

Abstract
This first part of a two-part review of pheochromocytoma and paragangliomas (PPGLs) addresses clinical presentation, diagnosis, management, treatment, and outcomes. In this first part, the epidemiology, prevalence, genetic etiology, clinical presentation, and biochemical and radiologic workup are discussed. In particular, recent advances in the genetics underlying PPGLs and the recommendation for genetic testing of all patients with PPGL are emphasized. Finally, the newer imaging methods for evaluating of PPGLs are discussed and highlighted.
Pheochromocytomas and paragangliomas (PPGLs) are rare neuroendocrine tumors of chromaffin tissue that may produce catecholamines.1 Pheochromocytomas are tumors that arise from chromaffin cells within the adrenal medulla, whereas paragangliomas (PGLs) arise from extra-adrenal chromaffin cells of the sympathetic or parasympathetic paravertebral ganglia within the chest, abdomen, and pelvis. Paragangliomas also may arise from the chromaffin cells of the parasympathetic ganglia of the head and neck.
Almost all pheochromocytomas and sympathetic extra-adrenal PGLs produce, store, metabolize, and secrete catecholamines (epinephrine, norepinephrine, dopamine) or their metabolites (metanephrines, normetanephrines). Approximately 80–85% of chromaffin cell tumors are pheochromocytomas (intra-adrenal), and 15–20% are PGLs (extra-adrenal).2–4
EPIDEMIOLOGY AND INCIDENCE
Approximately 75–80% of sympathetic PGLs arise in the abdomen and pelvis, typically in the paravertebral sympathetic ganglia at the junction of the inferior vena cava and left renal vein or organ of Zuckerkandl (aortic bifurcation near the origin of the inferior mesenteric artery). About 10% arise in the thorax, including mediastinal and pericardial locations.5–7 Approximately 5–10% of patients have multiple or bilateral tumors.3,8 Notably, PGLs also can arise from the parasympathetic ganglia located along the glossopharyngeal and vagal nerves in the neck and base of skull. Usually, PGLs arising from these parasympathetic nerves are not associated with catecholamine secretion.1,9
In the United States, the annual incidence of PPGLs is about 500 to 1600 cases per year,10 which likely is an underestimation given the relatively high prevalence (0.05–0.1%) of incidentally discovered PPGLs at autopsy.11,12 The prevalence of these rare tumors is estimated to be 1 to 2500 and 1 to 6500.10 Among adult patients with hypertension in outpatient clinics, the prevalence ranges from 0.1 to 0.6%,4,13,14 and among children with hypertension, the prevalence is approximately 1.7%.15
Pheochromocytomas comprise about 4–8% of all adrenal incidentalomas, and approximately 21.1–57.6% of all pheochromocytomas are discovered incidentally on imaging studies.16–22 Symptomatic patients typically present in their fourth or fifth decade of life, with a relatively equal male-to-female distribution.23 Most of these tumors are unifocal (90–95%) and benign. About 10% (range 2–50%) of all catecholamine-secreting tumors are malignant, but the likelihood of malignancy depends on the primary site (adrenal vs. extra-adrenal) and the presence of certain germline mutations (SDHB in particular).8,24,25
BRIEF REVIEW OF CATECHOLAMINE SYNTHESIS
Chromaffin cells synthesize norepinephrine and epinephrine from a tyrosine precursor. In brief, tyrosine is converted to DOPA, which is subsequently converted to dopamine. Dopamine then is converted to norepinephrine within storage vesicles. Norepinephrine leaks out of these vesicles and is converted in the adrenal gland to epinephrine by phenylethanolamine N-methyltransferase (PMNT) (Fig. 1). The metabolism of these catecholamines takes place within the same cells. Norepinephrine and epinephrine are O-methylated to normetanephrine and metanephrine within the same cells and slowly released into the bloodstream independently of sympathoadrenal activity. This independence provides the biologic underpinnings for the specificity of plasma and urinary metanephrines.26
FIG. 1.
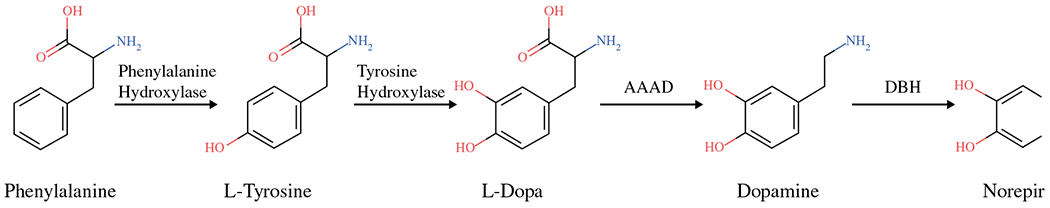
Chromaffin cells synthesize norepinephrine and epinephrine from phenylalanine, a tyrosine precursor. AAAD, L-aromatic amino acid decarboxylase; DBH, dopamine β-hydroxlase; L-DOPA, dihydroxyphenylalanine; PNMT, phenylethanolamine N-methyltransferase
PATHOPHYSIOLOGY
The systemic effects of PPGLs are due to the uncontrolled release of catecholamines leading to several physiologic changes and end-organ effects that can result in high cardiovascular morbidity and mortality, including arrhythmias, myocardial events, and stroke.27–29 Almost all pheochromocytomas and sympathetic extra-adrenal PGLs are biochemically active and secrete catecholamines (epinephrine, norepinephrine, dopamine) or their metabolites.9,10 Pheochromocytomas often secrete both norepinephrine and epinephrine, although the relative proportions may vary. Paragangliomas secrete norepinephrine exclusively because the PNMT enzyme necessary for epinephrine synthesis is found only in the adrenal medulla.
About half of patients present with paroxysmal signs and symptoms of catecholamine excess (e.g., headache, palpitations, sweating, flushing, fatigue, sustained or paroxysmal hypertension; see “Clinical Presentation” later).2,10 Patients with predominantly epinephrine-secreting tumors are more likely to have tachycardia and tachyarrhythmias in addition to hypertension.30 Exposure to prolonged or repeated norepinephrine release is associated with long periods of vasoconstriction and venous contraction, resulting in decreased circulating blood volume, which can lead to acute hypovolemia at the time of tumor removal and cessation of norepinephrine-induced vasoconstriction, highlighting the importance of preoperative alpha-blockade and volume expansion.
