

Abstract

The naphthaleman family, a set of uniquely designed visual molecular structures comprising multisubstituted naphthalenes, was synthesized utilizing regiocontrolled benzannulation as a key step. The naphthaleman family possesses a common naphthalene body with a head comprising the 3,4-methylenedioxy group, symmetrical or unsymmetrical right and left arms, and two alkynyl legs. The synthesis involves six C–C bond-forming reaction sequences. (i) syn-Stereoselective gem-dichlorocyclopropanation of methyl angelate (86%). (ii) Acylation with ArMgBr (three examples, 60–91% yield). (iii) Stereocontrolled introduction of the 3,4-methylenedioxyphenyl group (three examples, 67–92% yield). (iv) Crucial regiocontrolled benzannulation to construct a common body segment (71–73% yield). (v) Two Suzuki–Miyaura cross-couplings to install the right or left arms (first-stage route: four examples, 77–93% and second-stage route: four examples, 42–90% yield). (vi) Double alkynylation to insert two legs (first-stage route: four examples, 61–77% yield and second-stage route: sole example, 83% yield). The four core members were produced through both first-stage and second-stage routes, with the second-stage approach demonstrating superiority over the first-stage approach. One of the members was alternatively synthesized by switching the installation order of the right and left arms, and identical twin members were produced by high-performance liquid chromatography chiral separation. The most stable conformations of two naphthaleman family members were calculated by Spartan software.
Introduction
Aromatic compounds containing unique visual molecular structures have attracted the attention of many chemists over the past few decades, from not only a scientific but also an educational standpoint. The pioneering synthesis of “nanokid” and his family, introduced by Chanteau and Tour,1 caused quite sensation for both chemists and students due to their fascinating structures (Figure 1). The subsequent syntheses of nanocar,2 molecular motor,3 and penguinone4 as representative bewitched molecules with distinct aromatic structures have also attracted considerable attention.
Figure 1.
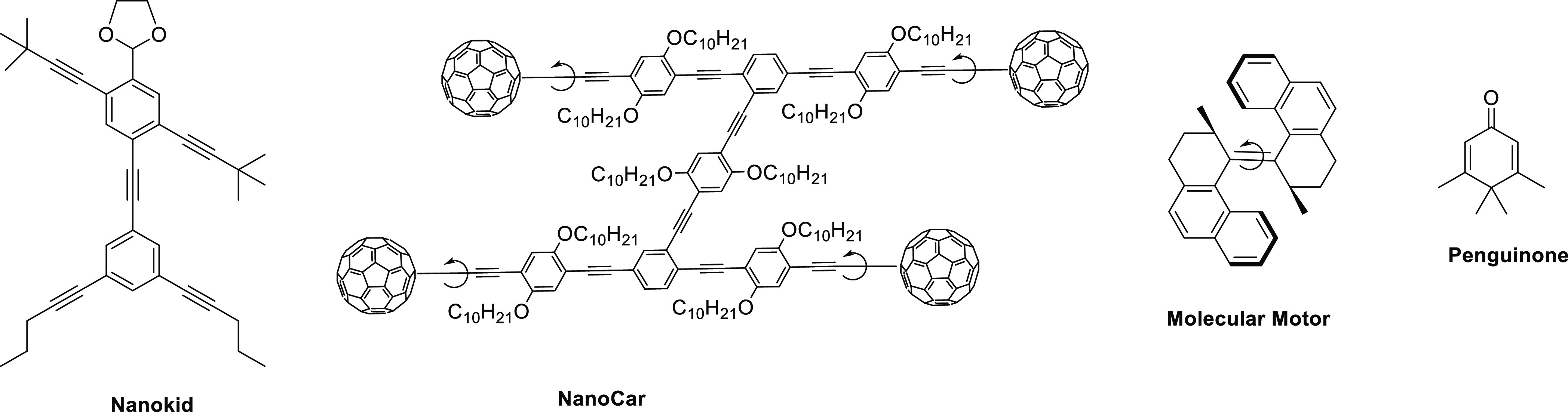
Representative visual compounds: nanokid, nanocar, molecular motor, and penguinone.
Inspired by these unique molecular structures and in close connection with our longstanding studies of the transformation of gem-dihalocyclopropanes,5 we present here the synthesis of a uniquely designed “naphthaleman family” with several charming members that are composed of fully substituted naphthalene structures (Figure 2). They resemble human forms and familiar objects and may evoke images of “a side-throwing pitcher”, “a football keeper”, “a tennis player”, “a flying bird”, or “a swimming frog”. The family comprises four naphthaleman core members 1a–1d, a clone of 1c by an alternative synthesis, and identical twins S-1d and R-1d by optical resolution.
Figure 2.
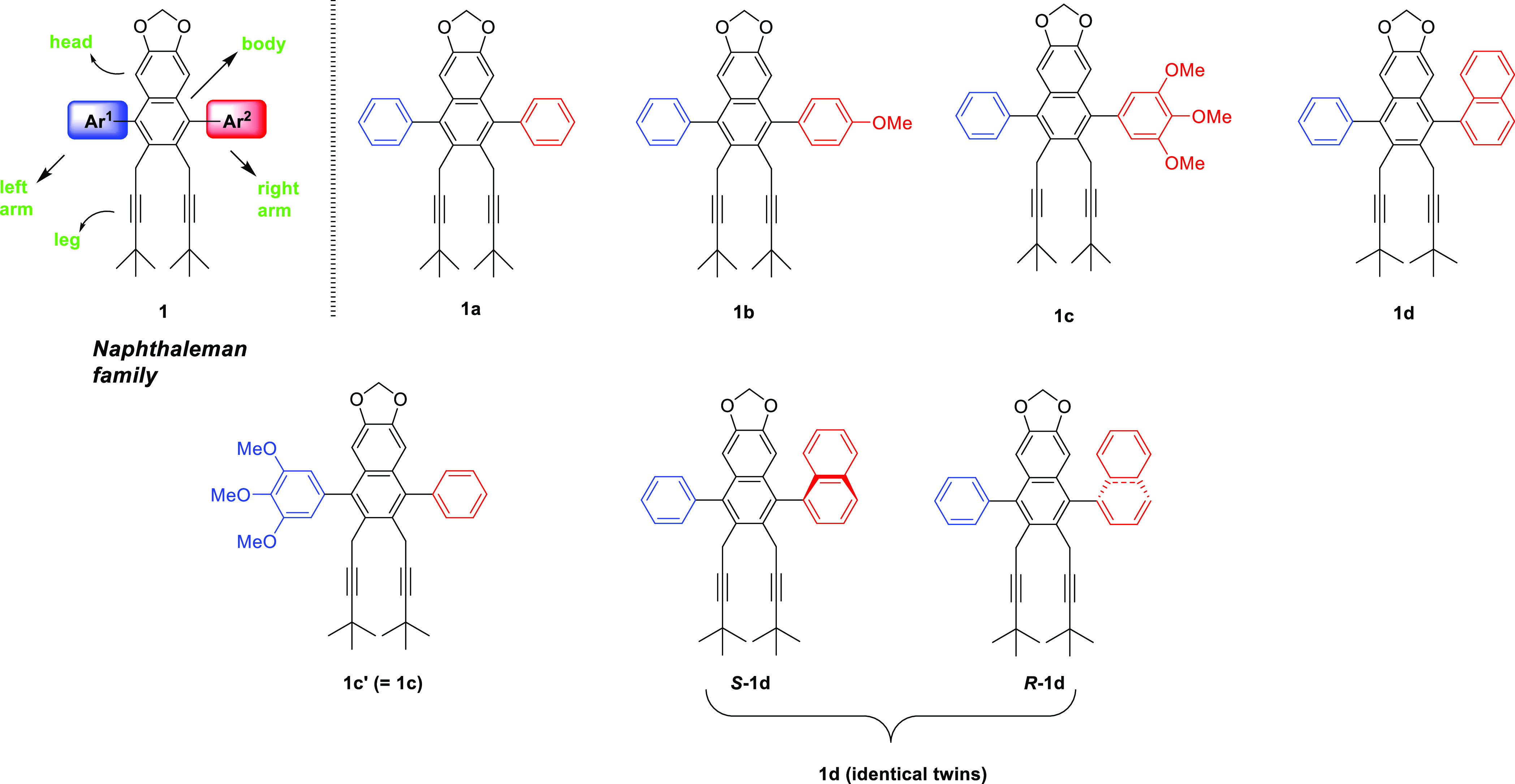
Naphthaleman family members.
Results and Discussion
Retrosyntheses
Regiocontrolled benzannulation strategies provide distinctive constructions for highly substituted and elaborated α-arylnaphthalenes, which have useful applications as reagents, catalysts, biologically active natural products, pharmaceuticals, and functionalized materials due to their core structural scaffolds.6 Our continuing investigations on nonregioselective,5a,5b regiocontrolled,5c chirality exchange,5d and large-scale5e benzannulations, and the relevant cyclopropane transformations6 or derivatizations,7 led us to envision the synthesis of a “naphthaleman family” 1 possessing a common naphthalene body with a head comprising the 3,4-methylenedioxy group, symmetrical or unsymmetrical right and left arms, and two alkynyl legs. Retrosynthetic pathways for the synthesis of 1 are classified as either the first-stage approach or the second-stage approach (Scheme 1).
Scheme 1. Two Retrosynthetic Approaches for Naphthaleman Family 1.
