

SUMMARY
The mechanisms of Myc-driven liver tumorigenesis are inadequately understood. Herein we show that Myc-driven hepatocellular carcinoma (HCC) is dramatically aggravated in mice with hepatocyte-specific Ptpn11/Shp2 deletion. However, Myc-induced tumors develop selectively from the rare Shp2-positive hepatocytes in Shp2-deficent liver, and Myc-driven oncogenesis depends on an intact Ras-Erk signaling promoted by Shp2 to sustain Myc stability. Despite a stringent requirement of Shp2 cell autonomously, Shp2 deletion induces an immunosuppressive environment, resulting in defective clearance of tumor-initiating cells and aggressive tumor progression. The basal Wnt/β-catenin signaling is upregulated in Shp2-deficient liver, which is further augmented by Myc transfection. Ablating Ctnnb1 suppresses Myc-induced HCC in Shp2-deficient livers, revealing an essential role of β-catenin. Consistently, Myc overexpression and CTNNB1 mutations are frequently co-detected in HCC patients with poor prognosis. These data elucidate complex mechanisms of liver tumorigenesis driven by cell-intrinsic oncogenic signaling in cooperation with a tumor-promoting microenvironment generated by disrupting the specific oncogenic pathway.
Graphical abstract
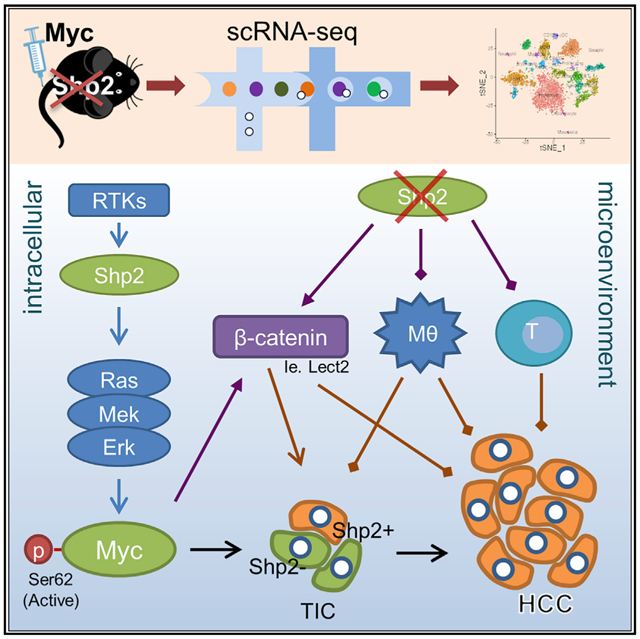
In brief
Chen et al. demonstrate that Shp2 is required cell intrinsically for Myc-driven hepatocarcinogenesis through maintaining an intact Ras-Erk pathway and Myc stability, although Shp2 deletion in neighboring hepatocytes generates a tumor-promoting immunosuppressive microenvironment by upregulation of β-catenin signaling.
INTRODUCTION
Hepatocellular carcinoma (HCC) represents the vast majority of primary liver cancer, a highly fatal disease with rising incidence and mortality worldwide. Liver tumorigenesis results from a series of proliferative, metabolic, and architectural alterations over chronic hepatic disorders (El-Serag, 2011; Llovet et al., 2016). HCC, characteristically resistant to chemotherapy, is often diagnosed at late stages with limited treatment options. Despite low therapeutic benefits, a multi-kinase inhibitor sorafenib has remained the frontline drug for advanced HCC for a decade. The U.S. Food and Drug Administration (FDA) has only recently approved a combination of atezolizumab and bevacizumab, which achieved significantly better overall and tumor progression-free survival rates than sorafenib in unresectable HCC (Finn et al., 2020). Future development of more efficacious mechanism-based therapeutics requires a deeper understanding of cell-intrinsic and cell-extrinsic tumorigenic signals intertwined in the liver.
Shp2/Ptpn11 is an SH2-containing tyrosine phosphatase acting immediately downstream of receptor tyrosine kinases (RTKs) and cytokine receptors (Feng and Pawson, 1994; Neel et al., 2003). Genetic and biochemical analyses revealed surprisingly a positive role of Shp2 in mediating the Ras/Erk pathway activation by RTKs (Feng, 1999; Neel et al., 2003). Although the biological significance for its promotion of Erk signaling is not fully understood, Shp2 is indeed the first identified oncogenic tyrosine phosphatase, with dominant active mutations in Ptpn11 detected in leukemia and solid tumors (Chan and Feng, 2007; Tartaglia and Gelb, 2005). Pharmaceutical inhibition of Shp2 robustly suppressed proliferation of cancer cells driven by oncogenic signals initiated by RTKs (Chen et al., 2016; Kerr et al., 2021). Consistently, deleting Shp2 in hepatocytes inhibited liver tumorigenesis driven by HGF receptor MET in combination with other oncoproteins (Liu et al., 2018). However, Shp2 deficiency also exacerbated HCC induced by a chemical carcinogen diethylnitrosamine (DEN) (Bard-Chapeau et al., 2011) and accelerated liver tumorigenesis driven by non-alcoholic steatohepatitis (NASH) in Pten-deficient liver (Luo et al., 2016). Collectively, these studies disclosed paradoxical anti- and pro-tumorigenic effects of Shp2 in the liver (Feng, 2012), with the underlying mechanisms yet to be elucidated.
Aberrant expression of c-Myc (Myc) is implicated in ~50% of all human cancers and also in a variety of experimentally induced tumors in animal models (Dang, 2012). In particular, Myc overexpression was detected in aggressive HCC and hepatoblastoma with poor prognosis (Abou-Elella et al., 1996). However, Myc alone was inefficient to drive HCC development in transgenic mice but required co-expression of another oncogene or simultaneous deletion of a tumor suppressor (Murakami et al., 1993; Ruiz de Galarreta et al., 2019). Genomic analysis has revealed frequent co-occurrence of Myc amplification and CTNNB1/β-catenin mutations in liver cancer patients as well as in mouse HCC models (Bisso et al., 2020; Cairo et al., 2008; Calvisi et al., 2001; Ruiz de Galarreta et al., 2019; The Cancer Genome Atlas Research Network, 2017). β-Catenin was shown to bind the Myc promoter and to drive Myc expression (Yochum et al., 2008). Myc was clearly positioned downstream of APC/β-catenin in the intestine by epistatic analysis (Sansom et al., 2007; Wong et al., 2015), but Myc deletion did not suppress the effect of APC loss in the liver, although β-catenin removal did (Reed et al., 2008). Hepatic zonation of ammonia metabolizing enzymes controlled by Wnt/β-catenin signaling was also found in-dependent of Myc (Burke et al., 2009). Therefore, the mechanism of cooperative tumorigenic effects between Myc and β-catenin in the liver-specific context is not fully understood.
By combining single-cell RNA-sequencing (scRNA-seq) with functional and molecular analyses in this study, we deciphered complex mechanisms of bi-directional Shp2 functions in liver tumorigenesis. Remarkably, we found that Shp2 was required cell-autonomously for Myc-driven HCC by maintaining an intact Ras/Erk pathway upstream of Myc. Meanwhile, Shp2 loss also induced a tumor-promoting microenvironment in the liver. Taken together, these results elucidate a new mosaic mechanism of tumor cell-intrinsic and -extrinsic factors that intertwine to drive a most severe tumor phenotype. This study clarifies previously documented contradictory roles of Shp2 tyrosine phosphatase in cancer, which will be instrumental for design of more efficacious therapy of malignant diseases.
RESULTS
Myc-driven carcinogenesis is drastically exacerbated in Shp2-deficient liver
Based on our recent results that deleting Shp2 in hepatocytes abrogated liver tumorigenesis induced by MET working upstream of Shp2 (Liu et al., 2018), we sought to examine the effect of Shp2 removal on Myc, a nuclear oncoprotein that acts downstream in oncogenic signaling. Accordingly, we performed hydrodynamic tail vein injection (HTVi) (Chen and Calvisi, 2014) to transfect human Myc (hMyc) and the sleeping beauty (SB) transposase into wild-type (WT) and hepatocyte-specific Shp2 knockout mice (SKO; Shp2f/f:albumin-Cre+) (Figure 1A). Shp2 was deleted in more than 90% hepatocytes in this SKO mouse line (Bard-Chapeau et al., 2006, 2011), directed by the Alb-Cre transgene (Postic and Magnuson, 2000). As expected, Myc alone was inefficient to induce liver tumors in WT mice on a C57BL/6 background (Figure 1B). Astonishingly, Myc transfection in SKO liver induced severe tumor development in only 4 weeks (Figures 1C-1G). Due to aggressive tumor progression, the tumor-bearing SKO mice rarely survived 6 weeks. Tumor cells expressed high levels of exogenous Myc, Ki67, and PCNA and were positive for HNF4α but not CK19 (Figures 1H-1J), which identified the tumors as HCC. Although the complex mechanisms of Myc activation remain to be fully elucidated, phosphorylation of Myc at S62 by Erk was shown to promote its stability and activity (Farrell and Sears, 2014; Hayes et al., 2016). Immunoblot analysis detected high levels of p-MycS62 in Myc+ tumors, compared to non-tumor liver tissues (Figure 1J). Histopathological analysis showed that tumors were primarily trabecular HCC, organized in pseudoglandular growth with inflammatory cell invasion (Figures 1K and 1L; Table S1). While Myc-induced tumors in FVB/N mice were classified as hepatoblastomas, we observed that the tumors had hepatoblastomalike high nucleus-to-cytoplasm ratios indicative of high proliferation yet maintained hepatocyte morphology (Table S1) (Chen and Calvisi, 2014; Liu et al., 2017). This extremely aggressive HCC model driven by Myc alone in SKO liver was unanticipated, as mouse HCC models with Myc were often induced in combination with β-catenin mutant or p53 deletion (Bisso et al., 2020; Dauch et al., 2016; Liu et al., 2017; Ruiz de Galarreta et al., 2019). Thus, loss of cytoplasmic phosphatase Shp2, while disrupting RTK-elicited tumorigenic signaling, dramatically aggravated hepato-oncogenesis driven by a single nuclear oncoprotein Myc.
Figure 1. Myc alone robustly drives tumorigenesis in Shp2-deficient liver.

(A) Experimental scheme.
(B) Representative liver images and H&E staining of liver sections 4 weeks after Myc transfection with SB-11 transposase via HTVi. Liver scale bars, 0.5 cm. Staining scale bars, 500 μm.
(C–G) Quantified tumor incidences (C), liver-to-body weight ratios (LW/BW) (D), maximal tumor diameters (E), visible tumor nodule numbers (F), and spleen-to-body weight ratios (SW/BW) (G). n = 22. Values are presented as means ± SD. Statistical significance was calculated using Students’ t test (**p < 0.01, ***p < 0.001).
(H and I) Representative immunostaining of liver sections for Myc and Ki67 (H) and HNF4α and CK19 (I) 4 weeks after Myc transfection. Scale bars, 250 μm (H) and 50 μm (I).
