

Abstract
Background
Gout is an inflammatory arthritis resulting from the deposition of monosodium urate crystals in and around joints. Non‐steroidal anti‐inflammatory drugs (NSAIDs) are commonly used to treat acute gout. This is an update of a Cochrane Review first published in 2014.
Objectives
To assess the benefits and harms of non‐steroidal anti‐inflammatory drugs (NSAIDs) (including cyclo‐oxygenase‐2 (COX‐2) inhibitors (COXIBs)) for acute gout.
Search methods
We searched the Cochrane Central Register of Controlled Trials (CENTRAL), MEDLINE, and Embase for studies to 28 August 2020. We applied no date or language restrictions.
Selection criteria
We considered randomised controlled trials (RCTs) and quasi‐RCTs comparing NSAIDs with placebo or another therapy for acute gout. Major outcomes were pain, inflammation, function, participant‐reported global assessment, quality of life, withdrawals due to adverse events, and total adverse events.
Data collection and analysis
We used standard methodological procedures as expected by Cochrane.
Main results
We included in this update 28 trials (3406 participants), including 5 new trials. One trial (30 participants) compared NSAIDs to placebo, 6 (1244 participants) compared non‐selective NSAIDs to selective cyclo‐oxygenase‐2 (COX‐2) inhibitors (COXIBs), 5 (712 participants) compared NSAIDs to glucocorticoids, 13 compared one NSAID to another NSAID (633 participants), and single trials compared NSAIDs to rilonacept (225 participants), acupuncture (163 participants), and colchicine (399 participants). Most trials were at risk of selection, performance, and detection biases.
We report numerical data for the primary comparison NSAIDs versus placebo and brief results for the two comparisons ‐ NSAIDs versus COX‐2 inhibitors and NSAIDs versus glucocorticoids.
Low‐certainty evidence (downgraded for bias and imprecision) from 1 trial (30 participants) shows NSAIDs compared to placebo. More participants (11/15) may have a 50% reduction in pain at 24 hours with NSAIDs than with placebo (4/15) (risk ratio (RR) 2.7, 95% confidence interval (CI) 1.1 to 6.7), with absolute improvement of 47% (3.5% more to 152.5% more). NSAIDs may have little to no effect on inflammation (swelling) after four days (13/15 participants taking NSAIDs versus 12/15 participants taking placebo; RR 1.1, 95% CI 0.8 to 1.5), with absolute improvement of 6.4% (16.8% fewer to 39.2% more). There may be little to no difference in function (4‐point scale; 1 = complete resolution) at 24 hours (4/15 participants taking NSAIDs versus 1/15 participants taking placebo; RR 4.0, 95% CI 0.5 to 31.7), with absolute improvement of 20% (3.3% fewer to 204.9% more). NSAIDs may result in little to no difference in withdrawals due to adverse events (0 events in both groups) or in total adverse events; two adverse events (nausea and polyuria) were reported in the placebo group (RR 0.2, 95% CI 0.0, 3.8), with absolute difference of 10.7% more (13.2% fewer to 38% more). Treatment success and health‐related quality of life were not measured.
Moderate‐certainty evidence (downgraded for bias) from 6 trials (1244 participants) shows non‐selective NSAIDs compared to selective COX‐2 inhibitors (COXIBs). Non‐selective NSAIDs probably result in little to no difference in pain (mean difference (MD) 0.03, 95% CI 0.07 lower to 0.14 higher), swelling (MD 0.08, 95% CI 0.07 lower to 0.22 higher), treatment success (MD 0.08, 95% CI 0.04 lower to 0.2 higher), or quality of life (MD ‐0.2, 95% CI ‐6.7 to 6.3) compared to COXIBs. Low‐certainty evidence (downgraded for bias and imprecision) suggests no difference in function (MD 0.04, 95% CI ‐0.17 to 0.25) between groups. Non‐selective NSAIDs probably increase withdrawals due to adverse events (RR 2.3, 95% CI 1.3 to 4.1) and total adverse events (mainly gastrointestinal) (RR 1.9, 95% CI 1.4 to 2.8).
Moderate‐certainty evidence (downgraded for bias) based on 5 trials (712 participants) shows NSAIDs compared to glucocorticoids. NSAIDs probably result in little to no difference in pain (MD 0.1, 95% CI ‐2.7 to 3.0), inflammation (MD 0.3, 95% CI 0.07 to 0.6), function (MD ‐0.2, 95% CI ‐2.2 to 1.8), or treatment success (RR 0.9, 95% CI 0.7 to 1.2). There was no difference in withdrawals due to adverse events with NSAIDs compared to glucocorticoids (RR 2.8, 95% CI 0.5 to 14.2). There was a decrease in total adverse events with glucocorticoids compared to NSAIDs (RR 1.6, 95% CI 1.0 to 2.5).
Authors' conclusions
Low‐certainty evidence from 1 placebo‐controlled trial suggests that NSAIDs may improve pain at 24 hours and may have little to no effect on function, inflammation, or adverse events for treatment of acute gout. Moderate‐certainty evidence shows that COXIBs and non‐selective NSAIDs are probably equally beneficial with regards to improvement in pain, function, inflammation, and treatment success, although non‐selective NSAIDs probably increase withdrawals due to adverse events and total adverse events. Moderate‐certainty evidence shows that systemic glucocorticoids and NSAIDs probably are equally beneficial in terms of pain relief, improvement in function, and treatment success. Withdrawals due to adverse events were also similar between groups, but NSAIDs probably result in more total adverse events. Low‐certainty evidence suggests no difference in inflammation between groups. Only low‐certainty evidence was available for the comparisons NSAID versus rilonacept and NSAID versus acupuncture from single trials, or one NSAID versus another NSAID, which also included many NSAIDs that are no longer in clinical use. Although these data were insufficient to support firm conclusions, they do not conflict with clinical guideline recommendations based upon evidence from observational studies, findings for other inflammatory arthritis, and expert consensus, all of which support the use of NSAIDs for acute gout.
Plain language summary
Non‐steroidal anti‐inflammatory drugs (NSAIDs) for acute gout
What is an acute gout flare, and what are NSAIDs?
Gout results from the deposition of monosodium urate crystals within and around joints and presents usually as self‐limited episodes of acute arthritis.
NSAIDs (non‐steroidal anti‐inflammatory drugs) are drugs that reduce pain and inflammation but may increase the risk of gastrointestinal ulcers and bleeding. Cyclo‐oxygenase (COX)‐2 selective inhibitors (COXIBs) are a subgroup of NSAIDs that lead to fewer stomach ulcers.
Study characteristics
This is an update of a Cochrane Review first published in 2014 and updated on 28 August 2020, which revealed 28 trials (3406 participants). Most participants were men (69% to 100%) aged 44 to 66 years, with acute gout lasting less than 48 hours.
One trial (30 participants) compared placebo to an NSAID, 13 trials (518 participants) compared an NSAID to another NSAID, 6 trials (1244 participants) compared NSAIDs to COXIBs, 5 trials (772 participants) compared glucocorticoids to NSAIDs, 1 trial compared interleukin‐1 inhibitors to NSAIDs (225 participants), 1 trial compared acupuncture plus infrared irradiation to NSAIDs (163 participants), and 1 trial compared colchicine to NSAIDs (399 participants). We present key results for the primary comparison, NSAIDs versus placebo, as follows.
Key results
NSAID versus placebo
Pain improvement by more than 50% after 24 hours
47 more people out of 100 who took NSAIDs reported more than 50% improvement in pain compared to those given placebo
‐ 73 people out of 100 who took NSAIDs reported more than 50% improvement in pain
‐ 26 people out of 100 who took placebo reported more than 50% improvement in pain
Swelling improvement by more than 50% after 24 hours
6 more people out of 100 who took NSAIDs had more than 50% improvement in swelling compared to those given placebo
‐ 86 people out of 100 who took NSAIDs reported more than 50% improvement in swelling
‐ 80 people out of 100 who took placebo reported more than 50% improvement in swelling
Function improvement at 24 hours
20 more people out of 100 who took NSAIDs had improvement in function compared to those given placebo
‐ 27 people out of 100 who took NSAIDs reported improvement in function
‐ 7 people out of 100 who took placebo reported improvement in function
Side effects
10 fewer people out of 100 who took NSAIDs reported side effects compared to those given placebo
‐ 3 people out of 100 who took NSAIDs had side effects
‐ 13 people out of 100 who took placebo had side effects
There were no withdrawals due to side effects.
Quality of the evidence
Low‐certainty evidence from 1 study suggests that NSAIDs may improve pain after 24 hours compared to placebo.
Moderate‐certainty evidence shows that NSAIDs probably were equal to COXIBs in reducing pain and inflammation but with more side effects, and NSAIDs probably are equally beneficial as glucocorticoids with regards to pain but are probably less beneficial with regards to swelling, with more side effects. Only low‐certainty evidence from single trials was available for the comparisons NSAID versus rilonacept and NSAID versus acupuncture, or one NSAID versus another NSAID, which included NSAIDs no longer in use.
Summary of findings
Summary of findings 1. NSAIDs compared to placebo for acute gout.
| NSAIDs compared to placebo for acute gout | ||||||
| Patient or population: acute gout Setting: Department of Rheumatology and Immunology of a general hospital Intervention: NSAIDs Comparison: placebo | ||||||
| Outcomes | Relative effect (95% CI) | Anticipated absolute effects* (95% CI) | Certainty of evidence (GRADE) | What happens | ||
| Without NSAIDs | With NSAIDs | Difference | ||||
| Pain: ≥ 50% improvement in pain (spontaneous pain at 24 hours) assessed with a 4‐point scale, 1 = no pain to 4 = worst pain Follow‐up: mean 11 days №. of participants: 30 (1 RCT) | RR 2.75 (1.13 to 6.72) | 26.7% | 73.3% (30.1 to 100) | 46.7% more (3.5 more to 152.5 more) | ⊕⊕⊝⊝ LOWa,b | Clinically important difference. Absolute change 47% more (3.5% more to 152.5% more) with NSAIDs. NNTB 3 (95% CI 2 to 12). NSAIDs may result in a decrease in pain (higher proportion of patients achieving ≥ 50% improvement in pain) |
| Inflammation: swelling assessed with a 4‐point scale, 1 = complete resolution to 4 = increase in inflammation at 4 days Follow‐up: mean 11 days №. of participants: 30 (1 RCT) |
RR 1.08 (0.79 to 1.49) | 80.0% | 86.4% (63.2 to 100) | 6.4% more (16.8 fewer to 39.2 more) | ⊕⊕⊝⊝ LOWa,b | The difference is not statistically significant (95% CI includes both improvement in swelling and no improvement). Absolute change 6.4% more patients (16.8% fewer to 39.2% more). NSAIDs may have no effect on inflammation (swelling) |
| Function: ≥ 50% improvement in pain with movement at 24 hours assessed with a 4‐point scale, 1 = complete resolution to 4 = increase in pain intensity Follow‐up: mean 11 days №. of participants: 30 (1 RCT) | RR 4.00 (0.50 to 31.74) | 6.7% | 26.7% (3.3 to 100) | 20.0% more (3.3 fewer to 204.9 more) | ⊕⊕⊝⊝ LOWa,b | The difference is not statistically significant (95% CI includes both improvement in function and no improvement). Absolute change 20% more (3.3% fewer to 204.9% more). NSAIDs may have no effect on function (pain with movement) |
| Participants' global assessment of treatment: not measured №. of participants: 30 (1 RCT) | not estimable | 0.0% | 0.0% (0 to 0) | 0.0% fewer (0 fewer to 0 fewer) | ⊕⊕⊝⊝ LOWa,b | No studies measured the outcome |
| Health‐related quality of life: not measured №. of participants: 30 (1 RCT) | not estimable | 0.0% | 0.0% (0 to 0) | 0.0% fewer (0 fewer to 0 fewer) | ⊕⊕⊝⊝ LOWa,b | No studies measured the outcome |
| Withdrawals due to adverse events assessed with reported withdrawals Follow‐up: mean 11 days №. of participants: 30 (1 RCT) | not estimable | 0.0% | 0.0% (0 to 0) | 0.0% fewer (0 fewer to 0 fewer) | ⊕⊕⊝⊝ LOWb | No events reported in both groups; NSAIDs may have no effect on withdrawals due to adverse events |
| Total adverse events assessed with reported adverse events Follow‐up: mean 11 days №. of participants: 30 (1 RCT) | RR 0.20 (0.01 to 3.85) | 13.3% | 2.7% (0.1 to 51.3) | 10.6% fewer (13.2 fewer to 38 more) | ⊕⊕⊝⊝ LOWa,b | Evidence suggests that NSAIDs result in little to no difference in adverse events. The difference is not statistically significant (95% confidence intervals include both an increase and a decrease in adverse events). Absolute change 10.6% fewer (13.2% fewer to 38% more) |
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; NNTB: number needed to treat for an additional beneficial outcome; NSAID: non‐steroidal anti‐inflammatory drug; RCT: randomised controlled trial; RR: risk ratio. | ||||||
| GRADE Working Group grades of evidence. High certainty: we are very confident that the true effect lies close to that of the estimate of the effect. Moderate certainty: we are moderately confident in the effect estimate: the true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. Low certainty: our confidence in the effect estimate is limited: the true effect may be substantially different from the estimate of the effect. Very low certainty: we have very little confidence in the effect estimate: the true effect is likely to be substantially different from the estimate of effect. | ||||||
aTrial at unclear risk of selection bias and reporting bias, hence downgraded for bias.
bEvidence from one small trial (30 participants) with wide confidence intervals, downgraded for imprecision.
Summary of findings 2. Non‐selective NSAIDs versus selective cyclo‐oxygenase‐2 inhibitors.
| NSAIDs compared to cyclo‐oxygenase (COX)‐2 inhibitors (COXIBs) for acute gout | ||||||
| Patient or population: acute gout Setting: hospital Intervention: NSAIDs Comparison: cyclo‐oxygenase (COX)‐2 inhibitors (COXIBs) | ||||||
| Outcomes | Relative effect (95% CI) | Anticipated absolute effects* (95% CI) | Certainty of evidence (GRADE) | What happens | ||
| Without NSAIDs | With NSAIDs | Difference | ||||
| Pain: mean change from baseline on 5‐point (0 to 4, 0 no pain) Likert scale Follow‐up: 7 days №. of participants: 1044 (6 RCTs) | ‐ | Mean change in pain from baseline without NSAIDs was 1.99 points | Mean change in pain from baseline with NSAIDs was 2.02 points | MD 0.03 points higher (0.07 lower to 0.14 higher) | ⊕⊕⊕⊝ MODERATEa | NSAIDs probably result in little to no difference in mean change from baseline pain compared to COXIBs |
| Inflammation (swelling): mean change from baseline on 4‐point (0 to 3, 0 no swelling) Likert scale Follow‐up: 7 days №. of participants: 1044 (6 RCTs) | ‐ | Mean change in inflammation from baseline without NSAIDs was 1.92 points | Mean change in inflammation from baseline with NSAIDs was 2 points | MD 0.08 points higher (0.07 lower to 0.22 higher) | ⊕⊕⊕⊝ MODERATEa | NSAIDs probably result in little to no difference in inflammation (swelling). The 95% confidence intervals included both an increase and a decrease in swelling |
| Function: mean change in pain with movement from baseline on 4‐point (0 to 3, 0 best function) Likert scale Follow‐up: 7 days №. of participants: 91 (1 RCT) | ‐ | Mean change in function from baseline without NSAIDs was 0 | Mean change in function from baseline with NSAIDs was 0.04 | MD 0.04 higher (0.17 lower to 0.25 higher) | ⊕⊕⊝⊝ LOWa,b | NSAIDs may result in little to no difference in function. The 95% confidence intervals included both an improvement and a reduction in function |
| Participants' global assessment of treatment success on 5‐point (0 very good) Likert scale Follow‐up: 7 days №. of participants: 730 (4 RCTs) | ‐ | Mean participant's global assessment of treatment success without NSAIDs was 1.36 points | Mean participant's global assessment of treatment success with NSAIDs was 1.44 points | MD 0.08 points higher (0.04 lower to 0.21 higher) | ⊕⊕⊕⊝ MODERATEa | NSAIDs probably do not increase participants' global assessment of treatment success. The 95% confidence intervals include both treatment success and no success |
| Health‐related quality of life on SF‐36 (mental health component) Scale from 0 to 100 (0 worst quality of life) Follow‐up: 7 days №. of participants: 222 (1 RCT) | ‐ | Mean health‐related quality of life without NSAIDs was 51.11 points | Mean health‐related quality of life with NSAIDs was 50.93 points | MD 0.18 point lower (6.7 lower to 6.34 higher) | ⊕⊕⊝⊝ LOWa,b | NSAIDs may result in little to no difference in health‐related quality of life. The 95% confidence intervals include both improvement and no improvement in quality of life |
| Withdrawals due to adverse events Follow‐up: 7 days №. of participants: 1243 (6 RCTs) | RR 2.34 (1.33 to 4.14) | 2.9% | 6.7% (3.8 to 11.9) | 3.9% more (1 more to 9 more) | ⊕⊕⊕⊝ MODERATEa | NSAIDs probably increase withdrawals due to adverse events. Absolute change 4% more (1% more to 9% more). NNTH 26 (NNTH 11 to 105)c |
| Total adverse events Follow‐up: 7 days №. of participants: 1232 (6 RCTs) | RR 1.94 (1.36 to 2.78) | 23.1% | 44.8% (31.4 to 64.2) | 21.7% more (8.3 more to 41.1 more) | ⊕⊕⊕⊝ MODERATEa | NSAIDs probably increase adverse events. Absolute change 22% more (8% more to 41% more). NNTH 5 (NNTH 3 to 13)c |
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; MD: mean difference; NNTH: number needed to treat for an additional harmful outcome; NSAID: non‐steroidal anti‐inflammatory drug; RCT: randomised controlled trial; RR: risk ratio; SF‐36: 36‐Item Short Form questionnaire. | ||||||
| GRADE Working Group grades of evidence High certainty: We are very confident that the true effect lies close to that of the estimate of the effect Moderate certainty: We are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different Low certainty: Our confidence in the effect estimate is limited: The true effect may be substantially different from the estimate of the effect Very low certainty: We have very little confidence in the effect estimate: The true effect is likely to be substantially different from the estimate of effect | ||||||
aXu 2016 was at high risk of bias regarding blinding of participants and personnel; Rubin 2004 and Willburger 2007 had unclear risk of bias for these domains. In three trials (Schumacher 2002; Schumacher 2012; Willburger 2007), the role of funding by the pharmaceutical industry might have biased the results. Only 1 out of 6 trials was at low risk of bias (Li 2013).
bNumber needed to treat for an additional beneficial outcome (NNTB) or number needed to treat for an additional harmful outcome (NNTH) = n/a when result is not statistically significant. NNT for dichotomous data was calculated using Cates NNT calculator (http://www.nntonline.net/ebm/visualrx/help.asp).
cEvidence from one single trial with a small number of participants (45 in each arm), downgraded for imprecision.
Summary of findings 3. NSAIDs compared to glucocorticoids for acute gout.
| NSAIDs compared to glucocorticoids for acute gout | ||||||
| Patient or population: acute gout Setting: hospital Intervention: NSAIDs Comparison: glucocorticoids | ||||||
| Outcomes | Relative effect (95% CI) | Anticipated absolute effects* (95% CI) | Certainty of evidence (GRADE) | What happens | ||
| Without NSAIDs | With NSAIDs | Difference | ||||
| Pain: mean decrease per hour on a VAS scale (0 to 100, 0 no pain) Follow‐up: mean 7 days №. of participants: 584 (3 RCTs) | ‐ | Mean pain without NSAIDs was 18.8 points | Mean pain without NSAIDs was 18.95 points | MD 0.15 points higher (2.71 lower to 3.02 higher) | ⊕⊕⊕⊝ MODERATEa | NSAIDs probably result in little to no difference in mean pain reduction on visual analogue scale per 1 to 6 hours. The 95% confidence intervals include both a reduction and an increase in pain |
| Inflammation: swelling mean difference in change from baseline to 14 days assessed with 4‐point Likert scale from 0 (no inflammation) to 3 (bulging beyond margin) Follow‐up: mean 14 days №. of participants: 69 (1 RCT) | ‐ | Mean change in swelling without NSAIDs was 0 | Mean change in swelling with NSAIDs was 0.33 | MD 0.33 higher (0.07 higher to 0.59 higher) | ⊕⊕⊝⊝ LOWa,b | NSAIDs may result in little to no difference in inflammation (swelling) |
| Function: walking disability during first 6 hours assessed with 0 to 100 VAS scale ‐ 0, no disability Follow‐up: mean 7 days №. of participants: 485 (2 RCTs) | ‐ | Mean function: walking disability without NSAIDs was 70 points | Mean function: walking disability with NSAIDs was 70.21 points | MD 0.21 points lower (2.22 lower to 1.8 higher) | ⊕⊕⊕⊝ MODERATEa | NSAIDs probably result in little to no difference in function. The 95% confidence intervals include both a reduction and an increase in function |
| Patients' global assessment of treatment success at day 3 to 4 assessed with 5‐point Likert scale Follow‐up: mean 4 days №. of participants: 129 (2 RCTs) | RR 0.92 (0.70 to 1.22) | 69.8% | 78.2% (64.3 to 95.7) | 8.4% more (5.6 fewer to 25.8 more) | ⊕⊕⊕⊝ MODERATEa | NSAIDs probably result in little to no difference in treatment success; 95% confidence intervals include both an increase and a decrease in the proportions of those reporting treatment success. Absolute change 8.4% more (5.6% fewer to 25.8% more). NNTB n/ac |
| Health‐related quality of life (HRQoL) Not reported | not estimable | 0.0% | 0.0% (0 to 0) | 0.0% fewer (0 fewer to 0 fewer) | ‐ | Not assessed |
| Withdrawals due to adverse events Follow‐up: mean 7 days №. of participants: 772 (5 RCTs) | RR 2.80 (0.55 to 14.22) | 0.8% | 2.2% (0.4 to 11.1) | 1.4% more (0.4 fewer to 10.4 more) | ⊕⊕⊕⊝ MODERATEa | NSAIDs probably result in little to no difference in withdrawals due to adverse events. The 95% confidence intervals include both an increase and a decrease in proportions of withdrawals. Absolute change 1.4% more (0.4% fewer to 10.4% more). NNTH n/ac |
| Total adverse events Follow‐up: mean 7 days №. of participants: 753 (5 RCTs) | RR 1.62 (1.03 to 2.55) | 52.1% | 84.5% (53.7 to 100) | 32.3% more (1.6 more to 80.8 more) | ⊕⊕⊕⊝ MODERATEa | NSAIDs probably result in more total adverse events. Absolute change 32% more (1.6% more to 80.8% more). NNTH 5 (4 to 19)c |
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; MD: mean difference; NNTB: number needed to treat for an additional beneficial outcome; NNTH: number needed to treat for an additional harmful outcome; NSAID: non‐steroidal anti‐inflammatory drug; RCT: randomised controlled trial; RR: risk ratio; VAS: visual analogue scale. | ||||||
| GRADE Working Group grades of evidence. High certainty: we are very confident that the true effect lies close to that of the estimate of the effect. Moderate certainty: we are moderately confident in the effect estimate: the true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. Low certainty: our confidence in the effect estimate is limited: the true effect may be substantially different from the estimate of the effect. Very low certainty: we have very little confidence in the effect estimate: the true effect is likely to be substantially different from the estimate of effect. | ||||||
aUnclear risk of bias for allocation concealment ‐ Janssens 2008a ‐ and for selective reporting ‐ Man 2007; Rainer 2016. Xu 2016 was at high risk of bias for blinding of participants and personnel and had unclear risk of bias for blinding of outcome assessment. Zhang 2014 had high risk of bias for blinding of participants and personnel and for blinding of outcomes assessment; random sequencing and allocation concealment were at unclear risk of bias.
bNumber needed to treat for an additional beneficial outcome (NNTB) or number needed to treat for an additional harmful outcome (NNTH) = n/a when result is not statistically significant. NNT for dichotomous data was calculated using Cates NNT calculator (http://www.nntonline.net/ebm/visualrx/help.asp) and for continuous data using the Wells calculator.
cEvidence from one single trial with a small number of participants (68 in both arms).
Background
Description of the condition
Gout is an inflammatory arthritis that is characterised by the deposition of monosodium urate (MSU) crystals within synovial fluid and other tissues. The natural history of articular gout is generally characterised by two different states: a preclinical state consisting of asymptomatic hyperuricaemia and a clinical state of gout defined by the presence of flares, with or without tophi and bone erosions (Bursill 2019). Gout often heralds its presence by an exquisitely painful acute monoarthritic flare of sudden onset; oligoarticular and polyarticular flares are less common and often occur in patients with poorly controlled disease or during hospitalisation (Dalbeth 2021). Gout occurs in the backdrop of hyperuricaemia, which is necessary but not sufficient to cause gout (Dalbeth 2021). Hyperuricaemia itself is most commonly caused by insufficient secretion of uric acid, rarely by overproduction, and sometimes by both (Dalbeth 2021). Lower limb joints, particularly the big toe, are the most commonly involved, followed by the mid‐tarsal, ankle, knee, and upper limb joints. Subsequent acute flares tend to be longer lasting and polyarticular and tend to affect upper limb joints, such as wrist or elbow (Dalbeth 2021).
Description of the intervention
Non‐steroidal anti‐inflammatory drugs (NSAIDs) including selective cyclooxygenase‐2 (COX‐2) inhibitors are commonly used to treat inflammatory conditions (Garner 2009; Garner 2010; Wienecke 2008). Published guidelines recommend their use for treating acute attacks, with maximum doses given for a short time (Jordan 2007; Khanna 2012; Zhang 2006). These guidelines state that all NSAIDs are equally effective.
How the intervention might work
NSAIDs inhibit inflammation by binding cyclo‐oxygenase (COX) enzymes. Evidence has shown that COX‐2 expression in monocytes is induced in response to MSU microcrystal formation (Pouliot 1998). Therefore, it is likely that NSAIDs exert their beneficial effects in gout by inhibiting the production of COX‐2‐mediated pro‐inflammatory prostaglandins. Most NSAIDs are non‐selective inhibitors; this means they inhibit both COX‐1 and COX‐2. Because non‐selective NSAIDs also act on COX‐1, they may decrease protective stomach prostaglandin levels, which explains the main adverse event of NSAIDs: ulcers and eventually bleeding. A newer class of NSAIDs are the COXIBs: they selectively inhibit COX‐2, which is not involved in the formation of prostaglandins for the stomach and, therefore, may have fewer adverse effects on the gastric mucosa; they are recommended for people at risk for development of ulcers. The main problem with the use of NSAIDs, including COXIBs, is the potential risk of cardiovascular and renal disease (Feenstra 2002; Kearney 2006; Marks 2011).
Why it is important to do this review
Acute gout is an extremely painful condition that has a significant impact on health‐related quality of life (HRQoL), as well as on productivity and ability to function (Rhody 2007; Singh 2006). Without treatment, flares resolve on average only after seven days (Bellamy 1987). Therefore, it is important to rapidly relieve the symptoms caused by acute gout. NSAIDs are known to be among the physician's first choice for treatment of acute gout, but due to potential adverse effects, their use is limited in people with comorbidities such as cardiovascular disease, renal impairment, and a history of peptic ulcer or gastrointestinal bleeding (Borer 2005). The benefits and harms of NSAIDs in treating acute gout were systematically reviewed in 2014 (van Durme CMPG 2014); it is important to update this review to include relevant new evidence.
Objectives
To assess the benefits and harms of non‐steroidal anti‐inflammatory drugs (NSAIDs) (including cyclooxygenase‐2 (COX‐2) inhibitors (COXIBs)) for acute gout.
Methods
Criteria for considering studies for this review
Types of studies
We considered all published randomised controlled trials (RCTs) or quasi‐randomised controlled clinical trials (CCTs) that compared NSAIDs to another therapy (active or placebo, including non‐pharmacological therapies) for acute gout. We included only trials that were published as full articles or were available as full trial reports.
Types of participants
We included studies of adults (aged 18 years or older) with a diagnosis of acute gout. We excluded populations that included a mix of people with acute gout and other musculoskeletal pain unless results for the acute gout population could be separately analysed.
Types of interventions
All trials that evaluated NSAIDs were included, other than those for NSAIDs that are no longer available (e.g. rofecoxib (trademark: Vioxx)).
Comparator treatments could be:
placebo;
no treatment;
paracetamol;
colchicine;
systemic or intra‐articular glucocorticoids;
interleukin‐1 (IL‐1) inhibitors;
non‐pharmacological treatments;
one NSAID versus another NSAID; or
combination therapy (any of the above in combination).
Types of outcome measures
For the purposes of this review, we included outcome measures that were considered to be of greatest importance to people with acute gout and the clinicians who care for them.
OMERACT (Outcome Measures in Rheumatology Clinical Trials) has proposed a set of recommended outcome measures to be used for evaluation of resolution of acute attacks (Grainger 2009; Schumacher 2009). Intense pain is the hallmark of an acute gout attack, hence pain has been proposed as an OMERACT outcome measure; it also has been a consistent outcome measure in clinical trials involving acute gouty arthritis, although the instruments and time intervals used to measure pain vary (Grainger 2009). Other proposed OMERACT outcome measures include joint swelling and tenderness, participant global assessment, and harms (Grainger 2009; Schumacher 2009).
It is recognised that interpreting the meaning of mean changes in continuous measures of pain (e.g. mean change on a 100‐mm visual analogue scale (VAS)) is hampered when participants report either very good or very poor pain relief (Moore 2010). For trials of interventions for chronic pain, the Initiative on Methods, Measurement, and Pain Assessment in Clinical Trials (IMMPACT) has recommended that dichotomous pain outcomes (the proportion of participants improved by 30% or greater and by 50% or greater) be reported (Dworkin 2008), although no recommendations have yet been published for acute pain. Therefore, we elected to include a dichotomous pain outcome measure (the proportion of participants reporting 30% or greater pain relief) as the primary benefit measure in this review. However, as most trials of interventions for acute gout report continuous measures, we included mean change in pain score as a secondary benefit measure.
Major outcomes
Pain: the proportion of participants who reported pain relief of 50% or greater; if not found, the following data were extracted: proportion of participants who achieved pain relief of 30% or greater, or proportion of participants achieving a pain score below 30/100 on VAS, or pain measured as a continuous outcome (e.g. VAS, numerical rating scale)
Inflammation (joint swelling, erythema, tenderness): if more than one measure was reported in an individual trial, we extracted only one according to the following hierarchy: swelling, erythema, and tenderness. We extracted data (when applicable) both for an index joint and for the total number of inflamed joints
Function of target joint (e.g. measured by the Health Assessment Questionnaire (HAQ))
Participants' global assessment of treatment success
HRQoL as reported by generic questionnaires (e.g. 36‐Item Short Form (SF‐36)) or by disease‐specific questionnaires (e.g. Gout Assessment Questionnaire (GAQ), Gout Impact Scale (GIS))
