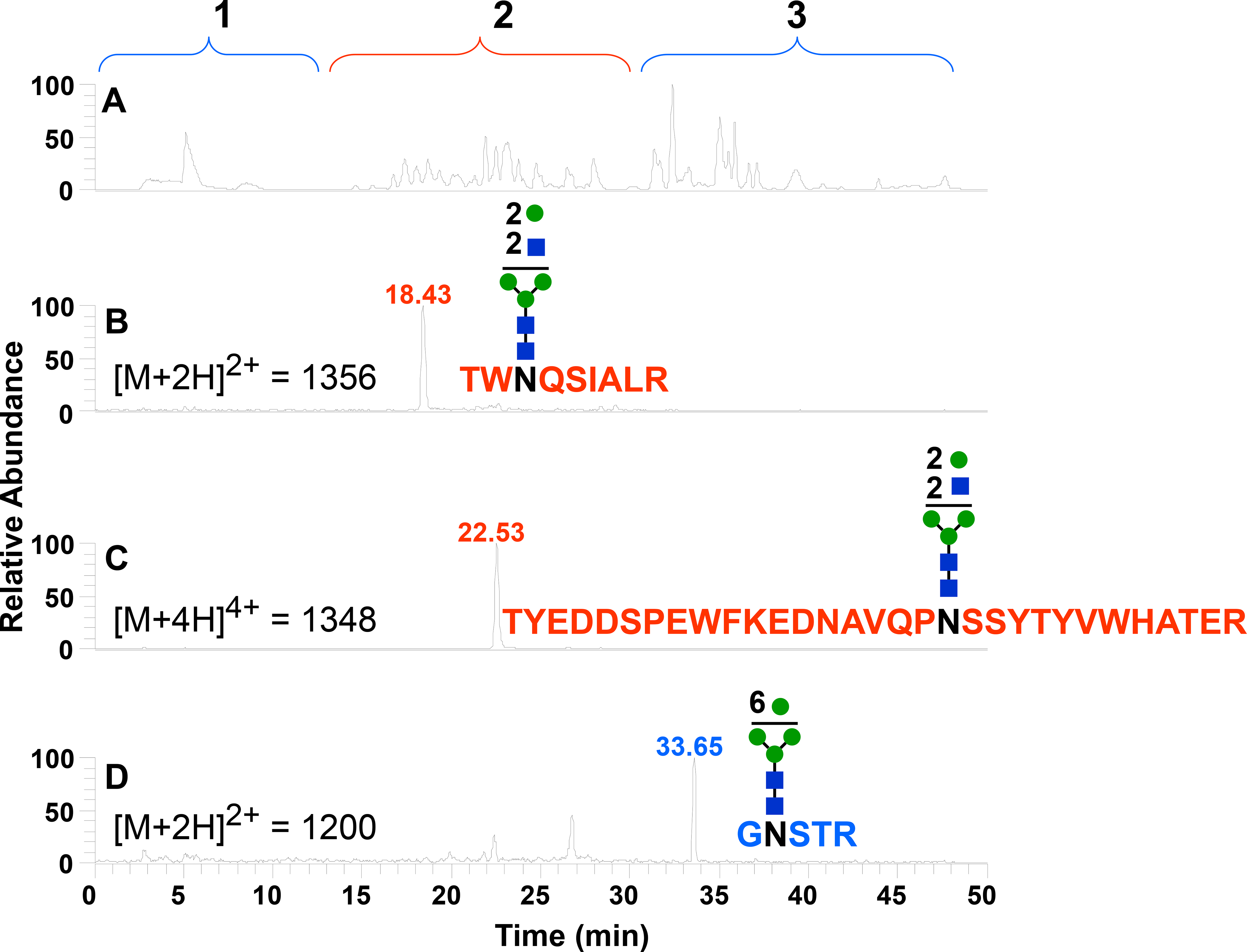
LC-MS chromatograms from a trypsin digestion of factor Va light chain. (A) Base peak plot. (B) Extracted ion chromatogram of TWNQSIALR glycosylated on Asn-2181. (C) Extracted ion chromatogram of TYEDDSPEWFKEDNAVQPNSSYTYVWHATER glycosylated on Asn-1675. (D) Extracted ion chromatogram of the hydrophilic GNSTR glycosylated on Asn-1982.
