

Abstract
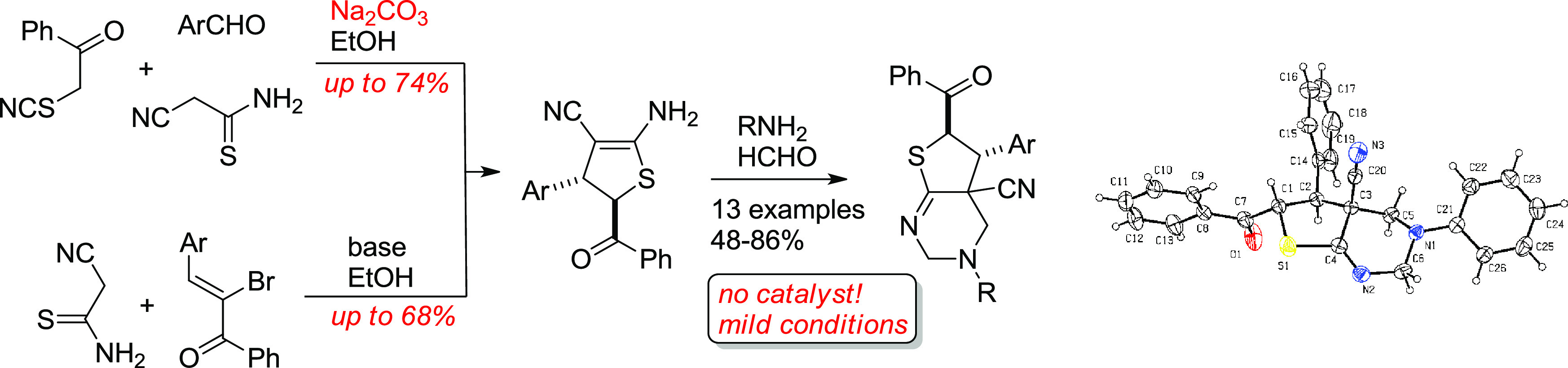
trans-2-Amino-4-aryl-5-benzoyl-4,5-dihydrothiophene-3-carbonitriles were prepared either by the reaction of 3-aryl-2-cyanothioacrylamides with α-thiocyanatoacetophenone or by the Michael-type addition of cyanothioacetamide to α-bromochalcones followed by intramolecular cyclization. The mechanism of the first reaction was studied using high-level quantum chemical calculations. Density functional theory (DFT) studies were carried out to determine the mechanism of the first reaction. A new approach toward the construction of the thieno[2,3-d]pyrimidine core system was demonstrated by the reaction of the prepared dihydrothiophenes with HCHO and RNH2 under noncatalyzed Mannich conditions.
Introduction
2-Aminothiophenes and related molecules are of particular interest, especially within the realm of medicinal chemistry (for reviews, see refs (1−7)). Many of the compounds based on the 2-aminothiophene structural motif show a broad range of biological properties and were recognized as potent pan-serotype dengue virus inhibitors,8 antitubercular agents,9−11 allosteric modulators of the A1 adenosine receptor (A1R),12 antiproliferative agents,13,14 antimicrobials,15 inhibitors of influenza virus polymerase,16 GluR6-antagonists,17 and protein-tyrosine phosphatase 1B (PTP1B) inhibitors18 (Figure 1). Some 2-aminothiophenes are traded drugs and were subjected to extensive pharmacological studies. Among them, strontium ranelate (Protelos/Osseor, useful for the treatment of osteoporosis,19−25 as a dental pulp-like cell proliferation agent,26 and as a radiopaque agent for calcium phosphate cement27), tinoridine (old but still useful anti-inflammatory drug with a strong antiperoxidative and hepatoprotective activity),28−30 and olanzapine31−33 (Zyprexa, used to treat certain mental disorders such as schizophrenia and bipolar disorder) should be noted (Figure 1). In addition, the 2-aminothiophene motif is present in drugs and bioactive molecules such as T-62 (a selective allosteric modulator of the adenosine A receptor),34 bentazepam (Tiadipona),35 and brotizolam (Lendormin);36−38 anxiolytic/anticonvulsant agents and skeletal muscle relaxants; and the anticancer drug raltitrexed (Tomudex)39−41 (Figure 1). 2-Aminothiophenes are able to act as starting points for the synthesis of a variety of thiophene-containing heterocycles and polycyclic hybrid molecules.3−6,42 Moreover, as shown recently, 2-aminothiophenes might find an application in the preparation of functional materials such as electrochemically color switching azomethines,43−46 liquid crystalline materials,47 oligothiophene-BODIPY hybrids as NIR dyes,48 organic photovoltaic cells,49 azodyes,50 nonlinear optical materials,51 and azomethine-bridged polythiopheneferrocenes.52
Figure 1.

Selected biologically active 2-aminothiophenes.
Such a diversity of available structures and applications is due to the synthetic availability of 2-aminothiophenes. The most widely used synthetic approach toward 2-aminothiophenes is based on the Gewald reaction of methylene active nitriles with elemental sulfur and methylene active ketones/aldehydes.1−6 However, this approach provides an access to aromatic 2-aminothiophenes only. Less is known on the chemistry of partially saturated analogs such as 2-amino-4,5-dihydrothiophenes (ADHTs). In general, 2,3(4,5)-dihydrothiophenes have a rich synthetic application, and dihydrothiophene ring systems have been incorporated into a variety of biologically active molecules (for reviews, see refs (53, 54)). For instance, ADHT 1 (Figure 2) was reported as a moderately active microbicide and fungicide,55 ADHT-thiazoline hybrids 2 showed good anticancer activity against colon carcinoma (HCT-116),56 and benzofurane-ADHT hybrid 3 showed an antinociceptive effect.57 Some esters of ADHT-3-carboxylic acids 4 were recognized as specific inhibitors of malaria agent Plasmodium falciparum dihydroorotate dehydrogenase.58
Figure 2.

Biologically active compounds featuring an ADHT scaffold.
However, the available methods for the synthesis of ADHTs are somewhat limited and the studies are somewhat hampered by the lack of common practical procedures and by the narrow scope of useful substrates. ADHTs can be prepared (Scheme 1) by the reaction of γ-haloacetoacetic acid derivatives with isothiocyanates,58,59 treatment of Michael adducts prepared from cyanoacetic esters and chalcone with elemental sulfur,60 recyclization of thiiranes,61,62 diastereoselective cycloadditions of aminothioisomünchnones with chiral 1,2-diaza-1,3-butadienes,63 condensation of S-acetylmercaptosuccinic anhydride with methyl cyanoacetate,64 reaction of cyanothioacetamide with β-nitrostyrene,65 reductive recyclization of functionalized thiazolidinones,66 and cyclocondensation of benzoylpyruvate with PhNCS67 or β-ketothioamides with oxalyl chloride.68 Very recently, the organotin-catalyzed reaction of arylmethylene malononitriles with α-mercaptoketones was reported to produce ADHTs as a mixture of diastereomers.69 Most of the reported procedures either require the use of expensive/exotic reagents and harsh reaction conditions or are accompanied by the formation of side products. The domino reaction of 1,3-thiazolidinedione, active methylene nitriles, amines, and aromatic aldehydes70−75 demonstrates a common approach to functionalized ADHTs. Another useful approach leading to trans-4,5-disubstituted ADHT-3-carbonitriles is the reaction of 2-cyanothioacrylamide 5 with pyridinium55,57,76,77 or sulfonium76,78,79 ylides. However, the ylide method suffers from some drawbacks such as the temperature-dependent cyclopropane or pyridine byproduct formation,80 elimination of foul-smelling dimethyl sulfide, and relatively low atom economy. Recent approaches consist of the Lewis acid catalyzed recyclizations of donor–acceptor cyclopropanes upon treatment with NH4SCN,81 thiourea,82 or tetrathiomolybdates83−85 (Scheme 1). These reactions are also not free from disadvantages such as long reaction times and difficult-to-obtain starting materials and catalysts. Given the practical significance of 2-aminothiophenes and ADHTs, the search for conceptually new rational methodologies for their synthesis, as well as an extension of the range, is of particular importance.
Scheme 1. The Reported Methods for the Preparation of ADHTs.

Earlier, we reported86 the unusual synthesis of highly functionalized ADHTs 6 as a mixture of trans isomers based on the tertiary base catalyzed reaction of α-thiocyanato acetophenone 7 with aldehydes and cyanothioacetamide 8 (or with 3-aryl-2-cyanothioacrylamides 5) (Scheme 2).
Scheme 2. Different Approaches toward the Synthesis of ADHTs 6.

Despite the availability of the starting reagents and low-cost, easy-to-handle synthesis, the reported procedures give only moderate (up to 54%) yields. In addition, the reaction mechanism still remains unclear. Herein, we report two modified superior procedures for the synthesis of ADHTs 6 affording higher yields across the range of substrates (Scheme 2). Also, we present the detailed quantum-chemical study to indicate the reaction mechanism. In addition, we also report the Mannich-type double aminomethylation reactions of ADHTs 6, providing an efficient approach to the synthesis of new functionalized thieno[2,3-d]pyrimidines 11.
Results and Discussion
To optimize the procedure for the preparation of ADHTs 6, we used 2-chlorobenzaldehyde/furfural, α-thiocyanatoacetophenone 7,87 and cyanothioacetamide 8(88) as the model reagents and examined the effect of catalyst and conditions (Scheme 3). We found that the use of 10% aq KOH instead of tertiary amines86 dramatically shortened the reaction time to within 1–2 min. However, it had no effect on the yields as the target product 6a was isolated in only a modest yield of 37% (Table 1, entry 1). Similarly, product 6b was prepared in 37% yield from furfural (Scheme 3 and Table 1, entry 5). The use of pre-prepared Knoevenagel products 5a,b had no advantages over the three-component approach since the yields were only slightly improved (38–40%; Table 1, entries 7 and 13). Potassium carbonate showed much better results as a catalyst to afford yields of ADHTs 6a,b up to 70% (Table 1, entries 2, 3, 8, 9, 14, and 15).
Scheme 3. Synthesis of ADHTs 6a–d.

Table 1. Optimization of the Conditions for the Synthesis of ADHTs 6a–d (Scheme 3)a.
| entry | reagents | conditions | product (yield, %) |
|---|---|---|---|
| 1 | 2-ClC6H4CHO, 7, 8 | 10% aq KOH, rt | 6a (37) |
| 2 | 2-ClC6H4CHO, 7, 8 | 10% aq K2CO3, 40–50 °C | 6a (64) |
| 3 | 2-ClC6H4CHO, 7, 8 | 10% aq K2CO3, rt | 6a (60) |
| 4 | 2-ClC6H4CHO, 7, 8 | 10% aqNa2CO3, 40–50 °C | 6a (68) |
| 5 | furfural, 7, 8 | 10% aq KOH, rt | 6b (37) |
| 6 | furfural, 7, 8 | 10% aqNa2CO3, 40–50 °C | 6b (62) |
| 7 | 5a, 7 | 10% aq KOH, rt | 6a (38) |
| 8 | 5a, 7 | 10% aq K2CO3, rt | 6a (61) |
| 9 | 5a, 7 | 10% aq K2CO3, 40–50 °C | 6a (70) |
| 10 | 5a, 7 | 10% aq K2CO3, reflux | 6a (31) |
| 11 | 5a, 7 | 10% aq Na2CO3, rt | 6a (57) |
| 12 | 5a, 7 | 10% aqNa2CO3, 40–50 °C | 6a (74) |
| 13 | 5b, 7 | 10% aq KOH, rt | 6b (40) |
| 14 | 5b, 7 | 10% aq K2CO3, rt | 6b (56) |
| 15 | 5b, 7 | 10% aq K2CO3, 40–50 °C | 6b (62) |
| 16 | 5b, 7 | 10% aq Na2CO3, rt | 6b (58) |
| 17 | 5b, 7 | 10% aq Na2CO3, 40–50 °C | 6b (63) |
| 18 | 5c, 7 | 10% aq Na2CO3, 40–50 °C | 6c (67) |
| 19 | 2-C3H4S-CHO, 7, 8 | 10% aq Na2CO3, 40–50 °C | 6c (69) |
| 20 | PhCHO, 7, 8 | 10% aq Na2CO3, 40–50 °C | 6d (64) |
Entries 1–6, 19, and 20: molar ratio aldehyde/7/8 = 1:1:1, EtOH. Entries 7–18: molar ratio 5/7 = 1:1, EtOH.
Finally, the best yields (62–74%) were achieved when aq Na2CO3 was taken as a catalyst and the reaction was conducted at 40–50 °C (Table 1, entries 4, 6, 12, and 17–20). As we found (see the discussion on the quantum-chemical calculations of the mechanism below), one of the possible reaction pathways include the Michael-type addition of phenacylthiocyanate 7 to thioacrylamide 5 followed by the intramolecular SN2 substitution of the SCN group.
Inspired by this, here we proposed a new approach to ADHTs 6 based on the tandem Michael addition–intramolecular SN2 substitution of thioamide 8 to easily available89 α-bromochalcones 9a,d (Scheme 4).
Scheme 4. Preparation of ADHTs 6 from α-Bromochalcones 9.

Optimization of the reaction of 8 with 9d showed that the yields of ADHTs 6 strongly depend on the reaction conditions and the base used (Table 2). The lowest yield of 6d was observed when the mixture of thioamide 8 and bromochalcone 9d was treated with excessive Et3N at 25 °C (Table 2, entry 1). The best results were achieved when the reaction mixture was gently refluxed with KOH (1 equiv) for 0.5 h (Table 2, entry 5). Compound 9a gave similar results (Table 2, entry 6).
Table 2. The Reaction Conditions and the Yields for the Synthesis of ADHTs 6a,d from α-Bromochalcones 9 and Cyanothioacetamide 8 (Scheme 4).
| entry | reagentsa | conditions | product (yield, %) |
|---|---|---|---|
| 1 | 8, 9d | Et3N (1.5 equiv), EtOH, rt, 6 days | 6d (25) |
| 2 | 8, 9d | Et3N 1.5 equiv, EtOH, reflux | 6d (46) |
| 3 | 8, 9d | K2CO3, EtOH, 40–50 °C | 6d (42) |
| 4 | 8, 9d | Na2CO3, EtOH, 40–50 °C | 6d (37) |
| 5 | 8, 9d | KOH (1 equiv), EtOH, reflux | 6d (61) |
| 6 | 8, 9a | KOH (1 equiv), EtOH, reflux | 6a (57) |
Molar ratio 8/9 = 1:1.
Quantum-Chemical Calculations of the Mechanism of the Reaction of 5 and 7
A quantum-chemical study of possible mechanisms of the reaction between thioacrylamides 5 and α-thiocyanatoacetophenone 7 was performed using the ORCA 5.0.1 software package.90−92 The search for the transition state, determination of reaction routes, and calculation of vibrational frequencies and Gibbs free energy were performed using DFT and the new Grimme composite approach r2SCAN-3c.93 This approach is a combination of r2SCAN functional with mTZVPP basis, including atom-pairwise dispersion correction based on tight binding partial charges D494 and geometrical counterpoise correction gCP.95 The found geometry of transition states was confirmed by the presence of an imaginary vibrational frequency corresponding to the reaction coordinate. All the calculations were performed with the correction for the influence of nonspecific solvation (EtOH) using the CPCM model.96 We used the Gabedit 2.5 software97 to generate the input files and the ChemCraft 1.8 software98 to visualize the molecular geometry and vibrational frequencies.
At a first glance at the overall reaction, it seems likely that the first step is the formation of the Michael adducts 10 (Scheme 5). However, further intramolecular cyclization of the Michael adducts 10 can proceed by two different mechanisms (Scheme 5, pathways A and B). In the first case (pathway A), one-step intramolecular nucleophilic substitution of the thiocyanate group at the carbon atom (C1) by the sulfur atom (S14) can occur. The literature data on the nucleofugality of pseudohalide NCS– anion are rather scarce, though some substitution reactions with thiocyanate as a leaving group were reported.99−106 It is likely that trans stereochemistry of the target ADHTs 6 should be determined at the formation of the Michael adducts 10 since bulky benzoyl and (het)aryl substituents would occupy a sterically favorable trans relationship. Therefore, two enantiomeric pairs of the Michael adducts 10A,10B and 10C,10D (Scheme 5) with an anti-periplanar orientation of (het)aryl and benzoyl groups appear to be the most stable.
Scheme 5. Possible Mechanisms of the Formation and Stereochemistry of ADHTs 6.

