

Abstract
Aim:
Waldenström macroglobulinemia (WM) is a low-grade B-cell lymphoma characterized by overproduction of monoclonal IgM. To date, there are no therapies that provide a cure for WM patients, and therefore, it is important to explore new therapies. Little is known about the efficiency of epigenetic targeting in WM.
Materials & methods:
WM cells were treated with BET inhibitors (JQ1 and I-BET-762) and venetoclax, panobinostat or ibrutinib.
Results:
BET inhibition reduces growth of WM cells, with little effect on survival. This finding was enhanced by combination therapy, with panobinostat (LBH589) showing the highest synergy.
Conclusion:
Our studies identify BET inhibitors as effective therapy for WM, and these inhibitors can be enhanced in combination with BCL2 or histone deacetylase inhibition.
Keywords: : BET inhibitors, epigenetics, panobinostat, venetoclax, Waldenström macroglobulinemia
Waldenström macroglobulinemia (WM) is a subtype of non-Hodgkin lymphoma characterized by the vast appearance of lymphoplasmacytic cells in the bone marrow (BM) [1]. WM is known for aberrant secretion of a monoclonal IgM protein which is associated with serum hyperviscosity syndrome, anemia and peripheral neuropathy [2]. Common treatment strategies for WM include rituximab-based combination therapies, which are considered standard care in the USA. The Bruton’s tyrosine kinase (BTK) inhibitor, ibrutinib, is the only US FDA-approved therapy for WM. Other therapies used (FDA-approved for B-cell lymphoma but not specifically for WM) include R-CHOP (cyclophosphamide, doxorubicin, vincristine and prednisone along with rituximab) [3], dexamethasone, bendamustine, fludarabine, chlorambucil and everolimus, while targeting IL-6 or histone deacetylases (HDACs) showed promising results in WM [4,5]. However, multiple adverse effects are linked to conventional WM targeted therapies. For example, administration of ibrutinib can cause a rash, joint pain, diarrhea, thrombocytopenia, atypical bleeding and pneumonia [6]. Thus, modifying conventional WM therapy by targeting additional cancer-related factors might decrease these side effects and increase the therapeutic window for more efficient targeting.
Changes in the epigenetic pattern of chromatin can cause altered expression of cancer-related genes and lead to tumorigenicity. Therefore, targeting epigenetic modulators by small molecule inhibitors to restore epigenetic homeostasis can be very effective in fighting cancer. HDAC inhibitors, such as the FDA-approved drug panobinostat (LBH589), have been proven to be a very powerful tool in fighting cancer because they reduce aberrant expression of cancer-promoting genes [7]. In addition to HDAC inhibitors, inhibition of bromodomain proteins (BRD2, BRD3, BRD4 and BRDT) showed a promising anticancerous effect in multiple cancer types [8,9]. This effect was even greater when used in combination with HDAC inhibitors or venetoclax, a well-established BCL-2 inhibitor and FDA-approved anticancer drug, proving that synergy between BET, HDAC and BCL-2 inhibition [10,11] is advantageous. Compounds such as JQ1 and I-BET-762 (iBET), which target BRD4, have been shown to be very effective in reducing BRD4-related activation of transcription and induce anticancerous effects [12,13]. BET inhibitors have shown reduced survival of neuroblastoma, glioblastoma, prostate cancer and diffuse large B cell lymphoma (DLBCL) [14–18]. However, their efficacy in WM has not been investigated.
Here, we investigated the efficacy of the BET inhibitors iBET and JQ1 in WM. Treatment of WM cells with iBET or JQ1 reduces cell proliferation in a dose-dependent manner, even in the presence of the tumor microenvironment (TME). Consistent with the reduction in cell proliferation, we found a reduction of MYC mRNA and protein expression following iBET/JQ1 treatment. This treatment also increased BCL-2 expression, suggesting that targeting BCL-2 may be effective in inducing WM cell death. Indeed, combined treatment of WM cells with JQ1 and venetoclax enhanced apoptosis of WM cells compared with JQ1 alone. More impressively, combined treatment of WM cells with JQ1 and panobinostat induced more cell death with a very low dose of panobinostat. Taken together, these results suggest that BET inhibitors may provide therapeutic efficacy in WM by targeting cell growth, and in combination with panobinostat, may provide efficacy by targeting cell viability. These studies lay the foundation for the investigation of these drugs in WM patients.
Materials & methods
Cells & reagents
The BCWM.1 cell line [19] was kindly provided by Dr S Treon (Dana Farber Cancer Institute, MA, USA); the MWCL-1 cell line [20] was kindly provided by Dr S Ansell (Mayo Clinic, MN, USA); and the RPCI-WM1 cell line [21] was kindly provided by Dr A Chanan-Khan (Mayo Clinic, FL, USA). Cells were maintained in Roswell Park Memorial Institute (RPMI) media supplemented with 10% fetal bovine serum (FBS) and 1% antibiotic–antimycotic. HS-5 stromal cells (ATCC CRL-11346) were purchased from American-Type Culture Collection (ATCC, VA, USA) and were maintained in Dulbecco’s modified eagle medium with 10% FBS and 1% antibiotic–antimycotic. The BET bromodomain inhibitors molibresib (iBET) and JQ1, the BCL-2-selective inhibitor venetoclax (ABT-199), the broad-spectrum HDAC inhibitor panobinostat (LBH589), and the BTK inhibitor ibrutinib were all purchased from Selleckchem (TX, USA).
Proliferation assay
Cell proliferation was assessed by XTT assay as previously described [22–24]. Briefly, 0.25 × 103 cells were cultured for 72 h with the drug(s) in a 96-well plate at 37°C. For the coculture experiment, 0.1 × 103 HS-5 cells were plated in 96-well plate and allowed to adhere overnight. Cells were pretreated with 10 mg/ml of mitomycin C for 3 h, followed by removal of media and washing with Dulbecco’s phosphate-buffered saline (PBS). A quantity of 0.5 × 103 serum-starved WM cells were added to each well (1:5 ratio of stromal cells/WM cells), followed by incubation for 72 h with drug(s) at 37°C. The XTT working solution (MD, USA) was added to each well and plates were incubated for 3 h at 37°C. Absorbance was determined using a SpectraMax microplate reader (Molecular Devices, CA, USA). Relative proliferation was determined by comparing proliferation of each treatment to that of control wells treated with dimethyl sulfoxide (DMSO).
Cell cycle analysis
Cell cycle analysis was performed using propidium iodide (PI) staining. Cells (0.5 × 106 cells/ml) were cultured in 12-well plates in the presence of either iBET, JQ1 or DMSO control for 72 h. Cells were harvested and washed once with ice cold PBS and resuspended in 1 ml PBS followed by addition of 9 ml fixation buffer (70% ethanol and 30% PBS) dropwise while vortexing cells. Cells were stored at -20°C for at least 2 h then washed with cold PBS and resuspended in PI staining solution (0.1% Triton-X in PBS, 2 mg/ml RNaseA and 20 µg/ml PI). Data were acquired immediately on a FACSCalibur flow cytometer (BD Biosciences, CA, USA) and analyzed using FlowJo software version 10.
Examination of cell morphology using scanning electron microscopy
Cell morphology was examined using scanning electron microscopy (SEM) as previously described [25]. Briefly, HS-5 cells (0.1 × 106 cells/well) were allowed to adhere to coverslips in 6-well plates overnight. Cells were treated with 10 mg/ml of mitomycin C for 3 h, followed by removal of media and washing with Dulbecco’s PBS. WM cells (0.5 × 106 cells/well) were then added at a ratio of 1:5 (HS-5:WM) and the co-culture continued for 2 days. Coverslips were removed from culture and fixed with 2% paraformaldehyde/2% gluteraldehyde in 0.1 M phosphate buffer overnight at 4°C. Samples were rinsed three-times with 0.1 M phosphate buffer followed by fixation with a 2% osmium tetroxide solution for 2 h at room temperature. Samples were rinsed with fresh 0.1 M phosphate buffer an additional three-times then dehydrated using an ethanol series (30, 50, 70, 95 and 100%) followed by an additional two steps in 100% ethanol, followed by dehydration overnight with hexamethyldisilane (Thermo Fisher Scientific, MA, USA). Samples were coated and imaged at the University Instrumentation Center at University of New Hampshire (NH, USA) using Tescan Lyra 3 SEM (Tescan, PA, USA).
Confocal microscopy for HS-5/WM cell coculture
HS-5 cells (10 × 104) were allowed to adhere to coverslips in 6-well plates overnight. Cell culture media was removed and cells were stained with Vybrant DiO cell-labeling solution (green) (Thermo Fisher Scientific) following manufacturer recommendations. Similarly, WM cells were stained with Vybrant DiI cell-labeling solution (red) (Thermo Fisher Scientific) following manufacturer recommendations. Cells were then co-cultured for 24 h and then cover slips were mounted on microscope slides and imaged using a Nikon A1R-HD confocal microscope (Nikon Instruments Inc., Melville, NY, USA).
