

Abstract
Fukutin-related protein (FKRP) is a glycosyltransferase involved in the functional glycosylation of α-dystroglycan (DG), a key component in the link between the cytoskeleton and the extracellular matrix (ECM). Mutations in FKRP lead to dystroglycanopathies with broad severity, including limb-girdle and congenital muscular dystrophy. Studies over the past five years have elucidated the function of FKRP, which has expanded the number of therapeutic opportunities for patients carrying FKRP mutations. These include small molecules, gene delivery and cell therapy. Here we summarize recent findings on the function of FKRP and describe available models for studying diseases and testing therapeutics. Lastly, we highlight preclinical studies that hold potential for the treatment of FKRP-associated dystroglycanopathies.
Keywords: muscular dystrophy, dystroglycanopathy, limb-girdle, congenital muscular dystrophy, α-dystroglycan, glycosylation
Dystroglycanopathies
Muscular dystrophies (MDs) are a group of more than 50 genetic diseases characterized by progressive muscle weakness and degeneration. Among these are dystroglycanopathies, a heterogeneous subgroup characterized by abnormal O-glycosylation (see Glossary) of α-dystroglycan (DG) [1]. α-DG has a fundamental role in linking the intracellular cytoskeleton with the extracellular matrix (ECM), but for this to occur, α-DG needs to be correctly glycosylated [2, 3]. This is a complex sequential process involving more than 20 proteins, among which are included fukutin related protein (FKRP), fukutin (FKTN), protein O-mannosyl transferase (POMT1), and like-acetylglucosaminyltransferase (LARGE1). Mutations in each of these genes result in some form of dystroglycanopathy [4]. In this review, we focus on dystroglycanopathies caused by mutations in FKRP.
FKRP was first identified in 2001 based on its homology to FKTN [5]. This led to the identification of mutations in the FKRP coding region of patients with MDs of previously unknown genetic etiology [5–7]. Recent improvements in sequencing technology and the use of targeted panels have greatly improved diagnostics, resulting in increased detection of mutant variants and better identification of patients affected by dystroglycanopathies [8, 9]. In the last decade, multiple animal models have been developed to study FKRP, and in the last five years, several studies aiming to treat the pathology have been reported. While significant advances have been achieved, curative treatments have not yet advanced to clinical trials. Here we describe available FKRP models and summarize experimental studies that may lead to the future treatment of patients with FKRP-associated dystroglycanopathies.
FKRP pathophysiology
FKRP is a ribitol-5-phosphate transferase that, in tandem with FKTN, adds ribitol-5-phosphate onto the M3 core O-mannosylation of α-DG (Box 1) [10–12]. The presence of ribitol-5-phosphate is a pre-requisite for the subsequent addition of xylose and glucuronic acid repeats, which are necessary for proper glycosylation of α-DG, and therefore, its binding to ECM ligands, in particular laminin, agrin, and perlecan in the sarcolemma, the neuromuscular junction and the central nervous system (CNS) (Figure 1). Additionally, in the CNS, α-DG also interacts with neurexin and slit, and in the retina, α-DG binds to pikachurin [13–15]. These interactions are critical for sarcolemma integrity, maintenance of the basal membrane, cell viability of muscle progenitor cells and regeneration, as well as for neuronal migration and synaptic structural maintenance [16, 17].
Box 1. Secondary dystroglycanopathies and M3 O-mannose glycosylation.
MDs associated with aberrant O-mannosylation of α-DG are called secondary or tertiary dystroglycanopathies [1]. DG is encoded by the DAG1 gene, which by post-translational processing is cleaved into α-DG and β-DG, a transmembrane protein that binds to dystrophin [97]. α-DG is heavily glycosylated, with N-linked and O-linked glycosylation, including O-mannosyl glycans. Among the several types of α-DG O-mannose glycans, FKRP is involved in M3 O-mannose glycosylation [4]. The M3 glycan core begins with protein O-mannosyl transferase 1/2 (POMT1/2) complex catalyzing the addition of a mannose group to serine/threonine residues on α-DG [98] (Figure 1). N-acetylglucosaminyltransferase 2 (POMGNT2) and β1,3-N-acetylgalactosaminyltransferase 2 (B3GALNT2) carry out further modification by adding a β1,4 linked N-acetylglucosamine (GlcNAc) and a β1,3-N-acetylgalactosamine (GalNAc), respectively. This O-mannosyl trisaccharide is the substrate for protein-O-mannose kinase (POMK) that phosphorylates the initial mannose at the 6-positon [99]. Isoprenoid synthase domain-containing (ISPD) modifies ribitol-5-phosphate by adding a cytidine 5′-diphosphate (CDP) group. Once in the Golgi apparatus, CDP-ribitol is then used by FKTN and FKRP to transfer a tandem ribitol-5-phosphate onto the 3-GalNAc-β3-GlcNAc-β4-(6-P)-Man-O-(Ser/Thr) modification on α-DG [10–12]. Finally, transmembrane protein 5 (TMEM5) and β −1,4-glucuronyltransferase 1 (B4GAT1) will sequentially transfer a β1,2-Xylose (Xyl) and a β1,4 glucuronic acid (GlcA) that will serve as a primer for like-acetylglucosaminyltransferase (LARGE1) to add α1,3-Xyl-β1,3-GlcA repeats allowing α-DG to bind to ECM ligands [100–103].
Figure 1: Core M3 α-DG glycosylation process and ECM binding.
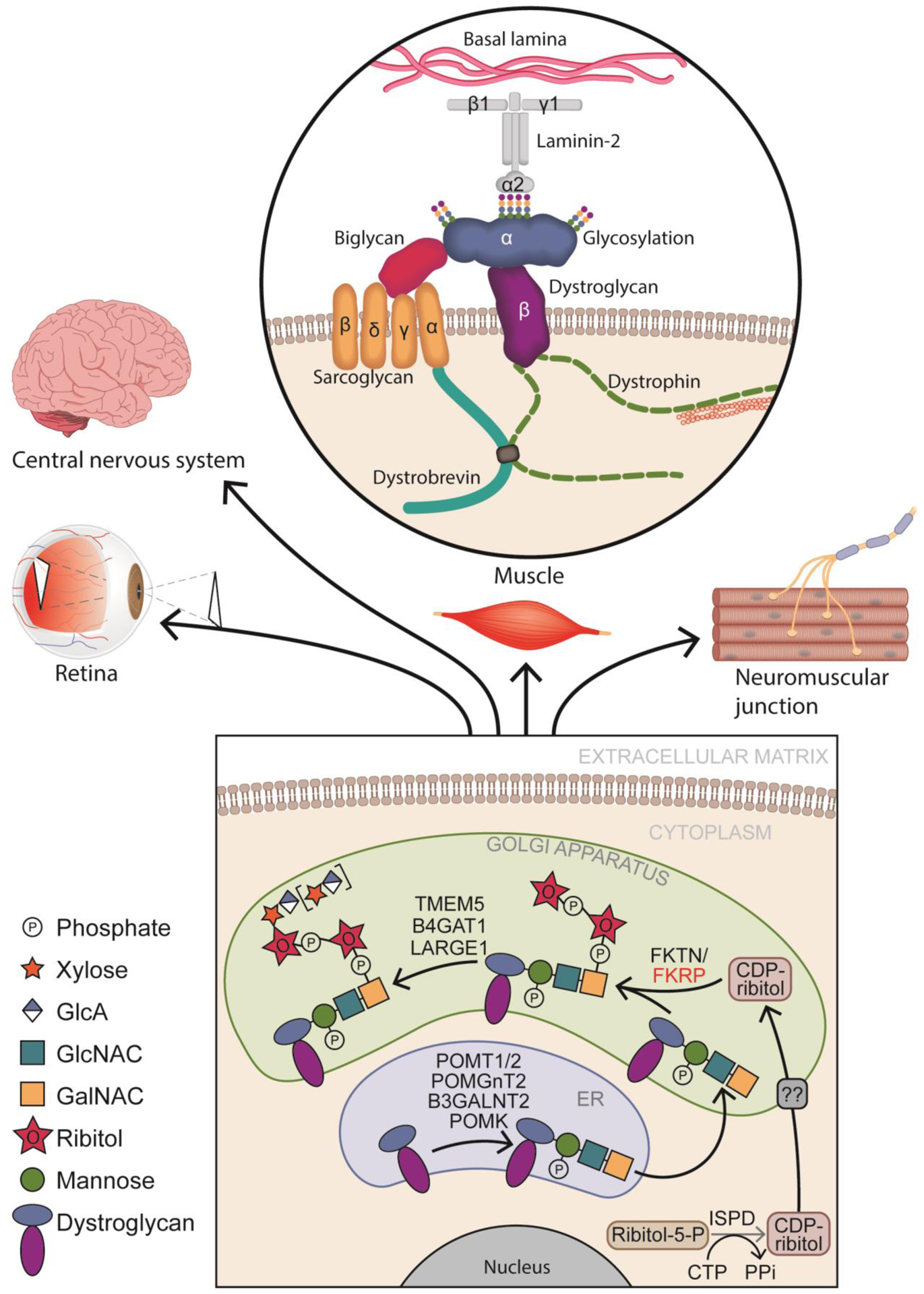
In the endoplasmic reticulum (ER), the glycosylation process begins when protein O-mannosyl transferase 1/2 (POMT1/2) complex catalyzes the addition of a mannose group to serine/threonine residues on α-dystroglycan (DG). Subsequently N-acetylglucosaminyltransferase 2 (POMGNT2) and β1,3-N-acetylgalactosaminyltransferase 2 (B3GALNT2) attach sequentially a N-acetylglucosamine (GlcNAc) and a N-acetylgalactosamine (GalNAc). Protein-O-mannose kinase (POMK) phosphorylates mannose. Once in the golgi apparatus, fukutin (FKTN) and fukutin related protein (FKRP) will use cytidine 5-diphosphate (CDP)-ribitol produced by isoprenoid synthase domain-containing protein (ISPD), to transfer tandem ribitol-5-phosphate onto the 3-GalNAc-β3-GlcNAc-β4-(6-P)-Man-O-(Ser/Thr) modification on α-DG. Afterwards, transmembrane protein 5 (TMEM5) and b-1,4-glucuronyltransferase 1 (B4GAT1) add a β1,2-Xylose (Xyl) and a β1,4 glucuronic acid (GlcA) allowing like-acetylglucosaminyltransferase 1 (LARGE1) to add α1,3-Xyl-β1,3-GlcA repeats. Glycosylated α-DG will bind to laminin α−2 in the muscle sarcolemma, as well as to other components of the extracellular matrix (ECM) in the retina, the central nervous system and at the neuromuscular junction.