In the second case (pathway B), a four-stage process is assumed, including the nucleophilic addition of the carbon atom (C3) to the nitrile fragment (C6-N7) of the thiocyanate group followed by the transfer of a proton from the nitrogen atom (N13) to the nitrogen atom (N7) and by the elimination of HNCS and transfer of the second proton from nitrogen atom (N13) to nitrogen atom (N7). Arguments in favor of the pathway B involving the nucleophilic attack of a carbanion on the carbon atom of the SCN group come from numerous examples of the reactions of organic thiocyanates with active methylene compounds.87,107
To determine the most plausible mechanism, a quantum-chemical study of the reaction trajectories was carried out. Molecular geometries and Gibbs energies for intermediates and transition states were calculated.
The optimized molecular structures of most stable conformations, all transition states, and intermediates are shown in Figures 3–6, and the calculated energy diagrams are shown in Figure 7. Since the starting anionic intermediate 10 contains two asymmetric carbon atoms (C1 and C2), it can exist as four diastereomers or two enantiomeric pairs: R,S/S,R and S,S/R,R. Using S,S- and R,S-structures, we have calculated possible reaction pathways for each diastereomeric channel. A preliminary conformational analysis was performed for the studied isomers, and the most stable conformations R1 (for the S,S-isomer) and R2 (for the R,S-isomer) were found (Figure 3). It is noteworthy that the difference in energy between the most stable conformations of these isomers is extremely small (less than 1.5 kJ/mol; the R,S-isomer is more stable).
Figure 3.

The most stable conformations R1 (for the S,S-isomer) and R2 (for the R,S-isomer). Geometry optimized at the r2SCAN-3c level.
Figure 6.

The optimized molecular structures of intermediates formed during the thiophene ring formation through the addition–elimination mechanism starting from S,S- (I1.1, I1.2, I1.3) and R,S-isomers (I2.1, I2.2, I2.3) (geometry optimized at the r2SCAN-3c level).
Figure 7.

Energy diagrams of the proposed cyclization mechanisms: intramolecular nucleophilic substitution of thiocyanate anion (S,S/R,R-diastereomeric channel, pathway A) or nucleophilic addition at the SCN nitrile fragment with subsequent elimination of thiocyanate anion (R,S/S,R-diastereomeric channel, pathway B). The calculated Gibbs energies are given relative to the energy of the most stable conformation of the S,S-isomer (R1).
First, proper conformational changes of the intermediate 10 are required for successful intramolecular nucleophilic substitution since S14, C1, and S4 atoms should occupy suitable relative positions in the molecule to permit a nucleophilic attack. The rotation barrier around the C1–C2 bond was calculated as 31.8 kJ/mol for the S,S-isomer and 34.0 kJ/mol for the R,S-isomer. The transition states of the indicated conformational changes (TS1c and TS2c) and the processes of subsequent nucleophilic substitution (TS1 and TS2) are shown in Figure 4.
Figure 4.

The optimized molecular structures of SN transition states (TS) for S,S-(TS1c, TS1) and R,S-isomers (TS2c, TS2) (geometry optimized at the r2SCAN-3c level).
As a result of calculations, it was found that intramolecular nucleophilic substitution (pathway A, Scheme 5) can be realized successfully only in the S,S/R,R-diastereomeric channel. In the R,S-isomer, S14, C1, and S4 atoms cannot occupy a configuration suitable for the SN2 process due to spatial difficulties caused by the bulky benzoyl group that prevents the sulfur S14 from approaching the C1 carbon from the rear side. Thereby, the process of synchronous substitution is disrupted, which leads to a sharp increase in the activation energy up to 298.8 kJ/mol. At the same time, the calculated activation energy of nucleophilic substitution for the S,S-isomer is even slightly lower (27.9 kJ/mol) than that of the previous conformational transition.
As for the alternative mechanism (pathway B, Scheme 5), the entire process can be presented as a sequence of the following four steps (the optimized molecular structures of transition states are shown in Figure 5):
-
1.
Nucleophilic addition of C3 atom to C6 atom of the SCN group through the formation of transition states TS1.1 (for the S,S-isomer) and TS2.1 (for the R,S-isomer), leading to the closure of the thiophene ring and the formation of anionic intermediates I1.1 and I2.1. According to the calculated data, the activation energy of this process is 54.5 kJ/mol for the S,S-isomer and 46.6 kJ/mol for the R,S-isomer. It should be noted that the process does not require preliminary conformational changes and can be realized directly from the most stable conformers R1 and R2.
-
2.
Transfer of H28 proton from N13 nitrogen atom to N7 atom with the formation of transition states TS1.2 (for the S,S-isomer) and TS2.2 (for the R,S-isomer) with the formation of intermediate products I1.2 and I2.2.
-
3.
The elimination of HNCS with the cleavage of the C3–C10 bond. This stage has the highest activation energy (63.4 kJ/mol for the S,S-isomer and 77.2 kJ/mol for the R,S-isomer) and therefore should be considered as the rate-limiting step.
-
4.
Transfer of the second proton H29 from N13 atom of the HNCS molecule to N7 atom with the formation of transition states TS1.4 (for the S,S-isomer) and TS2.4 (for the R,S-isomer), leading to the formation of final dihydrothiophenes 6.
Figure 5.

The optimized molecular structures of transition states formed during the thiophene ring formation through the addition–elimination mechanism starting from S,S- (TS1.1, TS1.2, TS1.3, TS1.4) and R,S-isomers (TS2.1, TS2.2, TS2.3, TS2.4) (geometry optimized at the r2SCAN-3c level).
Overall, the reaction of thioacrylamides 5 with α-thiocyanatoacetophenone 7 can proceed through the cyclization of the Michael adducts 10 by two alternative pathways: intramolecular SN2 substitution of the SCN group (pathway A, Scheme 5) and nucleophilic addition/elimination of HNCS sequence (pathway B, Scheme 5). For S,S/R,R-diastereomers, both variants are possible, but nucleophilic substitution (pathway A) seems to be more likely due to the lower activation energy. Moreover, from the stereochemical point of view, the intramolecular nucleophilic substitution for the S,S-isomer should lead to the formation of trans isomers of 6, while the realization of the alternative addition–elimination process should give only cis isomers that were not observed experimentally. In contrast, the intramolecular SN2 reaction cannot be realized with R,S/S,R-diastereomers of 10 due to steric hindrance. Therefore, in this case, cyclization can proceed only through the addition–elimination mechanism (pathway B) with the formation of trans isomers of dihydrothiophenes 6. The calculations performed are thus consistent with the experimental results pointing to the formation of trans isomers of dihydrothiophenes 6.
The Noncatalyzed Aminomethylation of ADHTs Leading to Thieno[2,3-d]pyrimidines
Thieno[2,3-d]pyrimidines are purine bioisosteres coming to the center of interest due to their high structural diversity and well-documented spectrum of biological activity (for reviews, see refs (108−115)). Much less is known on the preparation and reactions of partially saturated thieno[2,3-d]pyrimidines.116−120 To study the reactivity of the prepared ADHTs 6 under Mannich conditions, their behavior upon treatment with a series of primary amines and HCHO was examined here.
We found that ADHTs 6 react with RNH2 and excessive HCHO resulted in double aminomethylation to afford new 2,3,4,4a,5,6-hexahydrothieno[2,3-d]pyrimidines 11 (Scheme 6). To optimize the reaction conditions, p-toluidine and ADHT 6b were chosen as model reagents. We found that EtOH was a superior solvent compared to DMF, MeOH, or i-PrOH (Table 3, entries 1–2 and 4–5). This is probably connected with the better solubility of either products or starting ADHTs 6 in a solvent. However, the use of DMF or DMF–EtOH mixtures as solvents is also effective in the case of less soluble ADHT 6a. Doubling the amount of the amine component did not affect the yields of product 11e (Table 3, entry 3).
Scheme 6. The Aminomethylation of ADHTS 6.

Table 3. Optimization of the Conditions for the Synthesis of Thienopyrimidines 11.
| entry | reagentsa | conditions | product (yield, %) |
|---|---|---|---|
| 1 | 6b, 4-MeC6H4NH2 (1.05 equiv) | EtOH, reflux | 11e (69) |
| 2 | 6b, 4-MeC6H4NH2 (1.05 equiv) | DMF, reflux | 11e (44) |
| 3 | 6b, 4-MeC6H4NH2 (2 equiv) | DMF, reflux | 11e (43) |
| 4 | 6b, 4-MeC6H4NH2 (1.05 equiv) | MeOH, reflux | 11e (63) |
| 5 | 6b, 4-MeC6H4NH2 (1.05 equiv) | i-PrOH, reflux | 11e (61) |
At least 10-fold excess of aq 37% HCHO used in each entry.
With the optimized conditions, good yields (60–86%) of new 2,3,4,4a,5,6-hexahydrothieno[2,3-d]pyrimidines 11a–l were achieved (Scheme 6 and Figure 8). Also, when p-phenylenediamine was reacted with 2 equiv of ADHT 6d and excessive HCHO, polycyclic compound 12 was isolated in 48% yield. It is noteworthy that the reaction required no catalysts. It should be noted that ADHTs 6 play here an unusual role of 1,3-dinucleophilic β-enaminonitrile species. In general, the Mannich-type reactions of β-enaminocarbonyls and related enamines with HCHO and primary amines leading to tetrahydropyrimidines are well known;121−134 however, as far as we know, no cyclic enaminonitriles were reported as substrates in the Mannich reaction prior to our studies.
Figure 8.

The scope and yields of products 11.
The structure of the products was supported by IR, 1H NMR, 13C APT NMR, HPLC–MS, and elemental analysis.1H NMR spectra of compounds 11,12 revealed the presence of signals of the =NCH2NCH2– fragment: two doublets (or AB-quartet) of methylene protons C(4)H2 at δ 2.87–3.69 and δ 3.03–4.20 ppm (2J = 11.4–12.6 Hz) and two doublets (or AB-quartet) of methylene protons C(2)H2 at δ 4.05–4.79 ppm and δ 4.50–5.37 ppm (2J = 16.7–17.4 Hz). The doublet of C(5)H was observed at δ 4.04–4.64 ppm, and C(6)H appeared as doublet at δ 5.97–6.38 ppm (3J = 10.4–10.9 Hz). The IR spectra of compounds 11,12 revealed the absence of absorption bands of NH2 and conjugated C≡N groups. Instead, a weak band appears at ν 2235–2245 cm–1 (nonconjugated C≡N). The absorption bands corresponding to the vibrations of C=O and C=N groups are found at ν 1680–1690 and 1647–1664 cm–1. In addition, the structure of compound 11l was confirmed by X-ray studies (Figure 9).
Figure 9.