(J) Immunoblotting of liver lysates at 0 days (0D) and 4 weeks after HTVi of Myc, using antibodies as indicated. T, tumor tissue; N, non-tumor liver tissue.
(K) Representative H&E staining of SKO liver 4 weeks post-Myc injection. For trabecular HCC with features detailed in histopathological report, see Table S1. Scale bars, 2.5 mm (top) and 100 μm (bottom 1–4). Upper box, ×4.
(L) Immunostaining of Myc, F4/80 and DAPI in WT versus SKO (tumor) 4 weeks after HTVi of Myc. Scale bars, 100 μm.
scRNA-seq analysis reveals development of Myc-induced tumors from rare Shp2-positive hepatocytes
To dissect mechanisms underlying the aggressive HCC phenotype at single-cell resolution, we established a distinct liver tissue processing protocol, which allowed isolation of all hepatic cell types at a high quality suitable for scRNA-seq analysis (see STAR Methods). In order to interrogate tumor initiation and progression, we isolated hepatocytes and non-parenchymal cells (NPCs) from WT and SKO livers, at day 0 before HTVi (0D-Ctrl), 10 days after Myc or empty vector injection (10D-Myc or 10D-Vector), and 4 weeks after Myc transfection (4W-Myc) (Figure 2A). We performed droplet-based scRNA-seq using the 10x Genomics chromium platform (Figures 2B, 2C, S1A, and S1D). The data from 27,327 cells, after outlier filtering, were normalized and scaled for clustering and dimensional reduction and visualized using t-distributed stochastic neighbor embedding (tSNE) (Table S2). Hepatocytes and NPCs from WT and SKO livers were integrated and clustered for comparison at the different time points (Figures S1B, S1C, and S1E-S1J). In integrated WT and SKO liver data, we identified all major hepatic cell clusters in the 0D-Ctrl (Figures S1A and S1C), 10D-Vector (Figures S1D and S1F), 10D-Myc (Figures 2B and S1H), and the 4W-Myc data (Figures 2C and S1J).
Figure 2. scRNA-seq reveals tumor development from Myc-transfected Shp2-positive hepatocytes.

(A) Experimental design of scRNA-seq. Single hepatic cells were isolated from WT and SKO livers at day 0 (0D-Ctrl), day 10 (10D-Vector, 10D-Myc), and 4 weeks (4W-Myc). Hepatocytes and NPCs were separated for library preparation and data collection.
(B and C) tSNE plots show single-cell clusters from integrated scRNA-seq data at 10 days (B) and 4 weeks (C) after Myc injection. Integrated datasets for each time point include hepatocytes and NPCs in WT and SKO livers; cluster legend on right. Clusters marked with red dots exist only in 4-week data.
(D) tSNE plot shows clustering of hepatocytes in Myc-transfected SKO liver at 4 weeks (4W-Myc SKO). (1) Central vein zone; (2) OXPHOS and translation-related; (3) tumor; (4) portal vein zone; (5) Malat1hi hepatocytes.
(E) tSNE plot of data from (D) where blue dots show higher expression of Afp (α-fetoprotein) in tumor (cluster 3) than in non-tumor cells.
(F) tSNE plot of integrated WT and SKO 10D-Myc hepatocytes (left) and clusters of hepatocytes (right) include (1) peri-portal hepatocytes, (2) mid-zone hepatocytes, (3) peri-central hepatocytes, and (4) Malat1hi hepatocytes.
(G) Myc expression profile on tSNE plot of data from (F) showing that Myc+ hepatocytes were scattered among clusters.
(H) Post-data imputation by MAGIC of 4W-Myc data; red dots are tumor cells and green dots are non-tumor cells. Myc+ cells in the 4W-Myc data were largely Ptpn11/Shp2+.
(I) Corresponding tSNE plots showing Myc or Ptpn11/Shp2 MAGIC-imputed expression profiles in 4W-Myc hepatocyte data, separated by tumor and non-tumor. The tumor cell cluster shows high expression of both Myc and Shp2.
(J) Post-data imputation by MAGIC of 10D-Myc data. In both WT and SKO data, few Myc+ cells were Shp2+ at this early time point.
See also Figures S1 and S2.
In the 4W-Myc tumor-bearing SKO liver, hepatocytes clustered, separately from NPCs, into five subpopulations: (1) central vein zone; (2) oxidative phosphorylation OXPHOS and translation-related; (3) tumor; (4) portal vein zone; and (5) Malat1hi hepatocytes (Figure 2D). Expression of Alb and Cyp2e1 was detected in all clusters, with expected higher expression of Alb near the portal vein and Cyp2e1 near the central vein (Figure S2A), thus validating our data accuracy through zonation-based clustering. Cluster 1 was characterized by elevated expression of Cyp2e1 and Glul, while cluster 4 highly expressed Cyp2f2 and Alb (Figure S2A). Of note, cluster 3 represented Myc+ tumor cells featured by high expression of Afp and hMyc (Figures 2D, 2E, and S2B). In the integrated WT and SKO data, Myc+ and AFP+ cells overlapped in both datasets, indicating similar expression profiles in Myc+ tumor cells of the two genotypes (Figures S2C-S2E). Remarkably, the tumor cell cluster was absent in the 10D-Myc data, indicating heterogeneity of Myc-transfected hepatocytes at the pre-neoplastic stage (Figures 2F and 2G). However, Myc+ tumor cells in all tumor nodules in the 4W-Myc data formed a single cluster and were clearly separated from non-tumor hepatocytes, suggesting clonogenic tumor growth out of heterogeneous Myc-transfected cells.
To define molecular features of the tumor cells, we utilized MAGIC (van Dijk et al., 2018) for data imputation. Surprisingly, this analysis indicated that tumor cells with high exogenous hMyc expression (Mychi) also displayed high Ptpn11/Shp2 levels, compared to Myclow cells in tumor-bearing SKO livers at 4 weeks (Figure 2H). tSNE plots of the 4W-Myc SKO hepatocytes confirmed the closely associated expression of hMyc and Ptpn11 in the same cluster of tumor cells (Figures 2I). However, this correlation of positive Ptpn11 and hMyc expression was not detected in the 10D-Myc data, when Myc+ cells were not yet well clustered (Figure 2J), indicating no bias in Myc transfection into Shp2-positive or -negative hepatocytes in the SKO liver. As Shp2 was deleted in 90% of hepatocytes by Alb-Cre, the scRNA-seq data unveiled surprisingly that the resulting Myc+ tumors developed through a mechanism of forced selection from the rare Shp2+ hepatocytes that escaped from DNA excision in SKO liver.
Shp2 is required cell autonomously for Myc-induced HCC
In agreement with the scRNA-seq data, we detected higher Shp2 mRNA and protein levels in tumor than non-tumor tissue and hepatocyte lysates isolated from SKO livers (Figures 1J and S3A-S3C), indicating that Myc-induced tumors were indeed derived from Shp2-positive hepatocytes. To define a tumorigenic role of Shp2, we co-injected Myc and Shp2 expression constructs (Myc+Shp2) via HTVi. Similar to Myc alone, co-injection of Shp2 and Myc did not efficiently induce tumors in WT mice (Figure 3A), suggesting that Shp2 overexpression does not have a tumor-promoting effect in WT liver. However, co-transfection of Myc and Shp2 into SKO liver induced faster and more severe tumor growth than Myc alone, with numerous Shp2+Myc+ tumor nodules detected at 3 weeks (Figures 3A and S3D).
Figure 3. An intact Erk pathway promoted by Shp2 is required for Myc-induced HCC.

(A) Representative liver images and quantified tumor incidence rate 3 weeks after HTVi of Myc and Shp2. Scale bars, 0.5 cm.
(B) Representative liver images of WT and SKO mice 4 weeks after HTVi of Myc and CMV-Cre. Scale bars, 0.5 cm.
(C) Immunoblot analysis of Myc and Erk phosphorylation in PLC, MC38, and NR-PDAC cells treated with DMSO (Ctrl; 16 h), SHP099, or trametinib.
(D) Immunoblot analysis of PLC cells treated with Shp2 inhibitor SHP099 (20 μM) for 16 h (top) or Mek inhibitor trametinib (50 μM) for 4 h (bottom), using DMSO as control treatment. Timescale showing Myc protein levels after cycloheximide (CHX; 25 μg/mL) treatment. Quantified immunoblot demonstrates reduced Myc half-life in cells treated with Shp2 and Mek inhibitors. Related to Figures S4B and S4C.
(E and F) (E) Representative liver and H&E images of WT and SKO liver 4 weeks after HTVi of Ras+Myc (NRasG12V + Myc); n = 5. Scale bars, 0.5 cm. (F) Quantified liver-to-body weight ratios (LW/BW) and maximal tumor diameter.
(G) Comparison of tumor numbers in WT and SKO livers 4 weeks after HTVi of Myc alone or Ras+Myc. Average number of tumors listed per condition.
(H) Immunoblot analysis of liver, tumor (T) or non-tumor (NT) tissue lysates of WT or SKO mice transfected with Myc, NRasG12V+Myc, or tumors induced by diethylnitrosamine (DEN).
Values are presented as means ± SD. Statistical significance was calculated using Students’ t test (**p < 0.01, ***p < 0.001).
See also Figures S3 and S4.
Next, we injected adeno-associated virus (AAV)-thyroid hormone-binding globulin (TBG)-Cre virus to acutely delete Shp2 in hepatocytes and found AAV-Cre-induced Shp2 deletion suppressed Myc-induced tumor formation (Figure S3E). To further define a cell-autonomous requirement of Shp2, we co-transfected Myc and cytomegalovirus (CMV)-Cre, which allowed gene deletion selectively in the oncogene-transfected cells (Xu et al., 2019b). Remarkably, selective deletion of Shp2 by CMV-Cre in Myc-transfected cells abrogated HCC development in SKO liver (Figures 3B and S3F). Although Alb-Cre and AAV-Cre showed similarly effective Shp2 deletion in hepatocytes (Figure S3G), AAV-Cre induced acute and synchronized gene deletion, as compared to Alb-Cre, which mediates progressive gene ablation, resulting in induction of a tumor-permissive environment in the liver. Shp2+ hepatocytes were detected in the 0D-Ctrl time point without zonation-based bias (Figure S4A). Taken together, these results suggest that the severe tumor phenotype induced by Myc required cell-autonomous expression of Shp2, but it also required a hepatic microenvironment induced ironically by Shp2 loss in the majority of hepatocytes.