Annexin-V & PI double staining
Cell viability was assessed as previously described [22,23,26]. Briefly, 0.5 × 106 cells/ml were treated with either iBET, JQ1, ABT-199, ibrutinib, LBH589 alone or ABT-199/ibrutinib/LBH589 combined with JQ1, or controls for 72 h. Cells were stained with 5 mg Annexin-V-FITC (BD Biosciences, CA, USA) for 20 min at 4°C in darkness. Then cells were washed with annexin-V binding buffer and stained with 0.5 µg/ml PI. Cells were measured on a FACSCalibur flow cytometer. Data were analyzed using FlowJo software.
WM patient sample treatment
Mononuclear cells (MNCs) from WM patient BM aspirates (WM1 and WM2) were isolated using Ficoll-Paque™ PLUS Media (Cytiva, MA, USA) and 2 × 106 BM-MNCs were treated with the indicated doses of JQ1 for 24 h. Analysis of apoptotic cell death was performed on CD19+ cell population. MYD88 genotyping was performed for WM patient’s BM lymphoplasmacytic cells as previously described [27,28] WM1 patient was previously treated with rituximab while WM2 is an untreated patient. Subject participation was approved by the Harvard Cancer Center/Dana-Farber Cancer Institute Institutional Review Board, and all participants provided written consent for sample use.
Immunoblotting
Protein expression was determined by immunoblotting as previously described [22,23,29]. Whole-cell lysates were fractionated by SDS-PAGE using 10% SDS protein gels then transferred to a nitrocellulose membrane using a semidry electroblotting system (Bio-Rad, CA, USA). Antibodies specific for c-MYC (cat #: 9402S) and BCL-2 (cat #: 2872S) were purchased from Cell Signaling (MA, USA) and β-actin antibody (cat #: A3854) was purchased from Millipore Sigma (MO, USA). Protein expression was compared with β-actin, which was used as a loading control. Blots were developed using enhanced chemiluminescence (ECL) (Thermo Fisher Scientific).
RNA isolation & quantitative RT-PCR
Total RNA was isolated using TRIsure reagent (Bioline, London, UK), following the manufacturer’s recommended procedure. Reverse-transcription reactions were conducted by using Moloney Murine Leukemia Virus Reverse Transcriptase (Promega, WI, USA). Quantitative PCR was performed using the ViiA 7 Real-time PCR System (Life Technologies, NY, USA). To check the expression of IgM and MYC relative to the expression of the housekeeping gene, GAPDH, the following primers were used: GAPDH, 5′-CTCGACTTCAACAGCGACA-3′ (forward) and 5′-GTAGCCAAATTCGTTGTCATACC-3′ (reverse); IgM, 5′-CCCAACGGCAACAAAGAAA-3′ (forward) and 5′-GGACGAAGACGCTCACTTT-3′ (reverse); and MYC, 5′-GCTGCTTAGACGCTGGATTT-3′ (forward) and 5′-GAGTCGTAGTCGAGGTCATAGTT-3′ (reverse).
ELISA
ELISA plates were purchased from Thermo Fisher Scientific and were used to quantify IgM levels. Human IgM ELISA antibodies were purchased from Bethyl labs (TX, USA) and were used following the manufacturer’s recommendations. ELISA plates were developed using the Turbo TMB-ELISA (Thermo Fisher Scientific) and reactions were stopped using 1N H2SO4. Results were quantified on a SpectraMax microplate reader and data were analyzed using SoftMax Pro 7.0.2 software (Molecular Devices, CA, USA).
Analysis of drug interaction
The coefficient of drug interaction (CDI) was calculated as follows: CDI = AB/(A × B); where AB is the ratio of the combination group compared with the control group; A or B is the ratio of the single agent group to the control group. A CDI value of 0.1–0.3 indicates strong synergy; 0.3–0.7 indicates synergy; 0.7–0.85 indicates moderate synergy; and 0.85–0.9 indicates slight synergy. A CDI value = 1 indicates additive and >1 indicates antagonistic effects. All CDI values are presented in Table 1.
Table 1. . Coefficient of drug interaction values for drug combinations.
| Parameters | JQ1 + venetoclax | JQ1 + panobinostat | JQ1 + ibrutinib | |||
|---|---|---|---|---|---|---|
| Mean CDI ± SD | Description | Mean CDI ± SD | Description | Mean CDI ± SD | Description | |
| Cell viability | ||||||
| BCWM.1 | 0.721 ± 0.116 | Moderate synergy | 0.498 ± 0.058 | Synergy | 0.846 ± 0.023 | Moderate synergy |
| MWCL-1 | 0.793 ± 0.032 | Moderate synergy | 0.544 ± 0.169 | Synergy | 0.820 ± 0.052 | Moderate synergy |
| RPCI-WM1 | 0.594 ± 0.214 | Synergy | 0.506 ± 0.084 | Synergy | 0.887 ± 0.086 | Slight synergy |
| JQ1 + venetoclax | ||||||
|---|---|---|---|---|---|---|
| Mean CDI ± SD | Description | |||||
| Cell proliferation | ||||||
| BCWM.1 | 0.834 ± 0.130 | Moderate synergy | ||||
| MWCL-1 | 1.081 ±.262 | Additive | ||||
| RPCI-WM1 | 0.762 ± 0.305 | Moderate synergy | ||||
CDI: Coefficient of drug interaction; SD: Standard deviation.
Statistical analysis
A two-tailed t-test was used to determine statistical significance between two variables, and a two-way ANOVA was used when comparing more than two variables. Statistical significance on figures are indicated by *(p < 0.05); **(p < 0.01); ***(p < 0.001) and ****(p < 0.0001). Statistical analysis was conducted using GraphPad Prism software (GraphPad Software, CA, USA).
Results
BET inhibition reduces growth of WM cells
The antiproliferative effect of BET inhibition was evaluated in three WM-derived cell lines BCWM.1, MWCL-1 and RPCI-WM1. WM cells were treated with different concentrations (0.5, 1 and 5 mM) of either iBET (Figure 1A) or JQ1 (Figure 1B) for 72 h. We found a significant reduction in cell proliferation in all three cell lines, starting with the lowest dose of 0.5 mM for both iBET (p < 0.0001) and JQ1 (p < 0.0001). While a dose dependent effect was visible for both drugs in all three cell lines, JQ1 showed the strongest inhibitory effect on cell proliferation; approximately 70% reduction in cell proliferation was achieved at the highest dose (p < 0.0001). We investigated the effect of BET inhibition on cell cycle and found that iBET and JQ1 induced cell cycle arrest at G0/G1 phase (Figure 1C). Between both compounds used, JQ1 showed the stronger antiproliferative effect, at both lower (0.5 mM) and higher doses (5 mM) (Figure 1).
Figure 1. . BET bromodomain inhibitors reduce Waldenström macroglobulinemia cell growth.
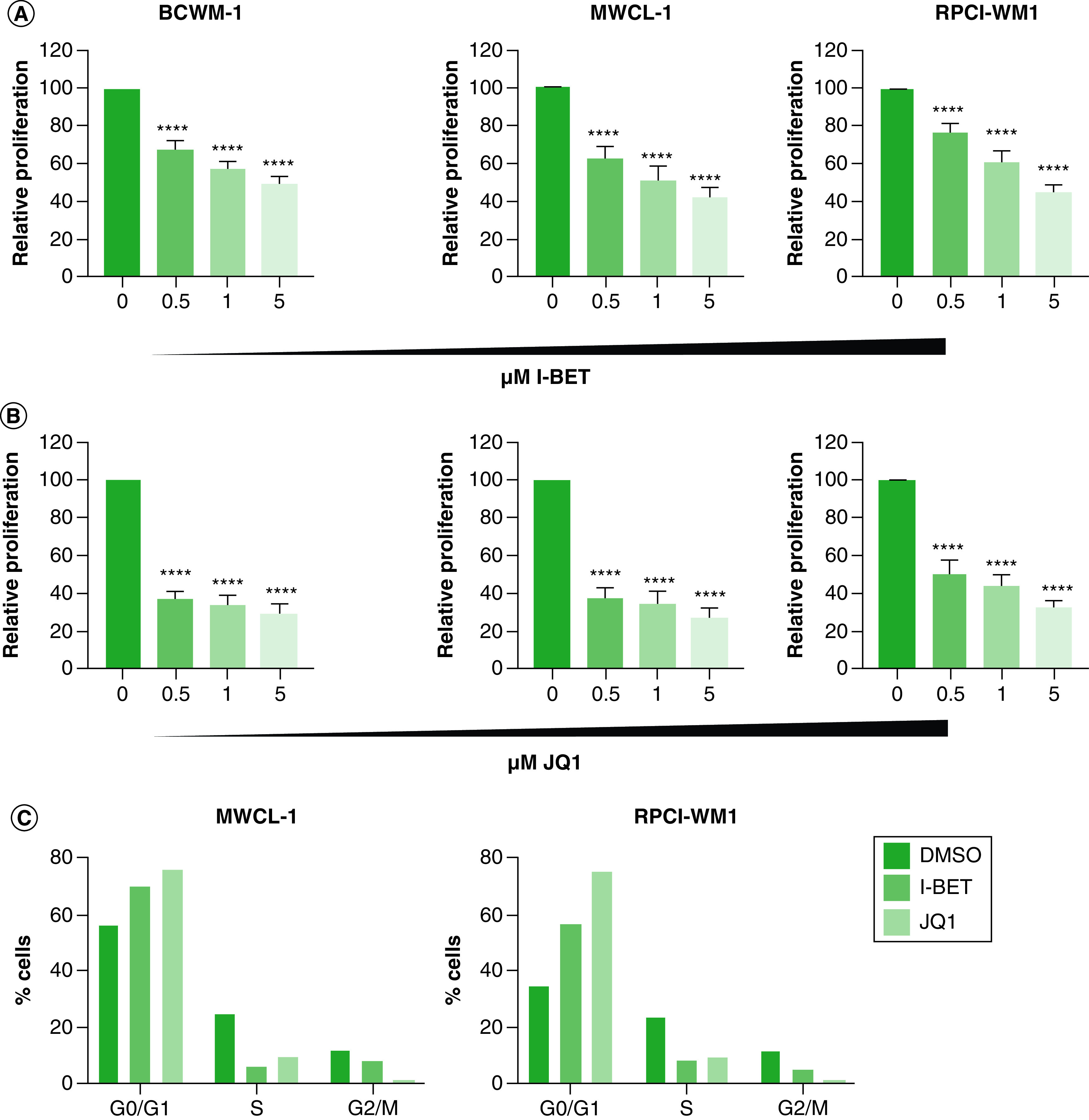
(A) WM cells (0.25 × 103) were treated with the indicated doses of iBET or DMSO control (0). (B) WM cells (0.25 × 103) were treated with the indicated doses of JQ1 or DMSO control. Cells were cultured at 37°C for 72 h, followed by the determination of cell proliferation using an XTT assay. Each experiment was repeated at least five-times. Bars represent the mean ± SE of at least three biological replicates of each experiment. (C) WM cells (0.5 × 106) were treated with 5 mM iBET, JQ1 or DMSO control for 72 h followed by cell-cycle analysis as described in the Materials & methods section. This experiment was repeated twice with similar results and shown is a representative experiment.
