

SUMMARY
Cardiac metabolism is a high-oxygen-consuming process, showing a preference for long-chain fatty acid (LCFA) as the fuel source under physiological conditions. However, a metabolic switch (favoring glucose instead of LCFA) is commonly reported in ischemic or late-stage failing hearts. The mechanism regulating this metabolic switch remains poorly understood. Here, we report that loss of PHD2/3, the cellular oxygen sensors, blocks LCFA mitochondria uptake and β-oxidation in cardiomyocytes. In high-fat-fed mice, PHD2/3 deficiency improves glucose metabolism but exacerbates the cardiac defects. Mechanistically, we find that PHD2/3 bind to CPT1B, a key enzyme of mitochondrial LCFA uptake, promoting CPT1B-P295 hydroxylation. Further, we show that CPT1B-P295 hydroxylation is indispensable for its interaction with VDAC1 and LCFA β-oxidation. Finally, we demonstrate that a CPT1B-P295A mutant constitutively binds to VDAC1 and rescues LCFA metabolism in PHD2/3-deficient cardiomyocytes. Together, our data identify an oxygen-sensitive regulatory axis involved in cardiac metabolism.
In brief
Angelini et al. describe a regulatory axis connecting long-chain fatty acid metabolism and oxygen sensing in cardiomyocytes. The findings may have relevance to ischemia and heart failure, in which a cardiac metabolic switch and hypoxia are extensively reported.
Graphical Abstract
INTRODUCTION
Cardiac metabolism is a high-oxygen- and fuel-source-consuming process in order to constantly keep cardiac contractile function at the most optimal range (Angelini et al., 2017; Pascual and Coleman, 2016). Under physiological conditions, the long-chain fatty acid (LCFA) β-oxidation represents the main source of ATP, while remaining fuel sources include glucose, lactate, or ketonic acids (Kerr et al., 2017; Lopaschuk et al., 2010; Pascual and Coleman, 2016). However, when oxygen and/or energy starvation occurs, as it usually happens in myocardial infarction (MI) and heart failure (HF), LCFA β-oxidation tends to decrease, in favor of glycolysis (Abdurrachim et al., 2015; Abozguia et al., 2009; Ashrafian and Frenneaux, 2007; Ashrafian et al., 2007; Nagoshi et al., 2011). This metabolic switch is commonly reported in HF as a cause, a consequence, or an exacerbating factor (Abdurrachim et al., 2015; Abozguia et al., 2009; Ashrafian and Frenneaux, 2007; Ashrafian et al., 2007; Doenst et al., 2010; Nagoshi et al., 2011; Turer et al., 2010). However, the underlying mechanisms remain roughly understood.
Located at the outer mitochondrial membrane (OMM), carnitine O-palmitoyltransferase 1b (CPT1B), the major CPT1 isoform expressed in heart, catalyzes the carnitine conjugation of LCFAs to facilitate their mitochondria uptake and plays a key role in LCFA β-oxidation in cardiomyocytes (Declercq et al., 1987; Hada et al., 2012; van der Leij et al., 2002). Although no disease-causing mutation has been reported in humans, CPT1B depletion in striated muscles leads to a fatal hypertrophic cardiomyopathy associated with lipotoxicity in mouse (Haynie et al., 2014). Moreover, CPT1B haplodeficiency exacerbates cardiac dysfunction when transversal aortic constriction was challenged in mouse (He et al., 2012). Surprisingly, administration of CPT1-specific inhibitors had recently revealed cardioprotective virtues in swine and mouse HF models (Chong et al., 2016; Lefort et al., 2017; Sepa-Kishi et al., 2016). Nevertheless, Malonyl-CoA, an indirect byproduct of cell metabolism, is also known as CPT1 natural feedback inhibitor, thus providing additional regulatory impact on CPT1B activity, and so on LCFA β-oxidation (Awan and Saggerson, 1993; Dyck and Lopaschuk, 2002; Onay-Besikci et al., 2003; Reszko et al., 2004). Such a complex dataset highlights the importance of CPT1B in myocardial metabolic and functional adaptation. However, it remains unclear whether CPT1B is involved in the regulation of cardiac metabolic switch.
During oxygen scarcity, mitochondrial double-membrane integrity is particularly affected (Beraud et al., 2009; Cho et al., 2014; Kaasik et al., 2007). On the OMM, voltage-dependent anion channels 1–3 (VDAC1–3) represent the main interphase between mitochondrial and cytosolic lumen in a wide range of cells and tissues (Lemasters and Holmuhamedov, 2006). VDACs work as two-way channels notably for ionic exchange (Cl−, but also K+, and Na+ in a lesser extend) or short-sized metabolite intermediates (ATP, citrate, glutamate, succinate, pyruvate) (Shoshan-Barmatz and Ben-Hail, 2012; Shoshan-Barmatz et al., 2006). In addition, VDAC1 can bind to Hexokinase-1, the first and key rate-limited enzyme of glycolysis, which had established VDAC1 involvement in metabolic imbalance (Krasnov et al., 2013; Pedersen, 2008). However, little is known about its potential role in LCFA β-oxidation.
Hypoxia response is an adaptive process that is transiently activated to sustain oxygen and energy needs during physiological or pathological stress (Brahimi-Horn et al., 2007; Brahimi-Horn and Pouysségur, 2007a; Giordano, 2005). In fact, the imbalance between normoxia and hypoxia is mainly linked to the activity of Prolyl-4-Hydroxylase domain proteins (PHDs), which represent a family protein of Fe2+ 2-oxoglutarate-dependent dioxygenases that are usually involved in the oxygen-dependent degradation of hypoxia inducible factor (HIF)-α subunit (Marxsen et al., 2004; Takeda et al., 2007). Independently from this canonical pathway, PHDs now emerge as key sensors of both cell redox status and metabolic activity, which makes them targets for therapeutics in ischemic diseases and heart failure (Fraisl et al., 2009; Martin-Puig et al., 2015; Ong and Hausenloy, 2012; Schreiber et al., 2019). PHD2 and PHD3 (PHD2/3) are the most abundant cardiac isoforms (Xie et al., 2015a). Transgenic mouse models had carried out interesting data concerning their role onsite. In fact, while individual deletion of PHD2 or PHD3 seemed to be cardio protective, triggering the loss of PHD2/3 potentiated cardiac injury induced by chronic stimulation of isoproterenol (Hölscher et al., 2011; Xie et al., 2015a, 2015b). Similarly, in PHD2flox/flox; PHD3−/− mice, the long-term depletion of PHD2/3 leads to a severe cardiomyopathy, which was accompanied by a net enrichment of genes involved in fatty acid β-oxidation, glycolysis, and neoglucogenesis in the hearts (Minamishima et al., 2009).
Here, we reported a regulatory complex involving PHD2/3, CPT1B, and VDAC1 that may orchestrate cardiac metabolism. The loss of PHD2/3 leads to early metabolic changes in mouse, while LCFA mitochondrial uptake and β-oxidation were dramatically impaired in response to PHD inhibition or depletion in primary cardiomyocytes. PHD2/3 interact with CPT1B, while CPT1B itself binds to VDAC1. Meanwhile, we found that VDAC1 positively impacts on LCFA β-oxidation in vitro and CPT1B-VDAC1 interaction is required for LCFA β-oxidation in cardiomyocytes. More interestingly, CPT1B-VDAC1 complex formation is PHD2/3 activity dependent, and we identified CPT1B-P295 residue as a prolyl-4-hydroxylation site required for CPT1B-VDAC1 binding. Finally, overexpression of an oxygen-insensitive CPT1B-P295A mutant was able to maintain CPT1B-VDAC1 interaction, as well as LCFA β-oxidation in PHD2/3-deficient cardiomyocytes. Our studies shed light on the regulation of LCFA metabolism in cardiomyocytes and provide strategies for treating cardiac metabolic diseases.