Shp2 promotes hepatocarcinogenesis by enhancing Erk-mediated Myc stability
We questioned why Shp2 was stringently required for HCC driven by Myc, which canonically acts downstream of Shp2 in cell signaling. To test a role of Shp2 in promoting Erk-Myc signaling (Farrell and Sears, 2014; Hayes et al., 2016), we treated HCC cells with a specific Shp2 inhibitor SHP099 or a Mek inhibitor trametinib. Indeed, pharmaceutic inhibition of either Shp2 or Mek suppressed phosphorylated (p-)Erk and p-MycS62 levels in HCC and other cancer cell lines, and it decreased the half-life of Myc protein in these tumor cells (Figures 3C, 3D, S4B, and S4C). Although Shp2 was shown to promote Erk activation two decades ago (Feng, 1999; Neel et al., 2003), this study provides in vivo evidence to demonstrate the significance of Shp2 function in enhancing Myc stability by Erk required for Myc-driven carcinogenesis. Consistent with the biochemical results in vitro, scRNA-seq data analysis showed significantly elevated expression of Myc target genes in Shp2-positive cells in both WT and SKO liver in the 10D-Myc data (Figure S4D). Myc-transfected cells at 10 days had increased OXPHOS and ATP production as compared to non-transfected cells (Figure S4E).
To validate a functional requirement of Shp2 for Ras-Erk-Myc signaling, we co-injected a dominant active NRasG12V mutant together with Myc (Ras+Myc) into WT and SKO mice (Figure 3E). Similar to Myc alone, co-injection of Ras and Myc drove more severe HCC development in SKO than WT mice (Figures 3E-3G; Table S1), reinforcing the notion that Shp2 deficiency induces a tumor-promoting environment. Nonetheless, compared to Myc alone, Ras and Myc induced drastically increased tumor burdens in SKO mice. The average number of tumor nodules changed from 14 by Myc alone to >400 induced by Ras and Myc in SKO liver in a comparison from equal plasmid DNA quantity introduced (Figure 3G). More importantly, unlike the Myc-induced tumors that were Shp2-positive, tumors induced by Ras and Myc were Shp2-negative but retained high p-Erk levels (Figure 3H). In addition, carcinogen DEN-induced tumors in SKO liver were also Shp2-negative (Figure 3H). Collectively, these data indicate that the cell-autonomous requirement of Shp2 in Myc-driven tumor was overridden by an oncogenic Ras mutant co-transfected with Myc.
Shp2 deficiency induces a hepatic immunosuppressive tumor environment
Despite the stringent cell-intrinsic requirement of Shp2 , as described above, the data that Myc alone did not efficiently induce HCC in WT mice strongly argues that an intact Erk-Myc pathway, enabled by Shp2, is necessary but not sufficient. Indeed, the severe HCC phenotype relied on the tumor-prone microenvironment in the Shp2-deficient liver. Thus, the focus of scRNA-seq data analysis turned to the hepatic immune cell populations. In the 0D-Ctrl data, before Myc injection, SKO liver had modestly higher numbers of almost all innate and adaptive immune cell types compared to WT livers (Figure S5A), suggesting increased basal levels of hepatic injuries and inflammation caused by Shp2 removal from hepatocytes. Next, we compared the 10D-Vector and 10D-Myc data to evaluate early hepatic response to Myc expression by negating any effects of the injection procedure. The 10D-Myc data yielded lower numbers of classical dendritic cells (cDCs), Kupffer cells, monocyte-derived macrophages (MDMs), neutrophils, and B cells in SKO livers than in WT livers (Figures 4A, 4B, and S5B). These data suggest a generally impaired innate immune cell response to initiation of Myc-induced tumors in the SKO liver at the early stage. The 4W-Myc data showed that aggressive tumor growth in SKO liver was accompanied by modest increases in NK2, CD11b+ cDCs, neutrophils, MDM, and Kupffer cells relative to the WT liver (Figures 4B and S5B).
Figure 4. scRNA-seq uncovers macrophage alterations in a tumor-permissive immune environment of SKO liver.

(A) Comparison of quantified immune cell populations between SKO and WT liver, based on 10D-Vector and 10D-Myc data. For each immune cell type, the relative cell abundance was calculated as the log-transformed ratio of SKO% to WT%.
(B) Quantified innate immune cell populations of SKO livers, relative to WT livers, between 10D-Vector, 10D-Myc, and 4W-Myc. Additional populations are depicted in Figure S5B.
(C) Differential gene expression analysis for monocyte-derived macrophages (MDMs) (blue) and Kupffer cells (purple) at all time points. Significantly expressed M1 (green) and M2 (red) genes are noted. Values in the heatmap indicate average log2 fold changes in SKO, relative to WT liver, for corresponding cell types at different time points.
(D) Differential gene expression analysis for MDMs (left) and Kupffer cells (right) in 10D-Myc data. Significantly differentially expressed phagocytosis genes are marked in red, with higher expression in WT than SKO livers.
See also Figure S5.
We further characterized macrophage polarization and activation status. Differential expression analysis showed that inflammatory M1 macrophage markers, such as CD38, Irf7, and Saa3, were significantly upregulated in 10D-Myc WT livers (Figure 4C). Moreover, we detected higher expression of phagocytosis-related genes, including Fcgr4, Lyn, and Fcgr1, in MDMs and Kupffer cells in the 10D-Myc data, with no significant change detected in 0D-Ctrl and 10D-Vector data (Figures 4D and S5C). These results suggest a swift hepatic response to the genesis of Myc+ tumor-initiating cells in the WT liver via M1 macrophage polarization and enhanced phagocytosis activity, which was markedly attenuated in SKO liver.
Clearance of Myc-transformed hepatocytes is impaired in Shp2-deficient liver
To validate the scRNA-seq data, we performed fluorescence-activated cell sorting (FACS) analysis at the early stage and identified significantly higher numbers of Kupffer (CD11b+Ly6c−F4/80+) and B cell populations, while lower numbers of monocytes (CD11b+Ly6chiF4/80−) and MDMs (CD11b+ Ly6c+ F4/80+), in WT than SKO livers (Figure 5A), with no significant difference observed for CD4, CD8, natural killer (NK)T, and NK cells (Figures 5A and S5D). We then performed qRT-PCR analysis of liver tissues at days 7, 10, and 14 to measure the expression of cytokines/chemokines involved in the recruitment of macrophages (Figures 5B and S5A). CCL9, a pro-tumorigenic chemokine secreted by inflammatory macrophages, was lower in SKO liver at all time points (Figure 5B). Consistently, the 10D-Myc data showed that CD11b+ cDCs, Kupffer cells, and MDMs from WT livers expressed higher CCL9 than SKO, which was not identified in 10D-Vector data (Figure S6B). In contrast, CCL17, a chemokine secreted by non-inflammatory macrophages that targets regulatory T (Treg) cells, was higher in SKO liver at all time points (Figure 5B). We did not observe changes in CCL4 and CCL5 expression in association with immune escape as reported previously (Ruiz de Galarreta et al., 2019; Spranger et al., 2015), but we detected significantly elevated expression of MIF (macrophage migration inhibitory factor) and Lect2 in Shp2-deficient hepatocytes at all time points (Figure S6C; Table S3). Lect2, a hepatokine that modulates macrophage, neutrophil, NK cell, DC, endothelial cell (EC), and hepatic stellate cell (HSC) activities, was identified as a β-catenin target and implicated in liver fibrosis and HCC (Anson et al., 2012; Ovejero et al., 2004; Xu et al., 2019a). PD-L1 and CCL2 were higher in SKO liver at 10 days and 4 weeks after Myc transfection (Figure 5B). Taken together, these data suggest that defects in a variety of innate immune cell functions are likely a major mechanism of immunosuppression in Shp2-deficient liver.
Figure 5. Myc-transformed cells are eliminated in WT livers by macrophages.

(A) FACS of immune cell populations 12 days after Myc transfection in isolated NPCs of WT and SKO livers. Initial gate, CD45+CD11b+: Kupffer cells, Ly6c−F4/80+; monocytes, Ly6chiF4/80−; neutrophils, Ly6chiF4/80−; MDMs, Ly6c+ F4/80+. Initial gate, CD45+: classical dendritic cells (cDCs), major histocompatibility complex class II (MHCII)+, CD11c+; B cells, TCRβ−CD19+; T cells, CD19− TCRβ+. Statistical analysis was done with Student’s t test (*p < 0.05, **p < 0.01). Values are presented as means ± SD. Additional cell population data are presented in Figure S5D.
(B) qRT-PCR analysis of cytokines CCL9, CCL17, CCL2, and PD-L1 in WT and SKO livers 7 days after vector injection or 7, 10, or 14 days after Myc injection. Statistical analysis used a Student’s t test. Values are presented as means ± SD *p < 0.05, **p < 0.01, ***p < 0.001). See also Figure S6A.
(C) Representative H&E and Myc immunostaining in consecutive sections at 7, 10, and 14 days after Myc injection. Arrows point to Myc+ cell clusters. Scale bars, 250 μm.
(D) Co-staining of Myc with F4/80 in WT and SKO livers at 10 days post-Myc transfection. Scale bars, 100 μm.
(E and F) (E) Representative H&E of microgranuloma structures in WT and SKO liver at 9 days post-Myc transfection. Scale bars, 50 μm. (F) Quantified microgranuloma areas per tissue area in WT and SKO livers at 9 and 14 days. Statistical analysis using Student’s t test (*p < 0.05, ***p < 0.001). Values are presented as means ± SD.
See also Figures S5 and S6.
We next examined the fate of Myc-transfected hepatocytes at the pre-neoplastic stage. The transfection efficiency was similar between WT and SKO livers, with comparable Myc+ colonies detected at days 7 and 10 (Figure 5C), which were populated by proliferative Mychi, rather than Myclo, cells (Figures S6D-S6F). However, the livers became different at day 14: Myc+ cells disappeared in WT liver, whereas Myc+ colonies were expanding in SKO liver (Figure 5C). Singular Myclo hepatocytes were rarely detected in either genotype at day 14 (Figures S6D and S6F). Immunostaining detected infiltration of inflammatory clusters of F4/80+ and CD8+ cells into Myc+ areas in WT rather than SKO livers (Figure 5D and S6G), consistent with the scRNA-seq data showing high M1 macrophage differentiation with increased phagocytic activity (Figures 4C and 4D). Indeed, we observed more and larger microgranuloma structures surrounding parenchymal cells in WT liver, demonstrating clearance of Myc-transfected hepatocytes (Figures 5E, 5F, and S6H). Thus, one critical mechanism underlying the severe tumor phenotype in SKO liver was defective elimination of Myc-transformed hepatocytes at the early stage, followed by an immunosuppressive niche conducive for tumor progression.
β-catenin signaling is aberrantly enhanced in Myc+ tumors
Next, we explored the mechanisms underlying induction of the tumor-permissive environment. Pathway enrichment analysis showed upregulation of several signaling and metabolic pathways, especially the Wnt/β-catenin pathway, in tumor cells in the 4W-Myc data (Figure 6A). Wnt pathway targets were highly expressed in the tumor cell cluster featured by high expression of Myc and Afp, but not genes driven by mTORC or Hippo/YAP signaling pathways (Figures 6B and S7A). Immunostaining detected higher levels of β-catenin and glutamine synthetase (GS) in tumor tissues as compared to non-tumor areas (Figures 6C and S7C), indicative of upregulated β-catenin activity in Myc-induced tumor cells. By analyzing the 0D-Ctrl and 10D-Myc data, we also found significantly higher expression of Wnt/β-catenin pathway genes in SKO liver and Shp2+ hepatocytes, relative to WT (Figures S5B and S7D), suggesting upregulation of both the basal and Myc-induced Wnt/β-catenin signaling in Shp2-deficient liver.