Study participant withdrawal due to adverse events (AEs)
Total number of adverse events
Minor outcomes
Serious adverse events
We planned to include outcomes at all time points measured in the included trials. We planned to pool available data into short‐term (up to two weeks), medium‐term (two to six weeks), and long‐term (more than six weeks) outcomes, but only short‐term data were available. When available, we chose to include the earliest time point for the outcome pain, swelling, and function, as this was more clinically relevant. For the other outcomes (participants' global assessment of treatment success and HRQoL), we chose the latest time point/end of treatment, as we also considered this to be more clinically relevant.
Search methods for identification of studies
Electronic searches
We searched a registry of all randomised controlled trials (RCTs) in gout, established by Cochrane Musculoskeletal to facilitate the updates of a series of reviews of interventions for gout, including this review update. The search for the gout registry was designed not to include terms for any interventions, to establish a registry of all randomised trials in this condition, regardless of the intervention. The following electronic databases were searched to establish the registry. The search strategy combined standard Cochrane search filters for 'gout' and 'randomised trial', with no language restrictions.
Cochrane Central Register of Controlled Trials (CENTRAL), in the Cochrane Library, via Ovid, to 28 August 2020 (Appendix 1).
MEDLINE via Ovid, 1948 to 28 August 2020 (Appendix 2).
Embase via Ovid, 1980 to 28 August 2020 (Appendix 3).
We also searched the clinical trials register clinicaltrials.gov and the World Health Organization (WHO) trials register for relevant trials, using the search term 'gout'. Details of search strategies used for the previous version of this review are given in van Durme CMPG 2014.
Searching other resources
We handsearched the bibliographies of all included papers for information on any other relevant studies.
Data collection and analysis
Selection of studies
Editorial staff from Cochrane Musculoskeletal initially screened titles and abstracts in the gout registry and retrieved full texts for all records that they identified as RCTs of an intervention for people with gout. Editorial staff annotated the population, intervention, and comparator for each full‐text article and assigned it to the appropriate gout review. They imported relevant records to Covidence to select studies eligible for inclusion in this review update (www.covidence.org).
Two review authors (CD and MW) independently screened each title and abstract for suitability for inclusion in the review. They decided independently of each other upon the eligibility of each article according to the pre‐determined selection criteria (see Criteria for considering studies for this review). If more information was required to establish whether inclusion criteria were met, we obtained the full text of the paper. We documented all reasons for excluding studies. We resolved disagreements by consensus after review of the full‐text article. A third review author (RL) resolved differences when necessary. We translated studies into English when necessary.
Data extraction and management
Two review authors (CD and MW) independently extracted data from included trials, including study design, characteristics of the study population, treatment regimen and duration, relevant outcomes, timing of outcome assessment, and duration of follow‐up. We extracted data using a standardised form.
We extracted raw data (means and standard deviations (SDs) for continuous outcomes, number of events or participants for dichotomous outcomes) for the outcome of interest. We resolved differences in data extraction by referring back to the original articles. When needed, we consulted a third review author (RL).
Assessment of risk of bias in included studies
Two review authors (CD and MW) assessed the risk of bias of included studies using the methods recommended by Cochrane for the following items (Higgins 2017): random sequence generation; allocation concealment; blinding of participants, care provider, and outcome assessor for each outcome measure; incomplete outcome data; selective reporting; and other sources of bias such as deviation from the study protocol in a way that did not reflect clinical practice, inappropriate administration of an intervention, presence of unequal co‐interventions, or funding by pharmacological industry.
We assessed these criteria as showing low, high, or unclear risk of bias. Review authors discussed disagreements at a consensus meeting. A third review author (RL) made the final decision when consensus could not be reached.
Measures of treatment effect
To assess benefit, we extracted, if available from published reports, raw data for outcomes of interest (means and SDs for continuous outcomes, numbers of events for dichotomous outcomes) as well as numbers of participants. If we needed to convert or impute reported data, we recorded this in the notes section of the Characteristics of included studies table. We plotted the results of each trial as point estimates with 95% confidence intervals (CIs). We planned to present point estimates as risk ratios (RRs) for dichotomous outcomes and as mean differences (MDs) for continuous outcomes. An RR greater than 1.0 indicates a beneficial effect of NSAIDs (Deeks 2020). RRs are considered clinically relevant if the 95% CI is smaller than 0.7 in favour of the intervention, or larger than 1.5 in favour of the control. This resembles an absolute difference of 25%.
For continuous data, we analysed MD results between intervention and comparator groups, with corresponding 95% CIs. The MD between groups was weighted by the inverse of the variance in the pooled treatment estimate. However, if different scales were used to measure the same conceptual outcome (e.g. functional status, pain), we calculated standardised mean differences (SMD) instead, with corresponding 95% CIs. SMDs were calculated by dividing the MD by the SD, resulting in a unit‐less measure of treatment effect (Deeks 2020). SMDs greater than zero indicate a beneficial effect in favour of NSAIDs for management of symptoms in acute gout attacks. We computed a 95% CI for the SMD when needed. The SMD can be interpreted as described by Cohen (Cohen 1988), that is, an SMD of 0.2 is considered to indicate a small beneficial effect, 0.5 a medium effect, and 0.8 a large effect of NSAIDs for management of symptoms in acute gout attacks. SMDs are considered to indicate a clinically relevant effect if they are larger than 0.5. Upon completion of the analysis, we had planned to translate the SMD back into an MD, using the control group SD at baseline to represent the population SD on a common scale (e.g. 0‐ to 10‐point pain scale), which can be better appraised by clinicians.
In the Effects of interventions section under Results and in the 'Comments' column of the 'Summary of findings' table, we provided the absolute percentage difference and the number needed to treat for an additional beneficial outcome (NNTB), or the number needed to treat for an additional harmful outcome (NNTH) (NNTB or NNTH only for dichotomous outcomes with a clinically significant difference). For dichotomous outcomes, the absolute percentage change was calculated from the difference in risk between intervention and control groups using GRADEpro (GRADEpro 2015), expressed as a percentage. We calculated the NNTB or the NNTH from the control group event rate and the RR by using the Visual Rx NNT calculator (Cates 2008).
Unit of analysis issues
We did not expect unit of analysis problems in this review. In the event that we had identified cross‐over trials in which reporting of continuous outcome data precluded paired analysis, we did not plan to include these data in a meta‐analysis, to avoid unit of analysis error. When carry‐over effects were thought to exist, and when sufficient data were found, we planned to include only data from the first period in the analysis (Higgins 2020a). When outcomes were reported at multiple follow‐up times, we planned to extract data at the following time points: short term (up to two weeks), medium term (more than two weeks to six weeks), and long term (more than six weeks). However, in the included trials, only short‐term outcomes were presented. If more than one time point was reported within the time frame (e.g. at one‐week follow‐up, at two‐week follow‐up), we planned to extract the later time point (i.e. two weeks).
Dealing with missing data
We contacted the study authors when important data were missing.
In case individuals were missing from reported results and no further information was forthcoming from the study authors, we assumed missing values to indicate a poor outcome. For dichotomous outcomes (e.g. number of withdrawals due to adverse events), we planned to calculate the withdrawal rate using the number of participants randomised in the group as the denominator (worst‐case analysis). For continuous outcomes (e.g. mean change in pain score), we planned to calculate the MD or the SMD based on the number of participants analysed at that time point. If the number of participants analysed was not presented for each time point, we planned to use the number of randomised participants in each group at baseline.
When possible, we computed missing SDs from other statistics such as standard errors, confidence intervals, or P values, according to the methods recommended in the Cochrane Handbook for Systematic Reviews of Interventions (Deeks 2020). If we could not calculate SDs, we planned to impute them (e.g. from other studies in the meta‐analysis).
Assessment of heterogeneity
We assessed studies for clinical homogeneity with respect to intervention groups (type of NSAID), control groups, timing of outcome assessment, and outcome measures. For any studies judged as clinically homogeneous, we planned to assess statistical heterogeneity using the I² statistic based on the following approximate guide (Deeks 2020): 30% to 60% may represent moderate heterogeneity, 50% to 90% may represent substantial heterogeneity, and 75% to 100% may represent considerable heterogeneity. In cases of considerable heterogeneity (defined as I² greater than 75%), we planned to explore the data further, including subgroup analyses, in an attempt to explain the heterogeneity.
Assessment of reporting biases
We planned an assessment of reporting biases through screening of the Clinical Trial Register at the International Clinical Trials Registry Platform of the WHO to determine whether the protocol of the RCT had been published before study participant recruitment was started (DeAngelis 2004).
Furthermore, we planned a comparison between the fixed‐effect estimate and the random‐effects model (to assess the possible presence of small‐sample bias), as well as a funnel plot (to assess the possible presence of reporting bias), if data were available (Page 2020). However, data were insufficient to permit these analyses.
Data synthesis
If we considered studies sufficiently homogeneous, we pooled data in a meta‐analysis using a random‐effects model, irrespective of I² statistic results. We performed analyses using Review Manager 5 (RevMan 2020), and we produced forest plots for all analyses for the following comparisons: NSAIDs versus placebo; non‐selective NSAIDs versus COXIBs; and NSAIDs versus glucocorticoids.
Subgroup analysis and investigation of heterogeneity
When sufficient data were available, we planned the following three subgroup analyses.
Disease severity (monoarticular versus polyarticular).
Presence or absence of comorbidities (such as cardiovascular or renal disease, history of peptic ulcer).
Duration of treatment: short term (up to two weeks) versus long term (longer than six weeks).
If trial data were available, we planned to extract major outcomes for the above subgroups within each trial (e.g. monoarticular versus polyarticular) and to informally compare the magnitude of effects between subgroups by assessing the overlap of CIs for the effect estimate (for the main benefit outcome only). Non‐overlap of CIs indicates statistically significant responses between subgroups. However, data were insufficient for any subgroup analyses to be performed.
Sensitivity analysis
When sufficient studies existed, we planned sensitivity analyses to assess the impact of any bias attributable to inadequate or unclear treatment allocation (including studies with quasi‐randomised designs) or to lack of blinding. However, data were insufficient for sensitivity analyses to be performed.
Interpreting results and reaching conclusions
We followed the guidelines in the Cochrane Handbook for Systematic Reviews of Interventions, Chapter 15 (Schunemann 2020a), when interpreting results, and we were aware of distinguishing lack of evidence of effect from lack of effect. We based our conclusions only on findings from the quantitative synthesis of studies included in this review. We avoided making recommendations for practice; our implications for research suggest priorities for future research and outline remaining uncertainties in this area.
Summary of findings and assessment of the certainty of the evidence
We produced 'Summary of findings' (SoF) tables using GRADEpro software (GRADEpro 2015). These tables include an overall grading of evidence based on the GRADE approach as recommended by Cochrane (Schünemann 2020). We produced a summary of available data on the following seven major outcomes: mean improvement in pain, reduction of inflammation measured by swelling, function of target joint, participant global assessment, HRQoL, number of withdrawals due to adverse events, and total adverse events. We have presented three SoF tables for the following comparisons: NSAIDs versus placebo; and two of the most clinically relevant comparisons with multiple trials that allowed pooling of outcomes (non‐selective NSAIDs versus COXIBs and NSAIDs versus glucocorticoids). We did not produce SoF tables for comparisons with single trials of only low‐certainty evidence (NSAID versus rilonacept, NSAID versus acupuncture), nor for comparison of one NSAID versus another NSAID, as these were mostly single‐trial comparisons and included many NSAIDs that are no longer in clinical use.
We originally intended to include in the SoF tables the proportions of participants who reported pain relief of 50% or greater. However, as this information was not included for most trials, we included a continuous measure of pain instead because this is how most trials measured pain.
Two people (CD and MW) used the five GRADE considerations (study limitations, consistency of effect, imprecision, indirectness, and publication bias) to independently assess the certainty of a body of evidence related to studies that contributed data to the meta‐analyses for prespecified outcomes, and we reported the certainty of evidence as high, moderate, low, or very low. We used methods and recommendations described in the Cochrane Handbook for Systematic Reviews of Interventions (Schünemann 2020). We justified all decisions to downgrade the certainty of studies by using footnotes, and we made comments to aid readers' understanding of the review when necessary. We provided the NNTB or the NNTH and absolute percentage change in the Comments column of the SoF table for dichotomous outcomes, as described in the Measures of treatment effect section above.
Results
Description of studies
Results of the search
Cochrane Musculoskeletal updated the search for all gout review updates on 28 August 2020, searching for studies from July 2011 to August 2020. Searches for this update yielded a total of 4511 new records from the following databases: MEDLINE (2125), Embase (1537), and Cochrane CENTRAL (849). We also identified 1 eligible study from the references of other studies. After removing duplicates, we excluded 3245 records based on title and abstract screening. We then assessed 571 full‐text articles for eligibility. Of these, we included 5 new studies in this review update (Li 2013; Rainer 2016; Roddy 2020; Xu 2016; Zhang 2014). We excluded 566 full‐text articles for the following reasons: 164 because of wrong study design, 65 because of wrong patient population, and 298 because of wrong intervention. Thirteen reports were duplicates from the same studies, and 2 studies used the wrong comparator. A total of 21 studies were already included in the previous version of this review, 2 studies are awaiting classification, and 1 study is ongoing.
A flow diagram summarising the study selection process is shown in Figure 1.
1.
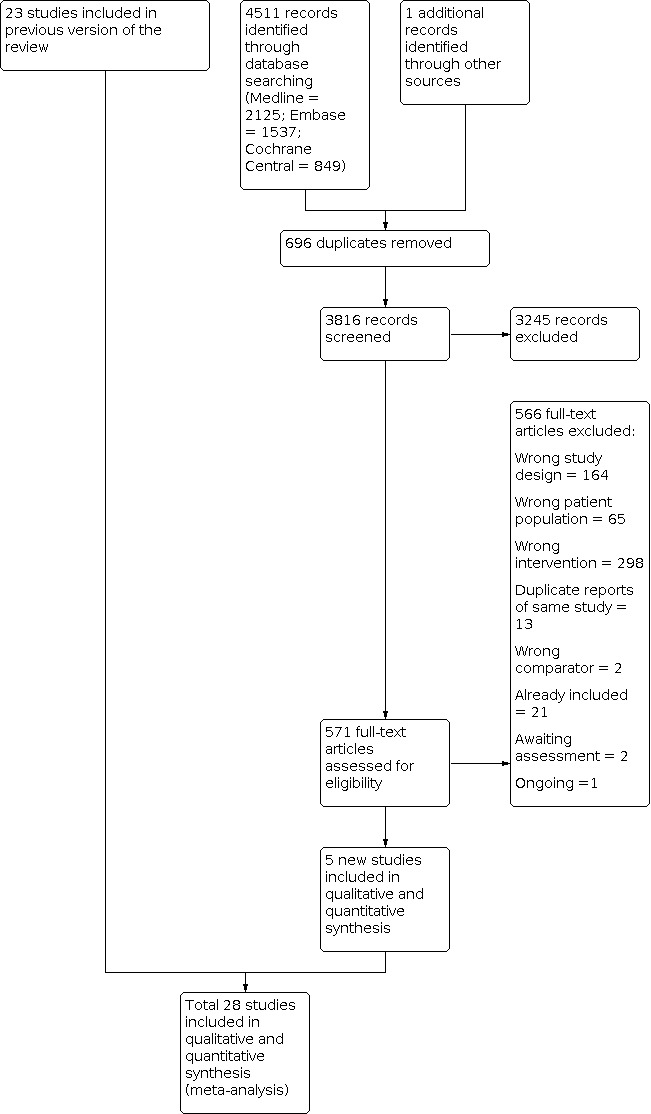
Study flow diagram.
Included studies
The 28 included trials involved 3406 participants (mean 122 participants; range 20 to 416, with study duration ranging from 90 hours to 14 days). A full description of the included studies is provided in the Characteristics of included studies table. Twenty‐six trials were reported in English, 1 in Portuguese (Klumb 1996), and 1 in German (Siegmeth 1976).
Diagnosis of gout and participant features
All included trials were RCTs. The diagnosis of gout was made on clinical grounds in 8 trials (Butler 1985; Douglas 1970; Eberl 1983; Lederman 1990; Maccagno 1991; Roddy 2020; Smyth 1973; Sturge 1977). Ten trials used the 1977 classification criteria of the American College of Rheumatology (ACR 1977) (Cheng 2004; Li 2013; Rubin 2004; Schumacher 2002; Schumacher 2012; Shrestha 1995; Terkeltaub 2013; Willburger 2007; Xu 2016; Zhang 2014), and 1 trial included only participants with gout confirmed by identification of MSU crystals in synovial fluid (Janssens 2008a). Eight trials used either clinical inclusion criteria (a clear history ‐ or the observation ‐ of at least two attacks of acute arthritis with abrupt onset and remission, history/observation of podagra, presence of tophi, history/observation of response to colchicine within 48 hours of therapy) or positive identification of MSU crystals in the synovial fluid for inclusion (Altman 1988; Axelrod 1988; Garcia de la Torre 1987; Klumb 1996; Lomen 1986; Man 2007; Rainer 2016; Siegmeth 1976). Zhou 2012 used the Criteria of Diagnosis and Therapeutic Effect of Diseases and Syndromes in Traditional Medicine (Traditional Chinese Medicine 1994).
All included studies recruited adults, and all but 1 study reported mean age of the study population (Lomen 1986); mean age of the whole study population ranged from 44 to 66 years. Twenty‐one trials included both males and females (Altman 1988; Cheng 2004; Douglas 1970; Garcia de la Torre 1987; Janssens 2008a; Lederman 1990; Li 2013; Maccagno 1991; Man 2007; Rainer 2016; Roddy 2020; Rubin 2004; Schumacher 2002; Schumacher 2012; Shrestha 1995; Smyth 1973; Sturge 1977; Terkeltaub 2013; Willburger 2007; Xu 2016; Zhang 2014). In these trials, the proportion of males varied between 69% and 97%. Five trials included only males (Axelrod 1988; Eberl 1983; Klumb 1996; Siegmeth 1976; Zhou 2012), and 2 trials did not describe gender distribution (Butler 1985; Lomen 1986).
Seven trials reported the mean duration of disease, which ranged from 5 to 17 years (Douglas 1970; Klumb 1996; Lomen 1986; Siegmeth 1976; Terkeltaub 2013; Xu 2016; Zhou 2012). Four trials included only participants with monoarthritis (Janssens 2008a; Lederman 1990; Lomen 1986; Maccagno 1991). Two trials included participants with monoarthritis and oligoarthritis (maximum three joints involved) (Schumacher 2012; Terkeltaub 2013). Nine trials included participants regardless of the number of joints involved: 66% to 96% of participants had monoarthritis, and 5% to 34% had more than one joint involved (Axelrod 1988; Eberl 1983; Klumb 1996; Man 2007; Roddy 2020; Rubin 2004; Schumacher 2002; Willburger 2007; Zhang 2014). Nine trials described affected sites: the first metatarsophalangeal joint was affected in 27% to 100%, the knee in 18% to 47%, the ankle in 19% to 27%, the thumb in 5%, the wrist in 5% to 14%, and the elbow in 3% to 10% of participants (Axelrod 1988; Eberl 1983; Garcia de la Torre 1987; Janssens 2008a; Klumb 1996; Li 2013; Roddy 2020; Schumacher 2002; Xu 2016).
Comparisons
Only 1 trial compared an NSAID (tenoxicam 40 mg) to placebo (Garcia de la Torre 1987).
Thirteen trials compared one NSAID to another NSAID (Altman 1988; Butler 1985; Cheng 2004; Douglas 1970; Eberl 1983; Klumb 1996; Lederman 1990; Lomen 1986; Maccagno 1991; Shrestha 1995; Siegmeth 1976; Smyth 1973; Sturge 1977). Although many of the studied NSAIDs are still registered, many are no longer commonly used in practice: NSAIDs studied included diclofenac (Cheng 2004), etodolac (Lederman 1990; Maccagno 1991), flufenamic acid (Douglas 1970), flurbiprofen (Butler 1985; Lomen 1986), ketorolac (Shrestha 1995), ketoprofen (Altman 1988; Siegmeth 1976), meclofenamate (Eberl 1983), meloxicam (Cheng 2004), nimesulide (Klumb 1996), and phenylbutazone (Butler 1985; Douglas 1970; Siegmeth 1976; Smyth 1973; Sturge 1977). The duration of treatment ranged from 5 days in Altman 1988, Lomen 1986, and Shrestha 1995 to 10 days in Butler 1985; follow‐up ranged from 24 hours in Maccagno 1991 to 14 days in Altman 1988 and Eberl 1983.
Six trials compared a non‐selective NSAID (indomethacin, 50 mg 3 times daily) to a selective COX‐2 inhibitor (etoricoxib 120 mg once daily; celecoxib 50, 200, or 400 mg twice daily, or lumiracoxib 400 mg once daily) (Li 2013; Rubin 2004; Schumacher 2002; Schumacher 2012; Willburger 2007; Xu 2016). Treatment was given for 4 days in Xu 2016, 7 days in Willburger 2007, and 8 days in Rubin 2004Schumacher 2002 and Schumacher 2012; follow‐up ranged from 4 days in Xu 2016 to 14 days in Schumacher 2012.
Four trials compared NSAIDs (naproxen 500 mg twice daily or indomethacin 50 mg 3 times daily) to oral glucocorticoids (prednisolone 30 or 35 mg once daily) (Janssens 2008a; Man 2007; Rainer 2016; Xu 2016). Drugs were given for 4 days in Xu 2016, Janssens 2008a, and Rainer 2016 and for 6 days in Man 2007; follow‐up ranged from 90 hours in Janssens 2008a and Xu 2016 to 14 days in Man 2007 and Rainer 2016.
One trial compared NSAIDs (diclofenac 75 mg twice daily for 7 days) to intramuscular glucocorticoids (betamethasone 7 mg once intramuscularly) (Zhang 2014). Participants were followed up for 7 days.
One trial compared an NSAID (indomethacin 50 mg 4 times daily) to adrenocorticotropin hormone (ACTH) (40 international units (IU) intramuscularly in a single dose) (Axelrod 1988). Participants were followed for 1 year, and every attack during that year was treated with either indomethacin or ACTH.
One trial compared an NSAID (indomethacin 50 mg 3 times daily for 3 days, followed by 25 mg 3 times daily for up to 9 days) to rilonacept (320 mg subcutaneously) and to NSAID plus rilonacept (Terkeltaub 2013).
One trial compared an NSAID (indomethacin 25 mg 3 times daily for 5 days) to acupuncture combined with infrared irradiation (Zhou 2012).
One trial compared an NSAID (naproxen 250 mg 3 times daily) to colchicine (500 mcg 3 times daily) (Roddy 2020).
Outcomes
Four trials included our primary benefit endpoint of proportion of participants improved by 50% or more (Eberl 1983; Garcia de la Torre 1987; Klumb 1996; Lomen 1986), and 20 trials included our primary harms endpoint of withdrawal due to adverse events (Altman 1988; Axelrod 1988; Butler 1985; Cheng 2004; Douglas 1970; Eberl 1983; Garcia de la Torre 1987; Janssens 2008a; Lederman 1990; Li 2013; Lomen 1986; Maccagno 1991; Man 2007; Rubin 2004; Schumacher 2002; Schumacher 2012; Shrestha 1995; Terkeltaub 2013; Willburger 2007; Xu 2016). Other endpoints were variably reported.
NSAID versus placebo (1 trial)
The primary outcomes of this trial were time to improvement and time to resolution; pain and the presence of inflammation were assessed as secondary outcomes. In addition, both of our primary outcomes were reported (Garcia de la Torre 1987).
One NSAID versus another NSAID (13 trials)
Only 3 trials reported proportions of participants improved by 50% or more (Eberl 1983; Klumb 1996; Lomen 1986). All trials used ordinal scales to report pain, with the exception of Klumb 1996, which used a VAS.
Seven trials assessed 'inflammation' as an outcome, but the method of assessment varied across trials (Cheng 2004; Douglas 1970; Eberl 1983; Lederman 1990; Lomen 1986; Maccagno 1991; Smyth 1973). Cheng 2004 used an inflammatory score that assessed tenderness, swelling, and restriction of function of the inflamed joint. Douglas 1970 reported the number of days needed for the redness, swelling, tenderness, or heat to resolve. Eberl 1983 reported numbers of participants who had no redness, swelling, or function restriction at the end of treatment. Lederman 1990 and Lomen 1986 assessed pain, swelling, erythema, and tenderness on a 5‐point scale.
Five trials assessed function (Altman 1988; Cheng 2004; Eberl 1983; Lederman 1990; Maccagno 1991). Altman 1988 and Cheng 2004 assessed function as part of a total 'inflammatory' score. The other 3 trials reported whether there was a limitation in motion of the index joint (absent/none or present).
Five trials included a measure of participants' global assessment (Altman 1988; Cheng 2004; Lederman 1990; Lomen 1986; Maccagno 1991), and no trials included a measure of HRQoL.
Twelve trials included the numbers of participants with AEs and provided a description of the AEs (Altman 1988; Butler 1985; Cheng 2004; Douglas 1970; Eberl 1983; Klumb 1996; Lederman 1990; Lomen 1986; Maccagno 1991; Shrestha 1995; Smyth 1973; Sturge 1977).
Non‐selective NSAIDs versus selective cyclo‐oxygenase‐2 inhibitors (6 trials)
None of these trials measured our primary benefit endpoint, but they all reported withdrawals due to AEs. All 6 trials measured pain as a primary outcome, using a Likert scale (Li 2013; Rubin 2004; Schumacher 2012; Willburger 2007; Xu 2016), or a 5‐point ordinal scale (Schumacher 2002). FIve trials measured inflammation and participants' global assessment as secondary outcomes (Li 2013; Rubin 2004; Schumacher 2002; Willburger 2007; Xu 2016). One trial assessed function (Xu 2016). Willburger 2007 was the only trial that measured HRQoL as a secondary outcome, using SF‐36 and EuroQoL Group Quality of Life Questionnaire based on 5 dimensions (EQ‐5D) questionnaires. Six trials included numbers of participants with AEs and provided a description of the AEs (Li 2013; Rubin 2004; Schumacher 2002; Schumacher 2012; Willburger 2007; Xu 2016).
NSAIDs versus oral glucocorticoids (4 trials) or intramuscular (IM) glucocorticoids (1 trial) or adrenocorticotropin hormone (1 trial)
Neither of the 4 trials comparing NSAID versus oral glucocorticoid included our primary benefit endpoint; all trials included numbers of withdrawals due to AEs (Janssens 2008a; Man 2007; Rainer 2016; Xu 2016). All 4 trials measured pain as mean pain reduction. Three trials measured function (Janssens 2008a; Rainer 2016; Xu 2016). Two trials included measures of inflammation (redness, tenderness, and swelling) (Rainer 2016; Xu 2016). Rainer 2016 and Xu 2016 assessed participants' global assessment. Only Rainer 2016 assessed HRQoL (SF‐36) but did not report the results of this outcome (these also were not obtained from study authors). All trials included the numbers of participants with AEs and provided a description of the AEs. Three trials reported withdrawals due to AEs (Janssens 2008a; Man 2007; Xu 2016).
The trial that compared NSAIDs to intramuscular glucocorticoids reported our main benefit outcome (number of patients without pain) after assessing pain on a Likert scale (Zhang 2014). Other outcomes that were assessed were measures of inflammation (swelling, tenderness) and patients' and physicians' assessments of global response to therapy.
The trial that compared NSAIDs to ACTH did not include any of our main benefit outcomes but did assess pain as the number of hours needed to achieve complete pain relief (Axelrod 1988). This trial also reported withdrawals due to AEs and numbers and types of adverse events.
NSAIDs versus rilonacept (interleukin‐1 inhibitors) (1 trial)
One trial compared NSAID to rilonacept (Terkeltaub 2013). This trial measured change in pain from baseline using both Likert and numerical scales and withdrawals due to adverse events but none of the other relevant measures in this review.
NSAIDs versus acupuncture (1 trial)
One trial compared NSAID to acupuncture. This trial measured only mean change in pain (Zhou 2012).
NSAIDs versus colchicine (1 trial)
Roddy 2020 compared NSAID to colchicine. This trial measured change in pain intensity from baseline as a primary outcome on a 0 to 10 numerical rating scale (NRS). No measure of inflammation was included. Quality of life was assessed using the EuroQoL Group Quality of Life Questionnaire based on 5 dimensions and a 5‐level scale (EQ‐5D‐5L). Adverse events and descriptions of adverse events were provided.
Excluded studies
We excluded 20 trials after detailed review. Reasons for exclusion are described in the Characteristics of excluded studies table.
Nine studies were not RCTs (Arnold 1988; Bach 1979; Cunovic 1973; Cuq 1973; Ecker‐Schlipf 2009; Janssens 2009; Navarra 2007; Steurer 2016; Werlen 1996).
We excluded 1 study because participants with renal insufficiency, history of gastrointestinal AEs to NSAIDs, peptic ulcer or gastritis, or any other contraindication to indomethacin were placed in the triamcinolone group (non‐randomised), and other participants were randomised (Alloway 1993). Data for randomised participants were not reported separately.
One trial did not include participants with acute gout (Kudaeva 2007). We excluded 3 trials because the NSAIDs used (feprazone, proquazone, and fenoprofen) are no longer available (Reardon 1980; Ruotsi 1978; Weiner 1979). We excluded 1 trial because the inclusion population consisted of patients with peptic haemorrhage ulcers who were having an acute gout flare (Xu 2015).
We excluded 2 trials because they compared two different doses of the same drug (Tumrasvin 1985; Valdes 1987).
We identified an additional trial comparing apremilast to indomethacin from the trial registry search, but the trial had been withdrawn (NCT00997581).
Studies awaiting classification
For one trial, only the conference abstract was available at the time of publication of this review (Katona 1988). Another study is written in Chinese and is awaiting translation (Yin 2005). We categorised trials as awaiting classification (see Characteristics of studies awaiting classification table).
Ongoing studies
One ongoing trial ‐ ChiCTR1800019612 ‐ is recruiting participants to study effects of NSAID plus ozone treatment of autologous blood versus ozone treatment of autologous blood.
Risk of bias in included studies
We judged most trials (26/28; 93%) as having unclear ‐ Altman 1988; Garcia de la Torre 1987; Janssens 2008a; Klumb 1996; Lomen 1986; Maccagno 1991; Man 2007; Rainer 2016; Rubin 2004; Schumacher 2002; Schumacher 2012; Siegmeth 1976; Smyth 1973; Terkeltaub 2013; Willburger 2007 ‐ or high risk of bias ‐ Axelrod 1988; Butler 1985; Cheng 2004; Douglas 1970; Eberl 1983; Lederman 1990; Roddy 2020; Sturge 1977; Xu 2016; Zhang 2014; Zhou 2012. We judged only 2 trials (8%) as having low risk of bias (Li 2013; Shrestha 1995).
A description of the risk of bias of included studies is presented in the Characteristics of included studies table. Summaries of the risk of bias of individual trials are shown in Figure 2 and of included trials as a group in Figure 3.
2.