RESULTS
PHDs interact with CPT1B and regulate LCFA uptake and oxidation in primary cardiomyocytes
PHD2 and 3 are highly enriched in the heart (Freeman et al., 2003). In order to uncover the potential roles of PHD2 and 3 in cardiac function, we first performed an unbiased screen to identify PHD3 interaction proteins in primary neonatal rat ventricular cardiomyocytes (NRVMs). Proteins co-immunoprecipitated with FLAG-PHD3 were separated by SDS-PAGE and visualized by Coomassie blue staining. Surprisingly, we found that CPT1B was one of the main interaction proteins pulled down with FLAG-PHD3 (Figure 1A). We then performed a co-immunoprecipitation experiment with FLAG-PHD3 followed by western blots to validate this interaction. As shown in Figure 1B, PHD3 was able to pull down CPT1B. Interestingly, PHD2 also interacted with CPT1B (Figure 1B). Finally, we performed a co-immunoprecipitation with endogenous CPT1B, and we were able to similarly pull down both PHD2 and PHD3 in NRVMs (Figure 1C), hence validating the interaction. These results demonstrated that CPT1B interacts with both PHD2 and PHD3 in primary cardiomyocytes, suggesting that they may be involved in cardiac LCFA metabolism.
Figure 1. PHD2/3 bind to CPT1B and regulate LCFA uptake and oxidation in primary cardiomyocytes.
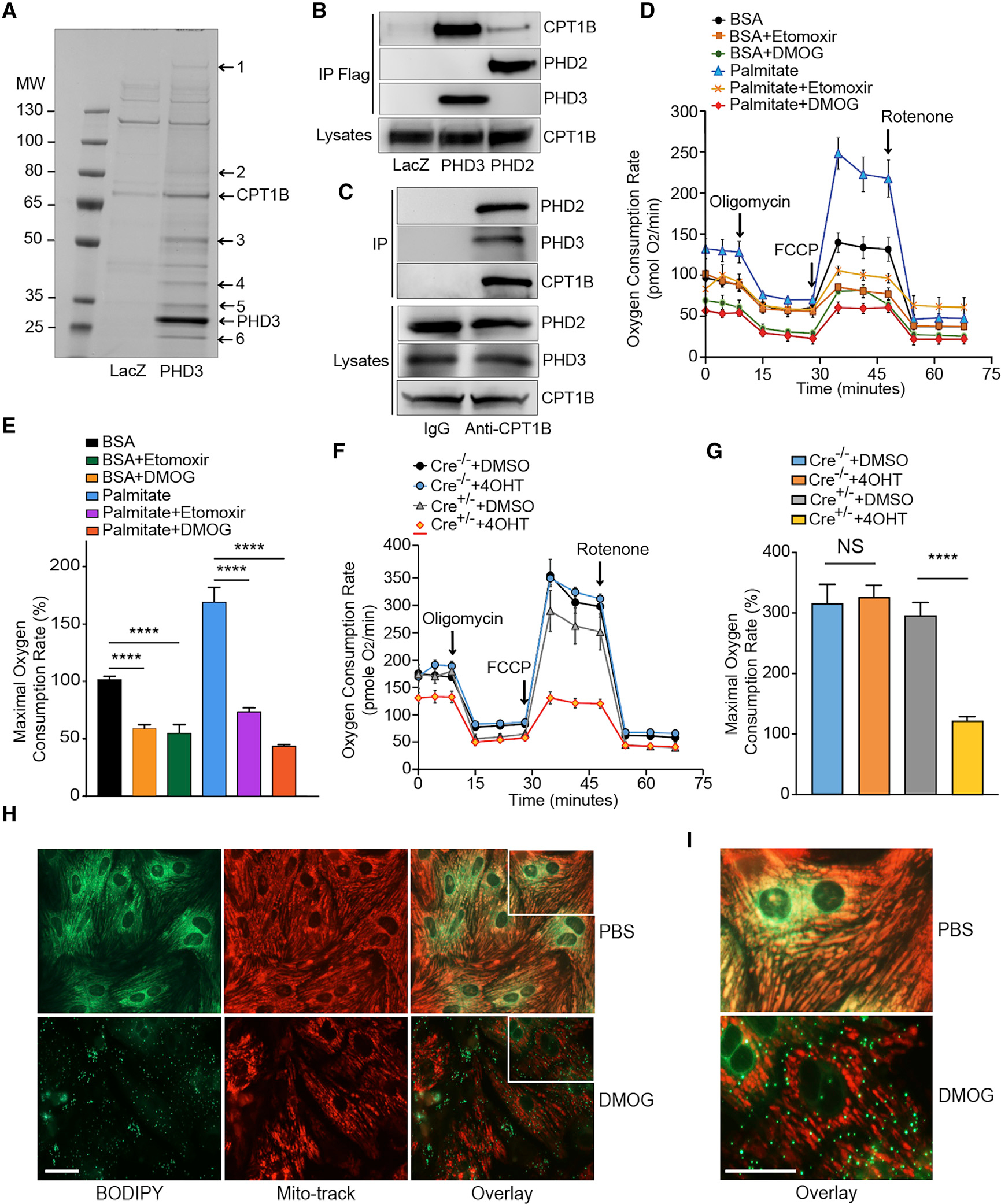
(A) Coomassie-stained SDS-PAGE of FLAG-PHD3 co-immunoprecipitated proteins in NRVMs. PHD3 or LacZ was overexpressed in NRVMs, and co-immunoprecipitation was performed with anti-FLAG agarose beads. Pulled-down proteins were separated with SDS-PAGE and visualized with G-250 Coomassie blue. Protein identities were determined by tandem mass spectrometry analysis (1. Dync1h1; 2. Na+/K+ ATPase α−1; 3. ATP synthase B; 4. GAPDH; 5. ANT; 6. MLP). The respective positions of PHD3 and CPT1B are labeled in the figure.
(B) Co-immunoprecipitation and western blot revealed PHD2 and 3 binding to CPT1B. FLAG-PHD2 and FLAG-PHD3 were immunoprecipitated by anti-FLAG beads, and the precipitated proteins were analyzed by western blot with anti-FLAG and anti-CPT1B antibodies.
(C) Co-immunoprecipitation and western blot revealed endogenous CPT1B binds to endogenous PHD2 and 3. The precipitated proteins were analyzed by western blot with anti-PHD2, PHD3, or CPT1B antibodies.
(D) Oxygen consumption rate (OCR) in primary cardiomyocytes after DMOG or Etomoxir (CPT1 inhibitor, Eto) treatment. OCR was real-time analyzed in living NRVMs by using a SeaHorse XFp Flux Analyzer. DMOG treatment leads to a drastic decrease in LC-FAO.
(E) Maximal respiration rate was estimated for each experimental group of cells.
(F and G) Similar experiment was conducted by using PHD2/3_HKO NMCMs. After 4 consecutive days of 4-hydroxyl-tamoxifen (4OHT) treatment to induce PHD2/3 disruption (or DMSO for untreated control cells), OCR was measured following the same protocol that described for NRVMs (F) and maximal respiration rate estimated afterward (G). A dramatic decrease in LC-FAO was found in PHD2/3-deficient cardiomyocytes, and maximal respiration rate was significantly decreased only in 4OHT treated PHD2/3_HKO NMCMs therefore in response to the loss of PHD2/3.
(H) Mitochondrial LCFA uptake in primary cardiomyocytes after DMOG treatment. Five days after seeding, NRVMs were cultured in serum free medium with 1 μM BODIPY 500/510 C12 in the presence of DMOG (1mM) or DMSO for 16 h (H, left panel). Mitochondria were labeled 5 with Mitotracker Red (H, middle panel).