The structure of compound 11l according to X-ray data. Thermal ellipsoids of nonhydrogen atoms are shown at the 30% probability level.
The independent part of the unit cell contains two molecules of compound 11l: A and B with a close conformation. The tetrahydropyrimidine ring has a distorted half-chair conformation with an almost planar fragment C6-N2-C4-C3 (torsion angle 4.7(3)° in molecule 11l-A and 3.2(3)° in molecule 11l-B). Atoms N1 and C5 deviate from this plane by 0.243(4) and −0.516(4) Å (molecule 11l-A) and by 0.276(4) and −0.496(4) Å (molecule 11l-B). The tetrahydrothiophene ring is in the twist conformation with the deviation of the C2 and C3 atoms from the plane of the rest of the ring atoms by −0.469(4) and 0.250(4) Å (11l-A) and by −0.258(4) and 0.396(4) Å (11l-B). The nitrogen atom N1 has a pyramidal configuration; the sum of the bond angles centered on the atom is 344.7° (11l-A) or 349.8° (11l-B). The substituents at C1, C2, and N1 atoms have an equatorial orientation (torsion angles C4-S1-C1-C7 139.98(15)° (11l-A) and 131.00(15)° (11l-B), S1-C1-C2-C14 −172.61(14)° (11l-A) and −163.25(14)° (11l-B), and N2-C6-N1-C21 178.98(18)° (11l-A) and 169.30(17)° (11l-B)). The nitrile group is in the axial position (torsion angle N2-C4-C3-C20–101.5(2)° (11l-A) and −100.4(2)° (11l-B)). Molecules 11l-A and 11l-B differ in the orientation of the phenyl substituent at the N1 atom, which is in the –ac conformation in molecule 11l-A and in the –sc conformation in molecule 11l-B relative to the idealized position of the lone pair (Lp) of electrons of the nitrogen atom (torsion angle C22-C21-N2-Lp(N1) −122° (11l-A) and −61° (11l-B)). The rotation of the substituent is facilitated by the repulsion between hydrogen atoms in the ortho positions (shortened intramolecular contacts H26...H6a 2.19 Å (11l-A) and 2.23 Å (11l-B), and H22...H5a 2.17 Å (11l-A, 11l-B) (sum of van der Waals radii 2.32 Å135)). The molecule also contains shortened intramolecular contacts between the hydrogen atom at C1 and hydrogen atoms in the ortho positions of the phenyl substituents at C2 and C7 (H1...H9 2.22 Å (11l-A) and 2.13 Å (11l-B), and H1...H15 2.29 Å (11l-A) and 2.24 Å (11l-B)).
Crystals of compound 11l consist of alternating layers of molecules 11l-A and 11l-B parallel to the plane (0 0 1). In this case, layers 11l-A and 11l-A have different structures. A common feature of molecules 11l-A and 11l-B is the formation of centrosymmetric dimers due to hydrogen bonds between the carbonyl group and methylene hydrogen atoms (C5-H5a...O1i [i: −x, 1 – y, 1 – z] (H...O 2.31 Å, C–H...O 152°) in layer A and C6-H6b...O1ii [ii: 1 – x, 1 – y, −z] (H...O 2.45 Å, C–H...O 141°)). Also in layer A, molecules are linked by hydrogen bonds C16-H16...N3ii [ii: 1 – x, −y, 1 – z] (H...N 2.58 Å, C–H...N 136°) and C–H...π contacts C6-H6b...C11iii [iii: −1 + x, y, z] (H...C 2.89 Å, C–H...C 135°) and C12-H12...C16iv [iv: 1 – x, 1 – y, 1 – z] (H...C 2.89 Å, C–H...C 158°). In layer B, the molecules are linked by intermolecular hydrogen bonds C1-H1...N3v [v: 2 – x, 1 – y, −z] (H...N 2.53 Å, C–H...N 158°) and C9-H9...N3v (H...N 2.52 Å, C–H...N 157°) and C–H...π contacts C13-H13...C26ii (H...C 2.87 Å, C–H...C 179°), C26-H26...C10vi [vi: −1 + x, 1 + y, z] (H...C 2.87 Å, C–H...C 154°), and C6-H6b...C24vii [vii: 1 – x, 2 – y, −z] (H...C 2.78 Å, C–H...C 133°).
The copies of IR and NMR spectra as well as LCMS and X-ray data for new compounds are given in the Supporting Information.
Conclusions
In summary, we have optimized synthetic procedures for the preparation of highly functionalized, useful building blocks, trans-2-amino-4-aryl-5-benzoyl-4,5-dihydrothiophene-3-carbonitriles 6 (ADHTs), starting from easily available α-thiocyanatoacetophenone 7 and cyanothioacetamide 8. The reaction mechanism was studied on the r2SCAN-3c level of theory. The reaction of α-thiocyanatoacetophenone 7 with 3-aryl-2-cyanothioacrylamides 5 proceeds through the formation of the corresponding Michael adduct that undergoes further cyclization. The cyclization can proceed by two different mechanisms: by intramolecular SN2 substitution of the SCN group (only for S,S/R,R-diastereomers of the Michael adduct 10) or through intramolecular nucleophilic addition to the SCN carbon atom followed by elimination of the HNCS molecule (only for S,R/R,S-diastereomers of the Michael adduct 10), as it was supported by quantum chemical calculations performed on the r2SCAN-3c level of theory.
In addition, a new approach for the preparation of ADHTs 6 was developed, starting from cyanothioacetamide 8 and available α-bromochalcones 9. We have also demonstrated that a small library of new 2,3,4,4a,5,6-hexahydrothieno[2,3-d]pyrimidine-4a-carbonitriles 11 could be synthesized by noncatalyzed double Mannich-type cyclization, starting from ADHTs 6, primary amines, and aq HCHO. The work demonstrates a new approach to the formation of pharmacologically interesting thieno[2,3-d]pyrimidines. The procedure has certain advantages such as mild reaction conditions, short reaction time, and a diversity of useful starting building blocks and provides pure target products in good yields. All the reported procedures preclude volatile, foul smelling, or toxic solvents or byproducts. The formation of both starting ADHTs 6 and thieno[2,3-d]pyrimidines 11 proceeds in an atom-economical way with a broad substrate scope under metal-free conditions.
Experimental Section
Solvents and starting reagents were purified according to common procedures. Melting points were determined on a Kofler hot stage and reported uncorrected. IR spectra were recorded on an IKS-29 spectrometer in Nujol mulls or a Thermo Nicolet Avatar 370 FT-IR spectrometer in KBr pellets. 1H and 13C NMR spectra were recorded on a Bruker DRX-500 (500.13 MHz for 1H and 125.74 MHz for 13C) or Bruker DPX-400 (400.40 MHz for 1H) spectrometer at room temperature in DMSO-d6. Chemical shifts are given in parts per million (ppm) with reference to TMS or to the residual solvent signals; coupling constants are given in Hz; and multiplicities are given as s (singlet), d (doublet), dd (doublet of doublets), m (multiplet), and br (broad). LC–MS data were obtained using the LC–MS system consisting of the high-performance liquid chromatograph Agilent 1100 equipped with diode-matrix and mass-selective detector Agilent LC/MSD SL (APCI ionization in positive and negative modes) and on a PE SCIEX API 150EX mass spectrometer following separation on a Shimadzu LC-10 AD liquid chromatography system equipped with a Shimadzu SP D-10AUV–vis detector (254 nm) and Sedex 75 ELSD detector (ES-API ionization). The elemental analysis (C, H, N) was performed using a Carlo Erba Strumentazione 1106 analyzer. The analytical results were within ±0.4% of the theoretical values. Thin-layer chromatography (TLC) was performed on Silufol UV-254 plates using EtOAc, EtOAc–hexane, or acetone–hexane 1:1 mixture as eluents; the spots were visualized with iodine vapors, UV light, or KMnO4–H2SO4 solution. Cyanothioacetamide 8 was prepared by bubbling H2S gas through a malononitrile solution in EtOH containing a catalytic amount of Et3N at 10–15 °C for 6–8 h.136 3-Aryl-2-cyanothioacrylamides 5 were prepared by the Knoevenagel condensation of cyanothioacetamide 8 with aromatic aldehydes in the presence of catalytic amounts of Et3N (EtOH, 20 °C).137 α-Bromochalcones 9a,d were prepared by dehydrobromination of the corresponding chalcone dibromide.138,139
α-Thiocyanatoacetophenone 7 was prepared as follows: the mixture of α-bromoacetophenone (23.5 g, 0.118 mol) and dry KSCN (12.6 g, 0.13 mol) in anhydrous acetone (70 mL) was boiled under vigorous stirring for 1 h and evaporated to one-half of the volume. The slurry obtained was cooled to 25 °C and treated with 50 mL of cold water. The precipitate formed was filtered off and washed with water and twice with cold 40% aq EtOH to give 20.6 g (98.5%) of thiocyanate 7 as colorless crystals, mp 74–75 °C [lit.:140 72.5–73.5 °C].
General Procedures for the Synthesis of 2-Amino-5-benzoyl-4-(het)aryl-4,5-dihydrothiophene-3-carbonitriles (6)
A. KOH-Catalyzed Reaction of Aldehydes, Cyanothioacetamide 8, and Phenacyl Thiocyanate 7 (Table 1, Entries 1 and 5)
To a suspension of 0.5 g (5 mmol) of cyanothioacetamide 8 in 20 mL of EtOH, 5 mmol of the corresponding aldehyde and one drop of 10% aq KOH were successively added with stirring. The mixture was stirred for 0.5 h, and then 0.89 g (5 mmol) of α-thiocyanatoketone 7 and an excess (4 mL) of 10% aq KOH were added. The mixture was stirred for 0.5 h, diluted with water (10 mL), and then kept for 0.5 h at 25 °C. The precipitate of dihydrothiophenes 6a or 6b was filtered off and purified by recrystallization from EtOH–acetone.
B. KOH-Catalyzed Reaction of Thioacrylamides 5a,b and Phenacyl Thiocyanate 7 (Table 1, Entries 7 and 13)
To a suspension of 2.5 mmol of α,β-unsaturated thioamide 5a,b (0.55 g of 5a or 0.45 g of 5b) and 0.44 g (2.5 mmol) of α-thiocyanatoketone 7 in 10 mL of EtOH, an excess (2.0 mL) of 10% aq KOH was added dropwise with vigorous stirring. The mixture immediately turned red, and the starting reagents dissolved. The reaction mixture quickly turned yellow, and a yellow solid precipitated within 30–60 s. The mixture was stirred for 0.5 h, diluted with 5 mL of water, and then allowed to stand for another 0.5 h. The precipitate was filtered off, washed with water and cold EtOH, and recrystallized from EtOH–acetone. The yields were 323 mg (38%, 6a) and 300 mg (40%, 6b).
C. Na2CO3-Catalyzed Reaction of Thioacrylamides 5a–c and Phenacyl Thiocyanate 7 (Table 1, Entries 12, 17, and 18)
To a suspension of 2.25 mmol of unsaturated thioamides 5a–c and 400 mg (2.26 mmol) of α-thiocyanatoketone 7 in 10 mL of alcohol, 2.4 mL of 10% aq Na2CO3 solution was added with stirring (Na2CO3 partially precipitated). The reaction mixture was stirred under gentle heating (40–50 °C) until the starting reagents had dissolved. The solution turned light brown, and CO2 was evolved. The mixture was allowed to cool to 25 °C and diluted with 3–4 mL of water (a yellow solid precipitated). After 72 h, the precipitate was filtered off, washed with water and cold EtOH, and purified (if appropriate) by recrystallization from EtOH–acetone.
D. Na2CO3-Catalyzed Reaction of Aldehydes, Cyanothioacetamide 8, and Phenacyl Thiocyanate 7 (Table 1, Entries 4, 6, 19, and 20)
To a suspension of 0.5 g (5 mmol) of cyanothioacetamide 8 in 10 mL of EtOH, 5 mmol of the corresponding aldehyde and one drop of 10% aq Na2CO3 were successively added with stirring. The mixture was stirred for 0.5 h, and then 0.89 g (5 mmol) of α-thiocyanatoketone 7 and an excess (4 mL) of 10% aq Na2CO3 were added. The mixture was stirred for 0.5 h, diluted with water (10 mL), and then kept for 0.5 h at 25 °C. The precipitate of dihydrothiophenes 6a–d was filtered off and purified (if appropriate) by recrystallization from EtOH–acetone.
E. Synthesis of ADHTs 6a,d from Cyanothioacetamide 8 and α-Bromochalcones 9a,d (Table 2, Entries 5 and 6)