Risk of bias graph: review authors' judgements about each risk of bias item presented as percentages across all included studies.
3.

Risk of bias summary: review authors' judgements about each risk of bias item for each included study.
Sequence generation (selection bias)
Fourteen trials reported an appropriate sequence generation (Cheng 2004; Douglas 1970; Janssens 2008a; Li 2013; Man 2007; Rainer 2016; Roddy 2020; Schumacher 2002; Schumacher 2012; Shrestha 1995; Smyth 1973; Willburger 2007; Xu 2016; Zhou 2012). For 12 trials, the method of sequence generation was unclear (Altman 1988; Butler 1985; Eberl 1983; Garcia de la Torre 1987; Klumb 1996; Lomen 1986; Maccagno 1991; Rubin 2004; Siegmeth 1976; Sturge 1977; Terkeltaub 2013; Zhang 2014). We judged 2 trials as having high risk of bias for the item sequence generation: 1 trial because participants were alternately assigned to one of the two treatment groups (Axelrod 1988), and the other trial because although stated as randomised with no description of the randomisation method, baseline characteristics were significantly different between the two treatment groups (Lederman 1990).
Allocation
For 17 trials, concealment of drug allocation was inappropriately described or was not described at all, and we judged them to be at unclear risk of bias (Altman 1988; Butler 1985; Cheng 2004; Douglas 1970; Eberl 1983; Garcia de la Torre 1987; Janssens 2008a; Klumb 1996; Lederman 1990; Lomen 1986; Maccagno 1991; Rubin 2004; Schumacher 2002; Siegmeth 1976; Terkeltaub 2013; Willburger 2007; Zhang 2014). We assigned 3 trials to be at high risk of allocation bias because the treatment was not concealed (Axelrod 1988; Sturge 1977; Zhou 2012). Eight trials were at low risk of selection bias, as the method of allocation concealment was clearly described (Li 2013; Man 2007; Rainer 2016; Roddy 2020; Schumacher 2012; Shrestha 1995; Smyth 1973; Xu 2016).
Blinding
We judged 8 trials as having unclear risk of performance bias regarding blinding of study personnel (Altman 1988; Eberl 1983; Lederman 1990; Lomen 1986; Maccagno 1991; Siegmeth 1976; Smyth 1973; Sturge 1977). For 6 trials (Axelrod 1988; Cheng 2004; Roddy 2020; Xu 2016; Zhang 2014; Zhou 2012), we considered risk of performance bias to be high because participants were not blinded. We judged 14 trials to be at low risk of performance bias, as the method of blinding participants and study personnel was adequately described (Butler 1985; Douglas 1970; Garcia de la Torre 1987; Janssens 2008a; Klumb 1996; Li 2013; Man 2007; Rainer 2016; Rubin 2004; Schumacher 2002; Schumacher 2012; Shrestha 1995; Terkeltaub 2013; Willburger 2007)
We judged 8 trials as having unclear risk of detection bias for self‐reported outcomes because blinding of participants was not described or was unclear (Altman 1988; Eberl 1983; Lederman 1990; Lomen 1986; Maccagno 1991; Siegmeth 1976; Smyth 1973; Sturge 1977). We assigned 6 trials high risk of bias because the trials were not blinded (Axelrod 1988; Cheng 2004; Roddy 2020; Xu 2016; Zhang 2014; Zhou 2012). We judged 14 trials to be at low risk of detection bias for self‐reported outcomes, as the method used to blind participants was adequately described (Butler 1985; Douglas 1970; Garcia de la Torre 1987; Janssens 2008a; Klumb 1996; Li 2013; Man 2007; Rainer 2016; Rubin 2004; Schumacher 2002; Schumacher 2012; Shrestha 1995; Terkeltaub 2013; Willburger 2007).
We judged 13 trials to be at unclear risk of detection bias for assessor‐reported outcomes because blinding of outcome assessors was not described or was unclear (Altman 1988; Butler 1985; Douglas 1970; Eberl 1983; Garcia de la Torre 1987; Klumb 1996; Lederman 1990; Lomen 1986; Maccagno 1991; Siegmeth 1976; Smyth 1973; Sturge 1977; Terkeltaub 2013). We assigned 5 trials high risk of detection bias for assessor‐reported outcomes because the trials were not blinded (Axelrod 1988; Roddy 2020; Xu 2016; Zhang 2014; Zhou 2012). We judged 10 trials to be at low risk of detection bias for assessor‐reported outcomes, as the method of blinding outcome assessors was adequately described (Cheng 2004; Janssens 2008a; Li 2013; Man 2007; Rainer 2016; Rubin 2004; Schumacher 2002; Schumacher 2012; Shrestha 1995; Willburger 2007).
Incomplete outcome data
We judged 12 trials as having unclear risk of bias for incomplete outcome data because they did not report if there were withdrawals or missing data, or how withdrawals or missing data (or both) were handled (Altman 1988; Axelrod 1988; Butler 1985; Eberl 1983; Garcia de la Torre 1987; Klumb 1996; Roddy 2020; Schumacher 2002; Smyth 1973; Sturge 1977; Willburger 2007; Xu 2016). We judged the remaining 16 trials to be at low risk of attrition bias (Cheng 2004; Douglas 1970; Janssens 2008a; Lederman 1990; Li 2013; Lomen 1986; Maccagno 1991; Man 2007; Rainer 2016; Rubin 2004; Schumacher 2012; Shrestha 1995; Siegmeth 1976; Terkeltaub 2013; Zhang 2014; Zhou 2012).
Selective reporting
Twenty trials were at low risk of reporting bias (Altman 1988; Axelrod 1988; Cheng 2004; Douglas 1970; Eberl 1983; Janssens 2008a; Lederman 1990; Li 2013; Lomen 1986; Maccagno 1991; Rubin 2004; Schumacher 2002; Schumacher 2012; Shrestha 1995; Siegmeth 1976; Smyth 1973; Sturge 1977; Terkeltaub 2013; Willburger 2007; Xu 2016). We assigned 6 trials unclear risk of selective reporting bias (Garcia de la Torre 1987; Klumb 1996; Man 2007; Rainer 2016; Zhang 2014; Zhou 2012). Man 2007 reported secondary outcomes, but not in the prespecified manner. Klumb 1996 did not provide a clear description of outcomes and provided inappropriate between‐group comparisons (only status scores). Garcia de la Torre 1987 and Rainer 2016 did not report all prespecified outcomes. Zhang 2014 reported an outcome that was not prespecified and did not report anywhere whether there was a statistically significant difference in this reported outcome. Other prespecified outcomes were reported, but again, it was not reported whether differences were statistically significant. Zhou 2012 did not report inflammation but named it in the methods section, so it is unclear if this was going to be a separate outcome.
We judged 1 trial as having high risk of bias for this criterion because it did not report one prespecified outcome ‐ pain measured on an ordinal scale (Butler 1985).
Other potential sources of bias
Two trials were judged to be at high risk of other bias (Douglas 1970; Eberl 1983). Eberl 1983 used a higher initial meclofenamate dose compared with the indomethacin dose used in the control group, which may have biased the results in favour of the meclofenamate group. In Douglas 1970, the mean age of participants was significantly higher in the flufenamic acid group (57.2 years) than in the phenylbutazone group (47.6 years).
In Sturge 1977, there was also a difference in age between the two groups: participants in the naproxen group were older (mean age 58.8 years, range 34 to 84) than those in the phenylbutazone group (mean age 50.4 years, range 30 to 73).
Four studies were subject to funding by manufacturers, but these relationships did not appear to affect reporting of study results, and it is unclear if there was any bias in the study design as a result of the funding relationships.
The rilonacept study was funded by Regeneron Pharmaceutics Inc. (manufacturers of rilonacept); employees of Regeneron Pharmaceutics Inc. participated in study design, data analysis, and writing of the manuscript (Terkeltaub 2013). It is unclear if this relationship resulted in any biased conduct in the trial.
For Schumacher 2002, Merck Research Laboratory provided funding to all participating investigators to cover the costs of patient procedures and investigations; one study author was on the Merck advisory board, one was a consultant for Merck, and four were employed by Merck and owned shares of Merck common stock.
Editorial support was funded by Pfizer for Schumacher 2012.
Four authors of Willburger 2007 were employed by Novartis Pharma; one author was a speaker for Novartis. It is unclear if this relationship resulted in any biased reporting of results in the trial.
Roddy 2020 reported a difference in length of treatment; naproxen was given for 4 days and colchicine for 7 days.
Effects of interventions
See: Table 1; Table 2; Table 3
NSAIDs versus placebo
Benefits
One trial of 30 participants compared an NSAID (tenoxicam 40 mg) with placebo (Garcia de la Torre 1987). All results are summarised in Table 1. NSAIDs may result in decreased pain (higher proportion of participants achieving ≥ 50% improvement in pain). Low‐certainty evidence downgraded for bias and imprecision suggests there may be a clinically significant improvement in the number of patients who achieve more than 50% reduction in overall pain (reported as 'spontaneous pain') at 24 hours (11/15 in the tenoxicam group, 4/15 in the placebo group; risk ratio (RR) 2.7, 95% confidence interval (CI) 1.1, 6.7), with absolute change of 47% more (3.5% more to 152.5% more) with NSAIDs and number needed to treat for an additional beneficial outcome (NNTB) of 3 (95% CI 2 to 12; Analysis 1.1). There was no difference in the number of participants who achieved more than 50% reduction in pain with movement at 24 hours (4/15 in the NSAIDs group versus 1/15 in the placebo group; RR 4.0, 95% CI 0.5 to 31.7) and at day 4 (13/15 in the NSAIDs group versus 14/15 in the placebo group; RR 0.9, 95% CI 0.7 to 1.2; Analysis 1.1).
1.1. Analysis.

Comparison 1: NSAIDs versus placebo, Outcome 1: Pain: ≥ 50% improvement in pain
Low‐certainty evidence downgraded for bias and imprecision suggests no reported between‐group differences in the proportions of participants with more than 50% improvement in joint swelling at 24 hours (5/15 in the NSAIDs group versus 2/15 in the placebo group; RR 2.5, 95% CI 0.6 to 10.9) or at day 4 (13/15 in the NSAIDs group versus 12/15 in the placebo group; RR 1.1, 95% CI 0.8 to 1.5), with absolute change of 6.4% more patients (16.8% fewer to 39.2% more).
NSAIDs may have no effect on function (≥ 50% improvement in pain with movement at 24 hours assessed on a 4‐point scale (1 = complete resolution to 4 = increased pain); RR 4.0 (95% CI 0.5 to 31.7)), with absolute change of 20% more (3.3% fewer to 204.9% more).
The trial did not measure global assessment of treatment success nor health‐related quality of life (HRQoL).
Harms
There were no withdrawals due to adverse events in either group in this trial and no significant between‐group differences in numbers of adverse events (0/15 in the NSAIDs group versus 2/15 in the placebo group; RR 0.2, 95% CI 0.0 to 3.8), with absolute change of 10.6% fewer (13.2% fewer to 38% more; Analysis 1.3).
1.3. Analysis.

Comparison 1: NSAIDs versus placebo, Outcome 3: Withdrawals due to adverse events
Non‐selective NSAIDs versus cyclo‐oxygenase‐2 inhibitors
Six trials including 1266 participants compared NSAIDs (indomethacin 50 mg 3 times daily or 75 mg twice daily) to COXIBs (etoricoxib 120 mg once daily; celecoxib 50, 200, or 400 twice daily; or lumiracoxib 400 mg once daily), and data could be pooled (Li 2013; Rubin 2004; Schumacher 2002; Schumacher 2012; Willburger 2007; Xu 2016). Two trials were at unclear risk of selection bias (Rubin 2004; Willburger 2007), 1 was at high risk of performance and detection bias (Xu 2016), and it is unclear whether funding in 3 trials provided by the manufacturer resulted in any bias (Schumacher 2002; Schumacher 2012; Willburger 2007). One trial was at low risk of bias (Li 2013). All results are summarised in Table 2.
Benefits
Six trials (1044 participants) showed no between‐group differences with respect to mean pain change from baseline on a 0 to 4 Likert scale (where 0 is no pain) at day 1 or 2 (mean difference (MD) 0.0, 95% CI ‐0.1 to 0.1; Analysis 2.1). There was no statistically or clinically significant difference between NSAIDs and COXIBs with regards to inflammation measured on a 0 to 3 Likert scale (0 is no swelling; MD 0.1, 95% CI ‐0.1 to 0.2; 6 trials with 1044 participants; moderate‐certainty evidence downgraded for bias; Analysis 2.2). Xu 2016 assessed function as pain with activity. There was no mean difference from baseline between the two groups (MD ‐0.0, 95% CI ‐0.2 to 0.2; low‐certainty evidence downgraded for bias and imprecision; Analysis 2.3). Moderate‐certainty evidence (downgraded for bias) from 4 trials (730 participants) showed no between‐group differences with respect to patients' global assessment of treatment success (MD 0.1, 95% CI ‐0.0 to 0.2; Analysis 2.4). One trial with 222 participants reported no between‐group differences with respect to HRQoL measured by the 36‐Item Short Form questionnaire (SF‐36) Mental Health component (MD ‐0.2, 95% CI ‐6.7 to 6.3; low‐certainty evidence downgraded for bias and imprecision; Analysis 2.5; Willburger 2007).
2.1. Analysis.

Comparison 2: NSAIDs versus cyclo‐oxygenase (COX)‐2 inhibitors (COXIBs), Outcome 1: Pain: mean change difference from baseline on a 5‐point Likert scale
2.2. Analysis.

Comparison 2: NSAIDs versus cyclo‐oxygenase (COX)‐2 inhibitors (COXIBs), Outcome 2: Inflammation: mean change difference in swelling from baseline
2.3. Analysis.

Comparison 2: NSAIDs versus cyclo‐oxygenase (COX)‐2 inhibitors (COXIBs), Outcome 3: Function: mean change difference in pain with activity from baseline
2.4. Analysis.

Comparison 2: NSAIDs versus cyclo‐oxygenase (COX)‐2 inhibitors (COXIBs), Outcome 4: Participants' global assessment of treatment success
2.5. Analysis.

Comparison 2: NSAIDs versus cyclo‐oxygenase (COX)‐2 inhibitors (COXIBs), Outcome 5: Health‐related quality of life measured by 36‐item Short Form
Harms
Moderate‐certainty evidence (downgraded for bias) from four trials (1266 participants) showed significantly fewer withdrawals due to adverse events among participants treated with COXIBs versus non‐selective NSAIDs (21/729 (3.0%) in the COXIB group versus 32/514 (7.0%) in the non‐selective NSAIDs group; RR 2.3, 95% CI 1.3 to 4.1), with absolute change of 4% more (1% more to 9% more) and number needed to treat for an additional harmful outcome (NNTH) of 26 (NNTH 11 to 105; Analysis 2.6). There were significantly fewer total adverse events among participants treated with COXIBs (168/727; 23%) compared with participants treated with NSAIDs (193/505; 45%) (RR 1.9, 95% CI 1.4 to 2.8; Analysis 2.7).
2.6. Analysis.

Comparison 2: NSAIDs versus cyclo‐oxygenase (COX)‐2 inhibitors (COXIBs), Outcome 6: Withdrawals due to adverse events
2.7. Analysis.

Comparison 2: NSAIDs versus cyclo‐oxygenase (COX)‐2 inhibitors (COXIBs), Outcome 7: Total adverse events
There were significantly fewer gastrointestinal adverse events with COXIBs compared with non‐selective NSAIDs (RR 2.4, 95% CI 1.6 to 3.4; 1232 participants, 6 studies; 43/727 (6%) in the COXIBs group versus 72/505 (14%) in the non‐selective NSAIDs group; Analysis 2.8). There were no significant between‐group differences in cardiovascular events (RR 2.4, 95% CI 1.0 to 5.7), other adverse events (RR 1.7, 95% CI 0.9 to 3.2), or serious adverse events (RR 2.3, 95% CI 0.5 to 11.2; Analysis 2.9; Analysis 2.10; Analysis 2.11).
2.8. Analysis.

Comparison 2: NSAIDs versus cyclo‐oxygenase (COX)‐2 inhibitors (COXIBs), Outcome 8: Gastrointestinal adverse events
2.9. Analysis.

Comparison 2: NSAIDs versus cyclo‐oxygenase (COX)‐2 inhibitors (COXIBs), Outcome 9: Cardiovascular adverse events
2.10. Analysis.

Comparison 2: NSAIDs versus cyclo‐oxygenase (COX)‐2 inhibitors (COXIBs), Outcome 10: Other adverse events
2.11. Analysis.

Comparison 2: NSAIDs versus cyclo‐oxygenase (COX)‐2 inhibitors (COXIBs), Outcome 11: Serious adverse events
NSAIDs versus oral glucocorticoids or intramuscular glucocorticoid or adrenocorticotropic hormone
Benefits
Four trials (712 participants) compared NSAIDs to oral glucocorticoids (Janssens 2008a; Man 2007; Rainer 2016; Xu 2016), 1 compared NSAIDs to adrenocorticotropin hormone (ACTH) (Axelrod 1988), and 1 compared NSAIDs to intramuscular glucocorticoid (Zhang 2014). Two trials that compared NSAIDs to oral glucocorticoids were at low risk of bias (Janssens 2008a; Man 2007), 1 at unclear risk of bias (Rainer 2016), and 3 at high risk of bias (Axelrod 1988; Xu 2016; Zhang 2014). Pain was assessed as mean decrease per hour on a visual analogue scale (VAS) (Janssens 2008a; Man 2007; Rainer 2016), and per time interval at zero to 90 hours (Janssens 2008a). One trial reported pain as mean decrease 4 hours following treatment on each day for 7 days on a Likert scale (Zhang 2014). All results are summarised in Table 3.
Moderate‐certainty evidence downgraded for bias showed no significant differences in mean pain reduction on a VAS scale (0 to 100; 0 no pain) between groups (MD 0.1, 95% CI ‐2.7 to 3.0; 3 trials, 584 participants; Analysis 3.1). One trial that compared NSAIDs (indomethacin 50 mg 4 times daily) to ACTH (40‐mg single dose intramuscularly) reported that complete pain relief was achieved significantly sooner in the ACTH group compared with the indomethacin group (mean ± SD; 24 ± 10 hours in indomethacin group versus 3 ± 1 hours in ACTH group; Axelrod 1988), but we were unable to verify this from the data presented.
3.1. Analysis.

Comparison 3: NSAIDs versus glucocorticoids, Outcome 1: Pain: mean reduction over visual analogue scale per hour during first 6 hours
Three trials reported measures of inflammation (Rainer 2016; Xu 2016; Zhang 2014). Xu 2016 and Zhang 2014 reported swelling as a measure of inflammation, and only Xu 2016 reported it as mean difference from baseline. There was a significant difference in mean change in swelling from day 0 to day 4 on a 4‐point Likert scale in favour of oral glucocorticoids (MD 0.3, 95% CI 0.1 to 0.6; low‐certainty evidence downgraded for bias and imprecision; Analysis 3.2).
3.2. Analysis.

Comparison 3: NSAIDs versus glucocorticoids, Outcome 2: Inflammation: swelling mean difference in change from day 0 to day 14
Two trials reported a measure of function (Janssens 2008a; Rainer 2016). Moderate‐certainty evidence downgraded for bias showed no significant difference in reduction in loss of function between groups (MD ‐0.2, 95% CI ‐2.2 to 1.8; Analysis 3.3).
3.3. Analysis.

Comparison 3: NSAIDs versus glucocorticoids, Outcome 3: Function: walking disability during first 6 hours
Two trials assessed patients' global assessment on a 5‐point Likert scale (Rainer 2016; Xu 2016). There were no differences between groups (RR 0.9, 95% CI 0.7 to 1.2), with absolute change of 8.4% more (5.6% fewer to 25.8% more; moderate‐certainty evidence downgraded for bias; Analysis 3.4).
3.4. Analysis.

Comparison 3: NSAIDs versus glucocorticoids, Outcome 4: Patient's Global Assessment of response to treatment: good to very good response at day 3 to 4
Rainer 2016 also assessed HRQoL using SF‐36 but did not present these results in the article.
Harms
Five trials with 772 participants reported withdrawals due to adverse events (Janssens 2008a; Man 2007; Rainer 2016; Xu 2016; Zhang 2014). Moderate‐certainty evidence downgraded for bias showed there was no statistically significant difference between NSAIDs (10/389) versus oral glucocorticoid (3/383) (RR 2.8, 95% CI 0.5 to 14.2), with absolute change of 1.4% more (0.4% fewer to 10.4% more; Analysis 3.5). The trial comparing NSAIDs to ACTH ‐ Altman 1988 ‐ reported significantly more withdrawals due to adverse events in the indomethacin group (10/50 in the indomethacin group versus 0/50 in the ACTH group; RR 21, 95% CI 1.3 to 348.9; Analysis 5.1).
3.5. Analysis.

Comparison 3: NSAIDs versus glucocorticoids, Outcome 5: Withdrawals due to adverse events
5.1. Analysis.

Comparison 5: NSAIDs versus adrenocorticotropin hormone (ACTH), Outcome 1: Withdrawals due to adverse events
A pooled analysis of 5 trials comparing NSAIDs with corticosteroids ‐ Janssens 2008a; Man 2007; Rainer 2016; Xu 2016; Zhang 2014 ‐ showed more total adverse events in the NSAID group (248/379 (65%)) than in the glucocorticoid group (195/374 (52%); RR 1.6, 95% CI 1.0 to 2.5; Analysis 3.6). There were no significant between‐group differences with respect to cardiovascular (RR 2.9, 95% CI 0.1 to 68.7), gastrointestinal (RR 1.8, 95% CI 0.9 to 3.7), or other adverse events (RR 1.1, 95% CI 0.7 to 1.8), nor serious adverse events (RR 12.4, 95% CI 0.7 to 214.6; Analysis 3.7; Analysis 3.8; Analysis 3.9; Analysis 3.10).
3.6. Analysis.

Comparison 3: NSAIDs versus glucocorticoids, Outcome 6: Total adverse events
3.7. Analysis.

Comparison 3: NSAIDs versus glucocorticoids, Outcome 7: Gastrointestinal adverse events
3.8. Analysis.

Comparison 3: NSAIDs versus glucocorticoids, Outcome 8: Cardiovascular adverse events
3.9. Analysis.

Comparison 3: NSAIDs versus glucocorticoids, Outcome 9: Other adverse events
3.10. Analysis.

Comparison 3: NSAIDs versus glucocorticoids, Outcome 10: Serious adverse events
In the trial of NSAID versus ACTH, significantly more adverse events were reported in the NSAIDs group compared to the ACTH group (49/50 (98%) in NSAID group versus 0/50 (0%) in ACTH group; RR 99, 95% CI 6.3 to 1562; Analysis 5.2).
5.2. Analysis.

Comparison 5: NSAIDs versus adrenocorticotropin hormone (ACTH), Outcome 2: Total adverse events
One NSAID versus another NSAID
Two trials including 121 participants that compared naproxen with etodolac could be pooled for two outcomes (Lederman 1990; Maccagno 1991). Lederman 1990 had high risk of bias and Maccagno 1991 had unclear risk of bias.
Benefits
There was no between‐group difference with respect to participants' global assessment of treatment success reported as proportions of people who considered themselves markedly improved at the end of treatment (53/60 (88%) in the etodolac group versus 53/61 (87%) in the naproxen group; RR 1.0, 95% CI 0.9 to 1.1; Analysis 4.1).
4.1. Analysis.

Comparison 4: Etodolac versus naproxen, Outcome 1: Participants' global assessment at end of therapy: markedly improved
Harms
There were no withdrawals due to adverse events. There was no between‐group difference with respect to numbers of adverse events (4/60 (7%) in the etodolac group versus 2/61 (3%) in the naproxen group; RR 1.7, 95% CI 0.4 to 7.9; Analysis 4.2).
4.2. Analysis.

Comparison 4: Etodolac versus naproxen, Outcome 2: Total adverse events
Four trials including 142 participants compared indomethacin to another NSAID (nimesulide (Klumb 1996), flurbiprofen (Lomen 1986), meclofenamate (Eberl 1983), or ketoprofen (Altman 1988)). These trials had unclear ‐ Altman 1988; Klumb 1996; Lomen 1986 ‐ or high ‐ Eberl 1983 risk of bias, and no between‐group differences in benefits or harms outcomes were reported in individual trials or in the limited number of pooled analyses that were possible (data not shown).
NSAIDs versus rilonacept (interleukin‐1 inhibitor)
Benefits
One trial at high risk of bias that included 225 participants found that NSAIDs provided greater pain relief from 24 to 72 hours than rilonacept (interleukin‐1 inhibitor), as measured on a 0 to 10 numerical rating scale (MD ‐2.1, 95% CI ‐3.14 to ‐1.1; Analysis 6.1). Combination therapy (NSAIDs plus rilonacept) versus NSAIDs did not provide greater pain relief from baseline to a mean of pain at 24 to 72 hours as measured on a 0 to 10 numerical rating scale (MD ‐0.5, 95% CI ‐1.5 to 2.4; Analysis 7.1).
6.1. Analysis.

Comparison 6: NSAIDs versus interleukin (IL)‐1 inhibitor, Outcome 1: Pain: mean pain reduction on numerical rating scale
7.1. Analysis.

Comparison 7: NSAIDs versus interleukin (IL)‐1 inhibitor plus NSAIDs, Outcome 1: Pain (change 24 to 72 hours numerical rating scale)
This trial did not measure inflammation, function, participants' global assessment of treatment success, nor HRQoL.
Harms
There was no between‐group difference with regards to withdrawals due to adverse events (2/77 (2%) in the NSAIDs group versus 1/75 (1%) in the rilonacept group; RR 1.9, 95% CI 0.2 to 21.0; Analysis 6.3) nor in total number of adverse events (23/77 (30%) for the NSAIDs group versus 27/75 (36%) for the rilonacept group; RR 0.8, 95% CI 0.5 to 1.3; Analysis 6.3). There were no serious adverse events.
6.3. Analysis.

Comparison 6: NSAIDs versus interleukin (IL)‐1 inhibitor, Outcome 3: Total adverse events
For combination therapy (NSAIDs plus rilonacept) versus NSAIDs, there were also no differences in study withdrawals due to adverse events (2/76 (3%) in the NSAIDs group versus 2/74 (3%) in the combination group; RR 1.0, 95% CI 0.1 to 6.7; Analysis 7.2), in risk of adverse events (23/76 (30%) in the NSAIDs group versus 34/74 (46%) in the combination group; RR 0.7, 95% CI 0.4 to 1.0), nor in risk of adverse events (0/76 (0%) in the NSAIDs group versus 3/74 (4%) in the combination group; RR 0.1, 95% CI 0.0 to 2.6; Analysis 7.3; Analysis 7.4).
7.2. Analysis.

Comparison 7: NSAIDs versus interleukin (IL)‐1 inhibitor plus NSAIDs, Outcome 2: Withdrawals due to adverse events
7.3. Analysis.

Comparison 7: NSAIDs versus interleukin (IL)‐1 inhibitor plus NSAIDs, Outcome 3: Total adverse events
7.4. Analysis.

Comparison 7: NSAIDs versus interleukin (IL)‐1 inhibitor plus NSAIDs, Outcome 4: Serious adverse events
NSAIDs versus acupuncture combined with infrared irradiation
Benefits
One trial at high risk of bias that included 163 participants found that acupuncture and infrared irradiation resulted in better benefit with respect to mean pain score after treatment compared with NSAIDs (MD 2.2, 95% CI 1.8 to 2.7; Analysis 8.1Zhou 2012).
8.1. Analysis.

Comparison 8: NSAIDs versus acupuncture combined with infrared irradiation, Outcome 1: Pain: mean score on visual analogue scale after treatment
This trial did not measure inflammation, function, participants' global assessment of treatment success, nor HRQoL.
Harms
Withdrawals due to adverse events and total adverse events were not reported.
NSAIDs versus colchicine
Benefits
Roddy 2020, which included 399 participants, found no difference in mean change in worst pain intensity over days 1 to 7 with NSAIDs compared to colchicine using a 0 to 10 NRS (MD 0.3, 95% CI ‐0.4 to 1.0). Quality of life was measured on the EuroQoL Group Quality of Life Questionnaire based on 5 dimensions and a 5‐level scale (EQ‐5D‐5L), and there was no difference between groups (MD 0.0, 95% CI ‐0.0 to 0.0; Analysis 9.1; Analysis 9.2).
9.1. Analysis.

Comparison 9: NSAIDs versus colchicine, Outcome 1: Mean change in pain over days 1 to 7
9.2. Analysis.

Comparison 9: NSAIDs versus colchicine, Outcome 2: Quality of life: EQ‐5D at day 7
Harms
Withdrawals due to adverse events were not reported. There was no difference with regards to total number of adverse events (91/200 (46%) in the NSAID group versus 101/199 (51%) in the colchicine group; RR 0.9, 95% CI 0.7 to 1.1). There were no differences in gastrointestinal adverse events (RR 0.8, 95% CI 0.7 to 1.0) nor in other adverse events (RR 1.0, 95% CI 0.2 to 4.9; Analysis 9.3; Analysis 9.4; Analysis 9.5).
9.3. Analysis.

Comparison 9: NSAIDs versus colchicine, Outcome 3: Total adverse events
9.4. Analysis.

Comparison 9: NSAIDs versus colchicine, Outcome 4: Gastrointestinal adverse events
9.5. Analysis.