****p < 0.0001.
DMSO: Dimethyl sulfoxide; iBET: I-BET-762; SM: Standard Error; WM: Waldenström macroglobulinemia.
The TME does not protect against BET inhibition
The TME plays an important role in the biology of many cancers, including WM [4,30–32]. In BM malignancies such as WM, BM stromal cells support the growth and survival of malignant cells and facilitate resistance to therapy [33]. Indeed, using SEM and confocal microscopy, we show that WM B cells associate with HS-5 stromal cells in coculture (Figure 2A). To determine if the TME can protect against BET inhibitor-mediated reduction in cell growth, we examined the role of the TME in BRD4-directed therapy in WM. We cocultured WM cell lines with human BM-derived stromal cells (HS-5) and then treated them with iBET and JQ1. As shown previously, cell proliferation was significantly reduced in both iBET (p < 0.0001) and JQ1 (p < 0.0001) treated WM cells in the absence of the TME. Interestingly, this effect was not compromised by the presence of the TME in BCWM.1 and MWCL-1 cells (Figure 2B). However, in RPCI-WM1 cells, the presence of the TME significantly reduced the effect of iBET (p = 0.0002) and JQ1 (p = 0.0009) treatments on growth inhibition. Although this effect was statistically significant, it was modest (5–10% increase in cell growth in the presence of the TME) (Figure 2). From our imaging experiments (Figure 2A), we were unable to image WM cells and stromal cells after treatment with BET inhibitors. This prompted us to examine the effect of BET inhibitors on BM stromal cell growth. We found that treatment of HS-5 cells with iBET or JQ1 significantly reduced HS-5 cell growth (Figure 2C) suggesting that BET inhibitors can target the TME in addition to malignant cells. Taken together, these results suggest that the presence of the TME (HS-5 cells) did not enhance WM cell proliferation in the presence of iBET or JQ1 potentially due to their reduced growth in the presence of BET inhibitors. Therefore, the TME is a target of BET inhibitors and therefore does not protect against BET inhibitor-mediated reduction in malignant cell growth.
Figure 2. . The tumor microenvironment does not protect against BET inhibitors.
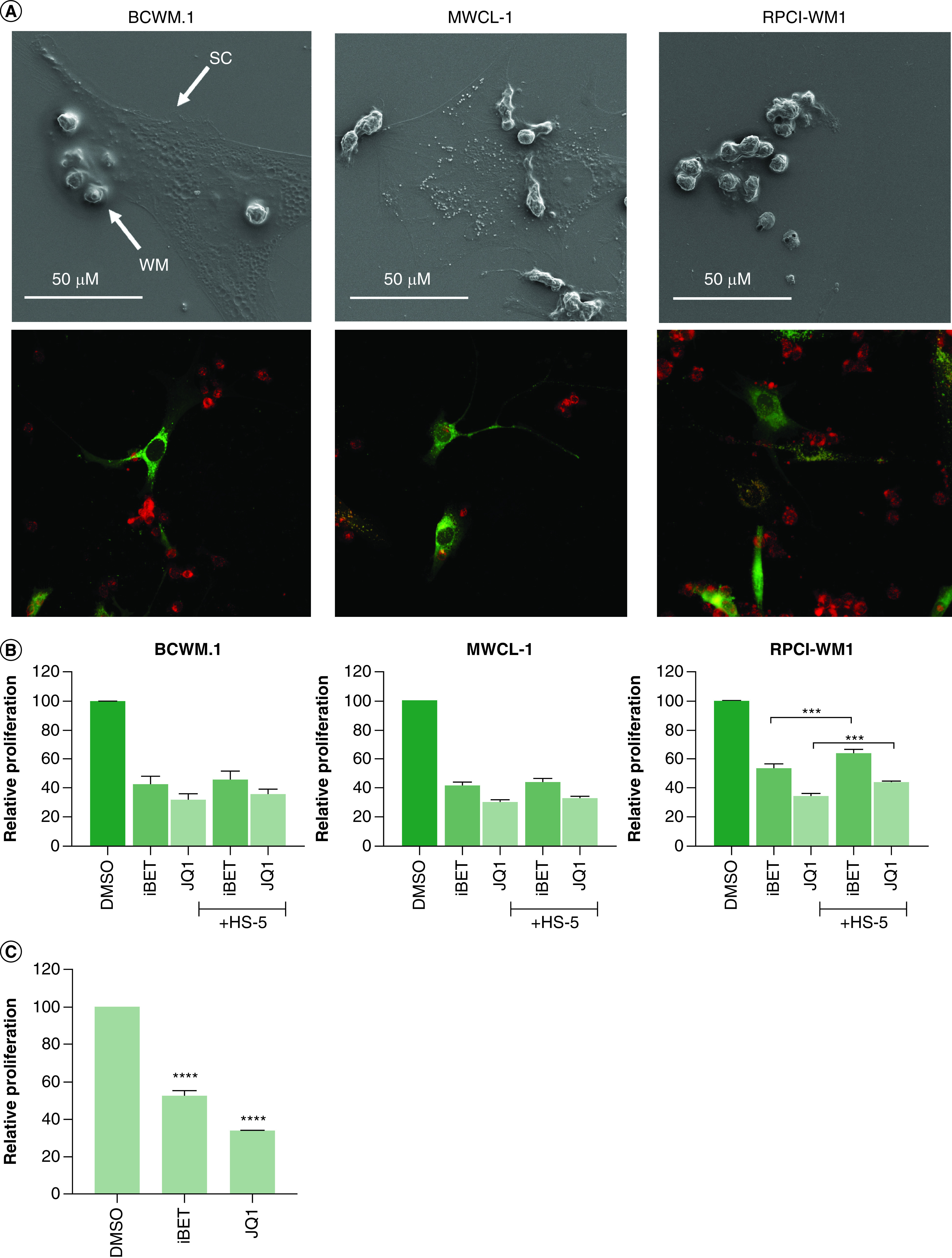
(A) HS-5 stromal cells (0.1 × 106) and WM cells (0.5 × 106) were co-cultured on coverslips as described in the Materials & methods section for 2 days. Coverslips were carefully removed and used for imaging using SEM (top panel) or confocal microscopy (lower panel). (B) Bone marrow stromal cells (HS-5; 0.1 × 103) were plated in 96-well plates and allowed to adhere overnight. Cells were fixed using 10 µg/ml of mitomycin C for 3 h, followed by washing with DPBS. WM cells (0.5 × 103) were added to each well and treated with either 5 mM of iBET, JQ1 or DMSO control. Cells were incubated at 37°C for 72 h, and cell proliferation was determined by XTT assay, as described in Materials & methods section. (C) HS-5 cells (0.25 × 103) were treated with 5 mM iBET, 5 mM JQ1 or DMSO control for 72 h, followed by the determination of cell proliferation using an XTT assay. These experiments were repeated at least three-times. Bars represent the mean ± SE of at least three biological replicates of each experiment.
***p < 0.001; ****p < 0.0001.
DMSO: Dimethyl sulfoxide; DPBS: Dulbecco’s phosphate-buffered saline; iBET: I-BET-762; SC: Stromal cells; SE: Standard error; WM: Waldenström macroglobulinemia.