In addition, elevated catecholamine levels can cause increased glycogenolysis and inhibition of insulin release by islet cells, resulting in hyperglycemia.31–33 Elevated catecholamines also can lead to stress-induced cardiomyopathy (Takotsubo cardiomyopathy) with severe left ventricular dysfunction.28,34
Finally, although rarely seen today, pheochromocytoma crisis (multisystem organ failure, high fever, encephalopathy, and severe hypertension and/or hypotension) can progress to severe metabolic acidosis and death if not recognized and treated with urgent operative intervention after a period of stabilization with appropriate blockade and supportive care.35 However, clinical judgment for emergent surgery must be used for those whose conditions deteriorate despite maximal medical therapy or extenuating circumstances such as hemorrhage or rupture of a pheochromocytoma.35,36
CLINICAL PRESENTATION
Most commonly, patients with PPGLs present with overt clinical signs and symptoms of catecholamine excess. Persistent, sometimes worsening, hypertension is the most common clinical feature and can be paroxysmal in nature. The classically described triad of symptoms (episodic headaches, diaphoresis, and palpitations) is found in less than 25% of patients with pheochromocytoma.1,37,38 Other symptoms may include anxiety, chest and abdominal pain, nausea and vomiting, and psychiatric manifestations. The variation in clinical symptoms also may be related to differences in secretion of epinephrine and norepinephrine and their individual effects on β1- and β2-adrenergic receptors. Epinephrine-secreting pheochromocytomas more frequently result in episodic hypertension and symptoms of palpitations, syncope, anxiety, and hyperglycemia. In contrast, norepinephrine-secreting tumors are more continuous, with headaches, sweating, and hypertension, which are less paroxysmal in nature.1,38
Pheochromocytomas also occur in patients without elevated blood pressures. The true prevalence is unknown, although it is reported in published series to reach 55% of patients37,39 Normotensive (“asymptomatic” or “silent”) pheochromocytomas often occur in incidentally identified adrenal tumors. Therefore, the absence of hypertension should not preclude biochemical evaluation of patients with incidental adrenal nodules or clinical symptoms otherwise suggestive of a pheochromocytoma.1,39
The pathophysiology of sporadic, “normotensive” pheochromocytomas is unknown, although it is suggested that the cardiovascular system may become desensitized to circulating catecholamines. In contrast, it is not uncommon for patients with hereditary pheochromocytomas to have minimal elevations in plasma and urinary catecholamines or in the setting of obvious catecholamine excess biochemically to be normotensive with few, if any, clinical signs and symptoms.1,37,38,40,41
Recent studies also have shown that cardiovascular complications secondary to chronic catecholamine stimulation can occur in up to 20% of patients with pheochromocytomas, and pheochromocytoma-induced cardiomyopathies are increasingly reported.42–44 A recent review of the literature suggested that two types of pheochromocytoma-related cardiomyopathies may exist: (1) Takotsubo (‘stress’) cardiomyopathy, associated with more acute changes and a higher rate of left ventricular recovery and (2) more chronic cardiomyopathies, including dilated cardiomyopathies and acute heart failure, with lower rates for recovery of left ventricular function.42 This variability highlights the importance of considering the diagnosis of PPGLs for patients with a newly diagnosed Takotsubo cardiomyopathy or unexplained dilated cardiomyopathy and, conversely, appropriate cardiac evaluation for patients with a diagnosis of PPGLs.42,44
BIOCHEMICAL WORKUP
Patients suspected of PPGLs should undergo biochemical testing to either confirm or rule out the disease. The metabolites of epinephrine and norepinephrine, metanephrines and normetaphrines,45 have been isolated aid in diagnosis and are superior to circulating catecholamines.46,47 However, the ideal test with both high sensitivity and specificity has been debated. The two best methods in current use are measurement of 24-h excretion of fractionated metanephrines and measurement of plasma-free metanephrines. Evidence for both tests with details of the testing methods has been extensively reviewed.1 The plasma test has very high sensitivity (90–100%), with slightly lower specificity (79–100%). The urinary test of fractionated metanephrines generally has been slightly lower in both sensitivity and specificity, but a head-to-head comparison between plasma and urinary metanephrines using mass spectrometry has never been completed. Therefore, at this writing, both plasma and urinary metanephrines should be considered for diagnosing or ruling out pheochromocytoma. The plasma test has significant advantages in terms of convenience.
Given the rarity of PPGLs and the pretest probability of 1%, the rate of false-positives will be high. Yu and Wei48 reviewed a large single-institution experience with testing for pheochromocytomas and found a false-positive rate for both plasma-free and urinary-fractionated metanephrines of 19–21%. The causes of the false-positive findings included physiologic variation, laboratory errors, and drug interference with measurement. Medications known to cause interference include phenoxybenzamine, tricyclic antidepressants, acetaminophen, labetalol, sotalol, alpha-methyldopa, buspirone, monoamine oxidase (MAO) inhibitors, sympathomimetics, cocaine, sulfasalazine, and levodopa.1,49 Patients should be instructed to hold acetaminophen 5 days before testing. Repeat testing may be prudent after medications have been adjusted and the patient is in supine position for testing.1 If medications cannot be adjusted, a clonidine suppression test may be performed to rule out false-positives.49
It should be emphasized that mild elevations seen with either the urinary or plasma test should be viewed with suspicion, as should abnormalities seen during unusual or stressful events (e.g., hospitalization for unrelated reasons). Repeated testing including both urinary and plasma tests often is advisable when the diagnosis is not clear.