FKRP is localized in the Golgi apparatus and contains transmembrane, N-terminal, and C-terminal catalytic domains [18]. It has been previously proposed that FKRP forms oligomers in vivo [19]. A recent report indicates that FKRP is a tetramer and that its dimerization is fundamental for its activity as a ribitol-5-phosphate transferase [20]. This study also demonstrated key regions necessary for the protein’s activity, among which, the zinc finger loop present in the catalytic domain (Figure 2).
Figure 2: FKRP mutations associated with dystroglycanopathies.
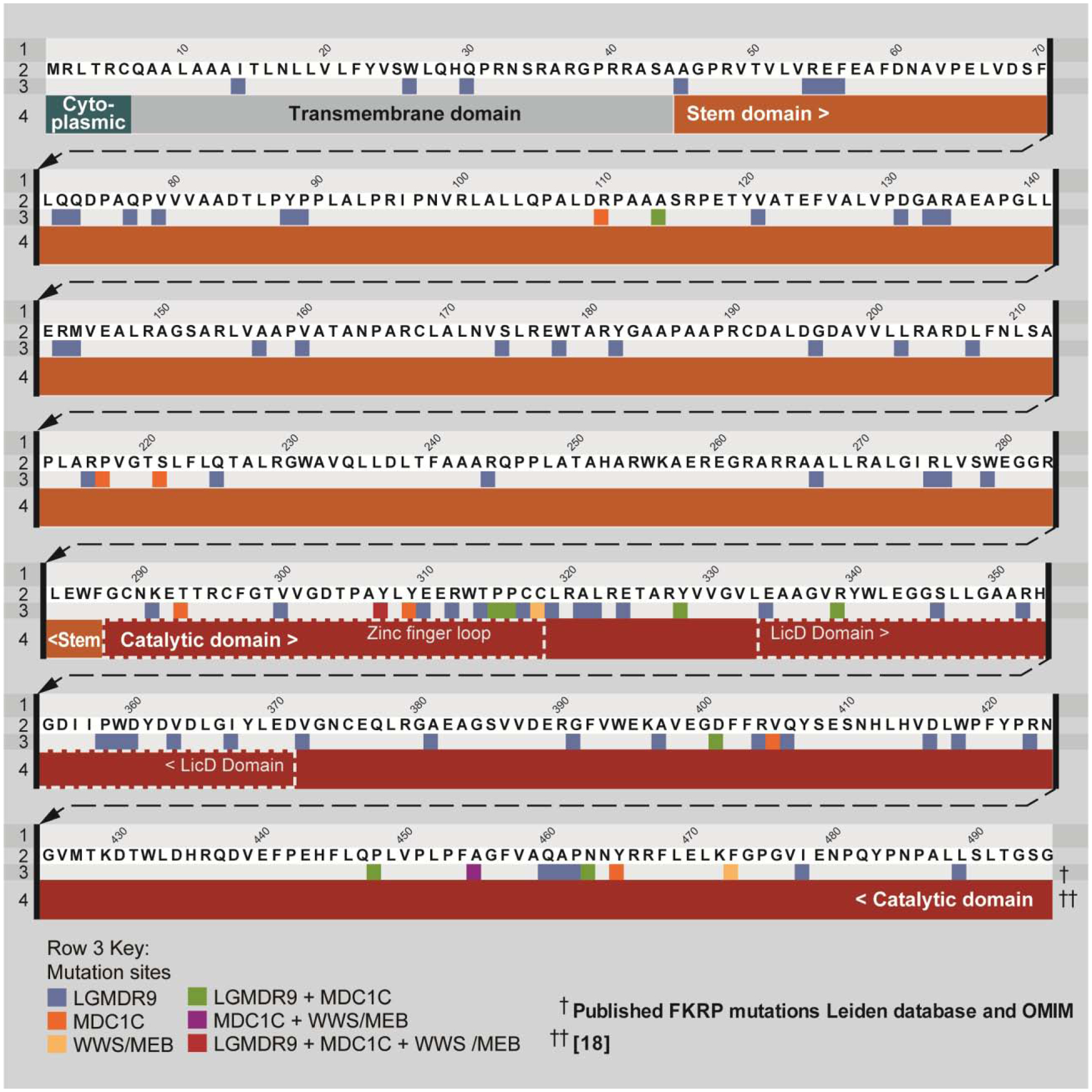
Linear representation of fukutin related protein (FKRP) protein and different unique pathological amino acid variants associated with misssense or framseshift mutations as described in the Leiden database. FKRP-related limb-girdle muscular dystrophy (LGMDR9) only in blue, FKRP-related congenital muscular dystrophy (MDC1C) in orange, Walker-Warburg Syndrome (WWS)/muscle eye brain disease (MEB) in yellow. Green shows mutations associated with LGMDR9 and MDC1C, purple mutations associated with MDC1C and WWS/MEB and the red residues are associated with LGMDR9, MDC1C and WWS/MEB.
FKRP and Associated Dystroglycanopathies
Numerous mutations have been identified in the FKRP gene. According to the Leiden databasei, missense mutations are the most common (63%), followed by frameshift (17%) and nonsense (13%) mutations. Interestingly, mutations in the catalytic domain are associated with more severe dystrophic phenotypes (Figure 2). Recessive mutations in FKRP are associated with a heterogeneous spectrum of dystroglycanopathies, which can range from limb-girdle muscular dystrophy (LGMDR9, previously known as LGMD2I) to several forms of congenital muscular dystrophy (MDC1C), including severe Walker-Warburg Syndrome (WWS) and muscle eye brain disease (MEB). Creatine kinase (CK), a common marker of muscle damage, has been reported to be elevated (1000–15000 U/L) in the serum of LGMDR9 and MDC1C patients, indicating muscle damage [5, 6, 21].
LGMDR9, the most common FKRP-associated dystroglycanopathy [22], manifests between the first and third decades of life. Loss of ambulation and cardiorespiratory issues are common but variable among patients [23]. The most frequently reported FKRP mutation for LGMDR9 is the 826C>A (L276I) [6, 24]. MDC1C is more severe, with symptoms developing in the first year of life, leading to rapid loss of ambulation, cardiorespiratory complications, and death before 10 years of age [5, 22]. At the end of the severity spectrum, WWS, a type of CMD, is accompanied by significant brain and eye abnormalities, with patients succumbing before reaching 3 years of age [7].
Muscle biopsies from these patients (both LGMDR9 and MDC1C) show a reduction in α-DG glycosylation, measured by VIA4 or IIH6C4 (IIH6) antibodies, and a decrease in laminin-α2 binding [25, 26]. The IIH6 antibody is specific for functional α-DG glycosylation, competing for the α-DG binding domain with laminin [2, 27]. While loss of IIH6 immunoreactivity has been used as a hallmark for the diagnosis of FKRP-associated dystroglycanopathies, this may vary among LGMDR9 patients [28–30], suggesting lack of clear correlation between changes in α-DG glycosylation and patient phenotype.