Figure 5C shows the extracted ion from the full MS scan for the glycopeptide on Asn-1675. This glycopeptide contains a miscleavage. Not surprisingly due to the size of the miscleaved peptide, the glycopeptide was also retained by the C8 trap and eluted from the C18 column in region 2 of the chromatogram. We identified the glycan composition to be the same as the glycopeptide described above: core plus two N-acetyl hexosamines and two hexoses. The hydrophobicity index for the peptide without the glycan from SSRCalc is 16.4.
Figure 5D shows the extracted ion chromatogram for the glycopeptide GNSTR (Asn-1982). GNSTR was not retained on the C8 trap, but was retained and eluted from the PGC column. The hydrophobicity index for this peptide sequence without glycan from SSRCalc is −4.3. This value is more comparable to indices for peptides that elute in region 1 (Table S-2). We are presuming that the glycan moiety increased retention on the PGC column for this peptide. The glycan composition shown in Figure 5D is the core plus six hexoses.
We summarize in Figure 6 the amino acid sequence for FVaLC found by using our system. Most of the amino acid sequence was detected from peptides that were retained by the C8 trap and eluted from the C18 column (Figure 6, red). Some of these peptides shown in red were weakly retained by the C8 trap and eluted from both columns during their respective gradients (Figure 6, red sequences with a blue underline). The tripeptide LAR was only weakly retained by the PGC column and eluted at 7.5 min (data not shown). The other hydrophilic peptides shown in blue in Figure 6 were well retained by the PGC column and eluted in region 3. Shown in black are the peptides that were not identified, which represent about 6% of the amino acid sequence. Overall, the PGC column allowed us to increase the protein amino acid sequence coverage by 16% versus using the C18 column alone, increasing the total amino acid sequence coverage for FVaLC to 94%.
Figure 6.

Amino acid sequence coverage for factor Va light chain. Amino acid position numbering is based on full length factor V. Peptides that were identified and came from the C18 column are shown in red. Peptides identified from the PGC column are shown in blue. Peptides that came from both the C18 and PGC columns due to some retention on the C8 trap are shown in red with a blue underline. Peptides that were not identified are shown in black. The amino acids in red and blue represent 94% of FVaLC.
CONCLUSION
Enzymatic digestion of proteins will produce a complex mixture of peptides. Some peptides will be hydrophilic and go unmeasured as they are barely or not at all retained on a C18 nanocolumn. We designed our system combining the complementary nature of the C18 and PGC phases to retain these hydrophilic peptides. The additional retention and separation provided by the PGC column for hydrophilic peptides allows the potential to discover PTMs that would otherwise be missed with a conventional LC-MS analysis using a C18 column. Hence, our system collects information on all the species in a complex mixture from a single sample preparation. As we have shown, the PGC column can be readily integrated into the LC-MS system with minimal changes. The mobile phases used with the PGC column are compatible with C18 and other reverse phase materials, making this combination a natural partnership. The only major change is a loss in throughput due to an increase in the analysis time by 25–50%. However, we believe this increase in time is a reasonable sacrifice for the increased ability to account for possible PTMs on short, hydrophilic peptides.
Supplementary Material
ACKNOWLEDGEMENTS
This work was supported by NIH grants P20 RR16462 and P01 HL46703. The authors thank Jolanta Krudysz-Amblo and Saulius Butenas for providing the purified human placenta-derived tissue factor and thank Mike Baynham, Dafydd Milton and Joanna Freeke from ThermoFisher Scientific for valuable information about the PGC column.