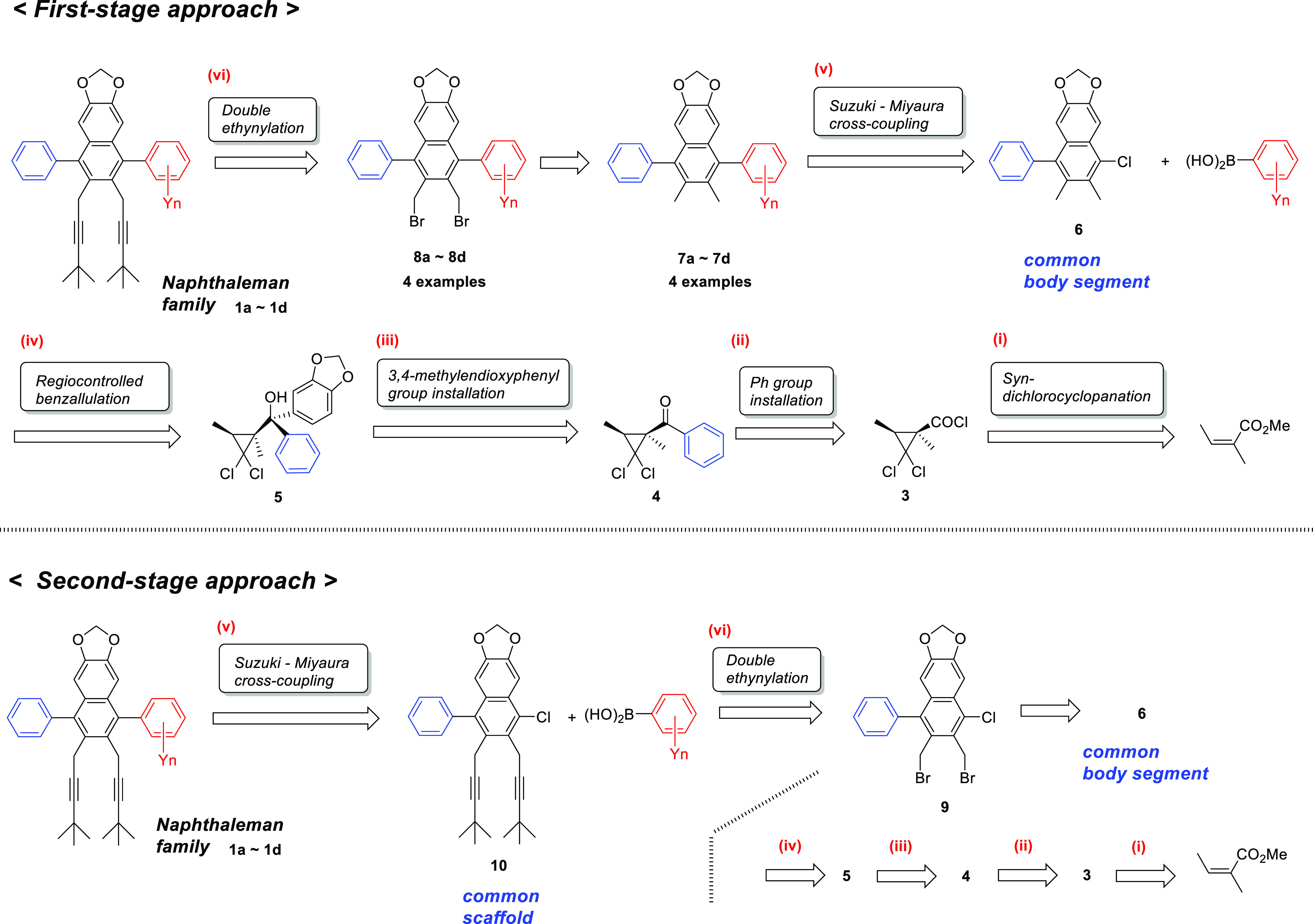
Common body segment 6 was conveniently prepared following the reported practical procedures.5e Three contiguous functional groups in 6 were effectively utilized for the construction of 1. Two contiguous benzylic positions were transformed to two legs by dibromination and successive ethynylation. On the other hand, the pendant 1-chloro group was coupled with four arms by Suzuki–Miyaura cross-couplings to produce naphthaleman family 1 with uniquely substituted head, hands, and legs.
The first stage approach consists of six C–C bond-forming reactions to construct the naphthaleman family 1. (i) Syn-stereoselective gem-dichlorocyclopropanation of methyl angelate leading to the formation of acid chloride 3. (ii) Acylation of 3 with PhMgBr leading to the formation of phenyl ketone 4. (iii) Stereocontrolled introduction of 3,4-methylenedioxyphenyl groups to 4 leading to the formation of [S*-(1S*,3S*)]-phenyl(3,4-methylenedioxypheny)-2,2-dichlorocyclopropylmethanol 5. (iv) Crucial regiocontrolled benzannulation of 5 affording 1-phenyl-4-chloronaphthelene 6 as a common body segment. (v) Suzuki–Miyaura cross-couplings using acceptor 6 leading to the formation of 1-aryl-4-phenyl-2,3-dimethylnaphthalenes 7a–7d. (vi) Double alkynylation of dibromide 8a–8d derived from 7a–7d producing the target naphthaleman family 1a–1d.
In contrast, the second-stage approach involves another sequence of six C–C bond-forming reactions. The first four steps (i)–(iv) are the same as those for the linear approach. The double alkynylation step of 6 precedes Suzuki–Miyaura cross-couplings using common scaffold 10. The convergent approach is apparently superior to the linear approach from the standpoint of total efficiency to produce family members 1a–1d.
Construction of the Common Body Segment
Scheme 2 shows the construction of the common “body” segment 6 for not only the linear approach but also the convergent approach. Methyl angelate was converted to acid chloride 3 by syn-stereoselective gem-dichlorocyclopropanation and two conventional transformations (hydrolysis and acid chloride formation) according to the reported method.5c Phenylation of 3 with PhMgBr at 0–5 °C afforded 2,2-dichlorocyclopropyl phenyl ketone 4 in 81% yield. Subsequent Cram rule-stereocontrolled 3,4-methylendioxyphenyl group addition to the predominant s-trans conformer of 4 proceeded smoothly to afford [S*-(1S*,3S*)]-stereodefined (3,4-methylendioxy)(phenyl)(2,2-dichlorocyclopropyl)methanol 5 in 92% yield.5b The key regiocontrolled benzannulation using 5 successfully produced the common body segment 6 in 72% yield with excellent regioselectivity.5c The regiocontrolled pathway can be rationalized by the reported chelation mechanism fixing the conformation during the cyclopropane cleavage.
Scheme 2. Construction of Common Body Segment 6.

First-Stage Approach for the Synthesis of the Naphthaleman Family
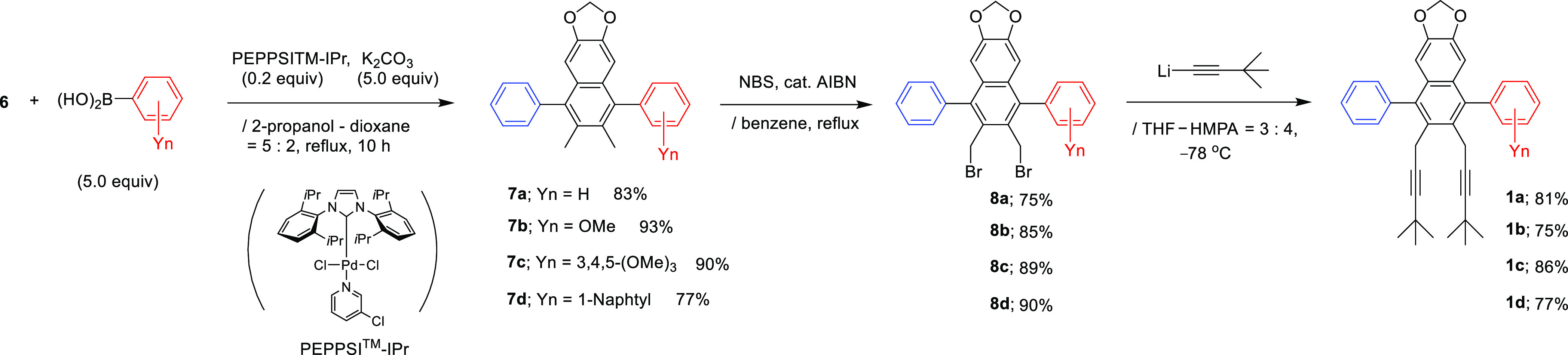
With the body segment 6 in hand, we next examined Suzuki–Miyaura cross-couplings of α-chloronaphthalenes 6 to install four “right arm” parts (Scheme 3). Despite the sterically congested and less reactive α-chloro position in 6, coupling with ArB(OH)2 (5.0 equiv) proceeded well to afford the desired full-body precursors 7a–7d in 77–93% yield using (1,3-diisopropylimidazol-2-ylidene)(3-chloropyridyl)palladium(II)dichloride (PEPPSI-IPr)8 (0.20 equiv)-K2CO3 (5.0 equiv) catalysis under somewhat harsh conditions (i.e., molar ratio, temperature, and time; optimization of PEPPSI-IPr-K2CO3 catalysis using 6: see the Supporting Information). The two leg parts were introduced by the following sequences: 7a–7d were dibrominated using NBS/cat, AIBN to afford 8a–8d in 75–90% yield, and subsequent double alkynylation with the lithium salt of 3,3-dimethylbut-1-yn provided the four members of the naphthaleman family 1a–1d in 75–86% yield.
Scheme 3. Syntheses of Naphthaleman Family 1 by the First-Stage Approach.