Comparison 9: NSAIDs versus colchicine, Outcome 5: Other adverse events
Discussion
Summary of main results
We studied 28 trials and included 5 new trials in this review update, with a total of 3406 participants with acute gout who received treatment with non‐steroidal anti‐inflammatory drugs (NSAIDs).
NSAIDs versus placebo
Low‐certainty evidence was based on 1 trial comparing tenoxicam (NSAID) to placebo. There was a gain in benefit, measured as more than 50% improvement in pain after 24 hours. This benefit was lost after 4 days. There was no difference in benefit, measured as more than 50% improvement in swelling at 24 hours or at day 4. There were no data on joint function, participants' global assessment, nor health‐related quality of life (HRQoL).
With regards to harms, there was no evidence of a difference in numbers of withdrawals, total numbers of adverse events, nor serious adverse events between NSAIDs and placebo.
Non‐selective NSAIDs versus cyclo‐oxygenase‐2 inhibitors
Overall, moderate‐certainty evidence is available from 6 trials that compared NSAIDs (indomethacin 50 mg 3 times daily or 75 mg twice daily) to cyclo‐oxygenase‐2 inhibitors (COXIBs) (etoricoxib 120 mg once daily; celecoxib 50, 200, or 400 twice daily; or lumiracoxib 400 mg once daily). With regards to benefit, assessed as mean differences from baseline in pain, inflammation, function, quality of life, and patients' global assessment, there were no differences between non‐selective NSAIDs and COXIBs.
With regards to harms, significantly fewer adverse events and fewer withdrawals due to adverse events were noted among people treated with COXIBs. Although more gastrointestinal adverse events were reported among people who received non‐selective NSAIDs, there was no significant difference in serious adverse events between those taking NSAIDs and those given COXIBs. One trial reported fewer cardiac events in the COXIBs group (etoricoxib) compared with the NSAIDs group (indomethacin) (Rubin 2004).
NSAIDs versus oral or intramuscular glucocorticoids
Overall, moderate‐certainty evidence is available from 4 trials comparing NSAIDs to oral glucocorticoids. With regards to benefit, assessed as mean decrease in pain per time interval, joint function (walking disability), and participants' global assessment of response, there were no statistically significant differences between groups. With regards to inflammation, a statistically significant difference favoured prednisolone. No data on HRQoL were provided.
With regards to harms, more total adverse events were reported with NSAIDs than with glucocorticoids. There were no differences in numbers of serious adverse events nor in gastrointestinal or cardiovascular adverse events.
Other comparisons
We are uncertain of the benefits or harms of the other comparisons, as only low‐certainty to very low‐certainty evidence is available from single trials for NSAID versus rilonacept, NSAID versus acupuncture, one NSAID versus another NSAID (most were single‐trial comparisons and included some NSAIDs that are no longer in use), or NSAIDs versus colchicine.
Overall completeness and applicability of evidence
Demographic data for participants in these studies seem representative of the average gout population. The age of trial participants ranged from 44 to 66 years. Twenty‐one trials included both females and males, and the proportion of males was higher than that of females, ranging from 69% to 97%. Nine trials included participants regardless of the number of joints involved. The proportion of participants with monoarthritis ranged from 66% to 96%.
One of the problems regarding applicability of evidence concerns external validity. This is especially important with regards to comorbidities, which are present in most people with gout and were excluded by most included trials. The short follow‐up duration of the included trials may have precluded the detection of certain adverse events that could have occurred after multiple short periods of drug use. Garcia de la Torre 1987 (comparing NSAIDs to placebo) excluded people with gastrointestinal or cardiac disease. In the comparison of COXIBs versus NSAIDs, all trials excluded people with a history of myocardial infarction or cerebral thrombotic ischaemic disease (or both) or a history of peptic ulcer haemorrhage (Li 2013; Rubin 2004; Schumacher 2002; Schumacher 2012; Willburger 2007; Xu 2016); 5 of the 6 trials also excluded people with other significant medical problems and those who had a concurrent medical condition that could confound or interfere with efficacy evaluations (Li 2013; Rubin 2004; Schumacher 2002; Schumacher 2012; Willburger 2007). Trials that compared NSAIDs to oral glucocorticoids also excluded people with common comorbid conditions such as coronary heart disease, heart failure, history of upper gastrointestinal disease, renal failure, or bleeding disorder. Man 2007 excluded people with a condition that could interfere with assessment without specifying which one, along with people with dementia and confusion. Also the trial comparing NSAIDs to colchicine excluded participants with ischaemic heart disease or impaired liver function (Roddy 2020).
The single trial comparing NSAIDs versus an interleukin (IL)‐1 inhibitor did not exclude people with significant comorbidities, resulting in a population with greater external validity (Terkeltaub 2013).
Quality of the evidence
Generation of an adequate randomisation sequence, concealment of treatment allocation, and blinding of outcome assessment were among the domains that were addressed most poorly, rendering many trials susceptible to selection and detection biases.
Three of the 4 (75%) studies comparing NSAIDs to COXIBs and 1 trial comparing NSAIDs to an IL‐1 inhibitor were sponsored and supported by the company manufacturing etoricoxib and lumiracoxib (1 trial did not mention any funding in the article). Although pharmaceutical industry sponsoring is very common, it has been shown that industry‐sponsored drug studies can lead to more favourable results than sponsorship from other sources (Lundh 2012).
We assessed the certainty of evidence according to the GRADE method.
For the comparison NSAIDs versus placebo, we downgraded the certainty of evidence to low for all outcomes due to study design flaws, making the results susceptible to selection and reporting biases, and because the evidence came from 1 study with 30 participants, we downgraded the results for imprecision.
For the comparison NSAIDs versus COXIBs, we downgraded the certainty of evidence to moderate for pain, inflammation, participants' global assessment of treatment success, study participant withdrawal due to adverse events, and total number of adverse events because of possible bias in study design. We downgraded the certainty of evidence to low for function and quality of life because of bias and imprecision, as evidence for these two outcomes came from a single trial with a small number of participants (45 in each arm).
For the comparison NSAIDs versus glucocorticoids, we downgraded the certainty of evidence to moderate for all outcomes (except inflammation) because of possible bias in study design and because participants in the NSAIDs group in Man 2007 were given an intramuscular injection of NSAIDs while the glucocorticoid group received placebo. We downgraded the certainty of evidence to low for inflammation because of bias and imprecision, as the evidence came from a single trial with a small number of participants (68 in both arms).
The other comparisons (NSAID versus rilonacept, NSAID versus acupuncture, one NSAID versus another NSAID) were not graded, as most were single‐trial comparisons and included many NSAIDs that are no longer in clinical use.
Potential biases in the review process
We believe that we have identified all relevant studies up until the date of the search. We devised a thorough search strategy and searched all major databases for relevant studies, and we applied no language restriction.
Two review authors assessed trials for inclusion in the review, extracted data, and assessed risk of bias independently; a third review author adjudicated in case of any discrepancies, minimising risk errors and bias in the review.
Agreements and disagreements with other studies or reviews
This review is an update of a systematic review we conducted in 2014 (van Durme CMPG 2014), and our conclusions are in agreement with those presented in the previous review.
We have not identified any other systematic review on the use of NSAIDs for acute gout.
In one Cochrane systematic review on the use of systemic glucocorticoids for acute gout (Janssens 2008b), review authors identified the same trial as we did comparing NSAIDs and systemic glucocorticoids (Man 2007), and they concluded that systemic glucocorticoids could be an alternative to NSAIDs for treatment of acute gout, although the evidence was graded as B (moderate risk of bias, moderate‐certainty evidence).
In another Cochrane systematic review on the use of NSAIDs for treatment of low back pain (Roelofs 2008), review authors similarly concluded that NSAIDs were probably equivalent to COXIBs with regards to benefits and harms based on evidence graded as strong by the review authors. With regards to COXIBs, review authors concluded that benefit was similar but that the total number of adverse events was less in the COXIBs group. Gastrointestinal and cardiovascular adverse events were not assessed separately. In our analysis, we also found similar benefit but less harm of COXIBs when compared to NSAIDs based on moderate‐certainty evidence. COXIBs were safer with regards to total adverse events and gastrointestinal and even cardiovascular events. The fact that COXIBs led to fewer cardiovascular events than NSAIDs in the reviewed trials could be due to the short follow‐up duration of included trials and to selection of participants, because 2 trials were published after the upheaval of COXIBs, potentially causing cardiovascular events (Schumacher 2012; Willburger 2007). As NSAIDs and COXIBs are most often used for short periods among people with gout, this issue seems to be less relevant here.
Authors' conclusions
Implications for practice.
Guidelines recommend the use of non‐steroidal anti‐inflammatory drugs (NSAIDs), cyclo‐oxygenase‐2 (COX‐2) inhibitors (COXIBs), low‐dose colchicine, or glucocorticoids for treatment of acute gout flares (FitzGerald 2020Qaseem 2017; Richette 2017). They do not rank any particular therapeutic class above the others but suggest that the choice of first‐line therapy should be individualised depending upon the presence of any comorbidities. Our review lends support to these guidelines. We found only low‐certainty evidence from 1 placebo‐controlled trial (Richette 2010). We downgraded the evidence due to potential selection and reporting biases and imprecision. This study indicated there may be short‐term benefit with NSAIDs during the first 24 hours, which was not evident after 4 days. However, this may be explained by the self‐limiting course of the disease with a mean duration of a few days. Although clinical experience and consensus views based on their effects in other inflammatory arthritis support the use of NSAIDs for acute gout, this low‐certainty single study provides inconclusive evidence to inform guidelines adequately (Richette 2010).
Moderate‐certainty evidence based on 6 trials showed that selective COX‐2 inhibitors and non‐selective NSAIDs were equally beneficial, although COXIBs were associated with significantly fewer total and gastrointestinal adverse events. We downgraded the evidence due to unclear risk of selection and detection bias. Moderate‐certainty evidence based on 5 trials showed that systemic glucocorticoids and NSAIDs are equally beneficial with regards to pain; there could be a beneficial effect of glucocorticoids with regards to reduction of swelling, but this is based on a single trial at high risk of bias for blinding of participants and personnel. Glucocorticoids seem to be associated with fewer adverse events. Researchers found no differences with regards to withdrawal due to adverse events. We found insufficient data regarding interleukin (IL)‐1 inhibitors for treatment of acute gout (1 trial at unclear risk of bias). A single trial at high risk of bias suggests that NSAIDs and colchicine are equally beneficial, but that there is more harm with colchicine with regards to gastrointestinal side effects, especially diarrhoea.
Implications for research.
Further data concerning the comparative benefits and harms of NSAIDs compared with colchicine and intra‐articular glucocorticoids are needed. As both COXIBs and glucocorticoids seem to be better tolerated than NSAIDs for the same efficacy, trials directly comparing COXIBs to glucocorticoids and glucocorticoids to colchicine are needed. Xu 2016 compared COXIBs and glucocorticoids and did not find any difference in benefit nor harm between these two dug classes. However as this trial was at high risk of bias for blinding of personnel and participants, this finding will need to be confirmed in further trials. Also the observation made by Zhang 2014 that glucocorticoids might act more quickly than NSAIDs when given intramuscularly needs to be confirmed, as this was an open‐label trial and thus was at high risk of bias. The single observation that an IL‐1 inhibitor (rilonacept) was not superior to NSAIDs (indomethacin) needs confirmation in other trials, although the cost of these new drugs might preclude their use in routine care. A recent systematic literature review pointed out that canakinumab may be more efficacious than NSAIDs for pain reduction (Zeng 2021), but this needs to be confirmed in larger randomised controlled trials. Another important implication for research should be analysis of the cost‐effectiveness of different drugs. The trial comparing NSAIDs to colchicine is the only trial that assessed cost‐effectiveness (Roddy 2020). Naproxen seemed to be slightly less costly and more effective than colchicine: at a willingness‐to‐pay of £20,000 per quality‐adjusted life‐year (QALY), naproxen had an 80% chance of being cost‐effective compared with colchicine.
Trial reporting should include methods of randomisation and treatment allocation concealment; blinding of study participants, study personnel, and outcome assessment; follow‐up numbers for all participants who entered the trial; and complete reporting of outcomes. Sample sizes should be reported and should have adequate power to answer the research question; ideally trials should assess both benefits and risks of an intervention. To enable comparison and pooling of the results of randomised controlled trials, we suggest that future trials report means with standard deviations for continuous measures, and numbers of events and total numbers analysed for dichotomous measures, and they should assess outcomes recommended by OMERACT (Outcome Measures in Rheumatology Clinical Trials) for studies of acute gout, including pain, joint swelling, joint tenderness, participants' global assessment, and activity limitations (Schumacher 2009). However, how these outcomes have to be assessed exactly still needs to be determined by OMERACT. Therefore, we suggest use of dichotomous measures to report pain as recommended by the International Measurement and Pain Assessment in Clinical Trials (IMMPACT) (the proportions of participants improved by 30% or greater and by 50% or greater) (Dworkin 2008).
What's new
| Date | Event | Description |
|---|---|---|
| 15 November 2021 | New citation required but conclusions have not changed | Five new trials added, conclusions not changed |
| 28 August 2020 | New search has been performed | Search updated, 5 new trials added, conclusions not changed |
History
Protocol first published: Issue 10, 2012 Review first published: Issue 9, 2014
| Date | Event | Description |
|---|---|---|
| 20 October 2014 | Amended | Typing error in abstract and in 'Summary of findings' table |
| 15 January 2014 | Amended | CMSG ID A080‐P |
Acknowledgements
The review authors thank Louise Falzon, Columbia University Medical Center, New York, for assisting with the design of the search strategy.
Thanks to Emma Murphy, Elizabeth Houlding‐Braunberger, Humaira Mahfuz, Nicholas Lebel, and Kaitlyn Brethour for uploading full‐text articles and tagging studies by intervention.
Appendices
Appendix 1. CENTRAL search strategy
1. exp gout/
2. gout$.tw.
3. Acute Disease/
4. acute.tw.
5. 1 or 2
6. 3 or 4
7. 5 and 6
8. exp Anti‐Inflammatory Agents/
9. exp Anti‐Inflammatory Agents, Non‐Steroidal/
10. Aspirin/
11. aspirin.tw.
12. flufenamic acid.tw.
13. mefenamic acid.tw.
14. tolfenamic acid.tw.
15. ibuprofen.tw.
16. ketoprofen.tw.
17. fenoprofen.tw.
18. oxaprozin.tw.
19. sulindac.tw.
20. flurbiprofen.tw.
21. diclofenac.tw.
22. naproxen.tw.
23. tenoxicam.tw.
24. piroxicam.tw.
25. droxicam.tw.
26. indomet?acin.tw.
27. feprazone.tw.
28. phenylbutazone.tw.
29. isoxicam.tw.
30. meclofenamate.tw.
31. ketorolac.tw.
32. lornoxicam.tw.
33. etoricoxib.tw.
34. celecoxib.tw.
35. meloxicam.tw.
36. lumiracoxib.tw.
37. etodolac.tw.
38. nimesulide.tw.
39. exp Cyclooxygenase 2 Inhibitors/
40. exp Cyclooxygenase Inhibitors/
41. Cyclooxygenase 2 inhibitor$.tw.
42. cox 2 inhibitor$.tw.
43. or/8‐42
44. 7 and 43
Appendix 2. MEDLINE search strategy
1. exp gout/
2. gout$.tw.
3. Acute Disease/
4. acute.tw.
5. 1 or 2
6. 3 or 4
7. 5 and 6
8. exp Anti‐Inflammatory Agents/
9. exp Anti‐Inflammatory Agents, Non‐Steroidal/
10. Aspirin/
11. aspirin.tw.
12. flufenamic acid.tw.
13. mefenamic acid.tw.
14. tolfenamic acid.tw.
15. ibuprofen.tw.
16. ketoprofen.tw.
17. fenoprofen.tw.
18. oxaprozin.tw.
19. sulindac.tw.
20. flurbiprofen.tw.
21. diclofenac.tw.
22. naproxen.tw.
23. tenoxicam.tw.
24. piroxicam.tw.
25. droxicam.tw.
26. indomet?acin.tw.
27. feprazone.tw.
28. phenylbutazone.tw.
29. isoxicam.tw.
30. meclofenamate.tw.
31. ketorolac.tw.
32. lornoxicam.tw.
33. etoricoxib.tw.
34. celecoxib.tw.
35. meloxicam.tw.
36. lumiracoxib.tw.
37. etodolac.tw.
38. nimesulide.tw.
39. exp Cyclooxygenase 2 Inhibitors/
40. exp Cyclooxygenase Inhibitors/
41. Cyclooxygenase 2 inhibitor$.tw.
42. cox 2 inhibitor$.tw.
43. or/8‐42
44. 7 and 43
45. randomized controlled trial.pt.
46. controlled clinical trial.pt.
47. randomized.ab.
48. placebo.ab.
49. drug therapy.fs.
50. randomly.ab.
51. trial.ab.
52. groups.ab.
53. or/45‐52
54. (animals not (humans and animals)).sh.
55. 53 not 54
56. 44 and 55
Appendix 3. Embase search strategy
1. exp gout/
2. gout$.tw.
3. acute disease/
4. acute.tw.
5. 1 or 2
6. 3 or 4
7. 5 and 6
8. exp nonsteroid antiinflammatory agent/
9. exp acetylsalicylic acid/
10. aspirin.tw.
11. flufenamic acid.tw.
12. mefenamic acid.tw.
13. tolfenamic acid.tw.
14. ibuprofen.tw.
15. ketoprofen.tw.
16. fenoprofen.tw.
17. oxaprozin.tw.
18. sulindac.tw.
19. flurbiprofen.tw.
20. diclofenac.tw.
21. naproxen.tw.
22. tenoxicam.tw.
23. piroxicam.tw.
24. droxicam.tw.
25. indomet?acin.tw.
26. feprazone.tw.
27. phenylbutazone.tw.
28. isoxicam.tw.
29. meclofenamate.tw.
30. ketorolac.tw.
31. lornoxicam.tw.
32. etoricoxib.tw.
33. celecoxib.tw.
34. meloxicam.tw.
35. lumiracoxib.tw.
36. etodolac.tw.
37. nimesulide.tw.
38. exp cyclooxygenase 2 inhibitor/
39. Cyclooxygenase 2 inhibitor$.tw.
40. cox 2 inhibitor$.tw.
41. or/8‐40
42. 7 and 41
43. (random$ or placebo$).ti,ab.
44. ((single$ or double$ or triple$ or treble$) and (blind$ or mask$)).ti,ab.
45. controlled clinical trial$.ti,ab.
46. RETRACTED ARTICLE/
47. or/43‐46
48. (animal$ not human$).sh,hw.
49. 47 not 48
50. 42 and 49
Data and analyses
Comparison 1. NSAIDs versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1.1 Pain: ≥ 50% improvement in pain | 1 | 90 | Risk Ratio (M‐H, Random, 95% CI) | 1.92 [0.43, 8.57] |
| 1.1.1 Pain with movement at 24 hours | 1 | 30 | Risk Ratio (M‐H, Random, 95% CI) | 4.00 [0.50, 31.74] |
| 1.1.2 Pain with movement at day 4 | 1 | 30 | Risk Ratio (M‐H, Random, 95% CI) | 0.93 [0.73, 1.18] |
| 1.1.3 'Spontaneous' pain at 24 hours | 1 | 30 | Risk Ratio (M‐H, Random, 95% CI) | 2.75 [1.13, 6.72] |
| 1.2 Inflammation: ≥ 50% improvement in joint swelling or tenderness | 1 | 90 | Risk Ratio (M‐H, Random, 95% CI) | 2.11 [0.52, 8.57] |
| 1.2.1 Joint swelling at 24 hours | 1 | 30 | Risk Ratio (M‐H, Random, 95% CI) | 2.50 [0.57, 10.93] |
| 1.2.2 Joint swelling at day 4 | 1 | 30 | Risk Ratio (M‐H, Random, 95% CI) | 1.08 [0.79, 1.49] |
| 1.2.3 Joint tenderness at 24 hours | 1 | 30 | Risk Ratio (M‐H, Random, 95% CI) | 6.00 [0.82, 44.00] |
| 1.3 Withdrawals due to adverse events | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 1.4 Total adverse events | 1 | 30 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.20 [0.01, 3.85] |
1.2. Analysis.

Comparison 1: NSAIDs versus placebo, Outcome 2: Inflammation: ≥ 50% improvement in joint swelling or tenderness
1.4. Analysis.

Comparison 1: NSAIDs versus placebo, Outcome 4: Total adverse events
Comparison 2. NSAIDs versus cyclo‐oxygenase (COX)‐2 inhibitors (COXIBs).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 2.1 Pain: mean change difference from baseline on a 5‐point Likert scale | 6 | Mean Difference (IV, Random, 95% CI) | 0.03 [‐0.07, 0.14] | |
| 2.2 Inflammation: mean change difference in swelling from baseline | 6 | Mean Difference (IV, Random, 95% CI) | 0.08 [‐0.07, 0.22] | |
| 2.3 Function: mean change difference in pain with activity from baseline | 1 | Mean Difference (IV, Random, 95% CI) | 0.04 [‐0.17, 0.25] | |
| 2.4 Participants' global assessment of treatment success | 4 | 730 | Mean Difference (IV, Random, 95% CI) | 0.08 [‐0.04, 0.21] |
| 2.5 Health‐related quality of life measured by 36‐item Short Form | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 2.5.1 Physical Health component | 1 | 222 | Mean Difference (IV, Random, 95% CI) | 0.49 [‐1.61, 2.60] |
| 2.5.2 Mental Health component | 1 | 222 | Mean Difference (IV, Random, 95% CI) | ‐0.18 [‐6.70, 6.34] |
| 2.6 Withdrawals due to adverse events | 6 | 1243 | Risk Ratio (M‐H, Random, 95% CI) | 2.34 [1.33, 4.14] |
| 2.7 Total adverse events | 6 | 1232 | Risk Ratio (M‐H, Random, 95% CI) | 1.94 [1.36, 2.78] |
| 2.8 Gastrointestinal adverse events | 6 | 1232 | Risk Ratio (M‐H, Random, 95% CI) | 2.37 [1.65, 3.40] |
| 2.9 Cardiovascular adverse events | 1 | 189 | Risk Ratio (M‐H, Random, 95% CI) | 2.40 [1.01, 5.67] |
| 2.10 Other adverse events | 6 | 1232 | Risk Ratio (M‐H, Random, 95% CI) | 1.73 [0.93, 3.21] |
| 2.11 Serious adverse events | 5 | 1152 | Risk Ratio (M‐H, Random, 95% CI) | 2.35 [0.49, 11.20] |
Comparison 3. NSAIDs versus glucocorticoids.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 3.1 Pain: mean reduction over visual analogue scale per hour during first 6 hours | 3 | 584 | Mean Difference (IV, Random, 95% CI) | 0.15 [‐2.71, 3.02] |
| 3.2 Inflammation: swelling mean difference in change from day 0 to day 14 | 1 | 69 | Mean Difference (IV, Fixed, 95% CI) | 0.33 [0.07, 0.59] |
| 3.3 Function: walking disability during first 6 hours | 2 | 485 | Mean Difference (IV, Random, 95% CI) | ‐0.21 [‐2.22, 1.80] |
| 3.4 Patient's Global Assessment of response to treatment: good to very good response at day 3 to 4 | 2 | 129 | Risk Ratio (M‐H, Random, 95% CI) | 0.92 [0.70, 1.22] |
| 3.5 Withdrawals due to adverse events | 5 | 772 | Risk Ratio (M‐H, Random, 95% CI) | 2.80 [0.55, 14.22] |
| 3.6 Total adverse events | 5 | 753 | Risk Ratio (M‐H, Random, 95% CI) | 1.62 [1.03, 2.55] |
| 3.7 Gastrointestinal adverse events | 4 | 663 | Risk Ratio (M‐H, Random, 95% CI) | 1.84 [0.90, 3.75] |
| 3.8 Cardiovascular adverse events | 1 | 90 | Risk Ratio (M‐H, Random, 95% CI) | 2.87 [0.12, 68.68] |
| 3.9 Other adverse events | 5 | 753 | Risk Ratio (M‐H, Random, 95% CI) | 1.13 [0.73, 1.76] |
| 3.10 Serious adverse events | 1 | 90 | Risk Ratio (M‐H, Random, 95% CI) | 12.45 [0.72, 214.59] |
Comparison 4. Etodolac versus naproxen.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 4.1 Participants' global assessment at end of therapy: markedly improved | 2 | 121 | Risk Ratio (M‐H, Random, 95% CI) | 1.03 [0.93, 1.14] |
| 4.2 Total adverse events | 2 | 121 | Risk Ratio (M‐H, Random, 95% CI) | 1.74 [0.38, 7.86] |
Comparison 5. NSAIDs versus adrenocorticotropin hormone (ACTH).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 5.1 Withdrawals due to adverse events | 1 | 100 | Risk Ratio (M‐H, Fixed, 95% CI) | 21.00 [1.26, 348.93] |
| 5.2 Total adverse events | 1 | 100 | Risk Ratio (M‐H, Fixed, 95% CI) | 99.00 [6.27, 1562.00] |
Comparison 6. NSAIDs versus interleukin (IL)‐1 inhibitor.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 6.1 Pain: mean pain reduction on numerical rating scale | 1 | 148 | Mean Difference (IV, Random, 95% CI) | ‐2.06 [‐3.06, ‐1.06] |
| 6.2 Withdrawals due to adverse events | 1 | 152 | Risk Ratio (M‐H, Random, 95% CI) | 1.95 [0.18, 21.03] |
| 6.3 Total adverse events | 1 | 152 | Risk Ratio (M‐H, Random, 95% CI) | 0.83 [0.53, 1.31] |
| 6.4 Other adverse events | 1 | 152 | Risk Ratio (M‐H, Random, 95% CI) | 0.83 [0.53, 1.31] |
| 6.5 Serious adverse events | 1 | 152 | Risk Ratio (M‐H, Random, 95% CI) | Not estimable |
6.2. Analysis.

Comparison 6: NSAIDs versus interleukin (IL)‐1 inhibitor, Outcome 2: Withdrawals due to adverse events
6.4. Analysis.

Comparison 6: NSAIDs versus interleukin (IL)‐1 inhibitor, Outcome 4: Other adverse events
6.5. Analysis.