Figure 6. Wnt/β-catenin signaling is aberrantly upregulated in Myc+ mouse tumors and in HCC patients.

(A) Pathway enrichment analysis with normalized enrichment score (NES) using differentially expressed genes between tumor (T) and non-tumor (NT) cells from 4W-Myc SKO data. Pathway with a positive NES was over-represented in tumor cells, and pathway with a negative NES was under-represented in tumor cells.
(B) tSNE plot and boxplot of scores defined for Wnt target gene sets in 4W-Myc SKO data. Tumor cells show higher expression of Wnt target genes. Statistical significance was calculated using a Wilcoxon test. See also Figure S7B.
(C) Representative staining for β-catenin and glutamine synthetase (GS) in WT and SKO livers, 4 weeks after Myc transfection. Scale bars, 250 μm. See also Figure S7C.
(D) tSNE plot and boxplot of scores defined for a set of Myc/β-catenin signature genes in 4 week SKO hepatocyte data. Tumor cluster shows higher expression of signature genes. Statistical significance was calculated using a Wilcoxon test.
(E) Kaplan-Meier survival analysis of HCC patients with MYC−CTNNB1+, MYC+CTNNB1−, and MYC+CTNNB1+ based on mRNA expression levels in TCGA database. Patients with MYC+CTNNB1+ show significantly worse survival.
(F) A total of 371 HCC patients in TCGA data were separated by Myc expression into columns and then distinguished by β-catenin mutational profiles within each column. Patients with higher Myc expression had higher β-catenin mutation rates. Myc copy number alterations are defined in Figure S8B.
See also Figures S7 and S8.
We examined a group of recently reported “Myc/β-catenin signature” genes (Bisso et al., 2020), whose induction required both Myc and β-catenin in a transgenic mouse HCC model. Remarkably, the expression of Myc/β-catenin signature genes was enhanced by Myc alone in tumor cells and also in the portal vein hepatocytes in SKO liver at 4 weeks (Figures 6D and S7E). Modest upregulation of the signature genes was observed in SKO livers, relative to WT, in the 10D-Myc data (Figures S7F and S7G). Biochemical analysis revealed increased β-catenin localization to the nucleus in tumor tissue, compared to non-tumor tissue, further supporting increased β-catenin transcriptional activity (Figure S7H). Thus, the basal level of Wnt/β-catenin signaling was elevated in Shp2-deficient liver, which was boosted by Myc overexpression, resulting in higher expression of the Myc/β-catenin signature genes whose upregulation required otherwise both Myc and β-catenin in another model (Bisso et al., 2020).
In support of the mouse data, bioinformatics analysis of The Cancer Genome Atlas (TCGA) and other patient datasets showed that HCC patients with upregulation of both Myc and β-catenin target genes had worse prognosis than did those with upregulation of only Myc or β-catenin target genes (Figures 6E and S8A). HCC patients with higher expression or increased copy numbers of MYC also had a higher frequency of mutations in CTNNB1 as well (Figures 6F and S8B). The human HCC data suggest a selective advantage of the two pathways altered concurrently, resulting in more aggressive disease progression. β-Catenin was known to bind to and activate the Myc promoter; studies in other cancers suggest that Myc may also induce β-catenin expression via a positive feedback loop (Cho et al., 2013; Cowling et al., 2007). To probe this mechanism, we examined the expression of endogenous Myc and did not find a consequent increase of the endogenous Myc expression in exogenous hMyc-overexpressing tumor cells (Figure S8C). These data did not support a role of β-catenin upstream of Myc in liver tumorigenesis.
β-catenin is required for Myc-driven hepatocarcinogenesis
Next, we examined the requirement of β-catenin in Myc-driven HCC. Co-injection of Myc with a dominant-negative mutant of TCF7l2 (dnTCF), which can inhibit endogenous β-catenin activity (Vacik et al., 2011), suppressed Myc-induced tumor formation in SKO liver (Figures 7A and 7B). This result further supports a notion that β-catenin is required for Myc-dependent HCC, despite being previously placed upstream of Myc in other cancer types. Co-injection of dnTCF with NRasG12V+Myc oncogenes did not reduce tumor burdens or tumor cell proliferation (Figures S8D-S8F). Therefore, β-catenin is particularly required for hepato-oncogenesis driven by Myc alone. We then examined changes in tumor initiation with Myc+dnTCF. At day 7, scattered Mychi cells proliferated in both livers (Figure S8G), suggesting that co-transfection with dnTCF did not inhibit initial cell transformation by Myc. However, subsequent growth of Myc+ cell nodules was suppressed by dnTCF when examined at day 12 (Figure S8G), suggesting that dnTCF abrogated tumor progression rather than initiation.
Figure 7. β-Catenin transcription activity is required for Myc-dependent tumorigenesis.

(A) Representative liver images with H&E and immunostaining of Myc and F4/80 for SKO livers 4 weeks after transfection of Myc or Myc+dnTCF (n = 5). Scale bars, 0.5 cm (liver), 250 μm (H&E), and 100 μm (immunostaining).
(B) Quantification of liver-to-body weight ratios (LW/BW), maximal tumor sizes, and tumor numbers in SKO livers (n = 6). Statistical analysis used a Student’s t test. (***p < 0.001).
(C and D) Representative liver images (C), quantification of liver-to-body weight ratios, maximal tumor diameters, and tumor numbers (D) in WT, SKO, BKO, and SBKO mice 4 weeks after Myc transfection (n = 6–9). Scale bars, 0.5 mm (top) and 250 μm (bottom). Statistical analysis used one-sided ANOVA with a Tukey posttest (**p < 0.01, ***p < 0.001).
(E and F) Representative liver images (E), quantification of LW/BW ratios, maximal tumor diameters, and tumor numbers (F) in WT, SKO, BKO, and SBKO mice 4 weeks after HTVi of Myc/ΔN90-β-catenin (n = 4–6). Scale bars, 0.5 mm. Statistical analysis used one-sided ANOVA with posttest Tukey (*p < 0.05, **p < 0.01). All graph values are presented as means ± SD.
See also Figures S8 and S9.
By breeding Shp2f/f mice with Ctnnb1f/f:Alb-Cre+ (BKO) mice, we generated a compound mutant SBKO (Shp2f/f:Ctnnb1f/f:Alb-Cre+) mouse line with hepatocyte-specific deletion of both Shp2 and β-catenin (Figures S9A-S9C). The BKO and SBKO mice were born at Mendelian frequency. At 2 months of age, SBKO mice developed healthy hepatic architecture, with no gross anatomical defect and no spontaneous tumor development in adults (Figures S9A-S9C and S9H). Similar to BKO mice as described previously (Liang et al., 2018; Tan et al., 2006), SBKO mice had smaller liver mass (Figures S9A and S9B), and SBKO livers exhibited modestly increased cell death, proliferation, and fibrosis (Figures S9D-S9G). Thus, the SBKO mice inherited pathological features from both SKO and BKO mouse lines.
Next, we transfected Myc into WT, SKO, BKO, and SBKO livers and examined the tumor loads. Additional removal of β-catenin from hepatocytes in SKO mice suppressed Myc-induced HCC development in SBKO livers, with tumor nodules barely detectable 4 weeks post-HTVi (Figures 7C and 7D). Furthermore, Myc injection did not induce tumors in BKO livers with only β-catenin loss in hepatocytes (Figures 7C and 7D), suggesting a requirement of β-catenin for Myc-driven HCC in mice. To rescue the tumor phenotype, we injected Myc together with an oncogenic truncated β-catenin mutant (ΔN90-β-catenin). Indeed, liver tumors developed in WT, SKO, BKO, and SBKO mice 4 weeks following transfection, with more severe tumor phenotypes in the three mutants than in WT mice (Figures 7E, 7F, and S9I). The tumors driven by Myc and ΔN90-β-catenin were developed at larger sizes and numbers than those by Myc alone, being histopathologically a mixture of HCC and hepatoblastoma (Figure 7F; Table S1). Taken together, these results clearly demonstrated a functional requirement of β-catenin for hepatocarcinogenesis driven by Myc, which provides an explanation for frequent co-detection of aberrant over-activation of Myc and β-catenin signaling in HCC patients.
DISCUSSION
By combining scRNA-seq with functional and mechanistic analyses, we have elucidated complex mechanisms of the Shp2-promoted Ras-Erk-Myc pathway in synergy with a tumor-permissive niche induced ironically by deleting Shp2 and disrupting the particular pathway in neighboring cells, resulting in most aggressive liver tumor development. We were astonished by the dramatic tumor phenotype observed for several reasons. First, Myc alone was sufficient to drive HCC using the HTVi approach, which is known to require two oncogenes for efficient induction of liver tumors. Second, the severe tumor burdens were observed in only 4 weeks after Myc transfection, as many combinations of oncogenes were shown to induce tumors in 8–12 weeks or longer. Third, this study showed rapid HCC induction by Myc alone in a tumor-resistant C57BL/6 mouse strain, whereas similar experiments were mainly performed in mice of the FVB background.
In this study, we developed a protocol of hepatic cell isolation for scRNA-seq that robustly covers all cell types in mouse liver. The application of this recent technology to the liver field was hurdled by difficulties in isolating single hepatic cells at high quality. Several groups recently reported scRNA-seq analysis for one or several cell types in the liver (Halpern et al., 2017; Hou et al., 2016; Ma et al., 2019; Xiong et al., 2019; Zheng et al., 2017a, 2019). These scRNA-seq data analyze all hepatic cell types, including hepatocytes, tumor cells, and NPCs, in a murine HCC model. Combining scRNA-seq with genetic manipulations and molecular/cellular assays elucidated a mosaic model of Shp2 gain and loss in driving and promoting HCC, respectively. The complex mechanisms were otherwise difficult to unveil without analysis at single-cell resolution.
A number of recent reports showed that chemical inhibition of Shp2 had a growth-inhibitory effect synergistic with other inhibitors of Ras-Erk signaling in a variety of cancer cell lines (Ahmed et al., 2019; Dardaei et al., 2018; Fedele et al., 2018; Mainardi et al., 2018; Ruess et al., 2018; Wong et al., 2018), making Shp2 an attractive drug target. However, Shp2 has been found to have paradoxically tumor-promoting and tumor-inhibiting effects in liver and other solid tumors (Bard-Chapeau et al., 2011; Liu et al., 2018; Luo et al., 2016; Yang et al., 2013), raising concerns on targeting Shp2 in oncological treatment. Following previous data revealing its essential role in oncogenic signaling initiated by RTK, this study has elucidated an indispensable Shp2 function in promoting Erk signaling and Myc stability for Myc-driven HCC development. Single-cell transcriptomics showed clearly that Myc-induced tumors only developed from the rare Shp2-positive cells in Shp2-deficient liver through forced selection, although Myc transfection was unbiased to Shp2 deletion status in hepatocytes. Of note, HCC development driven by oncogenic Ras and Myc did not require Shp2, with the resulting tumors being Shp2-negative. This study provides solid evidence for a stringent requirement of an intact Ras-Erk pathway in Myc-driven carcinogenesis in vivo, despite a body of literature on Ras-Erk-Myc signaling in cancer cells (Hayes et al., 2016; Vaseva et al., 2018).