Second-Stage Approach for the Synthesis of the Naphthaleman Family
After developing the first-stage approach, we turned our attention to a more straightforward approach. Treatment of body segment 6 with NBS/cat. AIBN under identical conditions afforded dibrominated product 9 in 93% yield (Scheme 4). A similar double ethynylation by the lithium salt of 3,3-dimethylbut-1-yn provided the common α-chloronaphthalene scaffold 10 in 89% yield. Suzuki–Miyaura cross-coupling of 10 with ArB(OH)2 using a similar PEPPSI-IPr catalysis, however, led to the formation of complex mixtures under the conditions described in Scheme 3 because the two alkynyl groups in the legs did not tolerate the identical conditions to give complex mixtures.
Scheme 4. Syntheses of Naphthaleman Family 1 by the Second-Stage Approach.
Fortuitously, the use of ArB(OH)2 (1.5 equiv) using a more reactive Pd(OAc)2 (0.04 equiv)–SPhos (0.06 equiv)–K3PO4 (2.0 equiv) catalysis9 instead of PEPPSI-IPr catalysis solved the problem with the toleration of the alkynyl moiety to produce the four family members of 1a–1d in 42–90%. Notably, these cross-couplings were implemented under more mild conditions (i.e., better molar ratio, lower catalyst loading, lower temperature, and shorter time).
Alternative Synthesis of Naphthaleman Family 1c by the Switching Route
To expand the scope of this project, we envisaged an alternative synthetic route for synthesizing the naphthaleman family 1c′ (= 1c) (Scheme 5). The 3,4,5-trimethoxyphenyl “left arm” group was initially installed by acylation with acid chloride 3 to afford ketone 11 in 60% yield. Next, the 3,4-methylenedioxyphenyl “right arm” group was added to 11 to afford alcohol 12 in 86% yield with high stereoselectivity. Alcohol 12 smoothly underwent regiocontrolled benzannulation to produce alternative body segment 13 in 73% yield. Notably, the reaction sequences follow the reported total synthesis of dehydrodesoxypodophyllotoxin, an unsymmetrically substituted lignan lactone.5c
Scheme 5. Synthesis of the Naphthaleman Family 1c′ (= 1c).
Sha’s group pointed out that the inherent reactivity order during the Friedel–Crafts-type reaction was 3,4,5-trimethoxyphenyl group >3,4-methylenedioxyphenyl group due to the favorable planar π-electron overlap of the 3,4-dimethoxy group.8 Nonetheless, the present regiocontrolled benzannulation proceeded smoothly toward the less-reactive 3,4-methylenedioxyphenyl group, probably because the chelation-controlled mechanism functioned effectively (see Scheme 2). Suzuki–Miyaura cross-coupling of 13 using PEPPSI-IPr-K2CO3 catalysis afforded clone precursor 7c (see Scheme 3). Subsequent dibromination and dialkynylation sequences using 7c successfully produced the target naphthaleman family 1c′ (= 1c). Eventually, the introduction order of the right and left arms was switched conversely.
Synthesis of “Identical Twins” by Optical Resolution
Family member 1d is composed of the body naphthalene connected with the right arm or left arm between the α- and α′- positions, in which axial chirality should emerge (Scheme 6). Actually, chiral high-performance liquid chromatography (HPLC) analysis revealed that 1d was formed as a racemate (two enantiomeric mixtures) (vide infra).
Scheme 6. Synthesis of “Identical Twins” of Naphthaleman 1d via an Alternative Route.

With this result in hand, we implemented an alternative shorter synthesis of 1d than the conventional route, as shown in Scheme 2; parent ester 2 was employed instead of acid chloride 3 for the acylation step (Scheme 6). Ketone 14 was obtained in good yield (67%) by the acylation of 2 with (1-naphthyl)MgBr, in which the use of a stereocongested (1-naphthyl)MgBr nucleophile completely prevented further undesirable addition. The addition of 3,4-methylenedioxyphenyl lithium to 14 also afforded alcohol 15 in better 76% yield, which was subjected to regiocontrolled benzannulation to produce novel body segment 16 in 71% yield. Suzuki–Miyaura cross-coupling of 16 with PhB(OH)2, catalyzed by Pd(OAc)2–SPhos–K2PO4, successfully furnished 7d in 70% yield. As described in the linear approach procedure of the First-Stage Approach for the Synthesis of the Naphthaleman Family, a formal synthesis of the desired family member 1d was completed. In a series of α-naphthyl(naphthalene) compounds (A), axial chirality induced geminal couplings of 1H NMR between Ha and Hb due to the inequivalency. Chiral HPLC analysis of 1d resulted in the separation of the identical twins (a couple of enantiomers) S-1d and R-1d with high resolution (a chart of the clear separation is addressed in the Supporting Information).
The most stable conformations of naphthaleman family members 1a and 1d were calculated by Spartan software (Wavefunction, Inc. ver. ’18 1.1.0) (Figure 3). In both compounds, the two legs were located not in parallel but rather spreading at ca. 130° due to the steric repulsion: not standup style but running style. In contrast, the two arms were located perpendicular and parallel to the naphthalene body, the result of which is in good accordance with the reported X-ray structure of the relevant and fundamental 4-chloro-2-methyl-1-phenylnaphthalene.5b
Figure 3.

Spartan calculation drawing of naphthaleman 1a and “identical twin” of naphthaleman 1d.
Conclusions
We developed the synthesis of the naphthaleman family, a set of uniquely designed visual molecular structures comprising multisubstituted naphthalenes, utilizing regiocontrolled benzannulation as the key step. The naphthaleman family possesses a common naphthalene body having a head comprising a 3,4-methylenedioxy group, symmetrical or unsymmetrical right and left arms, and two alkynyl legs. The synthetic approaches are classified as either a first-stage approach, catalyzed by PEPPSI-IPr–K2CO3 or a superior second-stage approach, catalyzed by Pd(OAc)2–SPhos–K2PO4. The four core members were produced through both routes, with the convergent approach demonstrating superiority over the linear approach (i.e., milder conditions, better molar ratio, and lower catalyst loading). One of the members was alternatively synthesized by switching the installation order of the right and left arms, and identical twin members were produced by chiral HPLC separation. The most stable conformations of two naphthaleman family members were calculated by Spartan software; the two legs were spreading and the two arms were located perpendicular and parallel to the naphthalene body.
We are now planning an asymmetric synthesis of twin members by utilizing chirality-transfer benzannulation (a distinctive chiral version) to provide the audience, particularly students, interested in this organic chemistry. The result will be presented in the future.
Experimental Section
[(1S*,3S*)-2,2-Dichloro-1,3-dimethylcyclopropyl]phenylmethanone (4)5c
Colorless oil; 1H NMR (500 MHz, CDCl3): δ 1.40 (d, J = 6.5 Hz, 3H), 1.64 (s, 3H), 1.68 (q, J = 6.5 Hz, 1H), 7.51–7.62 (m, 3H), and 7.98–8.00 (d, J = 7.0 Hz, 2H); 13C NMR (100 MHz, CDCl3): δ 11.7, 23.2, 35.3, 39.6, 68.1, 128.7, 130.0, 133.5, 134.4,
and 194.9; IR (neat) νmax: 2932, 1681, 1597, 1449,
1233, 1318, 1301, 1233, 1175, 984, 836, 792, and 710 cm–1.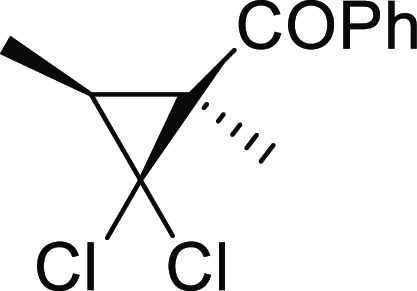
(S*)-[(1S*,3S*)-2,2-Dichloro-1,3-dimethylcyclopropyl](3,4-methylenedioxyphenyl)(phenyl)methanol (5)
nBuLi (1.58
M in hexane, 2.24 mL, 3.53 mmol) was added to a stirred solution of
3,4-methylenedioxy-1-bromobenzene (712 mg, 2.36 mmol) in tetrahydrofuran
(THF) (8 mL) at −78 °C under an Ar atmosphere, and the
mixture was stirred at the same temperature for 0.5 h. Ketone 4 (574 mg, 2.36 mmol) in THF (5 mL) was added to the mixture
at the same temperature, and the mixture was stirred for 2 h. The
mixture was poured into ice and maintained. NH4Cl aqueous
solution was extracted twice with Et2O. The combined organic
phase was washed with water and brine, dried (Na2SO4), and concentrated. The obtained crude oil was purified by
SiO2-column chromatography (hexane/AcOEt = 100:1 to 50:1)
to give the desired product 5 (788 mg, 91%, small amounts
of inseparable impurities were contained).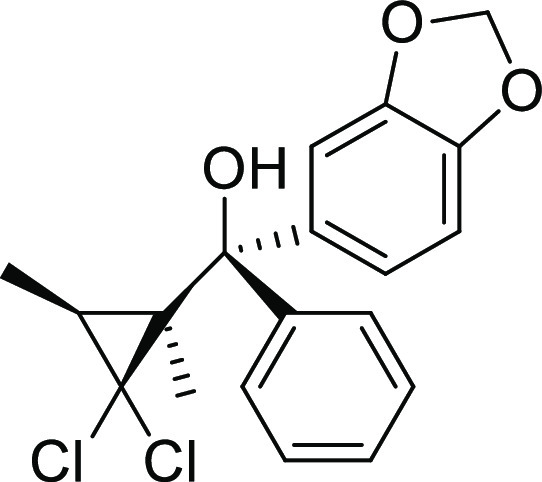
Colorless oil; 1H NMR (500 MHz, CDCl3): δ 1.19 (s, 3H), 1.53 (q, J = 6.9 Hz, 1H), 1.76 (d, J = 6.9 Hz, 3H), 2.78 (brs, 1H, −OH), 5.96 (s, 2H), 6.57 (dd, J = 1.8 Hz, 8.2 Hz, 1H), 6.71 (d, J = 8.2 Hz, 1H), 6.79 (d, J = 1.8 Hz, 1H), and 7.34–7.52 (m, 5H); 13C NMR (125 MHz, CDCl3): δ 11.1, 27.2, 36.3, 38.3, 73.8, 83.8, 101.0, 107.0, 108.9, 122.0, 127.9, 128.2, 128.8, 140.8, 144.6, 146.5, and 147.1; IR (neat) νmax: 3570, 2928, 1489, 1444, 1321, 1271, 1240, 1198, 1036, and 934 cm–1.