Comparison 6: NSAIDs versus interleukin (IL)‐1 inhibitor, Outcome 5: Serious adverse events
Comparison 7. NSAIDs versus interleukin (IL)‐1 inhibitor plus NSAIDs.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 7.1 Pain (change 24 to 72 hours numerical rating scale) | 1 | 148 | Mean Difference (IV, Random, 95% CI) | 0.46 [‐1.47, 2.39] |
| 7.2 Withdrawals due to adverse events | 1 | 150 | Risk Ratio (M‐H, Random, 95% CI) | 0.97 [0.14, 6.73] |
| 7.3 Total adverse events | 1 | 150 | Risk Ratio (M‐H, Random, 95% CI) | 0.66 [0.43, 1.00] |
| 7.4 Serious adverse events | 1 | 150 | Risk Ratio (M‐H, Random, 95% CI) | 0.14 [0.01, 2.65] |
Comparison 8. NSAIDs versus acupuncture combined with infrared irradiation.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 8.1 Pain: mean score on visual analogue scale after treatment | 1 | 160 | Mean Difference (IV, Random, 95% CI) | 2.22 [1.77, 2.67] |
Comparison 9. NSAIDs versus colchicine.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 9.1 Mean change in pain over days 1 to 7 | 1 | 344 | Mean Difference (IV, Fixed, 95% CI) | 0.30 [‐0.37, 0.97] |
| 9.2 Quality of life: EQ‐5D at day 7 | 1 | 399 | Mean Difference (IV, Fixed, 95% CI) | 0.01 [‐0.02, 0.04] |
| 9.3 Total adverse events | 1 | 399 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.90 [0.73, 1.10] |
| 9.4 Gastrointestinal adverse events | 1 | 399 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.82 [0.71, 0.96] |
| 9.5 Other adverse events | 1 | 399 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.00 [0.20, 4.87] |
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Altman 1988.
| Study characteristics | ||
| Methods |
Design: RCT
Sample size: not described
Analysis: not described whether ITT or PP Withdrawals: 4 (13.8%) from ketoprofen group (3 (10%) adverse reactions, 1 (3%) lack of co‐operation); 9 (29%) from indomethacin group (1 (3%) lack of benefit, 3 (10%) adverse reactions, 4 (13%) lost to follow‐up, 1 (3%) ineligible for the study) |
|
| Participants | 59 participants (29 in ketoprofen group, 30 in indomethacin group) Participant characteristics Mean age: 55.3 years (ketoprofen group); 57.4 years (indomethacin group) Male: 90% (ketoprofen group); 93% (indomethacin group) Mean disease duration: not described Mean number of affected joints: not described Affected joints: not described Inclusion criteria: acute gout, confirmed by identification of MSU crystals in synovial fluid or fulfilling 2/4 clinical criteria (clear history or observation of at least 2 attacks of acute arthritis with abrupt onset and remission, history/observation of podagra, presence of tophi, history/observation of response to colchicine within 48 hours of therapy); onset of inflammation within 48 hours before study entry, at least "moderate" pain or tenderness; total score of at least 9 for 5 symptoms on a scale of 0 (absent) to 3 (severe) Exclusion criteria: hypersensitivity to NSAIDs; pregnancy or lactation; had already received anti‐inflammatory therapy for acute arthritis, GI bleeding, or active peptic ulcer; impairment of renal/hepatic/cardiac function; other serious illness |
|
| Interventions |
Group 1: day 1: ketoprofen 3 × 50 mg capsules followed by up to 3 doses of 100 mg ≥ 3 hours apart (max 9 capsules/450 mg); days 2 to 7, 100 mg 3 times daily Group 2: day 1: indomethacin 3 × 25 mg capsules followed by up to 3 doses of 50 mg ≥ 3 hours apart (max 9 capsules/450 mg); days 2 to 7, 50 mg 3 times daily In both groups, drug therapy was discontinued if the participant had no clinical response, had "intolerable" AEs, or was in full remission on day 5 |
|
| Outcomes | Outcomes evaluated at prespecified time intervals: days 1, 2, 5, 8, and 15 Primary outcome • Composite score of pain, tenderness, restriction of motion, and swelling (each scored on a scale of 0 = absent to 3 = severe; overall score 0 to 12) Secondary outcomes • Clinical global improvement as graded by participants and physicians (marked, moderate, slight, no change, or worse) (on days 2, 5, 8, and 15) • Time to onset of pain relief recorded by participants in their diaries • AEs, with grading of severity and effect as definitely, probably, possibly, or not related to therapy |
|
| Notes | Data for 7/59 participants in the study were excluded from the benefit assessment: 2 participants in each group had inadequate drug intake, 2 participants in the indomethacin group had no follow‐up data and 1 had been misdiagnosed Funding: not reported Adverse events Group 1: ketoprofen Total adverse events: 15/29 Nature of events: occult blood in stool, diarrhoea, abdominal pain, sleepiness, dizziness, dyspepsia Group 2: indomethacin Total adverse events: 16/30 Nature of events: melena, diarrhoea, dyspepsia, abdominal pain, pruritic rash, dizziness, headache, nervousness, insomnia |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Method of randomisation not described Quote: "...each patient was randomly assigned to receive..." |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "under double blind conditions..."; no further description |
| Blinding of outcome assessment for self‐reported outcomes (detection bias) | Unclear risk | Not described |
| Blinding of outcome assessment for assessor‐reported outcomes (detection bias) | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Of 4 participants who did not complete the trial, there was 1 withdrawal from the ketoprofen group (3%) and 3 from the indomethacin group (17%) |
| Selective reporting (reporting bias) | Low risk | All prespecified outcomes reported |
| Other bias | Low risk | No other potential sources of bias identified |
Axelrod 1988.
| Study characteristics | ||
| Methods | Design: RCT Sample size: not described Analysis: not described whether ITT or PP Withdrawals: 10 (20%) from indomethacin group (AE ‐ abdominal discomfort) | |
| Participants | 100 (50 in indomethacin group, 50 in ACTH group)
Participant characteristics
Mean age (± SD): 63 ± 8 years (indomethacin group); 66 ± 10 years (ACTH group)
Male: 100% Mean disease duration: not described Mean number of affected joints: 90% monoarticular, 10% oligoarticular Affected joints: 100 (100%) participants MTP‐1, 10 (10%) participants knee (in combination with MTP‐1) Inclusion criteria: presented within 24 hours of onset of pain from an acute gout attack; diagnosis of gout determined by the clinical picture or the presence of intracellular urate crystals in aspirated materials (or both), as well as absence of organisms on Gram stain To remain in the study, each participant was required to present to investigators within 24 hours of onset of each attack of gout, for a period of 1 year Exclusion criteria: polyarticular gout; tophaceous gout; blood in the stool; current abdominal discomfort; major organ system or systemic infection; history of recurrent headaches; malignancy; autoimmune or endocrine disease; acute myocardial infarction; renal dysfunction as suggested by electrolyte, blood urea nitrogen, or serum creatinine determinations; pregnancy; receiving immunosuppressive drugs or concurrent treatment with steroids, colchicine, allopurinol, probenecid, or NSAIDs |
|
| Interventions | Group 1: indomethacin 50 mg orally 4 times daily with food, until pain abated Group 2: ACTH 40 IU intramuscularly in a single dose Subsequent documented acute gouty attacks were treated according to the therapeutic group assigned at study entry | |
| Outcomes | Outcomes evaluated at prespecified time intervals: day 1, on days 5 to 7 following each acute attack Primary outcome • Estimate of time from administration of therapy to complete relief of pain Secondary outcomes • Frequency of attacks during study period • AEs (not prespecified) | |
| Notes |
Funding: not reported Adverse events No side effects were noted in the ACTH group However, in the indomethacin group, 22 patients experienced abdominal discomfort or dyspepsia, 15 experienced headaches, and 12 developed difficulty with mentation |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | High risk | Alternate assignment of participants to the 2 treatment groups |
| Allocation concealment (selection bias) | High risk | Treatment not concealed |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Neither participants nor personnel blinded |
| Blinding of outcome assessment for self‐reported outcomes (detection bias) | High risk | Participants not blinded |
| Blinding of outcome assessment for assessor‐reported outcomes (detection bias) | High risk | Personnel not blinded |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Of 24 participants who did not complete the study, 10 (20%) were from the indomethacin group (refused to participate because of abdominal discomfort or headaches) and 14 (28%) were from the ACTH group (did not complete 1‐year follow‐up) |
| Selective reporting (reporting bias) | Low risk | We assumed that time to complete pain relief referred to time on ambulation |
| Other bias | Low risk | No other potential sources of bias identified |
Butler 1985.
| Study characteristics | ||
| Methods | Design: RCT Sample size: quote: "approximately 60 patients would be required in each group to detect a difference of two days in the mean intervals between onset of treatment and resolution of the attack at the 5% significance level with 90% power..." Analysis: not described whether ITT or PP Withdrawals: 5 (31.25%) in flurbiprofen group; 2 (11.8%) in phenylbutazone group; 2 incorrect diagnosis (not reported which study drug they used); 1 lack of benefit (flurbiprofen); 1 resolution of attack without treatment (not reported which study drug they used); 1 prolonged interval (> 24 hours) between start of attack and initiation of treatment; 2 given medication but no attack occurred | |
| Participants | 33 participants (16 in flurbiprofen group, 17 in phenylbutazone group) Participant characteristics Mean age: 52.8 years (phenylbutazone group); 56.2 years (flurbiprofen group) Male: not described Mean disease duration: not described Mean number of affected joints: not described Affected joints: not described Inclusion criteria: quote: "the diagnosis of acute gout was made on clinical grounds, supported in 15 cases by the demonstration of urate crystals within synovial fluid" Exclusion criteria: severe dyspepsia; GI bleeding; concomitant use of other NSAIDs or anticoagulants |
|
| Interventions |
Group 1: flurbiprofen 400 mg daily for 2 days, then 200 mg daily for 10 days Group 2: phenylbutazone 800 mg daily for 2 days, then 400 mg daily for 10 days |
|
| Outcomes | Outcomes evaluated at 10 days Primary outcome • Severity of pain at beginning and end of treatment (10 days) on a 5‐point scale Secondary outcomes • Time to resolution of symptoms • Requirement for additional analgesics • AEs (no further details prespecified) |
|
| Notes |
Funding: not reported Adverse events Group 1 (flurbiprofen) Total adverse events: 3/16 Nature of events: rash, dyspepsia, constipation, sleepiness, irritability Group 2 (phenylbutazone) Total adverse events: 5/17 Nature of events: dry skin, diarrhoea and dyspepsia, 'shaking of hand' |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Method of randomisation not described Quote: "patients were randomly allocated..." |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "the study drugs were supplied in identical unmarked study capsules" |
| Blinding of outcome assessment for self‐reported outcomes (detection bias) | Low risk | Participants blinded |
| Blinding of outcome assessment for assessor‐reported outcomes (detection bias) | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | 7 participants withdrawn: 5 from flurbiprofen group and 2 from phenylbutazone group. Reasons for withdrawal were 2 incorrect diagnosis (1 participant's request), 1 because of ongoing symptoms after 3 days on flurbiprofen, 1 resolution of attack without treatment, 1 prolonged interval between onset of attack and treatment, 2 participants had study medication given to them but no acute attack occurred |
| Selective reporting (reporting bias) | High risk | Pain assessed on an ordinal scale was not reported |
| Other bias | Low risk | No other potential sources of bias identified |
Cheng 2004.
| Study characteristics | ||
| Methods | Design: RCT Sample size: equivalence trial, with sample calculation based on an arbitrarily defined margin of equivalence between study drugs for pain Analysis: not described whether ITT or PP Withdrawals: 3; 1 from diclofenac group (4.8%) due to personal factors; 2 from meloxicam group (9.5%) (1 due to study drug ineffectiveness on day 2; 1 due to personal factors) | |
| Participants | 62 participants (20 in rofecoxib group, 21 in diclofenac group, 21 in meloxicam group) Participant characteristics Mean age: 52.2 years (rofecoxib group); 50.4 years (diclofenac group); 50.6 years (meloxicam group) Male: 53/60 (88%) Mean disease duration: not described Mean number of affected joints: not described Affected joints: not described Inclusion criteria: acute gout (onset < 48 hours) according to 1977 American College of Rheumatology criteria for classification of acute gout; physician‐assessed total inflammatory score ≥ 5 on a 0‐ to 9‐point scale (the sum of scores for restriction of function (0 to 3 points), tenderness (0 to 3 points), and swelling (0 to 3 points) of most severely affected joint); participant assessment of pain intensity as moderate, severe, or extreme; stable dose of any concomitant hypouricaemic agent for > 30 days; quote: "...patients had to be in otherwise good health..."; unresponsive acute gout; ≥ 3 joints involved; allergic reaction to any component of study drugs/other NSAIDs, or aspirin; history of asthma associated with nasal polyps; history of GI ulcer bleeding or perforation within 6 months; history of chronic analgesic or tranquilliser use or dependency within 3 months; uncontrolled hypertension, diabetes mellitus, renal disease, or neurological disorder; history of gastric, biliary, or small intestinal surgery resulting in clinical malabsorption; stroke or any significant cardiovascular, hepatic, or neoplastic disease or clinically significant abnormalities on pre‐study examination; significantly abnormal laboratory tests, such as serum aspartate aminotransferase levels > 2‐fold above upper limit of normal or serum creatinine level > 1.4 mg/dL; pregnant, possibly pregnant, and breastfeeding or using inadequate contraception; regularly consumed alcohol; use of NSAIDs or systemic corticosteroids within 48 hours before the study; on anticoagulant or antiplatelet drugs; use of colchicine (> 1 mg/d) within 8 days before study entry |
|
| Interventions |
Group 1: diclofenac sodium SR 75 mg daily for 7 days
Group 2: meloxicam 7.5 mg daily for 7 days Note: rofecoxib group not included in analysis as drug no longer in use |
|
| Outcomes | Outcomes evaluated at days 3 and 8 Primary outcome • Overall analgesic effect by participant and investigator global assessment of response to therapy: responses were no effect, poor, fair, good, or excellent (5‐point verbal scale) Secondary outcomes • Overall anti‐inflammatory effect using total inflammatory score (to determine tenderness, swelling, and restriction of function) using a 10‐point numerical score from 0 (no pain, swelling, or restriction of movement) to 9 (extreme pain to which participant winces and withdraws, swelling and bulging beyond the joint margin, and complete joint immobilisation) • Intensity of pain on a 5‐point verbal scale of none, slight, moderate, severe, or extreme at baseline (pre‐dose), and at 0.5, 1, 2, 6, and 12 hours after initial dosing of study drug • AEs: quote: "the tolerability of the study medications was determined using physical examination by the investigators, vital signs measured by assisting nurses, outpatient laboratory testing (estimated serum creatinine clearance [CCr] and serum aspartate aminotransferase [AST] level), and spontaneous reporting of any AEs by the patient" | |
| Notes |
Funding: drugs supplied by manufacturers (Merck, Novartis, Boehringer Ingelheim) Adverse events Group 1: diclofenac Total adverse events: 7/21 Nature of events: abdominal pain, oedema Group 2: meloxicam Total adverse events: 6/21 Nature of events: abdominal pain, oedema |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "patients were consecutively randomly assigned...according to a predetermined block randomisation table with block factor of 6" |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Participants not blinded; study personnel blinded Quote: "although patients were not blinded, all of the medications were prepackaged and sealed to maintain blinding of the investigators and study staff" |
| Blinding of outcome assessment for self‐reported outcomes (detection bias) | High risk | Participants not blinded |
| Blinding of outcome assessment for assessor‐reported outcomes (detection bias) | Low risk | Outcome assessors blinded |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 3 participants withdrawn: 1 from diclofenac group (4.8%); 2 from meloxicam group (9.5%). 1 in diclofenac group and 1 in meloxicam group withdrew due to personal factors; 1 in meloxicam group withdrew due to study drug ineffectiveness on day 2. |
| Selective reporting (reporting bias) | Low risk | All prespecified outcomes reported |
| Other bias | Low risk | No other potential sources of bias identified |
Douglas 1970.
| Study characteristics | ||
| Methods |
Design: RCT Sample size: equivalence trial, with sample calculation based on an arbitrarily defined margin of equivalence between study drugs for pain Analysis: ITT Withdrawals: 1 (not described if this was from phenylbutazone or flufenamic acid group) for use of phenylbutazone tablets a few days before the start of the trial |
|
| Participants | 25 participants (14 in phenylbutazone group, 11 in flufenamic acid group) Participant characteristics Mean age: 57.2 years (flufenamic acid group); 47.6 years (phenylbutazone group) Male: 22/25 (88%) Mean disease duration: 1 day to 26 years (mean 6 years) Mean number of affected joints: not described Affected joints: not described Inclusion criteria: acute gout (no further details of basis for diagnosis described) Exclusion criteria: had received phenylbutazone, flufenamic acid, or other NSAID in the preceding month; contraindication to phenylbutazone or flufenamic acid |
|
| Interventions |
Group 1: phenylbutazone 200 mg every 6 hours for 48 hours, then 100 mg every 6 hours until complete resolution of acute attack or dosage limit of 50 capsules used Group 2: flufenamic acid 200 mg every 6 hours for 48 hours, then 100 mg every 6 hours until complete resolution of acute attack or dosage limit of 50 capsules used |
|
| Outcomes | Outcomes evaluated day 0, 1, 2, 4, 7, 10, and 14 Primary outcome • Inflammatory index computed from the mean of combined scores for pain, heat, redness, local swelling, and tenderness (using an arbitrary 0 to 3 scale, with absent 0, slight 1, moderate 2, severe 3) Secondary outcomes • Function (graded as normal 0, slightly impaired 1, severely impaired 2) • Limb volume using a water displacement method • AEs |
|
| Notes |
Funding: not reported Adverse events Group 1 (phenylbutazone): 0 Group 2 (flufenamic acid): 0 |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Participants were allocated to 1 or other treatment group from a random series |
| Allocation concealment (selection bias) | Unclear risk | Unclear how allocation was concealed |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Both participants and personnel blinded Quote: "both drugs were supplied in identical gelatine capsules" |
| Blinding of outcome assessment for self‐reported outcomes (detection bias) | Low risk | Participants blinded |
| Blinding of outcome assessment for assessor‐reported outcomes (detection bias) | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 1 participant withdrawn from study (found to have taken phenylbutazone before study entry) |
| Selective reporting (reporting bias) | Low risk | All prespecified outcomes reported |
| Other bias | High risk | Mean age significantly higher in flufenamic acid group (57.2 years) vs phenylbutazone group (47.6 years) |
Eberl 1983.
| Study characteristics | ||
| Methods |
Design: RCT Sample size: not described Analysis: not described Withdrawals: 0 |
|
| Participants | 20 participants (10 in each group) Participant characteristics Mean age: 46.5 (range 31 to 71) years (meclofenamate group); 53 years (range 34 to 72) (indomethacin group) Male: 100% Mean disease duration: not described Mean number of affected joints: 19 (95%) participants with monoarticular, 1 (5%) participant with oligoarticular Affected joints: 7/21 (33%) big toe, 8/21 (38%) knee, 4/21 (19%) ankle, 1/21 (5%) thumb, 1/21 (5%) wrist Inclusion criteria: monoarticular acute gout of < 48 hours; quote: "...characterised by sudden onset with agonising pain in the afflicted joint accompanied by sensitivity to touch, reddening and local increase of temperature" Exclusion criteria: if drug treatment had already begun; acute gout overlying tophus; signs of GI or bone marrow disease; hypersensitivity to study drugs; concomitant anticoagulants; haemoglobin ≤ 9 g/L, haematocrit ≤ 30%, or white blood cell ≤ 3500/mm³ |
|
| Interventions |
Group 1: meclofenamate 200 mg, then 100 mg every 4 hours for the first 24 hours, then 100 mg every 8 hours for 6 days Group 2: indomethacin 25 mg, then 25 mg every 4 hours for the first 24 hours, then 50 mg every 8 hours for 6 days |
|
| Outcomes | Outcomes evaluated daily for 7 days, then on days 10 and 14 Primary outcome • Spontaneous pain, on a 4‐point scale (from 0 (no pain) to 3 (severe pain)) Secondary outcomes • Swelling on a 4‐point scale (from 0 (no pain) to 3 (severe pain)) • Tenderness on a 4‐point scale (from 0 (no pain) to 3 (severe pain)) • Limitation of mobility from 0 (no limitation) to 3 (severe limitation) • AEs |
|
| Notes |
Funding: not reported Adverse events Group 1 (meclofenamate) Total AEs 2/10 Nature of events: abdominal pain, nausea Group 2 (indomethacin) Total AEs: 5/10 Nature of events: abdominal pain, nausea, diarrhoea, headache, pruritis, dizziness |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Method of randomisation not described |
| Allocation concealment (selection bias) | Unclear risk | Allocation concealment not described |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Specific details about how blinding was assured not specified |
| Blinding of outcome assessment for self‐reported outcomes (detection bias) | Unclear risk | Unclear whether participants are blinded |
| Blinding of outcome assessment for assessor‐reported outcomes (detection bias) | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Withdrawals not described; probably no withdrawal due to adverse reaction because study authors state it was not necessary to discontinue medication because of intolerance |
| Selective reporting (reporting bias) | Low risk | All prespecified outcomes described |
| Other bias | High risk | Clinical diagnosis of acute gout Study author comment: other conditions like acute calcium pyrophosphate arthropathy could have mimicked acute gout and were not considered; higher initial meclofenamate dose vs indomethacin dose |
Garcia de la Torre 1987.
| Study characteristics | ||
| Methods |
Design: RCT Sample size: not performed Analysis: no data given Withdrawals: 1 in placebo group (unsatisfactory response to treatment) |
|
| Participants | 30 participants (15 in each group) Participant characteristics Mean (± SD) age: 55 years (± 11.9) Male: 29/30 (97%) Mean disease duration: not described Mean number of affected joints: not described Affected joints: 14/30 (47%) knee, 7/30 (23%) ankle, 4/30 (14%) wrist, 3/30 (10%) elbow, 2/30 (6%) MTP‐1 (2 participants) Inclusion criteria: acute gouty arthritis based on the simultaneous presence of spontaneous pain in a joint; pain induced by palpation or mobilisation in the same joint; joint swelling, local heat, redness over joint; plus at least 1 MSU crystal in synovial fluid or tophi or podagra or sUA > 7.0 mg/dL in men or > 6.0 mg/dL in women Exclusion criteria: adolescence; pregnancy; gastric peptic disease; renal, hepatic, or cardiac disease; treatment with anticoagulant drugs |
|
| Interventions |
Group 1: tenoxicam 40 mg once daily (before supper) for 4 days Group 2: placebo 4 days |
|
| Outcomes | Participants were reviewed every day for 4 days, then 1 week after stopping treatment. At all visits, the following data were collected Primary outcomes • Time to first improvement • Time to resolution Secondary outcomes (no list of secondary outcomes was provided; just a sentence stating that each clinical criterion was evaluated at each visit and evolution was analysed) • Spontaneous pain in joint (pain intensity: 1 = slight, 2 = moderate, 3 = severe; scale given verbally; pain improvement from baseline: 1 = complete resolution of pain, 2 = pain improvement > 50%, 3 = pain improvement ≤ 50%, 4 = increase in pain intensity) • Pain on palpation (with the same 2 scales as spontaneous pain) • Pain on joint movement (with the same 2 scales as spontaneous pain) • Heat (with the same 2 scales as spontaneous pain) • Swelling (with the same 2 scales as spontaneous pain) • Redness (with the same 2 scales as spontaneous pain) • Joint circumference (measured only at baseline and final visit) • AEs |
|
| Notes |
Funding: not reported Adverse events Observed in only 2 patients in the placebo group; 1 of them manifested polyuria, and the other nausea |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No details of how randomisation was done |
| Allocation concealment (selection bias) | Unclear risk | No details |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Placebo pills were the same as tenoxicam pills |
| Blinding of outcome assessment for self‐reported outcomes (detection bias) | Low risk | Participants were blinded |
| Blinding of outcome assessment for assessor‐reported outcomes (detection bias) | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | We do not know how many participants had data available for each item, so it is difficult to judge exclusion. We do not know what happened to the 1 excluded participant on day 3 in the assessments after day 3 |
| Selective reporting (reporting bias) | Unclear risk | No data reported 1 week after assessment (4 days showed no relevant differences, but what about rebound attacks?) |
| Other bias | Low risk | No other risk of bias identified |
Janssens 2008a.
| Study characteristics | ||
| Methods | Design: RCT Sample size: equivalence trial, with sample calculation based on an arbitrarily defined margin of equivalence between study drugs for pain Analysis: ITT, finally done PP. Quote: "analyses were done primarily for the per‐protocol group for sensitivity reasons (i.e., not missing difference) and repeated for the intention‐to‐treat group" Withdrawals: 1 (1%) in prednisone group; 1 (1%) in naproxen group (both incomplete data) | |
| Participants | 120 participants (60 in each group) Participant characteristics Mean age: 57.3 years (prednisolone group); 57.7 years (naproxen group) Male: 107/120 (89%) Mean duration of symptoms: not described Mean number of affected joints: 100% monoarthritis (quote: "participants were patients with a monoarticular gout arthritis...") Affected joints: 76/120 (63%) MTP‐1, 35 (29%) foot joints/ankle or knee, 9 (8%) elbow/wrist/hand Inclusion criteria: monoarticular acute gout, confirmed by identification of MSU crystals in synovial fluid Exclusion criteria: unstable comorbid condition (angina pectoris, myocardial infarction, heart failure, severe renal failure, renal transplant, or cancer); chronic rheumatic disease; current anticoagulant use; medical history of upper GI disease |
|
| Interventions |
Group 1: prednisolone 35 mg once daily and placebo naproxen twice daily for 5 days Group 2: naproxen 500 mg twice daily and placebo prednisolone once daily for 5 days |
|
| Outcomes | Outcomes evaluated at prespecified time intervals: 0 to 6, 6 to 18, 18 to 30, 30 to 42, 42 to 54, 54 to 66, 66 to 78, 78 to 90 hours Primary outcome • Pain as reported by the participant on a 100‐mm VAS Secondary outcomes • Disability related to use of affected joint scored on a 100‐mm VAS from 0 mm = absence of disability to 100 mm = completely unable to do something • Walking disability scored on a 100‐mm VAS from 0 mm = walking without any problem to 100 mm = completely unable to walk • AEs: quote: "patients were asked to select in the trial diary one or more of five categories of side‐effects: none; gastric pain, abdominal pain, or both; itch, dizziness, or both; dyspnoea, palpitations, or both; other" |
|
| Notes |
Funding: Rheumatology Research Fund Arnhem, Netherlands Adverse events Group 1 (prednisolone) Total adverse events: 20/60 Nature of adverse events: dyspnoea, itch, dizziness, palpitations, gastric and abdominal pain Group 2 (naproxen) Total adverse events: 22/60 Nature of adverse events: dyspnoea, itch, dizziness, palpitations, gastric and abdominal pain |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer‐generated randomisation procedure by an independent trial monitor Quote: "allocation sequence list with a block randomisation of four treatments, each treatment being given twice" |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "...double dummy design..."; participants and study personnel blinded to interventions |
| Blinding of outcome assessment for self‐reported outcomes (detection bias) | Low risk | Participants blinded to allocated study treatment |
| Blinding of outcome assessment for assessor‐reported outcomes (detection bias) | Low risk | Outcome assessment blinded to allocated study treatment |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Only 1 participant in each group had incomplete outcome data; quote: "both drop‐outs" had arthritis of leg or foot |
| Selective reporting (reporting bias) | Low risk | All outcomes reported |
| Other bias | Low risk | No other risk of bias identified |
Klumb 1996.
| Study characteristics | ||
| Methods |
Design: RCT Sample size: not reported Analysis: simple comparisons between groups; not clear if it was ITT or PP Withdrawals: not reported |
|
| Participants | 34 participants (20 in nimesulide group, 14 in indomethacin group) Participant characteristics Mean age: 54.55 ± 14.9 years (nimesulide group); 55 ± 10.8 years (indomethacin group) Male: 100% Mean duration of disease: 6.85 ± 5.71 years (nimesulide group); 7.42 ± 6.5 years (indomethacin group) Mean number of affected joints: 75% monoarthritis (nimesulide group); 78.6% monoarthritis (indomethacin group); 76% monoarthritis (both groups) Affected joints: 32% knee, 27% ankle, 27% 1st MTP, 12% wrist, 3% elbow Inclusion criteria: diagnosis of acute gouty arthritis confirmed by demonstration of sUA crystals in synovial fluid or hyperuricaemia associated with classic clinical history; no use of NSAIDs in the current crisis; attack duration < 72 hours; participant informed consent Exclusion criteria: chondrocalcinosis; hypersensitivity to drugs in investigation; renal or hepatic failure; post‐operative or post‐myocardial infarction state; active dyspeptic disease |
|
| Interventions | Group 1: nimesulide: first 24 hours 100 mg every 6 hours, followed by 100 mg every 8 hours during 72 hours, then 100 mg every 12 hours in the last 3 days. Total treatment duration: 7 days for all participants, except those with complete remission in a shorter period Group 2: indomethacin: first 24 hours 50 mg every 6 hours, followed by 50 mg every 8 hours during 72 hours, then 50 mg every 12 hours during 72 hours. Total treatment duration: 7 days for all participants, except those with complete remission in a shorter period | |
| Outcomes | Outcomes assessed at days 3 and 7
Outcomes (not exactly specified which was the primary outcome)
• Intensity of joint signs and symptoms on a 5‐point VAS (0 = absent, 1 = mild, 2 = moderate, 3 = intense, 4 = extreme) • Pain at rest on a 5‐point VAS (0 = absent, 1 = mild, 2 = moderate, 3 = intense, 4 = extreme) • Pain with active mobilisation on a 5‐point VAS (0 = absent, 1 = mild, 2 = moderate, 3 = intense, 4 = extreme) • Articular oedema and erythema on a 5‐point VAS (0 = absent, 1 = mild, 2 = moderate, 3 = intense, 4 = extreme) • Physicians' global assessment on a 5‐point VAS (0 = absent, 1 = mild, 2 = moderate, 3 = intense, 4 = extreme) • Patients' global assessment on a 5‐point VAS (0 = absent, 1 = mild, 2 = moderate, 3 = intense, 4 = extreme) • Erythrocyte sedimentation rate AEs • Numbers and types of AEs |
|
| Notes |
Funding: not reported Adverse events Group 1 (nimesulide) Total adverse events: 17/20 Nature of events: gastric pain, heartburn, diarrhoea, rash, oedema Group 2 (indomethacin) Total adverse events: 14/14 Nature of events: gastric pain, diarrhoea, rash, oedema, heartburn |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Participants were randomly distributed to 1 of 2 groups. No further specifications Quote: "os pacientes que preencheram os critérios de eligibilidade foram distribuídos randomicamente para um dos dois grupos de tratamento" |
| Allocation concealment (selection bias) | Unclear risk | Not reported |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Both drugs were administered in coded and identical capsules Quote: "as duas drogas em investigação foram administradas em cápsulas codificadas e idênticas" |
| Blinding of outcome assessment for self‐reported outcomes (detection bias) | Low risk | Participants blinded to treatment |
| Blinding of outcome assessment for assessor‐reported outcomes (detection bias) | Unclear risk | Not reported |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Not reported whether there were any missing data and consequently which type of analyses was used (ITT vs PP). From some tables, it is clear that the number of participants at each visit was the same, so there seemed to be no missing data (but not stated) |
| Selective reporting (reporting bias) | Unclear risk | Not clear, because outcomes were not clearly specified in the methods. Most often, comparisons were made between groups in terms of change variables, which were not shown, and just status scores at each visit |
| Other bias | Low risk | No other risk of bias identified |
Lederman 1990.
| Study characteristics | ||
| Methods | Design: RCT Sample size: not described Analysis: not described whether ITT or PP Withdrawals: 0 | |
| Participants | 60 participants (29 in etodolac group; 31 in naproxen group)
Participant characteristics
Mean age: 48 years (etodolac group); 49 years (naproxen group)
Male: 58/60 (97%) Mean disease duration: not described Mean number of affected joints: 100% monoarthritis (quote: "the patients had experienced acute pain...in a single joint for less than 48 hours") Affected joints: not described Inclusion criteria: monoarticular acute pain, tenderness, redness; heat for < 48 hours in the context of hyperuricaemia and history of at least 2 episodes of arthritis with acute onset and remission within 2 weeks; previous podagra or previous response to colchicine or presence of tophi (or a combination) Exclusion criteria: chondrocalcinosis; pregnancy or lactation; concurrent use of NSAIDs; other arthritic disorder; history of serious cardiovascular, hepatic, or renal disease |
|
| Interventions | Group 1: etodolac 300 mg twice daily for 7 days Group 2: naproxen 500 mg 3 times daily for 7 days | |
| Outcomes | Outcomes evaluated at prespecified time intervals: baseline, days 2, 4, and 7 (or when the participant withdrew from the study)
Primary outcomes
• Participants' overall evaluation on a 1 to 5 scale (1 = very good, 2 = good, 3 = fair, 4 = poor, 5 = very poor)
• Physicians' overall evaluation on a 1 to 5 scale (1 = very good, 2 = good, 3 = fair, 4 = poor, 5 = very poor)
Secondary outcomes
• Pain intensity, degree of swelling, degree of erythema, tenderness on a 1 to 5 scale (1 = none, 2 = mild, 3 = moderate, 4 = severe, 5 = very severe) • Range of motion on a 1 to 5 scale (1 = normal, 2 = mildly restricted, 3 = moderately restricted, 4 = severely restricted, 5 = immobilised) • Heat on a 1 to 4 scale (1 = none, 2 = mild, 3 = moderate, 4 = marked) |
|
| Notes |