(I) The same overlay of each experiment at a higher magnification.
Scale bars, 50 μm. For (D)–(G), results are shown ±SEM for an average of at least 4 different experiments: ****p < 0.0001.
To ascertain the potential role of PHD2/3 in LCFA β-oxidation in cardiomyocytes, we first treated NRVMs with Di-methyloxalylglycine (DMOG), a competitive pan-inhibitor of PHDs. LCFA β-oxidation was assessed by using a XFp96 Extracellular-Fluxanalyzer. DMOG-treated NRVMs exhibited a dramatic decrease of oxygen consumption rate (OCR) compared to control untreated cells at both the basal condition and in the presence of palmitate, thus leading to a clear and proportional decay of maximal respiration rate (Figures 1D and 1E). Compared with the response to Etomoxir, a CPT1 inhibitor, the impairment of mitochondrial oxidative phosphorylation (OXPHOS) due to DMOG was even more dramatic (Figures 1D and 1E). In order to validate the direct and specific impact of PHD2/3 on LCFA β-oxidation in cardiomyocytes, we isolated neonatal mouse ventricular cardiomyocytes (NMVMs) from PHD2/3flox/flox; CAG-CreER+/− pups and then added 4-hydroxyl tamoxifen (4OHT) in their culture medium for 4 consecutive days to induce PHD2/3 depletion (Xie et al., 2015a). Similar to DMOG-treated NRVMs, 4OHT-treated PHD2/3flox/flox; CAG-CreER+/− NMVMs had a dramatic decrease in basal and maximal respiration rates, whereas DMSO-treated PHD2/3flox/flox; CAG-CreER+/− NMVMs were not distinguishable from 4OHT or DMSO-treated PHD2/3flox/flox; CAG-CreER−/− control NMVMs (Figures 1F and 1G). Since CPT1-mediated carnitine conjugation of LCFAs is required for their mitochondrial uptake, we then utilized BODIPY 500/510 C12, a saturated 18-carbon FA analog, to track LCFA mitochondrial uptake in NRVMs (Rambold et al., 2015). As shown in Figures 1H and 1I, BODIPY 500/510 C12 was located in the mitochondria in NRVMs. In contrast, it was accumulated in lipid droplets (LDs) outside mitochondria in DMOG-treated NRVMs. In addition, NRVMs exposed to hypoxic condition exhibited exactly the same pattern as DMOG-treated cells, suggesting an impairment of LCFA mitochondrial uptake in response to PHDs inactivation (Figure S1).
Finally, we investigated how substrate fueling is specifically used in these 4OHT-treated PHD2/3flox/flox; CAG-CreER+/− NMVMs by using Mitofuel flex kit. Briefly, cells were pre-incubated with specific inhibitors for rate-limited steps of LCFA β-oxidation (etomoxir), glycolysis (UK-5099), and glutamine (BPTES). Inhibition of one pathway enabled us to measure the dependence for this pathway, whereas blocking the two others provided information concerning the fueling capacity of the remaining metabolic pathway. Here, first, we found that control NMVMs mainly (80%) use LCFA. In addition, they are also highly reliable on LCFA β-oxidation but with a poor (negative) flexibility for this fuel, which represents the combination between high yield of LCFA-dependent OCR and the drastic inhibition of OCR resulting from etomoxir incubation (Figure S2A). Then, not surprisingly, untreated cultured NMVMs only have a lower reliance for fueling from glucose/pyruvate and glutamine (above 10% for both of the source) (Figure S2B and S2C). By contrast, in the absence of PHD2/3, as shown above, LC-FAO is drastically impacted, which leads to a significant decrease in LCFA both dependency and total capacity (Figure S2A). As a result, we also noticed a tendency to increase for glycolysis reliance, as both the total capacity and the fuel dependency for glucose and pyruvate increased in PHD_HKO NMVMs. In addition, aside from LC-FAO/glycolysis imbalance, NMVMs lacking for PHD2/3 have a significant 4-fold increased dependency for glutamine metabolite, while the overall capacity remains unchanged (Figure S2C). Usually, increased glutamine works as a potential antioxidant, while sided increased glucosamine favors the production of UDP-GlcNAc and O-GlcNAc, which in turn are able to boost the rate of palmitate oxidation (Laczy et al., 2011). Therefore, this might be a short-term adaptation, in the attend to immediately relaunch the LC-FAO as soon as oxygen might be available again.
In summary, these results suggested that inhibition or depletion of PHD2/3 in primary cardiomyocytes inhibits LCFA uptake and subsequent β-oxidation in mitochondria.
Cardiomyocyte-specific depletion of PHD2/3 improves glucose metabolism in mice; however, it worsens hypertrophic cardiomyopathy
To maintain the high ATP demand for constant contraction, the heart has a metabolic flexibility, which allows it to switch between different energy substrates such as FA and glucose according to feeding, hormonal status, or substrate availability (Goldberg et al., 2012). Indeed, global depletion of PHD2/3 in PHD2/3flox/flox; CAG-CreER+/− (D2/3_KO) mice leads to significant increases in glucose infusion/disposal rate in mice, as we determined by hyperinsulinemic/euglycemic clamp experiments (Figure 2A). Interestingly, we found that hepatic glucose production after clamp was not significantly affected by PHD2/3 depletion; however, glucose uptake was dramatically increased in PHD2/3 knockout hearts (Figure 2B). To decipher more cardiac-restrictive consequences, we decided to use PHD2/3flox/flox; Myh6-CreER+/− (PHD2/3_HKO) mice instead, in which tamoxifen injection triggers cardiomyocyte-specific depletion of PHD2 and 3. Despite a less dramatic cardiac phenotype compared to PHD2/3_KO mice (data not shown), PHD2/3_HKO mice exhibited a slower growth curve and a significant decrease in blood glucose level at both control-chow (CC) and high-fat-diet (HFD)-feeding conditions (Figure S3A; Figure 2D). Glucose tolerance curves were also significantly improved in PHD2/3_HKO mice, compared with PHD2/3flox/flox; Myo-CreER−/− (PHD2/3_HWT) mice (Figures 2E and 2F).
Figure 2. Global and cardiac-specific loss of PHD2/3 impact cardiac metabolism.