1-Chloro-2,3-dimethyl-6,7-methylendioxy-4-phenylnaphthalene (6)
SnCl4 (261 mg, 1.0 mmol) was
added to a stirred solution of alcohol 5 (365 mg, 1.0
mmol) in 1,2-dichloroethane (20 mL, ca. 0.05 M) at 80 °C under
an Ar atmosphere, and the mixture was stirred at the same temperature
for 1 h. The mixture was filtered through the Celite with a glass
filter, and the filtrate was concentrated under reduced pressure.
Sat. NaHCO3 aqueous solution was added to the residue which
was extracted by CHCl3, and the separated organic phase
was washed with water and brine, dried (Na2SO4), and concentrated. The obtained crude solid was purified by recrystallization
(CHCl3) to give the desired product 6 (225
mg, 72%).
Colorless crystals; mp 160–161 °C; 1H NMR (500 MHz, CDCl3): δ 2.11 (s, 3H), 2.56 (s, 3H), 5.98 (s, 2H), 6.58 (s, 1H), 7.15–7.22 (m, 2H), 7.40–7.44 (m, 1H), 7.46–7.50 (m, 2H), and 7.66 (s, 1H); 13C NMR (125 MHz, CDCl3): δ 18.0, 18.8, 101.1, 101.3, 103.2, 126.5, 127.1, 128.5 (2C), 129.3, 130.0, 130.1 (2C), 131.5, 132.1, 136.9, 140.3, 147.1, and 147.7; IR (neat) νmax: 2907, 1611, 1501, 1464, 1397, 1310, 1242, 1200, 1041, and 945 cm–1.
2,3-Dimethyl-1,4-diphenyl-6,7-methylenedioxynaphthalene (7a)
PEPPSI-IPr (68 mg, 0.1 mmol) and K2CO3 (346 mg, 2.5 mmol) were successively added
to a stirred
solution of PhB(OH)2 (305 mg, 2,5 mmol) in i-PrOH (1.3 mL) at 80 °C under an Ar atmosphere, and the mixture
was stirred at the same temperature for 10 min. A solution of 1-chloronaphthalene 6 (155 mg, 0.5 mmol) in 1,4-dioxane (5 mL) was added to the
mixture, which was stirred at the same temperature for 10 h. The mixture
was quenched with water, which was filtered through the Celite with
a glass filter, and the filtrate was concentrated under reduced pressure.
The residue was extracted twice with CHCl3. The combined
organic phase was washed with water and brine, dried (Na2SO4), and concentrated. The obtained crude product was
purified by SiO2-column chromatography (hexane: AcOEt =
1:0 to 50:1) to give the desired product 7a (146 mg,
83%).
Colorless crystals; mp 277–278 °C; 1H NMR (500 MHz, CDCl3): δ 2.13 (s, 6H), 5.88 (s, 2H), 6.64 (s, 2H), 7.28–7.29 (m, 4H), and 7.39–7.55 (m, 6H); 13C NMR (125 MHz, CDCl3): δ 18.3 (2C), 100.8, 103.0 (2C), 127.0 (2C), 128.4 (2C), 128.6 (4C), 130.3 (4C), 131.5 (2C), 137.5 (2C), 141.2 (2C), and 146.5 (2C); IR (neat) νmax: 3023, 2903, 1497, 1464, 1238, 1038, 1017, 945, 862, and 752 cm–1.
2,3-Dimethyl-4-phenyl-6,7-methylenedioxy-1-(3-methoxyphenyl)naphthalene (7b)
Following the procedure for the preparation
of 7a, the reaction using PEPPSI-IPr (27 mg, 0.04 mmol),
K2CO3 (138 mg, 1.0 mmol), (4-MeO)C6H4B(OH)2 (212 mg, 1.0 mmol), and 6 (62 mg, 0.2 mmol) gave the desired product 7b (83 mg,
93%).
Colorless crystals; mp 265–266 °C; 1H NMR (400 MHz, CDCl3): δ 2.13 (s, 3H), 2.15 (s, 3H) 3.91 (s, 3H), 5.89 (s, 2H), 6.63 (s, 1H), 6.69 (s, 1H), 7.04–7.06 (m, 2H), 7.19–7.21 (m, 2H), 7.27–7.29 (m, 2H), and 7.40–7.52 (m, 3H); 13C NMR (100 MHz, CDCl3): δ 18.4 (2C), 55.3, 100.7, 102.9 (2C), 113.4, 114.1, 126.9, 127.7 (2C), 128.5 (2C), 130.2 (2C), 131.2 (2C), 131.4, 131.9, 133.2, 133.4, 137.1, 137.3, 141.1, 146.4, and 158.5; IR (neat) νmax: 2934, 1578, 1460, 1410, 1238, 1117, 1036, 972, 945, and 860 cm–1.
2,3-Dimethyl-4-phenyl-6,7-methylenedioxy-1-(3,4,5-trimethoxyphenyl)naphthalene (7c)
Following the procedure for the preparation
of 7a, the reaction using PEPPSI-IPr (27 mg, 0.04 mmol),
K2CO3 (138 mg, 1.0 mmol), 3,4,5-(MeO)3C6H2B(OH)2 (212 mg, 1.0 mmol), and 6 (62 mg, 0.2 mmol) gave the desired product 7c (80 mg, 90%).
Colorless crystals; mp 274–275 °C; 1H NMR (500 MHz, CDCl3): δ 2.18 (s, 3H), 2.18 (s, 3H) 3.85 (s, 6H), 3.97 (s, 3H), 5.91 (s, 2H), 6.51 (s, 2H), 6.64 (s, 1H), 6.74 (s, 1H), 7.27–7.28 (m, 2H), 7.42–7.45 (m, 1H), and 7.49–7.52 (m, 2H); 13C NMR (125 MHz, CDCl3): δ 18.3 (2C), 56.1 (2C), 61.1, 100.7, 102.9 (2C), 107.1 (2C), 126.9, 128.2, 128.5 (2C), 130.2 (2C), 131.5 (2C), 136.7, 137.5 (2C), 140.1 (2C), 146.5, and 153.3 (3C); IR (neat) νmax: 2934, 1578, 1460, 1410, 1238, 1117, 1036, 972, 945, and 860 cm–1.
2,3-Dimethyl-4-phenyl-6,7-methylenedioxy-1-naphthylnaphthalene (7d)
Following the procedure for the preparation
of 7a, the reaction using PEPPSI-IPr (27 mg, 0.04 mmol),
K2CO3 (138 mg, 1.0 mmol), naphthalene-1-ylboric
acid (103 mg, 0.6 mmol), and a solution of 6 (62 mg,
0.2 mmol) gave the desired product 7d (68 mg, 77%).
Colorless crystals; mp 203–204 °C; 1H NMR (500 MHz, CDCl3): δ 2.00 (s, 3H), 2.16 (s, 3H), 5.81 (d, Jgem = 1.0 Hz, 1H), 5.82 (d, Jgem = 1.0 Hz, 1H), 6.39 (s, 1H), 6.68 (s, 1H), 7.32–7.63 (m, 10H), and 7.91–8.00 (m, 2H); 13C NMR (125 MHz, CDCl3): δ 18.2, 18.3, 100.7, 102.9, 103.0 125.7, 125.9, 126.1, 126.2, 126.9, 127.5, 127.8, 128.2, 128.3, 128.5 (2C), 128.7, 130.3 (2C), 131.4, 132.6, 132.7, 133.8, 135.1, 137.7, 138.7, 141.1 (2C), and 146.4; IR (neat) νmax: 2897, 1615, 1499, 1461, 1234, 1117, 1040, 947, 859, and 779 cm–1.
2,3-Bis(bromomethyl)-1,4-diphenyl-6,7-methylenedioxynaphthalene (8a)
NBS (182 mg, 1.0 mmol) and AIBN (4 mg,
0.02 mmol) were successively added to a stirred solution of 2,3-dimethylnaphthalene 7a (157 mg, 0.45 mmol) in benzene (7.2 mL) at 20–25
°C under an Ar atmosphere, and the mixture was stirred at 80
°C for 1 h. The mixture was quenched with 1 M aqueous HCl, which
was extracted twice with CHCl3. The combined organic phase
was washed with water and brine, dried (Na2SO4), and concentrated. The obtained crude product was purified by SiO2-column chromatography (hexane/AcOEt = 1:0 to 50:1) to give
the desired product 8a (172 mg, 75%).
Colorless crystals; mp 261–262 °C; 1H NMR (300 MHz, CDCl3): δ 4.67 (s, 4H), 5.93 (s, 2H), 6.63 (s, 2H), 7.38–7.46 (m, 4H), and 7.47–7.61 (m, 6H); 13C NMR (75 MHz, CDCl3): δ 30.5, 101.3, 103.7, 128.0, 128.6, 129.9, 130.1, 130.4, 138.2, 140.7, and 148.1; IR (neat) νmax: 3058, 2915, 1497, 1472, 1250, 1200, 1071, 1038, 947, and 853 cm–1.