The mixture of 400 mg (4.0 mmol) of cyanothioacetamide 8, 4.0 mmol of the corresponding α-bromochalcone 9a,d, and EtOH (20 mL) was treated with 2.4 mL (4 mmol) of 10% aq KOH. The resulting red solution was slowly brought to reflux under vigorous stirring and kept for 0.5 h. The mixture was cooled; after 24 h, the precipitate was filtered off, washed with 50% EtOH and ether, and purified by recrystallization from acetone/n-BuOH (2:1) to give 6a,d as yellow crystalline solids.
trans-2-Amino-5-benzoyl-4-(2-chlorophenyl)-4,5-dihydrothiophene-3-carbonitrile (6a)
Recrystallization from 1:1 acetone/EtOH gave bright yellow crystals, mp 243–245 °C (dec.). IR (Nujol, cm–1) νmax 3415, 3310, 3200 (NH2), 2195 (C≡N), 1675 (C=O).1H NMR (400 MHz, DMSO-d6) δ 4.83 (1H, d, 3J = 2.9 Hz, H-4), 5.19 (1H, d, 3J = 2.9 Hz, H-4), 7.00 (2H, br s, NH2), 7.20–7.52 (7H, m, H–Ar), 7.85 (2H, d, 3J = 7.1 Hz, H-2 and H-6 benzoyl). 13C NMR (126 MHz, DMSO-d6) δ 46.9 (C-4), 55.9 (C-5), 71.2 (C-3), 119.0 (C≡N), 128.0 (C–Ar), 129.0 (C–Ar), 129.3 (C–Ar), 129.9 (C–Ar), 131.8 (C–Ar), 132.1 (C–Ar), 133.5 (C–Ar), 134.0 (C–Ar), 135.1 (C–Ar), 140.5 (C–Ar), 161.3 (C-2), 193.0 (C=O). Anal. Calcd for C18H13ClN2OS: C, 63.43; H, 3.84; N, 8.22. Found C, 63.31; H, 3.93; N, 8.18.
trans-2-Amino-5-benzoyl-4-(2-furyl)-4,5-dihydrothiophene-3-carbonitrile (6b)
Recrystallization from 1:1 EtOH/1,4-dioxane gave dark yellow crystals, mp 208–210 °C. IR (KBr, cm–1) νmax 3405, 3290, 3170 (NH2), 2200 (C≡N), 1670 (C=O).1H NMR (400 MHz, DMSO-d6) δ 4.90 (1H, d, 3J = 3.0 Hz, H-4), 5.17 (1H, d, 3J = 3.0 Hz, H-5), 6.29–6.30 (1H, m, H-3 furyl), 6.34–6.35 (1H, m, H-4 furyl), 6.97 (2H, br s, NH2), 7.45–7.63 (4H, m, 3 H–Ar and H-5 furyl overlapped), 7.92 (2H, d, 3J = 7.1 Hz, H-2 and H-6 benzoyl).13C NMR (126 MHz, DMSO-d6) δ 46.0 (C-4), 54.9 (C-5), 71.0 (C-3), 104.0 (C-4 furyl), 109.1 (C-3 furyl), 118.7 (C≡N), 128.3 (C–Ar), 131.0 (C–Ar), 133.6 (C–Ar), 135.1 (C–Ar), 142.9 (C-5 furyl), 161.1 (C-2), 163.2 (C-2 furyl), 192.7 (C=O). Anal. Calcd for C16H12N2O2S: C, 64.85; H, 4.08; N, 9.45. Found C, 64.80; H, 4.16; N, 9.43.
trans-2-Amino-5-benzoyl-4-(2-thienyl)-4,5-dihydrothiophene-3-carbonitrile (6c)
Recrystallization from 1:1 acetone/EtOH gave yellow crystals, mp 211–213 °C. IR (KBr, cm–1) νmax 3405, 3290, 3175 (NH2), 2202 (C≡N), 1670 (C=O).1H NMR (400 MHz, DMSO-d6) δ 4.84 (1H, d, 3J = 3.0 Hz, H-4), 5.24 (1H, d, 3J = 3.0 Hz, H-5), 7.07 (2H, br s, NH2), 7.10–7.11 (1H, m, H-3 thienyl), 7.40–7.41 (1H, m, H-4 thienyl), 7.51–7.52 (1H, m, H-5 thienyl), 7.55–7.65 (3H, m, H-3, H-4, H-5 benzoyl), 7.90 (2H, d, 3J = 7.3 Hz, H-2 and H-6 benzoyl).13C NMR (126 MHz, DMSO-d6) δ 50.8 (C-4), 55.5 (C-5), 71.3 (C-3), 119.1 (C≡N), 123.1 (C–Ar), 125.0 (C–Ar), 128.6 (C–Ar), 130.8 (C–Ar), 131.1 (C–Ar), 133.0 (C–Ar), 135.1 (C–Ar), 144.6 (C–Ar), 160.9 (C-2), 193.0 (C=O). Anal. Calcd for C16H12N2OS2: C, 61.51; H, 3.87; N, 8.97. Found C, 61.57; H, 4.00; N, 8.90.
trans-2-Amino-5-benzoyl-4-phenyl-4,5-dihydrothiophene-3-carbonitrile (6d)
Recrystallization from 1:2 n-BuOH/acetone gave yellow crystals, mp 207–209 °C. IR (KBr, cm–1) νmax 3412, 3300, 3184 (NH2), 2201 (C≡N), 1670 (C=O).1H NMR (400 MHz, DMSO-d6) δ 4.75 (1H, d, 3J = 3.0 Hz, H-4), 5.21 (1H, d, 3J = 3.0 Hz, H-5), 7.13 (2H, br s, NH2), 7.27–7.32 (1H, m, H–Ph), 7.37–7.38 (4H, m, H–Ph), 7.49–7.59 (2H, m, H-3, H-5 benzoyl), 7.62–7.66 (1H, m, H-4 benzoyl), 7.90 (2H, d, 3J = 7.5 Hz, H-2 and H-6 benzoyl).13C NMR (126 MHz, DMSO-d6) δ 50.8 (C-4), 56.0 (C-5), 70.9 (C-3), 118.2 (C≡N), 127.3 (C-4 Ph), 127.4 (C-3, C-5 Ph), 128.61 (C-2, C-6 Ph), 128.62 (C-3, C-5 benzoyl), 128.8 (C-2, C-6 benzoyl), 133.6 (C-4 benzoyl), 134.5 (C-1 benzoyl), 141.8 (C-1 Ph), 161.1 (C-2), 193.0 (C=O). LCMS (m/z, ES-API) 307.5 [M + H]+, 613.3 [2 M + 1]+, 919.3 [3 M + 1]+. Anal. Calcd for C18H14N2OS: C, 70.56; H, 4.61; N, 9.14. Found C, 70.60; H, 4.70; N, 9.08.
General Procedure for the Synthesis of 2,3,4,4a,5,6-Hexahydrothieno[2,3-d]pyrimidine-4a-carbonitriles (11a–i)
The corresponding ADHTs 6a-d (0.6–0.8 mmol) and a primary amine (1.05 equiv, 0.65–0.85 mmol) were dissolved in EtOH (10–12 mL) (for less soluble ADHTs 6a,b, DMF (2 mL) or DMF–EtOH mixture (2 + 8 mL) is also useful), and an excess (1.0 mL) of 37% aq HCHO was added to the resulting solution. The reaction mixture was heated to reflux under vigorous stirring for 2–3 min (in some cases, a colorless crystalline solid started to separate). The reaction mixture was allowed to stand for 24 h at 20 °C, and the crystals were filtered off, washed with EtOH and hexane, and purified (if appropriate) by recrystallization to give thieno[2,3-d]pyrimidines 11 as colorless crystals.
6-Benzoyl-3-benzyl-5-(2-chlorophenyl)-2,3,4,4a,5,6-hexahydrothieno[2,3-d]pyrimidine-4a-carbonitrile (11a)
Recrystallization from acetone gave colorless crystals, yield 63%, mp 208–210 °C. IR (KBr, cm–1) νmax 2235 (C≡N), 1680 (C=O), 1657 (C=N).1H NMR (500 MHz, DMSO-d6) δ 2.87 (1H, d, 2J = 11.4 Hz, H-4), 3.03 (1H, d, 2J = 11.4 Hz, H-4), 3.65 (1H, d, 2J = 13.7 Hz, CH2Ph), 3.77 (1H, d, 2J = 13.7 Hz, CH2Ph), 4.11 (1H, d, 2J = 17.1 Hz, H-2), 4.49–4.56 (2H, m, two doublets overlapped: 1H, d, H-2 and 1H, d, H-5), 6.30 (1H, d, 3J = 10.9 Hz, H-6), 7.20–7.22 (1H, m, H-4 Ph), 7.26–7.37 (6H, m, H–Ar), 7.50 (1H, d, 3J = 7.3, H–Ar), 7.55–7.58 (2H, m, H–Ar), 7.68–7.71 (1H, m, H–Ar), 8.03 (1H, d, 3J = 7.8, H–Ar), 8.07 (2H, d, 3J = 7.8, H-2 and H-6 benzoyl).13C NMR (126 MHz, DMSO-d6) δ 47.1 (C-5), 49.7 (C-6), 52.2 (C-4a), 53.1 (CH2Ph), 56.3 (C-4), 70.6 (C-2), 116.8 (C≡N), 127.3 (C–Ar), 127.5 (C–Ar), 128.27 (C–Ar), 128.31 (C–Ar), 128.9 (C–Ar), 129.1 (C–Ar), 129.75 (C–Ar), 129.79 (C–Ar), 130.0 (C–Ar), 131.3 (C–Ar), 134.2 (C–Ar), 134.68 (C–Ar), 134.73 (C–Ar), 137.0 (C–Ar), 161.7 (C=N), 192.9 (C=O). LCMS (m/z, ES-API) 120.7 [PhCH2N = CH2 + H]+, 472.6 [M + H]+, 945.8 [2M + H]+. Anal. Calcd for C27H22ClN3OS: C, 68.71; H, 4.70; N, 8.90. Found C, 68.70; H, 4.76; N, 8.88.
6-Benzoyl-5-(2-chlorophenyl)-3-phenyl-2,3,4,4a,5,6-hexahydrothieno[2,3-d]pyrimidine-4a-carbonitrile (11b)
Recrystallization from DMF/H2O 1:1 gave a snow-white fine crystalline powder, yield 75%, mp 261–263 °C. IR (KBr, cm–1) νmax 2235 (C≡N), 1682 (C=O), 1651 (C=N).1H NMR (400 MHz, DMSO-d6) δ 3.64 (1H, d, 2J = 12.6 Hz, H-4), 3.91 (1H, d, 2J = 12.6 Hz, H-4), 4.63 (1H, d, 3J = 10.6 Hz, H-5), 4.79 (1H, d, 2J = 17.4 Hz, H-2), 5.35 (1H, d, 2J = 17.4 Hz, H-2), 6.38 (1H, d, 3J = 10.6 Hz, H-6), 6.83–6.87 (1H, m, H-4 Ph), 6.91 (2H, d, 3J = 7.9, H–Ph), 7.20–7.24 (2H, m, H–Ar), 7.39–7.41 (2H, m, H–Ar), 7.56–7.60 (3H, m, H–Ar), 7.70–7.74 (1H, m, H-4 benzoyl), 8.07–8.11 (3H, m, H–Ar).13C NMR (126 MHz, DMSO-d6) δ 47.2 (C-5), 49.5 (C-6), 50.9 (C-4a), 51.4 (C-4), 66.7 (C-2), 116.2 (C–Ar), 116.3 (C≡N), 120.4 (C–Ar), 127.5 (C–Ar), 128.9 (C–Ar), 129.1 (C–Ar), 129.2 (C–Ar), 129.8 (C–Ar), 129.9 (C–Ar), 130.1 (C–Ar), 131.3 (C–Ar), 134.4 (C–Ar), 134.7 (C–Ar), 134.8 (C–Ar), 146.9 (C-1 NPh), 161.7 (C=N), 192.9 (C=O). Anal. Calcd for C26H20ClN3OS: C, 68.19; H, 4.40; N, 9.17. Found C, 68.15; H, 4.48; N, 9.16.
6-Benzoyl-5-(2-chlorophenyl)-3-(4-methoxyphenyl)-2,3,4,4a,5,6-hexahydrothieno[2,3-d]pyrimidine-4a-carbonitrile (11c)
Recrystallization from acetone gave a beige crystalline solid, yield 72%, mp 218–220 °C. IR (Nujol mulls, cm–1) νmax 2240 (C≡N), 1685 (C=O), 1650 (C=N).1H NMR (400 MHz, DMSO-d6) δ 3.55 (1H, d, 2J = 12.5 Hz, H-4), 3.65 (1H, d, 2J = 12.5 Hz, H-4), 3.68 (3H, s, MeO), 4.60 (1H, d, 3J = 10.6 Hz, H-5), 4.64 (1H, d, 2J = 17.0 Hz, H-2), 5.23 (1H, d, 2J = 17.0 Hz, H-2), 6.36 (1H, d, 3J = 10.6 Hz, H-6), 6.85 (4H, AB-q, 3J = 8.9, H-Ar 4-MeOC6H4) 7.33–7.50 (3H, m, H–Ar), 7.59–7.70 (4H, m, H–Ar), 8.09 (2H, d, 3J = 7.8, H-2 and H-6 benzoyl).13C NMR (126 MHz, DMSO-d6) δ 47.2 (C-5), 50.1 (C-6), 51.5 (C-4a), 52.1 (C-4), 55.4 (MeO), 68.9 (C-2), 114.0 (C–Ar), 116.7 (C≡N), 119.0 (C–Ar), 128.7 (C–Ar), 129.0 (C–Ar), 129.2 (C–Ar), 129.7 (C–Ar), 129.9 (C–Ar), 130.2 (C–Ar), 131.3 (C–Ar), 134.4 (C–Ar), 134.7 (C–Ar), 134.8 (C–Ar), 141.3 (C-1 NAr), 154.0 (C–OMe), 162.1 (C=N), 193.0 (C=O). Anal. Calcd for C27H22ClN3O2S: C, 66.45; H, 4.54; N, 8.61. Found C, 66.39; H, 4.60; N, 8.56.
6-Benzoyl-5-(2-furyl)-3-phenyl-2,3,4,4a,5,6-hexahydrothieno[2,3-d]pyrimidine-4a-carbonitrile (11d)
Recrystallization from EtOH–acetone 1:3 gave a beige crystalline solid, yield 60%, mp 187–189 °C. IR (KBr, cm–1) νmax 2241 (C≡N), 1688 (C=O), 1664 (C=N). 1H NMR (500 MHz, DMSO-d6) δ 3.69 (1H, d, 2J = 12.5 Hz, H-4), 4.20 (1H, d, 2J = 12.5 Hz, H-4), 4.34 (1H, d, 3J = 10.4 Hz, H-5), 4.73 (d, 2J = 17.1 Hz, H-2), 5.37 (d, 2J = 17.1 Hz, H-2), 5.97 (1H, d, 3J = 10.4 Hz, H-6), 6.47–6.48 (1H, m, H-4 furyl), 6.64–6.65 (1H, m, H-3 furyl), 6.88–6.91 (1H, m, H-4 Ph), 6.99 (2H, d, 3J = 7.8 Hz, H-2, H-6 Ph), 7.26–7.29 (2H, m, H-3, H-5 Ph), 7.59–7.62 (2H, m, H-3, H-5 benzoyl), 7.70–7.71 (1H, m, H-5 furyl), 7.73-7.76 (1H, m, H-4 benzoyl), 8.11 (2H, d, 3J = 8.3 Hz, H-2, H-6 benzoyl). 13C NMR (126 MHz, DMSO-d6) δ 45.4 (C-5), 47.6 (C-6), 49.6 (C-4a), 51.1 (C-4), 66.8 (C-2), 109.0 (C-3 furyl), 110.8 (C-4 furyl), 116.4 (C-2, C-6 NPh and C≡N overlapped), 120.5 (C-4 NPh), 128.8 (C–Ar), 129.21 (C–Ar), 129.25 (C–Ar), 134.75 (C–Ar), 134.79 (C–Ar), 143.7 (C-5 furyl), 147.0 (C-1 NPh), 147.9 (C-2 furyl), 161.5 (C=N), 192.6 (C=O). Anal. Calcd for C24H19N3O2S: C, 69.71; H, 4.63; N, 10.16. Found C, 69.69; H, 4.70; N, 10.18.
6-Benzoyl-5-(2-furyl)-3-(4-methylphenyl)-2,3,4,4a,5,6-hexahydrothieno[2,3-d]pyrimidine-4a-carbonitrile (11e)
Recrystallization from EtOH–DMF 4:1 gave colorless crystals, yield 69%, mp 199–201 °C. IR (Nujol mulls, cm–1) νmax 2240 (C≡N), 1690 (C=O), 1650 (C=N).1H NMR (400 MHz, DMSO-d6) δ 2.21 (3H, s, Me), 3.64 (1H, d, 2J = 12.4 Hz, H-4), 4.10 (1H, d, 2J = 12.4 Hz, H-4), 4.33 (1H, d, 3J = 10.6 Hz, H-5), 4.67 (d, 2J = 17.4 Hz, H-2), 5.30 (d, 2J = 17.4 Hz, H-2), 5.97 (1H, d, 3J = 10.6 Hz, H-6), 6.47–6.48 (1H, m, H-4 furyl), 6.64–6.65 (1H, d, 3J = 3.3 Hz, H-3 furyl), 6.90 (2H, d, 3J = 8.5 Hz, H-Ar), 7.08 (2H, d, 3J = 8.5 Hz, H-Ar), 7.59–7.63 (2H, m, H-3, H-5 benzoyl), 7.70–7.71 (1H, m, H-5 furyl), 7.73–7.76 (1H, m, H-4 benzoyl), 8.10 (2H, d, 3J = 7.9 Hz, H-2, H-6 benzoyl).13C NMR (126 MHz, DMSO-d6) δ 20.0 (Me), 45.5 (C-5), 47.8 (C-6), 50.1 (C-4a), 51.5 (C-4), 67.0 (C-2), 109.2 (C-3 furyl), 110.8 (C-4 furyl), 116.6 (C≡N), 116.7 (C-2, C-6 NAr), 128.8 (C–Ar), 129.0 (C–Ar), 129.2 (C–Ar), 129.3 (C–Ar), 134.7 (C–Ar), 134.8 (C–Ar), 143.8 (C-5 furyl), 144.9 (C-1 NAr), 147.6 (C-2 furyl), 161.6 (C=N), 192.5 (C=O). LCMS (m/z, ES-API) 120.1 [ArN=CH2 + H]+. Anal. Calcd for C25H21N3O2S: C, 70.24; H, 4.95; N, 9.83. Found C, 70.26; H, 5.06; N, 9.77.
6-Benzoyl-3-benzyl-5-(2-thienyl)-2,3,4,4a,5,6-hexahydrothieno[2,3-d]pyrimidine-4a-carbonitrile (11f)
Recrystallization from EtOH–acetone 1:3 gave colorless needles, yield 79%, mp 238–240 °C. IR (KBr, cm–1) νmax 2245 (C≡N), 1686 (C=O), 1659 (C=N). 1H NMR (500 MHz, DMSO-d6) δ 2.89 (1H, d, 2J = 11.4 Hz, H-4), 3.11 (1H, d, 2J = 11.4 Hz, H-4), 3.64 (1H, d, 2J = 13.5 Hz, CH2Ph), 3.83 (1H, d, 2J = 13.5 Hz, CH2Ph), 4.09 (1H, d, 2J = 16.6 Hz, H-2), 4.34 (1H, d, 3J = 10.4 Hz, H-5), 4.59 (1H, d, 2J = 16.6 Hz, H-2), 5.99 (1H, d, 3J = 10.4 Hz, H-6), 6.99–7.01 (1H, m, H-4 thienyl), 7.23–7.25 (1H, m, H-3 thienyl), 7.29–7.32 (5H, m, Ph), 7.45–7.46 (1H, dd, 3J = 5.2 Hz,4J = 1.0 Hz, H-5 thienyl), 7.56–7.60 (2H, m, H-3 and H-5 benzoyl), 7.70–7.73 (1H, m, H-4 benzoyl), 8.07 (2H, d, 3J = 8.3 Hz, H-2, H-6 benzoyl).13C NMR APT (126 MHz, DMSO-d6) δ 46.8* (C-5), 50.0* (C-6), 51.2 (C-4a), 53.0 (CH2Ph), 56.3 (C-4), 70.7 (C-2), 117.1 (C≡N), 126.4* (CH Ar), 127.1* (CH Ar), 127.2* (CH Ar), 127.4* (CH Ar), 128.27* (CH Ar), 128.29* (CH Ar), 128.7* (CH Ar), 129.2* (CH Ar), 134.7* (C-4 benzoyl), 134.8 (C Ar), 135.3 (C Ar), 137.0 (C Ar), 161.6 (C=N), 192.5 (C=O). *Signals in antiphase. LCMS (m/z, APCI) 120.1 [PhCH2N=CH2 + H]+, 365.0 [M–PhH + H]+, 444.1 [M + 1]+. Anal. Calcd for C25H21N3OS2: C, 67.69; H, 4.77; N, 9.47. Found C, 67.73; H, 4.82; N, 9.40.