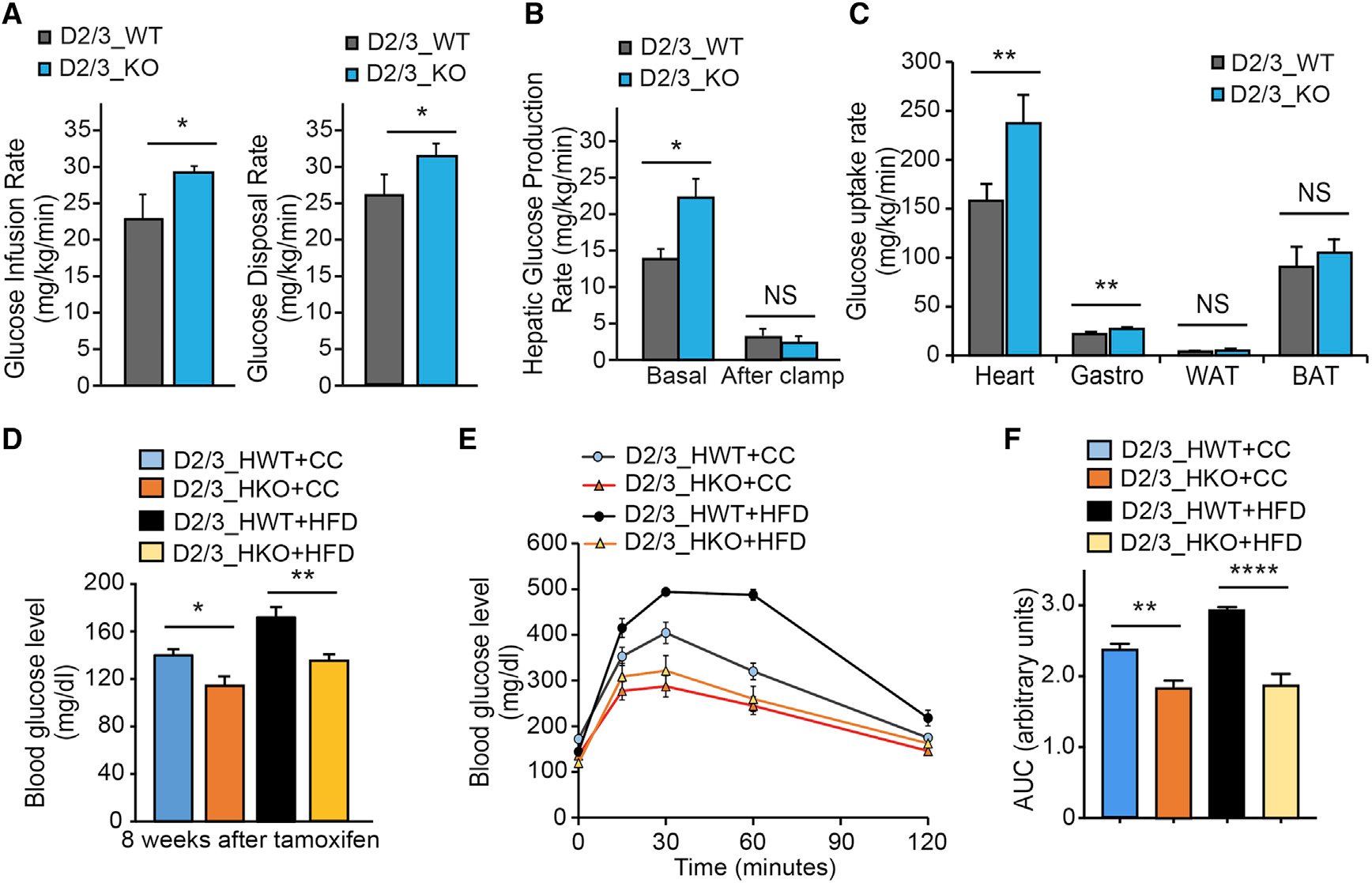
(A–C) Hyperinsulinemic-euglycemic clamp in conscious D2/3_KO and D2/3_WT mice at 4 weeks after tamoxifen injection (n = 6 per each group). Glucose metabolism is significantly increased in D2/3_KO mice (A). Hepatic glucose production rate is shown in (B). Tissue glucose uptake is shown in (C), showing a predominant increase of glucose uptake in the heart of D2/D3_KO mice, and also in Gastrocnemius muscle at a lower range (C).
(D–F) Cardiomyocyte-specific disruption of PHD2/3 was triggered by tamoxifen injection (20 mg/kg/day) for 4 consecutive days in D2/3_HKO mice. Mice from both genotypes were split in two groups randomly and fed with normal chow (CC) or high-fat diet (HFD) for 8–12 weeks (n = 6–11 mice per group). The glucose blood level was monitored at the 8-week time point following PHD2/3 disruption (D). D2/3_HKO mice showed a significantly lower blood glucose level, at both CC and HFD-feeding conditions. (E) Glucose tolerance test at 8 weeks following CC or HFD in D23_HKO and D2/3_HWT mice. Similarly, as expected, HFD-D2/3_HWT mice had a pre-diabetic-specific slower response to glucose, whereas D2/3_HKO mice exhibited a faster response to glucose, which remained equivalent regardless CC or HFD feeding. (F) Area under the curve from data shown in (E), depicting significant difference among the groups of mice.
Data are shown as averages ± SEM. Statistically significant differences are indicated by asterisks: *p < 0.05, **p < 0.01, and ***p < 0.001.
Since defects in cardiac fatty acid β-oxidation (FAO) used to be depicted as worsening cardiac function (Haynie et al., 2014; Kolwicz et al., 2012), we decided to submit PHD2/3_HKO mice to HFD feeding, in order to ascertain whether the defect in LCFA β-oxidation can fasten PHD2/3-deficiency-induced cardiomyopathy. Compared with HFD-D2/3_HWT group, as monitored by echocardiography, HFD-PHD2/3_HKO mice started to exhibit cardiac contractile dysfunction at 4 weeks following combination of PHD2/3 depletion and HFD feeding, which was characterized by a decrease in ejection fraction and fractional shortening, whereas CC-fed D2/3_HKO remained non-differentiable from control littermates at this early time point (Figure 3A; Figure S3B). By contrast, at 12 weeks, both CC and HFD PHD2/3_HKO groups exhibit significant and comparable decreases in ejection fraction or fractional shortening and increases in heart weight to tibia length ratio or left ventricular internal diameters (LVIDs) (Figure 3B; Figures S3C–S3F). While comparing the cardiomyocyte area by WGA staining, we noticed a 1.5-fold increase of the cardiomyocyte cell-surface area in response to PHD loss triggering under the control-chow diet, whereas HFD in PHD_HKO mice leads to a 2.5-fold increase of cardiomyocyte size at 12 weeks after tamoxifen treatment (Figures 3C and 3D). In addition, although Oil red O staining revealed lipid accumulation in CC-PHD2/3_HKO mouse hearts, there were significantly more numerous and bigger lipid droplets in the hearts of HFD-PHD2/3_HKO mice (Figures 3E and 3F). Therefore, a defect in LCFA β-oxidation accelerates the onset of PHD2 and 3 deficiency-associated cardiac defects.
Figure 3. Cardiac-specific depletion of PHD2/3 worsens hypertrophic cardiomyopathy.