2,3-Bis(bromomethyl)-4-phenyl-6,7-methylenedioxy-1-(3-methoxyphenyl)naphthalene (8b)
Following the procedure for the preparation
of the reaction of 8a, the reaction of 7b (76 mg, 0.2 mmol) using NBS (82 mg, 0.46. mmol) and AIBN (2 mg,
0.01 mmol) gave the desired product 8b (96 mg, 85%).
Colorless crystals; mp 266–267 °C; 1H NMR (300 MHz, CDCl3): δ 3.92 (s, 3H), 4.66 (s, 2H), 4.70 (s, 2H), 5.92 (s, 2H), 6.62 (s, 1H), 6.68 (s, 1H), 7.03–7.12 (m, 2H), and 7.30–7.60 (m, 7H); 13C NMR (75 MHz, CDCl3): δ 30.6, 30.7, 55.3, 101.3, 103.7, 103.8, 114.0, 127.9, 128.5, 129.9, 130.1, 130.2, 130.5, 131.0, 138.2, 140.5, 140.6, 148.1, and 159.2; IR (neat) νmax: 2835, 1613, 1497, 1458, 1286, 1234, 1104, 1036, 942, and 865 cm–1.
2,3-Bis(bromomethyl)-4-phenyl-6,7-methylenedioxy-1-(3,4,5-trimethoxyphenyl)naphthalene (8c)
Following the procedure for the preparation
of 8a, the reaction of 7c (89 mg, 0.2 mmol)
using NBS (82 mg, 0.46. mmol) and AIBN (2 mg, 0.01 mmol) gave the
desired product 8c (107 mg, 89%).
Colorless crystals; mp 215–216 °C; 1H NMR (300 MHz, CDCl3): δ 3.89 (s, 6H), 3.99 (s, 3H), 4.67 (s, 2H), 4.71 (s, 2H), 5.96 (s, 2H), 6.62 (s, 2H), 6.66 (s, 1H), 6.78 (s, 1H), 7.36–7.44 (m, 2H), and 7.48–7.61 (m, 3H); 13C NMR (75 MHz, CDCl3): δ 25.1, 30.4, 30.8, 56.2, 61.0, 101.4, 103.7, 106.9, 128.0, 128.6, 129.8, 130.0, 130.1, 130.2, 130.3, 133.4, 137.3, 138.0, 140.5, 140.7, 148.2, and 153.2; IR (neat) 2835, 1610, 1500, 1457, 1285, 1234, 1105, 1036, 942, and 868 cm–1.
2,3-Bis(bromomethyl)-4-phenyl-6,7-methylenedioxy-1-naphthylnaphthalene (8d)
Following the procedure for the preparation
of the reaction of 8a, the reaction of 7d (81 mg, 0.2 mmol) using NBS (82 mg, 0.46. mmol) and AIBN (2 mg,
0.01 mmol) gave the desired product 8d (107 mg, 90%).
Colorless crystals; mp 186–187 °C; 1H NMR (300 MHz, CDCl3): δ 4.34 (d, J = 12.0 Hz, 1H), 4.62 (d, J = 10.0 Hz, 1H), 4.75 (d, J = 10.0 Hz, 1H), 4.79 (d, J = 12.0 Hz, 1H), 5.86 (s, 1H), 5.88 (s, 1H), 6.36 (s, 1H), 6.67 (s, 1H), 7.28–7.76 (m, 10H), and 7.93–8.09 (m, 2H); 13C NMR (75 MHz, CDCl3): δ 30.3, 30.5, 101.3, 103.7, 103.8, 125.5, 126.0, 126.2, 126.5, 128.0, 128.1, 128.3, 128.56, 128.61, 129.9, 130.0, 130.3, 130.5, 130.7, 131.1, 132.4, 133.7, 135.4, 138.1, 138.7, 141.0, and 148.2; IR (neat) νmax: 2899, 1615, 1499, 1457, 1343, 1239, 1153, 1038, 945, and 859 cm–1.
2,3-Bis(4,4-dimethylpent-2-ynyl)-1,4-diphenyl-6,7-methylenedioxynaphthalene (1a)
nBuLi (1.3
mL 2.0 mmol) was added to a stirred solution of 3,3-dimethylbut-1-yne
(164 mg, 2.0 mmol) in THF (0.2 mL) at −78 °C under an
Ar atmosphere, and the mixture was stirred at the same temperature
for 0.5 h. A solution of 8a (51 mg, 0.1 mmol) in hexamethylphosphoramide
(HMPA) (0.4 mL) and THF (0.3 mL) was added to the mixture, which was
stirred at the same temperature for 1 h. The mixture was quenched
with aqueous sat. NH4Cl solution was extracted twice with
CHCl3. The combined organic phase was washed with water
and brine, dried (Na2SO4), and concentrated.
The obtained crude product was purified by SiO2-column
chromatography (hexane/AcOEt = 50:1 → 10:1) to give the desired
product 1a (42 mg, 81%).
2,3-Bis(4,4-dimethylpent-2-ynyl)-4-phenyl-6,7-methylenedioxy-1-(3-methoxyphenyl)naphthalene (1b)
Following the procedure for the preparation
of 1a, the reaction using nBuLi (1.3 mL, 2.0 mmol) and 3,3-dimethylbut-1-yne (164 mg, 2.0 mmol), 8b (54 mg, 0.1 mmol), gave the desired product 1b (41 mg, 75%).
2,3-Bis(4,4-dimethylpent-2-ynyl)-4-phenyl-6,7-methylenedioxy-1-(3,4,5-trimethoxyphenyl)naphthalene (1c)
Following the procedure for the preparation
of 1a, the reaction of nBuLi
(0.6 mL 1.0 mmol), 3,3-dimethylbut-1-yne (82 mg, 1.0 mmol), 1c (51 mg, 0.1 mmol), gave the desired product 1c (52 mg, 86%).
2,3-Bis(4,4-dimethylpent-2-ynyl)-4-phenyl-6,7-methylenedioxy-1-naphthylnaphthalene (1d)
Following the procedure for the preparation
of the reaction of 1a, the reaction of nBuLi (0.6 mL, 1.0 mmol) and 3,3-dimethylbut-1-yne (82 mg, 1.0
mmol), 8d (54 mg, 0.1 mmol), gave the desired product 1d (44 mg, 77%).
6,7-Bis(bromomethyl)-5-chloro-8-phenylnaphtho[2,3-d][1,3]dioxole (9)
Following the procedure
for
the preparation of the reaction of 8a, the reaction of 6 (155 mg, 0.5 mmol) using NBS (196 mg, 1.1 mmol) and AIBN
(8.2 mg, 0.05 mmol) gave the desired product 9 (218 mg,
93%).
Colorless crystals; mp 162–164 °C; 1H NMR (500 MHz, CDCl3): δ 4.53 (s, 2H), 5.11 (s, 2H), 6.04 (s, 2H), 6.58 (s, 1H), 7.33–7.35 (m, 2H), 7.50–7.56 (m, 3H), and 7.70 (s, 1H); 13C NMR (125 MHz; CDCl3): δ 28.7, 29.9, 101.8, 101.9, 104.0, 128.2, 128.4, 128.6 (2C), 129.7 (2C), 130.0, 130.7, 131.3, 132.5, 137.4, 139.7, 148.9, and 149.4; IR (neat) νmax: 3062, 3005, 2906, 1456, 1240, 1199, 1039, and 947 cm–1.
HRMS (DART) m/z: calcd for C19H13Br1Cl1O2 [M]+, 386.9787; found, 386.9773.
5-Chloro-6,7-bis(4,4-dimethylpent-2-yn-1-yl)-8-phenylnaphtho[2,3-d][1,3]dioxole (10)
nBuLi (42 mL, 66 mmol) was added to a stirred solution of 3,3-dimethylbut-1-yne
(5.99 g, 73 mmol) in THF (15 mL) at −78 °C under an Ar
atmosphere, and the mixture was stirred at the same temperature for
0.5 h. A solution of bis(bromomethyl)naphthalene 9 (3.43
g, 7.3 mmol) in HMPA (7.3 mL) and THF (22 mL) was added to the mixture,
which was stirred at the same temperature for 1 h, followed by stirring
at 20–25 °C for 1 h. The mixture was quenched with water,
which was extracted twice with hexane. The combined organic phase
was washed with water and brine, dried (Na2SO4), and concentrated. The obtained crude solid was purified by SiO2-column chromatography (hexane/AcOEt = 50:1) to give the desired
product 10 (2.16 g, 67%).
Paled yellow crystals; mp 159–161 °C; 1H NMR (500 MHz; CDCl3): δ 1.14 (s, 9H), 1.16 (s, 9H), 3.41 (s, 2H), 4.03 (s, 2H), 6.00 (s, 2H), 6.63 (s, 1H), 7.32–7.33 (m, 2H), 7.44–7.50 (m, 3H), and 7.70 (s, 1H); 13C NMR (125 MHz; CDCl3): δ 21.0, 21.5, 27.3, 27.4, 31.1 (3C), 31.2 (3C), 75.0, 76.6, 89.3, 89.7, 101.3, 101.5, 103.5, 127.2, 127.5, 128.3 (2C), 130.1, 130.3 (2C), 130.4, 131.6, 132.5, 137.5, 139.1, 147.7, and 148.1; IR (neat) νmax: 2966, 2899, 2866, 1483, 1456, 1238, and 758 cm–1.
HRMS (DART) m/z: calcd for C31H32ClO2 [M + H]+, 471.2091; found, 471.2065.