6-Benzoyl-3-(4-methylphenyl)-5-(2-thienyl)-2,3,4,4a,5,6-hexahydrothieno[2,3-d]pyrimidine-4a-carbonitrile (11g)
Recrystallization from EtOH–acetone 1:1 gave a beige fine crystalline solid, yield 86%, mp 214–216 °C. IR (KBr, cm–1) νmax 2241 (C≡N), 1684 (C=O), 1651 (C=N).1H NMR (500 MHz, DMSO-d6) δ 2.20 (3H, s, Me), 3.64 (1H, d, 2J = 12.4 Hz, H-4), 3.90 (1H, d, 2J = 11.4 Hz, H-4), 4.47 (1H, d, 3J = 10.4 Hz, H-5), 4.67 (1H, d, 2J = 17.4 Hz, H-2), 5.31 (1H, d, 2J = 17.4 Hz, H-2), 6.05 (1H, d, 3J = 10.4 Hz, H-6), 6.85 (2H, d, 3J = 8.3, H-2, H-6 ArN), 7.05–7.09 (3H, m, H-4 thienyl, H-3, H-5 ArN overlapped), 7.42–7.43 (1H, m, H-3 thienyl), 7.55 (1H, d, 3J = 5.4 Hz, H-5 thienyl), 7.58–7.61 (2H, m, H-3 and H-5 benzoyl), 7.72–7.75 (1H, m, H-4 benzoyl), 8.11 (2H, d, 3J = 7.9 Hz, H-2, H-6 benzoyl).13C NMR (126 MHz, DMSO-d6) δ 20.1 (Me), 46.8 (C-5), 49.7 (C-6), 50.2 (C-4), 51.6 (C-4a), 67.4 (C-2), 116.6 (C≡N), 116.8 (C–Ar), 126.5 (C–Ar), 127.3 (C–Ar), 127.6 (C–Ar), 128.8 (C–Ar), 129.2 (C–Ar), 129.6 (C–Ar), 129.7 (C–Ar), 134.8 (C–Ar), 134.9 (C–Ar), 135.3 (C–Ar), 144.8 (C–Ar), 161.6 (C=N), 192.6 (C=O). LCMS (m/z, ES-API) 120.2 [ArN=CH2 + H]+, 444.0 [M + H]+. Anal. Calcd for C25H21N3OS2: C, 67.69; H, 4.77; N, 9.47. Found C, 67.70; H, 4.81; N, 9.45.
6-Benzoyl-3-(4-methylphenyl)-5-phenyl-2,3,4,4a,5,6-hexahydrothieno[2,3-d]pyrimidine-4a-carbonitrile (11h)
Recrystallization from EtOH–acetone 1:2 gave a beige fine crystalline solid, yield 86%, mp 224–226 °C. IR (KBr, cm–1) νmax 2239 (C≡N), 1682 (C=O), 1655 (C=N).1H NMR (400 MHz, DMSO-d6) δ 2.18 (3H, s, Me), 3.64 (2H, AB-q, 2J = 12.6 Hz, H-4), 4.19 (1H, d, 3J = 10.6 Hz, H-5), 4.66 (1H, d, 2J = 17.3 Hz, H-2), 5.29 (1H, d, 2J = 17.3 Hz, H-2), 6.28 (1H, d, 3J = 10.6 Hz, H-6), 6.80 (2H, d, 3J = 8.3 Hz, H-2, H-6 ArN), 7.04 (2H, d, 3J = 8.3 Hz, H-3, H-5 ArN), 7.33–7.42 (3H, m, H-Ph), 7.56–7.60 (2H, m, H-3 and H-5 benzoyl), 7.64 (2H, d, 3J = 7.3 Hz, H-2 and H-6 Ph), 7.70–7.74 (1H, m, H-4 benzoyl), 8.10 (2H, d, 3J = 8.1 Hz, H-2, H-6 benzoyl).13C NMR (126 MHz, DMSO-d6) δ 20.0 (Me), 48.4 (C-5), 50.4 (C-6), 51.6 (C-4 and C-4a overlapped), 67.5 (C-2), 116.7 (C≡N), 116.8 (C–Ar), 128.5 (C–Ar), 128.7 (C–Ar), 128.78 (C–Ar), 128.80 (C–Ar), 129.1 (C–Ar), 129.59 (C–Ar), 129.63 (C–Ar), 133.7 (C–Ar), 134.6 (C–Ar), 135.0 (C–Ar), 144.9 (C–Ar), 162.3 (C=N), 193.0 (C=O). LCMS (m/z, ES-API) 120.3 [ArN=CH2 + H]+, 426.9 [M-120 + HCO2H + HCO2NH4], 438.0 [M + H]+. Anal. Calcd for C27H23N3OS: C, 74.11; H, 5.30; N, 9.60. Found C, 74.13; H, 5.34; N, 9.63.
6-Benzoyl-3-(4-methoxyphenyl)-5-phenyl-2,3,4,4a,5,6-hexahydrothieno[2,3-d]pyrimidine-4a-carbonitrile (11i)
Recrystallization from EtOH–acetone 1:3 gave a pale yellow crystalline solid, yield 67%, mp 213–215 °C. IR (KBr, cm–1) νmax 2245 (C≡N), 1682 (C=O), 1655 (C=N).1H NMR (400 MHz, DMSO-d6) δ 3.52 (1H, d, 3J = 12.5 Hz, H-4), 3.62 (1H, d, 3J = 12.5 Hz, H-4), 3.67 (3H, s, MeO), 4.18 (1H, d, 3J = 10.6 Hz, H-5), 4.63 (1H, d, 2J = 17.1 Hz, H-2), 5.19 (1H, d, 2J = 17.1 Hz, H-2), 6.26 (1H, d, 3J = 10.6 Hz, H-6), 6.83 (2H, d, 3J = 8.8 Hz, 4-MeOC6H4), 6.89 (2H, d, 3J = 8.8 Hz, 4-MeOC6H4), 7.34–7.41 (3H, m, H-Ph), 7.56–7.60 (2H, m, H-3, H-5 benzoyl), 7.64 (2H, d, 3J = 7.3 Hz, H-2, H-6 Ph), 7.70–7.73 (1H, m, H-4 benzoyl), 8.09 (2H, d, 3J = 7.3 Hz, H-2, H-6 benzoyl).13C APT NMR (126 MHz, DMSO-d6) δ 48.4* (C-5), 50.4 (C-4a), 51.6* (C-6), 52.7 (C-4), 55.2* (MeO), 68.4 (C-2), 114.5* (CH Ar), 116.7 (C≡N), 119.2* (CH Ar), 128.5* (CH Ar), 128.6* (CH Ar), 128.7* (CH Ar), 128.8* (CH Ar), 129.1* (CH Ar), 133.6 (C-1 Ph), 134.6* (C-4 benzoyl), 135.0 (C-1 Ph), 141.0 (C-OMe), 154.2 (C-1 Ar), 162.2 (C-2), 193.0 (C=O). *Signals in antiphase. LCMS (m/z, ES-API) 136.1 [ArN=CH2 + H]+, 453.9 [M + H]+, 906.7 [2 M + 1]+. Anal. Calcd for C27H23N3O2S: C, 71.50; H, 5.11; N, 9.26. Found C, 71.47; H, 5.17; N, 9.20.
6-Benzoyl-3-phenethyl-5-phenyl-2,3,4,4a,5,6-hexahydrothieno[2,3-d]pyrimidine-4a-arbonitrile (11j)
Recrystallization from EtOH–acetone 1:1 gave colorless needles, yield 65%, mp 167–169 °C. IR (KBr, cm–1) νmax 2237 (C≡N), 1684 (C=O), 1647 (C=N).1H NMR (500 MHz, DMSO-d6) δ 2.66–2.78 (4H, m, CH2CH2Ph), 2.92 (1H, d, 3J = 11.4 Hz, H-4), 3.03 (1H, d, 3J = 11.4 Hz, H-4), 4.04 (1H, d, 3J = 10.6 Hz, H-5), 4.11 (1H, d, 2J = 16.9 Hz, H-2), 4.64 (1H, d, 2J = 16.9 Hz, H-2), 6.19 (1H, d, 3J = 10.6 Hz, H-6), 7.15–7.21 (5H, m, Ph), 7.32–7.39 (3H, m, Ph), 7.55–7.59 (4H, m, Ph), 7.69–7.72 (1H, m, H-4 benzoyl), 8.07 (2H, d, 3J = 7.8 Hz, H-2, H-6 benzoyl).13C NMR (126 MHz, DMSO-d6) δ 32.5 (CH2CH2Ph), 48.6 (C-5), 51.0 (C-6), 51.7 (C-4a), 53.8 (CH2Ph), 54.3 (C-4), 70.6 (C-2), 117.2 (C≡N), 125.8 (C–Ar), 128.1 (C–Ar), 128.4 (C–Ar), 128.60 (C–Ar), 128.64 (C–Ar), 128.67 (C–Ar), 128.73 (C–Ar), 129.1 (C–Ar), 133.7 (C–Ar), 134.6 (C–Ar), 135.0 (C–Ar), 139.7 (C–Ar), 162.1 (C=N), 193.0 (C=O). LCMS (m/z, ES-API) 105.3 [PhCHCH3]+, 134.1 [PhCH2CH2N=CH2 + H]+, 452.0 [M + H]+. Anal. Calcd for C28H25N3OS: C, 74.47; H, 5.58; N, 9.31. Found C, 74.44; H, 5.67; N, 9.27.
6-Benzoyl-3-benzyl-5-phenyl-2,3,4,4a,5,6-hexahydrothieno[2,3-d]pyrimidine-4a-carbonitrile (11k)
Recrystallization from EtOH–acetone 1:1 gave colorless needles, yield 73%, mp 188–190 °C. IR (KBr, cm–1) νmax 2243 (C≡N), 1689 (C=O), 1655 (C=N).1H NMR (400 MHz, DMSO-d6) δ 2.90 (2H, br s, H-4), 3.61 (1H, d, 2J = 14.0 Hz, CH2Ph), 3.78 (1H, d, 2J = 14.0 Hz, CH2Ph), 4.05–4.09 (2H, m: 1H, d, H-2 and 1H, d, H-5 overlapped), 4.56 (1H, d, 2J = 16.7 Hz, H-2), 6.21 (1H, d, 3J = 10.8 Hz, H-6), 7.20–7.22 (1H, m, H-4 Ph), 7.25–7.35 (7H, m, Ph), 7.55–7.58 (4H, m, Ph), 7.68–7.72 (1H, m, H-4 benzoyl), 8.06 (2H, d, 3J = 7.5 Hz, H-2, H-6 benzoyl).13C APT NMR (126 MHz, DMSO-d6) δ 48.7* (C-5), 51.3 (C-4a), 51.6* (C-6), 53.3 (CH2Ph), 56.4 (C-4), 70.8 (C-2), 117.2 (C≡N), 127.2* (CH Ph), 128.3* (CH Ph), 128.4* (CH Ph), 128.6* (CH Ph), 128.7* (CH Ph), 128.8* (CH Ph), 129.1* (CH Ph), 133.7 (C Ph), 134.6* (C-4 benzoyl), 135.0 (C Ph), 137.1 (C Ph), 162.4 (C=N), 193.0 (C=O). *Signals in antiphase. LCMS (m/z, ES-API) 120.4 [PhCH2N=CH2 + H]+, 438.5 [M + H]+, 875.5 [2 M + H]+. Anal. Calcd for C27H23N3OS: C, 74.11; H, 5.30; N, 9.60. Found C, 74.10; H, 5.36; N, 9.57.
6-Benzoyl-3,5-diphenyl-2,3,4,4a,5,6-hexahydrothieno[2,3-d]pyrimidine-4a-carbonitrile (11l)
Recrystallization from EtOH–acetone 1:1 gave large colorless cubes, yield 70%, mp 214–216 °C. IR (KBr, cm–1) νmax 2239 (C≡N), 1682 (C=O), 1666 (C=N).1H NMR (400 MHz, DMSO-d6) δ 3.66 (1H, d, 2J = 12.5 Hz, H-4), 3.76 (1H, d, 2J = 12.5 Hz, H-4), 4.21 (1H, d, 3J = 10.6 Hz, H-5), 4.72 (1H, d, 2J = 17.3 Hz, H-2), 5.37 (1H, d, 2J = 17.3 Hz, H-2), 6.29 (1H, d, 3J = 10.6 Hz, H-6), 6.84–6.90 (3H, m, Ph), 7.21–7.25 (2H, m, Ph), 7.36–7.43 (3H, m, Ph), 7.56–7.60 (2H, m, H-3 and H-5 benzoyl), 7.64 (2H, d, 3J = 7.3 Hz, H-2 and H-6 Ph), 7.70–7.74 (1H, m, H-4 benzoyl), 8.10 (2H, d, 3J = 7.8 Hz, H-2, H-6 benzoyl).13C APT NMR (126 MHz, DMSO-d6) δ 48.5* (C-5), 50.3 (C-4), 51.2 (C-4a), 51.6* (C-6), 67.0 (C-2), 116.3* (C-2, C-6 NPh), 116.6 (C≡N), 120.5* (C-4 N-Ph), 128.5* (CH Ph), 128.7* (CH Ph), 128.8* (CH Ph), 129.0* (CH Ph), 129.1* (CH Ph), 129.2* (CH Ph), 133.6 (C-1 Ph), 134.6* (C-4 benzoyl), 135.0 (C Ph), 147.0 (C-1 N-Ph), 162.4 (C=N), 193.0 (C=O). *Signals in antiphase. LCMS (m/z, ES-API) 129.4, 141.3, 149.6, 158.4, 214.4, 424.5 [M + H]+, 847.0 [2 M + H]+. Anal. Calcd for C26H21N3OS: C, 73.73; H, 5.00; N, 9.92. Found C, 73.70; H, 5.06; N, 9.97.
X-ray Studies of the Crystal of 11l
Single crystals of 6-benzoyl-3,5-diphenyl-2,3,4,4a,5,6-hexahydrothieno[2,3-d]pyrimidine-4a-carbonitrile C26H21N3OS (11l), M = 423.52, were prepared by recrystallization from EtOH/acetone = 1:1. The crystals are triclinic at 298 K: a = 11.9327(7) Å, b = 12.7829(6) Å, c = 16.4847(6) Å, α = 79.805(4)°, β = 88.022(4)°, γ = 62.234(6)°, V = 2186.53(19) Å3, T = 839(2), space group P1 (no. 2), Z = 4, μ(Mo Kα) = 0.171 mm–1, dcalc = 1.29 g/cm3, F(000) = 888, 16,692 reflections measured, 9840 unique (Rint = 0.0200) that were used in all calculations. The final wR2 was 0.1243 (all data), and R1 was 0.0582 (>2sigma(I)). The unit cell parameters and the intensities of 16,692 reflections were measured on an Xcalibur 3 diffractometer (Mo Kα, graphite monochromator, CCD detector, ω-scanning, 2θmax 57.52°). The structure was solved by the direct method with the SHELX-97 software package.141 The hydrogen atoms were placed geometrically and refined with a riding model with Uiso = 1.2 Ueq for the supporting atom. The structure was refined on F2 by the full-matrix least-squares method with an anisotropic approximation for the nonhydrogen atoms to wR2 0.124 at 9840 reflections (R1 0.058 at 6619 reflections with F > 4σ(F), S = 1.03). A full set of crystallographic data has been deposited in the Cambridge Crystallographic Data Center (CCDC 1063909).
3,3′-(1,4-Phenylene)-bis(6-benzoyl-5-phenyl-2,3,4,4a,5,6-hexahydrothieno[2,3-d]pyrimidine-4a-carbonitrile) (12)
The mixture of ADHT 6d (151 mg, 0.49 mmol) and p-phenylenediamine (27 mg, 0.25 mmol) was dissolved in hot EtOH (10 mL). Then an excess (0.8 mL) of 37% aq HCHO was added, and the mixture was refluxed for 5 min under vigorous stirring. The precipitate formed upon cooling was filtered off after 4 h and triturated with boiling acetone for 2–3 min. Beige powder, yield 90 mg (48%), mp 164–166 °C. IR (Nujol mulls, cm–1) νmax 2245 (C≡N), 1685 (C=O), 1650 (C=N).1H NMR (400 MHz, DMSO-d6) δ 3.57 (4H, AB-q, 2J = 11.9 Hz, H-4, H-4′), 4.17 (2H, d, 3J = 10.6 Hz, H-5, H-5′), 4.61 (2H, d, 2J = 17.3 Hz, H-2, H-2′), 5.20 (2H, d, 2J = 17.3 Hz, H-2, H-2′), 6.26 (2H, d, 3J = 10.6 Hz, H-6, H-6′), 6.84 (4H, br s, 1,4-NC6H4N), 7.34–7.39 (6H, m, Ph), 7.56–7.63 (8H, m, Ph), 7.70–7.73 (2H, m, H-4, H-4′ benzoyl), 8.09 (4H, d, 3J = 7.5 Hz, H-2, H-6, H-2′, H-6′ benzoyl). Due to the poor solubility, the authors were unable to record 13C NMR spectra of 12. LCMS (m/z, ES-API) 120.3 [H2C=NC6H4N + H]+, 426.8 [M + 2MeCN + 2H]+2, 451.0 [thienopyrimidine-3-yl-C6H4-N=CH2 + H]+, 768.8 [M + H]+. Anal. Calcd for C46H36N6O2S2: C, 71.85; H, 4.72; N, 10.93. Found 71.74; H, 4.76; N, 11.00.
Acknowledgments
The authors are indebted to Dr. Oleg Shishkin (SSI ″Institute for Single Crystals″, Kharkov), who has passed away, for X-ray studies.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.1c04141.
Copies of 1H, 13C NMR, FTIR, LCMS, and X-ray data of the synthesized compounds (PDF)
The authors declare no competing financial interest.
Notes
The research was funded by the Russian Scientific Foundation, Project No. 17-73-20017.
Supplementary Material
References
- Gewald K. Methods for the synthesis of 2-aminothiophenes and their reactions. Chem. Heterocycl. Compd. 1976, 1077. 10.1007/BF00945583. [DOI] [Google Scholar]
- Unverferth K. Wirkstoffe durch Gewald-Reaktion. Pharmazie 1990, 45, 545. [Google Scholar]
- Sabnis R. W. The Gewald Synthesis. Sulphur Rep. 1994, 16, 1. 10.1080/01961779408048964. [DOI] [Google Scholar]