Models
Several models have been established to recapitulate the different phenotypes related to human FKRP mutations. These include FKRP mutant animal models developed in the last decade, as well as the use of patient-specific induced pluripotent stem cells (iPSC), which have emerged as an attractive tool to study human disease (Figure 3).
Figure 3: Available in vitro and in vivo models to study FKRP mutations.
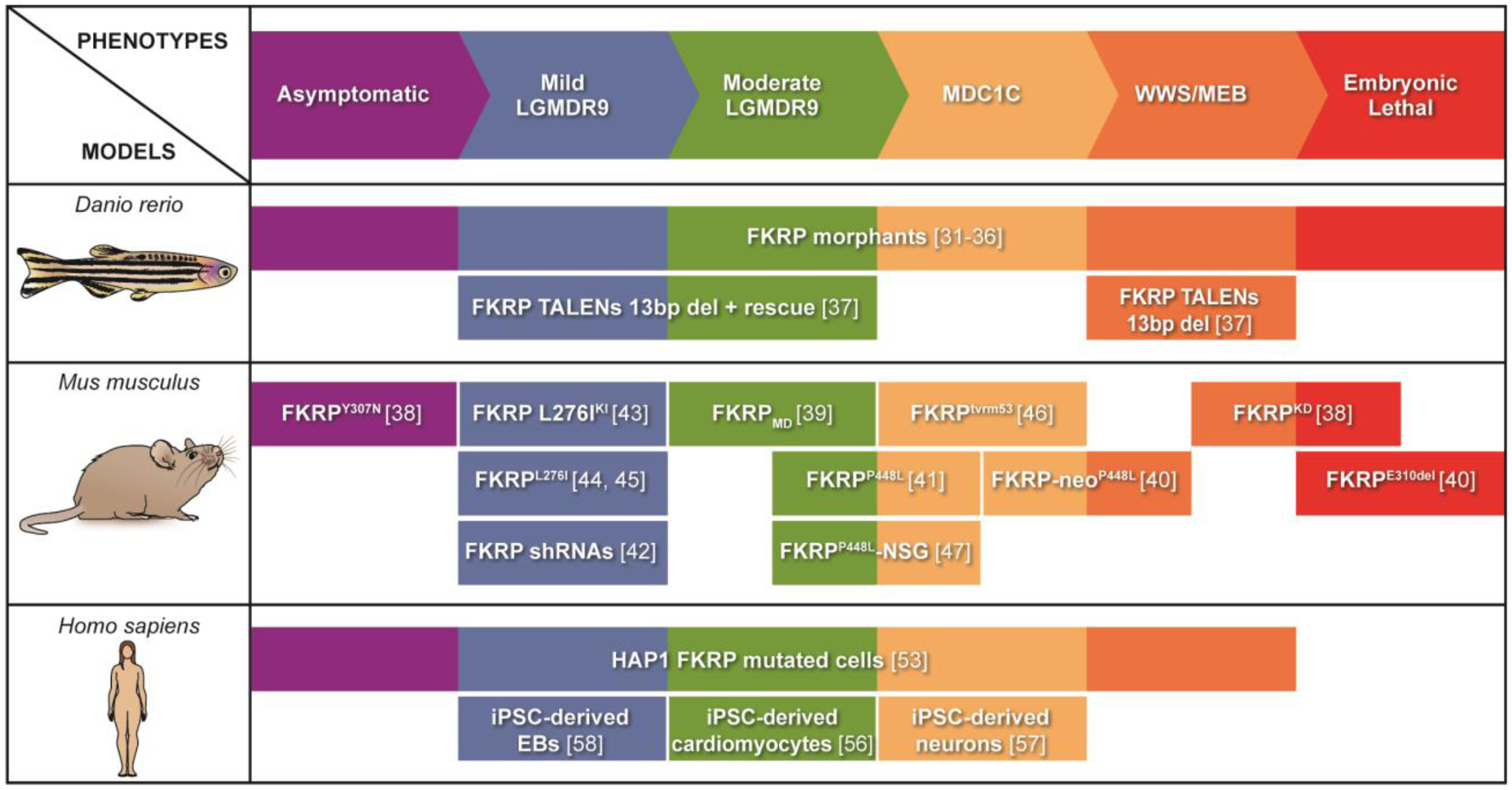
Biological models of fukutin related protein (FKRP)-associated dystroglycanopathies have been developed using zebrafish (danio rerio), mouse (mus musculus), and human cells (homo sapiens). Several zebrafish and mouse FKRP models recapitulate the large spectrum of diseases. The recent use of patient-specific induced pluripotent stem cells (iPSCs) to recapitulate in vitro disease phenotype allowed for the generation of LGMDR9 iPSC-derived EBs and cardiomyocytes, as well as MDC1 iPSC-derived-neurons.
Zebrafish models
Zebrafish (Danio rerio) has been widely used to model human genetic disease due to its rapid development and ease of genetic manipulation and screening [31]. Similarities between zebrafish and human skeletal muscles have triggered studies on MDs [32], and several groups have investigated FKRP function in zebrafish embryos. Morpholino knockdowns show muscle, ocular, vascular, and neuronal defects, recapitulating the broad spectrum of human phenotypes (Figure 3). At the cellular level, embryos show reduced IIH6 immunoreactivity and laminin binding [33–37]. It has been shown that these phenotypes can be partially rescued by co-injecting the human FKRP mRNA, demonstrating functional homology [38]. Recently, TALENs were used to generate two FKRP mutant zebrafish models, which recapitulate WWS and LGMDR9 phenotypes [39]. In the WWS model, a 13 base pair deletion induced a frameshift loss of function that resulted in reduced IIH6 immunoreactivity, severe neuronal, muscular and vasculature defects as well as early death. The LGMDR9 model was obtained by introducing the human FKRP mRNA sequence carrying the L276I mutation in WWS zebrafish [39].
Mouse models
Various FKRP mutant mouse models have been generated with the goal of recapitulating the full spectrum of phenotypes observed in human patients (Figure 3). The first generated FKRP mutant mouse model was the FKRP-neoY307N (FKRPKD), which recapitulates the MEB phenotype [40]. Of note, addition of the neomycin selection cassette (neo) in intron 2 was required to decrease FKRP expression and achieve the desired phenotype. Later on, a model for LGMDR9 (FKRPMD) was derived from this FKRPKD by rescuing FKRP expression in the CNS [41]. In the last decade, Qi Long Lu and colleagues have generated several mouse models that are broadly used by the FKRP scientific community. Among these, FKRP-neoP448L and FKRPP448L mice, which carry the P448L mutation that is associated with MDC1C in humans [42, 43]. As mentioned above, the conservation of the neomycin selection cassette aggravates disease phenotype. Whereas the FKRP-neoP448L mice show MD as well as brain and eye abnormalities, displaying a phenotype between MDC1C and embryonic lethality [42], the FKRPP448L mouse model (following excision of the neomycin cassette), shows a less severe phenotype, which was found restricted to skeletal muscles, thus recapitulating moderate LGMDR9 to mild MDC1C (Figure 3) [43].
Several mouse models carrying the common L276I mutation associated with LGMDR9 have been generated [44–47]. Knock-in, shRNAs and homologous recombination strategies have been used to alter FKRP expression. All these strains have phenotypes resembling the features of mild LGMDR9 with late onset. To study ocular diseases, the FKRPtvrm53 mouse line was generated by chemical mutagenesis using N-ethyl-N-nitrosourea to produce a missense mutation (I356T). These mice display a severe MDC1C phenotype with ocular and muscle defects [48]. Recently, we documented the generation of an immunodeficient FKRP mutant mouse model [49]. This strain was generated by crossing the FKRPP448L mouse [43] with the NSG immunodeficient mouse model [50]. Similar to the immunocompetent counterpart, the FKRPP448L-NSG mice show hypoglycosylation of α-DG in muscle cells, early signs of regeneration, and weaker muscles compared to wild type, recapitulating a moderate LGMDR9 phenotype. This is the first and only FKRP mutant mouse model suitable for the transplantation of human cells [49].
Human models
Studies involving animal models are informative and have been vital for expanding our understanding on the mechanisms underlining FKRP’s function and pathogenesis, but they do not recapitulate all the aspects of human disease, and therefore, promising treatments tested solely in vivo could fail when translated to patients due to differences in physiology. To address this, some investigators have used muscle biopsies from patients with FKRP mutations, but the difficulty of obtaining these samples is a limiting factor [28, 51, 52]. To bypass these hurdles, several groups have used patient fibroblast [53, 54] or modified haploid cell lines, such as the HAP1 overexpressing FKRP variants [55]. Although informative, due to differences in tissue type, the pathophysiology of the disease may not always be recapitulated. A powerful tool for generating large numbers of patient-specific cell types is the reprogramming of somatic cells into iPSCs using the expression of pluripotency-associated transcription factors OCT3/4, SOX2, KL4 and MYC [56]. Using lineage-specific in vitro differentiation, iPSCs can give rise to desired cell types, allowing for disease modeling and drug screening [57].