BET inhibition has a modest effect on cell survival
While a reduction in cell proliferation is an effective therapeutic outcome, it would not provide a cure unless cell viability is reduced. Therefore, we investigated the effect of BET inhibitors on WM cell survival. We found a significant increase (one to threefold) in the percent of cells with positive staining for annexin-V, suggesting that iBET and/or JQ1 may kill WM cells (Figure 3A). However, when we examined the viable cells (annexin-V negative/PI negative), although there was a significant reduction in cell viability (Figure 3B), approximately 70–90% of WM cells remained viable after treatment with iBET/JQ1. Using primary cells from WM patients, we found a similar effect of JQ1 on cell viability (Figure 3C). Treatment of total BM MNCs with JQ1 induced a dose-dependent increase in annexin-V-positive CD19+ cells (Figure 3D). This also resulted in a reduction in cell viability in both patient samples used; however, the majority of CD19+ cells remained viable (∼80% for WM1 and 70% for WM2) (Figure 3E). Interestingly, WM1 was previously treated with rituximab while WM2 was previously untreated; suggesting the effects of JQ1 are independent of treatment status. Consistent with the effect of BET inhibitors on cell proliferation, we found a significant reduction in MYC mRNA expression in WM cells treated with iBET/JQ1 (Figure 4A). This also resulted in decreased MYC protein expression upon treatment with iBET (Figure 4B). To understand why a majority of cells were viable following iBET/JQ1 treatment, we examined the expression of the anti-apoptotic protein BCL-2. There was an increase in BCL-2 protein levels in cells treated with iBET/JQ1 (Figure 4C). Taken together, these data suggest that BET inhibitors reduce cell proliferation via regulation of MYC while increasing BCL-2 levels, which partially protects the cells against apoptotic death in response to these inhibitors.
Figure 3. . BET inhibitors induce modest cell death.
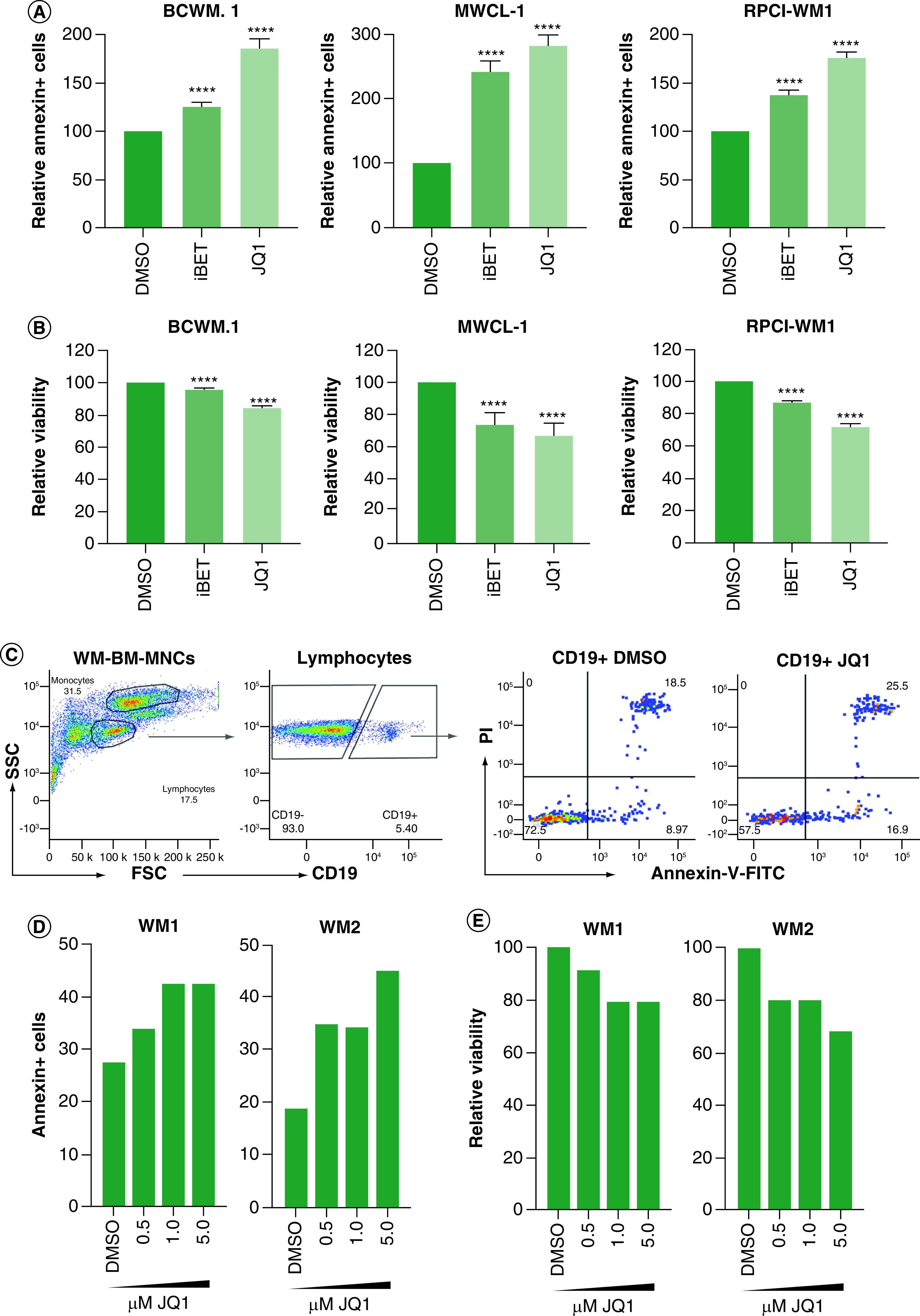
WM cells (0.5 × 106) were treated with 5 mM of iBET or JQ1 or DMSO control for 72 h. Cells were harvested and viability was determined by annexin-V/PI staining. (A) Percent annexin-V-positive cells were determined by flow cytometry. (B) Percent viable cells as determined by flow cytometry. Each experiment was repeated at least three-times. Bars represent the mean ± SE of all experiments. (C) MNCs (2 × 106 cells) from BM biopsy specimens from WM patients were treated with the indicated doses of JQ1 or DMSO control as described in Materials & methods section. Cells were harvested and viability was determined by annexin-V/PI and CD19 staining. Gating was done on lymphocytes from total MNCs followed by gating on CD19+ cells. Annexin-V/PI was analyzed on CD19+ cells. (D) Annexin-V-positive cells were determined by flow cytometry in two WM patient samples (WM1 and WM2). (E) Percent viable cells were determined for primary WM patient samples.
****p < 0.0001.
BM: Bone marrow; DMSO: Dimethyl sulfoxide; iBET: I-BET-762; MNC: Mononuclear cell; PI: Propidium iodide; SE: standard error; WM: Waldenström macroglobulinemia.
Figure 4. . BET inhibitors reduce MYC but induce BCL-2 expression.

(A) WM cells (2 × 106) were treated with 5 mM of either iBET or JQ1 or DMSO control for 24 h. Cells were harvested and RNA was extracted and used to determine MYC expression by qRT-PCR. This experiment was repeated at least three-times and data were presented as mean ± SE of all experiments. (B) WM cells (5 × 106) were treated with either 5 mM of iBET, JQ1 or DMSO control for 24 h. Cells were harvested and lysed and lysates were used to determine protein levels by western blot. (C) WM cells (5 × 106) were treated with either 5 mM of iBET, JQ1 or DMSO control for 24 h. Cells were lysed and lysates were used to determine BCL-2 protein levels by Western blot. Western blot experiments were repeated at least three-times with similar results.
DMSO: Dimethyl sulfoxide; iBET: I-BET-762; SE: Standard error; WM: Waldenström macroglobulinemia.
Reduced IgM expression & secretion in response to iBET/JQ1
Because WM is characterized by the overproduction of a monoclonal IgM protein which is associated with significant disease symptoms, we examined the effect of BET inhibitors on IgM levels [34]. In WM cells treated with iBET/JQ1 for 24 h, we found a significant decrease in IgM expression by qRT-PCR (Figure 5A). This decrease in IgM mRNA levels resulted in a decrease in IgM secretion in WM cells treated with BET inhibitors (Figure 5B).
Figure 5. . BET inhibitor reduces IgM levels.