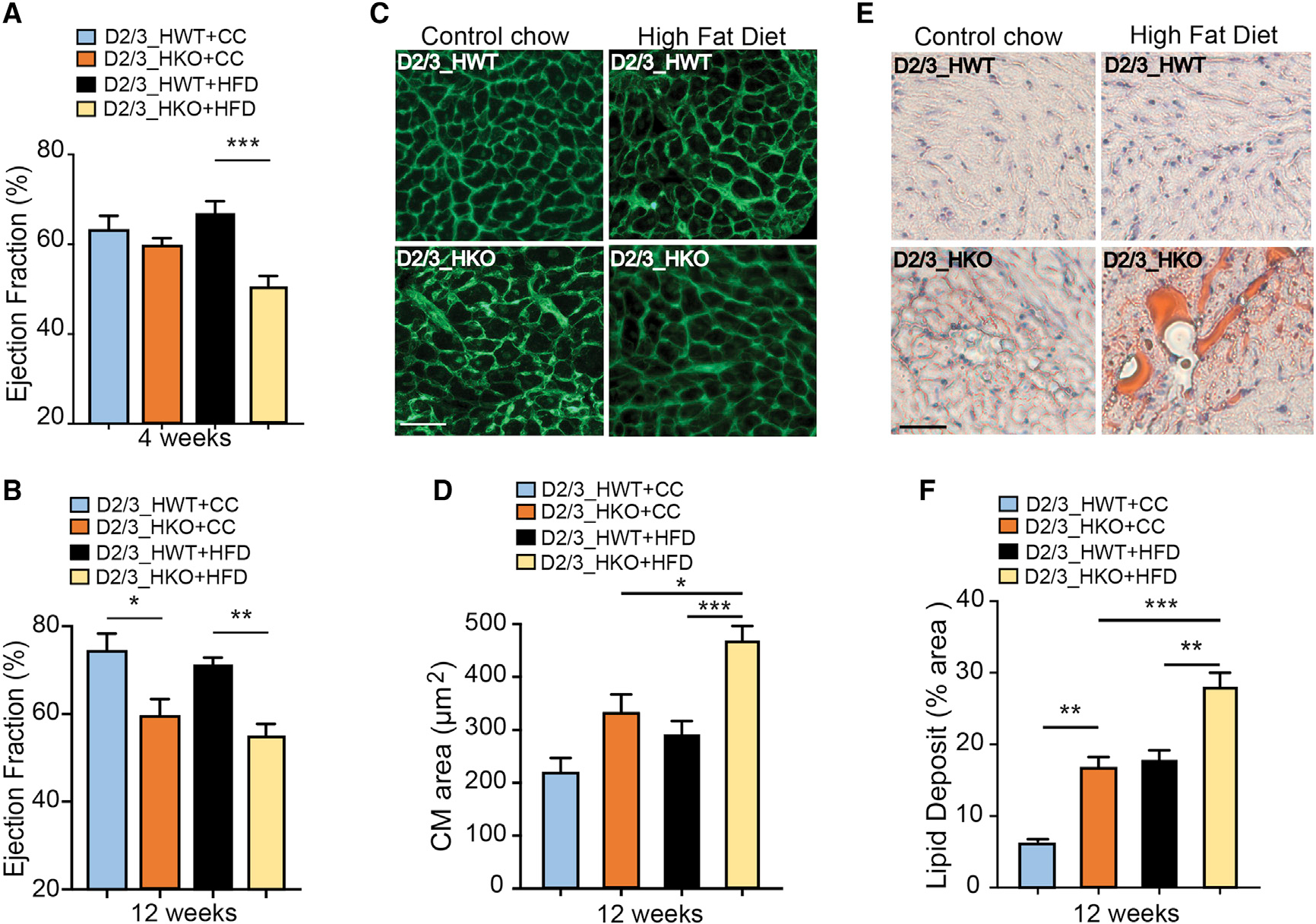
Following tamoxifen injection, D2/3_HKO and D2/3_HWT mice were fed with HFD or CC for 12 weeks (n = 6–11 mice group).
(A and B) Echocardiography analyses were performed at 4 weeks (top panel) and 12 weeks (bottom panel) after HFD or CC. HFD fastens the impairment of cardiac function in D2/3_HKO, showing significant decrease of ejection fraction at 4 weeks after tamoxifen infusion, instead of 12 weeks for CC-fed D2/3_HKO mice.
(C and D) Representative pictures of WGA-stained mid-myocardial cross section (top panel), and the resulting quantification (bottom panel; n = 4 per group). Scale bar, 40 μm.
(E and F) Representative pictures of Oil Red O-stained cardiac cross-sections at 12 weeks after HFD or CC and subsequent estimation of lipid deposit area (bottom panel, n = 4 per group). Scale bar, 40 μm.
Data are presented as means ± SEM. Statistical differences after single-factor ANOVA and post hoc analysis were indicated with asterisks: *p < 0.05 versus the control chow or HFD PHD_HWT control group; **0.01 < p < 0.05 versus the corresponding control group; ***p < 0.001 versus the corresponding control group.
Loss of PHD2/3 leads to an accumulation of carnitine-conjugated LCFA in mouse heart
To demonstrate the underlying mechanism by which loss of PHD2/3 activity in cardiomyocytes inhibits LCFA mitochondrial uptake and β-oxidation, we first ascertained whether knockout of PHDs might affect CPT1B expression or activity. Surprisingly, the CPT1B protein level seemed to be transitorily increased in PHD_HKO heart during the first week following tamoxifen injection but stable and equivalent to control level afterward (Figure 4A). In addition, we noticed a transient 2-fold increase in CPT1B cardiac transcript at 1 weeks following tamoxifen injection (Figure 4B). Further, the CPT1B protein level remains also equivalent between WT and KO cardiac lysates at 4 weeks following tamoxifen (Figures 4C and D). While assayed from cardiac mitochondrial fractions, CPT1B activity did not show any significant difference after 4 weeks (Figure 4E). Therefore, it was more likely that steps downstream of CPT1B might be affected in the PHD2/3-knockout hearts. We then performed a LC-MS/MS experiment to assay lipid composition in the heart of PHD2/3_HKO, for both carnitine-conjugated (Figure 4D) and free LCFA (Figure S4A). Interestingly, compared to PHD2/3_HWT samples, PHD2/3-depleted hearts were significantly 3- to 5-folded enriched in carnitine-conjugated LCFA (palmitoyl-carnitine, myristoyl-carnitine, lauryl-carnitine and decanoyl-carnitine) and 6-folded in acetyl-carnitine level. However, both genotypes remain comparable concerning the relative content of free LCFA, carnitine-conjugated short-chain FAs, malonyl-carnitine or carnitine (Figure 4F; Figure S4A). Furthermore, metabolomics has also been performed from mitochondria isolated from the hearts of PHD_HWT and PHD_HKO mice. As shown in Figure S4B, in PHD_HKO mitochondria, we found a significantly decreased abundance of free LCFAs such as palmitoleic acid, implicating that mitochondrial uptake of carnitine-conjugated LCFA might be blocked in PHD2/3 knockout cardiomyocytes. Although we observed a significant increase in carnitine-conjugated long-chain fatty acids in mitochondria, the increases are much smaller than the increases observed in total hearts (Figure S4C; Figure 4F). In addition, carnitine-conjugated medium or short-chain fatty acids are also accumulated in mitochondria (Figure S4C). Since the transport of these short fatty acids is mainly CPT/carnitine independent, the accumulation of carnitine-conjugated fatty acids in mitochondria is thus more likely the result of the interruption of LC-FAO within the mitochondria.
Figure 4. Loss of PHD2/3 transiently increases CPT1B expression, leading to an increase in carnitine-conjugated LFCA in the heart of D2/3_HKO mice.