Several iPSC patient-specific lines have been established for LGMDR9 and MDC1C, among them, an iPSC line from a LGMDR9 patient with the L276I mutation [58]. Cardiomyocytes derived from this iPSC line showed ion channel dysfunction and abnormal calcium dynamics, providing a platform to further elucidate the role of FKRP in cardiac function [58]. Recently, the derivation of iPSCs from an MDC1C patient with CNS involvement was reported [59]. These cells were differentiated into cortical neurons and used for drug screening. LGMDR9 patient-specific iPSCs have also been generated to study ECM defects in vitro [60]. Both these studies demonstrated reduced functional α-DG glycosylation, indicating that iPSC-derivatives can be used to study FKRP-associated diseases.
Experimental Treatments
At present, there are no active clinical trials for FKRP-associated dystroglycanopathies, but several promising therapies are underway (Figure 4). Here we describe therapeutic strategies that show rescue of the dystrophic pathology in experimental models. Detailed information is provided in Table 1.
Figure 4: Potential treatments for FKRP-associated dystroglycanopathies.
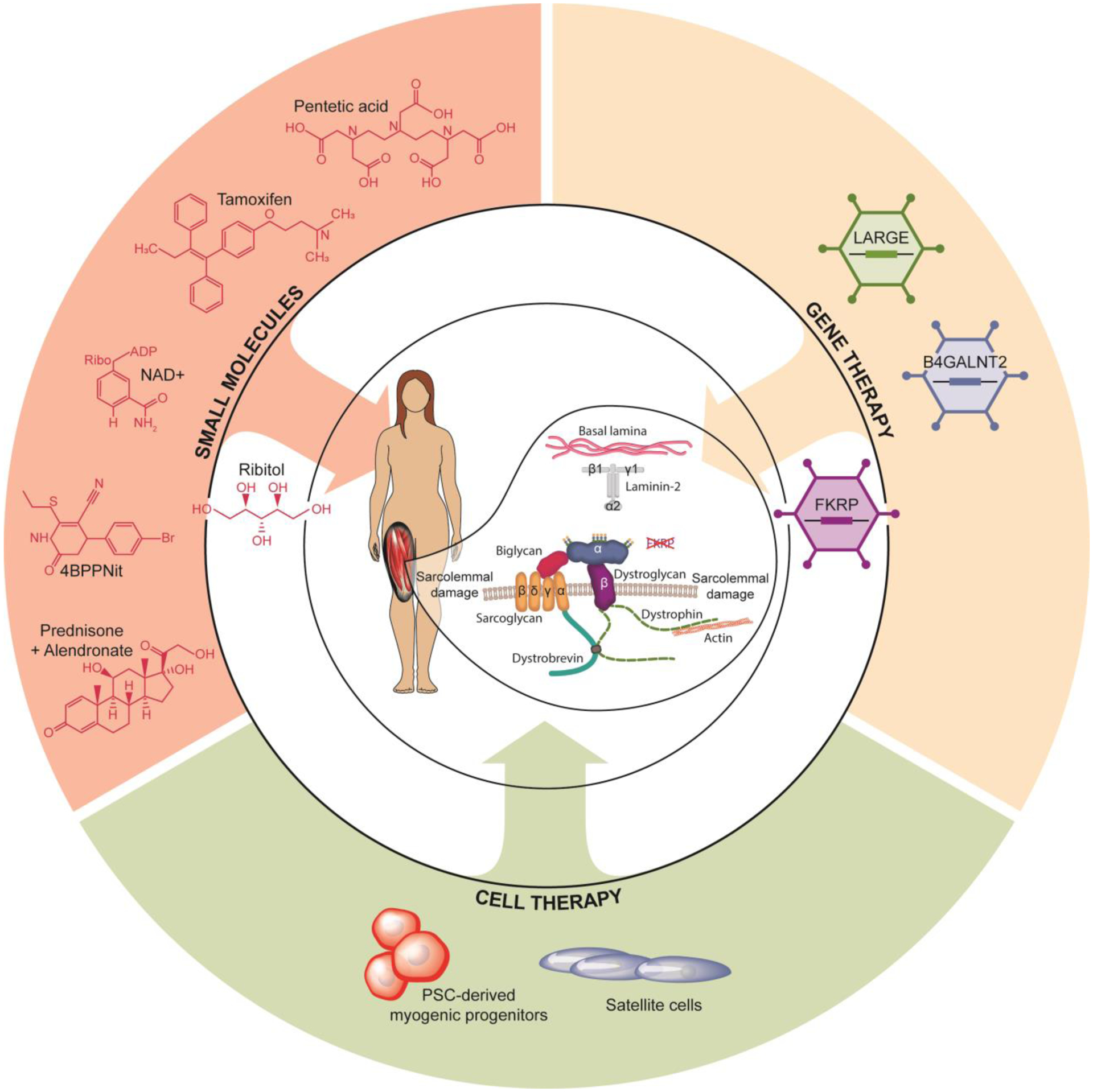
Preclinical studies aiming to develop treatments for fukutin related protein (FKRP)-related disorders include small molecules, gene and cell therapy. Small molecules, like ribitol, nicotinamide adenine dinucleotide (NAD+), 4-(4-bromophenyl)-6-ethylsulfanyl-2-oxo-3,4-dihydro-1H-pyridine-5-carbonitrile (4BBNit), pentetic acid, prednisolone combine with alendronate and the use of tamoxifen have shown the ability to improve disease phenotype. Gene therapy studies with FKRP, like-acetylglucosaminyltransferase (LARGE1) and Beta-1,4 N-acetylgalactosaminyltransferase 2 (B4GALNT2) have shown promising results. Cell therapy studies using engineered satellite cells and the tranplantation of pluripotent stem cell (PSC)-derived myogenic progenitors have also susccesfully rescued the FKRP pathology.
Table 1.
Reported therapeutic strategies for FKRP-associated dystroglycanopathies.
| Treatment | Model | Results | Refs | |
|---|---|---|---|---|
| Small molecules and metabolites | Prednisolone + Alendronate | FKRPP448L mouse |
|
[61] |
| Tamoxifen and Raloxifen | FKRPP448L mouse |
|
[62] | |
| Pantetic Acid | Zebrafish Tg [hsp70l-hFKRP(L276I)-IRES-eGFP] |
|
[39] | |
| Ribitol | FKRPP448L mouse |
|
[66] | |
| iPSC-derived human embryoid bodies |
|
[60] | ||
| NAD+ | Zebrafish FKRP morphants |
|
[36] | |
| 4BPPNit | iPSC-derived cortical neurons |
|
[59] | |
| Gene Therapy | AAV9-CAG-FKRP | FKRPP448L mouse | IP and IV injection:
|
[69, 72, 73] |
| FKRP L276IKI mouse | IV injection:
|
[45] | ||
| rAAV2/9-Des-mFkrp | FKRPL276Imouse | IM and IV injection:
|
[47] | |
| AAV9-CAG-LARGE1 | FKRPP448L mouse | IP injection:
|
[70] | |
| AAVrh74-MCK-GALGT2 | FKRPP448L mouse | IM injection:
|
[71] | |
| Cell therapy | pLenti-CAG (hFKRP)-Rsv satellite cells | FKRP L276IKI mouse | TA IM injection of cells:
|
[81] |
| Pax3/7 iPSC-derived myogenic progenitors | FKRPP448L-NSG mouse | TA IM injection of cells:
|
[49] | |
Abbreviations: CNF, centrally nucleated fibers; DIA, diaphragm; EF, ejection fraction; FS, fractional shortening; GAS, gastrocnemius; GLU, gluteus; IP, intraperitoneal; IV, intravenous; TA, tibialis anterior; HA, heart; QUA, quadriceps; PSO, psoas.
Small molecules
The combinatorial use of corticosteroids and bisphosphonates have been tested in the FKRPP448L mouse model [61, 62]. Corticosteroids are anti-inflammatory drugs used as a palliative treatment for Duchenne MD (DMD) as they enhance muscle strength and function [63], whereas bisphosphonates prevent bone density loss, a well-known side effect of extended corticosteroid treatment [64]. Results in FKRPP448L mice showed decreased muscle degeneration, but no improvement in functional outcomes [61]. Later on, the same group tested the therapeutic effect of the selective estrogen receptor modulators tamoxifen and raloxifene [62], which have been shown to inhibit fibrosis and improve muscle strength in a DMD mouse model [65]. One-year treatment in FKRPP448L mice resulted in decreased infiltration and muscle degeneration, as well as improved muscle and respiratory function. While the dose for raloxifene was above that recommended in humans, tamoxifen dosage was consistent with upper levels currently used in the clinic for other diseases.