GENETIC BASIS AND TESTING OF PHEOCHROMOCYTOMAS AND PARAGANGLIOMAS
Pheochromocytoma and paragangliomas are among the most common inherited tumors, with up to 30–40% arising from an attributable germline mutation.50 The old axiom of pheochromocytomas characterized by the rule of tens (10% familial) is no longer accurate because many new tumor syndromes have been identified and characterized during the past 15 years.51 Currently, it is recommended that all patients with a diagnosis of PPGL should be referred for genetic evaluation regardless of their age or family history.52 If an inherited syndrome is identified, the management of the patient and affected family members will be determined by the specific mutation.53
To date, more than 20 genes have been identified as playing a role in tumor development, as either a germline (inherited) or somatic (non-inherited) mutation. The specific gene mutation provides insight into the pathophysiology and biologic behavior of the PPGL. Table 1 groups genes by their function and transcriptional profile.54 The first group includes genes that contribute to dysregulation of hypoxia-inducible factor (HIF), which includes von Hippel-Lindau (VHL) disease, the succinate dehydrogenase subunits (SDHA, SDHB, SDHC, SDHD), succinate dehydrogenase complex assembly factor 2 (SDHAF2), egl-9 prolyl hydroxylase 1 and 2 (EGLN1/2), malate dehydrogenase 2 (MDH2), and fumarate hydratase (FH). These tumors are characterized by an upregulation of HIF without hypoxia. In general, these tumors tend to be more aggressive than those associated with other genetic alterations. Particularly, the SDHB mutation has been associated with an increased risk of malignant behavior.55 These tumors (except VHL) are more commonly extra-adrenal and have a noradrenergic phenotype.
TABLE 1.
Clustering of inherited pheochromocytoma and paraganglioma syndromes
| Cluster 1: pseudohypoxia | Cluster 2: kinase signaling |
|---|---|
| VHL | RET |
| SDHA, SDHB, SDHC, SDHD | NF1 |
| SDHAF2 | TMEM127 |
| FH | MAX |
| EGLN1/2 (PHD1) | |
| MDH2 | |
| Generally, more aggressive | Generally, more pheochromocytomas with an adrenergic phenotype |
Cluster 3 (Wnt signaling) represents only genes with nonfamilial associations
The second group includes genes that affect the kinase-signaling pathway involving PI3K/mTOR. These genes include the RET proto-oncogene (multiple endocrine neoplasia [MEN] 2A and 2B), neurofibromin 1 (NF1), and the more recently associated genes, namely, transmembrane protein 127 (TMEM127) and Myc-associated factor X (MAX). The tumors in this cluster tend to be predominantly adrenal in origin with an adrenergic phenotype.
VHL
The VHL syndrome, arising from mutations in the VHL gene, is characterized by a variety of tumors including cerebellar and spinal hemangioblastomas, retinal hemangiomas, clear cell renal carcinomas, pancreatic neuroendocrine tumors, and cysts and cystadenomas of the pancreas, epididymis, and broad ligament.56 Central nervous system hemangioblastomas are the dominant cause of mortality.53 Pheochromocytomas and rarely PGLs are seen in about 20% of patients, usually with a young age of onset. Up to one half of pediatric patients presenting with PPGLs are found to have VHL.57 Tumors are frequently of adrenal origin and produce dominantly norepinephrine instead of epinephrine.58 Metastatic pheochromocytomas are rare (5%).
MEN2
The MEN2 syndromes are caused by activating mutations in the RET proto-oncogene. Both MEN2A and MEN2B are characterized by medullary thyroid cancer, with a penetrance near 100%. Patients with MEN2B and medullary thyroid cancer present at an earlier age with aggressive characteristics. Patients with MEN2A also have hyperparathyroidism, whereas patients with MEN2B frequently have neuromas and a Marfanoid habitus. Both syndromes are characterized by pheochromocytomas, with a penetrance of about 50% for both types.53 Frequently, bilateral disease is observed. Therefore, a cortical-sparing approach to surgery is favored by some surgeons to avoid steroid dependence and the risk of adrenal crisis. Metastatic pheochromocytomas and extra-adrenal PGLs are quite rare (about ≤ 1%).
NF1
Neurofibromatosis type 1 is characterized by the development of multiple neuromas, with up to 15% developing malignant peripheral nerve sheath tumors.59 The penetrance of pheochromocytomas in these cases is only 1–5%. Almost all these tumors are adrenal in origin. Metastatic pheochromocytomas are reported 7–12% of the time. Virtually all patients with NF1 and a pheochromocytoma will show cutaneous manifestations on the physical exam.58
PARAGANGLIOMA SYNDROMES TYPES 1 TO 5 (SDH COMPLEX)
The PGL syndromes arise from genes encoding the succinate dehydrogenase (SDH) enzyme. The SDH complex, composed of four subunits (SDHA, SDHB, SDHC, and SDHD), oxidizes succinate to fumarate as part of the electron transport chain within the mitochondrial inner membrane. Mutations cause destabilization of the SDH complex, resulting in stabilization of HIF leading to tumor formation. The fifth gene identified as causing PGL is SDHAF2, which flavinates SDHA. These syndromes are more commonly associated with PGL (including head and neck PGLs) than the previously described syndromes, but pheochromocytomas are seen in all five types. Both the SDHD and SDHAF2 germline mutations leading to PPGLs are almost always inherited paternally and have evidence of complicated maternal imprinting.58 The SDHB mutations are distinguished by a higher rate of malignancy, reported to be 30–70%.58 The morbidity of SDHB inherited tumors is frequently related to metastatic disease rather than catecholamine and metabolite production.
GENETIC TESTING
Previously, genetic testing was recommended for patients who were young at diagnosis or had a family history or evidence of multifocal disease. Currently, the American College of Medical Genetics and Genomics and the National Society of Genetic Counselors recommend that all patients with PPGLs undergo genetic testing regardless of age or family history.52 Likewise, the Endocrine Society and the European Society of Endocrinology recommend that genetic testing be considered for all patients.1,60 A recent study suggested that preoperative genetic testing affects the surgical approach at the initial operation. In particular, patients with an SDHB mutation, which typically represents more aggressive and malignant disease, were more likely to undergo an open rather than a minimally invasive approach.61
Next generation sequencing (NGS), introduced in 2005, has become the gold standard for genetic testing because it is more efficient and cost-effective than previous sequencing techniques. A consensus statement on NGS testing for patients with inherited PPGL details the variety of associated genes and standardizes reporting.62 With NGS, it is relatively straightforward to test the 10 to 15 most common predisposing genes. The genes typically tested are EGLN1, FH, KIF1B, MAX, MEN1, NF1, RET, SDHA, SDHAF2, SDHB, SDHC, SDHD, TMEM127, and VHL. The genes typically are sequenced and evaluated for exon deletions and duplications. Due to the complexity of interpreting the results and potential health and life insurance implications, it is ideally recommended that patients meet with a genetics counselor before genetic testing.