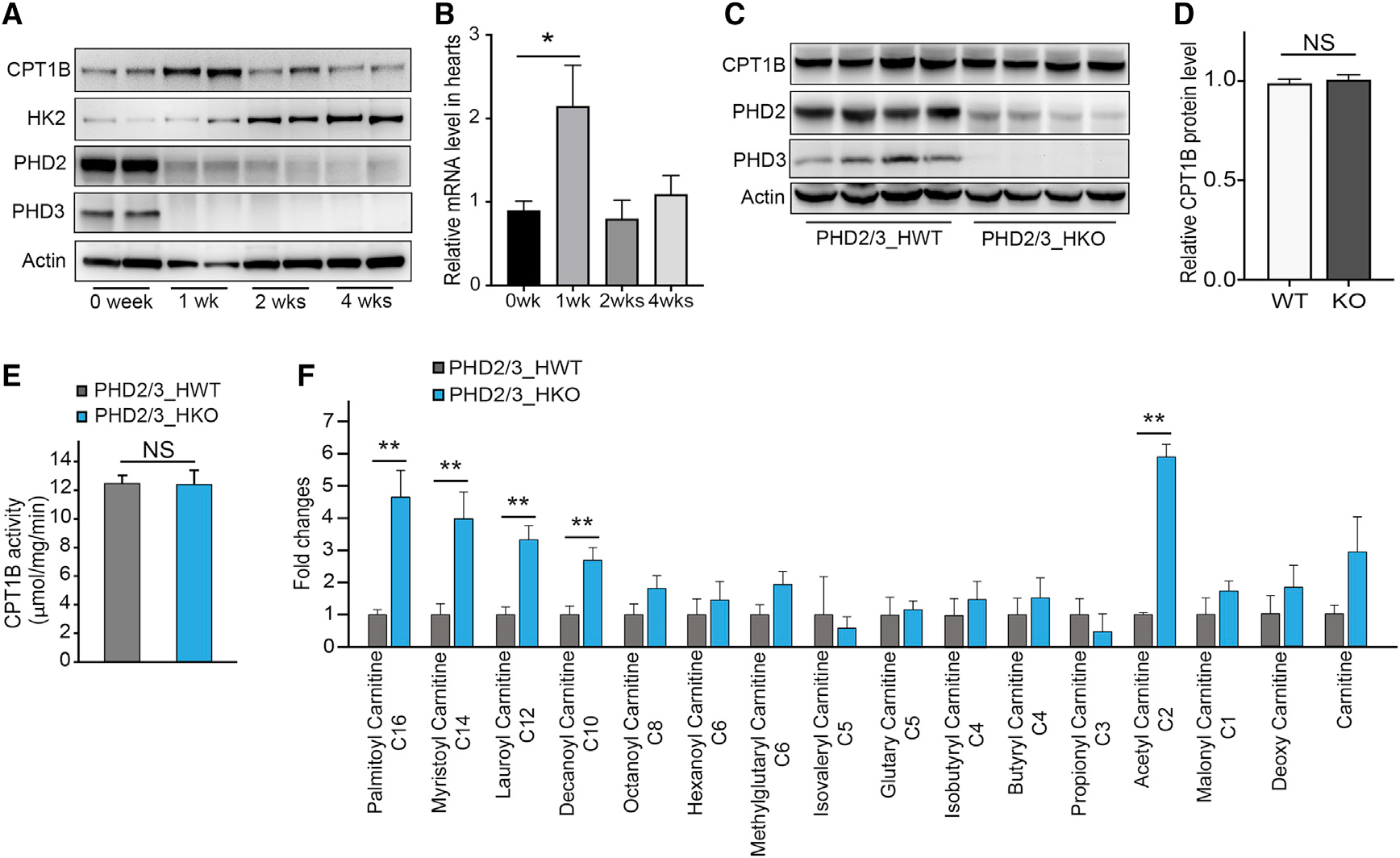
(A) Representative western blot for D2/3_HKO cardiac lysates collected at 0, 1, 2, and 4 weeks after tamoxifen infusion. The CPT1B protein level was transiently increased at 1 week after tamoxifen treatment, whereas the Hexokinase-2 (HK-2) protein level started to gradually increase.
(B) Relative level of CPT1B level of transcript over the time after PHD2/3 disruption (n = 4 per group).
(C) Western blot for PHD2/3_HWT and PHD2/3_HKO cardiac lysates collected after 4 weeks of tamoxifen infusion.
(D) Quantitative analysis of CPT1B protein level was performed and normalized with actin protein level (n = 4 per group).
(E) CPT1B catalytic activity in D2/3_HWT and D2/3_HKO mouse hearts by DTNB colorimetric assay (n = 10 per group), showing no difference regardless the genotype.
(F) Liquid chromatography-mass spectrometry analysis of relative abundance of carnitine-conjugated LCFA in the heart of D2/3_HKO mouse (n = 4 per group), suggesting an accumulation of carnitine-conjugated LCFA and acetyl-carnitine in the heart of PHD2/3_HKO mice.
Data are presented as means ± SEM. Statistically significant difference is shown by asterisks: *p < 0.05, **p < 0.01.
VDAC1 binds to CPT1B and is involved in LCFA β-oxidation in cardiomyocytes
CPT1 is an integral outer mitochondrial membrane protein with its catalytic C-terminal domain exposed on the cytosolic side (Fraser et al., 1997), which converts long-chain acyl-CoA esters to acyl-carnitine esters. Carnitine/acylcarnitine translocase (CACT), an inner mitochondrial membrane protein, will then transport acyl-carnitines into mitochondrial matrix for β-oxidation (Ogawa et al., 2000). However, it remains unclear how acyl-carnitines are transported into mitochondrial intermembrane space. In an attempt to identify CPT1B interacting proteins, we performed a co-immunoprecipitation experiment with lysates from NRVMs infected with FLAG-CPT1B, and the co-precipitated proteins were then separated on SDS-PAGE for LC-MS/MS analysis. Interestingly, VDAC1 was among the potential interaction partner (data not shown). To define CPT1B and VDAC1 interaction, we co-transfected HEK293 cells with plasmids encoding full-length or three different carboxyl-terminal-truncated CPT1Bs with VDAC1-GFP. We found that CPT1B1–616 and CPT1B1–451 were able to pull down VDAC1 similarly to full-length CPT1B, whereas CPT1B1–219 had completely loss its binding capacity to VDAC1 (Figure 5A), suggesting that the CPT1B domain consisting of residues 220–451 is responsible for its binding to VDAC1. This hypothesis was further confirmed by in vitro pull-down assay (Figure 5B). In addition, both full-length VDAC1 and C-terminal-truncated VDAC1 (VDAC1-ΔC) could specifically pull down CPT1B (Figure 5C). More interestingly, CPT1B1–616, which lacks the carboxyl-terminal region hosting essential residues for catalytic activity, was able to compete with full-length CPT1B for binding to VDAC1 and block CPT1B-FL/VDAC1 complex formation (Figure 5D), further validating the specific interaction between CPT1B and VDAC1.
Figure 5. CPT1B binds to VDAC1, which may be involved in LCFA oxidation.
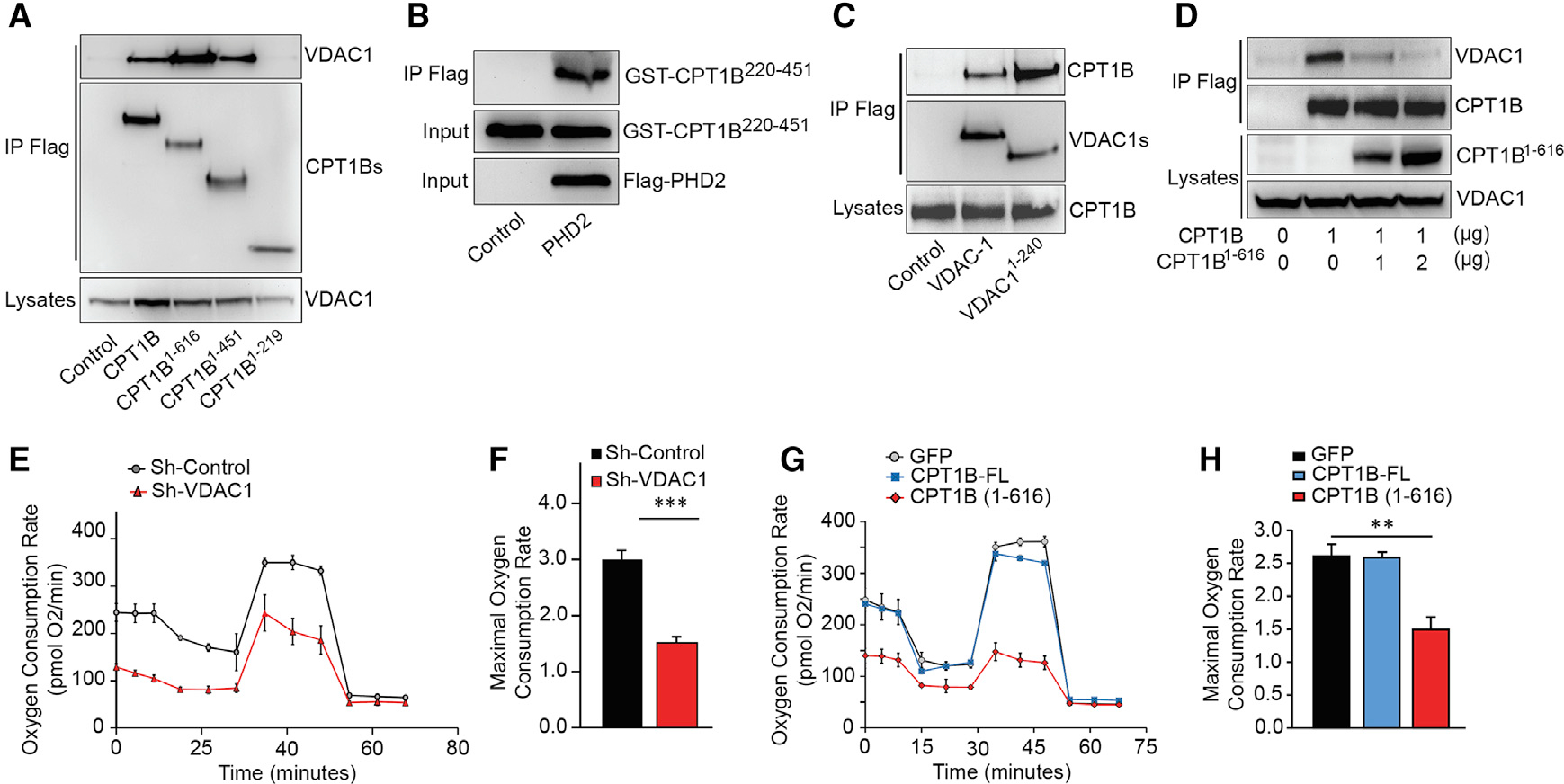
(A) CPT1B/VDAC1 binding domain identification. Full-length and C-terminal-truncated CPT1B were overexpressed with VDAC1 in 293T cells. Immunoblot for VDAC1 after anti-FLAG co-immunoprecipitation demonstrates that both CPT1B1–616 and CPT1B1–451 bind to VDAC1, whereas CPT1B1–219 lost its binding capacity to VDAC1.