Several compounds [39] that had previously been shown to ameliorate the dystrophic pathology in DMD models were tested in the zebrafish L276I model. Among these, the authors found pentetic acid, a calcium and magnesium chelator, to be the most efficacious in rescuing the FKRP zebrafish pathology (Table 1). Using a library of FDA approved drugs, they identified 20 different compounds that attenuated the MD pathology by decreasing eye and head malformations, pericardiac edema, and increasing muscle birefringence and overall viability. Most of these hits were steroids and nonsteroidal anti-inflammatory drugs, or antibacterial compounds [39].
The recent identification of FKRP as a ribitol-5-phosphate transferase opened the possibility of manipulating the levels of CDP-ribitol and ribitol-5-phosphate (Figure 1; Box 1) to increase α-DG glycosylation [10–12]. Studies by Cataldi et al. investigating the effect of oral ribitol supplementation in FKRPP448L mice showed increased levels of CDP-ribitol and ribitol-5-phosphate in the heart and quadriceps, confirming the capacity of ribitol to increase downstream substrates [66]. Of note, continuous ribitol treatment from the start of pregnancy, led to increased α-DG functional glycosylation in all analyzed muscles, which was accompanied by reduced fibrosis and improved muscle function [66]. Recently, ribitol was also reported to enhance IIH6 immunoreactivity in a human iPSC-derived basal lamina embryoid body model, but no significant changes were noted in laminin binding [60].
Due to the reported capacity of nicotinamide adenine dinucleotide (NAD+) to improve dystrophic pathology in a DG model [67], the effect of this coenzyme was tested in FKRP zebrafish morphants [36]. The study reported decreased muscle degeneration and improved muscle organization and function when treatment occurred at gastrulation. However, there were no improvements in the vascular system and brain.
To identify new drugs, a drug screening was performed on immortalized mouse myoblasts [59]. They identified 4-(4-bromophenyl)-6-ethylsulfanyl-2-oxo-3,4-dihydro-1H-pyridine-5-carbonitrile (4BPPNit) as a candidate due to its capacity to increase IIH6 immunoreactivity. Using MDC1C iPSC-derived cortical neurons, the group confirmed 4BPPNit increased α-DG functional glycosylation and laminin binding.
Gene therapy
Gene therapy has been extensively investigated for the treatment of MDs [68]. Several preclinical studies have investigated the use of recombinant adeno-associated virus (AAV) to deliver functional FKRP as well as other genes involved in glycosylation, such as LARGE1 and Beta-1,4 N-acetylgalactosaminyltransferase 2 (B4GALNT2 previously GALGT2) [69–71] (Figure 4).
Initial studies tested intraperitoneal delivery of the human FKRP gene using an AAV9 vector with a non-muscle specific promoter (AAV9-FKRP) in FKRPP448L mice (Table 1). After 4 months, the authors observed increased IIH6 immunoreactivity and laminin binding, reduced muscle degeneration, and enhanced muscle function [69]. Similar rescue was observed when AAV9-FKRP was delivered to neonate and adult FKRP L276IKI mice [45]. Subsequent studies demonstrated that systemic delivery of AAV9-FKRP in FKRPP448L mice promoted long-term one year rescue of α-DG functional glycosylation. Moreover, the authors observed improvement in respiratory function, muscle performance, and partial correction of metabolic alterations [72, 73]. Another group investigated delivery of the mouse FKRP gene using a muscle-specific promoter in FKRPL276I mice (Table 1). Intramuscular (IM) and systemic delivery resulted in increased IIH6 immunoreactivity, laminin binding, and decreased muscle regeneration. Because IM delivery of high doses of the vector led to decreased α-DG functional glycosylation, the group concluded that gene therapy could be effective, but that controlled expression of FKRP is critical to avoid toxicity [47].
Several investigators have explored the overexpression of other genes involved in α-DG glycosylation as a strategy to rescue the FKRP phenotype. As described above, LARGE1 is a key enzyme involved in α-DG glycosylation (Figure 1). Previous studies provided evidence that LARGE1 overexpression can compensate for the loss of other glycosyltransferases, such as POMGNT1, restoring α-DG glycosylation in vitro and in vivo [74, 75]. In the context of FKRP deficiency, overexpression of LARGE1 using an AAV9 vector in FKRPP448L mice led to increased α-DG functional glycosylation, but levels were higher than those observed in wild type mice [70]. While treated mice showed less muscle degeneration, no changes were detected in CK levels. Because LARGE1 overexpression resulted in hyperglycosylation of α-DG, further studies are necessary to understand the implications in muscle function.
Another approach is overexpression of B4GALNT2, a glycosyltransferase involved in the generation of cytotoxic T-cell glycan in adult skeletal muscle [76], which had provided encouraging results in other forms of MD, including DMD, α-sarcoglycan-associated LGMD, and laminin α2-deficient CMD [77–79]. One study assessed B4GALNT2 overexpression using an AAVrh74 vector with a muscle creatine kinase (MCK) promoter [71], which following IM delivery, resulted in improved muscle pathology and increased number of embryonic myosin heavy chain positive fibers in treated mice when compared to untreated counterparts, suggesting that B4GALNT2 overexpression may enhance regeneration [71] (Table 1). In any case, no striking increase in α-DG glycosylation was observed and therefore, it remains to be defined the mechanism underlining these effects and whether B4GALNT2 overexpression improves muscle function.
More recently, investigators have begun exploring gene therapy combined with the use of small molecules. Specifically, FKRPP448L mice were subjected to overexpression of isoprenoid synthase domain-containing (ISPD), which is involved in the generation of CDP-ribitol (Figure 1), along with ribitol treatment [80]. This combinatorial treatment did not result in functional improvement when compared to ribitol alone, but a significant increase in α-DG functional glycosylation was observed in the heart.
Cell therapy
Since skeletal muscle is a highly regenerative tissue, cell-based therapeutic approaches focusing on the delivery of muscle stem cells/early progenitor cells to replace diseased muscle tissue with healthy myofibers as well as satellite cells are highly attractive. To date, two studies have been reported on the use of cell transplantation for FKRP-associated dystroglycanopathies (Figure 4). One utilized transplantation of satellite cells isolated from FKRP L276IKI mice engineered to overexpress wild type human FKRP [81]. Following IM injection of these cells into the tibialis anterior (TA) muscles of FKRP L276IKI mice, the authors confirmed increased levels of FKRP protein, which were accompanied by enhanced α-DG functional glycosylation in the TA, however there was no evidence of cell engraftment. The group also documented rescue of α-DG glycosylation in the heart of treated mice. These counterintuitive results led the authors to hypothesize that FKRP may circulate in exosomes, therefore having the capacity to contribute to α-DG glycosylation at distant sites [81]. Nevertheless, the mechanism, as well as the long-term effects for this strategy remain to be further elucidated.
The second study used PSC-derived myogenic progenitors, which have been extensively validated for their in vivo regenerative capacity in several MD mouse models [82–86]. To determine the ability of these progenitors to treat FKRP-associated dystroglycanopathies, Azzag and colleagues injected mouse and human iPSC-derived myogenic progenitors into TA muscles of FKRPP448L-NSG mice [49]. These transplantation studies revealed robust myofiber engraftment that was accompanied by increased α-DG functional glycosylation, and improved muscle function compared to untreated controls. Importantly, the authors reported the presence of donor-derived satellite cells, which correlated with the persistence of donor-derived fibers 4 months post-transplantation, indicating the long-term contribution of these cells, which is critical to ensure long-lasting therapeutic benefit [49].
Concluding Remarks
Significant progress has been achieved in recent years, but several questions remain to be addressed as well as challenges to be circumvented for the development of therapies. For instance, studies to date have focused on the activity of FKRP glycosyltransferase on α-DG, but the recent discovery of ribitol-5-phosphate post-translational modifications in mammals [11, 12] may allow for the identification of other proteins that might be modified by FKRP (see Outstanding Questions). While weak ECM binding is a well-established hallmark for disease progression, other mechanisms have been proposed to contribute to FKRP pathogenesis, including endoplasmic reticulum stress and unfolded protein response [34, 36, 39, 87]. Moreover, certain mutations have been associated with FKRP mislocalization [55, 88, 89]. Increased understanding of the pathophysiological mechanisms underlining FKRP-associated dystroglycanopathies will be essential for the development of effective therapies.
OUTSTANDING QUESTIONS.
Does FKRP participate in the glycosylation of other glycoproteins? If so, is this function tissue specific?
Are there other mechanisms underlining LGMDR9 pathophysiology in patients displaying high IIH6 immunoreactivity?
Do FKRP mutations trigger endoplasmic reticulum stress and unfolded protein response? Are these involved in disease progression?