Although surgical resection typically is the optimal therapy for these tumors, efficacious treatment for metastatic disease is suboptimal, consisting of 131I-MIBG (FDA approved), chemotherapy, targeted therapy, and potentially Lu-177 DOTATATE (currently in trial), which is discussed in part 2 of this review. By understanding the genetic background of these tumors, both germline and somatic, personalized targeting of these pathways should be possible in the future. In addition, germline testing allows the surgeon and clinician to personalize surgical approaches and determine the extent of adrenalectomy.
RADIOLOGIC EVALUATION
In general, the radiologic workup for PPGLs should be performed only after biochemical confirmation of disease. Anatomic imaging with computed tomography (CT), magnetic resonance imaging (MRI), or both is critical for surgical planning and should be performed for every patient. Functional imaging methods, which use radiotracers dependent on catecholamine metabolism and secretion, glucose metabolism, or tumor somatostatin receptor status, have been demonstrated to have variable sensitivities and specificities related to location and extent of disease as well as the genetic status of the patient. Figure 2 shows a patient with a pheochromocytoma who underwent anatomic and functional imaging.
FIG. 2.
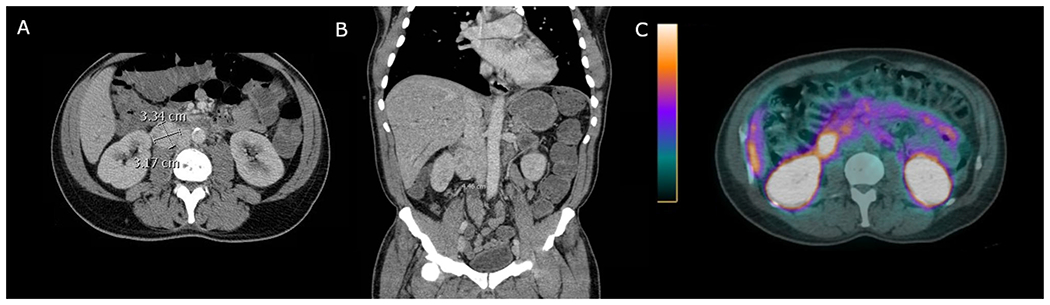
A patient with a 3.34-cm right paraganglioma shown by a computed tomography (CT) axial slice and b CT coronal slice. The patient underwent a c 68-gallium dotatate scan, which showed high avidity
ANATOMIC IMAGING
Often, CT is the first imaging performed for patients with PPGLs, and in the setting of a seemingly sporadic small (< 3 cm) tumor confined to the adrenal may be the only imaging required.63 If adrenal protocol CT is obtained, unlike lipid-rich adenomas, pheochromocytomas will measure more than 10 Hounsfield on non-contrasted images and have marked enhancement on arterial phase images as well as delayed venous washout.64,65 The patterns of enhancement may vary depending on the size of the lesion and the presence of intra-tumor hemorrhage or necrosis. The imaging characteristics of paragangliomas are similar to those of pheochromocytomas, commonly identified as a hypervascular, para-aortic mass. A finding of bilateral adrenal or extra-adrenal lesions on CT should raise concern for a hereditary syndrome.
For PPGLs, MRI is not as commonly used. Typically, an MRI shows increased signal intensity on T2-weighted imaging (light bulb sign), and as with CT, variable patterns of post-contrast enhancement. Unlike lipid-rich adenomas, no signal dropout is observed on chemical shift imaging.66 With PGLs, a “salt and pepper” appearance may be described on T1- and T2-weighted images secondary to areas of hyperintense foci (slow flow or hemorrhage) interspersed with areas of signal void (high-velocity flow through serpiginous vessels).67
For head and neck PGLs, a combination of both CT and MRI techniques may be useful. Contrast-enhanced three-dimensional magnetic resonance angiography has been reported to improve tumor detection and differentiation from other neoplasms such as schwannoma, plasmacytoma, meningioma, and vascular malformations,68 although CT may permit better evaluation of temporal bone extension at the jugular foramen and hypotympanum.66,69
FUNCTIONAL IMAGING
Historically, 123I-metaiodobenzylguanidine (MIBG) has been the primary functional imaging method for patients with PPGLs. As a guanethidine precursor, MIBG is taken up by presynaptic adrenergic neural cells after intravenous administration. As reported, 123I-MIBG has a sensitivity of nearly 90% and specificity of 70–100% for isolated pheochromocytomas.70,71 However, its sensitivity in the detection of extra-adrenal, metastatic, and recurrent PPGLs generally is low70–72 and it has been largely replaced by newer techniques. Currently, 23I-MIBG is most commonly indicated for the pre-therapy evaluation of patients with metastatic PPGLs who may be candidates for 131I-MIBG internal radiotherapy.64,73 The routine use of MIBG scanning is not recommended because it can be misleading as often as it is helpful.74 It still may be useful in highly selected cases such as for patients with negative genetic screens and those with bilateral adrenal lesions, both of which are possibly pheochromocytomas based on CT and/or MRI findings. However, this is an uncommon scenario, found in only 1 of 340 patients reported by Rao et al.74 Although physiologic uptake is expected in the salivary glands, heart, liver, spleen, and urinary collecting system, it is important to remember that patients undergoing 123I-MIBG need to be pretreated with supersaturated potassium iodide solution to prevent uptake by the thyroid.