Funding: Wyeth‐Ayerst Laboratories, Philadelphia, PA, USA Adverse events Group 1: etodolac Total adverse events: 1/29 Nature of event: intermittent dizziness and tremors Group 2: naproxen No adverse events in this group |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | High risk | Method of randomisation not described Quote: "patients were randomly assigned to receive...", but baseline prespecified outcome values were statistically higher (P < 0.05) in the etodolac group than in the naproxen group |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | No details provided on how blinding was assured |
| Blinding of outcome assessment for self‐reported outcomes (detection bias) | Unclear risk | Not described |
| Blinding of outcome assessment for assessor‐reported outcomes (detection bias) | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No withdrawals Quote: "no patient withdrew from the study for any reason other than remission of gout symptoms" |
| Selective reporting (reporting bias) | Low risk | All prespecified outcomes reported |
| Other bias | Low risk | No other risk of bias identified |
Li 2013.
| Study characteristics | ||
| Methods | Design: RCT Sample size: 179 participants (90 in etoricoxib group, 89 in indomethacin group) Analysis: ITT Withdrawals: 1 (0.5%) lost to follow‐up in the etoricoxib group, 0 in the indomethacin group | |
| Participants | 179 participants (90 in etoricoxib group, 89 in indomethacin group) Participant characteristics Mean age: 52 ± 15 years (etoricoxib group); 53 ± 14 years (indomethacin group) Male: 85 (96%) (etoricoxib group), 81 (91%) (indomethacin group) Mean disease duration: not described Mean number of affected joints: monoarticular: 75 (83%) participants (etoricoxib group), 73 (82%) participants (indomethacin group); polyarticular: 14 (17%) participants (etoricoxib group), 27 (18%) participants (indomethacin group) Affected joints: MTP 46 (52%) (etoricoxib group 53%, indomethacin group 60%); intertarsal joint 6 (8%) (etoricoxib group 6%, indomethacin group 8%); ankle 21 (27%) (etoricoxib group 19%, indomethacin group 24%); knee 10 (13%) (etoricoxib group 2%, indomethacin group 3%); wrist 3 (4%) (etoricoxib group 4%, indomethacin group 5%); elbow 1 (1%) (etoricoxib group 1%, indomethacin group 1%); other 2 (3%) (etoricoxib group 4% indomethacin group 5%) Inclusion criteria: adults with acute gout attack (< 48 hours from onset), diagnosed according to American College of Rheumatology classification criteria, and with total score of 5 (of a max possible score of 10) on 3 symptom questions for pain (0‐ to 4‐point Likert scale), tenderness (0‐ to 3‐point scale), and swelling (0‐ to 3‐point scale); eligible participants also had at least 1 blood count, blood chemistry, and urinalysis performed 1 year before randomisation without abnormalities that would contraindicate the use of any study medication Exclusion criteria: concurrent medical/arthritic disease that could confound evaluation of benefit or that contraindicated use of study medication; history contraindicating use of indomethacin; polyarticular gout involving > 4 joints; history of cancer during previous 5 years or history of cerebrovascular events, myocardial infarction, or coronary bypass in the previous year; received corticosteroids within 1 month before randomisation; received anticoagulants, ticlopidine, clopidogrel, or digoxin; use of NSAIDs 48 hours before baseline assessments or analgesics, including aspirin, within 6 hours before baseline assessments or during the trial |
|
| Interventions |
Group 1: 1 tablet of etoricoxib 120 mg or placebo from bottle A once daily in the morning for 5 days Group 2: 1 capsule of indomethacin 75 mg or placebo from bottle B twice daily (morning and evening) for 5 days |
|
| Outcomes | Outcomes evaluated at prespecified time intervals: baseline, 4 hours after initial dose on day 1, 4 hours after daily dose on days 2 and 5 Primary outcome • Patients' assessment of pain in the affected joint from baseline, as reported by patients on a 0‐ to 4‐point (0 = no pain, 4 = extreme pain) Likert scale Secondary outcome • Investigators' assessment of tenderness of the study joint on a 4‐point Likert scale (0 = no pain, 1 = patient states there is pain, 2 = patient winces, 3 = patient withdraws) • Investigators' assessment of swelling of study joint on a 4‐point Likert scale (0 = no swelling to 3 = bulging beyond joint margins) • Patients' and investigators' global assessment of response to therapy on a 5‐point Likert scale (0 = excellent, 4 = poor) • Proportion of participants who discontinued treatment because of lack of efficacy • AEs |
|
| Notes |
Funding: Merck Sharp & Dohme, a subsidiary of Merck & Company, Inc. Adverse events Group 1: etoricoxib Total AEs: 13/89 Serious AEs: 0/89 Nature of total adverse events: gastric dilation, diarrhoea, stomachache, dizziness, chills, fever, leg swelling Group 2: indomethacin Total AEs: 19/89 Serious AEs: 1/89 Nature of total adverse events: abdominal distension, stomach upset, stomachache, digestive tract upset, nausea, cough, somnolence, right‐hand numbness, dizziness, leg swelling Nature of serious adverse events: 16‐day duration of gouty arthritis in a formerly unaffected joint |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer‐generated allocation schedule |
| Allocation concealment (selection bias) | Low risk | Study sponsor who provided the computer‐generated allocation schedule was blinded to group allocations |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "patients took one tablet of etoricoxib or placebo from bottle A once daily and one tablet of indomethacin or placebo from bottle B twice daily" |
| Blinding of outcome assessment for self‐reported outcomes (detection bias) | Low risk | Participants blinded |
| Blinding of outcome assessment for assessor‐reported outcomes (detection bias) | Low risk | Outcome assessors blinded to treatment |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 1 withdrawal in the etoricoxib group due to loss to follow‐up |
| Selective reporting (reporting bias) | Low risk | All prespecified outcomes were reported |
| Other bias | Low risk | None apparent |
Lomen 1986.
| Study characteristics | ||
| Methods |
Design: RCT Sample size: not described Analysis: not described whether ITT or PP Withdrawals: 3 in flurbiprofen group (2 lack of benefit, 1 AEs) |
|
| Participants | 29 participants (14 in flurbiprofen group, 15 in indomethacin group) Participant characteristics Mean age: not described Male: not described. Quote: "there were no significant differences in demographic characteristics....among patients" Mean disease duration: 5.8 years (flurbiprofen group); 6.9 years (indomethacin group) Mean number of affected joints: 100% monoarthritis (quote: "...patients presenting with an acute attack of monoarticular gouty arthritis...") Affected joints: not described Inclusion criteria: monoarticular gout of < 48 hours' duration defined as abrupt onset of excruciating pain in the involved joint accompanied by tenderness, erythema, and heat; diagnosis confirmed by synovial fluid analysis for urate crystals or presence of hyperuricaemia and 2 out of 4 of the following clinical criteria: clear history or observation of at least 2 attacks of acute arthritis with abrupt onset and remission, history/observation of podagra, presence of tophi, history of decrudescence after colchicine within 48 hours Exclusion criteria: chondrocalcinosis; active peptic ulcer; serious concomitant disease |
|
| Interventions |
Group 1: flurbiprofen 100 mg 4 times daily on day 1, then 50 mg 4 times daily for a max of 5 days if necessary Group 2: indomethacin 50 mg 4 times daily on day 1, then 25 mg 4 times daily for a max of 5 days if necessary |
|
| Outcomes | Outcomes evaluated at prespecified time intervals: baseline, then every 24 hours for 5 days Primary outcome • Pain on motion and at rest graded by participants according to the Keele Scale as none, slight, moderate, severe, and extreme (5‐point scale) Secondary outcomes • Swelling and erythema graded by investigators as mild, moderate, or severe • Local heat graded by investigators as normal or elevated • Participants' and investigators' subjective assessment of improvement compared with pretreatment condition graded as much better, better, the same, worse, or much worse • Skin temperature determined by an electronic tape‐on surface thermistor probe placed on the most inflamed area at the same place each day • AEs (not prespecified) |
|
| Notes |
Funding: not reported Adverse events Group 1 (flurbiprofen) Total AEs: 5/14 Nature of events: abdominal pain, oedema of feet, diarrhoea, nausea Serious AEs: 1/14 Nature of event: massive upper gastrointestinal bleeding Group 2 (indomethacin) Total AEs: 2/15 Nature of events: abdominal pain, oedema of feet, diarrhoea, nausea Serious AE: 0/15 |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Method of randomisation not described Quote: "...in accordance with a standardised randomisation scheme..." |
| Allocation concealment (selection bias) | Unclear risk | Unclear how allocation was concealed |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Specific details about how blinding was assured were not specified |
| Blinding of outcome assessment for self‐reported outcomes (detection bias) | Unclear risk | Specific details about how blinding was assured were not specified |
| Blinding of outcome assessment for assessor‐reported outcomes (detection bias) | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 3 from flurbiprofen group withdrew: 1 after 96 hours because of lack of benefit, 1 at 24 hours because of GI AEs, and 1 at 48 hours because of lack of response |
| Selective reporting (reporting bias) | Low risk | All prespecified outcomes described |
| Other bias | Low risk | No other potential sources of bias identified |
Maccagno 1991.
| Study characteristics | ||
| Methods |
Design: RCT Sample size: not described Analysis: not described whether ITT or PP Withdrawals: 0 |
|
| Participants | 61 participants (31 in etodolac group, 30 in naproxen group) Participant characteristics Mean age: 55 years (etodolac group); 54 years (naproxen group) Males: 47/61 (77%) Mean disease duration: not described Mean number of affected joints: 100% monoarthritis (quote: "...patients had to have experienced...in a single joint...") Affected joints: not described Inclusion criteria: monoarticular acute pain, tenderness, redness, heat; onset of attacks < 48 hours; diagnosis of gout confirmed by presence of hyperuricaemia and history of at least 2 episodes of arthritis with acute onset and remission within 2 weeks or presence of tophi, podagra, or good response to colchicine or a combination Exclusion criteria: chondrocalcinosis; low‐grade synovitis secondary to chronic gout; pregnancy or lactation; women of childbearing potential |
|
| Interventions |
Group 1: etodolac 300 mg twice daily for 7 days Group 2: naproxen 500 mg twice daily for 7 days |
|
| Outcomes | Outcomes evaluated at prespecified time intervals: baseline, days 2, 4, and 7 Primary outcome • Pain intensity, swelling, tenderness, erythema on a 1 to 5 scale (1 = none, 2 = mild, 3 = moderate, 4 = severe, 5 = very severe) Secondary outcomes • Heat on a 1 to 4 scale (1 = none, 2 = mild, 3 = moderate, 4 = marked) • Physicians' and participants' overall evaluation on a 1 to 5 scale (1 = very good, 2 = good, 3 = fair, 4 = poor, 5 = very poor) • Range of motion on a 1 to 5 scale (1 = normal, 2 = mildly restricted, 3 = moderately restricted, 4 = severely restricted, 5 = immobilised) • AEs (not prespecified) |
|
| Notes |
Funding: grant from Wyeth‐Ayerst Laboratories, Philadelphia, PA, USA Adverse events Group 1: etodolac Total adverse events: 3/31 Nature of events: dyspepsia, abdominal pain Group 2: naproxen Total adverse events: 2/30 Nature of events: headache, abdominal pain |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Method of randomisation not described Quote: "...patients were allocated at random to receive..." |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Specific details about how blinding was assured were not specified |
| Blinding of outcome assessment for self‐reported outcomes (detection bias) | Unclear risk | Not described |
| Blinding of outcome assessment for assessor‐reported outcomes (detection bias) | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | None Quote: "...all patients were included in the analysis" |
| Selective reporting (reporting bias) | Low risk | All prespecified outcomes reported |
| Other bias | Low risk | No other potential sources of bias identified |
Man 2007.
| Study characteristics | ||
| Methods |
Design: RCT Sample size: not described Analysis: ITT Withdrawals: 0 |
|
| Participants | 90 participants (46 in indomethacin group, 44 in prednisolone group) Participant characteristics Mean age: 66 years (indomethacin group); 64 years (prednisolone group) Male: 74/90 (82%) Mean disease duration: not described Mean number of affected joints: monoarthritis: 45 (98%) participants (indomethacin group), 41 (93%) participants (prednisolone group); > 1 joint involved: 1 (2%) participant (indomethacin group), 3 (7%) participants (prednisolone group) Affected joints: not described Inclusion criteria: clinical diagnosis of acute arthritis suggestive of gout, defined as presence of pain and warmth in a joint; presented within 3 days of onset of pain and also had ≥ 1 of the following: metatarsal‐phalangeal joint involvement or knee/ankle joint involvement and aspirate containing crystals or typical gouty arthritis with either gouty tophi present or previous joint aspiration confirming the diagnosis of gout Exclusion criteria: clinical suspicion of sepsis/other joint disease; if follow‐up was deemed to be impossible, comorbidity that would interfere with assessment; concurrent presence of dementia/confusion/active GI symptoms/renal insufficiency with serum creatinine level > 200 mol/L/bleeding disorder/treatment with warfarin; allergy to study drugs; joint aspirate that excluded the diagnosis of gout |
|
| Interventions |
Group 1: prednisolone: intramuscular placebo, oral prednisolone 30 mg, paracetamol 1 g, and placebo indomethacin, followed by prednisolone 30 mg for 5 days, paracetamol 1 g every 4 hours as required, and placebo indomethacin 50 mg 3 times daily for 2 days and 25 mg 3 times daily for 3 days Group 2: indomethacin: intramuscular diclofenac 75 mg, oral indomethacin 50 mg, paracetamol 1 g, and placebo prednisolone, followed by indomethacin 50 mg 3 times daily for 2 days and 25 mg 3 times daily for 3 days; and paracetamol 1 g every 4 hours as required and placebo prednisolone for 5 days |
|
| Outcomes | Outcomes evaluated at prespecified time intervals: by telephone interview at 24 hours or physical review if in hospital and review after 5 and 14 days. Pain scores and AEs were recorded every 30 minutes for 2 hours after drug administration Participants in the indomethacin group received diclofenac 75 mg intramuscularly in addition to indomethacin 50 mg orally; those in the prednisolone group received prednisone 30 mg orally and intramuscular placebo Primary outcomes • Pain as reported by participants on a 10‐cm VAS (0 = absence of pain to 10 = the most severe pain the participant had ever experienced) • AEs: quote: "patients were asked to select in the trial diary one or more of five categories of side‐effects: none; gastric pain, abdominal pain, or both; itch, dizziness, or both; dyspnoea, palpitations, or both; other" Secondary outcomes • Time to complete resolution of pain, stiffness, and joint swelling • Supplementary paracetamol • Treatment failure defined as non‐resolution of symptoms or recurrence of symptoms at day 14 • Relapse rate |
|
| Notes | With regards to pain scores, quote: "patients were aware of their previous scores at all stages of recording" Funding: no funding received Adverse events Group 1: prednisolone Total adverse events: 12/44 Nature of events: rash, dizziness, drowsiness, dry mouth, nausea Group 2: indomethacin Total adverse events: 29/46 Nature of events: epigastric pain, nausea, diarrhoea, vomiting, indigestion, dizziness, drowsiness |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer‐generated random sequence |
| Allocation concealment (selection bias) | Low risk | Allocation concealed in sealed envelopes |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Both participants and study personnel blinded Quote: "...preparations and identical placebos were all pre packed..." |
| Blinding of outcome assessment for self‐reported outcomes (detection bias) | Low risk | Participants blinded |
| Blinding of outcome assessment for assessor‐reported outcomes (detection bias) | Low risk | Outcome assessors blinded |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No withdrawals in the present study |
| Selective reporting (reporting bias) | Unclear risk | Secondary endpoints not reported in the prespecified way |
| Other bias | Low risk | None apparent |
Rainer 2016.
| Study characteristics | ||
| Methods |
Design: RCT Sample size: equivalence trial, with sample calculation based on a predefined margin of equivalence between study drugs for pain, based on previous study finding that a clinically relevant difference in pain score on a 100‐mm VAS is greater than 13 mm Analysis: PP and ITT Withdrawals: 18 in the indomethacin group, 19 in the prednisolone group |
|
| Participants | 416 participants (208 in indomethacin group, 208 in prednisolone group) Participant characteristics Mean age: 64.37 years (indomethacin group); 65.91 years (prednisolone group) Male: 164/208 (78.8%) (indomethacin group), 145 (69.7%) (prednisolone group) Mean disease duration: 2.74 days (indomethacin group); 2.91 (prednisolone group) Affected joints: only metatarsophalangeal 1 joint involvement 86/208 (41.3%) (indomethacin group), 94/208 (45.2%) (prednisolone group) Number (%) of gouty tophi: 27 (13%) (indomethacin group); 34 (16.3%) (prednisolone group) Inclusion criteria: presented to the emergency department (ED) within 3 days of symptom onset; considered to have gout by a specialist emergency physician and to have fulfilled the following 2 criteria for the diagnosis of acute gout: • First: rapid onset of severe pain, swelling, tenderness, and erythema of affected joint, which was maximal by 6 to 12 hours • Second: at least 1 of the following clinical findings: ‐ A: metatarsophalangeal (MTP) joint involvement (podagra) ‐ B: knee, ankle, wrist, or elbow joint involvement with: ‐ B1: tophi ‐ B2: previous joint aspiration confirming a diagnosis of gout ‐ B3: hyperuricaemia ‐ B4: clinical history of 1 or more clinical gouty arthritis attacks If criteria B1 to B4 were not met, confirmation was sought by microscopic examination of aspirated fluid from the most affected joint for the presence of MSU crystals Exclusion criteria: corticosteroids or indomethacin within 24 hours before recruitment; history of bleeding disorder or anticoagulant use; allergic to a study drug; suspected septic arthritis or another joint disease; no monosodium crystals found after joint aspiration; unstable cardiac condition (angina pectoris, acute, myocardial infarction, heart failure); significant comorbidities that could interfere with assessment (dementia, confusion, or active gastrointestinal symptoms); serum creatinine level > 200 μmol/L (> 2.26 mg/dL) or estimated glomerular filtration rate < 30 mL/min/1.73 m² |
|
| Interventions |
Group 1: indomethacin: 50 mg (two 25‐mg tablets) of oral indomethacin 3 times a day and 6 tablets of oral placebo prednisolone once a day for 2 days, followed by 25 mg of indomethacin 3 times a day and 6 tablets of placebo prednisolone once a day for 3 days. Paracetamol 1 g was also prescribed to be taken every 6 hours if needed Group 2: prednisolone: initially 30 mg (three 10‐mg tablets) of oral prednisolone once a day and 2 tablets of placebo indomethacin 3 times a day for 2 days, followed by 30 mg (3 tablets) of prednisolone once a day and 1 tablet of placebo indomethacin 3 times a day for 3 days. Paracetamol 1 g was also prescribed to be taken every 6 hours if needed |
|
| Outcomes | Outcomes evaluated at prespecified time intervals: 0, 30 minutes, 60 minutes, 90 minutes, and 120 minutes after intake of first tablets, assessed by a study investigator, and once a day for 14 days after patient's visit to the ED documented in a trial diary Primary outcome • Pain at rest and with activity in the worst affected joint, measured on a 100‐mm VAS (0 mm (complete absence of pain) to 100 (the most severe pain a patient had ever experienced)) Secondary outcomes •Tenderness assessed using a 5‐point Likert scale • Joint swelling assessed using a 3‐point Likert scale • Joint redness assessed using a 5‐point Likert scale • Time to symptom resolution • Use of paracetamol: recorded as yes or no • Length of stay at the ED • Patient satisfaction with analgesia and ED services (addressed on days 5 and 14) using a scale of 0 (complete dissatisfaction) to 100 (complete satisfaction) • Adherence to study medication: assessed by checking completeness of data in diaries and by counting unused drugs on day 5. Adherence was defined as 100% of prescribed drugs taken on a given day • Health care utilisation (visits to GP or ED) • Functional activity assessed with the physical component of the SF‐36 (data sought but not found) • SF‐36 • Adverse events prespecified as dizziness, sleepiness, nausea, vomiting, abdominal pain, indigestion, rash, dry mouth, and any other symptom. Major (or serious) AE required hospitalisation |
|
| Notes |
Funding: Health and Health Services Research Grant Committee of the Hong Kong Government Adverse events Group 1: indomethacin Total AEs: 82/208 Nature of adverse events: dizziness, sleepiness, rash, dry mouth Gastrointestinal AEs: 67/208 Nature of adverse events: vomiting, abdominal pain, nausea, indigestion Serious AEs: 0/208 Group 2 prednisolone Total AEs: 96/208 Nature of adverse events: dizziness, sleepiness, rash, dry mouth Gastrointestinal AEs: 30/208 Nature of adverse events: vomiting, abdominal pain, nausea, indigestion Serious AEs: 0/208 |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer‐generated randomisation |
| Allocation concealment (selection bias) | Low risk | Sealed envelopes and packages |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Double dummy (number of tablets taken by each group was checked with authors of the article) |
| Blinding of outcome assessment for self‐reported outcomes (detection bias) | Low risk | Participants blinded |
| Blinding of outcome assessment for assessor‐reported outcomes (detection bias) | Low risk | Outcome assessors blinded |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | In the indomethacin group, 18 patients dropped out, 19 in the prednisolone group; reasons for dropout are given. 1 patient in the indomethacin group had incomplete data, 2 in the prednisolone group, and therefore were excluded from PP analysis |
| Selective reporting (reporting bias) | Unclear risk | Swelling, length of stay, and SF‐36 were not reported |
| Other bias | Low risk | None apparent |
Roddy 2020.
| Study characteristics | ||
| Methods |
Design: RCT Sample size: superiority trial of naproxen or colchicine (2‐tailed hypothesis testing), with sample calculation based on a predefined margin of equivalence between study drugs for pain, based on previous study finding that a clinically relevant difference in pain score on a 100‐mm VAS is greater than 13 mm Analysis: PP and ITT Withdrawals: 6 in the naproxen group, 4 in the colchicine group; reasons for withdrawal were not specified |
|
| Participants | 399 participants (200 in naproxen group, 199 in colchicine group) Participant characteristics Mean (SD) age: 58.7 (14.4) years (naproxen group); 60.0 (13.4) years (colchicine group) Male: 173/200 (86.5%) (naproxen group); 174/199 (87.4%) (colchicine group) Mean disease duration: not described Mean number of affected joints: not specified; only number of body parts affected is mentioned Affected joints: first metatarsophalangeal joint involvement 142/200 (72.4%) (naproxen group), 135/199 (69.2%) (colchicine group); other foot joints 58/200 (29.6%) (naproxen group), 48/199 (24.6%) (colchicine group); other lower limb 46/200 (23.5%) (naproxen group), 47/199 (24.1%) (colchicine group); upper limb 23/200 (11.7%) (naproxen group), 31/199 (15.9%) (colchicine group) Inclusion criteria: aged 18 years and older; consulting for a current gout flare. Clinical diagnosis was made by the GP without joint aspiration, blood tests, imaging, or diagnostic criteria Exclusion criteria: unstable medical condition (e.g. ischaemic heart disease, impaired liver function); known stage 4/5 chronic kidney disease (estimated glomerular filtration rate/creatinine clearance < 30 mL/min); recent surgery or gastrointestinal bleed; history of gastric ulcer; current anticoagulant use; allergy to aspirin or NSAID; previous inability to tolerate naproxen or low‐dose colchicine; other contraindication to either study drug described in the 'Summary of product characteristics'; prescription of naproxen or colchicine in previous 24 hours; pregnancy or lactation; potentially vulnerable patients; participation in the CONTACT trial during previous gout flare or involvement in another clinical trial in the last 90 days or other research within 30 days |
|
| Interventions | Group 1: single initial dose of oral naproxen 750 mg (three 250‐mg tablets), followed by 250 mg (1 tablet) every 8 hours for up to 7 days. Co‐prescription of a proton pump inhibitor was done at the GP's discretion Group 2: 500 mcg (1 tablet) of colchicine every 8 hours for 4 days. Participants prescribed a statin were advised to omit the statin during colchicine treatment |
|
| Outcomes | Outcome measures were collected by self‐complete daily diary (days 1 to 7) and a questionnaire at week 4 Primary outcome • Mean change in pain intensity from baseline measured over first 7 days (worst pain experience in the last 24 hours using a 0 to 10 numerical rating scale (NRS)) Secondary outcomes • Time to treatment effect • Complete pain resolution (reporting 0 or 1 on NRS) • Self‐reported side effects (nausea, vomiting, headache, skin rash, dyspepsia, abdominal pain, constipation, diarrhoea) • Patient global assessment of treatment response (completely better/much better/somewhat better/about the same/somewhat worse/much worse) • Use of corticosteroids, paracetamol, NSAIDs, or opiates for gout pain • Treatment adherence • Relapsed/recurrent gout flare • Quality of life (EQ‐5D‐5L) • Attendance at GP, emergency department, or primary care out‐of‐hours service • Absence from work/education |
|
| Notes |
Funding: National Institute for Health Research School for Primary Care Research (NIHR SPCR) UK Adverse events Group 1 naproxen Total adverse events: 91/200 Nature of events: headache, rash Gastrointestinal events: 89/200 Nature of events: nausea, vomiting, abdominal pain, dyspepsia, diarrhoea Group 2 colchicine Total adverse events: 101/199 Nature of events: headache, rash Gastrointestinal events: 76/199 Nature of events: nausea, vomiting, abdominal pain, dyspepsia |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Web access to a secure remote allocation system or a telephone randomisation service |
| Allocation concealment (selection bias) | Low risk | Clinicians did not know which treatment a participant would receive before randomisation |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Open label |
| Blinding of outcome assessment for self‐reported outcomes (detection bias) | High risk | Open label |
| Blinding of outcome assessment for assessor‐reported outcomes (detection bias) | High risk | Open label |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | 6 in the naproxen group, 4 in the colchicine group; reasons for withdrawal were not specified |
| Selective reporting (reporting bias) | Low risk | All prespecified outcomes were reported |
| Other bias | Unclear risk | There was a difference in length of treatment: naproxen was given for 4 days and colchicine was given for 7 days |
Rubin 2004.
| Study characteristics | ||
| Methods | Design: RCT Sample size: quote: "a sample size of 87 patients per group had 90% power to demonstrate comparability if the true (not the observed) mean difference between the etoricoxib and indomethacin groups was 0.1" Analysis: modified ITT approach, which included all treated participants who had measurements at baseline and at least once during treatment Withdrawals: 11 in etoricoxib group (5 (4.9%) AEs, 5 (4.9%) lack of benefit, 1 (1.0%) loss to follow‐up); 14 in indomethacin group (5 (5.8%) AEs, 7 (8.1%) lack of benefit, 1 (1.2%) loss to follow‐up, 1 (1.2%) protocol deviation (not specified what type of protocol deviation)) | |
| Participants | 189 participants (103 in etoricoxib group, 86 in indomethacin group) Participant characteristics Mean age: 51.1 years (etoricoxib group); 52.2 years (indomethacin group) Male: 176/189 (93%) Mean disease duration: not described Mean number of affected joints: monoarticular: 81 (79%) participants (etoricoxib group 63%, indomethacin group 73%); polyarticular: 22 (21%) participants (etoricoxib group 23%, indomethacin group 27%); 144 (76%) participants from whole study population had monoarthritis; 44 (24%) participants from whole study population had polyarthritis Affected joints: not described Inclusion criteria: adults with acute gout attack (< 48 hours from onset), diagnosed according to 1977 American College of Rheumatology classification criteria, and with total score of 5 (of a max possible score of 10) on 3 symptom questions for pain (0‐ to 4‐point Likert scale), tenderness (0‐ to 3‐point scale), and swelling (0‐ to 3‐point scale), with the pain score of at least 2 = moderate, 3 = severe, or 4 = extreme on the Likert scale; eligible participants also had at least 1 blood count, blood chemistry, and urinalysis performed within 1 year before randomisation without abnormalities that would contraindicate the use of any study medication Exclusion criteria: concurrent medical/arthritic disease that could confound evaluation of benefit or that contraindicated use of study medication; previous gout non‐responsive to NSAIDs; polyarticular gout involving > 4 joints; history of allergy to NSAIDs |
|
| Interventions |
Group 1: etoricoxib 120 mg daily for 8 days Group 2: indomethacin 50 mg 3 times daily for 8 days |
|
| Outcomes | Outcomes evaluated at prespecified time intervals: baseline, then daily for days 2 to 8, 4 hours after daily dose of study medication Primary outcome • Pain in affected joint from days 2 to 5, as reported by participant on a 0‐ to 4‐point (0 = no pain, 4 = extreme pain) Likert scale over days 2 to 5 measured at baseline, then 4 hours after study medication Secondary outcome • Participant assessment of pain in the study joint (all secondary endpoints measured over days 2 to 8) • Participant and investigator global assessment of response to therapy on a 5‐point Likert scale (0 = excellent, 4 = poor) • Investigators' assessment of tenderness and swelling of study joint • Proportion of participants who discontinued treatment because of lack of benefit • AEs (throughout treatment period and for 14 days after completion) |
|
| Notes |
Funding: Merck Co. Adverse events Group 1: etoricoxib Total adverse events: 45/103 Nature of events: gastrointestinal effects, cardiovascular effects including hypertension, oedema Serious adverse event: 1/103 Nature of event: renal failure Group 2: indomethacin Total adverse events: 49/86 Nature of events: gastrointestinal effects, cardiovascular effects including hypertension, oedema |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Method of randomisation not described |
| Allocation concealment (selection bias) | Unclear risk | Methods of allocation not described |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Participants and personnel blinded, as double dummy was used |
| Blinding of outcome assessment for self‐reported outcomes (detection bias) | Low risk | Participants blinded |
| Blinding of outcome assessment for assessor‐reported outcomes (detection bias) | Low risk | Outcome assessors blinded |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 11 withdrawals in etoricoxib group (5 AEs, 5 lack of benefit, 1 loss to follow‐up); 14 withdrawals in indomethacin group (5 AEs, 7 lack of benefit, 1 loss to follow‐up, 1 protocol deviation) |
| Selective reporting (reporting bias) | Low risk | All prespecified outcomes reported |
| Other bias | Low risk | No other potential sources of bias identified |
Schumacher 2002.
| Study characteristics | ||
| Methods | Design: RCT Sample size: to have comparable benefit, a power calculation revealed the requirement of 62 participants per group Analysis: ITT Withdrawals: 8 (11%) in etoricoxib group (3 (4.0%) lack of benefit, 2 (2.7%) clinical adverse experience, 1 (1.3%) laboratory adverse experience, 2 (2.7%) other reasons); 15 (20%) in indomethacin group (2 (2.7%) lack of benefit, 8 (10.7%) clinical adverse experience, 5 (6.7%) other reasons) | |
| Participants | 150 participants (75 in each group) Participant characteristics Mean age: 48.5 years (etoricoxib group); 49.5 years (indomethacin group) Male: 73 (97%) (etoricoxib group); 69 (92%) (indomethacin group) Mean disease duration: not described Mean number of affected joints: monoarthritis: 46 (61%) participants (etoricoxib group 53%, indomethacin group 71%); polyarthritis: 29 (39%) participants (etoricoxib group 22%, indomethacin group 29%) Affected joints: 55 (37%) big toe, 32 (21%) ankle, 27 (18%) knee, 36 (24%) others Inclusion criteria: adults with acute gout attack (< 48 hours from onset), diagnosed as per ACR 1977 classification criteria with total score of 5 (of a max possible score of 10) on 3 symptom questions for pain (0‐ to 4‐point scale), tenderness (0‐ to 3‐point scale), and swelling (0‐ to 3‐point scale); no abnormalities on blood count, blood chemistry, and urinalysis done within 1 year before randomisation Exclusion criteria: concurrent medical/arthritic disease that could confound evaluation of benefit or that contraindicated use of study medication; unstable medical condition; contraindication to use of indomethacin; cancer in the past 5 years; cerebrovascular events, myocardial infarction, or coronary bypass in the past 1 year; concurrent use of anticoagulants, digoxin, ticlopidine, or clopidogrel; corticosteroid use within 1 month of study entry; use of NSAIDs within 48 hours or aspirin/analgesics within 6 hours of study entry/during the trial |
|
| Interventions |
Group 1: etoricoxib 120 mg per day for 8 days Group 2: indomethacin 50 mg 3 times daily for 8 days Participants permitted to continue low‐dose aspirin (≤ 325 mg daily) and colchicine (≤ 1.2 mg daily) if taken at a stable dose for > 30 days before randomisation |
|
| Outcomes | Outcomes evaluated at prespecified time intervals: baseline, then daily on days 2 to 8 Primary outcome • Pain in affected joint on a 0‐ to 4‐point scale (none, mild, moderate, severe, extreme) at baseline and 4 hours after study medication (on each day until end of treatment) Secondary outcomes (all secondary endpoints measured over days 2 to 8) • Investigator assessment of tenderness (scale of 0 to 3: 'no pain' to 'patient states there is pain, winces and withdraws'); swelling (0 to 3: 'none' to 'bulging beyond joint margins'); erythema (present or absent) • Participant and investigator global assessment of response to therapy on a 5‐point Likert scale (0 = excellent, 4 = poor) • Investigators' assessment of tenderness and swelling of study joint • Proportion of participants who discontinued treatment because of lack of benefit • AEs (for quote: "intensity, seriousness, and relation to study drug while blinded to the treatment allocation") |
|
| Notes |
Funding: Merck Research Laboratories, Rahway, NJ, USA Adverse events Group 1: etoricoxib Total adverse events: 17/75 Nature of adverse events: dizziness, headache, sleepiness, gastric events Group 2: indomethacin Total adverse events: 35/75 Nature of adverse events: dizziness, headache, sleepiness, gastric events Serious adverse events: 4/75 Nature of events: vomiting, headache, drug overdose, laryngeal neoplasm |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Randomisation done by computer‐generated allocation schedule |
| Allocation concealment (selection bias) | Unclear risk | Unclear how allocation was concealed |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Double‐dummy design Quote: "patients took one tablet of etoricoxib or placebo from bottle A...and one capsule of indomethacin or placebo from bottle B three times a day" |