Will ribitol treatment be effective for the treatment of all FKRP pathological mutations?
Is it possible to enhance the production of CDP-ribitol and ribitol-5-phosphate by using additional small molecules?
Several experimental studies have shown improvement of disease phenotype. These include the use of small molecules, gene delivery and cell therapy (Figure 4). Although each of these treatments have the potential to treat FKRP-associated dystroglycanopathies, all have specific challenges to be circumvented. The small molecules discovered in zebrafish models should be further validated in mouse and/or human models. Ribitol treatment holds promise as it could be an economically accessible treatment. One aspect to consider is that FKRP residual functional activity is required, warranting further investigation of its therapeutic effect on patients carrying different mutations. FKRP gene replacement has been extensively studied, however challenges remain to be addressed, among which i) the immune response associated with pre-existing immunity, ii) the immunotoxicity associated with large vector doses [90] and iii) the timing of treatment since several studies suggest that age may potentially influence efficiency, with earlier interventions being more efficacious [91, 92]. Importantly, as discussed above, FKRP expression may have to be controlled, as high levels might be deleterious. For cell therapy, challenges include efficient cell delivery to all target muscles as well as high cost at present, in particular if one considers the use of an autologous cell product, which would involve gene editing of patient-specific iPSC prior to the generation of myogenic progenitors and subsequent transplantation. One option would be the transplantation of allogeneic iPSC-derived myogenic progenitors, which would allow the same iPSC muscle product to be used for multiple patients. Several institutions worldwide have established universal donor iPSC banks, also referred as the HLA haplobank model [93–96], which allows for the selection of matched donors to generate graft material that will require only limited immunosuppression.
To date, two interventional clinical trials involving patients with FKRP mutations have been conducted. Ten years ago, the myostatin inhibitor MYO-029 was testedii on a subset of adult MD patients, including LGMDR9, but this trial did not progress beyond phase II. More recently, a trial for the glucocorticoid deflazacort, used for DMD patients, was registerediii for LGMDR9, but this trial is no longer recruiting. Most of the treatments discussed here have been established recently, and although no new interventional clinical trials are currently underway, initiatives like the Global FKRP registryiv intend to facilitate and accelerate this process. The potential displayed by all these treatments to rescue α-DG functional glycosylation offers hope for the development of effective treatments for FKRP-associated dystroglycanopathies.
HIGHLIGHTS.
FKRP is a ribitol-5-phosphate transferase involved in α-DG glycosylation. Mutations in FKRP have been associated with a broad spectrum of dystroglycanopathies.
Several FKRP mutant models have been generated to better understand disease pathogenesis of FKRP-associated dystroglycanopathies as well as to develop therapeutics.
Several strategies, including the use of small molecules, gene therapy and cell therapy, have shown improvement of disease phenotype as evidenced by rescue of α-DG functional glycosylation.
ACKNOWLEDGMENTS
R.C.R.P. is supported by the National Institutes of Health (NIH) grants R01 AR055299 and R01 AR071439. C.O.C. was supported by PINN MICITT Costa Rica. We are thankful to Cynthia Faraday for graphical design.
GLOSSARY
- Agrin
extracellular matrix proteoglycan involved in synaptic formation and maintenance in the brain and muscle.
- Creatine Kinase (CK)
is an enzyme expressed in various tissues, like brain and muscle. High levels of CK in the blood can be used as an indirect indicator of muscle damage.
- Extracellular matrix (ECM)
network of proteins and polysaccharides that provide structural scaffolding and biochemical cues to support surrounding cells.
- Frameshift mutations
mutations that add or delete extra bases of DNA causing a shift in the reading frame. This leads to the DNA sequence after the mutation to be disrupted or read incorrectly.
- Glycosylation
is a common modification in which a carbohydrate is attached to an organic molecule donor. This commonly occurs on proteins and is important for protein function and stability.
- Missense mutations
mutations in which there is a change in a single base pair causing a substitution of a different amino acid in the resulting protein.
- Morphants
organisms treated with morpholinos to modify gene expression.
- Morpholino
synthetic nucleotides that recognize and bind short sequences (18–25 nucleotides) of RNA commonly used to prevent splicing or translation of RNA.
- Myogenic progenitors
cells which can differentiate to form muscle in vitro and in vivo.
- Nonsense mutations
mutations in which the substitution of a single base pair causes the change of a normal codon into a stop codon, resulting in a truncated protein.
- Perlecan
proteoglycan found in the extracellular matrix of muscle and other tissues.
- Pluripotent stem cells (PSCs)
undifferentiated cells endowed with the ability to self-renew as well as to differentiate into any cell type of the body.
- Ribitol
sugar alcohol formed by the reduction of ribose.
- Sarcolemma
specialized plasma membrane surrounding skeletal and cardiac muscle fibers.
- Satellite cells
adult muscle stem cells. These cells regenerate muscle in response to injury or disease.
- TALENs
engineered enzymes that can be used to cut specific DNA sequences for gene editing.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Martin PT (2005) The Dystroglycanopathies: The New Disorders of O-Linked Glycosylation. Seminars in Pediatric Neurology 12 (3), 152–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ervasti JM and Campbell KP (1993) A role for the dystrophin-glycoprotein complex as a transmembrane linker between laminin and actin. The Journal of Cell Biology 122 (4), 809–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ibraghimov-Beskrovnaya O et al. (1992) Primary structure of dystrophin-associated glycoproteins linking dystrophin to the extracellular matrix. Nature 355, 696–702. [DOI] [PubMed] [Google Scholar]
- 4.Sheikh MO et al. (2017) Recent advancements in understanding mammalian O-mannosylation. Glycobiology 27 (9), 806–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brockington M et al. (2001) Mutations in the Fukutin-Related Protein Gene (FKRP) Cause a Form of Congenital Muscular Dystrophy with Secondary Laminin a2 Deficiency and Abnormal Glycosylation of a-Dystroglycan. Am J Hum Genet 69, 1198–1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brockington M et al. (2001) Mutations in the fukutin-related protein gene (FKRP) identify limb girdle muscular dystrophy 2I as a milder allelic variant of congenital muscular dystrophy MDC1C. Human Molecular Genetics 10 (25), 2851–2859. [DOI] [PubMed] [Google Scholar]
- 7.Beltran-Valero de Bernabe D et al. (2004) Mutations in the FKRP gene can cause muscle-eye-brain disease and Walker-Warburg syndrome. Journal of Medical Genetics 41 (5), e61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nallamilli BRR et al. (2018) Genetic landscape and novel disease mechanisms from a large LGMD cohort of 4656 patients. Annals of Clinical and Translational Neurology 5 (12), 1574–1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Thuriot F et al. (2020) Molecular diagnosis of muscular diseases in outpatient clinics: A Canadian perspective. Neurology Genetics 6 (2), e408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Praissman JL et al. (2016) The functional O-mannose glycan on alpha-dystroglycan contains a phospho-ribitol primed for matriglycan addition. Elife 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gerin I et al. (2016) ISPD produces CDP-ribitol used by FKTN and FKRP to transfer ribitol phosphate onto alpha-dystroglycan. Nat Commun 7, 11534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kanagawa M et al. (2016) Identification of a Post-translational Modification with Ribitol-Phosphate and Its Defect in Muscular Dystrophy. Cell Rep 14 (9), 2209–2223. [DOI] [PubMed] [Google Scholar]
- 13.Barresi R and Campbell KP (2006) Dystroglycan: from biosynthesis to pathogenesis of human disease. Journal of Cell Science 119 (2), 199–207. [DOI] [PubMed] [Google Scholar]
- 14.Sato S et al. (2008) Pikachurin, a dystroglycan ligand, is essential for photoreceptor ribbon synapse formation. Nature Neuroscience 11 (8), 923–931. [DOI] [PubMed] [Google Scholar]
- 15.Wright KM et al. (2012) Dystroglycan Organizes Axon Guidance Cue Localization and Axonal Pathfinding. Neuron 76 (5), 931–944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kanagawa M and Toda T (2018) Ribitol-phosphate-a newly identified posttranslational glycosylation unit in mammals: structure, modification enzymes and relationship to human diseases. J Biochem 163 (5), 359–369. [DOI] [PubMed] [Google Scholar]