Positron emission tomography (PET)/CT using a variety of radiotracers has been evaluated in patients with PPGLs. In a cohort of patients at the National Institutes of Health (NIH), Timmers et al.72 compared 18F-3,4-dihydroxyphenylalanine (18F-DOPA), 18F-dopamine (18F-FDA) PET, 18F-fluordeoxyglucose (18F-FDG), and 123I-MIBG for the detection of tumors in patients with localized, metastatic, and hereditary PPGLs. They identified similar sensitivities (on the order of 80%) among these techniques for patients with localized PPGLs, but found that 18F-FDA was superior to 18F-DOPA and 123I-MIBG in localizing metastatic disease. Notably, at this writing, 18F-FDA is available only at the NIH. For patients with SDHB mutations, 18F-FDG PET/CT demonstrated the highest sensitivity in detecting metastases.
Because 18F-FDG is dependent on glucose uptake and metabolism, the authors proposed that this finding may be explained by SDHB-associated alteration in oxygen metabolism resulting in increased glycolysis and intracellular glucose requirements. Subsequent work by this group and others has further illustrated the existence of radiographic genotype–phenotype correlations in PPGL patients.75,76 Specifically, a higher degree of 18F-FDG uptake is observed in SDHx- and VHL-associated PPGLs than in MEN2- or NF1-related tumors. These findings correlate with well-described mutational and pathophysiologic clusters (clusters 1 and 2, as previously described) of PPGLs into pseudohypoxia-driven (SDHx and VHL) and kinase-signaling (MEN2 and NF1) subtypes.77 Whereas 18F-FDG PET/CT has a high sensitivity for SDHx and VHL (pseudohypoxia cluster)-associated tumors, 18F-DOPA PET/CT appears to have improved performance in sporadic as well as in MEN2 and NF1 (kinase signaling cluster)-related PPGLs72,78 and is the more appropriate functional imaging choice for these patients. Furthermore, 18F-DOPA PET/CT is the preferred functional method for non-SDHx head and neck PGL.78,79
Like many other neuroendocrine neoplasms, PPGLs express somatostatin receptors that can be exploited for tumor detection. Although 111In-pentetreotide (octreoscan) currently has a very limited, if any, role in the imaging of these tumors, a growing body of literature has consistently demonstrated the superiority of (68Ga)-DOTATATE PET/CT in the detection of PPGLs compared with other functional imaging methods.80–84 Given these findings as well as its growing availability within the United States, future recommendations are likely to incorporate (68Ga)-DOTATATE PET/CT as the primary functional imaging technique for PPGLs.
CONCLUSION
The PPGLs are rare neuroendocrine tumors with a variable presentation that require specialized management and treatment by experienced and expert endocrine surgeons, endocrinologists, and anesthesiologists. A biochemical workup is required for any patient suspected of PPGL. The genetic etiology of PPGLs has a strong impact on subsequent management and operative approach. Once the diagnosis has been confirmed, a radiologic workup is initiated to identify the PPGL, although a large number of PPGLs currently are being discovered incidentally. In the second part of this review, the perioperative management, surgical approach, pathologic features of malignancy, optimal surveillance, and management of advanced disease are discussed.
ACKNOWLEDGMENT
This report was reviewed by the SSO Endocrine and Head and Neck Disease Site Working Group, and all members expressed support for its publication.
DISCLOSURE
Dr. Yang’s research is supported by the National Heart Lung and Blood Institute (NHLBI) of the National Institutes of Health (K08HL145139). The remaining authors have no conflicts of interest.
REFERENCES
- 1.Lenders JW, Duh QY, Eisenhofer G, et al. Pheochromocytoma and paraganglioma: an endocrine society clinical practice guideline. J Clin Endocrinol Metab. 2014;99:1915–42. [DOI] [PubMed] [Google Scholar]
- 2.Lenders JW, Eisenhofer G, Mannelli M, Pacak K. Phaeochromocytoma. Lancet. 2005;366:665–75. [DOI] [PubMed] [Google Scholar]
- 3.Whalen RK, Althausen AF, Daniels GH. Extra-adrenal pheochromocytoma. J Urol. 1992;147:1–10. [DOI] [PubMed] [Google Scholar]
- 4.Pacak K, Linehan WM, Eisenhofer G, Walther MM, Goldstein DS. Recent advances in genetics, diagnosis, localization, and treatment of pheochromocytoma. Ann Intern Med. 2001;134:315–29. [DOI] [PubMed] [Google Scholar]
- 5.Dailey L, Laplantine E, Priore R, Basilico C. A network of transcriptional and signaling events is activated by FGF to induce chondrocyte growth arrest and differentiation. J Cell Biol. 2003;161:1053–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ramlawi B, David EA, Kim MP, et al. Contemporary surgical management of cardiac paragangliomas. Ann Thorac Surg. 2012;93:1972–6. [DOI] [PubMed] [Google Scholar]
- 7.Brown ML, Zayas GE, Abel MD, Young WF Jr, Schaff HV. Mediastinal paragangliomas: the Mayo Clinic experience. Ann Thorac Surg. 2008;86:946–51. [DOI] [PubMed] [Google Scholar]
- 8.Bravo EL. Pheochromocytoma: new concepts and future trends. Kidney Int. 1991;40:544–56. [DOI] [PubMed] [Google Scholar]
- 9.McNicol AM. Update on tumours of the adrenal cortex, phaeochromocytoma, and extra-adrenal paraganglioma. Histopathology. 2011;58:155–68. [DOI] [PubMed] [Google Scholar]