| Blinding of outcome assessment for self‐reported outcomes (detection bias) | Low risk | Participants blinded |
| Blinding of outcome assessment for assessor‐reported outcomes (detection bias) | Low risk | Outcome assessor blinded |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | 8 withdrawals in etoricoxib group (3 lack of benefit, 3 AEs, 2 other reasons) and 15 withdrawals in indomethacin groups (2 lack of benefit, 8 AEs, 5 other reasons) |
| Selective reporting (reporting bias) | Low risk | All prespecified outcomes reported |
| Other bias | Unclear risk | Merck Research Laboratory provided funding to all participating investigators to cover the costs of patient procedures and investigations. 1 study author was on the Merck advisory board, 1 study author was a consultant for Merck, and 4 study authors were employed by Merck and owned shares of Merck common stock |
Schumacher 2012.
| Study characteristics | ||
| Methods |
Design: RCT
Sample size: sample size of 100 participants per group would provide about 90% power to demonstrate superior benefit of high‐dose regimen
Analysis: ITT
Withdrawals: 62 (21%) in celecoxib group, 24 (24%) in indomethacin group Celecoxib 50 mg twice daily group: 24 (24%) withdrawals (9 (9%) lack of benefit, 5 (5%) AEs not related to study drug, 1 (1%) loss to follow‐up, 9 (9%) other reasons) Celecoxib 400/200 mg twice daily group: 20 (20%) withdrawals (6 (6%) lack of benefit, 1 (1%) AE not related to study drug, 9 (9%) other reasons, 4 (4%) no longer willing to participate) Celecoxib 400/200 mg twice dailygroup: 16 (16%) withdrawals (4 (4%) lack of benefit, 1 (1%) AE not related to study drug, 1 (1%) loss to follow‐up, 9 (9%) other reasons, 1 (1%) no longer willing to participate) Indomethacin 50 mg 3 times daily group: 24 (24%) withdrawals (6 (6%) AEs related to study drug, 2 (2%) lack of benefit, 3 (3%) AEs not related to study drug, 2 (2%) losses to follow‐up, 11 (11%) other reasons) |
|
| Participants | 400 participants (101 in celecoxib 50 mg twice daily group; 99 in celecoxib 400/200 mg twice daily group; 98 in celecoxib 800/400 mg twice daily group; 102 in indomethacin group) Participant characteristics Mean age: 52.4 years (celecoxib 50 mg twice daily group); 52.3 years (celecoxib 400/200 mg twice daily group); 51.0 years (celecoxib 800/400 mg twice daily group); 49.6 years (indomethacin group) Male: 90% in all groups Mean disease duration: not described Mean number of affected joints: 82 (81.2%) monoarticular/19 (19%) oligoarticular (celecoxib 50 mg twice daily group), 78 (79%) monoarticular/21 (21%) oligoarticular (celecoxib 400/200 mg twice daily group), 72 (74%) monoarticular/26 (27%) oligoarticular (celecoxib 800/400 mg twice daily group), 78 (77%) monoarticular/24 (23%) oligoarticular male (indomethacin group) Affected joints: not described Inclusion criteria: adults with acute gout attack (< 48 hours from onset), diagnosed as per ACR 1977 classification criteria with moderate, severe, or extreme pain in an index joint identified by investigator over previous 24 hours on 5‐point (0 to 4) Likert scale and, in the opinion of the investigator, to be candidates for daily therapy with NSAID or analgesics or both Exclusion criteria: polyarticular gout (> 4 joints affected); chronic joint damage or persistent inflammation from gout, or any other form of arthritis (except for mild or moderate osteoarthritis that did not affect the index joint); current use of NSAIDs/analgesics (or taken within 5 half‐lives of appropriate agent), oral or injectable corticosteroids (< 2 weeks before study start), acetylsalicylic acid (> 325 mg/d), intra‐articular injections of hyaluronic acid (in the index joint), anticoagulants, and colchicine (> 1.2 mg/d); history of gout that was unresponsive to NSAID; known allergy or hypersensitivity to COX‐2 inhibitors/NSAIDs or acetylsalicylic acid; previous myocardial infarction; any significant uncontrolled disease/condition that would have contraindicated study participation; known laboratory abnormalities; positive pregnancy test |
|
| Interventions |
Group 1: celecoxib 50 mg with 50 mg 12 hours later on day 1, followed by 50 mg twice daily for 7 days Group 2: celecoxib 400 mg with 200 mg 12 hours later on day 1, followed by 200 mg twice daily for 7 days Group 3: celecoxib 800 mg with 400 mg 12 hours later on day 1, followed by 400 mg twice daily for 7 days Group 4: indomethacin 50 mg 3 times daily |
|
| Outcomes | Outcomes evaluated at prespecified time intervals: baseline, then daily before morning dose of study drug on days 2 to 14 Primary outcome • Change in pain intensity (on a Likert scale) in the index joint from baseline to day 2 Secondary outcomes • Investigator assessment of tenderness (0 to 3 scale: 0 = no tenderness; 1 = participant complained of pain to touch; 2 = participant complained of pain and winced; 3 = participant complained of pain, winced, and withdrew); swelling (0 to 3 scale: 0 = none; 1 = palpable; 2 = visible; 3 = bulging beyond joint margin), erythema (present or absent), warmth (present or absent) • Changes from baseline in pain intensity on days 1 to 13 • Time‐weighted mean change in participants' assessments of pain intensity over 8, 12, and 24 hours after first dose of study medication • Incidence of and time to withdrawal due to lack of benefit • AEs | |
| Notes |
Funding: Pfizer Inc. Adverse events Total AEs Groups 1, 2, and 3 (all doses celecoxib): 88 Group 4 (indomethacin): 44 Nature of events: headache, gout flares Serious AEs Groups 1, 2, and 3 (all doses celecoxib): 0/298 Group 4 (indomethacin): 0/102 GI AEs Sum of AE due to diarrhoea, dyspepsia, upper abdominal pain, nausea Groups 1, 2, and 3 (all doses celecoxib): 16/298 Group 4 (indomethacin): 14/102 P value not reported Cardiovascular AEs None reported Withdrawals due to AEs Groups 1, 2, and 3 (all doses celecoxib): 9 Group 4 (indomethacin): 9 For outcome Investigator assessment of tenderness, swelling, erythema, warmth, SD from Rubin 2004 was imputed, as this was the most representative study |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer‐generated schedule |
| Allocation concealment (selection bias) | Low risk | Randomised 1:1:1:1 using an interactive telephone system |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Double‐blind, double‐dummy design |
| Blinding of outcome assessment for self‐reported outcomes (detection bias) | Low risk | Participants blinded |
| Blinding of outcome assessment for assessor‐reported outcomes (detection bias) | Low risk | Outcome assessors blinded |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Numbers of withdrawals and reasons for withdrawals were described |
| Selective reporting (reporting bias) | Low risk | All prespecified outcomes reported |
| Other bias | Unclear risk | Editorial support was funded by Pfizer |
Shrestha 1995.
| Study characteristics | ||
| Methods |
Design: RCT
Sample size: to show a clinically important pain score decrease difference of 1 pain unit (20% difference) at 90 minutes, the calculated sample size was 10 participants in either group
Analysis: PP Withdrawals: 0 |
|
| Participants | 20 participants (10 in each group) Participant characteristics Mean age (± SD): 53 ± 13 in ketorolac group; 48 ± 8 in indomethacin group Males: 9/10 (90%) in ketorolac group; 10/10 (100%) in indomethacin group Mean disease duration: not described Mean number of affected joints: not described Affected joints: not described Inclusion criteria: acute gout as diagnosed meeting ACR 1977 criteria for diagnosis of gout with fulfilment of 1 major criterion (MSU crystals in synovial fluid aspirate or confirmed tophus) or any 6/12 minor criteria (max inflammation developed within 1 day, ≥ 1 attack of acute arthritis, monoarthritis, redness over joints, pain or swelling of first MTP joint, unilateral tarsal joint attack, suspected tophus, hyperuricaemia, asymmetrical swelling in a joint on radiographic examination, subcortical cysts without erosion on radiographic examination, and joint fluid culture negative during an attack) Exclusion criteria: history of adverse reaction to indomethacin or ketorolac; evidence of a septic joint; history of GI bleeding, peptic ulcer disease, renal insufficiency, or congestive heart failure; pregnancy | |
| Interventions | Group 1: ketorolac 60 mg intramuscular and oral placebo, followed by oral indomethacin 50 mg 3 times daily for 2 days, then twice daily for 5 days Group 2: indomethacin 50 mg oral and intramuscular placebo, followed by oral indomethacin 50 mg 3 times daily for 2 days, then twice daily for 5 days | |
| Outcomes | Outcomes evaluated at baseline and at 30, 60, 90, and 120 minutes (observed as inpatient) and at 6, 12, and 24 hours (by return mail) after treatment Primary outcome • Pain as rated on a Wong‐Baker pain scale (with 6 cartoon faces ranging from 1 with a smile (score = 0) to 1 with tears coming from both eyes (score = 5)) Secondary outcome • AEs | |
| Notes |
Funding: not reported Adverse events No adverse event was reported in either group |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "randomisation was performed with the Latin squares method to yield an equal number of patients in every patient block" |
| Allocation concealment (selection bias) | Low risk | Quote: "...the medications, which were prepackaged in envelopes, without investigator observation..." |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "the nurses who dispensed the medications knew the patients' group assignments but were instructed not to reveal this information to the attending physicians or to the investigator....Group assignments were not disclosed to the physicians...or the patients" |
| Blinding of outcome assessment for self‐reported outcomes (detection bias) | Low risk | Participants blinded |
| Blinding of outcome assessment for assessor‐reported outcomes (detection bias) | Low risk | Assessors blinded |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No withdrawals |
| Selective reporting (reporting bias) | Low risk | All prespecified outcomes reported |
| Other bias | Low risk | No other potential sources of bias identified |
Siegmeth 1976.
| Study characteristics | ||
| Methods | Design: RCT Sample size: not described Analysis: not described Withdrawals: 0 | |
| Participants | 46 participants (23 in each group) Participant characteristics Mean age: 59.9 (range 36 to 78) years Male: 100% Mean disease duration: 4.8 years (ketoprofen group); 5.6 years (phenylbutazone group) Mean number of affected joints: not described Affected joints: not described Inclusion criteria: acute gout defined as (1) hyperuricaemia 7 mg%; (2a) crystal identification; (2b) typical history of a podagra; (2c) at least 2 arthritis episodes during maximally 2 weeks before the trial; (2d) presence of tophi. For the diagnosis of acute gout, the following were needed: 1 and 2a, or 1 and 2 out of 3 criteria (2b, 2c, 2d) Exclusion criteria: chronic gout and kidney disease, GI disorder, hepatic disorder, haematological disorder | |
| Interventions | Group 1: ketoprofen 2 intramuscular injections 50 mg each day for 7 days Group 2: phenylbutazone 2 intramuscular injections 300 mg each day for 7 days | |
| Outcomes | Outcomes evaluated at days 1 and 7
Primary outcome
• Pain as rated on an ordinal scale (0 to 3 scale; 0 = absent, 1 = moderate, 2 = strong, 3 = very strong)
Secondary outcomes
• Sleep deprivation • Inflammation defined as redness and swelling as rated on an ordinal scale (0 to 3 scale; 0 = absent, 1 = moderate, 2 = strong, 3 = very strong) • Uric acid concentrations • Tolerance |
|
| Notes |
Funding: not reported Adverse events Ketoprofen appears to be slightly better tolerated with respect to systemic and local side effects compared to phenylbutazone |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Not described; stated only that both study drugs were provided by intramuscular injection |
| Blinding of outcome assessment for self‐reported outcomes (detection bias) | Unclear risk | Not reported |
| Blinding of outcome assessment for assessor‐reported outcomes (detection bias) | Unclear risk | Not reported |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No withdrawals |
| Selective reporting (reporting bias) | Low risk | All outcomes reported |
| Other bias | Low risk | None apparent |
Smyth 1973.
| Study characteristics | ||
| Methods |
Design: RCT Sample size: not described Analysis: not described Withdrawals: not described |
|
| Participants | 28 participants (14 in each group) Participant characteristics Mean age: 63 years (phenylbutazone group); 57 years (indomethacin group) Male: 24/28 (84%) Mean disease duration: not described Mean number of affected joints: not described Affected joints: not described Inclusion criteria: acute gout, quote: "diagnosis was established on generally accepted grounds" Exclusion criteria: not described |
|
| Interventions |
Group 1: phenylbutazone 200 mg every 6 hours for 4 doses, then 200 mg every 8 hours for 3 doses, then 100 mg every 6 hours until 1 day after all signs of inflammation had subsided Group 2: indomethacin 50 mg every 6 hours for 4 doses, then 50 mg every 8 hours for 3 doses, then 25 mg every 6 hours until 1 day after all signs of inflammation had subsided |
|
| Outcomes | Outcomes evaluated daily until acute attack resolved Primary outcome • Volumes of affected and unaffected extremities using a water displacement method Secondary outcomes • Clinical estimation of degree of pain, tenderness, redness, heat, swelling, and joint effusion (using an arbitrary 1 to 4 scale) • Subsidence of pain sufficient to resume normal activities • AEs: not prespecified |
|
| Notes | 31 acute attacks of gout (in 28 participants) evaluated. 16 attacks were treated with phenylbutazone and 15 attacks with indomethacin (1 participant had 2 attacks treated with indomethacin, and 2 participants had 2 attacks treated with phenylbutazone) | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "choice of therapy was determined by blind selection of containers coded according to a restricted series of random numbers" |
| Allocation concealment (selection bias) | Low risk | Coded containers used |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Unclear how participants and personnel were blinded |
| Blinding of outcome assessment for self‐reported outcomes (detection bias) | Unclear risk | Not clearly described |
| Blinding of outcome assessment for assessor‐reported outcomes (detection bias) | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Not described |
| Selective reporting (reporting bias) | Low risk | All prespecified outcomes reported |
| Other bias | Low risk | None |
Sturge 1977.
| Study characteristics | ||
| Methods |
Design: RCT Sample size: not described Analysis: not described whether ITT or PP Withdrawals: not described |
|
| Participants | 45 participants (22 in naproxen group, 23 in phenylbutazone group); 4 participants received both drugs in different attacks Participant characteristics Mean age (range): 58.8 years (34 to 84) in naproxen group; 50.4 years (30 to 73) in phenylbutazone group Males: 20/22 (91%) in naproxen group; 23/23 (100%) in phenylbutazone group Mean disease duration: not described Mean number of affected joints: not described Affected joints: not described Inclusion criteria: acute gout as diagnosed, quote: "by the investigating physician on generally acceptable clinical grounds" Exclusion criteria: not described |
|
| Interventions |
Group 1: naproxen 750 mg followed by 250 mg 3 times daily, until affected joint was pain free Group 2: phenylbutazone 200 mg 4 times daily followed by 200 mg 3 times daily, until affected joint was pain free |
|
| Outcomes | Outcomes evaluated at quote: "follow‐up" Primary outcome • Time to resolution of attack as assessed by absence of pain, swelling, tenderness, and ability to walk without a limp Secondary outcome • AEs |
|
| Notes |
Funding: not reported Adverse events Group 1: naproxen Total adverse events: 1/22 Nature of event: ankle oedema Group 2: phenylbutazone Total adverse events: 2/23 Nature of events: diarrhoea, flatulence, palpitations |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Method of randomisation not described |
| Allocation concealment (selection bias) | High risk | Allocation not concealed |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Not described |
| Blinding of outcome assessment for self‐reported outcomes (detection bias) | Unclear risk | Not described |
| Blinding of outcome assessment for assessor‐reported outcomes (detection bias) | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Not described |
| Selective reporting (reporting bias) | Low risk | All prespecified outcomes reported |
| Other bias | Unclear risk | Participants in naproxen group older (aged 58.8 years (34 to 84)) than those in phenylbutazone group (aged 50.4 years (30 to 73)) |
Terkeltaub 2013.
| Study characteristics | ||
| Methods | Design: RCT Sample size: sample size of 75 participants per group was calculated to provide at least 90% power for pair‐wise comparisons Analysis: ITT Withdrawals: 8 (10.5%) in indomethacin group (2 AEs, 2 lack of benefit, 2 requested by participant, 1 loss to follow‐up, 1 other reasons), 11 (14.7%) in rilonacept group (2 protocol non‐compliance, 1 AE, 2 lack of benefit, 2 requested by participant, 2 losses to follow‐up, 2 other reasons), 8 (12.2%) in rilonacept plus indomethacin group (1 AEs, 2 lack of benefit, 3 requested by participant, 1 death, 1 other reasons) | |
| Participants | 225 participants (76 in indomethacin group; 75 in rilonacept group, 74 in rilonacept plus indomethacin group) Participant characteristics Mean age (± SD): 51.3 ± 10.9 years (indomethacin group); 51.0 ± 10.8 years (rilonacept group); 48.6 ± 10.0 years (rilonacept plus indomethacin group) Males: 71 (94.7%) in indomethacin group; 67 (91.8%) in rilonacept group; 71 (95.9%) in rilonacept plus indomethacin group Mean disease duration (± SD): 8.8 ± 6.7 (indomethacin group); 10.2 ± 9.9 years (rilonacept group); 11.0 ± 7.9 years (rilonacept plus indomethacin group) Mean number of affected joints: not described Affected joints: not described Inclusion criteria: aged 18 to 70 years; acute gout based on 1977 ACR criteria; previously demonstrated symptomatic relief with NSAIDs for treatment of gout flare; presentation within 48 hours of an acute gout flare; pain in the gouty index joint of at least moderate severity using a 5‐point Likert scale; score of at least 1 on a 0 to 3 scale for assessments of swelling and tenderness at the gouty index joint; presentation of acute gout flare in max 3 joints Exclusion criteria: quote: "included but were not limited to" treatment with short‐acting NSAIDs within 48 hours of randomisation; use of colchicine at a dose exceeding 0.6 mg twice daily within 7 days of randomisation; history of NSAID intolerance or absolute contraindication; active or recurrent infection; estimated creatinine clearance < 60 mL/min using the Cockcroft‐Gault method; history of bleeding disorder, GI bleeding, or perforation; poorly controlled hypertension; other cardiovascular risk factors |
|
| Interventions |
Group 1: indomethacin: subcutaneous placebo at baseline, oral indomethacin 50 mg 3 times daily for 3 days followed by 25 mg 3 times daily for up to 9 days Group 2: indomethacin + rilonacept: subcutaneous rilonacept 320 mg at baseline plus oral indomethacin 50 mg 3 times daily for 3 days followed by 25 mg 3 times daily for up to 9 days Group 3: rilonacept: subcutaneous rilonacept 320 mg at baseline plus oral placebo 3 times daily for 3 days, then oral placebo 3 times daily for up to 9 days |
|
| Outcomes | Outcomes were evaluated at baseline and at 4, 8, 12, and 24 hours, then daily until the flare ended Primary outcome • Change in participant‐reported pain in the index joint using 5‐point Likert scale (1 = no pain to 5 = extreme pain) and an 11‐point numerical scale (0 = no pain to 10 = extreme pain) to the composite endpoint of mean of participant‐reported pain values at 24 hours (day 2), 48 hours (day 3), and 72 hours (day 4) Secondary outcomes • Change from baseline in participant‐reported pain in the index joint at 24 hours (day 2), 48 hours (day 3), and 72 hours (day 4) • Proportion of participants requiring rescue medication (indomethacin in the rilonacept group, blinded placebo rescue in the other 2 groups) • Analysis of high‐sensitivity C‐reactive protein at baseline and on days 4, 8, and 31 • AEs |
|
| Notes |
Funding: Regeneron Pharmaceuticals, Inc. ClinicalTrials.gov registration number NCT00855920. Adverse events Group 1: indomethacin Total adverse events: 23/77 Nature of events: headache, dizziness Group 2: indomethacin + rilonacept Total adverse events: 34/73 Nature of events: headache, dizziness Serious adverse events: 3/73 Nature of events: hypertensive cardiomyopathy, myocardial infarction, ulcerative colitis, tubulointerstitial nephritis, pyoderma gangrenosum Group 3: rilonacept Total adverse events: 27/75 Nature of events: headache, dizziness |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "randomly allocated 1:1:1 to treatment" |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Double‐dummy, placebo pills given at the same time as indomethacin |
| Blinding of outcome assessment for self‐reported outcomes (detection bias) | Low risk | Participants blinded |
| Blinding of outcome assessment for assessor‐reported outcomes (detection bias) | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 8 (10.5%) withdrawals in indomethacin group (2 AEs, 2 lack of benefit, 2 requested by participant, 1 loss to follow‐up, 1 other reasons), 11 (14.7%) in rilonacept group (2 protocol non‐compliance, 1 AE, 2 lack of benefit, 2 requested by participant, 2 losses to follow‐up, 2 other reasons), 9 (12.2%) in rilonacept plus indomethacin group (1 AEs, 2 lack of benefit, 3 requested by participant, 1 death, 1 other reasons) |
| Selective reporting (reporting bias) | Low risk | All prespecified outcomes reported |
| Other bias | Unclear risk | Study funded by Regeneron Pharmaceuticals, Inc. Editorial support in preparation of the manuscript was funded by Regeneron Pharmaceuticals, Inc. 4 study authors were employees and stockholders at Regeneron Pharmaceuticals, Inc. Regeneron holds patents related to the content of this manuscript |
Willburger 2007.
| Study characteristics | ||
| Methods | Design: RCT Sample size: estimated sample size to show non‐inferiority of lumiracoxib to indomethacin with respect to the primary benefit variable with 95% power was a total of 105 participants in each treatment group Analysis: PP Withdrawals: 2 (1.7%) in lumiracoxib group (2 AEs); 10 (8.5%) in indomethacin group (7 (6%) AEs, 1 (0.9%) no longer needed study drug, 1 (0.9%) withdrew consent, 1 (0.9%) unsatisfactory therapeutic effect) | |
| Participants | 235 participants (118 in lumiracoxib, 117 in indomethacin group) Participant characteristics Mean age (± SD): 56.8 ± 14.06 years in lumiracoxib group; 56.1 ± 13.29 years in indomethacin group Males: 71/118 (69%) in lumiracoxib group; 70/117 (69%) in indomethacin group Mean disease duration: not described Mean number of affected joints: monoarthritis: 94 (78%) participants (lumiracoxib group), 93 (80%) participants (indomethacin group); oligoarthritis: 24 (20%) participants (lumiracoxib group), 24 (21%) participants (indomethacin group) Affected joints: not described Inclusion criteria: adults with acute gout attack according to 1977 ACR classification criteria; involvement of ≤ 4 joints; onset within 48 hours and at least moderate pain intensity (3 on a 5‐point Likert scale) before randomisation; had analgesia with ibuprofen ≤ 400 mg, paracetamol ≤ 1 g, aspirin ≤ 600 mg, or ≤ 2 tablets of other non‐prescription aspirin‐based or paracetamol‐based medications; or 8 hours after ibuprofen ≤ 600 mg or diclofenac ≤ 50 mg; or 12 hours after naproxen > 500 mg prerandomisation Exclusion criteria: acute attack of gout with onset ≥ 48 hours before evaluation; > 4 joints involved; rheumatoid arthritis, infectious arthritis, pseudo‐gout, or other acute form of inflammatory arthritis; clinically significant hepatic or renal disease; previous or active peptic ulceration or GI bleeding; history of cardiac or cerebrovascular disease; other significant medical problems; use of NSAIDs in previous 24 hours or treatment with etoricoxib in previous 48 hours; use of systemic or intra‐articular steroids in previous 4 weeks; allergic‐type reaction after taking aspirin, paracetamol, or any NSAIDs (including selective COX‐2 inhibitors); pregnancy, lactation, or inadequate contraception |
|
| Interventions |
Group 1: lumiracoxib 400 mg daily for 7 days Group 2: indomethacin 50 mg 3 times daily for 7 days |
|
| Outcomes | Outcomes evaluated at prespecified time intervals: baseline, then 4 hours after treatment, with first dose of study medication from days 1 to 7 Primary outcome • Change in pain intensity in affected joint from baseline over days 2 to 5 on a 5‐point Likert scale (0 = none, 1 = mild, 2 = moderate, 3 = severe, 4 = extreme) Secondary outcomes • Mean change in pain intensity from baseline over days 2 to 7 • Patients' global assessment of response to therapy over days 2 to 5 and days 2 to 7 on a 5‐point Likert scale (0 = excellent, 4 = poor) • Physicians' global assessment of response to therapy over days 2 and 5 and days 2, 5, and end of study (day 7 + 1 day if necessary) on a 5‐point Likert scale (0 = excellent, 4 = poor) • Physicians' assessment of tenderness (on days 2 and 5 and at end of study) on a 4‐point Likert scale (0 = no pain, 1 = "there is pain", 2 = "there is pain" and wincing, 3 = "there is pain" and wincing and withdrawal) • Physicians' assessment of swelling on a 4‐point Likert scale (0 = no swelling, 1 = palpable, 2 = visible, 3 = bulging beyond joint margins) • Physicians' assessment of erythema (present, absent, or not assessable) of study joint over days 2 and 5 and on days 2 and 5 and at end of study • Use of rescue medication (paracetamol ≤ 3 g/d was permitted) • HRQoL, as assessed by SF‐36 and EQ‐5D questionnaires at end of study • C‐reactive protein at end of study • AEs |
|
| Notes |
Funding: Novartis Pharma AG Adverse events Group 1: lumiracoxib Total adverse events: 12/118 Nature of events: abdominal pain, headache, diarrhoea, nausea, eyelid edema, nasopharyngitis, hyperglycaemia, sleep disorder Group 2: indomethacin Total adverse events: 26/117 Nature of events: vertigo, abdominal pain, headache, diarrhoea, nausea, eyelid edema, nasopharyngitis, hyperglycaemia, sleep disorder, flatulence, fatigue, dizziness |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "computer‐generated randomisation list using a validated system that automates the random assignment of treatment groups to randomisation numbers in a block formation" |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "a double‐dummy design was used to blind the identity of the study drugs, which could not be disguised due to their different forms, and their different regimens..." |
| Blinding of outcome assessment for self‐reported outcomes (detection bias) | Low risk | Participants blinded |
| Blinding of outcome assessment for assessor‐reported outcomes (detection bias) | Low risk | Quote: "...personnel involved in monitoring... were all blinded..." |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | 2 withdrawals in lumiracoxib group (AEs); 10 withdrawals in indomethacin group (7 AE, 1 consent withdrawal, 1 unsatisfactory therapeutic effect, 1 no further requirement for study drug |
| Selective reporting (reporting bias) | Low risk | All prespecified outcomes reported |
| Other bias | Unclear risk | 4 study authors were employed by Novartis Pharma; 1 study author was a speaker for Novartis |
Xu 2016.
| Study characteristics | ||
| Methods | Design: RCT Sample size: not described Analysis: PP Withdrawals: 8 (19.5%) in prednisolone group (4 due to non‐compliance, 2 due to AEs, 1 due to lack of efficacy, 1 was loss to follow‐up); 2 (4.3%) in etoricoxib group (1 due to AEs, 1 due to lack of efficacy); 9 (20%) in indomethacin group (3 due to non‐compliance; 3 due to AEs, 1 due to lack of efficacy, 2 were losses to follow‐up) | |
| Participants | 132 participants (41 in prednisolone group, 46 in etoricoxib group, 45 in indomethacin group) Participant characteristics Mean age (± SD): 44.03 ± 15.37 years in prednisolone group; 44.43 ± 15.08 years in etoricoxib group; 43.81 ± 12.29 years in indomethacin group Males: 41/41 (100%) in prednisolone group; 46/46 (100%) in etoricoxib group; 44/45 (97.2%) in indomethacin group Mean disease duration: 2.0 (range 0 to 8) years in prednisolone group; 2.0 (range 0 to 8) years in etoricoxib group; 2.0 (range 0 to 17) years in indomethacin group No. (%) of affected joints: • Metatarsophalangeal joint 1: 13 (39.4%) in prednisolone group, 16 (36.4%) in etoricoxib group, 15 (41.7%) in indomethacin group • Other foot joints, ankle, or knee: 19 (57.6%) in prednisolone group, 27 (61.4%) in etoricoxib group, 21 (58.3%) in indomethacin group • Elbow, wrist, or hand: 1 (13%) in prednisolone group, 1 (2.3%) in etoricoxib group, none in indomethacin group Inclusion criteria: adults with acute gout attack according to 1977 ACR classification criteria; gout attacks within 72 hours of screening; degree of pain was at least moderate (2/5 on Likert scale) at baseline Exclusion criteria: chronic gouty arthritis stage; clinical suspicion of joint infection or other joint disease; polyarticular gout involving more than 4 joints; coronary heart disease, heart failure, gastrointestinal haemorrhage, or history of peptic ulcer; digestive tract operation history, inflammatory bowel disease, or malignant tumour; using NSAIDs or corticosteroids within 72 hours before baseline assessment; allergic to any of the study drugs; abnormal liver function with transaminase levels > 2 times upper limit of normal; renal insufficiency with serum creatinine levels > 200 μmol/L |
|
| Interventions |
Group 1: prednisolone 35 mg qd for for 4 days Group 2: etoricoxib 120 mg qd for 4 days Group 3: indomethacin 50 mg tid for 4 days |
|
| Outcomes | Outcomes evaluated at prespecified time intervals: baseline, then 4 hours after treatment with first dose of study medication from days 1 to 7 Primary outcome • Reduction in pain in the index joint (most painful joint at baseline) on a 5‐point Likert scale (0 = none, 1 = mild, 2 = moderate, 3 = severe, 4 = extreme) Secondary outcomes • Change in physicians' assessment of tenderness on a 3‐point Likert scale (0 = no pain on palpation, 1 = patient states 'there is pain', 2 = patient states 'there is pain' and withdraws affected limb) • Change in physician assessment of erythema (3‐point Likert scale: 0 = absent, 1 = not assessable, 2 = present) • Change in physician assessment of swelling (4‐point LIkert scale: 0 = no swelling, 1 = palpable, 2 = visible, 3 = bulging beyond joint margins) • Change in joint activity from baseline on a 4‐point Likert scale (0 = no restriction, 1 = moderately restricted, 2 = significantly restricted, not engaging in general activities, 3 = unbearable, cannot take care of themselves) • Patients' global response to treatment on a 5‐point Likert scale, assessed at end of study on day 4 (0 = very good, 1 = good, 2 = fair, 3 = poor, 4 = very poor) • AEs (total adverse events, gastric/abdominal pain, dizziness, oedema, fatigue or drowsiness, dry mouth) |
|
| Notes |
Funding: not reported Chinese Clinical Trials Register (#ChiCTR‐IPR‐15006269) Adverse events Group 1: prednisolone Total adverse events: 2/33 Nature of adverse events: abdominal pain Group 2: etoricoxib Total adverse events: 3/44 Nature of adverse events: dizziness, oedema Group 3: indomethacin Total adverse events: 11/36 Nature of adverse events: abdominal pain, dizziness, oedema, fatigue, drowsiness, dry mouth |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer‐generated tables and sequential sealed envelopes prepared by a statistician independent to the trial |
| Allocation concealment (selection bias) | Low risk | Sequential sealed envelopes prepared by a statistician independent to the trial |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Open label |
| Blinding of outcome assessment for self‐reported outcomes (detection bias) | High risk | Open label |
| Blinding of outcome assessment for assessor‐reported outcomes (detection bias) | High risk | Open label |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Withdrawals were reported: 8 (19.5%) in prednisolone group (4 (50%), 2 AEs (25%), 1 (12.5%) lack of efficacy, 1 (12.5%) loss to follow‐up); 2 (4.3%) in etoricoxib group (1 (50%) AEs, 1 (50%) lack of efficacy); 9 (20%) in indomethacin group (3 (33,3%) non‐compliance; 3 (33.3%) AEs, 1 (11%) lack of efficacy, 2 (22%) losses to follow‐up) |
| Selective reporting (reporting bias) | Low risk | All prespecified outcomes were reported |
| Other bias | Low risk | None |
Zhang 2014.
| Study characteristics | ||
| Methods | Design: RCT Sample size: not described Analysis: not described if ITT or PP, probably ITT as there were no withdrawals and both numbers of patients are the same in all analyses Withdrawals: none | |
| Participants | 60 participants (30 in each group) Participant characteristics Mean age: 52.3 years (betamethasone group); 54.2 years (diclofenac group) Male: 29/29 (96.7%) Mean duration of symptoms: not described Number (%) of affected joints: first metatarsophalangeal joint 18 (60.0%) (betamethasone group 19%, diclofenac group 63.3%); insteps 6 (20.0%) (betamethasone group 7%, diclofenac group 23.3%); ankles 3 (10.0%) (betamethasone group 3%, diclofenac group 10.0%); heels 1 (3.33%) (betamethasone group 0, diclofenac group 0); knees 2 (6.7%) (betamethasone group 1%, diclofenac group 3.33%) Inclusion criteria: acute attack of gout within 24 hours and with at least moderate pain intensity (2 on a 5‐point Likert scale); diagnosis based on 1977 Criteria of American College of Rheumatology for primary gout Exclusion criteria: history of ischaemic heart disease, heart failure, gastrointestinal haemorrhage, or active gastroduodenal ulceration less than 90 days before being screened; inflammatory bowel disease, gastric surgery, erosive oesophagitis, gastric outlet obstruction, or active malignant disease; allergy to diclofenac sodium and compound betamethasone; serum alanine transaminase or aspartate transaminase concentrations more than twice and serum creatinine concentrations more than upper limit of normal (according to central laboratory definition) |
|
| Interventions |
Group 1: compound betamethasone (Diprospan) 7 mg IM once only Group 2: diclofenac 75 mg twice daily for 7 days |
|
| Outcomes | Outcomes evaluated at prespecified time intervals: baseline, 4 hours after treatment, each day during study duration (7 days) Primary outcome • Pain intensity of affected joints, using a 5‐point Likert scale (0 = none, 1 = mild, 2 = moderate, 3 = severe, 4 = extreme) Secondary outcomes • Patients' global assessment of response to therapy on a 5‐point Likert scale (0 = very good, 1 = good, 2 = fair, 3 = poor, 4 = very poor) • Physicians' assessment of joint tenderness on palpation or passive movement of affected joints on a 4‐point Likert scale (0 = no pain, 1 = patient states "there is pain", 2 = patient states "there is pain" and winces, 3 = patient states "there is pain" and winces and withdraws), of joint swelling on a 4‐point Likert scale (0 = no swelling, 1 = palpable, 2 = visible, 3 = bulging beyond joint margins) • Physicians' assessment of global response to therapy on a 5‐point Likert scale (0 = very good, 1 = good, 2 = fair, 3 = poor, 4 = very poor) • AEs were recorded |
|
| Notes |
Funding: Independent Innovation Foundation of Shandong University (2012TS136), National Natural Science Foundation of China (81201287), and Beijing Nova Program of China (Z121107002512071) Adverse events Group 1: compound betamethasone Total adverse events: 13/30 Nature of adverse events: hyperglycaemia, sleep disorder, headache Group 2: diclofenac Total adverse events: 4/30 Nature of adverse events: abdominal pain, nausea, flatulence, headache, dizziness, eyelid oedema |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Open label |
| Blinding of outcome assessment for self‐reported outcomes (detection bias) | High risk | Open label |
| Blinding of outcome assessment for assessor‐reported outcomes (detection bias) | High risk | Open label (all patients were followed up by the same physician) |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No withdrawals |