(B) Recombinant PHD2 interacts with GST-CPT1B220–451 in vitro.
(C) Both VDAC1 and C-terminal-truncated VDAC1 bind to CPT1B. Immunoblot for CPT1B following co-immunoprecipitation with full-length or C-terminal-truncated (1–240) VDAC1 in 293T cells, which validates CPT1B/VDAC1 complex formation.
(D) Competitive co-immunoprecipitation between CPT1B and C-truncated CPT1B. FLAG-tagged CPT1B was co-overexpressed in NRVMs, aside with a gradient expression level of GFP-tagged CPT1B1–616, which significantly decreases the amount of pull-down VDAC1.
(E and F) VDAC1 knockout induced by Sh-RNA interference leads to a decrease in respiratory capacity (E), leading to a significant decrease in Maximal oxygen consumption rate (F).
(G) Oxidative respiration was challenged in isolated NRVMs overexpressing CPT1B, CPT1B1–616, or GFP (control), showing a significant decrease of OCR in CPT1B1–616-overexpressing NRVMs.
(H) Maximal OCR rate was significantly decreased in response to CPT1B1–616 overexpression, whereas full-length CPT1B overexpression did not lead to any significant change. Representative pictures from 4 different experiments were shown per each result, as an average ± SEM. Statistically significant difference is represented by asterisks: **p < 0.01, ***p < 0.001.
VDACs form a beta barrel spanning the mitochondria outer membrane and have been recognized as crucial channels for ions or metabolite intermediates (Hiller et al., 2008; Shoshan-Barmatz and Ben-Hail, 2012; Shoshan-Barmatz et al., 2006). It is intriguing to examine whether VDAC1 plays a role in LCFA metabolism in cardiomyocytes. First, we transfected NRVMs with a specific oligo-thiophosphate nucleotide, which is considered as a natural blocker of VDACs channels (Bohr et al., 2017; Stein and Colombini, 2008; Tan, 2012). In NRVMs, this biochemical blocking strategy led to a net decay of OCR (Figures S5A and S5B). In addition, we conducted the more specific depletion of VDAC1 by shRNA interference, and we also found that NRVMs oxidative capacity was dramatically decreased at both basal and maximal rate, compared to shRNA-control cells (Figures 5E and 5F). When we overexpressed CPT1B1–616 in NRVMs, OCR was significantly decreased according to SeaHorse Flux analysis (Figures 5G and 5H), suggesting CPT1B1–616 that might act as a negative regulator of LCFA β-oxidation. Taken together, these results demonstrated that VDAC1 plays a crucial role in LCFA β-oxidation, strongly suggesting that CPT1B and VDAC1 binding may facilitate long-chain acyl-carnitine mitochondria translocation in cardiomyocytes.
PHD-mediated CPT1B-P295 hydroxylation regulates its specific interaction with VDAC1 and LCFA β-oxidation in cardiomyocytes
Since loss of PHD2/3 activity had no effects on CPT1B protein level and activity, we hypothesized that PHD2/3 might impact on CPT1B/VDAC1 complex formation. By performing co-immunoprecipitation experiment, we demonstrated that interaction between CPT1B and overexpressed or endogenous VDAC1 was dramatically inhibited in cardiomyocytes in the presence of DMOG (Figures 6A and 6B). Hence, we wondered whether PHD2/3 might regulate CPT1B/VDAC1 binding via catalyzing CPT1B prolyl hydroxylation. To verify this hypothesis, we first performed an in vitro hydroxylation assay for recombinant GST-CPT1B220–451, which interacts with PHD2. Liquid chromatography-tandem mass spectrometry (LC-MS/MS) analysis identified that CPT1B-P295 was hydroxylated by recombinant PHD2 in vitro (Figure 6C). Furthermore, we provided data suggesting that P295 was hydroxylated in NRVMs (Figure S6). We then generated plasmid encoding for missense mutant CPT1B-P295A or CPT1B-P295R, and then immunoprecipitation was performed afterward. Although the interaction remained equivalent to wild-type CPT1B, the CPT1B-P295A mutant depicted a similar binding efficiency to VDAC1 even after DMOG treatment (Figure 6D). In contrast, the CPT1B-P295R mutant showed a decreased binding with VDAC1 (Figure S7A).
Figure 6. CPT1B binds to VDAC1 in an oxygen- and PHD-dependent manner, which can be involved in LCFA metabolism in primary cardiomyocytes.