- 10.Chen H, Sippel RS, O’Dorisio MS, et al. The North American Neuroendocrine Tumor Society consensus guideline for the diagnosis and management of neuroendocrine tumors: pheochromocytoma, paraganglioma, and medullary thyroid cancer. Pancreas. 2010;39:775–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lo CY, Lam KY, Wat MS, Lam KS. Adrenal pheochromocytoma remains a frequently overlooked diagnosis. Am J Surg. 2000;179:212–5. [DOI] [PubMed] [Google Scholar]
- 12.McNeil AR, Blok BH, Koelmeyer TD, Burke MP, Hilton JM. Phaeochromocytomas discovered during coronial autopsies in Sydney, Melbourne, and Auckland. Aust N Z J Med. 2000;30:648–52. [DOI] [PubMed] [Google Scholar]
- 13.Anderson GH Jr, Blakeman N, Streeten DH. The effect of age on prevalence of secondary forms of hypertension in 4429 consecutively referred patients. J Hypertens. 1994;12:609–15. [DOI] [PubMed] [Google Scholar]
- 14.Ariton M, Juan CS, AvRuskin TW. Pheochromocytoma: clinical observations from a Brooklyn tertiary hospital. Endocr Pract. 2000;6:249–52. [DOI] [PubMed] [Google Scholar]
- 15.Wyszynska T, Cichocka E, Wieteska-Klimczak A, Jobs K, Januszewicz P. A single pediatric center experience with 1025 children with hypertension. Acta Paediatr. 1992;81:244–6. [DOI] [PubMed] [Google Scholar]
- 16.Mantero F, Terzolo M, Arnaldi G, et al. A survey on adrenal incidentaloma in Italy. Study Group on Adrenal Tumors of the Italian Society of Endocrinology. J Clin Endocrinol Metab. 2000;85:637–44. [DOI] [PubMed] [Google Scholar]
- 17.Mansmann G, Lau J, Balk E, Rothberg M, Miyachi Y, Bornstein SR. The clinically inapparent adrenal mass: update in diagnosis and management. Endocr Rev. 2004;25:309–40. [DOI] [PubMed] [Google Scholar]
- 18.Bryant J, Farmer J, Kessler LJ, Townsend RR, Nathanson KL. Pheochromocytoma: the expanding genetic differential diagnosis. J Natl Cancer Inst. 2003;95:1196–204. [DOI] [PubMed] [Google Scholar]
- 19.Baguet JP, Hammer L, Mazzuco TL, et al. Circumstances of discovery of phaeochromocytoma: a retrospective study of 41 consecutive patients. Eur J Endocrinol. 2004;150:681–6. [DOI] [PubMed] [Google Scholar]
- 20.Kopetschke R, Slisko M, Kilisli A, et al. Frequent incidental discovery of phaeochromocytoma: data from a German cohort of 201 phaeochromocytoma. Eur J Endocrinol. 2009;161:355–61. [DOI] [PubMed] [Google Scholar]
- 21.Motta-Ramirez GA, Remer EM, Herts BR, Gill IS, Hamrahian AH. Comparison of CT findings in symptomatic and incidentally discovered pheochromocytomas. AJR Am J Roentgenol. 2005;185:684–8. [DOI] [PubMed] [Google Scholar]
- 22.Noshiro T, Shimizu K, Watanabe T, et al. Changes in clinical features and long-term prognosis in patients with pheochromocytoma. Am J Hypertens. 2000;13:35–43. [DOI] [PubMed] [Google Scholar]
- 23.Guerrero MA, Schreinemakers JM, Vriens MR, et al. Clinical spectrum of pheochromocytoma. J Am Coll Surg. 2009;209:727–32. [DOI] [PubMed] [Google Scholar]
- 24.Plouin PF, Chatellier G, Fofol I, Corvol P. Tumor recurrence and hypertension persistence after successful pheochromocytoma operation. Hypertension. 1997;29:1133–9. [DOI] [PubMed] [Google Scholar]
- 25.Hamidi O, Young WF Jr, Iniguez-Ariza NM, et al. Malignant pheochromocytoma and paraganglioma: 272 patients over 55 years. J Clin Endocrinol Metab. 2017;102:3296–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Eisenhofer G, Huynh TT, Hiroi M, Pacak K. Understanding catecholamine metabolism as a guide to the biochemical diagnosis of pheochromocytoma. Rev Endocr Metab Disord. 2001;2:297–311. [DOI] [PubMed] [Google Scholar]
- 27.Plouin PF, Duclos JM, Soppelsa F, Boublil G, Chatellier G. Factors associated with perioperative morbidity and mortality in patients with pheochromocytoma: analysis of 165 operations at a single center. J Clin Endocrinol Metab. 2001;86:1480–6. [DOI] [PubMed] [Google Scholar]
- 28.Prejbisz A, Lenders JW, Eisenhofer G, Januszewicz A. Cardiovascular manifestations of phaeochromocytoma. J Hypertens. 2011;29:2049–60. [DOI] [PubMed] [Google Scholar]
- 29.Zelinka T, Petrak O, Turkova H, et al. High incidence of cardiovascular complications in pheochromocytoma. Horm Metab Res. 2012;44:379–84. [DOI] [PubMed] [Google Scholar]
- 30.Brunjes S, Johns VJ Jr, Crane MG. Pheochromocytoma: postoperative shock and blood volume. N Engl J Med. 1960;262:393–6. [DOI] [PubMed] [Google Scholar]
- 31.La Batide-Alanore A, Chatellier G, Plouin PF. Diabetes as a marker of pheochromocytoma in hypertensive patients. J Hypertens. 2003;21:1703–7. [DOI] [PubMed] [Google Scholar]
- 32.Hamaji M Pancreatic alpha- and beta-cell function in pheochromocytoma. J Clin Endocrinol Metab. 1979;49:322–5. [DOI] [PubMed] [Google Scholar]
- 33.Turnbull DM, Johnston DG, Alberti KG, Hall R. Hormonal and metabolic studies in a patient with a pheochromocytoma. J Clin Endocrinol Metab. 1980;51:930–3. [DOI] [PubMed] [Google Scholar]
- 34.Gilsanz FJ, Luengo C, Conejero P, Peral P, Avello F. Cardiomyopathy and phaeochromocytoma. Anaesthesia. 1983;38:888–91. [DOI] [PubMed] [Google Scholar]
- 35.Scholten A, Cisco RM, Vriens MR, et al. Pheochromocytoma crisis is not a surgical emergency. J Clin Endocr Metab. 2013;98:581–91. [DOI] [PubMed] [Google Scholar]
- 36.Newell KA, Prinz RA, Pickleman J, et al. Pheochromocytoma multisystem crisis: a surgical emergency. Arch Surg. 1988;123:956–9. [DOI] [PubMed] [Google Scholar]
- 37.Adler JT, Meyer-Rochow GY, Chen H, et al. Pheochromocytoma: current approaches and future directions. Oncologist. 2008;13:779–93. [DOI] [PubMed] [Google Scholar]