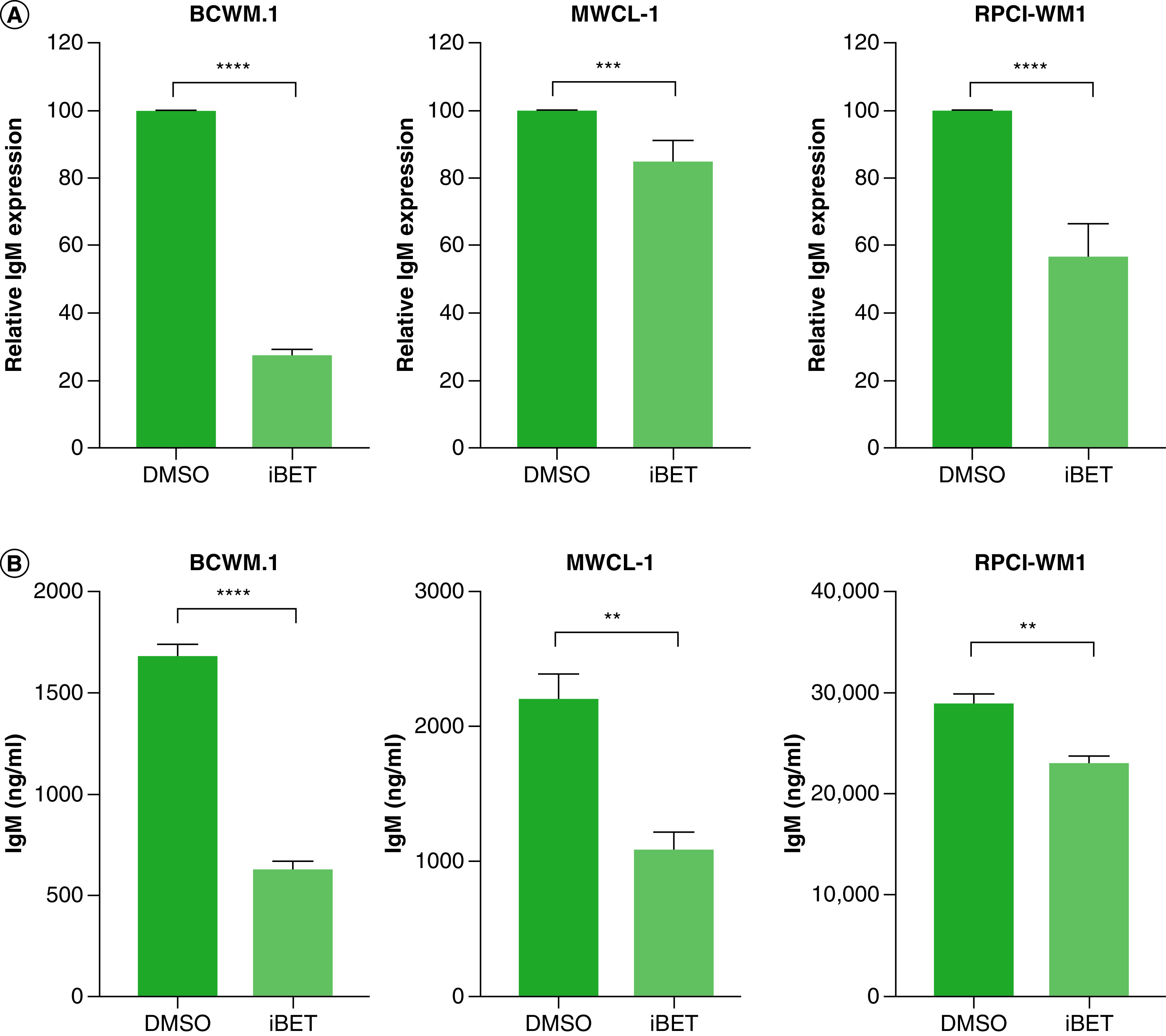
(A) WM cells (2 × 106) were treated with 5 mM of iBET or DMSO control for 24 h. Cells were harvested and RNA was extracted and used in qRT-PCR to determine IgM expression. (B) WM cells (2 × 106) were treated with either 5 mM iBET or DMSO control for 72 h. Supernatant was used to quantify IgM secretion by ELISA. Each experiment was repeated at least three-times. Bars represent the mean ± SE of all experiments.
**p < 0.01; ***p < 0.001; ****p < 0.0001.
DMSO: Dimethyl sulfoxide; iBET: I-BET-762; qRT: quantitative reverse-transcription; SE: Standard error; WM: Waldenström macroglobulinemia.
Combined treatment with BET inhibitors synergizes with HDAC & BCL2 but not BTK inhibition in WM
We hypothesized that combining targeting of BRD4 with BCL-2 therapy may overcome the resistance to apoptosis observed in WM cells treated with iBET/JQ1 (Figure 3B). Therefore, we treated WM cells with either 5 mM JQ1, 5 mM venetoclax (ABT-199) or a combination of both and examined the effect on WM cell survival. We found a significant increase in annexin-V-positive cells in cells treated with a combination of JQ1 and ABT-199 compared with either treatment alone (Figure 6A). This also resulted in a significantly lower viable cell population in cells treated with both JQ1 and ABT-199 compared with either drug along (Figure 6A); additionally, there was a moderate synergistic effect in BCWM.1 and MWCL-1 cells and a synergistic effect in RPCI-WM1 cells (Table 1). Despite this improved killing of WM cells, 40–60% of the cells remained viable after treatment with both drugs.
Figure 6. . Combined targeting of BET proteins and BCL-2/histone deacetylase enhances cell death in Waldenström macroglobulinemia cells.

(A & B) WM cells (0.5 × 106) were treated with either 500 nM of ABT-199, 5 mM of JQ1, a combination of venetoclax (ABT-199) and JQ1, or DMSO control for 72 h. Cells were harvested and stained with annexin-V/PI followed by data acquisition using flow cytometry. Annexin-V-FITC positive cells are shown in (A) and viable cells are shown in (B). (C & D) WM cells (0.5 × 106) were treated with either 25 nM of panobinostat (LBH589), 5 mM of JQ1, a combination of LBH589 and JQ1, or DMSO control for 72 h. Cells were harvested and stained with annexin-V/PI followed by data acquisition using flow cytometry. Annexin-V-FITC positive cells are shown in (C) and viable cells are shown in (D). Each experiment was repeated at least three-times. Bars represent the mean ± SE of all experiments.
****p < 0.0001.
DMSO: Dimethyl sulfoxide; PI: Propidium iodide; SE: Standard error; WM: Waldenström macroglobulinemia.
Panobinostat (LBH589) is a pan HDAC inhibitor that was shown to be effective in WM at very low concentrations [5]. Therefore, we examined the efficacy of combined targeting of BET and HDAC proteins in WM. Cells were treated with either 5 mM JQ1 or 25 nM panobinostat (LBH589) alone or a combination of both and examined for the effect on WM cell survival. At 25 nM of LBH589, there was no effect on apoptotic cells or overall cell viability (Figure 6B). However, combined treatment of WM cells with JQ1 and LBH589 significantly increased apoptotic cells and decreased cell viability compared with cells treated with JQ1 alone (Figure 6B). A calculation of CDI indicated that this drug combination was synergistic in all cells (Table 1). Furthermore, cell viability was reduced to 30–40%, suggesting that combined targeting of BET proteins and HDAC may provide therapeutic efficacy in WM.
To date, the only FDA-approved therapy for WM is ibrutinib, a BTK inhibitor. Ibrutinib is also approved for chronic lymphocytic leukemia (CLL) and mantle cell lymphoma [35–37]. To examine the role of ibrutinib in combination with BET inhibition, we treated WM cells with either 5 mM JQ1, 5 mM ibrutinib or a combination of both and examined the effect on WM cell survival. Interestingly, ibrutinib treatment alone had no effect on cell viability (Figure 7A). However, in BCWM.1 and MWCL-1 cells (but not in RPCI-WM1 cells), the addition of ibrutinib to JQ1 therapy induced a moderate synergistic effect that led to enhanced killing of WM cells. However, approximately 70% of WM cells remained viable, suggesting that ibrutinib may not be the optimal drug in combination with JQ1. Since ibrutinib works well as a therapeutic option in WM patients [37], we examined the effect of ibrutinib/JQ1 combination on cell proliferation. Indeed, ibrutinib alone induced a 20% reduction in cell proliferation in BCWM.1 and MWCL-1 cells (Figure 7B). Furthermore, combined treatment of WM cells with ibrutinib and JQ1 resulted in a significant decrease in cell proliferation, although this effect was moderately synergistic in BCWM.1 and RPCI-WM1 cells and additive in MWCL-1 cells (Table 1).
Figure 7. . Combined targeting of BET proteins and Bruton’s tyrosine kinase has a modest effect on cell viability in Waldenström macroglobulinemia cells.