- 17.Nickolls AR and Bonnemann CG (2018) The roles of dystroglycan in the nervous system: insights from animal models of muscular dystrophy. Dis Model Mech 11 (12). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Esapa CT et al. (2002) Functional requirements for fukutin-related protein in the Golgi apparatus. Human Molecular Genetics 11 (26), 3319–3331. [DOI] [PubMed] [Google Scholar]
- 19.Alhamidi M et al. (2011) Fukutin-related protein resides in the Golgi cisternae of skeletal muscle fibres and forms disulfide-linked homodimers via an N-terminal interaction. PLoS One 6 (8), e22968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kuwabara N et al. (2020) Crystal structures of fukutin-related protein (FKRP), a ribitol-phosphate transferase related to muscular dystrophy. Nat Commun 11 (1), 303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mercuri E et al. (2003) Phenotypic Spectrum Associated with Mutations in the Fukutin-Related Protein Gene. Ann Neurol 53 (4), 537–542. [DOI] [PubMed] [Google Scholar]
- 22.Richard I et al. (2016) 216th ENMC international workshop: Clinical readiness in FKRP related myopathies January 15–17, 2016 Naarden, The Netherlands. Neuromuscul Disord 26 (10), 717–724. [DOI] [PubMed] [Google Scholar]
- 23.Margeta M et al. (2009) Cardiac pathology exceeds skeletal muscle pathology in two cases of limb-girdle muscular dystrophy type 2I. Muscle Nerve 40 (5), 883–889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Murphy LB et al. (2020) Global FKRP Registry: observations in more than 300 patients with Limb Girdle Muscular Dystrophy R9. Ann Clin Transl Neurol 7 (5), 757–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brown SC et al. (2004) Abnormalities in alpha-Dystroglycan Expression in MDC1C and LGMD2I Muscular Dystrophies. American Journal of Pathology 164 (2), 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lin YC et al. (2007) A novel FKRP gene mutation in a Taiwanese patient with limb-girdle muscular dystrophy 2I. Brain and Development 29 (4), 234–238. [DOI] [PubMed] [Google Scholar]
- 27.Ervasti JM and Campbell KP (1991) Membrane Organization of the Dystrophin-Glucoprotein Complex. Cell 66, 1121–1131. [DOI] [PubMed] [Google Scholar]
- 28.Yamamoto LU et al. (2008) Muscle Protein Alterations in LGMD2I Patients With Different Mutations in the Fukutin-related Protein Gene. Journal of Histochemistry & Cytochemistry 56 (11), 995–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jimenez-Mallebrera C et al. (2009) A comparative study of alpha-dystroglycan glycosylation in dystroglycanopathies suggests that the hypoglycosylation of alpha-dystroglycan does not consistently correlate with clinical severity. Brain Pathol 19 (4), 596–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Alhamidi M et al. (2017) Limb girdle muscular dystrophy type 2I: No correlation between clinical severity, histopathology and glycosylated alpha-dystroglycan levels in patients homozygous for common FKRP mutation. Neuromuscul Disord 27 (7), 619–626. [DOI] [PubMed] [Google Scholar]
- 31.Santoriello C and Zon LI (2012) Hooked! Modeling human disease in zebrafish. Journal of Clinical Investigation 122 (7), 2337–2343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Widrick JJ et al. (2019) Discovery of Novel Therapeutics for Muscular Dystrophies using Zebrafish Phenotypic Screens. Journal of Neuromuscular Diseases 6 (3), 271–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Thornhill P et al. (2008) Developmental defects in a zebrafish model for muscular dystrophies associated with the loss of fukutin-related protein (FKRP). Brain 131 (6), 1551–1561. [DOI] [PubMed] [Google Scholar]
- 34.Lin YY et al. (2011) Zebrafish Fukutin family proteins link the unfolded protein response with dystroglycanopathies. Hum Mol Genet 20 (9), 1763–1775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kawahara G et al. (2010) Zebrafish models for human FKRP muscular dystrophies. Hum Mol Genet 19 (4), 623–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bailey EC et al. (2019) NAD+ improves neuromuscular development in a zebrafish model of FKRP-associated dystroglycanopathy. Skelet Muscle 9 (1), 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wood AJ et al. (2011) Abnormal vascular development in zebrafish models for fukutin and FKRP deficiency. Human Molecular Genetics 20 (24), 4879–4890. [DOI] [PubMed] [Google Scholar]
- 38.Kawahara G et al. (2010) Zebrafish models for human FKRP muscular dystrophies. Human Molecular Genetics 19 (4), 623–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Serafini PR et al. (2018) A limb-girdle muscular dystrophy 2I model of muscular dystrophy identifies corrective drug compounds for dystroglycanopathies. JCI Insight 3 (18), e120493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ackroyd MR et al. (2011) Fukutin-Related Protein Alters the Deposition of Laminin in the Eye and Brain. Journal of Neuroscience 31 (36), 12927–12935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Whitmore C et al. (2014) The transgenic expression of LARGE exacerbates the muscle phenotype of dystroglycanopathy mice. Human Molecular Genetics 23 (7), 1842–1855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chan YM et al. (2010) Fukutin-related protein is essential for mouse muscle, brain and eye development and mutation recapitulates the wide clinical spectrums of dystroglycanopathies. Hum Mol Genet 19 (20), 3995–4006. [DOI] [PubMed] [Google Scholar]
- 43.Blaeser A et al. (2013) Mouse models of fukutin-related protein mutations show a wide range of disease phenotypes. Hum Genet 132 (8), 923–934. [DOI] [PubMed] [Google Scholar]
- 44.Wang CH et al. (2011) Post-Natal knockdown of fukutin-related protein expression in muscle by long-termRNA interference induces dystrophic pathology [corrected]. Am J Pathol 178 (1), 261–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Qiao C et al. (2014) Muscle and heart function restoration in a limb girdle muscular dystrophy 2I (LGMD2I) mouse model by systemic FKRP gene delivery. Mol Ther 22 (11), 1890–1899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Krag TO and Vissing J (2015) A New Mouse Model of Limb-Girdle Muscular Dystrophy Type 2I Homozygous for the Common L276I Mutation Mimicking the Mild Phenotype in Humans. J Neuropathol Exp Neurol 74 (12), 1137–1146. [DOI] [PubMed] [Google Scholar]
- 47.Gicquel E et al. (2017) AAV-mediated transfer of FKRP shows therapeutic efficacy in a murine model but requires control of gene expression. Hum Mol Genet 26 (10), 1952–1965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Krebs MP et al. (2017) Mouse models of human ocular disease for translational research. PLOS ONE 12 (8), e0183837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Azzag K et al. (2020) Efficient engraftment of pluripotent stem cell-derived myogenic progenitors in a novel immunodeficient mouse model of limb girdle muscular dystrophy 2I. Skeletal Muscle 10 (1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Shultz LD et al. (2005) Human lymphoid and myeloid cell development in NOD/LtSz-scid IL2R gamma null mice engrafted with mobilized human hemopoietic stem cells. J Immunol 174 (10), 6477–6489. [DOI] [PubMed] [Google Scholar]
- 51.Boito CA et al. (2005) Clinical and Molecular Characterization of Patients With Limb-Girdle Muscular Dystrophy Type 2I. Archives of neurology 62, 1894–1899. [DOI] [PubMed] [Google Scholar]
- 52.Torelli S et al. (2005) Sub-cellular localisation of fukutin related protein in different cell lines and in the muscle of patients with MDC1C and LGMD2I. Neuromuscul Disord 15 (12), 836–843. [DOI] [PubMed] [Google Scholar]
- 53.Willer T et al. (2012) ISPD loss-of-function mutations disrupt dystroglycan O-mannosylation and cause Walker-Warburg syndrome. Nature Genetics 44 (5), 575–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Stevens E et al. (2013) Flow Cytometry for the Analysis of a-Dystroglycan Glycosylation in Fibroblasts from Patients with Dystroglycanopathies. Plos One 8 (7), e68958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Henriques SF et al. (2019) Functional and cellular localization diversity associated with Fukutin-related protein patient genetic variants. Hum Mutat 40 (10), 1874–1885. [DOI] [PubMed] [Google Scholar]
- 56.Takahashi K et al. (2007) Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 131 (5), 861–72. [DOI] [PubMed] [Google Scholar]
- 57.Xia G et al. (2018) Human iPSC Models to Study Orphan Diseases: Muscular Dystrophies. Current Stem Cell Reports 4 (4), 299–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.El-Battrawy I et al. (2018) Ion Channel Dysfunctions in Dilated Cardiomyopathy in Limb-Girdle Muscular Dystrophy. Circulation: Genomic and Precision Medicine 11 (3), e001893. [DOI] [PubMed] [Google Scholar]