Although Shp2 is required cell-autonomously, Shp2 deficiency in the liver triggered an immunosuppressive environment, contributed by aberrantly elevated β-catenin signaling. The basal Wnt/β-catenin signaling level was upregulated in Shp2-deficient liver, which was further augmented by Myc overexpression. Myc+ tumors in SKO liver featured higher expression of Myc/β-catenin signature genes detected in tumors induced by overexpression of both Myc and β-catenin (Bisso et al., 2020). In contrast to previous data positioning Myc downstream of β-catenin (Sansom et al., 2007; Wong et al., 2015), this study provides genetic evidence on the functional requirement of β-catenin for Myc-driven hepatocarcinogenesis. Although the underlying mechanism remains to be deciphered, the data presented herein suggest both cell-intrinsic and cell-extrinsic mechanisms for upregulation of Wnt/β-catenin signaling in this Myc-induced liver tumor model. It is possible that the expression of β-catenin and its critical downstream target molecules is regulated directly by Myc in hepatocytes, but we do not rule out another possibility that Myc and β-catenin work in mutually enhancing parallel pathways. Further investigation is warranted to elucidate the complex mechanisms.
Although this study unveiled a critical role of oncogenic β-catenin in induction of immunosuppression in agreement with previous reports (Ruiz de Galarreta et al., 2019; Spranger et al., 2015), conflicting data exist with regard to the underlying mechanisms. In contrast to the claimed roles of CCL4 and CCL5 (Ruiz de Galarreta et al., 2019; Spranger et al., 2015), our results suggest involvement of Lect2, CCL2, CCL9, CCL17, and MIF in suppressing various innate immune cell functions and blocking tumor initiation. In particular, Lect2 is a β-catenin target gene product that has been implicated in liver fibrosis and HCC (Anson et al., 2012; Ovejero et al., 2004; Xu et al., 2019a). This hepatokine was shown to modulate activities of HSCs and infiltrated inflammatory and immune cells, including macrophages, neutrophils, NK cells, DCs, and ECs in the liver, although the underlying mechanisms are underexplored (Meex and Watt, 2017). Upregulated PD-L1 expression may also induce immune escape during tumor progression driven by cooperative activities of Myc and β-catenin. Further mechanistic analysis is required to elucidate the immunosuppressive tumor environment induced by aberrant β-catenin signaling in Shp2-deficient liver. Consistent with the mouse data, both Myc amplification and CTNNB1 mutations were detected simultaneously in many HCC samples, and more than 50% of hepatoblastoma cases were found to have Myc overexpression and also mutations in CTNNB1 (Zheng et al., 2017b). At the early stage, Shp2-deficient liver exhibited significantly decreased numbers and activity of Kupffer and MDMs, with impaired capacity to eliminate Myc-transformed hepatocytes. Consistently, we recently reported a robust HCC-preventing effect by activation of Kupffer and NK cells (Lee et al., 2017; Wen et al., 2019).
This study clarifies conflicting data on Shp2 in cancer and suggests a potential of targeting Shp2 in treatment of Myc-dependent HCCs, in addition to tumors driven by RTKs (Chen et al., 2016). However, inhibiting Shp2 also led to aberrant β-catenin signaling and a tumor-promoting niche. This mosaic mechanism of liver tumorigenesis presents a challenge to the idea of precision medicine in cancer therapy. The intertwined cell-intrinsic and cell-extrinsic mechanisms disclosed herein may also illustrate the rapid and aggressive growth of relapsed tumors from the few residual tumor cells that survived the targeted treatment of primary tumors. Specific targeting of cell-intrinsic oncogenic pathways must be combined with simultaneous correction of the secondary immunosuppressive tumor environment to achieve lasting therapeutic effect.
Limitations of the study
One major unanswered question is how Shp2 deletion in hepatocytes generates a tumor-permissive microenvironment in the liver, which aggravates and accelerates Myc-driven hepatocarcinogenesis. We hypothesize that distinct cell types and factors are involved in defective clearance of the tumor-initiating cells at the early stage and accelerated tumor progression at the late stage, respectively. It remains to be elucidated how β-catenin is functionally required for Myc-driven hepatocarcinogenesis and whether Myc regulates directly the expression of β-catenin and its critical downstream target molecules in hepatocytes.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Gen-Sheng Feng (gfeng@health.ucsd.edu).
Materials availability
Plasmids used in this study were acquired from Dr. Xin Chen (UCSF). Mouse lines used and generated in this study are available upon request to the authors.
Data and code availability
Single cell RNA-sequencing data have been deposited at GEO and are publicly available as of the date of publication. Accession numbers are listed in the key resources table. Original western blot images and Microscopy data reported in this paper will be shared by the lead contact upon request. This paper analyzes existing publically available data. These accession numbers for the datasets are listed in the key resources table.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animal models
All animals used in this study were C57BL/6J background. The animal protocol (S09108) was approved by the IACUC at University of California San Diego. The SKO (Shp2hep−/−) and BKO (B-cateninhep−/−) mouse lines were generated using Albumin-Cre as previously described (Bard-Chapeau et al., 2006). The SBKO line was generated by breeding SKO and BKO mouse lines together, maintaining heterozygous expression of Albumin-cre. Tumor models were generated by hydrodynamic tail vein injection (HTVi) using a ratio of 10:1 for DNA plasmid to sleeping beauty transposase. For the Ras/Myc model, Ras and Myc were used at a ratio of 95:5 for a total 10 μg/ml. Plasmids (Bcat: PT3-EF1a-ΔN90-β-catenin; PT3-EF1a-C-Myc; PT/Caggs-Nras-V12, pCMV/SB11) were kindly provided by Dr. X Chen at University of California San Francisco. Plasmid PCKO-dnTCF7l2 was kindly provided by Dr. Gregory Lemke at Salk Institute for Biological Sciences (Vacik et al., 2011). PT3-EF1a-eGFP and PT3-EF1a-dnTCF7l2 were cloned for use. Plasmid DNAs were diluted in phosphate-buffered saline (PBS) and injected at 0.1 mL/g body weight through the tail vein in 4-6 s. AAV-TBG-GFP (Addgene; 105535) and AAV-TBG-Cre (Addgene; 107787) were purchased from Addgene and injected at the titer of 2 × 1011 pfu/mouse.
In vitro models
PLC/PRF/5 and NR-PDAC cells were cultured in DMEM (GIBCO SH30910.03) containing 10% Fetal Bovine Serum (Hyclone; 10313021), non-essential amino acid (GIBCO; 11140050) and Penicillin-Streptomycin (GIBCO; 1514022). BXPC-3 cells were cultured in RPMI-1640 containing 10% Fetal Bovine Serum and Penicillin-Streptomycin. Cells were pre-treated with SHP099 (Chemietek; CT-SHP099) or Trametinib (APExBIO; A3018), followed by cycloheximide (Sigma-Aldrich; 239764) treatment. Batches of cells were harvested every 15 minutes after addition of cycloheximide, followed by cell lysis and immunoblotting to determine residual Myc protein level.
METHOD DETAILS
Immunoblotting and immunostaining
Protein was extracted from liver lysate or cells using RIPA buffer (30mg liver per 400ul RIPA) and denatured using an SDS-based sampling buffer with dithiothreitol (DTT) and bromophenol blue. Tumor and non-tumor tissues were extracted separately as noted. Cytosolic and Nuclear fractions were separated using nuclear extraction kit (ab113474). Denatured protein samples were fractioned on SDS-PAGE gels and transferred to 0.45 μm nitrocellulose membrane (Bio-Rad; 1620115). Membranes were washed 3x for 10 minutes in TBST, blocked with 5% fat free milk, and incubated with primary antibodies at 4°C overnight. Next, membranes were washed 3x for 10 minutes and appropriate horseradish peroxidase conjugated secondary antibodies were applied for 2 hours at room temperature, before detection with Chemiluminescent Substrate on a BioRad Chemilluminescence Imager.
FFPE sections and Hematoxylin and Eosin (H&E) stained sections were generated by the Tissue Technology Shared Resource and Histopathology division of the Comparative Phenotyping core at UCSD. Hematoxylin and eosin (H&E) stained liver sections were assessed by Dr. Nissi Varki (UCSD) at the above core, for histopathological evaluation of liver tissues. Immunohistochemical staining was performed on FFPE sections (5 μm thickness) and counterstained with Hematoxylin (EMS; 2617303). FFPE sections were deparaffined using histoclear, blocked with 3% hydrogen peroxide for 10 minutes, steamed in antigen retrieval for 30 minutes (cooled for 10-20 minutes), blocked in 5% NGS in TBST, incubated with primary antibody at 4°C overnight, incubated with secondary antibody with HRP for 2 hours at room temperature, and exposed using DAB.
Immunofluorescence was performed on fresh frozen tissue sections fixed with 4% PFA at 4°C overnight. Slides were then washed 3x for 10 minutes using TBST, blocked in 3% BSA, incubated with primary antibody at 4°C overnight, washed 3x for 10 minutes each, incubated with secondary antibody for 2 hours at room temperature, and mounted with VECTASHIELD mounting medium with DAPI (VWR; H-1200). Other staining kits include Pico-Sirius Red Stain Kit (American MasterTech; KTPSRPT), TUNEL kit (Takara; MK500), and RNAScope 2.5 HD Assay- Red (ACDBio; 322360, 3223650).
RNA extraction and quantitative real-time PCR
cDNA was acquired from fresh frozen liver through RNA isolation via Trizol reagent (Thermo Fisher Scientific; 15596018) and reverse transcription using High-Capacity cDNA Reverse Transcription Kit from Thermo Fisher Scientific (4368814). Real-time qPCR was performed using DyNAmo Flash SYBR Green qPCR Master Mix from Thermo Fisher Scientific (F415) on Stratagene mx3005p instrument. Relative expression of mRNA was calculated by 2−ΔΔCt method and normalized to GAPDH or BACTIN. Primer list in Table S4.
Flow cytometry analysis
Non-parenchymal cells were isolated from the liver via two-step perfusion with Ca2+-free HBSS buffer and then with collagenase H in HBSS buffer containing Ca2+. Non-parenchymal cells were separated by centrifugation and cleared of erythrocytes by ACK lysis buffer. Cells were first stained for L/D dye and then stained for panel antibodies for 30 minutes at 4°C. Cells were fixed using 1% PFA in PBS overnight at 4°C, resuspended in PBS, and analyzed on a BD LSRFortessa X-20 HTS flow cytometer at the Human Embryonic Stem Cell Core at UCSD. Data was analyzed using FlowJo 10.6.2 (BD).