Footnotes
The authors declare no competing financial interest.
ASSOCIATED CONTENT
The Supporting Information is available free of charge on the ACS Publications website.
Two tables provided. The first table provides the m/z list of neutral losses used to select for site-specific glycoprofiling and glycan identification. The second table provides a list of hydrophobicity indices calculated for the peptides shown in Figure 3. (PDF)
REFERENCES
- (1).Yates JR; Ruse CI; Nakorchevsky A Proteomics by mass spectrometry: approaches, advances, and applications. Annu. Rev. Biomed. Eng. 2009, 11, 49–79. [DOI] [PubMed] [Google Scholar]
- (2).Zhou H; Watts JD; Aebersold R A systematic approach to the analysis of protein phosphorylation. Nat. Biotechnol. 2001, 19, 375–378. [DOI] [PubMed] [Google Scholar]
- (3).Carr SA; Annan RS; Huddleston MJ Mapping posttranslational modifications of proteins by MS-based selective detection: application to phosphoproteomics. Methods Enzymol. 2005, 405, 82–115. [DOI] [PubMed] [Google Scholar]
- (4).Hoopmann MR; Moritz RL Current algorithmic solutions for peptide-based proteomics data generation and identification. Curr. Opin. Biotechnol. 2013, 24, 31–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Guo DC; Mant CT; Taneja AK; Parker JM; Hodges RS Prediction of peptide retention times in reversed-phase high-performance liquid chromatography I. Determination of retention coefficients of amino acid residues of model synthetic peptides. J. Chromatogr. 1986, 359, 499–517. [Google Scholar]
- (6).Olsen J; Blagoev B; Gnad F; Macek B; Kumar C; Mortensen P; Mann M Global, in vivo, and site-specific phosphorylation dynamics in signaling networks. Cell 2006, 127, 635–648. [DOI] [PubMed] [Google Scholar]
- (7).Spiro R Protein glycosylation: nature, distribution, enzymatic formation, and disease implications of glycopeptide bonds. Glycobiology 2002, 12, 43R–56R. [DOI] [PubMed] [Google Scholar]
- (8).Pearson JD; McCroskey MC Perfluorinated acid alternatives to trifluoroacetic acid for reversed-phase high-performance liquid chromatography. J. Chromatogr. A 1996, 746, 277–281. [DOI] [PubMed] [Google Scholar]
- (9).Petritis K; Brussaux S; Guenu S; Elfakire C; Dreux M Ion-pair reversed-phase liquid chromatography-electrospray mass spectrometry for the analysis of underivatized small peptides. J. Chromatogr. A 2002, 957, 173–185. [DOI] [PubMed] [Google Scholar]
- (10).Kim J; Petritis K; Shen Y; Camp DG 2nd; Moore RJ; Smith RD Phosphopeptide elution times in reversed-phase liquid chromatography. J. Chromatogr. A 2007, 1172, 9–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Mirza UA; Chait BT Effects of anions on the positive ion electrospray ionization mass spectra of peptides and proteins. Anal. Chem. 1994, 66, 2898–2904. [DOI] [PubMed] [Google Scholar]
- (12).Eshraghi J; Chowdhury SK Factors affecting electrospray ionization of effluents containing trifluoroacetic acid for high-performance liquid chromatography/mass spectrometry. Anal. Chem. 1993, 65, 3528–3533. [DOI] [PubMed] [Google Scholar]
- (13).Previs MJ; VanBuren P; Begin KJ; Vigoreaux JO; LeWinter MM; Matthews DE Quantification of protein phosphorylation by liquid chromatography-mass spectrometry. Anal. Chem. 2008, 80, 5864–5872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Gatlin C; Eng J; Cross S; Detter J; Yates JR III Automated Identification of amino acid sequence variations in proteins by HPLC/microspray tandem mass siectrometry. Anal. Chem. 2000, 72, 757–763. [DOI] [PubMed] [Google Scholar]