- Sabnis R. W.; Rangnekar D. W.; Sonawane N. D. 2-Aminothiophenes by the Gewald reaction. J. Heterocycl. chem. 1999, 36, 333. 10.1002/jhet.5570360203. [DOI] [Google Scholar]
- Puterová Z.; Krutošíková A.; Végh D. Gewald reaction: synthesis, properties and applications of substituted 2-aminothiophenes. ARKIVOC 2010, 2010, 209. 10.3998/ark.5550190.0011.105. [DOI] [Google Scholar]
- Huang Y.; Dömling A. The Gewald multicomponent reaction. Mol. Diversity 2011, 15, 3. 10.1007/s11030-010-9229-6. [DOI] [PubMed] [Google Scholar]
- Bozorov K.; Nie L. F.; Zhao J.; Aisa H. A. 2-Aminothiophene scaffolds: Diverse biological and pharmacological attributes in medicinal chemistry. Eur. J. Med. Chem. 2017, 140, 465. 10.1016/j.ejmech.2017.09.039. [DOI] [PubMed] [Google Scholar]
- Hung K.; Liu Y.; Simon O.; Zhang L.; Lu P.; Yeung B. K. S.; Sarko C.; Yokokawa F. Synthesis of a potent pan-serotype dengue virus inhibitor having a tetrahydrothienopyridine core. Synlett 2020, 10.1055/a-1323-4036. [DOI] [Google Scholar]
- Wilson R.; Kumar P.; Parashar V.; Vilcheze C.; Veyron-Churlet R.; Freundlich J. S.; Barnes S. W.; Walker J. R.; Szymonifka M. J.; Marchiano E.; Shenai S.; Colangeli R.; Jacob W. R. Jr.; Neiditch M. B.; Kremer L.; Alland D. Antituberculosis thiophenes define a requirement for Pks13 in mycolic acid biosynthesis. Nat. Chem. Biol. 2013, 9, 499. 10.1038/nchembio.1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thanna S.; Knudson S. E.; Grzegorzewicz A.; Kapil S.; Goins C. M.; Ronning D. R.; Jackson M.; Slayden R. A.; Sucheck S. J. Synthesis and evaluation of new 2-aminothiophenes against Mycobacterium tuberculosis. Org. Biomol. Chem. 2016, 14, 6119. 10.1039/C6OB00821F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartkoorn R. C.; Ryabova O. B.; Chiarelli L. R.; Riccardi G.; Makarov V.; Cole S. T. Mechanism of Action of 5-Nitrothiophenes against Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2014, 58, 2944. 10.1128/AAC.02693-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valant C.; Aurelio L.; Devine S. M.; Ashton T. D.; White J. M.; Sexton P. M.; Christopoulos A.; Scammells P. J. Synthesis and Characterization of Novel 2-Amino-3-benzoylthiophene Derivatives as Biased Allosteric Agonists and Modulators of the Adenosine A1 Receptor. J. Med. Chem. 2012, 55, 2367. 10.1021/jm201600e. [DOI] [PubMed] [Google Scholar]
- Romagnoli R.; Prencipe F.; Oliva P.; Cacciari B.; Balzarini J.; Liekens S.; Hamel E.; Brancale A.; Ferla S.; Manfredini S.; Zurlo M.; Finotti A.; Gambari R. Synthesis and biological evaluation of new antitubulin agents containing 2-(3′,4′,5′-trimethoxyanilino)-3,6-disubstituted-4,5,6,7-tetrahydrothieno[2,3-c]pyridine scaffold. Molecules 2020, 25, 1690. 10.3390/molecules25071690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W.; Lv D.; Qiu N.; Zhang L.; Hu C.; Hu Y. Design, synthesis and biological evaluation of novel 3,4,5-trisubstituted aminothiophenes as inhibitors of p53–MDM2 interaction. Part 2. Bioorg. Med. Chem. 2013, 21, 2886. 10.1016/j.bmc.2013.03.070. [DOI] [PubMed] [Google Scholar]
- Rossetti A.; Bono N.; Candiani G.; Meneghetti F.; Roda G.; Sacchetti A. Synthesis and Antimicrobial Evaluation of Novel Chiral 2-Amino-4,5,6,7-tetrahydrothieno[2,3-c]pyridine Derivatives. Chem. Biodiversity 2019, 16, e1900097 10.1002/cbdv.201900097. [DOI] [PubMed] [Google Scholar]
- Lepri S.; Nannetti G.; Muratore G.; Cruciani G.; Ruzziconi R.; Mercorelli B.; Palú G.; Loregian A.; Goracci L. Optimization of Small-Molecule Inhibitors of Influenza Virus Polymerase: From Thiophene-3-Carboxamide to Polyamido Scaffolds. J. Med. Chem. 2014, 57, 4337. 10.1021/jm500300r. [DOI] [PubMed] [Google Scholar]
- Briel D.; Rybak A.; Kronbach C.; Unverferth K. Substituted 2-Aminothiopen-derivatives: A potential new class of GluR6-Antagonists. Eur. J. Med. Chem. 2010, 45, 69. 10.1016/j.ejmech.2009.09.025. [DOI] [PubMed] [Google Scholar]
- Andersen H. S.; Olsen O. H.; Iversen L. F.; Sørensen A. L. P.; Mortensen S. B.; Christensen M. S.; Branner S.; Hansen T. K.; Lau J. F.; Jeppesen L.; Moran E. J.; Su J.; Bakir F.; Judge L.; Shahbaz M.; Collins T.; Vo T.; Newman M. J.; Ripka W. C.; Møller N. P. H. Discovery and SAR of a Novel Selective and Orally Bioavailable Nonpeptide Classical Competitive Inhibitor Class of Protein-Tyrosine Phosphatase 1B. J. Med. Chem. 2002, 45, 4443. 10.1021/jm0209026. [DOI] [PubMed] [Google Scholar]
- Pilmane M.; Salma-Ancane K.; Loca D.; Locs J.; Berzina-Cimdina L. Strontium and strontium ranelate: Historical review of some of their functions. Mater. Sci. Eng. C. 2017, 78, 1222. 10.1016/j.msec.2017.05.042. [DOI] [PubMed] [Google Scholar]
- Meunier P. J.; Roux C.; Seeman E.; Ortolani S.; Badurski J. E.; Spector T. D.; Cannata J.; Balogh A.; Lemmel E.-M.; Pors-Nielsen S.; Rizzoli R.; Genant H. K.; Reginster J.-Y. The effects of strontium ranelate on the risk of vertebral fracture in women with postmenopausal osteoporosis. N. Engl. J. Med. 2004, 350, 459. 10.1056/NEJMoa022436. [DOI] [PubMed] [Google Scholar]
- Atkins G. J.; Welldon K. J.; Halbout P.; Findlay D. M. Strontium ranelate treatment of human primary osteoblasts promotes an osteocyte-like phenotype while eliciting an osteoprotegerin response. Osteoporos. Int. 2009, 20, 653. 10.1007/s00198-008-0728-6. [DOI] [PubMed] [Google Scholar]
- Reginster J. Strontium Ranelate in Osteoporosis. Curr. Pharm. Design 2002, 8, 1907. 10.2174/1381612023393639. [DOI] [PubMed] [Google Scholar]
- Deeks E. D.; Dhillon S. Spotlight on Strontium Ranelate. Drugs Aging 2010, 27, 771. 10.2165/11206440-000000000-00000. [DOI] [PubMed] [Google Scholar]
- Hamdy N. A. T. Strontium ranelate improves bone microarchitecture in osteoporosis. Rheumatology 2009, 48, iv9. 10.1093/rheumatology/kep274. [DOI] [PubMed] [Google Scholar]
- Han W.; Fan S.; Bai X.; Ding C. Strontium ranelate, a promising disease modifying osteoarthritis drug. Expert Opin. Invest. Drugs 2017, 26, 375. 10.1080/13543784.2017.1283403. [DOI] [PubMed] [Google Scholar]
- Bakhit A.; Kawashima N.; Hashimoto K.; Noda S.; Nara K.; Kuramoto M.; Tazawa K.; Okiji T. Strontium ranelate promotes odonto–/osteogenic differentiation/mineralization of dental papillae cells in vitro and mineralized tissue formation of the dental pulp in vivo. Sci. Rep. 2018, 8, 9224. 10.1038/s41598-018-27461-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu T.; Yang S.; Lu T.; He F.; Zhang J.; Shi H.; Lin Z.; Ye J. Strontium ranelate simultaneously improves the radiopacity and osteogenesis of calcium phosphate cement. Biomed. Mater. 2019, 14, 035005 10.1088/1748-605X/ab052d. [DOI] [PubMed] [Google Scholar]
- Shimada O.; Yasuda H. Hydroxyl radical scavenging action of tinoridine. Agents Actions 1986, 19, 208. 10.1007/BF01966208. [DOI] [PubMed] [Google Scholar]
- Yasuda H.; Izumi N.; Shimada O.; Kobayakawa T.; Nakanishi M. The protective effect of tinoridine against carbon tetrachloride hepatotoxicity. Toxicol. Appl. Pharmacol. 1980, 52, 407. 10.1016/0041-008X(80)90335-X. [DOI] [PubMed] [Google Scholar]
- Kalariya P. D.; Patel P. N.; Kavya P.; Sharma M.; Garg P.; Srinivas R.; Kumar Talluri M. V. N. Rapid structural characterization of in vivo and in vitro metabolites of tinoridine using UHPLC–QTOF–MS/MS and in silico toxicological screening of its metabolites. J. Mass Spect. 2015, 50, 1222. 10.1002/jms.3640. [DOI] [PubMed] [Google Scholar]
- Li Y.; Du C.; Jiaxiang N.; Liqiang Y.; Qi F. Olanzapine versus placebo for people with schizophrenia. Cochrane Database Syst Rev. 2019, 2019, CD013310. 10.1002/14651858.CD013310. [DOI] [Google Scholar]
- Meftah A. M.; Deckler E.; Citrome L.; Kantrowitz J. T. New discoveries for an old drug: a review of recent olanzapine research. Postgrad. med. 2020, 132, 80. 10.1080/00325481.2019.1701823. [DOI] [PubMed] [Google Scholar]
- Del Fabro L.; Delvecchio G.; D’Agostino A.; Brambilla P. Effects of olanzapine during cognitive and emotional processing in schizophrenia: A review of functional magnetic resonance imaging findings. Hum. Psychopharmacol. Clin. Exp. 2019, 34, e2693 10.1002/hup.2693. [DOI] [PubMed] [Google Scholar]
- Li X.; Conklin D.; Pan H. L.; Eisenach J. C. Allosteric adenosine receptor modulation reduces hypersensitivity following peripheral inflammation by a central mechanism. J. Pharmacol. Exp. Therap. 2003, 305, 950. 10.1124/jpet.102.047951. [DOI] [PubMed] [Google Scholar]
- Gonzalez López F.; Mariño E. L.; Dominguez-Gil A. Pharmacokinetics of tiadipone: a new anxiolytic. Int. J. Clin. Pharmacol. Ther. Toxicol. 1986, 24, 482. [PubMed] [Google Scholar]
- Langley M. S.; Clissold S. P. Brotizolam. Drugs 1988, 35, 104. 10.2165/00003495-198835020-00002. [DOI] [PubMed] [Google Scholar]
- Lavon O.; Bejel S. Safety of brotizolam in hospitalized patients. Eur. J. Clin. Pharmacol. 2018, 74, 939. 10.1007/s00228-018-2447-z. [DOI] [PubMed] [Google Scholar]
- Yan X.; Huang S.; Ma C.; Shen Y.; Gu N.; Chen H.; Wu W.; Li S.; Hong Z.; Li H. A randomized, double-blind, double-dummy, multicenter, controlled trial on brotizolam intervention in outpatients with insomnia. Int. J. Psychiatry Clin. Pract. 2013, 17, 239. 10.3109/13651501.2012.735242. [DOI] [PubMed] [Google Scholar]
- Köhne C. H.; Thuss-Patience P.; Friedrich M.; Daniel P. T.; Kretzschmar A.; Benter T.; Bauer B.; Dietz R.; Dörken B. Raltitrexed (Tomudex): an alternative drug for patients with colorectal cancer and 5-fluorouracil associated cardiotoxicity. Br. J. Cancer 1998, 77, 973. 10.1038/bjc.1998.160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham D.; Zalcberg J.; Maroun J.; James R.; Clarke S.; Maughan T. S.; Vincent M.; Schulz J.; González-Barón M.; Facchini T. T. Facchini. Efficacy, tolerability and management of raltitrexed (Tomudex) monotherapy in patients with advanced colorectal cancer: a review of phase II/III trials. Eur. J. Cancer 2002, 38, 478. 10.1016/S0959-8049(01)00413-0. [DOI] [PubMed] [Google Scholar]
- Morey J.; Llinás P.; Bueno-Costa A.; León A. J.; Piña M. N. Raltitrexed-Modified Gold and Silver Nanoparticles for Targeted Cancer Therapy: Cytotoxicity Behavior In Vitro on A549 and HCT-116 Human Cancer Cells. Materials 2021, 14, 534. 10.3390/ma14030534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dotsenko V. V.; Buryi D. S.; Lukina D. Y.; Krivokolysko S. G. Recent advances in the chemistry of thieno[2,3-b]pyridines 1. Methods of synthesis of thieno[2,3-b]pyridines. Russ. Chem. Bull. 2020, 69, 1829. 10.1007/s11172-020-2969-2. [DOI] [Google Scholar]
- Tremblay M. H.; Gellé A.; Skene W. G. Ambipolar azomethines as potential cathodic color switching materials. New J. Chem. 2017, 41, 2287. 10.1039/C6NJ01732K. [DOI] [Google Scholar]
- Tremblay M. H.; Al Ahmad A.; Skene W. G. End-group functionalization of a conjugated azomethine with ureas for property tailoring. New J. Chem. 2020, 44, 18813. 10.1039/C9NJ04722K. [DOI] [Google Scholar]
- Bolduc A.; Dufresne S.; Skene W. G. EDOT-containing azomethine: an easily prepared electrochromically active material with tuneable colours. J. Mater. Chem. 2010, 20, 4820. 10.1039/B923821B. [DOI] [Google Scholar]
- Bolduc A.; Skene W. G. Direct preparation of electroactive polymers on electrodes and their use in electrochromic devices. Polym. Chem. 2014, 5, 1119. 10.1039/C3PY01370G. [DOI] [Google Scholar]