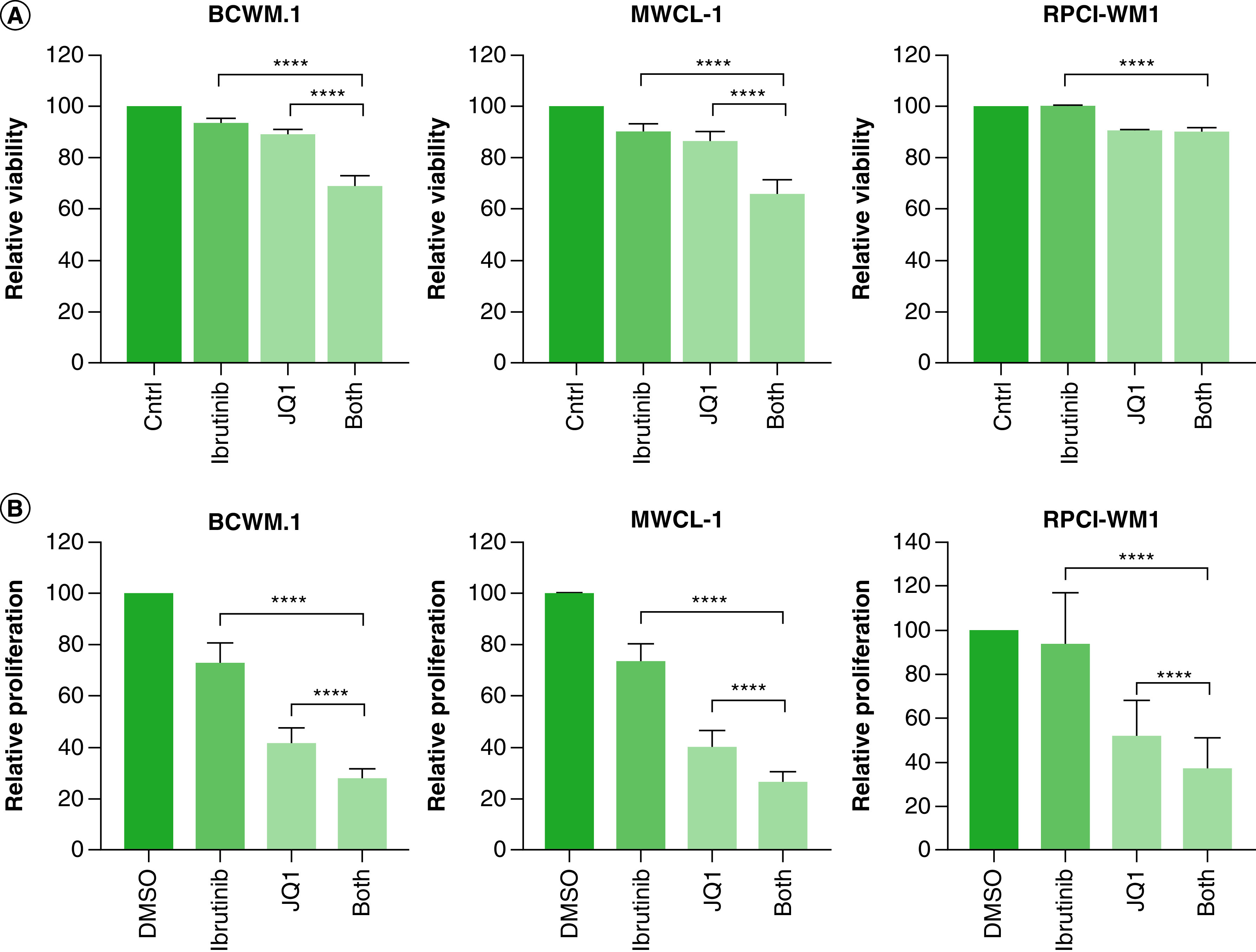
(A) WM cells (0.5 × 106) were treated with either 5 mM of ibrutinib, 5 mM of JQ1, combination of ibrutinib and JQ1 or DMSO control for 72 h, followed by determination of cell viability by annexin-V/PI double staining. (B) WM cells (0.25 × 103) were treated with either 5 mM of ibrutinib, 5 mM of JQ1, combination of ibrutinib and JQ1 or DMSO control for 72 h, followed by the determination of cell proliferation using an XTT assay as described in Materials & methods section. Each experiment was repeated at least three-times. Bars represent the mean ± SE of all experimental.
****p < 0.0001.
DMSO: Dimethyl sulfoxide; PI: Propidium iodide; SE: Standard error; WM: Waldenström macroglobulinemia.
Taken together, these results suggest that combined treatment with JQ1/ABT-199 overcomes the increase in BCL-2 observed in JQ1 treated cells. However, combined treatment with JQ1/LBH589 provides a synergistic effect on cell survival and therefore may provide therapeutic efficacy in WM patients.
Discussion
It is well established that cancer cell development and maintenance is associated with DNA modification and epigenetic abnormalities. In the past decade, the reversible nature of these modifications makes their epigenetic targeting an attractive therapeutic option. However, there is very limited investigation into the efficacy of epigenetic therapies in WM. To this point, ibrutinib is the only FDA-approved therapy for WM patients. However, this BTK inhibitor does not target epigenetic modifications and the heterogeneous nature of the disease leads to resistance of ibrutinib, which makes evaluating new therapeutic approaches essential for WM [3]. In this study, we show the therapeutic potential of treating of WM cells with a combination of BET inhibitors and either a BCL-2 inhibitor or an HDAC inhibitor, where synergistic activity was observed with the addition of HDAC inhibition to BET inhibition.
Bromodomain containing proteins have been studied in different B-cell malignancies [17,38,39]. Their function received attention due to their indirect effect on MYC gene transcriptional regulation [40]. The human c-MYC gene is upregulated in a majority of cancers including lymphomas [41–44] and is involved in multiple cellular functions including cell proliferation, cellular metabolism, DNA damage response and translation machinery [45]. BET inhibitors function as small-molecules for targeting MYC expression and their inhibition of cell proliferation has been reported in several lymphomas including DLBCL and multiple myeloma [17,18,46]. In the current study, we show an antiproliferative effect of iBET and JQ1 in WM cells (Figure 1) wherein these molecules displayed a moderate effect on WM cell survival (Figure 3). As expected, we saw a decrease in MYC expression, which is associated with the antiproliferative effect. However, the antiproliferative effect of BET inhibitors was associated with an increase in the anti-apoptotic protein BCL-2 (Figure 4). This is likely why BET inhibition had a modest effect on WM cell viability (Figure 3). Inhibition of BCL-2 has previously been shown to reduce WM cell viability [47], and clinical studies using the BCL-2 inhibitor ABT-199 showed major responses in three out of four WM patients [48]. Indeed, combined targeting of WM cells with JQ1 and ABT-199 reduced cell viability beyond that observed by either drug alone (Figure 6). However, when we examined the synergy between the two drugs using CDI, we only observed a moderate synergy in two out of three WM cell lines (CDI 0.594–0.721) (Table 1). These results are similar to those observed in DLBCL, where combined treatment of BET inhibitors and ABT-199 significantly enhanced apoptotic death in DLBCL cell lines [18]. Furthermore, we recently reported the efficacy of combined treatment of double-hit and triple-hit DLBCL with BET and BCL-2 inhibitors [18]. Consistent with our results, combined treatment of CLL cells with JQ1 and venetoclax synergistically induced apoptosis; however, in CLL, JQ1 treatment did not change BCL-2 protein levels [49]. Despite this, the synergy between BET inhibitors and BCL-2 inhibitors appears to be an effective therapeutic option for B-cell malignancies, including WM.
We also showed that BET inhibitors significantly reduce IgM at both the transcriptional level and secretion in WM (Figure 5), which suggests a role for bromodomain-containing proteins in immunoglobulin gene regulation. This finding is consistent with a report showing that JQ1 significantly reduced immunoglobulin gene expression in human B cells through an Oct-2-dependent mechanism [50]. Overall, these findings warrant further investigation into the mechanisms by which bromodomain-containing proteins regulate immunoglobulin at expression in WM and in similar diseases.
The TME plays an important role in the initiation and progression of cancer. In the presence of the TME, malignant B cells are more resistant to therapy and have increased growth and survival [51–57]. Therefore, it is important to evaluate the role of the TME when evaluating novel therapies in WM. Here, we show that the efficacy of iBET and JQ1 in WM was not compromised by the presence of the TME. Interestingly, we found that BET inhibitors reduced HS-5 cell proliferation, therefore, providing a mechanism for the lack of support of WM cells from BET-inhibitor therapy. This suggests that despite the crosstalk between the TME and WM cells [26,29,58,59], BET inhibition might provide a broad influence on the epigenetic regulatory elements of both tumor cells and cells in TME.
Because bromodomain-containing proteins are the readers of post-transcriptional modifications, they are known for both their recognition of acetylated histones as well as their ability to recruit the other proteins needed for transcription initiation and eventually gene expression regulation [60]. Therefore, the interplay between bromodomain proteins and HDAC inhibitors has gained attention as a potential combination therapy for cancer. The synergistic effect of HDAC inhibitors and BET inhibitors on the induction of cell cycle arrest and apoptosis in malignant cells has been demonstrated [61,62]. HDAC inhibitors have been examined in WM and were found to induce significant preclinical activity both as monotherapy and in combination with the proteasome inhibitor bortezomib [5]. We found that integrating HDAC inhibition to BET inhibition significantly increased the efficacy of JQ1-induced apoptosis in WM. These findings are consistent with the synergistic apoptotic effect of JQ1 and panobinostat on acute myeloid leukemia blast progenitor cells, although, in this study, JQ1 treatment led to a reduction in both c-MYC and BCL-2 expression [62]. In comparison with the combination of BET inhibition with BCL-2 inhibition, the synergistic effect of JQ1 and HDAC inhibition was more pronounced than the inhibitory effect of JQ1 and BCL-2 inhibition alone. This was evidenced by fewer WM cells surviving at much lower doses (25 nM) of LBH-589. This suggests that this combination may be effective at a lower dose, thereby reducing potential side effects on patients.