- 38.Pacak K Preoperative management of the pheochromocytoma patient. J Clin Endocrinol Metab. 2007;92:4069–79. [DOI] [PubMed] [Google Scholar]
- 39.Haissaguerre M, Courel M, Caron P, et al. Normotensive incidentally discovered pheochromocytomas display specific biochemical, cellular, and molecular characteristics. J Clin Endocrinol Metab. 2013;98:4346–54. [DOI] [PubMed] [Google Scholar]
- 40.Eisenhofer G, Walther MM, Huynh TT, et al. Pheochromocytomas in von Hippel-Lindau syndrome and multiple endocrine neoplasia type 2 display distinct biochemical and clinical phenotypes. J Clin Endocrinol Metab. 2001;86:1999–2008. [DOI] [PubMed] [Google Scholar]
- 41.Eisenhofer G, Klink B, Richter S, Lenders JW, Robledo M. Metabologenomics of phaeochromocytoma and paraganglioma: an integrated approach for personalised biochemical and genetic testing. Clin Biochem Rev. 2017;38:69–100. [PMC free article] [PubMed] [Google Scholar]
- 42.Batisse-Lignier M, Pereira B, Motreff P, et al. Acute and chronic pheochromocytoma-induced cardiomyopathies: different prognoses? A systematic analytical review. Med Baltim. 2015;94:e2198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gagnon N, Mansour S, Bitton Y, Bourdeau I. Takotsubo-like cardiomyopathy in a large cohort of patients with pheochromocytoma and paraganglioma. Endocr Pract. 2017;23:1178–92. [DOI] [PubMed] [Google Scholar]
- 44.Zhang R, Gupta D, Albert SG. Pheochromocytoma as a reversible cause of cardiomyopathy: analysis and review of the literature. Int J Cardiol. 2017;249:319–23. [DOI] [PubMed] [Google Scholar]
- 45.Armstrong MD, Mc MA, Shaw KN. 3-Methoxy-4-hydroxy-D-mandelic acid, a urinary metabolite of norepinephrine. Biochim Biophys Acta. 1957;25:422–3. [DOI] [PubMed] [Google Scholar]
- 46.Bravo EL, Tarazi RC, Gifford RW, Stewart BH. Circulating and urinary catecholamines in pheochromocytoma: diagnostic and pathophysiologic implications. N Engl J Med. 1979;301:682–6. [DOI] [PubMed] [Google Scholar]
- 47.Lenders JW, Keiser HR, Goldstein DS, et al. Plasma metanephrines in the diagnosis of pheochromocytoma. Ann Intern Med. 1995;123:101–9. [DOI] [PubMed] [Google Scholar]
- 48.Yu R, Wei M. False-positive test results for pheochromocytoma from 2000 to 2008. Exp Clin Endocrinol Diabetes. 2010;118:577–85. [DOI] [PubMed] [Google Scholar]
- 49.Eisenhofer G, Goldstein DS, Walther MM, et al. Biochemical diagnosis of pheochromocytoma: how to distinguish true- from false-positive test results. J Clin Endocrinol Metab. 2003;88:2656–66. [DOI] [PubMed] [Google Scholar]
- 50.Vicha A, Musil Z, Pacak K. Genetics of pheochromocytoma and paraganglioma syndromes: new advances and future treatment options. Curr Opin Endocrinol Diabetes Obes. 2013;20:186–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Alrezk R, Suarez A, Tena I, Pacak K. Update of pheochromocytoma syndromes: genetics, biochemical evaluation, and imaging. Front Endocrinol Lausanne. 2018;9:515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hampel H, Bennett RL, Buchanan A, et al. A practice guideline from the American College of Medical Genetics and Genomics and the National Society of Genetic Counselors: referral indications for cancer predisposition assessment. Genet Med. 2015;17:70–87. [DOI] [PubMed] [Google Scholar]
- 53.Neumann HP, Young WF Jr, Krauss T, et al. 65 Years of the double helix: genetics informs precision practice in the diagnosis and management of pheochromocytoma. Endocr Relat Cancer. 2018;25:T201–19. [DOI] [PubMed] [Google Scholar]
- 54.Jochmanova I, Pacak K. Genomic landscape of pheochromocytoma and paraganglioma. Trends Cancer. 2018;4:6–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Assadipour Y, Sadowski SM, Alimchandani M, et al. SDHB mutation status and tumor size but not tumor grade are important predictors of clinical outcome in pheochromocytoma and abdominal paraganglioma. Surgery. 2017;161:230–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lonser RR, Glenn GM, Walther M, et al. von Hippel–Lindau disease. Lancet. 2003;361:2059–67. [DOI] [PubMed] [Google Scholar]
- 57.Bausch B, Wellner U, Bausch D, et al. Long-term prognosis of patients with pediatric pheochromocytoma. Endocr Relat Cancer. 2014;21:17–25. [DOI] [PubMed] [Google Scholar]
- 58.Lefebvre M, Foulkes WD. Pheochromocytoma and paraganglioma syndromes: genetics and management update. Curr Oncol. 2014;21:e8–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Miettinen MM, Antonescu CR, Fletcher CDM, et al. Histopathologic evaluation of atypical neurofibromatous tumors and their transformation into malignant peripheral nerve sheath tumor in patients with neurofibromatosis 1: a consensus overview. Hum Pathol. 2017;67:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Plouin PF, Amar L, Dekkers OM, et al. European Society of Endocrinology Clinical Practice Guideline for long-term followup of patients operated on for a phaeochromocytoma or a paraganglioma. Eur J Endocrinol. 2016;174:G1–10. [DOI] [PubMed] [Google Scholar]
- 61.Nockel P, El Lakis M, Gaitanidis A, et al. Preoperative genetic testing in pheochromocytomas and paragangliomas influences the surgical approach and the extent of adrenal surgery. Surgery. 2018;163:191–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Group NGSiPS, Toledo RA, Burnichon N, et al. Consensus statement on next-generation-sequencing-based diagnostic testing of hereditary phaeochromocytomas and paragangliomas. Nat Rev Endocrinol. 2017;13:233–47. [DOI] [PubMed] [Google Scholar]