- Romiszewski J.; Puterová-Tokarová Z.; Mieczkowski J.; Gorecka E. Optical properties of thiophene-containing liquid crystalline and hybrid liquid crystalline materials. New J. Chem. 2014, 38, 2927. 10.1039/C4NJ00298A. [DOI] [Google Scholar]
- Collado D.; Casado J.; Rodriguez Gonzalez S.; Lopez Navarrete J. T.; Suau R.; Perez-Inestrosa E.; Pappenfus T. M.; Raposo M. M. M. Enhanced functionality for donor-acceptor oligothiophenes via inclusion of BODIPY: synthesis, electrochemistry, photophysics and model chemistry. Chem. – Eur. J. 2011, 17, 498. 10.1002/chem.201001942. [DOI] [PubMed] [Google Scholar]
- Stylianakis M. M.; Mikroyannidis J. A.; Kymakis E. A facile, covalent modification of single-wall carbon nanotubes by thiophene for use in organic photovoltaic cells. Sol. Energy Mater. Sol. Cells 2010, 94, 267. 10.1016/j.solmat.2009.09.013. [DOI] [Google Scholar]
- Sabnis R. W. The Gewald reaction in dye chemistry. Coloration Technol. 2016, 132, 49. 10.1111/cote.12182. [DOI] [Google Scholar]
- Borbone F.; Caruso U.; Diana R.; Panunzi B.; Roviello A.; Tingoli M.; Tuzi A. Second order nonlinear optical networks with excellent poling stability from a new trifunctional thiophene based chromophore. Org. Electron. 2009, 10, 53. 10.1016/j.orgel.2008.10.004. [DOI] [Google Scholar]
- Rodlovskaya E. N.; Vasnev V. A. Thiophene-containing monomers for the synthesis of new polythiopheneferrocenes. Russ. Chem. Bull. 2020, 69, 1148. 10.1007/s11172-020-2881-9. [DOI] [Google Scholar]
- Shvekhgeimer M.-G.; Dihydrothiophenes A. Synthesis and properties. Chem. Heterocycl. Compd. 1998, 34, 1101. 10.1007/BF02319487. [DOI] [Google Scholar]
- Benetti S.; De Risi C.; Pollini G. P.; Zanirato V. Synthetic routes to chiral nonracemic and racemic dihydro- and tetrahydrothiophenes. Chem. Rev. 2012, 112, 2129. 10.1021/cr200298b. [DOI] [PubMed] [Google Scholar]
- Darwish E. S. Facile synthesis of heterocycles via 2-picolinium bromide and antimicrobial activities of the products. Molecules 2008, 13, 1066. 10.3390/molecules13051066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mabkhot Y. N.; Kheder N. A.; Barakat A.; Choudhary M. I.; Yousuf S.; Frey W. Synthesis, antimicrobial, anti-cancer and molecular docking of two novel hitherto unreported thiophenes. RSC Adv. 2016, 6, 63724. 10.1039/C6RA09883E. [DOI] [Google Scholar]
- Dawood K. M.; Abdel-Gawad H.; Ellithey M.; Mohamed H. A.; Hegazi B. Synthesis, anticonvulsant, and anti-inflammatory activities of some new benzofuran-based heterocycles. Arch. Pharm. Chem. Life Sci. 2006, 339, 133. 10.1002/ardp.200500176. [DOI] [PubMed] [Google Scholar]
- Xu M.; Zhu J.; Diao Y.; Zhou H.; Ren X.; Sun D.; Huang J.; Han D.; Zhao Z.; Zhu L.; Xu Y.; Li H. Novel Selective and Potent Inhibitors of Malaria Parasite Dihydroorotate Dehydrogenase: Discovery and Optimization of Dihydrothiophenone Derivatives. J. Med. Chem. 2013, 56, 7911. 10.1021/jm400938g. [DOI] [PubMed] [Google Scholar]
- Ibrahim N. S.; Sadek K. U.; Aziz S. I.; Elnagdi M. H. A novel synthesis of heterocyclic β-enaminoamides: The reaction of halomethyl acet-p-toluidide with isothiocyanates and with isocyanates. Z. Naturforsch. 1985, 40, 129. 10.1515/znb-1985-0126. [DOI] [Google Scholar]
- Shestopalov A. M.; Nikishin K. G. Stereoselective method of synthesis of substituted trans-4,5-dihydro-2-aminothiophenes. Chem. Heterocycl. Compd. 1998, 34, 1089. 10.1007/BF02251559. [DOI] [Google Scholar]
- Yamagata K.; Tomioka Y.; Yamazaki M.; Matsuda T.; Noda K. Studies on heterocyclic enaminonitriles. II. Synthesis and aromatization of 2-amino-3-cyano-4,5-dihydrothiophenes. Chem. Pharm. Bull. 1982, 30, 4396. 10.1248/cpb.30.4396. [DOI] [Google Scholar]
- Wamhoff H.; Thiemig H. A. Heterocyclische β-Enaminoester, 38. Vergleichende Untersuchungen an heterocyclischen β-Enaminonitrilen. Synthese heterokondensierter Pyrimidine. Chem. Ber. 1985, 118, 4473. 10.1002/cber.19851181118. [DOI] [Google Scholar]
- Avalos M.; Babiano R.; Cintas P.; Clemente F. R.; Gordillo R.; Jiménez J. L.; Palacios J. C.; Raithby P. R. Diastereoselective cycloadditions of 1,3-thiazolium-4-olates with chiral 1,2-diaza-1,3-butadienes. J. Org. Chem. 2000, 65, 5089. 10.1021/jo991841v. [DOI] [PubMed] [Google Scholar]
- Kikionis S.; McKee V.; Markopoulos J.; Igglessi-Markopoulou O. Regioselective ring opening of thiomalic acid anhydrides by carbon nucleophiles. Synthesis and X-ray structure elucidation of novel thiophenone derivatives. Tetrahedron 2009, 65, 3711. 10.1016/j.tet.2009.02.081. [DOI] [Google Scholar]
- Chunikhin K. S.; Rodinovskaya L. A.; Shestopalov A. M. Synthesis of 2-amino-4-aryl-3-cyano-5-hydroxyimino-4,5-dihydrothiophenes. Chem. Heterocycl. Compd. 2007, 43, 1247. 10.1007/s10593-007-0190-y. [DOI] [Google Scholar]
- Zeng F.; Liu P.; Shao X.; Li Z.; Xu X. Catalyst-free and selective synthesis of 2-aminothiophenes and 2-amino-4,5-dihydrothiophenes from 4-thiazolidinones in water. RSC Adv. 2016, 6, 59808. 10.1039/C6RA11151C. [DOI] [Google Scholar]
- Kabirifard H.; Ghahremani S.; Afsharpoor A. A simple and versatile protocol for the preparation of functionalized heterocycles utilizing 4-benzoyl-5-phenylamino-2,3-dihydrothiophene-2,3-dione. J. Sulfur Chem. 2015, 36, 591. 10.1080/17415993.2015.1074687. [DOI] [Google Scholar]
- Britsun V. N.; Borisevich A. N.; Samoylenko L. S.; Chernega A. N.; Lozynskii M. O. Oxalylation of the 3-oxo-N-phenyl-3-R-propanethioamides. Russ. Chem. Bull. 2005, 54, 770. 10.1007/s11172-005-0318-0. [DOI] [Google Scholar]
- Suzuki I.; Sakamoto Y.; Seo Y.; Ninomaru Y.; Tokuda K.; Shibata I. Synthesis of 5-membered sulfur heterocycles via tin-catalyzed annulation of mercapto ketones with activated alkenes. J. Org. Chem. 2020, 85, 2759. 10.1021/acs.joc.9b03055. [DOI] [PubMed] [Google Scholar]
- Sun J.; Zhang L. L.; Xia E. Y.; Yan C. G. Synthesis of dihydrothiophenes or spirocyclic compounds by domino reactions of 1,3-thiazolidinedione. J. Org. Chem. 2009, 74, 3398. 10.1021/jo900215a. [DOI] [PubMed] [Google Scholar]
- Sun J.; Xia E. Y.; Zhang L. L.; Yan C. G. Triethylamine-catalyzed domino reactions of 1,3-thiazolidinedione: a facile access to functionalized dihydrothiophenes. Eur. J. Org. Chem. 2009, 2009, 5247. 10.1002/ejoc.200900845. [DOI] [PubMed] [Google Scholar]
- Sun J.; Xia E. Y.; Yao R.; Yan C. G. Convenient synthesis of polyfunctional dihydrothiophenes with tandem reaction of 1,3-thiazolidinedione, aldehyde, arylamine and ethyl cyanoacetate. Mol. Diversity 2011, 15, 115. 10.1007/s11030-010-9278-x. [DOI] [PubMed] [Google Scholar]
- Gao L.; Xia S.; Wu N.; Tao S.; Feng Y.; Rong L. Efficient preparation of 5-amino-4-cyano-N-(cyclopropylcarbamoyl)-3-aryl-2,3-dihydrothiophene-2-carboxamide derivatives without using any other catalysts. Synth. Commun. 2013, 43, 2590. 10.1080/00397911.2012.721919. [DOI] [Google Scholar]
- Kordnezhadian R.; Shekouhy M.; Khalafi-Nezhad A. Microwave-accelerated diastereoselective catalyst-free one-pot four-component synthesis of 2-(N-carbamoylacetamide)-substituted 2,3-dihydrothiophenes in glycerol. Mol. Diversity 2020, 24, 737. 10.1007/s11030-019-09985-w. [DOI] [PubMed] [Google Scholar]
- Khazaei A.; Veisi H.; Safaei M.; Ahmadian H. Green synthesis of 5-arylidene-2,4-thiazolidinedione, 5-benzylidene rhodanine and dihydrothiophene derivatives catalyzed by hydrated ionic liquid tetrabutylammonium hydroxide in aqueous medium. J. Sulfur Chem. 2014, 35, 270. 10.1080/17415993.2013.860142. [DOI] [Google Scholar]
- Dawood K. M. An efficient route to trans-4,5-dihydrothiophenes and thiazoles via nitrogen and sulfur ylides. Synth. Commun. 2001, 31, 1647. 10.1081/SCC-100103983. [DOI] [Google Scholar]
- Shestopalov A. M.; Bogomolova O. P.; Litvinov V. P. Stereoselective synthesis of trans-2,3-disubstituted 5-amino-4-cyano-2,3-dihydrothiophenes. Synthesis 1991, 277. 10.1055/s-1991-26445. [DOI] [Google Scholar]
- Samet A. V.; Shestopalov A. M.; Nesterov V. N.; Semenov V. V. Reactions of sulfur ylides with α,β-unsaturated thioamides: Synthesis of dihydrothiophenes and cyclopropanes. Russ. Chem. Bull. 1998, 47, 127. 10.1007/BF02495519. [DOI] [Google Scholar]
- Samet A. V.; Shestopalov A. M.; Nesterov V. N.; Semenov V. V. An improved stereoselective synthesis of 5-acyl-2-amino-4-aryl-3-cyano-4,5-dihydrothiophenes. Synthesis 1997, 1997, 623. 10.1055/s-1997-1396. [DOI] [Google Scholar]
- Shestopalov A. M.; Litvinov V. P.; Rodinovskaya L. A.; Sharanin Y. A. Stereoselective synthesis of trans-4,5-substituted 1,4,5,6-tetrahydropyridine-2-(olates)thiolates. Synthesis 1991, 402. 10.1055/s-1991-26477. [DOI] [Google Scholar]
- Jacob A.; Barkawitz P.; Andreev I. A.; Ratmanova N. K.; Trushkov I. V.; Werz D. B. (3+2)-Cycloaddition of donor-acceptor cyclopropanes with thiocyanate: A facile and efficient synthesis of 2-amino-4,5-dihydrothiophenes. Synlett 2021, 32, 901. 10.1055/a-1385-2385. [DOI] [Google Scholar]
- Xie M. S.; Zhao G. F.; Qin T.; Suo Y. B.; Qu G. R.; Guo H. M. Thiourea participation in [3+2] cycloaddition with donor–acceptor cyclopropanes: a domino process to 2-amino-dihydrothiophenes. Chem. Commun. 2019, 55, 1580. 10.1039/C8CC09595G. [DOI] [PubMed] [Google Scholar]
- Gopinath P.; Chandrasekaran S. Synthesis of functionalized dihydrothiophenes from doubly activated cyclopropanes using tetrathiomolybdate as the sulfur transfer reagent. J. Org. Chem. 2011, 76, 700. 10.1021/jo102059p. [DOI] [PubMed] [Google Scholar]
- Kasai N.; Maeda R.; Furuno H.; Hanamoto T. A practical synthesis and applications of (E)-diphenyl-β-(trifluoromethyl) vinylsulfonium triflate. Synthesis 2012, 44, 3489. 10.1055/s-0032-1316792. [DOI] [Google Scholar]
- Ishikawa T.; Kasai N.; Yamada Y.; Hanamoto T. Difluoromethyl vinyl sulfonium salt: a one-pot access to difluoromethyl-containing cyclopropanes. Tetrahedron 2015, 71, 1254. 10.1016/j.tet.2014.12.102. [DOI] [Google Scholar]
- Dotsenko V. V.; Krivokolysko S. G.; Chernega A. N.; Litvinov V. P. A new stereoselective synthesis of 2-amino-4,5-dihydrothiophene-3-carbonitrile derivatives. Russ. Chem. Bull. 2007, 56, 1431. 10.1007/s11172-007-0217-7. [DOI] [Google Scholar]
- A review on the chemistry of thiocyanatoacetophenone:; Gouda M. A. 1-Phenyl-2-thiocyanatoethanone as synthons in heterocyclic synthesis. Synth. Commun. 2013, 43, 2547. 10.1080/00397911.2012.730649. [DOI] [Google Scholar]
- The chemistry of cyanothioacetamide has been reviewed recently:; Dyachenko V. D.; Dyachenko I. V.; Nenajdenko V. G. Cyanothioacetamide: a polyfunctional reagent with broad synthetic utility. Russ. Chem. Rev. 2018, 87, 1. 10.1070/RCR4760. [DOI] [Google Scholar]
- Kamal R.; Kumar R.; Kumar V.; Bhardwaj V. Synthetic utilization of α,β-chalcone dibromide in heterocyclic chemistry and stereoselective debromination. ChemistrySelect 2019, 4, 11578. 10.1002/slct.201902262. [DOI] [Google Scholar]
- Neese F. The ORCA program system. WIREs Comput. Mol. Sci. 2012, 2, 73. 10.1002/wcms.81. [DOI] [Google Scholar]
- Neese F. Software update: the ORCA program system, version 4.0. WIREs Comput. Mol. Sci. 2018, 8, e1327. 10.1002/wcms.1327. [DOI] [Google Scholar]
- Neese F.; Wennmohs F.; Becker U.; Riplinger C. The ORCA quantum chemistry program package. J. Chem. Phys. 2020, 152, 224108. 10.1063/5.0004608. [DOI] [PubMed] [Google Scholar]