Since ibrutinib is the only approved therapy for WM patients, we investigated the potential synergistic effect of JQ1 and ibrutinib in WM. Although ibrutinib has shown efficacy in WM patients [37], at 5 mM, there was no effect on cell survival and a limited effect on cell proliferation; RPCI-WM1 cells were the most resistant to ibrutinib (Figure 7). In combination with JQ1, ibrutinib only resulted in moderate to slight synergy (Table 1). In mantle cell lymphoma, another B cell malignancy in which ibrutinib is approved, a synergic effect of JQ1 and ibrutinib was reported [63]. Therefore, in WM patients undergoing ibrutinib therapy, the addition of JQ1 may enhance the efficacy of ibrutinib; however, epigenetic targeting is likely to provide a better therapeutic outcome.
Conclusion
In summary, this is the first study to report the efficacy of BET inhibitors in WM. We examined different combinations with BET inhibitors and found that combined targeting of WM cells with JQ1 and ibrutinib is less effective than JQ1 and panobinostat or JQ1 and venetoclax. These results provide a framework to examine BET inhibitors as monotherapy and in combination with BCL-2 or HDAC inhibitors in WM.
Summary points.
Waldenström macroglobulinemia (WM) is a B cell lymphoma which is characterized by overexpression of monoclonal IgM.
To date therapy for WM is limited with ibrutinib being the only US FDA approved drug for treatment.
Targeting epigenetic regulators by inhibiting BET proteins increased therapeutic efficacy and showed high success rate in many types of cancer; however, studies to show therapeutic success in WM cells are still lacking.
Treatment of WM cell lines with JQ1 and I-BET-762 showed high anti-proliferative and pro-apoptotic effect which was even amplified when cells were treated with panobinostat or venetoclax concluding synergy.
This study provides insights for new therapeutic targets in WM.
Acknowledgments
The FACS Calibur used is managed by the University Instrumentation Center (UIC) at the University of New Hampshire (UNH) and was purchased by the UIC. The Tescan Lyra3 GMU field-emission scanning electron microscope/focused ion beam (FE-SEM/FIB) used is managed by the UIC at the UNH and was purchased with funds awarded to UNH from the US National Science Foundation (NSF) (MRI grant 1337897; T Gross, Mechanical Engineering, UNH, PI) and funds from UNH. EDS/EBSD components on this system were purchased with startup funds from UNH at the discretion of M Knezevic (Mechanical Engineering, UNH).
Footnotes
Financial & competing interests disclosure
This research was supported by an NIH COBRE Center of Integrated Biomedical and Bioengineering Research (CIBBR, P20 GM113131) through an Institutional Development Award (IDeA) from the National Institute of General Medical Sciences. This work was supported in part by a grant from the International Waldenström Macroglobulinemia Foundation and the Leukemia & Lymphoma Society (IWMF-LLS). The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
Ethical conduct of research
The authors state that they have obtained appropriate institutional review board approval or have followed the principles outlined in the Declaration of Helsinki for all human or animal experimental investigations. In addition, for investigations involving human subjects, informed consent has been obtained from the participants involved.
References
Papers of special note have been highlighted as: • of interest; •• of considerable interest
- 1.Kapoor P, Paludo J, Vallumsetla N, Greipp PR. Waldenström macroglobulinemia: what a hematologist needs to know. Blood Rev. 29(5), 301–319 (2015). [DOI] [PubMed] [Google Scholar]; •• Discusses aspects of Waldenström macroglobulinemia (WM) disease.
- 2.Ansell SM, Kyle RA, Reeder CB et al. Diagnosis and management of Waldenström macroglobulinemia: mayo stratification of macroglobulinemia and risk-adapted therapy (mSMART) guidelines. Mayo Clin. Proc. 85(9), 824–833 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Treon SP, Gustine J, Meid K et al. Ibrutinib monotherapy in symptomatic, treatment-naïve patients with Waldenström macroglobulinemia. J. Clin. Oncol. 36(27), 2755–2761 (2018). [DOI] [PubMed] [Google Scholar]; • Discusses the role of ibrutinib in WM.
- 4.Han W, Matissek SJ, Jackson DA, Sklavanitis B, Elsawa SF. Targeting IL-6 receptor reduces IgM levels and tumor growth in Waldenström macroglobulinemia. Oncotarget 10(36), 3400–3407 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]; • Highlights the efficacy of targeting the tumor microenvironment.
- 5.Sun JY, Xu L, Tseng H et al. Histone deacetylase inhibitors demonstrate significant preclinical activity as single agents, and in combination with bortezomib in Waldenström’s macroglobulinemia. Clin. Lymphoma Myeloma Leuk. 11, 152-156 (2011). [DOI] [PubMed] [Google Scholar]; • Efficacy of epigenetic targeting in WM using histone deacetylase inhibitors.
- 6.De Weerdt I, Koopmans SM, Kater AP, Van Gelder M. Incidence and management of toxicity associated with ibrutinib and idelalisib: a practical approach. Haematologica 102(10), 1629–1639 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tate CR, Rhodes LV, Segar HC et al. Targeting triple-negative breast cancer cells with the histone deacetylase inhibitor panobinostat. Breast Cancer Res. 14(3), 1–15 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dawson MA, Kouzarides T, Huntly BJP. Targeting epigenetic readers in cancer. N. Engl. J. Med. 367(7), 647–657 (2012). [DOI] [PubMed] [Google Scholar]
- 9.Shi J, Vakoc CR. The Mechanisms behind the therapeutic activity of BET bromodomain inhibition. Mol. Cell 54(5), 728–736 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu S, Li F, Pan L et al. BRD4 inhibitor and histone deacetylase inhibitor synergistically inhibit the proliferation of gallbladder cancer in vitro and in vivo. Cancer Sci. 110(8), 2493–2506 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kim SR, Lewis JM, Cyrenne BM et al. BET inhibition in advanced cutaneous T cell lymphoma is synergistically potentiated by BCL2 inhibition or HDAC inhibition. Oncotarget 9(49), 29193–29207 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nicodeme E, Jeffrey KL, Schaefer U et al. Suppression of inflammation by a synthetic histone mimic. Nature 468(7327), 1119–1123 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Filippakopoulos P, Qi J, Picaud S et al. Selective inhibition of BET bromodomains. Nature 468(7327), 1067–1073 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Puissant A, Frumm SM, Alexe G et al. Targeting MYCN in neuroblastoma by BET bromodomain inhibition. Cancer Discov. 3(3), 309–323 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]; •• MYC as a target of BET inhibitors.
- 15.Cheng Z, Gong Y, Ma Y et al. Inhibition of BET bromodomain targets genetically diverse glioblastoma. Clin. Cancer Res. 19(7), 1748–1759 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Asangani IA, Dommeti VL, Wang X et al. Therapeutic targeting of BET bromodomain proteins in castration-resistant prostate cancer. Nature 510(7504), 278–282 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Trabucco SE, Gerstein RM, Evens AM et al. Inhibition of bromodomain proteins for the treatment of human diffuse large B-cell lymphoma. Clin. Cancer Res. 21(1), 113–122 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li W, Gupta SK, Han W et al. Targeting MYC activity in double-hit lymphoma with MYC and BCL2 and/or BCL6 rearrangements with epigenetic bromodomain inhibitors. J. Hematol. Oncol. 12(1), 1–13 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]; •• MYC as a target of BET inhibitors in diffuse large B-cell lymphoma.