6,7-Bis(4,4-dimethylpent-2-yn-1-yl)-5,8-diphenylnaphtho[2,3-d][1,3]dioxole (1a)
A mixture of α-chloronaphthalene 10 (221 mg, 0.50 mmol), PhB(OH)2 (91 mg, 0.75 mmol),
K3PO4 (212 mg, 1.0 mmol), Pd(OAc)2 (3.4 mg, 0.02 mmol), and SPhos (12 mg, 0.03 mmol) in toluene (1.6
mL) was stirred at 80–85 °C for 2 h. After cooling down,
water was added to the mixture, which was extracted twice with AcOEt.
The combined organic phase was washed with water and brine, dried
(Na2SO4), and concentrated. The obtained crude
solid was purified by SiO2-column chromatography (hexane/AcOEt
= 50:1) to give desired product 1a (228 mg, 89%).
Paled yellow crystals; mp 209–210 °C; 1H NMR (500 MHz; CDCl3): δ 1.13 (s, 18H), 3.48 (s, 4H), 5.89 (s, 2H), 6.70 (s, 2H), and 7.41–7.52 (m, 10H); 13C NMR (125 MHz; CDCl3): δ 21.1 (2C), 27.4 (2C), 29.7, 31.1 (6C), 89.2 (2C), 100.9 (2C),103.1 (2C), 127.3 (2C), 128.3 (4C), 129.0 (2C), 130.4 (4C), 131.8 (2C), 138.2 (2C), 139.8 (2C), and 146.9 (2C); IR (neat) νmax: 2969, 1541, 1501 1263, 1240, 1044, 955, 858, 754, and 702 cm–1.
HRMS (DART) m/z: calcd for C37H37O2 [M + H]+, 513.2794; found, 513.2818.
6,7-Bis(4,4-dimethylpent-2-yn-1-yl)-5-(4-methoxyphenyl)-8-phenylnaphtho[2,3-d][1,3]dioxole (1b)
Following the
procedure for the preparation of 1a, the reaction using
α-chloronaphthalene 10 (221 mg, 0.50 mmol), (4-MeO)C6H4B(OH)2 (114 mg, 0.75 mmol), K3PO4 (212 mg, 1.0 mmol), Pd(OAc)2 (3.4
mg, 0.02 mmol), and SPhos (12 mg, 0.03 mmol) gave the desired product 1b (244 mg, 90%).
Paled yellow crystals; mp 111–112 °C; 1H NMR (500 MHz; CDCl3): δ 1.13 (s, 9H), 1.14 (s, 9H), 3.47 (s, 2H), 3.50 (s, 2H), 3.92 (s, 3H), 5.89 (s, 2H), 6.69 (s, 1H), 6.75 (s, 1H), 7.05 (d, J = 8.6 Hz, 2H). 7.34 (d, J = 8.6 Hz, 2H), and 7.41–7.52 (m, 5H); 13C NMR (125 MHz; CDCl3): δ 21.1 (2C), 27.4, 31.1 (6C), 55.3, 89.1, 100.9,103.1 (2C), 103.2 (2C), 113.7 (2C), 114.1, 127.3, 127.7, 128.2 (2C), 129.0, 129.3, 130.4 (2C), 131.5 (2C), 131.8, 132.0, 132.2, 137.9, 138.1, 139.8, 146.8, and 158.8; IR (neat) νmax: 2966, 1520 1460, 1287, 1239, 1177, 1039, 947, 864, and 764 cm–1.
HRMS (DART) m/z: calcd for C38H39O3 [M + H]+, 543.2899; found, 543.2876.
6,7-Bis(4,4-dimethylpent-2-yn-1-yl)-5-phenyl-8-(3,4,5-trimethoxyphenyl)naphtho[2,3-d][1,3]dioxole (1c)
Following the
procedure for the preparation of 1a, the reaction using
α-chloronaphthalene 10 (221 mg, 0.50 mmol), 3,4,5-(MeO)3C6H2B(OH)2 (159 mg, 0.75
mmol), K3PO4 (212 mg, 1.0 mmol), Pd(OAc)2 (3.4 mg, 0.02 mmol), and SPhos (12 mg, 0.03 mmol) gave the
desired product 1c (127 mg, 42%).
Paled yellow crystals; mp 83–84 °C; 1H NMR (500 MHz; CDCl3): δ 1.13 (s, 9H), 1.14 (s, 9H), 3.53 (s, 2H), 3.55 (s, 2H), 3.88 (s, 6H), 4.00 (s, 3H), 5.92 (s, 2H), 6.65 (s, 2H), 6.69 (s, 1H), 6.83 (s, 1H), 7.39–7.42 (m, 2H), and 7.46–7.52 (m, 3H); 13C NMR (125 MHz; CDCl3): δ 20.9, 21.2, 27.4 (2C), 31.1 (3C), 31.2 (3C), 56.1 (2C), 61.0, 89.0, 89.2, 100.9,103.2 (2C), 103.2 (2C), 107.3 (2C), 127.3, 128.3 (2C), 128.8, 129.0, 130.4 (2C), 131.7 (2C), 132.0, 135.3, 137.0, 137.9, 138.4, 139.6, 146.9, 147.0, and 153.1; IR (neat) νmax: 2969, 2901, 1582, 1501, 1460, 1410, 1360, 1240, 1130, and 947 cm–1.
HRMS (DART) m/z: calcd for C40H43O5 [M + H]+, 603.3110; found, 603.3100.
6,7-Bis(4,4-dimethylpent-2-yn-1-yl)-5-(naphthalen-1-yl)-8-phenylnaphtho[2,3-d][1,3]dioxole (1d)
Following the
procedure for the preparation of 1a, the reaction using
α-chloronaphthalene 10 (221 mg, 0.50 mmol), naphthalene-1-lyboric
acid (129 mg, 0.75 mmol), K3PO4 (212 mg, 1.0
mmol), Pd(OAc)2 (3.4 mg, 0.02 mmol), and SPhos (12 mg,
0.03 mmol) gave the desired product 1d (247 mg, 88%).
Paled yellow crystals; mp 125–126 °C; 1H NMR (500 MHz; CDCl3): δ 1.04 (s, 9H), 1.14 (s, 9H), 3.26 (d, 4H), 5.82 (d, Jgem = 1.2 Hz, 1H), 5.85 (d, Jgem = 1.2 Hz, 1H), 6.36 (s, 1H), 6.73 (s, 1H) 7.29–7.33 (m, 1H), 7.36–7.38 (m, 1H), 7.44–7.55 (m, 7H), 7.60–7.63 (m, 1H), and 7.95–7.98 (m, 2H); 13C NMR (125 MHz; CDCl3): δ 15.3, 21.0, 21.1, 27.2, 27.4, 31.0 (3C), 31.1 (3C), 65.9, 88.9, 89.4, 100.8, 103.2, 103.2, 125.5, 125.8, 126.1, 126.5, 127.3, 127.9, 128.1, 128.3 (2C), 128.4, 129.0, 129.4, 130.4, 130.5, 132.1, 132.7, 132.8, 133.7, 135.9, 137.4, 138.4, 139.8, and 146.9 (2C); IR (neat) νmax: 2898, 1615, 1459 1341, 1233, 1153, 1040, 947, 906, 858, and 780 cm–1; HRMS (DART) m/z: calcd for C41H39O2 [M + H]+, 563.2950; found, 563.2927.
HPLC analysis (Daicel Chiralcel OD-H column, hexane/2-propanol = 100:1, 1.0 mL/min, 254 nm UV detector), tR = 5.76 min and tR = 11.85 min.
(1S*,3S*)-2,2-Dichloro-1,3-dimethylcyclopropyl](3,4,5-trimethoxyphenyl)methanone (11)5c
To a stirred
solution of (3,4,5-trimethoxyphenyl)magnesium bromide prepared from
Mg (48 mg, 2.0 mmol) and 1,2,3-trimethoxy-5-bromobenzene (494 mg,
2.0 mmol) in THF (1.0 mL), acid chloride 3 (201 mg, 1.0
mmol) in THF (1.0 mL) was added at 0–5 °C. The mixture
was stirred at the same temperature for 1 h. Sat. NH4Cl
aqueous solution was added to the mixture, which was extracted twice
with ether. The combined organic phase was washed with water and brine,
dried (Na2SO4), and concentrated. The obtained
crude oil was purified by SiO2-column chromatography (hexane/AcOEt
= 10:1) to give the desired product 11 (200 mg, 91%).
Colorless oil; 1H NMR (400 MHz; CDCl3): δ 1.41 (d, J = 6.6 Hz, 3H), 1.68 (q, J = 6.6 Hz, 1H), 1.68 (s, 3H), 3.87 (s, 3H), 3.95 (s, 6H), and 7.28 (s, 2H); 13C NMR (100 MHz; CDCl3): δ 11.6, 23.6, 35.2, 39.5, 56.2, 61.0, 68.6, 105.2, 107.1, 142.9, 153.0, and 193.8; IR (neat) 3570, 2928, 1489, 1444, 1321, 1271, 1240, 1198, 1036, and 934 cm–1.