- Grimme S.; Hansen A.; Ehlert S.; Mewes J.-M. r2SCAN-3c: A “Swiss army knife” composite electronic-structure method. J. Chem. Phys. 2021, 154, 064103 10.1063/5.0040021. [DOI] [PubMed] [Google Scholar]
- Caldeweyher E.; Ehlert S.; Hansen A.; Neugebauer H.; Spicher S.; Bannwarth C.; Grimme S. A generally applicable atomic-charge dependent London dispersion correction. J. Chem. Phys. 2019, 150, 154122. 10.1063/1.5090222. [DOI] [PubMed] [Google Scholar]
- Kruse H.; Grimme S. A geometrical correction for the inter- and intra-molecular basis set superposition error in Hartree-Fock and density functional theory calculations for large systems. J. Chem. Phys. 2012, 136, 154101. 10.1063/1.3700154. [DOI] [PubMed] [Google Scholar]
- Tomasi J.; Mennucci B.; Cammi R. Quantum mechanical continuum solvation models. Chem. Rev. 2005, 105, 2999. 10.1021/cr9904009. [DOI] [PubMed] [Google Scholar]
- Allouche A. R. Gabedit – A graphical user interface for computational chemistry softwares. J. Comput. Chem. 2011, 32, 174. 10.1002/jcc.21600. [DOI] [PubMed] [Google Scholar]
- Chemcraft – graphical software for visualization of quantum chemistry computations. Avail. URL: https://www.chemcraftprog.com
- Wu F. Y.; Li Y.; Feng H.; Wu Q.; Jiang B.; Shi F.; Tu S. J. Stereoselective synthesis of functionalized cyclopropane derivatives via α-thiocyanate ketone-based three-component reaction. Synthesis 2011, 2011, 2459. 10.1055/s-0030-1260096. [DOI] [Google Scholar]
- Rappoport Z.; Avramovitch B.; Karni M.; Apeloig Y. Thiocyanate ion promoted E⇄Z isomerization of α-bromo-β,β-diactivated electrophilic olefins: An apparent high nucleofugality of NCS–. Israel J. Chem. 1989, 29, 267. 10.1002/ijch.198900036. [DOI] [Google Scholar]
- Ceccon A.; Papa I.; Fava A. Structural effects on concurrent first- and second-order processes in borderline nucleophilic substitution. The isotopic exchange between substituted benzhydryl thiocyanates and ionic thiocyanate in acetonitrile and acetone. J. Am. Chem. Soc. 1966, 88, 4643. 10.1021/ja00972a021. [DOI] [Google Scholar]
- Fava A.; Iliceto A.; Ceccon A. Concurrent operation of unimolecular and bimolecular processes in borderline nucleophilic substitution reactions. The isotopic exchange between benzhydryl thiocyanates and NaSCN in acetonitrile. Tetrahedron Lett. 1963, 4, 685. 10.1016/S0040-4039(01)90697-5. [DOI] [Google Scholar]
- Oae S.; Yamada N.; Fujimori K.; Kikuchi O. Nucleophilic substitution on 4-methylbenzyl thiocyanate with nucleophiles. Bull. Chem. Soc. Jap. 1983, 56, 248. 10.1246/bcsj.56.248. [DOI] [Google Scholar]
- Al-Khalil S. I.; Bowman W. R. Radical-nucleophilic substitution (SRN1) reactions of α-nitro-thiocyanates. Tetrahedron Lett. 1983, 24, 2517. 10.1016/S0040-4039(00)81970-X. [DOI] [Google Scholar]
- Hanefeld W. Untersuchungen an 1,3-Thiazinen, VII. Reaktionen von 3-thiocyanato-propionyl-derivaten mit aminen. Arch. Pharm. 1977, 310, 344. 10.1002/ardp.19773100411. [DOI] [PubMed] [Google Scholar]
- Sá M. M.; Fernandes L.; Ferreira M.; Bortoluzzi A. J. Synthesis of allylic thiocyanates and novel 1,3-thiazin-4-ones from 2-(bromomethyl) alkenoates and S-nucleophiles in aqueous medium. Tetrahedron Lett. 2008, 49, 1228. 10.1016/j.tetlet.2007.12.029. [DOI] [Google Scholar]
- Erian A. W.; Sherif S. M. The chemistry of thiocyanic esters. Tetrahedron 1999, 55, 7957. 10.1016/S0040-4020(99)00386-5. [DOI] [Google Scholar]
- Ibrahim Y. A.; Elwahy A. H. M.; Kadry A. M. Thienopyrimidines: synthesis, reactions, and biological activity. Adv. Heterocycl. Chem. 1996, 65, 235. 10.1016/S0065-2725(08)60297-4. [DOI] [Google Scholar]
- Litvinov V. P. Thienopyrimidines: synthesis, properties, and biological activity. Russ. Chem. Bull. 2004, 53, 487. 10.1023/B:RUCB.0000035630.75564.2b. [DOI] [Google Scholar]
- Litvinov V. P. The chemistry of thienopyrimidines. Adv. Heterocycl. Chem. 2006, 92, 83. 10.1016/S0065-2725(06)92003-0. [DOI] [Google Scholar]
- Aly A. A.; Ishak E. A.; Ramadan M.; Germoush M. O.; El-Emary T. I.; Al-Muaikel N. S. Recent report on thieno[2,3-d]pyrimidines. Their preparation including microwave and their utilities in fused heterocycles synthesis. J. Heterocycl. Chem. 2013, 50, 451. 10.1002/jhet.1118. [DOI] [Google Scholar]
- Bozorov K.; Zhao J. Y.; Elmuradov B.; Pataer A.; Aisa H. A. Recent developments regarding the use of thieno[2,3-d]pyrimidin-4-one derivatives in medicinal chemistry, with a focus on their synthesis and anticancer properties. Eur. J. Med. Chem. 2015, 102, 552. 10.1016/j.ejmech.2015.08.018. [DOI] [PubMed] [Google Scholar]
- Elrazaz E. Z.; Serya R. A. T.; Ismail N. S. M.; Abou El Ella D. A.; Abouzid K. A. M. Thieno[2,3-d]pyrimidine based derivatives as kinase inhibitors and anticancer agents. Future J. Pharm. Sci. 2015, 1, 33. 10.1016/j.fjps.2015.09.001. [DOI] [Google Scholar]
- Wilding B.; Klempier N. Newest developments in the preparation of thieno[2,3-d]pyrimidines. Org. Prep. Proced. Int. 2017, 49, 183. 10.1080/00304948.2017.1320513. [DOI] [Google Scholar]
- Ali E. M. H.; Abdel-Maksoud M. S.; Oh C.-H. Thieno[2,3-d]pyrimidine as a promising scaffold in medicinal chemistry: Recent advances. Bioorg. Med. Chem. 2019, 27, 1159. 10.1016/j.bmc.2019.02.044. [DOI] [PubMed] [Google Scholar]
- Elmongy E. I. Thieno[2,3-d]pyrimidine derivatives: Synthetic approaches and their FLT3 kinase inhibition. J. Heterocycl. Chem. 2020, 57, 2067. 10.1002/jhet.3934. [DOI] [Google Scholar]
- Kobayashi K.; Murayama I.; Ono R.; Fujiwara D.; Hiyoshi H.; Umezu K. Synthesis of 5-hydroxythieno[2,3-d]pyrimidin-6(5H)-one derivatives by the reaction of 2-(4-chloropyrimidin-5-yl)-2-hydroxyalkanoate with sodium hydrogensulfide. Heterocycles 2018, 96, 1248. 10.3987/COM-18-13911. [DOI] [Google Scholar]
- Maruoka H.; Yamagata K.; Yamazaki M. Synthesis of 5,6-dihydrothieno (and furo) pyrimidines bearing an active methine group at the 4-position. J. Heterocycl. Chem. 2001, 38, 269. 10.1002/jhet.5570380140. [DOI] [Google Scholar]
- Dotsenko V. V.; Krivokolysko S. G.; Litvinov V. P. The Mannich reaction in the synthesis of N,S-containing heterocycles 9. A new approach to thieno[2, 3-d]pyrimidines. Russ. Chem. Bull. 2009, 58, 1524. 10.1007/s11172-009-0206-0. [DOI] [Google Scholar]
- Maruoka H.; Okabe F.; Yamagata K. Synthesis and reactions of N-methyl-4-(methylthio) thieno[2,3-d]pyrimidinium salts. J. Heterocycl. Chem. 2008, 45, 1503. 10.1002/jhet.5570450543. [DOI] [Google Scholar]
- Mondal M. A.; Mondal S.; Khan A. A. A mechanistic insight into the acid catalyzed, one-pot synthesis of isoindole-fused quinazolin-4-ones. J. Chem. Sci. 2020, 132, 63. 10.1007/s12039-020-01768-3. [DOI] [Google Scholar]
- Khan A. T.; Khan M. M.; Das D. K.; Lal M. Silica-supported perchloric acid (HClO4–SiO2): An efficient catalyst for one-pot synthesis of functionalized tetrahydropyrimidine derivatives. J. Heterocycl. Chem. 2012, 49, 1362. 10.1002/jhet.1017. [DOI] [Google Scholar]
- Das B.; Kanth B. S.; Shinde D. B.; Kamble V. T. Efficient synthesis of tetrahydropyrimidines and pyrrolidines by a multicomponent reaction of dialkyl acetylenedicarboxylates (= dialkyl but-2-ynedioates), amines, and formaldehyde in the presence of iodine as a catalyst. Helv. Chim. Acta 2011, 94, 2087. 10.1002/hlca.201100185. [DOI] [Google Scholar]
- Nagarapu L.; Vulupala H. R.; Venkatanarsimhaji C.; Bantu R.; Reddy A. R. Molecular iodine-catalyzed synthesis of 1,2,3,6-tetrahydropyrimidines. Synth. Commun. 2012, 42, 2131. 10.1080/00397911.2011.554061. [DOI] [Google Scholar]
- Muthusaravanan S.; Perumal S.; Almansour A. I. Facile catalyst-free pseudo five-component domino reactions in the expedient synthesis of 5-aroyl-1,3-diarylhexahydropyrimidines. Tetrahedron Lett. 2012, 53, 1144. 10.1016/j.tetlet.2011.12.097. [DOI] [Google Scholar]
- Karim E.; Kishore K.; Vishwakarma J. N. A facile one-pot synthesis of novel substituted 1,2,3,4-tetrahydropyrimidines. J. Heterocycl. Chem. 2003, 40, 901. 10.1002/jhet.5570400524. [DOI] [Google Scholar]
- Chen Z.-P.; Wang H.-B.; Wang Y.-Q.; Zhu Q.-H.; Xie Y.; Liu S.-W. Synthesis of fused pyrrolo[3,4-d]tetrahydropyrimidine derivatives by proline-catalyzed multicomponent reaction. Tetrahedron 2014, 70, 4379. 10.1016/j.tet.2014.04.075. [DOI] [Google Scholar]
- Kiss P.; Holly S. Herstellung von Pyrimido[6,1-a]isochinolinen mit Formaldehyd. Chem. Ber. 1981, 114, 61. 10.1002/cber.19811140107. [DOI] [Google Scholar]
- Vishwakarma J. N.; Dutta M. C.; Chanda K.; Das B.; Laskar M. A.; Nongkhlaw R. L. Synthesis and anti-bacterial activities of novel 5-isonicotinoyl-1,2,3,4-tetrahydropyrimidines and bis-(5-isonicotinoyl-1,2,3,4-tetrahydropyrimidines). ARKIVOC 2010, 2009, 131. 10.3998/ark.5550190.0010.d11. [DOI] [Google Scholar]
- Yıldırım M.; Celikel D.; Dürüst Y.; Knight D. W.; Kariuki B. M. A rapid and efficient protocol for the synthesis of novel nitrothiazolo[3,2-c]pyrimidines via microwave-mediated Mannich cyclisation. Tetrahedron 2014, 70, 2122. 10.1016/j.tet.2014.02.003. [DOI] [Google Scholar]
- Youssif S.; Agili F. One-pot synthesis of fused 2-thiouracils: pyrimidopyrimidines, pyridopyrimidines and imidazolopyrimidines. Z. Naturforsch. 2008, 63, 860. 10.1515/znb-2008-0709. [DOI] [Google Scholar]
- Alekszi-Kaszás A.; Nemes P.; Tóth G.; Halász J.; Scheiber P. Mannich condensations of activated cyclic enamines. Synth. Commun. 2018, 48, 2099. 10.1080/00397911.2018.1484488. [DOI] [Google Scholar]
- Zhao Y.; Guo X.; Du Y.; Shi X.; Yan S.; Liu Y.; You J. Synthesis of fused-tetrahydropyrimidines: one-pot methylenation–cyclization utilizing two molecules of CO2. Org. Biomol. Chem. 2020, 18, 6881. 10.1039/D0OB01504K. [DOI] [PubMed] [Google Scholar]
- Vadivelu M.; Raheem A. A.; Sugirdha S.; Bhaskar G.; Karthikeyan K.; Praveen C. Gold catalyzed synthesis of tetrahydropyrimidines and octahydroquinazolines under ball milling conditions and evaluation of anticonvulsant potency. ARKIVOC 2019, 2018, 90. 10.24820/ark.5550190.p010.372. [DOI] [Google Scholar]
- Zefirov Y. V.; Zorkii P. M. Van der Waals radii and their application in chemistry. Russ. Chem. Rev. 1989, 58, 421. 10.1070/RC1989v058n05ABEH003451. [DOI] [Google Scholar]
- Dotsenko V. V.; Krivokolysko S. G.; Polovinko V. V.; Litvinov V. P. On the regioselectivity of the reaction of cyanothioacetamide with 2-acetylcyclohexanone, 2-acetylcyclopentanone, and 2-acetyl-1-(morpholin-4-yl)-1-cycloalkenes. Chem. Heterocycl. Compd. 2012, 48, 309. 10.1007/s10593-012-0991-5. [DOI] [Google Scholar]
- Dyachenko V. D.; Krivokolysko S. G.; Litvinov V. P. Synthesis of arylmethylenecyanothioacetamides in a Michael reaction. Mendeleev Commun. 1998, 8, 23. 10.1070/MC1998v008n01ABEH000816. [DOI] [Google Scholar]
- Cromwell N. H. α,β-Unsaturated Aminoketones. II. α- and β-Morpholinobenzalacetophenones. J. Am. Chem. Soc. 1940, 62, 2897. 10.1021/ja01868a003. [DOI] [Google Scholar]
- Bickel C. L. The preparation and alkaline cleavage of o-chlorodibenzoylmethane. J. Am. Chem. Soc. 1946, 68, 865. 10.1021/ja01209a046. [DOI] [Google Scholar]
- Lund H. Electroörganic preparations. IX. Polarography and reduction of substituted aryl alkyl ketones. Acta Chem. Scand. 1960, 14, 1927. 10.3891/acta.chem.scand.14-1927. [DOI] [PubMed] [Google Scholar]
- Sheldrick G. M. A short history of SHELX. Acta Cryst. A 2008, 64, 112. 10.1107/S0108767307043930. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.