- 59.Kim J et al. (2019) A new patient-derived iPSC model for dystroglycanopathies validates a compound that increases glycosylation of alpha-dystroglycan. EMBO Rep 20 (11), e47967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Nickolls AR et al. (2020) Human embryoid bodies as a 3D tissue model of the extracellular matrix and alpha-dystroglycanopathies. Dis Model Mech, dmm042986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wu B et al. (2016) Glucocorticoid Steroid and Alendronate Treatment Alleviates Dystrophic Phenotype with Enhanced Functional Glycosylation of alpha-Dystroglycan in Mouse Model of Limb-Girdle Muscular Dystrophy with FKRPP448L Mutation. Am J Pathol 186 (6), 1635–1648. [DOI] [PubMed] [Google Scholar]
- 62.Wu B et al. (2018) Long-Term Treatment of Tamoxifen and Raloxifene Alleviates Dystrophic Phenotype and Enhances Muscle Functions of FKRP Dystroglycanopathy. Am J Pathol 188 (4), 1069–1080. [DOI] [PubMed] [Google Scholar]
- 63.Waldrop MA and Flanigan KM (2019) Update in Duchenne and Becker muscular dystrophy. Curr Opin Neurol 32 (5), 722–727. [DOI] [PubMed] [Google Scholar]
- 64.Wood CL and Straub V (2018) Bones and muscular dystrophies: what do we know? Current Opinion in Neurology 31 (5), 583–591. [DOI] [PubMed] [Google Scholar]
- 65.Dorchies OM et al. (2013) The anticancer drug tamoxifen counteracts the pathology in a mouse model of duchenne muscular dystrophy. Am J Pathol 182 (2), 485–504. [DOI] [PubMed] [Google Scholar]
- 66.Cataldi MP et al. (2018) Ribitol restores functionally glycosylated alpha-dystroglycan and improves muscle function in dystrophic FKRP-mutant mice. Nat Commun 9 (1), 3448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Goody MF et al. (2012) NAD+ Biosynthesis Ameliorates a Zebrafish Model of Muscular Dystrophy. PLoS Biology 10 (10), e1001409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Crudele JM and Chamberlain JS (2019) AAV-based gene therapies for the muscular dystrophies. Human Molecular Genetics, 28 (R1), R102–R107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Xu L et al. (2013) Adeno-associated virus 9 mediated FKRP gene therapy restores functional glycosylation of alpha-dystroglycan and improves muscle functions. Mol Ther 21 (10), 1832–1840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Vannoy CH et al. (2014) Adeno-associated virus-mediated overexpression of LARGE rescues alpha-dystroglycan function in dystrophic mice with mutations in the fukutin-related protein. Hum Gene Ther Methods 25 (3), 187–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Thomas PJ et al. (2016) B4GALNT2 (GALGT2) Gene Therapy Reduces Skeletal Muscle Pathology in the FKRP P448L Mouse Model of Limb Girdle Muscular Dystrophy 2I. Am J Pathol 186 (9), 2429–2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Vannoy CH et al. (2018) Dose-Dependent Effects of FKRP Gene-Replacement Therapy on Functional Rescue and Longevity in Dystrophic Mice. Molecular Therapy - Methods & Clinical Development 11, 106–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Vannoy CH et al. (2019) Metabolomics Analysis of Skeletal Muscles from FKRP-Deficient Mice Indicates Improvement After Gene Replacement Therapy. Sci Rep 9 (1), 10070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Barresi R et al. (2004) LARGE can functionally bypass alpha-dystroglycan glycosylation defects in distinct congenital muscular dystrophies. Nat Med 10 (7), 696–703. [DOI] [PubMed] [Google Scholar]
- 75.Yu M et al. (2013) Adeno-associated viral-mediated LARGE gene therapy rescues the muscular dystrophic phenotype in mouse models of dystroglycanopathy. Hum Gene Ther 24 (3), 317–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Xia B et al. (2002) Overexpression of the CT GalNAc transferase in skeletal muscle alters myofiber growth, neuromuscular structure, and laminin expression. Dev Biol 242 (1), 58–73. [DOI] [PubMed] [Google Scholar]
- 77.Nguyen HH et al. (2002) Overexpression of the cytotoxic T cell GalNAc transferase in skeletal muscle inhibits muscular dystrophy in mdx mice. Proc Natl Acad Sci U S A 99 (8), 5616–5621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Xu R et al. (2007) Overexpression of the cytotoxic T cell (CT) carbohydrate inhibits muscular dystrophy in the dyW mouse model of congenital muscular dystrophy 1A. Am J Pathol 171 (1), 181–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Xu R et al. (2009) Overexpression of Galgt2 reduces dystrophic pathology in the skeletal muscles of alpha sarcoglycan-deficient mice. Am J Pathol 175 (1), 235–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Cataldi MP et al. (2020) ISPD Overexpression Enhances Ribitol-Induced Glycosylation of a-Dystroglycan in Dystrophic FKRP Mutant Mice. Molecular Therapy - Methods & Clinical Development 17, 271–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Frattini P et al. (2017) Autologous intramuscular transplantation of engineered satellite cells induces exosome-mediated systemic expression of Fukutin-related protein and rescues disease phenotype in a murine model of limb-girdle muscular dystrophy type 2I. Hum Mol Genet 26 (19), 3682–3698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Darabi R et al. (2008) Functional skeletal muscle regeneration from differentiating embryonic stem cells. Nat Med 14 (2), 134–143. [DOI] [PubMed] [Google Scholar]
- 83.Darabi R et al. (2012) Human ES- and iPS-derived myogenic progenitors restore DYSTROPHIN and improve contractility upon transplantation in dystrophic mice. Cell Stem Cell 10 (5), 610–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Filareto A et al. (2013) An ex vivo gene therapy approach to treat muscular dystrophy using inducible pluripotent stem cells. Nat Commun 4, 1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Selvaraj S et al. (2019) Gene Correction of LGMD2A Patient-Specific iPSCs for the Development of Targeted Autologous Cell Therapy. Mol Ther 27 (12), 2147–2157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Selvaraj S et al. (2019) Pluripotent Stem Cell-Based Therapeutics for Muscular Dystrophies. Trends Mol Med 25 (9), 803–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Boito CA et al. (2007) Biochemical and ultrastructural evidence of endoplasmic reticulum stress in LGMD2I. Virchows Arch 451 (6), 1047–1055. [DOI] [PubMed] [Google Scholar]
- 88.Matsumoto H et al. (2004) Subcellular localization of fukutin and fukutin-related protein in muscle cells. J Biochem 135 (6), 709–712. [DOI] [PubMed] [Google Scholar]
- 89.Keramaris-Vrantsis E et al. (2007) Fukutin-related protein localizes to the Golgi apparatus and mutations lead to mislocalization in muscle in vivo. Muscle Nerve 36 (4), 455–465. [DOI] [PubMed] [Google Scholar]
- 90.Shirley JL et al. (2020) Immune Responses to Viral Gene Therapy Vectors. Mol Ther 28 (3), 709–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Gray SJ (2016) Timing of Gene Therapy Interventions: The Earlier, the Better. Molecular Therapy 24 (6), 1017–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Vannoy CH et al. (2017) Efficacy of Gene Therapy Is Dependent on Disease Progression in Dystrophic Mice with Mutations in the FKRP Gene. Molecular Therapy - Methods & Clinical Development 5, 31–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Solomon S et al. (2015) Banking on iPSC--is it doable and is it worthwhile. Stem Cell Rev 11 (1), 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Turner M et al. (2013) Toward the development of a global induced pluripotent stem cell library. Cell Stem Cell 13 (4), 382–4. [DOI] [PubMed] [Google Scholar]
- 95.de Rham C and Villard J (2014) Potential and limitation of HLA-based banking of human pluripotent stem cells for cell therapy. J Immunol Res 2014, 518135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Taylor CJ et al. (2012) Generating an iPSC bank for HLA-matched tissue transplantation based on known donor and recipient HLA types. Cell Stem Cell 11 (2), 147–52. [DOI] [PubMed] [Google Scholar]
- 97.Holt KH et al. (2000) Biosynthesis of dystroglycan: processing of a precursor propeptide. FEBS Lett 468, 79–83. [DOI] [PubMed] [Google Scholar]
- 98.Manya H et al. (2004) Demonstration of mammalian protein O-mannosyltransferase activity: Coexpression of POMT1 and POMT2 required for enzymatic activity. Proc Natl Acad Sci 101 (2), 500–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Yoshida-Moriguchi T et al. (2013) SGK196 Is a Glycosylation-Specific O-Mannose Kinase Required for Dystroglycan Function. Science 341 (6148), 896–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Inamori K et al. (2012) Dystroglycan Function Requires Xylosyl- and Glucuronyltransferase Activities of LARGE. Science 335, 93–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Willer T et al. (2014) The glucuronyltransferase B4GAT1 is required for initiation of LARGE-mediated alpha-dystroglycan functional glycosylation. Elife 3, e03941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Praissman JL et al. (2014) B4GAT1 is the priming enzyme for the LARGE-dependent functional glycosylation of alpha-dystroglycan. Elife 3, e03943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Manya H et al. (2016) The Muscular Dystrophy Gene TMEM5 Encodes a Ribitol beta1,4-Xylosyltransferase Required for the Functional Glycosylation of Dystroglycan. J Biol Chem 291 (47), 24618–24627. [DOI] [PMC free article] [PubMed] [Google Scholar]