- 63.Castinetti F, Kroiss A, Kumar R, Pacak K, Taieb D. 15 Years of paraganglioma: imaging and imaging-based treatment of pheochromocytoma and paraganglioma. Endocr Relat Cancer. 2015;22:T135–45. [DOI] [PubMed] [Google Scholar]
- 64.Ctvrtlik F, Koranda P, Schovanek J, Skarda J, Hartmann I, Tudos Z. Current diagnostic imaging of pheochromocytomas and implications for therapeutic strategy. Exp Ther Med. 2018;15:3151–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Baez JC, Jagannathan JP, Krajewski K, et al. Pheochromocytoma and paraganglioma: imaging characteristics. Cancer Imaging. 2012;12:153–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Elsayes KM, Menias CO, Siegel CL, Narra VR, Kanaan Y, Hussain HK. Magnetic resonance characterization of pheochromocytomas in the abdomen and pelvis: imaging findings in 18 surgically proven cases. J Comput Assist Tomogr. 2010;34:548–53. [DOI] [PubMed] [Google Scholar]
- 67.Lee KY, Oh YW, Noh HJ, et al. Extraadrenal paragangliomas of the body: imaging features. AJR Am J Roentgenol. 2006;187:492–504. [DOI] [PubMed] [Google Scholar]
- 68.Neves F, Huwart L, Jourdan G, et al. Head and neck paragangliomas: value of contrast-enhanced 3D MR angiography. AJNR Am J Neuroradiol. 2008;29:883–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Olsen WL, Dillon WP, Kelly WM, Norman D, Brant-Zawadzki M, Newton TH. MR imaging of paragangliomas. AJR Am J Roentgenol. 1987;148:201–4. [DOI] [PubMed] [Google Scholar]
- 70.Bhatia KS, Ismail MM, Sahdev A, et al. 123I-metaiodobenzyl-guanidine (MIBG) scintigraphy for the detection of adrenal and extra-adrenal phaeochromocytomas: CT and MRI correlation. Clin Endocrinol Oxf. 2008;69:181–8. [DOI] [PubMed] [Google Scholar]
- 71.Wiseman GA, Pacak K, O’Dorisio MS, et al. Usefulness of 123I-MIBG scintigraphy in the evaluation of patients with known or suspected primary or metastatic pheochromocytoma or paraganglioma: results from a prospective multicenter trial. J Nucl Med. 2009;50:1448–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Timmers HJ, Chen CC, Carrasquillo JA, et al. Comparison of 18F-fluoro-L-DOPA, 18F-fluoro-deoxyglucose, and 18F-fluorodopamine PET, and 123I-MIBG scintigraphy in the localization of pheochromocytoma and paraganglioma. J Clin Endocrinol Metab. 2009;94:4757–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Timmers HJ, Taieb D, Pacak K. Current and future anatomical and functional imaging approaches to pheochromocytoma and paraganglioma. Horm Metab Res. 2012;44:367–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Rao D, van Berkel A, Piscaer I, et al. Impact of 123 I-MIBG scintigraphy on clinical decision making in pheochromocytoma and paraganglioma. J Clin Endocrinol Metab. 2019;104:3812–20. [DOI] [PubMed] [Google Scholar]
- 75.Timmers HJ, Chen CC, Carrasquillo JA, et al. Staging and functional characterization of pheochromocytoma and paraganglioma by 18F-fluorodeoxyglucose (18F-FDG) positron emission tomography. J Natl Cancer Inst. 2012;104:700–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Tiwari A, Shah N, Sarathi V, et al. Genetic status determines (18)F-FDG uptake in pheochromocytoma/paraganglioma. J Med Imaging Radiat Oncol. 2017;61:745–52. [DOI] [PubMed] [Google Scholar]
- 77.Nolting S, Grossman AB. Signaling pathways in pheochromocytomas and paragangliomas: prospects for future therapies. Endocr Pathol. 2012;23:21–33. [DOI] [PubMed] [Google Scholar]
- 78.Taieb D, Timmers HJ, Hindie E, et al. EANM 2012 guidelines for radionuclide imaging of phaeochromocytoma and paraganglioma. Eur J Nucl Med Mol Imaging. 2012;39:1977–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Guichard JP, Fakhry N, Franc J, Herman P, Righini CA, Taieb D. Morphological and functional imaging of neck paragangliomas. Eur Ann Otorhinolaryngol Head Neck Dis. 2017;134:243–8. [DOI] [PubMed] [Google Scholar]
- 80.Janssen I, Blanchet EM, Adams K, et al. Superiority of [68 Ga]-DOTATATE PET/CT to other functional imaging modalities in the localization of SDHB-associated metastatic pheochromocytoma and paraganglioma. Clin Cancer Res. 2015;21:3888–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Janssen I, Chen CC, Millo CM, et al. PET/CT comparing (68)Ga-DOTATATE and other radiopharmaceuticals and in comparison with CT/MRI for the localization of sporadic metastatic pheochromocytoma and paraganglioma. Eur J Nucl Med Mol Imaging. 2016;43:1784–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Jing H, Li F, Wang L, et al. Comparison of the 68 Ga-DOTA-TATA PET/CT, FDG PET/CT, and MIBG SPECT/CT in the evaluation of suspected primary pheochromocytomas and paragangliomas. Clin Nucl Med. 2017;42:525–9. [DOI] [PubMed] [Google Scholar]
- 83.Chang CA, Pattison DA, Tothill RW, et al. (68)Ga-DOTATATE and (18)F-FDG PET/CT in paraganglioma and pheochromocytoma: utility, patterns, and heterogeneity. Cancer Imaging. 2016;16:22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Han S, Suh CH, Woo S, Kim YJ, Lee JJ. Performance of (68)Ga-DOTA-conjugated somatostatin receptor targeting peptide PET in detection of pheochromocytoma and paraganglioma: a systematic review and meta-analysis. J Nucl Med. 2018;60:369–76. [DOI] [PubMed] [Google Scholar]