| Selective reporting (reporting bias) | Unclear risk | Number of patients with no pain was not a prespecified outcome; there is no statistical analysis of whether this was statistically significant. Other prespecified outcomes were reported but it is not reported whether differences were statistically significant |
| Other bias | Low risk | No other risk of bias identified |
Zhou 2012.
| Study characteristics | ||
| Methods |
Design: RCT Sample size: not done Analysis: per protocol Withdrawals: 2 in indomethacin group (no reason given), 1 in acupuncture group (no reason given) |
|
| Participants | 163 participants (80 in each group)
Participant characteristics
Mean age: 45 ± 8.4 years (indomethacin group); 46.0 ± 8.2 years (acupuncture group)
Male: 100% Mean disease duration: 14.0 ± 1.6 years (indomethacin group); 13.6 ± 3.6 years (acupuncture group) Mean number of affected joints: not described Affected joints: not described Inclusion criteria: adults with acute gout attack within 7 days from inclusion according to Criteria of Diagnosis and Therapeutic Effects of Diseases and Syndromes in Traditional Chinese Medicine: (1) redness, swell, and pain suddenly occurring in a single joint of metatarsus and finger, pain gradually aggravating like tiger bite, mild in daylight and severe at night, after repeated attack, possibly complicated by headache, fever, and other symptoms; (2) commonly seen in middle‐aged males, possibly with family history of gout, often induced by tiredness, eating and drinking too much at 1 meal, eating food with a great number of purines, drinking, and affection by exopathic wind and cold, etc.; (3) at beginning, a single joint attacked with first MTP joint most seen, followed by redness, swell, and pain in ankle, heel, finger, and other small joints, even though exudate in articular cavity. After repeated attacks, it is possibly complicated by occurrence of gouty stone around joints, auricle, and helix, and between metatarsal bones and finger bones; (4) blood uric acid and urine uric acid increased, during attack total white cell count possibly increased; (5) if necessary, B‐ultrasonic examination of kidney, routine examination of urine, examinations of renal function were made to understand renal lesion after gout. X‐roentgenogram: irregular drilling‐like circular defect on cartilage margin close to joint bone substances could be showed. Body temperature below 38°C Exclusion criteria: quote: "history of diabetes, rheumatoid arthritis, and the diseases of heart, liver, kidney and hematopoietic system" |
|
| Interventions |
Group 1: acupuncture once daily for 5 days Group 2: indomethacin 25 mg 3 times daily for 5 days |
|
| Outcomes | Study authors did not state whether outcomes were assessed at prespecified moments Primary outcome • Pain degree on a numerical rating scale (0 = no pain; 1 to 3 = mild pain; 4 to 6 = moderate pain; 7 to 10 = severe pain) Secondary outcomes • Inflammation of joints: redness and swelling, not assessed how • Assessment of uric acid concentrations in blood, liver function, and erythrocyte sedimentation rate before and after treatment |
|
| Notes |
Funding: not reported Adverse events: not reported |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "patients were divided according to random number table in visiting sequence into an acupuncture and an indomethacin group" |
| Allocation concealment (selection bias) | High risk | No allocation concealment |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Not blinded |
| Blinding of outcome assessment for self‐reported outcomes (detection bias) | High risk | Not blinded |
| Blinding of outcome assessment for assessor‐reported outcomes (detection bias) | High risk | Not blinded |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 2 withdrawals in indomethacin group (no reason given), 1 withdrawal in acupuncture group (no reason given) |
| Selective reporting (reporting bias) | Unclear risk | Inflammation not reported but only named in the methods section, so unclear if this was going to be a separate outcome |
| Other bias | Low risk | No other risk of bias identified |
ACR: American College of Rheumatology. ACTH: adrenocorticotropin hormone. AE: adverse event. CI: confidence interval. COX‐2: cyclo‐oxygenase‐2. EQ‐5D‐5L: EuroQoL Group Quality of Life Questionnaire based on 5 dimensions and 5‐level scale. GI: gastrointestinal. HRQoL: health‐related quality of life. ITT: intention to treat. IU: international unit. max: maximum. MSU: monosodium urate. MTP: metatarsophalangeal. NRS: numerical rating scale. NSAID: non‐steroidal anti‐inflammatory drug. PP: per protocol. RCT: randomised controlled trial. SD: standard deviation. SE: standard error. SF‐36: 36‐item Short Form. sUA: serum uric acid. VAS: visual analogue scale.
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Alloway 1993 | Randomised controlled trial, but participants with renal insufficiency, history of gastrointestinal adverse events to non‐steroidal anti‐inflammatory drugs, peptic ulcer or gastritis, or any other contraindication to indomethacin were included in the trial but placed in the triamcinolone group (non‐randomised); other participants were randomised; data for randomised participants were not reported separately |
| Arnold 1988 | Review |
| Bach 1979 | Observational study |
| Cunovic 1973 | Observational study |
| Cuq 1973 | Observational study |
| Ecker‐Schlipf 2009 | Wrong study type ‐ summary of the article Janssens 2008a |
| Janssens 2009 | Wrong study type ‐ Dutch translation of Janssens 2008a |
| Kudaeva 2007 | Wrong population ‐ not acute gout (participants with acute arthritis for > 3 weeks) |
| Navarra 2007 | Meta‐analysis of different randomised trials of etoricoxib sponsored by the drug company Merck |
| NCT00997581 | Study withdrawn |
| Reardon 1980 | Study drug (feprazone) no longer available |
| Ruotsi 1978 | Study drug (proquazone) no longer available |
| Steurer 2016 | Wrong study type ‐ summary of Janssens 2008a and Rainer 2016 |
| Tumrasvin 1985 | Wrong comparator ‐ comparison of 2 different doses of the same drug |
| Valdes 1987 | Wrong comparator ‐ comparison of 2 different doses of the same drug |
| Weiner 1979 | Study drug (fenoprofen) no longer available |
| Werlen 1996 | Observational study |
| Xu 2015 | Wrong population: patients with peptic ulcer haemorrhage and acute gout |
Characteristics of studies awaiting classification [ordered by study ID]
Katona 1988.
| Methods | Open comparative study |
| Participants | 30 patients with gout |
| Interventions | Group 1: ketoprofen Group 2: tiaprofenic acid at a dose of 200 mg intramuscularly twice daily for 5 days |
| Outcomes | • Paint intensity • Inflammation • Beginning of effect • Duration of analgesic effect • Patients' assessment of response |
| Notes | Conference abstract. Full‐text article not available (study author contacted but did not respond) |
Yin 2005.
| Methods | Randomised controlled trial |
| Participants | 100 participants with acute gout |
| Interventions | Group 1: electroacupuncture Group 2: indomethacin and benzbromaron Group 3: combination of electroacupuncture combined with indomethacin and benzbromaron |
| Outcomes | • Clinical therapeutic effect • Effective rate • Change in blood uric acid level |
| Notes | Awaiting translation |
Characteristics of ongoing studies [ordered by study ID]
ChiCTR1800019612.
| Study name | Therapeutic effect and mechanism of ozonated autohaemotherapy for patients with gout |
| Methods |
Study design: randomised controlled 2‐arm parallel trial Setting: Sir Run Run Hospital, Nanjing Medical University, China Sample size: 40 |
| Participants |
Inclusion criteria
Exclusion criteria
|
| Interventions |
Group 1 Ozone treatment of autologous blood Group 2 Ozone treatment of autologous blood and etoricoxib |
| Outcomes |
Primary outcomes • Uric acid symptom Secondary outcome • Blood lipid, blood glucose, BMI, liver function, kidney function, HbA1c |
| Starting date | 19 November 2018 |
| Contact information | Liu Yu drliuyu@njmu.edu.cn |
| Notes | Status: recruiting |
BMI: body mass index. HbA1c: glycosylated haemoglobin. NSAIDs: non‐steroidal anti‐inflammatory drugs.
Differences between protocol and review
We planned to report outcomes in the short term (up to two weeks), medium term (two to six weeks), and long term (more than six weeks), but only short‐term data were available.
We originally intended to present the results from all the 'Summary of findings' tables in the Plain Language Summary. However, due to the word count allowed, this was not possible, so we presented the results for the two most clinically relevant comparisons: NSAIDs versus placebo and NSAIDs versus COXIBs.
When available, we included the earliest time point for the outcomes of pain, swelling, and function, as this is more clinically relevant. For the other outcomes (participants' global assessment of treatment success and health‐related quality of life), we chose the latest time point, as we also considered this more clinically relevant.
The primary and secondary outcomes have been replaced in the review by a list of major outcomes (i.e. those presented in the 'Summary of findings' tables). This was done to implement GRADE and the use of 'Summary of findings' tables.
Other sources of bias have been defined in the methods of the review after the protocol was published (e.g. deviation from the study protocol in a way that did not reflect clinical practice, or inappropriate administration of an intervention). We also noted the presence of co‐administration and funding by manufacturers. Detection bias was assessed separately for self‐reported and assessor‐reported outcomes.
Contributions of authors
CD wrote the current version of the review.
JPP established the gout registry, which was used to select studies for this review update.
MW, RB, DvH, NS, SC, and RL provided comments and suggestions on draft versions of this review update.
All review authors approved the final version.
Sources of support
Internal sources
-
Maastricht University Medical Center, Maastricht, Netherlands
In kind support
-
Flinders University, Adelaide, Australia
In kind support
-
University of Medicine & Dentistry of New Jersey/Robert Wood Johnson Medical School, USA
In kind support
-
Cabrini Hospital, Melbourne, Australia
In kind support
-
Monash University, Melbourne, Australia
In kind support
-
Leiden University Medical Center, Leiden, Netherlands
In kind support
-
Atrium Medical Centre, Heerlen, Netherlands
In kind support
-
University of Amsterdam, Amsterdam, Netherlands
In kind support
External sources
-
National Health and Medical Research Council (NHMRC), Australia
Funding for Cochrane Musculoskeletal ‐ Australia and Cochrane Australia
Declarations of interest
CD received payment from Amgen and Grunenthal for lectures. Her institution received funding from Roche in 2019 for attending the BSR. Lecture fees and travel expenses were not related to the submitted work (lectures were about biosimilars and gout education for general practitioners, and travel expenses were for attending the annual meeting of the BSR). To the best of my knowledge, none of these manufacturers produces any of the products included as interventions in this review.
MW has no declaration of interest for this review; he has received research grants for unrelated work in a different disease area from Janssen Research (Philadelphia, PA, USA).
RL received honoraria for lectures and advisory meetings from AbbVie, Novartis, Eli‐Lilly, Pfizer, UCB, Galapagos, and Gilead.
JPP is a Managing Editor of Cochrane Musculoskeletal, but he was not involved in editorial decisions regarding this review.
SC: is an Assistant Managing Editor of Cochrane Musculoskeletal, but she was not involved in editorial decisions regarding this review.
DvH received payments from AbbVie, Amgen, Astellas, AstraZeneca, BMS, Boehringer Ingelheim, Celgene, Cyxone, Daiichi, Eisai, Eli‐Lilly, Galapagos, Gilead, Glaxo‐Smith‐Kline, Janssen, Merck, Novartis, Pfizer, Regeneron, Roche, Sanofi, Takeda, and UCB Pharma.
RB is the Co‐ordinating Editor of Cochrane Musculoskeletal but was not involved in editorial decisions regarding this review. She is a recipient of a National Health and Medical Research Council (NHMRC) Cochrane Collaboration Round 7 Funding Program Grant, which supports the activities of Cochrane Musculoskeletal Australia and Cochrane Australia, but the funders do not participate in the conduct of reviews. She has no declarations of interest.
New search for studies and content updated (no change to conclusions)
References
References to studies included in this review
Altman 1988 {published data only}
- Altman RD, Honig S, Levin JM, Lightfoot RW. Ketoprofen versus indomethacin in patients with acute gouty arthritis: a multicenter, double blind comparative study. Journal of Rheumatology 1988;15:1422-6. [PubMed] [Google Scholar]
Axelrod 1988 {published data only}
- Axelrod D, Preston S. Comparison of parenteral adrenocorticotropic hormone with oral indomethacin in the treatment of acute gout. Arthritis & Rheumatism 1988;31:803-5. [DOI] [PubMed] [Google Scholar]
Butler 1985 {published data only}
- Butler RC, Goddard DH, Higgens CS, Hollingworth P, Pease CT, Stodell MA, et al. Double-blind trial of flurbiprofen and phenylbutazone in acute gouty arthritis. British Journal of Clinical Pharmacology 1985;20:511-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
Cheng 2004 {published data only}
- Cheng T-T, Lai H-M, Chiu C-K, Chem Y-C. A single-blind, randomized, controlled trial to assess the efficacy and tolerability of rofecoxib, diclofenac sodium, and meloxicam in patients with acute gouty arthritis. Clinical Therapeutics 2004;26:399-406. [DOI] [PubMed] [Google Scholar]
Douglas 1970 {published data only}
- Douglas G, Thompson M. A comparison of phenylbutazone and flufenamic acid in the treatment of acute gout. Annals of Physical Medicine 1970;10:275-80. [DOI] [PubMed] [Google Scholar]
Eberl 1983 {published data only}
- Eberl R, Dunky A. Meclofenamate sodium in the treatment of acute gout. Results of a double-blind study. Arzneimittel-Forschung 1983;33(4A):641-3. [PubMed] [Google Scholar]
Garcia de la Torre 1987 {published data only}
- Garcia de la Torre I. A comparative, double-blind, parallel study with tenoxicam vs placebo in acute gouty arthritis [Estudio doble-ciego paralelo, comparativo con tenoxicam vs placebo en artritis gotosa aguda]. Investigación Médica Internacional 1987;14:92-7. [Google Scholar]
Janssens 2008a {published data only}
- Janssens HJ, Janssen M, de Lisdonk EH, Riel PL, Weel C. Use of oral prednisolone or naproxen for the treatment of gout arthritis: a double-blind, randomised equivalence trial. Lancet 2008;371:1854-60. [DOI] [PubMed] [Google Scholar]
Klumb 1996 {published data only}
- Klumb EM, Pinheiro GRC, Ferrari A, Albuquerque EMN. The treatment of acute gout arthritis. Double-blind randomized comparative study between nimesulid and indomethacin [O tratamento da crise aguda de gota. Estudo duplo-cego, randômico, comparativo entre o nimesulide e a indometacina]. Revista Brasileira de Medicina 1996;53(6):540-6. [Google Scholar]
Lederman 1990 {published data only}
- Lederman R. A double-blind comparison of etodolac (Lodine) and high doses of naproxen in the treatment of acute gout. Advances in Therapy 1990;7(6):344-54. [Google Scholar]
Li 2013 {published data only}
- Li T, Chen S, Dai Q, Han X, Li Z, Wu D, et al. Etoricoxib versus indomethacin in the treatment of Chinese patients with acute gouty arthritis: a randomized double-blind trial. Chinese Medical Journal 2013;126:1867-71. [PubMed] [Google Scholar]
Lomen 1986 {published data only}
- Lomen PL, Turner LF, Lamborn KR, Winbald MA, Sack RL, Brinn EL. Flurbiprofen in the treatment of acute gout. A comparison with indomethacin. American Journal of Medicine 1986;80(3A):134-9. [DOI] [PubMed] [Google Scholar]
Maccagno 1991 {published data only}
- Maccagno A, Di Giorgio E, Romanowicz A. Effectiveness of etodolac ('Lodine') compared with naproxen in patients with acute gout. Current Medical Research and Opinion 1991;12:423-9. [DOI] [PubMed] [Google Scholar]
Man 2007 {published data only}
- Man CY, Cheung ITF, Cameron PA, Rainer TH. Comparison of oral prednisolone/paracetamol and oral indomethacin/paracetamol combination therapy in the treatment of acute goutlike arthritis: a double-blind, randomized, controlled trial. Annals of Emergency Medicine 2007;49:670-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Rainer 2016 {published data only}
- Rainer TH, Cheng CH, Janssens HJEM, Man CY, Tam LS, Choi YF, et al. Oral prednisolone in the treatment of acute gout. A pragmatic, multicenter, double-blind, randomized trial. Annals of Internal Medicine 2016;164:464-71. [DOI] [PubMed] [Google Scholar]
Roddy 2020 {published data only}
- Roddy E, Clarkson K, Blaogevic-Bucknall M, Mehta R, Oppong R, Avery A, et al. Open-label randomised pragmatic trial (CONTACT) comparing naproxen and low-dose colchicine for the treatment of gout flares in primary care. Annals of Rheumatic Diseases 2020;79:276-84. [DOI] [PMC free article] [PubMed] [Google Scholar]
Rubin 2004 {published data only}
- Rubin BR, Burton R, Navarra S, Antigua J, Londoño J, Pryhuber KG, et al. Efficacy and safety profile of treatment with etoricoxib 120 mg once daily compared with indomethacin 50 mg three times daily in acute gout: a randomised controlled trial. Arthritis & Rheumatism 2004;50:598-606. [DOI] [PubMed] [Google Scholar]
Schumacher 2002 {published data only}
- Schumacher HR Jr, Boice JA, Daikh DI, Mukhopadhyay S, Malmstrom K, Ng J, et al. Randomised double blind trial of etoricoxib and indometacin in treatment of acute gouty arthritis. BMJ 2002;324:1488-92. [DOI] [PMC free article] [PubMed] [Google Scholar]
Schumacher 2012 {published data only}
- Schumacher HR, Berger MF, Li-Yu J, Perez-Ruiz F, Burgos-Vargas R, Li C. Efficacy and tolerability of celecoxib in the treatment of acute gouty arthritis: a randomized controlled trial. Journal of Rheumatology 2012;39:1859-66. [DOI] [PubMed] [Google Scholar]
Shrestha 1995 {published data only}
- Shrestha M, Morgan DL, Moreden JM, Singh R, Nelson M, Hayes JE. Randomized double-blind comparison of the analgesic efficacy of intramuscular ketorolac and oral indomethacin in the treatment of acute gouty arthritis. Annals of Emergency Medicine 1995;26:682-6. [DOI] [PubMed] [Google Scholar]
Siegmeth 1976 {published data only}
- Siegmeth W, Placheta P. Double-blind trial: ketoprofen versus phenylbutazone in acute gouty arthritis (author's translation). Wiener Klinische Wochenschrift 1976;88:535-7. [PubMed] [Google Scholar]
Smyth 1973 {published data only}
- Smyth CJ, Percy JS. Comparison of indomethacin and phenylbutazone in acute gout. Annals of the Rheumatic Diseases 1973;32:351-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
Sturge 1977 {published data only}
- Sturge RA, Scott JT, Hamilton EB, Liyanage SP, Dixon AS, Davies J, et al. Multicentre trial of naproxen and phenylbutazone in acute gout. Annals of the Rheumatic Diseases 1977;36:80-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
Terkeltaub 2013 {published data only}
- Terkeltaub RA, Schumacher HR, Carter JD, Baraf HSB, Evans RR, Wang J, et al. Rilonacept in the treatment of acute gouty arthritis: a randomised, controlled clinical trial using indomethacin as the active comparator. Arthritis Research & Therapy 2013;15:R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
Willburger 2007 {published data only}
- Willburger RE, Mysler E, Derbot J, Jung T, Thurston H, Kreiss A, et al. Lumiracoxib 400 mg once daily is comparable to indomethacin 50 mg three times daily for the treatment of acute flares of gout. Rheumatology 2007;46(7):1126-32. [DOI] [PubMed] [Google Scholar]
Xu 2016 {published data only (unpublished sought but not used)}
- Xu L, Liu S, Guan M, Xue Y. Comparison of prednisolone, etoricoxib, and indomethacin treatment of acute gouty arthritis: an open-label, randomized, controlled trial. Medical Science Monitor 2016;22:810-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Zhang 2014 {published and unpublished data}
- Zhang YK, Yang H, Zhang JY, Song LJ, Fan YC. Comparison of intramuscular compound betamethasone and oral diclofenac sodium in the treatment of acute attacks of gout. International Journal of Clinical Practice 2014;68(5):633-8. [DOI] [PubMed] [Google Scholar]
Zhou 2012 {published data only}
- Zhou L, Xu QF, Zhang WS. Comparative observation of therapeutic effects of acupuncture combined with infrared irradiation and western medicine on acute gouty arthritis. World Journal of Acupuncture 2012;22(1):30-4. [PubMed] [Google Scholar]
References to studies excluded from this review
Alloway 1993 {published data only}
- Alloway JA, Moriarty MJ, Hoogland YT, Nashel DJ. Comparison of triamcinolone acetonide with indomethacin in the treatment of acute gouty arthritis. Journal of Rheumatology 1993;20:111-3. [PubMed] [Google Scholar]
Arnold 1988 {published data only}
- Arnold MH, Preston SJ, Buchanan WW. Comparison of the natural history of untreated acute gouty arthritis vs acute gouty arthritis treated with non-steroidal-anti-inflammatory drugs. British Journal of Clinical Pharmacology 1988;26:488-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Bach 1979 {published data only}
- Bach GL. The acute gout attack [Der akute gichtaanfall]. Medizinische Welt 1979;30(45):1696-8. [PubMed] [Google Scholar]
Cunovic 1973 {published data only}
- Cunovic S. Treatment of acute uric arthritis using perclusone [Lijecenje akutnog napadaja urickog artritisa Perclusone-om]. Reumatizam 1973;20(4):156-9. [PubMed] [Google Scholar]
Cuq 1973 {published data only}
- Cuq P. Treatment of acute attacks of gout with naproxen-C 1674. Scandinavian Journal of Rheumatology Supplement 1973;2:64-8. [DOI] [PubMed] [Google Scholar]
Ecker‐Schlipf 2009 {published data only}
- Ecker-Schlipf B. Gouty arthritis: prednisolone is as effective as naproxen. Medizinische Monatsschrift für Pharmazeuter 2009;32:315-6. [Google Scholar]
Janssens 2009 {published data only}
- Janssens HJEM, Janssen M, de Lisdonk EH, Riel PLCM, Weel C. Use of oral prednisolone or naproxen for the treatment of acute gout arthritis: a double blind, randomised equivalent trial. Nederlands Tijdschrift voor Geneeskunde 2009;153:B393. [Google Scholar]
Kudaeva 2007 {published data only}
- Kudaeva FM, Eliseev MS, Barskova VG, Nasonova VA. Comparison of the time to analgetic and anti-inflammatory effect in the treatment of gouty arthritis with nimesulide and sodium diclofenac. Terapevticheskii Arkhiv 2007;79(5):35-40. [PubMed] [Google Scholar]
Navarra 2007 {published data only}
- Navarra S, Rubin BR, Yu Q, Smugar SS, Tershakovec AM. Association of baseline disease and patient characteristics with response to etoricoxib and indomethacin for acute gout. Current Medical Research and Opinion 2007;23:1685-91. [DOI] [PubMed] [Google Scholar]
NCT00997581 {published data only}
- Wortmann R. Apremilast therapy for acute gouty arthritis. ClinicalTrials.gov NCT00997581.
Reardon 1980 {published data only}
- Reardon JA, Stockman A, Darlington LG, Scott JT. Double-blind trial of feprazone and phenylbutazone in acute gout. Current Medical Research and Opinion 1980;6:445-8. [DOI] [PubMed] [Google Scholar]
Ruotsi 1978 {published data only}
- Ruotsi A, Vainio U. Treatment of acute gouty arthritis with proquazone and indomethacin. A comparative, double-blind trial. Scandinavian Journal of Rheumatology Supplement 1978;21:15-7. [DOI] [PubMed] [Google Scholar]
Steurer 2016 {published data only}
- Steurer J. Oral steroids are effective in the treatment of acute gout attacks [Orale Steroide sind in der Behandlung akuter Gicht-Attacken wirksam.]. Praxis 2016;15:589-90. [DOI] [PubMed] [Google Scholar]
Tumrasvin 1985 {published data only}
- Tumrasvin T, Deesomchok U. Piroxicam in treatment of acute gout high dose versus low dose. Journal of the Medical Association of Thailand 1985;68(3):111-16. [PubMed] [Google Scholar]
Valdes 1987 {published data only}
- Valdes EF. Use of tenoxicam in patients with acute gouty arthritis. European Journal of Rheumatology & Inflammation 1987;9(2):133-6. [PubMed] [Google Scholar]
Weiner 1979 {published data only}
- Weiner GI, White SR, Weitzner RI, Rubinstein HM. Double-blind study of fenoprofen versus phenylbutazone in acute gouty arthritis. Arthritis & Rheumatism 1979;22:425-6. [DOI] [PubMed] [Google Scholar]
Werlen 1996 {published data only}
- Werlen D, Gabay C, Vischer TL. Corticosteroid therapy for the treatment of acute attacks of crystal-induced arthritis: an effective alternative to nonsteroidal antiinflammatory drugs. Revue du Rhumatisme (English ed.) 1996;63:248-54. [PubMed] [Google Scholar]
Xu 2015 {published data only}
- Xu Z, Zhang R, Yao J, Shi R, Tang Q, Wang L. Peptic ulcer hemorrhage combined with acute gout: analyses of treatment in 136 cases. International Journal of Clinical and Experimental Medicine 2015;8(4):6193-9. [PMC free article] [PubMed] [Google Scholar]
References to studies awaiting assessment
Katona 1988 {published data only}
- Katona G, Burgos-Vargas R. Clinical experiences with the intramuscular injection of tiaprofenic acid in rheumatic diseases, with particular emphasis on time of onset and duration of the analgesic effect. Drugs 1988;35 Suppl 1:72-80. [DOI] [PubMed] [Google Scholar]
Yin 2005 {published data only}
- Yin Y, Zhang H-X, Zhang T-F. Clinical observation on electroacupuncture combined with medicine for treatment of acute gouty arthritis. Chinese Acupuncture & Moxibustion 2005;25:683-5. [PubMed] [Google Scholar]
References to ongoing studies
ChiCTR1800019612 {published data only}
- Therapeutic effect and mechanism of ozonated autohaemotherapy for patients with gout. Ongoing study. 19 November 2018. Contact author for more information.
Additional references
ACR 1977
- American College of Rheumatology. 1977 criteria for the classification of acute arthritis of primary gout. www.rheumatology.org/practice/clinical/classification/gout.asp (accessed 1 September 2014).
Bellamy 1987
- Bellamy N, Downie WW, Buchanan WW. Observations on spontaneous improvement in patients with podagra: implications for the therapeutic trials of non-steroidal anti-inflammatory drugs. British Journal of Clinical Pharmacology 1987;24:33-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Borer 2005
- Borer JS, Simon LS. Cardiovascular and gastrointestinal effects of COX-2 inhibitors and NSAIDs: achieving a balance. Arthritis Research & Therapy 2005;7 Suppl 4:S14-S22. [DOI] [PMC free article] [PubMed] [Google Scholar]
Bursill 2019
- Bursill D, Taylor W, Terkeltaub R, et al. Gout, Hyperuricaemia and Crystal-Associated Disease Network (G-CAN) consensus statement regarding labels and definitions of disease states of gout. Annals of Rheumatic Diseases 2019;78(11):1592-600. [DOI] [PMC free article] [PubMed] [Google Scholar]
Cates 2008
- Visual Rx [Computer program]. Version 3. Dr. Christopher Cates' EBM website. 2008. http://www.nntoline.net.
Cohen 1988
- Cohen J. Statistical Power Analysis for the Behavorial Sciences. Hillsdale, NJ: Lawrence Erlbaum Associates, 1988. [Google Scholar]
Dalbeth 2021
- Dalbeth N, Gosling A, Gaffo A, Abhishek A. Gout. Lancet 2021;397:1843-55. [DOI] [PubMed] [Google Scholar]
DeAngelis 2004
- DeAngelis CD, Drazen JM, Frizelle FA, Haug C, Hoey J, Horton R, et al. Clinical trial registration: a statement from the International Committee of Medical Journal Editors. JAMA 2004;292(11):1363-4. [DOI] [PubMed] [Google Scholar]
Deeks 2020
- Deeks JJ, Higgins JT, Altman DG. Chapter 10. Analysing data and undertaking meta-analyses. In: Higgins JT, Thomas J, Chandler J, Cumpston M, Li T, Page MJ, et al, editor(s). Cochrane Handbook for Systematic Reviews of Interventions Version 6.1 (updated September 2020), Cochrane. Available from training.cochrane.org/handbook.
Dworkin 2008
- Dworkin RH, Turk DC, Wyrwich KW, Beaton D, Cleeland CS, Farrar JT, et al. Interpreting the clinical importance of treatment outcomes in chronic pain clinical trials: IMMPACT recommendations. Journal of Pain 2008;9(2):105-21. [PMID: ] [DOI] [PubMed] [Google Scholar]
Feenstra 2002
- Feenstra J, Heerdink ER, Grobbee DE, Stricker BHC. Association of non-steroidal anti-inflammatory drugs with first occurrence of heart failure and with relapsing heart failure: the Rotterdam Study. Archives of Internal Medicine 2002;162:265-70. [DOI] [PubMed] [Google Scholar]
FitzGerald 2020
- FitzGerald JD, Dalbeth N, Mikuls T, et al. 2020 American College of Rheumatology Guideline for the Management of Gout. Arthritis and Rheumatology 2020;72(6):879-95. [DOI] [PubMed] [Google Scholar]
Garner 2009
- Garner SE, Fidan D, Frankish RR, Judd M, Shea B, Towheed T, et al. Celecoxib for rheumatoid arthritis. Cochrane Database of Systematic Reviews 2009, Issue 1. Art. No: CD003831. [DOI: 10.1002/14651858.CD003831] [DOI] [PubMed] [Google Scholar]
Garner 2010
- Garner SE, Fidan D, Frankish RR, Judd M, Towheed T, Tugwell P, et al. Rofecoxib for rheumatoid arthritis. Cochrane Database of Systematic Reviews 2010, Issue 7. Art. No: CD003685. [DOI: 10.1002/14651858.CD003685.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
GRADEpro 2015 [Computer program]
- GRADEpro GDT [Computer program]. Version accessed 8 September 2020. Hamilton (ON): McMaster University (developed by Evidence Prime), 2015. Available at gradepro.org.
Grainger 2009
- Grainger R, Taylor WJ, Dalbeth N, Perez-Ruiz F, Singh JA, Waltrip RW, et al. Progress in measurement instruments for acute and chronic gout studies. Journal of Rheumatology 2009;36(10):2346-55. [DOI] [PubMed] [Google Scholar]
Higgins 2017
- Higgins JT, Altman DG, Sterne JAC (editors). Chapter 8. Assessing risk of bias in included studies. In: Higgins JT, Churchill R, Chandler J, Cumpston MS (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.2.0 (updated June 2017), Cochrane. Available from www.cochrane-handbook.org.
Higgins 2020a
- Higgins JT, Eldridge S. Chapter 23. Including variants on randomized trials. Cochrane Handbook for Systematic Reviews of Interventions Version 6.1 (updated September 2020). Available from training.cochrane.org/handbook.
Janssens 2008b
- Janssens HJ, Lucassen PL, Van de Laar FA, Janssen M, Van de Lisdonk EH. Systemic corticosteroids for acute gout. Cochrane Database of Systematic Reviews 2008, Issue 2. Art. No: CD005521. [DOI: 10.1002/14651858.CD005521.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Jordan 2007
- Jordan KM, Cameron JS, Snaith M, Zhang W, Doherty M, Seckl J, et al. British Society for Rheumatology and British Health Professionals in Rheumatology guideline for the management of gout. Rheumatology 2007;46(8):1372-4. [DOI] [PubMed] [Google Scholar]
Kearney 2006
- Kearney P, Baigent C, Godwin J, Halls H, Emberson J, Patrono C. Do selective cyclo-oxygenase-2 inhibitors and traditional non-steroidal anti-inflammatory drugs increase the risk of atherothrombosis? BMJ 2006;332:1302-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Khanna 2012
- Khanna D, Khanna PP, Fitzgerald JD, Singh MK, Bae S, Neogi T, et al. 2012 American College of Rheumatology guidelines for management of gout. Part 2: therapy and antiinflammatory prophylaxis in acute gouty arthritis. Arthritis Care & Research 2012;64(10):1447-61. [DOI] [PMC free article] [PubMed] [Google Scholar]
Lundh 2012
- Lundh A, Sismondo S, Lexchin J, Busuioc OA, Bero L. Industry sponsorship and research outcome. Cochrane Database of Systematic Reviews 2012, Issue 12. Art. No: MR000033. [DOI: 10.1002/14651858.MR000033.pub2] [DOI] [PubMed] [Google Scholar]
Marks 2011
- Marks JL, Colebach AN, Buchbinder R, Edwards CJ. Pain management for rheumatoid arthritis and cardiovascular or renal comorbidity. Cochrane Database of Systematic Reviews 2011, Issue 10. Art. No: CD008952. [DOI: 10.1002/14651858.CD008952.pub2] [DOI] [PubMed] [Google Scholar]
Moore 2010
- Moore RA, Eccleston C, Derry S, Wiffen P, Bell RF, Straube S, et al. "Evidence" in chronic pain - establishing best practice in the reporting of systematic reviews. Pain 2010;150(3):386-9. [PMID: ] [DOI] [PubMed] [Google Scholar]
Page 2020
- Page MJ, Higgins JT, Sterne JAC. Chapter 13: Assessing risk of bias due to missing results in a synthesis. In: Higgins JPT, Thomas J, Chandler J, Cumpston M, Li T, Page MJ, et al, editor(s). Cochrane Handbook for Systematic Reviews of Interventions Version 6.1 (updated September 2020), Cochrane. Available from www.training.cochrane.org/handbook.
Pouliot 1998
- Pouliot M, James MJ, McColl SR, Naccache PH, Cleland LG. Monosodium urate microcrystals induce cyclooxygenase-2 in human monocytes. Blood 1998;91:1769-76. [PubMed] [Google Scholar]
Qaseem 2017
- Qaseem A, Harris RP, Forciea MA, et al. Management of acute and recurrent gout: a clinical practice guideline from the American College of Physicians. Annals of Internal Medicine 2017;166(1):58-68. [DOI] [PubMed] [Google Scholar]
RevMan 2020 [Computer program]
- Review Manager (RevMan) [Computer program]. Version 5.4. Copenhagen: The Nordic Cochrane Centre, The Cochrane Collaboration, 2020.
Rhody 2007
- Rhody E, Zhang W, Doherty M. Is gout associated with reduced quality of life? A case-control study. Rheumatology 2007;46:1441-4. [DOI] [PubMed] [Google Scholar]
Richette 2010
- Richette P, Bardin T. Gout. Lancet 2010;375:318-28. [DOI] [PubMed] [Google Scholar]
Richette 2017
- Richette P, Doherty M, Pascual E, et al. 2016 updated EULAR evidence-based recommendations for the management of gout. Annals of Rheumatic Diseases 2017;76:29-42. [DOI] [PubMed] [Google Scholar]
Roelofs 2008
- Roelofs PD, Deyo RA, Koes BW, Scholten RJ, Tulder MW. Non-steroidal anti-inflammatory drugs for low back pain. Cochrane Database of Systematic Reviews 2008, Issue 1. Art. No: CD000396. [DOI: 10.1002/14651858.CD000396.pub3] [DOI] [PMC free article] [PubMed] [Google Scholar]
Schumacher 2009
- Schumacher HR, Taylor W, Edwards L, Grainger R, Schlesinger N, Dalbeth N, et al. Outcome domains for studies of acute and chronic gout. Journal of Rheumatology 2009;36(10):2342-5. [DOI] [PubMed] [Google Scholar]
Schünemann 2020
- Schünemann HJ, Higgins JT, Vist GE, Glasziou P, Akl EA, Skoetz N, et al. Chapter 14. Completing ‘Summary of findings’ tables and grading the certainty of the evidence. Cochrane Handbook for Systematic Reviews of Interventions Version 6.1 (updated September 2020), Cochrane. Available from training.cochrane.org/handbook.
Schunemann 2020a
- Schünemann HJ, Vist GE, Higgins JT, Santesso N, Deeks JJ, Glasziou P, et al. Chapter 15. Interpreting results and drawing conclusions. Cochrane Handbook for Systematic Reviews of Interventions Version 6.2 (updated September 2020), Cochrane. Available from www.training.cochrane.org/handbook.
Singh 2006
- Singh JA, Strand V. Gout is associated with more comorbidities, poorer health-related quality of life and higher health-care utilization in US veterans. Annals of Rheumatic Diseases 2006;67:1310-6. [DOI] [PubMed] [Google Scholar]
Traditional Chinese Medicine 1994
- State Administration of Traditional Chinese Medicine. Criteria of Diagnosis and Therapeutic Effect of Diseases and Syndromes in Traditional Chinese Medicine. Nanjing: Nanjing University Press, 1994. [Google Scholar]
Wienecke 2008
- Wienecke T, Gotzsche PC. Paracetamol versus nonsteroidal anti-inflammatory drugs for rheumatoid arthritis. Cochrane Database of Systematic Reviews 2008, Issue 4. Art. No: CD003789. [DOI: 10.1002/14651858.CD003789.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Zeng 2021
- Zeng L, Qasim A, Neogi T, et al. Efficacy and safety of pharmacologic interventions in patients experiencing a gout flare: a systematic review and network meta-analysis. Arthritis Care and Research 2021;73(5):755-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
Zhang 2006
- Zhang W, Doherty M, Bardin T, Pascual E, Barskova V, Conaghan P, et al. EULAR evidence based recommendations for gout. Part II: management. Report of a task force of the EULAR standing committee for international clinical studies including therapeutics (ESCISIT). Annals of Rheumatic Diseases 2006;65(10):1312-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
References to other published versions of this review
van Durme CMPG 2014
- Durme CMPG, Wechalekar MD, Buchbinder R, Schlesinger N, Heijde D, Landewé RBM. Non‐steroidal anti‐inflammatory drugs for acute gout. Cochrane Database of Systematic Reviews 2014, Issue 9. Art. No: CD010120. [DOI: 10.1002/14651858.CD010120.pub2] [DOI] [PubMed] [Google Scholar]