- (15).Le Maux S; Nongonierma AB; FitzGerald RJ Improved short peptide identification using HILIC–MS/MS: Retention time prediction model based on the impact of amino acid position in the peptide sequence. Food Chem. 2015, 173, 847–854. [DOI] [PubMed] [Google Scholar]
- (16).Piovesana S; Cerrato A; Antonelli M; Benedetti B; Capriotti AL; Cavaliere C; Montone CM; Laganà A A clean-up strategy for identification of circulating endogenous short peptides in human plasma by zwitterionic hydrophilic liquid chromatography and untargeted peptidomics identification. J. Chromatogr. A 2020, 1613, 460699. [DOI] [PubMed] [Google Scholar]
- (17).Malerod H; Rogeberg M; Tanaka N; Greibrokk T; Lundanes E Large volume injection of aqueous peptide samples on a monolithic silica based zwitterionic-hydrophilic interaction liquid chromatography system for characterization of posttranslational modifications. J. Chromatogr. A 2013, 1317, 129–37. [DOI] [PubMed] [Google Scholar]
- (18).Piovesana S; Capriotti AL; Cerrato A; Crescenzi C; La Barbera G; Laganà A; Montone CM; Cavaliere C Graphitized carbon black enrichment and UHPLC-MS/MS allow to meet the challenge of small chain peptidomics in urine. Anal. Chem. 2019, 91, 11474–11481. [DOI] [PubMed] [Google Scholar]
- (19).Yamaki S; Isobe T; Okuyama T; Shinoda T High-performance liquid chromatography of peptides on a microspherical carbon column. J. Chromatogr. A 1996, 729, 143–153. [DOI] [PubMed] [Google Scholar]
- (20).An HJ; Peavy TR; Hedrick JL; Lebrilla CB Determination of N-glycosylation sites and site heterogeneity in glycoproteins. Anal. Chem. 2003, 75, 5628–5637. [DOI] [PubMed] [Google Scholar]
- (21).Alley WR Jr.; Mechref Y; Novotny MV Use of activated graphitized carbon chips for liquid chromatography/mass spectrometric and tandem mass spectrometric analysis of tryptic glycopeptides. Rapid Commun. Mass Spectrom. 2009, 23, 495–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Lewandrowski U; Sickmann A Online dual gradient reversed-phase/porous graphitized carbon nano-HPLC for proteomic applications. Anal. Chem. 2010, 82, 5391–5396. [DOI] [PubMed] [Google Scholar]
- (23).Lam MPY; Lau E; Siu SO; Ng DCM; Kong RPW; Chiu PCN; Yeung WSB; Lo C; Chu IK Online combination of reversed-phase/reversed-phase and porous graphitic carbon liquid chromatography for multicomponent separation of proteomics and glycoproteomics samples. Electrophoresis 2011, 32, 2930–2940. [DOI] [PubMed] [Google Scholar]
- (24).Zhao Y; Szeto SSW; Kong RPW; Law CH; Li GH; Quan Q; Zhang ZJ; Wang YQ; Chu IK Online two-dimensional porous graphitic carbon/reversed phase liquid chromatography platform applied to shotgun proteomics and glycoproteomics. Anal. Chem. 2014, 86, 12172–12179. [DOI] [PubMed] [Google Scholar]
- (25).Zhao Y; Law HCH; Zhang ZJ; Lam HC; Quan Q; Li GH; Chu IK Online coupling of hydrophilic interaction/strong cation exchange/reversed-phase liquid chromatography with porous graphitic carbon liquid chromatography for simultaneous proteomics and N-glycomics analysis. J. Chromatogr. A 2015, 1415, 57–66. [DOI] [PubMed] [Google Scholar]
- (26).Stavenhagen K; Plomp R; Wuhrer M Site-specific protein N- and O-glycosylation analysis by a C18-porous graphitized carbon–liquid chromatography-electrospray ionization mass spectrometry approach using pronase treated glycopeptides. Anal. Chem. 2015, 87, 11691–11699. [DOI] [PubMed] [Google Scholar]