Liver cell isolation for single-cell RNA-seq
Mouse liver was perfused with collagenase H (Roche; 11074059001). Next, the liver was dissociated and passed through a 100μm cell strainer. Cells were centrifuged at 50 g for 3 minutes, in order to pellet hepatocytes from non-parenchymal cells. To remove dead cells and debris, the pelleted hepatocytes were resuspended in 45% Percoll (Sigma-Aldrich) and centrifuged at 50 g for 10 min, without brake. Meanwhile, the Dead Cell Removal Kit (Miltenyi Biotech; 130-090-101) was used to eliminate dead cells in non-parenchymal cells, after spinning down at 250 g for 5 minutes. Hepatocytes and non-parenchymal cells were further washed with PBS and counted with hemocytometer. Of note, for tumor-bearing livers, after perfusion the tumors were first dissected out, minced, and incubated in perfusion buffer with collagenase H for 1 hour at 37°C. Uniquely, 30% Percoll was used to purify tumor cells, instead of 45%.
Single-cell RNA library construction and sequencing
The isolated hepatocytes and non-parenchymal cells were loaded onto 10x Chromium Controller, separately, and then partitioned into nanoliter-scale Gel Beads-In-Emulsion (GEMs). Cells in GEMs were lysed, and RNAs released from cells were immediately captured by barcoded beads in the same GEMs, followed by reverse transcription, amplification, fragmentation, adaptor ligation and index PCR. For each library, the volume of single cell suspension was calculated in order to generate 5000 GEMs per sample. Libraries were constructed using Chromium Single Cell 3′ Reagent Kits (V2 chemistry, 10x Genomics). Sequencing was performed on Illumina HiSeq 4000 at IGM Genomics Center, University of California, San Diego, with the following read length: Read 1, 26bp, including 16bp cell barcode and 12bp unique molecular identifier (UMI); Read 2, 98bp transcript insert; i7 sample index, 8bp. The single cell RNA-seq data have been deposited into the GEO repository, with a GEO accession number assigned (GSE157561).
Single-cell RNA-seq data preprocessing
Obtained reads were mapped to mouse reference genome GRCm38 using CellRanger package (v3.0.2). The human MYC sequence and annotation were manually added into GRCm38 genome, in order to track the expression of plasmid we injected. For each sample, an expression matrix was generated after mapping, UMI counting and cell barcode calling, with each row representing a gene and each column representing a cell. Next, low quality cells and genes were filtered for downstream analysis. In brief, for each sample, genes expressed in less than 3 cells were removed; cells failed to meet following criteria were removed: 1) the number of genes detected in each cell should be more than 200 but less than 5000; 2) the UMIs of mitochondrial genes should be less than 20% of total UMI; 3) the total UMIs of a cell should be less than 30000 to avoid non-singlet (i.e., transcriptome representing more than one cell). The number of cells passing these filters was listed in Table S2. After filtering, the raw expression matrix was normalized by the total expression, multiplied by scale factor 10,000, and log-transformed. Next, we regressed on total number of UMIs per cell as well as mitochondrial gene percentage. The z-scored residuals calculated by normalization and scaling were stored for downstream analysis.
Dataset integration and subpopulation detection
We integrated all cells collected from the same time point or condition, i.e., 0 day, 10 days after vector injection, 10 days after Myc injection, and 4 weeks after Myc injection. For each time point or condition, the integrated datasets included hepatocytes and non-parenchymal cells from both WT and SKO liver. The single cell datasets integration was performed using Seurat R package (v2.4). In brief, canonical correlation analysis (CCA) was used to identify common source of variation between prepared datasets and generate vectors that projected each dataset into the maximally correlated subspaces. Aligning these CCA subspaces returned a dimension reduction for downstream analysis. Next, we performed SNN graph-based clustering and visualized integrated datasets with t-SNE. We used 30-40 CCs for these calculation and tuned resolution parameter in order to get reasonable number of subpopulations (Figures S1A, S1D, 2B, and 2C). The genes used for cell type assignment are well-known markers. We plotted their specific expression in corresponding subpopulations (Figures S1C, S1F, S1H, and S1J). Besides, for hepatocytes collected at time point or condition of interest, we extracted these cells out from integrated datasets and re-clustered with higher resolution in order to further identify subpopulations within hepatocytes.
MAGIC data imputation
To study gene-gene relationship in our datasets, we used a data imputation method called Markov affinity-based graph imputation of cells (MAGIC). “Dropout” is the most challenging technique problem of single cell RNA-sequencing technique. Many expressed mRNAs cannot be captured by current protocols. Therefore, the gene-gene relationship is lost in raw datasets. MAGIC was developed to fill in missing transcripts, via sharing information across similar cells. We performed MAGIC and calculated Pearson correlation coefficient between two genes after data imputation.
Differential expression analysis and gene set enrichment analysis
After cell type assignment, we performed differential expression analysis for specified comparison. 1) Identify subpopulation specific genes. We performed differential expression test for each gene between cells in subpopulation of interest and cells not in this subpopulation. 2) For each assigned cell type, identify differential expression genes between cells from WT and SKO. 3) For hepatocytes grouped by Shp2 and Myc expression, we first performed MAGIC imputation and set threshold based on expression density plots after imputation. Hepatocytes with higher Shp2 imputation levels than threshold were assigned as Shp2+ cells, while hepatocytes with lower Shp2 imputation levels were assigned as Shp2− cells. We assigned Myc+ cells and Myc− cells using the same strategy. We then performed differential expression test between Shp2+ Myc+, Shp2− Myc+, and Shp2−Myc− hepatocytes. 4) Test for differential expressed genes between SKO tumor cells and non-tumor hepatocytes. Wilcoxon Rank Sum test was used in all comparisons described above. Next, Gene-Set Enrichment Analysis (GSEA) was applied to identify pathways that are significantly enriched in these differential expression gene lists. We first ordered genes by log fold-change of the average expression between two groups. Then gene sets collected by Msigdb (Subramanian et al., 2005), including Hallmark, BioCarta, KEGG, Pathway Interaction Database, and Reactome, were used for GSEAPreranked analysis.
Defining gene set signature score
We defined signature score per cell for gene set we are interested as:
We calculated signature scores for gene sets: 1) Myc/β-catenin Signature, consisting of 125 genes defined by Bisso et al. (2020). 2) Wnt target, consisting of 155 genes collected from Wnt target database (https://web.stanford.edu/group/nusselab/cgi-bin/wnt/target_genes) and The Molecular Signatures Database (MSigDB) gene set HALLMARK_WNT_BETA_CATENIN_SIGNALING. 3) Hippo-Yap pathway, consisting of 32 genes collected from MsigDB gene sets REACTOME_YAP1_AND_WWTR1_TAZ_ STIMULATED_GENE_EXPRESSION and REACTOME_SIGNALING_BY_HIPPO. 4) mTORC1 signaling pathway, consisting of 23 genes collected from MsigDB gene set REACTOME_MTORC1_MEDIATED_SIGNALING. 5) Myc target, consisting of 243 genes collected from MsigDB gene sets HALLMARK_MYC_TARGETS_V1 and HALLMARK_MYC_TARGETS_V2. After calculation, we made boxplot and performed Wilcoxon test to check significant difference of signature scores between groups. The heatmap of genes from indicated gene sets was made using scaled expression values in 150 randomly selected cells from each group.
TCGA data analysis
The TCGA dataset was downloaded from TCGA-LIHC project, including RNA-sequencing results and clinical information for 371 HCC patients. And the CTNNB1 mutation and MYC copy number variation for these patients were obtained from cBioPortal. We first divided all patients into three groups based on their MYC expression levels: Low (MYC expression level of the patient is lower than 30% percentile); High (MYC expression level of the patient is higher than 70% percentile); Mid (other patients). We plotted different types of CTNNB1 mutation in three groups respectively. For survival analysis, all patients were divided into four groups based on MYC and CTNNB1 activities: MYC+CTNNB1+ (both MYC and CTNNB1 activity of this patient are higher than third quantile); MYC+CTNNB1− (only MYC activity of this patient is higher than third quantile); MYC−CTNNB1+ (only CTNNB1 activity of this patient is higher than third quantile); MYC−CTNNB1− (other patients). The MYC or CTNNB1 activity for each patient was defined as aggregated expression levels of MYC downstream targets or CTNNB1 downstream targets. And the aggregated expression levels were calculated as:
CTNNB1 downstream targets are described above. We used log2 fold change calculated based on the comparison between WT mice and β-catenin KO mice RNA-seq dataset (Liang et al., 2018) as weight for CTNNB1 target genes. MYC downstream targets included 158 genes collected from Msigdb DANG_MYC_TARGETS_UP and DANG_MYC_TARGETS_DN. We used log2 fold change calculated based on the comparison between Myc overexpression mice and WT mice as weight for MYC target genes (Bisso et al., 2020). Next, we performed survival analysis using survival and survminer R package. The similar patients clustering and survival analysis was done for tumor samples in GSE14520.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analysis was performed using GraphPad Prism 5.0. Cells with positive staining were scored in at least three fields at × 200 magnification per mouse using ImageJ/FIJI. Statistical significance between means of multiple groups was calculated by: two-way ANOVA with posttest Bonferroni (multiple paired groups), one-sided ANOVA with posttest Tukey (multiple groups), or Student’s t test (two groups) as required. Bar graph values are presented as means ± standard deviation. Dot plot values presented with median and quartile markers. Survival Curves were plotted by Kaplan-Meier analysis and compared using log-rank test.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibody | ||