(R*)-[(1S*,3S*)-2,2-Dichloro-1,3-dimethylcyclopropyl](3,4-methylenedioxyphenyl)(3,4,5-trimethoxyphenyl)methanol (12)5c
nBuLi (1.58 M in hexane, 1.40 mL, 2.21 mmol) was added
to a stirred solution of 3,4-methylenedioxy-1-bromobenzene (578 mg,
2.34 mmol) in THF (5 mL) at −78 °C under an Ar atmosphere,
and the mixture was stirred at the same temperature for 0.5 h. Ketone 11 (520 mg, 1.56 mmol) in THF (5 mL) was added to the mixture
at the same temperature, and the mixture was stirred for 2 h. The
mixture was poured into ice and maintained. NH4Cl aqueous
solution was extracted twice with Et2O. The combined organic
phase was washed with water and brine, dried (Na2SO4), and concentrated. The obtained crude oil was purified by
SiO2-column chromatography (hexane/AcOEt = 100:1 to 50:1)
to give the desired product 12 (610 mg, 86%).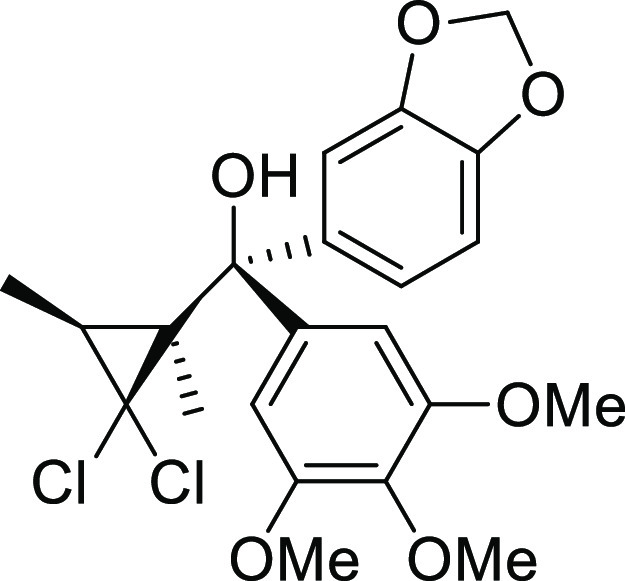
Colorless amorphous solid; 1H NMR (400 MHz; CDCl3): δ 1.19 (s, 3H), 1.56 (q, J = 6.6 Hz, 1H), 1.76 (d, J = 6.6 Hz, 3H), 2.73 (brs, 1H, -OH), 3.86 (s, 6H), 3.92 (s, 3H), 5.96 (d, J = 1.5 Hz, 1H), 5.97 (d, J = 1.5 Hz, 1H), 6.61 (dd, J = 2.0 Hz, J = 8.3 Hz, 1H), 6.72 (d, J = 8.3 Hz, 1H), 6.77 (s, 2H), and 6.81 (d, J = 2.0 Hz, 1H); 13C NMR (100 MHz; CDCl3): δ 11.1, 27.1, 36.4, 38.5, 56.3, 60.9, 74.3, 83.9, 101.1, 106.2, 107.1, 108.8, 122.0, 139.9, 140.5, 146.6, 147.2, and 152.7; IR (KBr) νmax: 3569, 1591, 1487, and 1238 cm–1.
5-Chloro-6,7-dimethyl-8-(3,4,5-trimethoxyphenyl)naphtho[2,3-d][1,3]dioxole (13)5c
Following the procedure for the preparation of α-chloronaphthalene 6, the reaction of alcohol 12 (610 mg, 1.34 mmol)
and SnCl4 (1 M solution: 1.34 mL, 1.34 mmol) gave the desired
product 13 (368 mg, 73%).
Colorless crystals; mp 181–186 °C; 1H NMR (400 MHz; CDCl3): δ 2.20 (s, 3H), 2.25 (s, 3H), 3.71 (s, 3H), 3.85 (s, 6H), 5.91 (s, 2H), 6.51 (s, 2H), 6.65 (s, 1H), and 6.75 (s, 1H); 13C NMR (100 MHz, CDCl3): δ 14.4, 16.1, 56.1 (2C), 60.8, 101.2, 103.2, 104.7, 106.3 (2C), 121.9, 125.7, 128.7, 130.8, 132.3, 136.2, 136.4, 138.1, 147.2, 149.5, and 153.1 (2C); IR (KBr) νmax: 1464, 1252, and 1107 cm–1.
Suzuki–Miyaura Cross-Coupling of 13 Affording 7c
Following the procedure for the preparation of α-chloronaphthalene 7b, the reaction of alcohol 13 (80 mg, 0.20 mmol) gave the desired product 7c (65 mg, 73%).
((1S*,3S*)-2,2-Dichloro-1,3-dimethylcyclopropyl)(naphthalen-1-yl)methanone (14)
nBuLi (1.58
M in hexane, 47 mL, 75 mmol) was added to a stirred solution of 1-bromonaphthalene
(15.5 g, 75 mmol) in THF (50 mL) at −78 °C under an Ar
atmosphere, and the mixture was stirred at the same temperature for
0.5 h. Ester 4 (9.85 g, 50 mmol) in THF (50 mL) was added
to the mixture at the same temperature, and the mixture was stirred
for 2 h. The mixture was poured into ice and maintained. NH4Cl aqueous solution was extracted twice with Et2O. The
combined organic phase was washed with water and brine, dried (Na2SO4), and concentrated. The obtained crude oil
was purified by SiO2-column chromatography (hexane/AcOEt
= 50:1) to give the desired product 14 (11.1 g, 76%).
Colorless oil; 1H NMR (500 MHz; CDCl3): δ 1.46 (d, J = 6.9 Hz, 3H), 1.72–1.76 (m, 4H), 7.53–7.56 (m, 1H), 7.60–7.65 (m, 2H), 7.89–7.91 (m, 1H), 8.05–8.07 (m, 1H), 8.10–8.11 (m, 1H), and 8.92–8.94 (m, 1H); 13C NMR (125 MHz, CDCl3): δ 11.9, 24.3, 36.4, 41.3, 69.1, 124.5, 125.8, 126.5, 128.5, 128.6, 131.2, 131.3, 131.5, 134.1, 134.2, and 197.9; IR (neat) νmax: 2899, 1610, 1465, 1240, 1041, 948, 908, 798, 777, and 734 cm–1; HRMS (DART) m/z: calcd for C16H15Cl2O1 [M + H]+, 293.0500; found, 293.0522.
(S*)-Benzo[d][1,3]dioxol-5-yl((1S*,3S*)-2,2-dichloro-1,3-dimethylcyclopropyl)(naphthalen-1-yl)methanol (15)
Following the procedure for the preparation
of AACM 5, the reaction of ketone 14 (1.47
g, 5.0 mmol), nBuLi (1.58 M in hexane,
4.7 mL, 7.5 mmol), and 3,4-methylenedioxy-1-bromobenzene (1.51 g,
7.5 mmol) gave the desired product 15 (1.40 g, 67%).
Amorphous solid; 1H NMR (500 MHz; CDCl3): δ
1.24 (d, J = 12.0 Hz, 3H), 1.57 (q, J = 6.9 Hz 1H), 1.83 (t, J = 8.0 Hz 3H), 5.90–5.94
(m, 1H), 6.00 (d, Jgem = 1.2 Hz, 1H), 6.04 (d, Jgem = 1.2 Hz, 1H), and 7.16–7.90 (m, 10H); 13C NMR (125 MHz, CDCl3): δ 11.4, 26.9, 36.5,
39.6, 74.7, 84.7, 101.1, 106.6, 108.3, 119.4, 122.9, 124.5, 125.1,
126.8, 128.7, 129.0, 129.7, 131.5, 135.3, 137.7, 139.9, 146.3, and
148.0; IR (neat) νmax: 3576, 1600, 1002, 966, 908,
810, 794, 781, and 655 cm–1; HRMS (DART) m/z: calcd for C23H19Cl2O2 [M – OH]+, 397.0762; found, 397.0776.
5-Chloro-6,7-dimethyl-8-(naphthalen-1-yl)naphtho[2,3-d][1,3]dioxole (16)
Following the
procedure for the preparation of α-chloronaphthalene 6, the reaction of alcohol 15 (208 mg, 0.5 mmol) and
SnCl4 (1 M solution: 500 μL, 0.5 mmol) gave the desired
product 16 (128 mg, 71%).
Amorphous solid; 1H NMR (500 MHz; CDCl3): δ 1.99 (s, 3H), 2.60 (s, 3H), 5.91 (d, Jgem = 1.2 Hz, 1H), 5.93 (d, Jgem = 1.2 Hz, 1H), 6.34 (s, 1H), and 7.27–7.95 (m, 8H); 13C NMR (125 MHz, CDCl3): δ 17.9, 18.6, 101.1, 101.3, 103.3, 125.6, 125.9, 126.0, 126.2, 126.7, 127.8 (2C), 128.3, 129.8, 130.1, 131.6, 132.5, 133.3, 133.7, 134.6, 137.9, 147.3, and 147.8; IR (neat) νmax: 2985, 2931, 1298, 1228, 1103, 1060, 970, 947, 837, 810, 732, and 619 cm–1; HRMS (DART) m/z: calcd for C23H18Cl1O2 [M]+, 361.0995; found, 361.1002.
Acknowledgments
This research was partially supported by Grant-in-Aids for Scientific Research on Basic Areas (B) “18350056”, Priority Areas (A) “17035087” and “18037068”, (C) 15K05508, and Exploratory Research “17655045” from the Ministry of Education, Culture, Sports, Science and Technology (MEXT). We thank Taichi Yoshida and Dr. Yoshinori Nishii in Kwansei Gakuin University for some of their helpful discussions. One of the authors (Y.T.) offers his warmest congratulations to Professor Ben L. Feringa (University of Groningen, The Netherlands) on being awarded the 2016 Nobel Prize in Chemistry. Dedication to the late Professors Teruaki Mukaiyama and Kenji Mori.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.1c04413.