- 19.Ditzel Santos D, Ho AW, Tournilhac O et al. Establishment of BCWM.1 cell line for Waldenström’s macroglobulinemia with productive in vivo engraftment in SCID-hu mice. Exp. Hematol. 35(9), 1366–1375 (2007). [DOI] [PubMed] [Google Scholar]
- 20.Hodge LS, Novak AJ, Grote DM et al. Establishment and characterization of a novel Waldenström macroglobulinemia cell line, MWCL-1. Blood 117(19), 190–197 (2011). [DOI] [PubMed] [Google Scholar]
- 21.Chitta KS, Paulus A, Ailawadhi S et al. Development and characterization of a novel human Waldenström macroglobulinemia cell line: RPCI-WM1, Roswell Park Cancer Institute- Waldenström Macroglobulinemia 1. Leuk. Lymphoma 54(2), 387–396 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Han W, Jackson DA, Matissek SJ et al. Novel molecular mechanism of regulation of CD40 ligand by the transcription factor GLI2. J. Immunol. 198(11), 4481–4489 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jackson DA, Smith TD, Amarsaikhan N et al. Modulation of the IL-6 receptor α underlies GLI2-mediated regulation of Ig secretion in Waldenström macroglobulinemia cells. J. Immunol. 195(6), 2908–2916 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Han W, Ibarra G, Gupta M, Yin Y, Elsawa SF. Elevated GLI3 expression in germinal center diffuse large B cell lymphoma. Leuk. Lymphoma 59(11), 2743–2745 (2018). [DOI] [PubMed] [Google Scholar]
- 25.Duvvuru MK, Han W, Chowdhury PR, Vahabzadeh S, Sciammarella F, Elsawa SF. Bone marrow stromal cells interaction with titanium; Effects of composition and surface modification. PLoS ONE 14(5), e0216087 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Elsawa SF, Novak AJ, Ziesmer SC et al. Comprehensive analysis of tumor microenvironment cytokines in Waldenstrom macroglobulinemia identifies CCL5 as a novel modulator of IL-6 activity. Blood 118(20), 5540–5549 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Treon SP, Xu L, Yang G et al. MYD88 L265P somatic mutation in Waldenström’s macroglobulinemia. N. Engl. J. Med. 367(9), 826–833 (2012). [DOI] [PubMed] [Google Scholar]
- 28.Xu L, Hunter ZR, Yang G et al. MYD88 L265P in Waldenström macroglobulinemia, immunoglobulin M monoclonal gammopathy, and other B-cell lymphoproliferative disorders using conventional and quantitative allele-specific polymerase chain reaction. Blood 121(11), 2051–2058 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Elsawa SF, Almada LL, Ziesmer SC et al. GLI2 transcription factor mediates cytokine cross-talk in the tumor microenvironment. J. Biol. Chem. 286(24), 21524–21534 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Leleu X, Jia X, Runnels J et al. The Akt pathway regulates survival and homing in Waldenstrom macroglobulinemia. Blood 110(13), 4417–4426 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jalali S, Ansell SM. Bone marrow microenvironment in Waldenstrom’s macroglobulinemia. Best Pract. Res. Clin. Haematol. 29(2), 148–155 (2016). [DOI] [PubMed] [Google Scholar]
- 32.Roccaro AM, Sacco A, Husu EN et al. Dual targeting of the PI3K/Akt/mTOR pathway as an antitumor strategy in Waldenstrom macroglobulinemia. Blood 115(3), 559–569 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Agarwal A, Ghobrial IM. The bone marrow microenvironment in Waldenström macroglobulinemia. Clin. Lymphoma Myeloma Leuk. 13(2), 218–221 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dimopoulos MA, Galani E, Matsouka C. Waldenstrom’s macroglobulinemia. Hematol. Oncol. Clin. North Am. 13(6), 1351–1366 (1999). [DOI] [PubMed] [Google Scholar]
- 35.Byrd JC, Furman RR, Coutre SE et al. Targeting BTK with ibrutinib in relapsed chronic lymphocytic leukemia. N. Engl. J. Med. 369(1), 32–42 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang ML, Rule S, Martin P et al. Targeting BTK with ibrutinib in relapsed or refractory mantle-cell lymphoma. N. Engl. J. Med. 369(6), 507–516 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Treon SP, Tripsas CK, Meid K et al. Ibrutinib in previously treated Waldenström’s macroglobulinemia. N. Engl. J. Med. 372(15), 1430–1440 (2015). [DOI] [PubMed] [Google Scholar]
- 38.Dawson MA, Prinjha RK, Dittmann A et al. Inhibition of BET recruitment to chromatin as an effective treatment for MLL-fusion leukaemia. Nature 478(7370), 529–533 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ott CJ, Kopp N, Bird L et al. BET bromodomain inhibition targets both c-Myc and IL7R in high-risk acute lymphoblastic leukemia. Blood 120(14), 2843–2852 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Delmore JE, Issa GC, Lemieux ME et al. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell 146(6), 904–917 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]; •• MYC as a target of BET inhibitors.
- 41.Nguyen L, Papenhausen P, Shao H. The role of c-MYC in B-cell lymphomas: diagnostic and molecular aspects. Genes (Basel) 8(4), 2–22 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Escot C, Theillet C, Lidereau R et al. Genetic alteration of the c-myc protooncogene (MYC) in human primary breast carcinomas. Proc. Natl Acad. Sci. USA 83(13), 4834–4838 (1986). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jenkins RB, Qian J, Lieber MM, Bostwick DG. Detection of c-myc oncogene amplification and chromosomal anomalies in metastatic prostatic carcinoma by fluorescence in situ hybridization. Cancer Res. 57(3), 524–531 (1997). [PubMed] [Google Scholar]
- 44.Erisman MD, Rothberg PG, Diehl RE, Morse CC, Spandorfer JM, Astrin SM. Deregulation of c-myc gene expression in human colon carcinoma is not accompanied by amplification or rearrangement of the gene. Mol. Cell. Biol. 5(8), 1969–1976 (1985). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Miller DM, Thomas SD, Islam A, Muench D, Sedoris K. c-Myc and cancer metabolism. Clin. Cancer Res. 18(20), 5546–5553 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Matthews GM, Gandolfi S, Bruggentheis J et al. BET bromodomain degradation as a therapeutic strategy in multiple myeloma. Blood 128(22), 1062–1062 (2016). [Google Scholar]
- 47.Cao Y, Hunter ZR, Liu X et al. CXCR4 WHIM-like frameshift and nonsense mutations promote ibrutinib resistance but do not supplant MYD88L265P-directed survival signalling in Waldenström macroglobulinaemia cells. Br. J. Haematol. 168(5), 701–707 (2015). [DOI] [PubMed] [Google Scholar]
- 48.Davids MS, Seymour JF, Gerecitano JF et al. Phase I study of ABT-199 (GDC-0199) in patients with relapsed/refractory non-hodgkin lymphoma: responses observed in diffuse large B-cell (DLBCL) and follicular lymphoma (FL) at higher cohort doses. Clin. Adv. Hematol. Oncol. 12(8), 18–19 (2014). [PubMed] [Google Scholar]
- 49.Carrà G, Nicoli P, Lingua MF et al. Inhibition of bromodomain and extra-terminal proteins increases sensitivity to venetoclax in chronic lymphocytic leukaemia. J. Cell. Mol. Med. 24(2), 1650–1657 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Shim JM, Lee JS, Russell KE et al. BET proteins are a key component of immunoglobulin gene expression. Epigenomics 9(4), 393–406 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Burger JA, Ghia P, Rosenwald A, Caligaris-Cappio F. The microenvironment in mature B-cell malignancies: a target for new treatment strategies. Blood 114(16), 3367–3375 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Eliopoulos N, Zhao J, Bouchentouf M et al. Human marrow-derived mesenchymal stromal cells decrease cisplatin renotoxicity in vitro and in vivo and enhance survival of mice post-intraperitoneal injection. Am. J. Physiol. Renal Physiol. 299(6), F1288-F1298 (2010). [DOI] [PubMed] [Google Scholar]
- 53.Lis R, Touboul C, Mirshahi P et al. Tumor associated mesenchymal stem cells protects ovarian cancer cells from hyperthermia through CXCL12. Int. J. Cancer 128(3), 715–725 (2011). [DOI] [PubMed] [Google Scholar]
- 54.Secchiero P, Zorzet S, Tripodo C et al. Human bone marrow mesenchymal stem cells display anti-cancer activity in SCID mice bearing disseminated non-Hodgkin’s lymphoma xenografts. PLoS ONE 5(6), e11140 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Trédan O, Galmarini CM, Patel K, Tannock IF. Drug resistance and the solid tumor microenvironment. J. Natl Cancer Inst. 99(19), 1441–1454 (2007). [DOI] [PubMed] [Google Scholar]
- 56.Villanueva J, Herlyn M. Melanoma and the tumor microenvironment. Curr. Oncol. Rep. 10(5), 439–446 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yañez R, Oviedo A, Aldea M, Bueren JA, Lamana ML. Prostaglandin E2 plays a key role in the immunosuppressive properties of adipose and bone marrow tissue-derived mesenchymal stromal cells. Exp. Cell Res. 316(19), 3109–3123 (2010). [DOI] [PubMed] [Google Scholar]
- 58.Elsawa S, Ansell S. Cytokines in the microenvironment of Waldenström’s macroglobulinemia. Clin. Lymphoma Myeloma. 9(1), 43–45 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Han W, Jackson DA, Matissek SJ et al. Novel molecular mechanism of regulation of CD40 ligand by the transcription factor GLI2. J. Immunol. 198(11), 4481–4489 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Taverna SD, Li H, Ruthenburg AJ, Allis CD, Patel DJ. How chromatin-binding modules interpret histone modifications: lessons from professional pocket pickers. Nat. Struct. Mol. Biol. 14(11), 1025–1040 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Shahbazi J, Liu PY, Atmadibrata B et al. The bromodomain inhibitor jq1 and the histone deacetylase inhibitor panobinostat synergistically reduce n-myc expression and induce anticancer effects. Clin. Cancer Res. 22(10), 2534–2544 (2016). [DOI] [PubMed] [Google Scholar]
- 62.Fiskus W, Sharma S, Qi J et al. Highly active combination of BRD4 antagonist and histone deacetylase inhibitor against human acute myelogenous leukemia cells. Mol. Cancer Ther. 13(5), 1142–1154 (2014). [DOI] [PubMed] [Google Scholar]
- 63.Sun B, Shah B, Fiskus W et al. Synergistic activity of BET protein antagonist-based combinations in mantle cell lymphoma cells sensitive or resistant to ibrutinib. Blood 126(13), 1565–1574 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]