| AFP | Cell Signaling Technologies | Cat#: 4448; RRID:AB_10897327 |
| β-actin | Sigma | Cat#: A5316; RRID:AB_476743 |
| β-catenin (H-102) | Santa Cruz | Cat#: SC-7199; RRID:AB_634603 |
| CD8 | eBioscience | Cat#: 14-0032-81; RRID:AB_467052 |
| CK19 | DSHB | Cat#: Troma-III; RRID:AB_2133570 |
| Clec4f | BioLegend | Cat#: 156804; RRID:AB_2814082 |
| Erk1/2 | Cell Signaling Technologies | Cat#: 4695; RRID:AB_390779 |
| F4/80 | eBioscience | Cat#: 14-4801-82; RRID:AB_467558 |
| Gapdh | Cell Signaling Technologies | Cat#: 5172 |
| Glutamine Synthetase | Cell Signaling Technologies | Cat#: 3886 |
| Histone 3 | Cell Signaling Technologies | Cat#: 4499P; RRID:AB_10544537 |
| HNF4a | Santa Cruz | Cat#: sc-8987; RRID:AB_2116913 |
| Ki67 | eBioscience | Cat#: 14-5698-80; RRID:AB_10853185 |
| MYC | abcam | Cat#: ab32072; RRID:AB_731658 |
| p-c-Myc (S62) | abcam | Cat#: ab51156; RRID:AB_869189 |
| p-c-Myc (T58) | abcam | Cat#: ab28842; RRID:AB_731667 |
| PCNA | Santa Cruz | Cat#: 7907; RRID:AB_2160375 |
| p-Erk1/2 | Cell Signaling Technologies | Cat#: 4370; RRID:AB_2315112 |
| p-β-catenin (S33/37/T41) | Cell Signaling Technologies | Cat#: 9561; RRID:AB_331729 |
| Shp2 | Santa Cruz | Cat#: sc-7384; RRID:AB_628252 |
| FACS antibodies | ||
| Life/Dead Aqua Stain | ThermoFisher | Cat#: L34957 |
| CD45 – PerCP | Biolegend | Cat#: 147706; RRID:AB_2563538 |
| Ly6c – PE | Biolegend | Cat#: 128008; RRID:AB_1186132 |
| F4/80 - BV421 | Biolegend | Cat#: 123124; RRID:AB_893475 |
| CD11c – APC | Biolegend | Cat#: 117309; RRID:AB_313778 |
| MHCII - APC Cy7 | Biolegend | Cat#: 107628; RRID:AB_2069377 |
| CD11b - BV605 | Biolegend | Cat#: 101257; RRID:AB_2565431 |
| TCRβ - BV421 | Biolegend | Cat#: 109226; RRID:AB_1027649 |
| NK1.1 – APC | Biolegend | Cat#: 108710; RRID:AB_313397 |
| CD8a - PE Cy7 | Biolegend | Cat#: 100722; RRID:AB_312761 |
| CD19 - APC Cy7 | Biolegend | Cat#: 115530; RRID:AB_830707 |
| CD4 - BV605 | Biolegend | Cat#: 100451; RRID:AB_2564591 |
| CD69 - BV711 | Biolegend | Cat#: 104537; RRID:AB_2566120 |
| Commercial assays | ||
| Pico-Sirius Red Stain Kit | American MasterTech | Cat#: KTPSRPT |
| Mayer’s Hematoxylin | Electron Microscopy Services | Cat#: 2617303 |
| TUNEL | Takara | Cat#: MK500; RRID:AB_2800362 |
| RNAScope 2.5 HD Assay - Red | ACDBio | Cat#: 322360, 3223650 |
| Nuclear Extraction Kit | abcam | Cat#: ab113474 |
| Dead Cell Removal Kit | Miltenyi Biotech | Cat#: 130-090-101 |
| Oligonucleotides | ||
| See Table S4 | ||
| Software and algorithms | ||
| FlowJo 10.6.2 | BD | https://www.flowjo.com |
| Graphpad Prism 5.0 | Graphpad | https://www.graphpad.com/scientific-software/prism/ |
| ImageJ / FIJI | ImageJ, FIJI team | https://imagej.net/software/fiji |
| Cellranger v3.0.2 | 10x Genomics | https://www.10xgenomics.com/ |
| STAR | Dobin et al., 2013 | https://github.com/alexdobin/STAR |
| R | The R Foundation | https://www.r-project.org/ |
| Seurat | Butler et al., 2018; Stuart et al., 2019 | https://github.com/satijalab/seurat |
| Python 3 | Python Software Foundation | https://www.python.org/ |
| MAGIC | van Dijk et al., 2018 | https://github.com/KrishnaswamyLab/MAGIC |
| Experimental models: Organisms | ||
| Mouse: SKO (Albumin-Cre; Shp2 f/f) | Bard-Chapeau et al., 2006 | N/A |
| Mouse: BKO(Albumin-Cre; β-catenin f/f) | Liang et al., 2018 | N/A |
| Mouse: SBKO(Albumin-Cre; Shp2 f/f; β-catenin f/f) | This paper | N/A |
| Deposited data | ||
| Single Cell RNA-sequencing data | GEO Repository | GSE157561 |
| Public datasets analyzed | ||
| TCGA-LIHC project | cBioPortal | NA |
| GSE14520 dataset | GEO Repository | GSE14520 |
Highlights.
Myc alone drives a severe HCC phenotype in Shp2-deficient liver
Shp2 is required cell autonomously for control of Myc stability
Shp2 deficiency induces a tumorigenic environment in the liver
Aberrantly elevated β-catenin signaling promotes Myc-driven HCC
ACKNOWLEDGMENTS
This work was supported by NCI grants R01CA236074 and R01CA239629 to G.-S.F. and by NIH training grant T32CA009523 to W.S.C. We thank Drs. X. Chen (UCSF), G. Lemke (The Salk Institute), and N. Varki and K. Willert (UCSD) for reagents or histopathological analysis. We also thank the Genomics, Histology, Flow Cytometry, and Animal Facility at UCSD for technical assistance.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2021.109974.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Abou-Elella A, Gramlich T, Fritsch C, and Gansler T (1996). c-myc amplification in hepatocellular carcinoma predicts unfavorable prognosis. Mod. Pathol 9, 95–98. [PubMed] [Google Scholar]
- Ahmed TA, Adamopoulos C, Karoulia Z, Wu X, Sachidanandam R, Aaronson SA, and Poulikakos PI (2019). SHP2 drives adaptive resistance to ERK signaling inhibition in molecularly defined subsets of ERK-dependent tumors. Cell Rep. 26, 65–78.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anson M, Crain-Denoyelle AM, Baud V, Chereau F, Gougelet A, Terris B, Yamagoe S, Colnot S, Viguier M, Perret C, and Couty JP (2012). Oncogenic β-catenin triggers an inflammatory response that determines the aggressiveness of hepatocellular carcinoma in mice. J. Clin. Invest 122, 586–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bard-Chapeau EA, Yuan J, Droin N, Long S, Zhang EE, Nguyen TV, and Feng GS (2006). Concerted functions of Gab1 and Shp2 in liver regeneration and hepatoprotection. Mol. Cell. Biol 26, 4664–4674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bard-Chapeau EA, Li S, Ding J, Zhang SS, Zhu HH, Princen F, Fang DD, Han T, Bailly-Maitre B, Poli V, et al. (2011). Ptpn11/Shp2 acts as a tumor suppressor in hepatocellular carcinogenesis. Cancer Cell 19, 629–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bisso A, Filipuzzi M, Gamarra Figueroa GP, Brumana G, Biagioni F, Doni M, Ceccotti G, Tanaskovic N, Morelli MJ, Pendino V, et al. (2020). Cooperation between MYC and β-catenin in liver tumorigenesis requires Yap/Taz. Hepatology 72, 1430–1443. [DOI] [PubMed] [Google Scholar]
- Burke ZD, Reed KR, Phesse TJ, Sansom OJ, Clarke AR, and Tosh D (2009). Liver zonation occurs through a β-catenin-dependent, c-Myc-independent mechanism. Gastroenterology 136, 2316–2324.e1-3. [DOI] [PubMed] [Google Scholar]
- Butler A, Hoffman P, Smibert P, Papalexi E, and Satija R (2018). Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol 36, 411–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cairo S, Armengol C, De Reyniès A, Wei Y, Thomas E, Renard CA, Goga A, Balakrishnan A, Semeraro M, Gresh L, et al. (2008). Hepatic stem-like phenotype and interplay of Wnt/β-catenin and Myc signaling in aggressive childhood liver cancer. Cancer Cell 14, 471–484. [DOI] [PubMed] [Google Scholar]
- Calvisi DF, Factor VM, Loi R, and Thorgeirsson SS (2001). Activation of β-catenin during hepatocarcinogenesis in transgenic mouse models: Relationship to phenotype and tumor grade. Cancer Res. 61, 2085–2091. [PubMed] [Google Scholar]
- Chan RJ, and Feng GS (2007). PTPN11 is the first identified proto-oncogene that encodes a tyrosine phosphatase. Blood 109, 862–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, and Calvisi DF (2014). Hydrodynamic transfection for generation of novel mouse models for liver cancer research. Am. J. Pathol 184, 912–923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YN, LaMarche MJ, Chan HM, Fekkes P, Garcia-Fortanet J, Acker MG, Antonakos B, Chen CH, Chen Z, Cooke VG, et al. (2016). Allosteric inhibition of SHP2 phosphatase inhibits cancers driven by receptor tyrosine kinases. Nature 535, 148–152. [DOI] [PubMed] [Google Scholar]
- Cho JH, Dimri M, and Dimri GP (2013). A positive feedback loop regulates the expression of polycomb group protein BMI1 via WNT signaling pathway. J. Biol. Chem 288, 3406–3418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowling VH, D’Cruz CM, Chodosh LA, and Cole MD (2007). c-Myc transforms human mammary epithelial cells through repression of the Wnt inhibitors DKK1 and SFRP1. Mol. Cell. Biol 27, 5135–5146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang CV (2012). MYC on the path to cancer. Cell 149, 22–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dardaei L, Wang HQ, Singh M, Fordjour P, Shaw KX, Yoda S, Kerr G, Yu K, Liang J, Cao Y, et al. (2018). SHP2 inhibition restores sensitivity in ALK-rearranged non-small-cell lung cancer resistant to ALK inhibitors. Nat. Med 24, 512–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dauch D, Rudalska R, Cossa G, Nault JC, Kang TW, Wuestefeld T, Hohmeyer A, Imbeaud S, Yevsa T, Hoenicke L, et al. (2016). A MYC-aurora kinase A protein complex represents an actionable drug target in p53-altered liver cancer. Nat. Med 22, 744–753. [DOI] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Serag HB (2011). Hepatocellular carcinoma. N. Engl. J. Med 365, 1118–1127. [DOI] [PubMed] [Google Scholar]
- Farrell AS, and Sears RC (2014). MYC degradation. Cold Spring Harb. Perspect. Med 4, a014365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fedele C, Ran H, Diskin B, Wei W, Jen J, Geer MJ, Araki K, Ozerdem U, Simeone DM, Miller G, et al. (2018). SHP2 inhibition prevents adaptive resistance to MEK inhibitors in multiple cancer models. Cancer Discov. 8, 1237–1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng GS (1999). Shp-2 tyrosine phosphatase: Signaling one cell or many. Exp. Cell Res 253, 47–54. [DOI] [PubMed] [Google Scholar]
- Feng GS (2012). Conflicting roles of molecules in hepatocarcinogenesis: Paradigm or paradox. Cancer Cell 21, 150–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng GS, and Pawson T (1994). Phosphotyrosine phosphatases with SH2 domains: Regulators of signal transduction. Trends Genet. 10, 54–58. [DOI] [PubMed] [Google Scholar]
- Finn RS, Qin S, Ikeda M, Galle PR, Ducreux M, Kim TY, Kudo M, Breder V, Merle P, Kaseb AO, et al. ; IMbrave150 Investigators (2020). Atezolizumab plus bevacizumab in unresectable hepatocellular carcinoma. N. Engl. J. Med 382, 1894–1905. [DOI] [PubMed] [Google Scholar]