Characterization of all new products (1H and 13C NMR spectra), characterization of all new products for 1H NMR (500 MHz, CDCl3) and 13C NMR (125 MHz, CDCl3) spectra (PDF), Chiral HPLC analysis of compound 1d, and calculation data of 1a and 1d using Spartan software (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Chanteau S. H.; Tour J. M. Synthesis of Anthropomorphic Molecules: The NanoPutians. J. Org. Chem. 2003, 68, 8750–8766. 10.1021/jo0349227. [DOI] [PubMed] [Google Scholar]
- a Shirai Y.; Osgood A. J.; Zhao Y.; Kelly K. F.; Tour J. M. Directional Control in Thermally Driven Single-Molecule Nanocars. Nano Lett. 2005, 5, 2330–2334. 10.1021/nl051915k. [DOI] [PubMed] [Google Scholar]; b Shirai Y.; Morin J.-F.; Sasaki T.; Guerrero J. M.; Tour J. M. Recent progress on nanovehicles. Chem. Soc. Rev. 2006, 35, 1043–1055. 10.1039/b514700j. [DOI] [PubMed] [Google Scholar]
- a Koumura N.; Zijlstra R. W. J.; van Delden R. A.; Harada N.; Feringa B. L. Light-driven monodirectional molecular rotor. Nature 1999, 401, 152–155. 10.1038/43646. [DOI] [PubMed] [Google Scholar]; b Kudernac T.; Ruangsupapichat N.; Parschau M.; Maciá B.; Katsonis N.; Harutyunyan S. R.; Ernst K.-H.; Feringa B. L. Electrically driven directional motion of a four-wheeled molecule on a metal surface. Nature 2011, 479, 208–211. 10.1038/nature10587. [DOI] [PubMed] [Google Scholar]
- May P. W.Molecules with Silly or Unusual Names: Imperial College Press; London, 2008; p 35. [Google Scholar]
- Selected examples;; a Seko S.; Tanabe Y.; Suzukamo G. A Novel Synthesis of α- and β-Halo-naphthalenes via Regioselective Ring Cleavage of Aryl(gem-Dihalocyclopropyl)methanols and Its Application to Total Synthesis of Lignan Lactones, Justicidin E and Taiwanin C. Tetrahedron Lett. 1990, 31, 6883–6886. 10.1016/S0040-4039(00)97197-1. [DOI] [Google Scholar]; b Tanabe Y.; Seko S.; Nishii Y.; Yoshida T.; Utsumi N.; Suzukamo G. Novel Method for the Synthesis of α- and β-Halonaphthalenes via Regioselective Benzannulation of Aryl(gem-dihalocyclopropyl)methanols. Application to Total Synthesis of Lignan Lactones, Justicidin E and Taiwanin C1. J. Chem. Soc., Perkin Trans. 1 1996, 2157–2165. 10.1039/P19960002157. [DOI] [Google Scholar]; c Nishii Y.; Yoshida T.; Asano H.; Wakasugi K.; Morita J.-i.; Aso Y.; Yoshida E.; Motoyoshiya J.; Aoyama H.; Tanabe Y. Regiocontrolled Benzannulation of Diaryl(gem-dichlorocyclopropyl)methanols for the Synthesis of Unsymmetrically Substituted α-Arylnaphthalenes: Application to Total Synthesis of Natural Lignan Lactones. J. Org. Chem. 2005, 70, 2667–2678. 10.1021/jo047751u. [DOI] [PubMed] [Google Scholar]; d Nishii Y.; Wakasugi K.; Koga K.; Tanabe Y. Chirality Exchange from sp3 Central Chirality to Axial Chirality: Benzannulation of Optically Active Diaryl-2,2-dichrolocyclopropylmethanols to Axially Chiral α-Arylnaphthalenes. J. Am. Chem. Soc. 2004, 126, 5358–5359. 10.1021/ja0319442. [DOI] [PubMed] [Google Scholar]; e Tanabe Y.; Moriguchi K.; Kono T.; Seko S. Gram-Scale Robust Synthesis of 1-Chloro-2,3-dimethyl-4-phenylnaphthalene: A Promising Scaffold with Three Contiguous Reaction Positions. Synthesis 2020, 52, 3811–3817. 10.1055/s-0040-1706471. [DOI] [Google Scholar]; f Moriguchi K.; Sasaki R.; Morita J.-i.; Kamakura Y.; Tanaka D.; Tanabe Y. Ipso-type regiocontrolled benzannulation for the synthesis of uniquely substituted α-arylnaphthalenes: application to the first total synthesis of chaihunaphthone. ACS Omega 2021, 6, 18135–18156. 10.1021/acsomega.1c02000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radical approaches;; a Tanabe Y.; Wakimura K.-i.; Nishii Y. Sequential and Highly Stereospecific Intermolecular Radical Additions of 2,3-cis-Disubstituted 1,1-Dibromo- and 1-Bromocyclopropanes with Electron-Deficient Olefins. Tetrahedron Lett. 1996, 37, 1837–1840. 10.1016/0040-4039(96)00156-6. [DOI] [Google Scholar]; b Nishii Y.; Fujiwara A.; Wakasugi K.; Miki M.; Yanagi K.; Tanabe Y. Stereoselective Bifurcating-type Radical Cyclization of gem-Dibromocyclopropanes for the Synthesis of Uniquely Fused 5-3-5-Type Tricyclic Compounds. Chem. Lett. 2002, 31, 30–31. 10.1246/cl.2002.30. [DOI] [Google Scholar]; Anionic approaches; c Nishii Y.; Wakasugi K.; Tanabe Y. Dimethylation and Hydrodechlorination of gem-Dichlorocyclopropanes with Grignard Reagents Promoted by Fe(III) or Co(II) Catalyst. Synlett 1998, 67–69. 10.1055/s-1998-1581. [DOI] [Google Scholar]; Natural and the related product syntheses; d Fujiwara T.; Okabayashi T.; Takahama Y.; Matsuo N.; Tanabe Y. Ring-Closing Strategy Utilizing Nitrile α-Anions: Chiral Synthesis of (+)-Norchrysanthemic Acid and Expeditious Asymmetric Total Synthesis of (+)-Grandisol. Eur. J. Org. Chem. 2018, 6018–6027. 10.1002/ejoc.201801160. [DOI] [Google Scholar]; Selected Pyrethroid syntheses; e Nishii Y.; Wakimura K.-i.; Tsuchiya T.; Nakamura S.; Tanabe Y. Synthesis and Stereostructure-Activity Relationship of a Synthetic Pyrethroid, 2-Chloro-1-methyl-3-phenylcyclopropylmethyl 3-Phenoxybenzyl Ether. J. Chem. Soc., Perkin Trans. 1 1996, 1243–1249. 10.1039/P19960001243. [DOI] [Google Scholar]; f Nishii Y.; Maruyama N.; Wakasugi K.; Tanabe Y. Synthesis and Stereostructure-activity Relationship of Three Asymmetric Center Pyrethroids [2]: 2-Methyl-3-phenylcyclopropylmethyl 3-Phenoxybenzyl Ether and Cyanohydrin Ester. Bioorg. Med. Chem. 2001, 9, 33–39. 10.1016/S0968-0896(00)00217-0. [DOI] [PubMed] [Google Scholar]; g Kawamoto M.; Moriyama M.; Ashida Y.; Matsuo N.; Tanabe Y. Total Syntheses of All Six Chiral Natural Pyrethrins: Accurate Determination of the Physical Properties, Their Insecticidal Activities, and Evaluation of Synthetic Methods. J. Org. Chem. 2020, 85, 2984–2999. 10.1021/acs.joc.9b02767. [DOI] [PubMed] [Google Scholar]
- a O’Brien C. J.; Kantchev E. A. B.; Valente C.; Hadei N.; Chass G. A.; Lough A.; Hopkinson A. C.; Organ M. G. Easily prepared air- and moisture-stable Pd-NHC (NHC = N-heterocyclic carbene) complexes: a reliable, user-friendly, highly active palladium precatalyst for the Suzuki-Miyaura reaction. Chem.—Eur. J. 2006, 12, 4743–4748. 10.1002/chem.200600251. [DOI] [PubMed] [Google Scholar]; b Nasielski J.; Hadei N.; Achonduh G.; Kantchev E. A. B.; O’Brien C. J.; Lough A.; Organ M. G. Structure-Activity Relationship Analysis of Pd-PEPPSI Complexes in Cross-Couplings: A Close Inspection of the Catalytic Cycle and the Precatalyst Activation Model. Chem.—Eur. J. 2010, 16, 10844–10853. 10.1002/chem.201000138. [DOI] [PubMed] [Google Scholar]; c Barder T. E.; Walker S. D.; Martinelli J. R.; Buchwald S. L. New Catalysts for Suzuki-Miyaura Coupling Processes: Scope and Studies of the Effect of Ligand Structure. J. Am. Chem. Soc. 2005, 127, 4685–4696. 10.1021/ja042491j. [DOI] [PubMed] [Google Scholar]; d Altman R. A.; Buchwald S. L. Pd-Catalyzed Suzuki-Miyaura Reactions of Aryl Halides Using Bulky Biarylmonophosphine Ligands. Nat. Protoc. 2007, 2, 3115–3121. 10.1038/nprot.2007.411. [DOI] [PubMed] [Google Scholar]
- Sha C.-K.; Young J.-J.; Yeh C.-P.; Chang S.-C.; Wang S.-L. A Friedel-Crafts cyclization approach toward cephalotaxine. J. Org. Chem. 1991, 56, 2694–2696. 10.1021/jo00008a023. [DOI] [Google Scholar]
- Charlton J. L.; Oleschuk C. J.; Chee G.-L. Hindered Rotation in Arylnaphthalene Lignans. J. Org. Chem. 1996, 61, 3452–3457. 10.1021/JO952048E. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.