- (27).Krudysz-Amblo J; Jennings ME II; Mann KG; Butenas S Carbohydrates and activity of natural and recombinant tissue factor. J. Biol. Chem. 2010, 285, 3371–3382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Krudysz-Amblo J; Jennings ME II; Matthews DE; Mann KG; Butenas S Differences in the fractional abundances of carbohydrates of natural and recombinant human tissue factor. Biochim. Biophys. Acta 2011, 1810, 398–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Parhami-Seren B; Butenas S; Krudysz-Amblo J; Mann KG Immunologic quantitation of tissue factors. J. Thromb. Haemost. 2006, 4, 1747–1755. [DOI] [PubMed] [Google Scholar]
- (30).Nesheim M; Katzmann J; Tracy PB; Mann KG Factor V. Meth. Enzymol. 1981, 80, 249–274. [DOI] [PubMed] [Google Scholar]
- (31).Bapiro TE; Richards FM; Jodrell DI Understanding the complexity of porous graphitic carbon (PGC) chromatography: Modulation of mobile-stationary phase interactions overcomes loss of retention and reduces variability. Anal. Chem. 2016, 88, 6190–6194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Go EP; Irungu J; Zhang Y; Dalpathado DS; Liao H; Sutherland LL; Alam M; Haynes BF; Desaire H Glycosylation site-specific analysis of HIV envelope proteins (JR-FL and CON-S) reveals major differences in glycosylation site occupancy, glycoform profiles, and antigenic epitopes’ accessibility. J. Proteome Res. 2008, 7, 1660–1674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Iacob R; Perdivara I; Przybylski M; Tomer K Mass spectrometric characterization of glycosylation of hepatitis C virus E2 envelope glycoprotein reveals extended microheterogeneity of N-glycans. J. Am. Soc. Mass Spectrom. 2008, 19, 428–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Wang Y; Wu S; Hancock W Monitoring of glycoprotein products in cell culture lysates using lectin affinity chromatography and capillary HPLC coupled to electrospray linear ion trap-Fourier transform mass spectrometry (LTQ/FTMS). Biotechnol. Prog. 2006, 22, 873–880. [DOI] [PubMed] [Google Scholar]
- (35).Mackman N Role of tissue factor in hemostasis and thrombosis. Blood Cells Mol. Dis. 2006, 36, 104–107. [DOI] [PubMed] [Google Scholar]
- (36).Krokhin OV Sequence-specific retention calculator. Algorithm for peptide retention prediction in ion-pair RP-HPLC: Application to 300- and 100-Å pore size C18 sorbents. Anal. Chem. 2006, 78, 7785–7795. [DOI] [PubMed] [Google Scholar]
- (37).Apffel A; Fischer S; Goldberg G; Goodley PC; Kuhlmann FE Enhanced sensitivity for peptide mapping with electrospray liquid chromatography-mass spectrometry in the presence of signal suppression due to trifluoroacetic acid-containing mobile phases. J. Chromatogr. A 1995, 712, 177–190. [DOI] [PubMed] [Google Scholar]
- (38).Liu T; Qian WJ; Gritsenko MA; Camp DG II; Monroe ME; Moore RJ; Smith RD Human plasma N-glycoproteome analysis by immunoaffinity subtraction, hydrazide chemistry, and mass spectrometry. J. Proteome Res. 2005, 4, 2070–2080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Ma C; Liu D; Li D; Zhang J; Xu XQ; Zhu H; Wan XF; Miao CH; Konkle BA; Onigman P; Xiao W; Li L Comprehensive N- and O-glycosylation mapping of human coagulation factor V. J. Thromb. Haemost. 2020, 18, 1884–1892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Kornfeld R; Kornfeld S Assembly of asparagine-linked oligosaccharides. Annu. Rev. Biochem. 1985, 54, 631–664. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.