- Halpern KB, Shenhav R, Matcovitch-Natan O, Toth B, Lemze D, Golan M, Massasa EE, Baydatch S, Landen S, Moor AE, et al. (2017). Single-cell spatial reconstruction reveals global division of labour in the mammalian liver. Nature 542, 352–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayes TK, Neel NF, Hu C, Gautam P, Chenard M, Long B, Aziz M, Kassner M, Bryant KL, Pierobon M, et al. (2016). Long-term ERK inhibition in KRAS-mutant pancreatic cancer is associated with MYC degradation and senescence-like growth suppression. Cancer Cell 29, 75–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou Y, Guo H, Cao C, Li X, Hu B, Zhu P, Wu X, Wen L, Tang F, Huang Y, and Peng J (2016). Single-cell triple omics sequencing reveals genetic, epigenetic, and transcriptomic heterogeneity in hepatocellular carcinomas. Cell Res. 26, 304–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerr DL, Haderk F, and Bivona TG (2021). Allosteric SHP2 inhibitors in cancer: Targeting the intersection of RAS, resistance, and the immune microenvironment. Curr. Opin. Chem. Biol 62, 1–12. [DOI] [PubMed] [Google Scholar]
- Lee J, Liao R, Wang G, Yang BH, Luo X, Varki NM, Qiu SJ, Ren B, Fu W, and Feng GS (2017). Preventive Inhibition of liver tumorigenesis by systemic activation of innate immune functions. Cell Rep. 21, 1870–1882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang Y, Feng Y, Zong M, Wei XF, Lee J, Feng Y, Li H, Yang GS,Wu ZJ, Fu XD, and Feng GS (2018). β-Catenin deficiency in hepatocytes aggravates hepatocarcinogenesis driven by oncogenic β-catenin and MET. Hepatology 67, 1807–1822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu P, Ge M, Hu J, Li X, Che L, Sun K, Cheng L, Huang Y, Pilo MG, Cigliano A, et al. (2017). A functional mammalian target of rapamycin complex 1 signaling is indispensable for c-Myc-driven hepatocarcinogenesis. Hepatology 66, 167–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu JJ, Li Y, Chen WS, Liang Y, Wang G, Zong M, Kaneko K, Xu R, Karin M, and Feng GS (2018). Shp2 deletion in hepatocytes suppresses hepatocarcinogenesis driven by oncogenic β-catenin, PIK3CA and MET. J. Hepatol 69, 79–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llovet JM, Zucman-Rossi J, Pikarsky E, Sangro B, Schwartz M, Sherman M, and Gores G (2016). Hepatocellular carcinoma. Nat. Rev. Dis. Primers 2, 16018. [DOI] [PubMed] [Google Scholar]
- Luo X, Liao R, Hanley KL, Zhu HH, Malo KN, Hernandez C, Wei X, Varki NM, Alderson N, Chu C, et al. (2016). Dual Shp2 and Pten deficiencies promote non-alcoholic steatohepatitis and genesis of liver tumor-initiating cells. Cell Rep. 17, 2979–2993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma L, Hernandez MO, Zhao Y, Mehta M, Tran B, Kelly M, Rae Z, Hernandez JM, Davis JL, Martin SP, et al. (2019). Tumor cell biodiversity drives microenvironmental reprogramming in liver cancer. Cancer Cell 36, 418–430.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mainardi S, Mulero-Sánchez A, Prahallad A, Germano G, Bosma A, Krimpenfort P, Lieftink C, Steinberg JD, de Wit N, Gonçalves-Ribeiro S, et al. (2018). SHP2 is required for growth of KRAS-mutant non-small-cell lung cancer in vivo. Nat. Med 24, 961–967. [DOI] [PubMed] [Google Scholar]
- Meex RCR, and Watt MJ (2017). Hepatokines: Linking nonalcoholic fatty liver disease and insulin resistance. Nat. Rev. Endocrinol 13, 509–520. [DOI] [PubMed] [Google Scholar]
- Murakami H, Sanderson ND, Nagy P, Marino PA, Merlino G, and Thorgeirsson SS (1993). Transgenic mouse model for synergistic effects of nuclear oncogenes and growth factors in tumorigenesis: Interaction of c-myc and transforming growth factor α in hepatic oncogenesis. Cancer Res. 53, 1719–1723. [PubMed] [Google Scholar]
- Neel BG, Gu H, and Pao L (2003). The “Shp”ing news: SH2 domain-containing tyrosine phosphatases in cell signaling. Trends Biochem. Sci 28, 284–293. [DOI] [PubMed] [Google Scholar]
- The Cancer Genome Atlas Research Network (2017). Comprehensive and integrative genomic characterization of hepatocellular carcinoma. Cell 169, 1327–1341.e23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ovejero C, Cavard C, Périanin A, Hakvoort T, Vermeulen J, Godard C, Fabre M, Chafey P, Suzuki K, Romagnolo B, et al. (2004). Identification of the leukocyte cell-derived chemotaxin 2 as a direct target gene of β-catenin in the liver. Hepatology 40, 167–176. [DOI] [PubMed] [Google Scholar]
- Postic C, and Magnuson MA (2000). DNA excision in liver by an albumin-Cre transgene occurs progressively with age. Genesis 26, 149–150. [DOI] [PubMed] [Google Scholar]
- Reed KR, Athineos D, Meniel VS, Wilkins JA, Ridgway RA, Burke ZD, Muncan V, Clarke AR, and Sansom OJ (2008). B-catenin deficiency, but not Myc deletion, suppresses the immediate phenotypes of APC loss in the liver. Proc. Natl. Acad. Sci. USA 105, 18919–18923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruess DA, Heynen GJ, Ciecielski KJ, Ai J, Berninger A, Kabacaoglu D, Görgülü K, Dantes Z, Wörmann SM, Diakopoulos KN, et al. (2018). Mutant KRAS-driven cancers depend on PTPN11/SHP2 phosphatase. Nat. Med 24, 954–960. [DOI] [PubMed] [Google Scholar]
- Ruiz de Galarreta M, Bresnahan E, Molina-Sánchez P, Lindblad KE, Maier B, Sia D, Puigvehi M, Miguela V, Casanova-Acebes M, Dhainaut M, et al. (2019). β-Catenin activation promotes immune escape and resistance to anti-PD-1 therapy in hepatocellular carcinoma. Cancer Discov. 9, 1124–1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sansom OJ, Meniel VS, Muncan V, Phesse TJ, Wilkins JA, Reed KR, Vass JK, Athineos D, Clevers H, and Clarke AR (2007). Myc deletion rescues Apc deficiency in the small intestine. Nature 446, 676–679. [DOI] [PubMed] [Google Scholar]
- Spranger S, Bao R, and Gajewski TF (2015). Melanoma-intrinsic β-catenin signalling prevents anti-tumour immunity. Nature 523, 231–235. [DOI] [PubMed] [Google Scholar]
- Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM, Hao Y, Stoeckius M, Smibert P, and Satija R (2019). Comprehensive integration of single-cell data. Cell 177, 1888–1902.e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, and Mesirov JP (2005). Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan X, Behari J, Cieply B, Michalopoulos GK, and Monga SP (2006). Conditional deletion of β-catenin reveals its role in liver growth and regeneration. Gastroenterology 131, 1561–1572. [DOI] [PubMed] [Google Scholar]
- Tartaglia M, and Gelb BD (2005). Germ-line and somatic PTPN11 mutations in human disease. Eur. J. Med. Genet 48, 81–96. [DOI] [PubMed] [Google Scholar]
- Vacik T, Stubbs JL, and Lemke G (2011). A novel mechanism for the transcriptional regulation of Wnt signaling in development. Genes Dev. 25, 1783–1795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Dijk D, Sharma R, Nainys J, Yim K, Kathail P, Carr AJ, Burdziak C, Moon KR, Chaffer CL, Pattabiraman D, et al. (2018). Recovering gene interactions from single-cell data using data diffusion. Cell 174, 716–729.e27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaseva AV, Blake DR, Gilbert TSK, Ng S, Hostetter G, Azam SH, Ozkan-Dagliyan I, Gautam P, Bryant KL, Pearce KH, et al. (2018). KRAS suppression-induced degradation of MYC is antagonized by a MEK5-ERK5 compensatory mechanism. Cancer Cell 34, 807–822.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen L, Xin B, Wu P, Lin CH, Peng C, Wang G, Lee J, Lu LF, and Feng GS (2019). An efficient combination immunotherapy for primary liver cancer by harmonized activation of innate and adaptive immunity in mice. Hepatology 69, 2518–2532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong C, Chen C, Wu Q, Liu Y, and Zheng P (2015). A critical role for the regulated Wnt-Myc pathway in naive T cell survival. J. Immunol 194, 158–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong GS, Zhou J, Liu JB, Wu Z, Xu X, Li T, Xu D, Schumacher SE, Puschhof J, McFarland J, et al. (2018). Targeting wild-type KRAS-amplified gastroesophageal cancer through combined MEK and SHP2 inhibition. Nat. Med 24, 968–977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong X, Kuang H, Ansari S, Liu T, Gong J, Wang S, Zhao XY, Ji Y, Li C, Guo L, et al. (2019). Landscape of intercellular crosstalk in healthy and NASH liver revealed by single-cell secretome gene analysis. Mol. Cell 75, 644–660.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu M, Xu HH, Lin Y, Sun X, Wang LJ, Fang ZP, Su XH, Liang XJ, Hu Y, Liu ZM, et al. (2019a). LECT2, a ligand for Tie1, plays a crucial role in liver fibrogenesis. Cell 178, 1478–1492.e20. [DOI] [PubMed] [Google Scholar]
- Xu Z, Xu M, Liu P, Zhang S, Shang R, Qiao Y, Che L, Ribback S, Cigliano A, Evert K, et al. (2019b). The mTORC2-Akt1 cascade is crucial for c-Myc to promote hepatocarcinogenesis in mice and humans. Hepatology 70, 1600–1613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang W, Wang J, Moore DC, Liang H, Dooner M, Wu Q, Terek R, Chen Q, Ehrlich MG, Quesenberry PJ, and Neel BG (2013). Ptpn11 deletion in a novel progenitor causes metachondromatosis by inducing hedgehog signalling. Nature 499, 491–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yochum GS, Cleland R, and Goodman RH (2008). A genome-wide screen for β-catenin binding sites identifies a downstream enhancer element that controls c-Myc gene expression. Mol. Cell. Biol 28, 7368–7379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q, He Y, Luo N, Patel SJ, Han Y, Gao R, Modak M, Carotta S, Haslinger C, Kind D, et al. (2019). Landscape and dynamics of single immune cells in hepatocellular carcinoma. Cell 179, 829–845.e20. [DOI] [PubMed] [Google Scholar]
- Zheng C, Zheng L, Yoo JK, Guo H, Zhang Y, Guo X, Kang B, Hu R, Huang JY, Zhang Q, et al. (2017a). Landscape of infiltrating T cells in liver cancer revealed by single-cell sequencing. Cell 169, 1342–1356.e16. [DOI] [PubMed] [Google Scholar]
- Zheng K, Cubero FJ, and Nevzorova YA (2017b). c-MYC-making liver sick: Role of c-MYC in hepatic cell function, homeostasis and disease. Genes (Basel) 8, 123. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Single cell RNA-sequencing data have been deposited at GEO and are publicly available as of the date of publication. Accession numbers are listed in the key resources table. Original western blot images and Microscopy data reported in this paper will be shared by the lead contact upon request. This paper analyzes existing publically available data. These accession numbers for the datasets are listed in the key resources table.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.