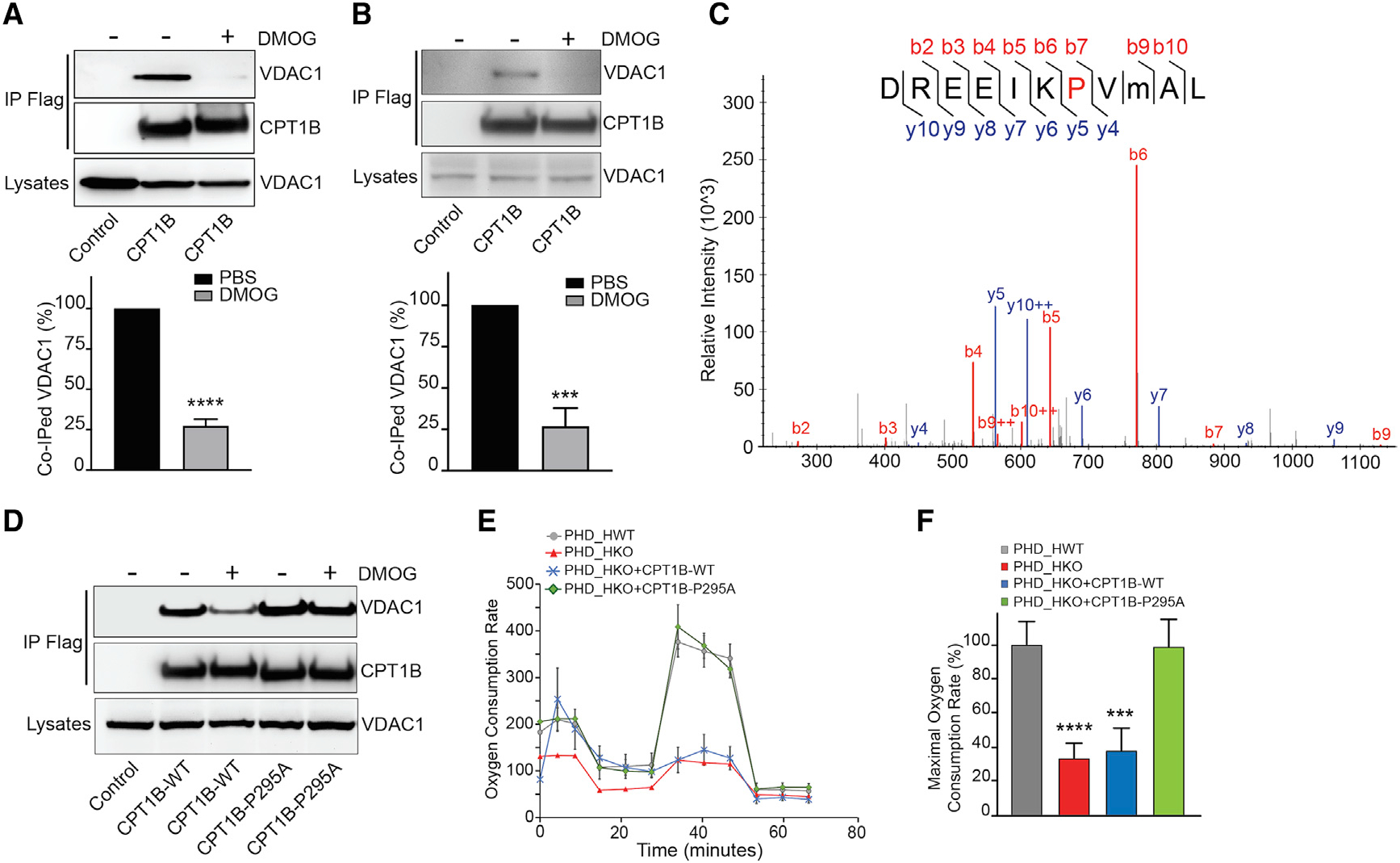
(A and B) Representative co-immunoprecipitation of CPT1B and overexpressed VDAC1-GFP in 293T cells (A) or endogenous VDAC1 in NRVMs (B), and resulted quantification (bottom panel, n = 4).
(C) LC-MS/MS spectrum for prolyl-hydroxylation of GST-CPT1B220–451 catalyzed by recombinant PHD2 in vitro. P295 residue was identified as the hydroxylation site in CPT1B.
(D) Co-immunoprecipitation of VDAC1-GFP with FLAG-CPT1B-WT or CPT1B-P295A from PBS- or DMOG-pre-treated 293T cells, showing a better preservation of CPT1B-295A binding capacity to VDAC1 in DMOG pre-treated 293T cells.
(E) OCR measurement by Sea Horse flux analyzer in D2/3_HKO NMCMs following PHD2/3 depletion and CPT1B-WT or CPT1B-P295 overexpression.
(F) Estimated maximal oxygen consumption rate.
Diagrams are shown as an average of 4 experiments ± SEM. Statistically significant difference is indicated by asterisks: *p < 0.05, ***p < 0.001, ****p < 0.0001.
Since CPT1B-P295A prevented CPT1B/VDAC1 dissociation induced by PHD inhibition, we decided to verify whether or not this specific mutation should be able to preserve FAO in PHD2/3-depleted cardiomyocytes. First, we had ascertained that CPT1B-WT and CPT1B-295A constructs were both efficiently expressed and fully functional in the same range. To do so, we generated a transgenic mouse model CPT1Bflox/flox; Myo-CreER+/− (CPT1B_HKO) that ensures the tamoxifen-inducible cardiomyocyte-specific disruption of CPT1B gene. NMVMs were isolated from neonatal pups of this mouse line. After being cultured with 4OH-Tamoxifen for 4 consecutive days, as we could expect, the loss of CPT1B protein led to a clear decrease of OCR for in CPT1B_HKO NMVMs after 4OH-tamoxifen treatment and overexpression of either CPT1B-WT or CPT1B-P295A in these CPT1B-deficient NMVMs successfully restored the impaired OCR (Figures S7B and S7C).
Finally, we performed the same lentiviral-driven rescue by using 4OH-tamoxifen pre-treated PHD2/3_HKO NMVMs instead. As shown previously, FAO decreased dramatically in 4OH-tamoxifen-pre-cultured D2/3_HKO NMVMs. Interestingly, although depicting a higher basal respiration rate, CPT1B-WT did not provide significant improvement of FAO, hence leading to a comparable diagram from 4OH-tamoxifen-treated PHD2/3_HKO NMVMs (Figures 6E and 6F). By contrast, CPT1B-P295A overexpression was able to upkeep FAO and respiratory parameters at the same range as the control PHD2/3_HWT NMVMs (Figures 6E and 6F). More interestingly, CPT1B-295A could also restore BODIPY 500/510 C12 mitochondria uptake in NRVMs in the presence of DMOG (Figure S7D). Taken together, these data suggest that PHD2/3-dependent P295-hydroxylation takes actively part in cardiomyocyte FAO capacity by promoting the transitory CPT1B/VDAC1 complex formation.
DISCUSSION
For decades, the contribution of cardiac metabolism switch to the development of cardiac diseases has been put to the test and extensively explored. In this paper, we described a regulatory axis that may create a link between cardiac metabolism and oxygen supply in cardiomyocytes. We found that PHD2/3 catalyzes the hydroxylation of the CPT1B-P295 residue, which facilitates LCFA mitochondrial uptake by promoting CPT1B/VDAC1 complex formation (Figure 7). The loss of PHDs activity dissociates the CPT1B/VDAC1 complex, leading to a dramatic decrease in LCFA β-oxidation and improvement in glucose metabolism; however, it foreruns and exacerbates cardiac dysfunction in mice fed with HFD.
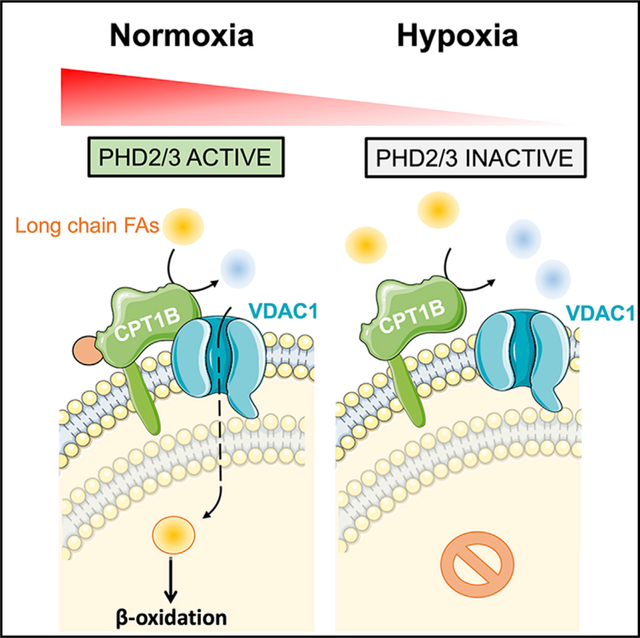
Figure 7. Working model: PHD2/3, CPT1B, and VDAC1 may work together in cardiomyocytes under normoxic condition.
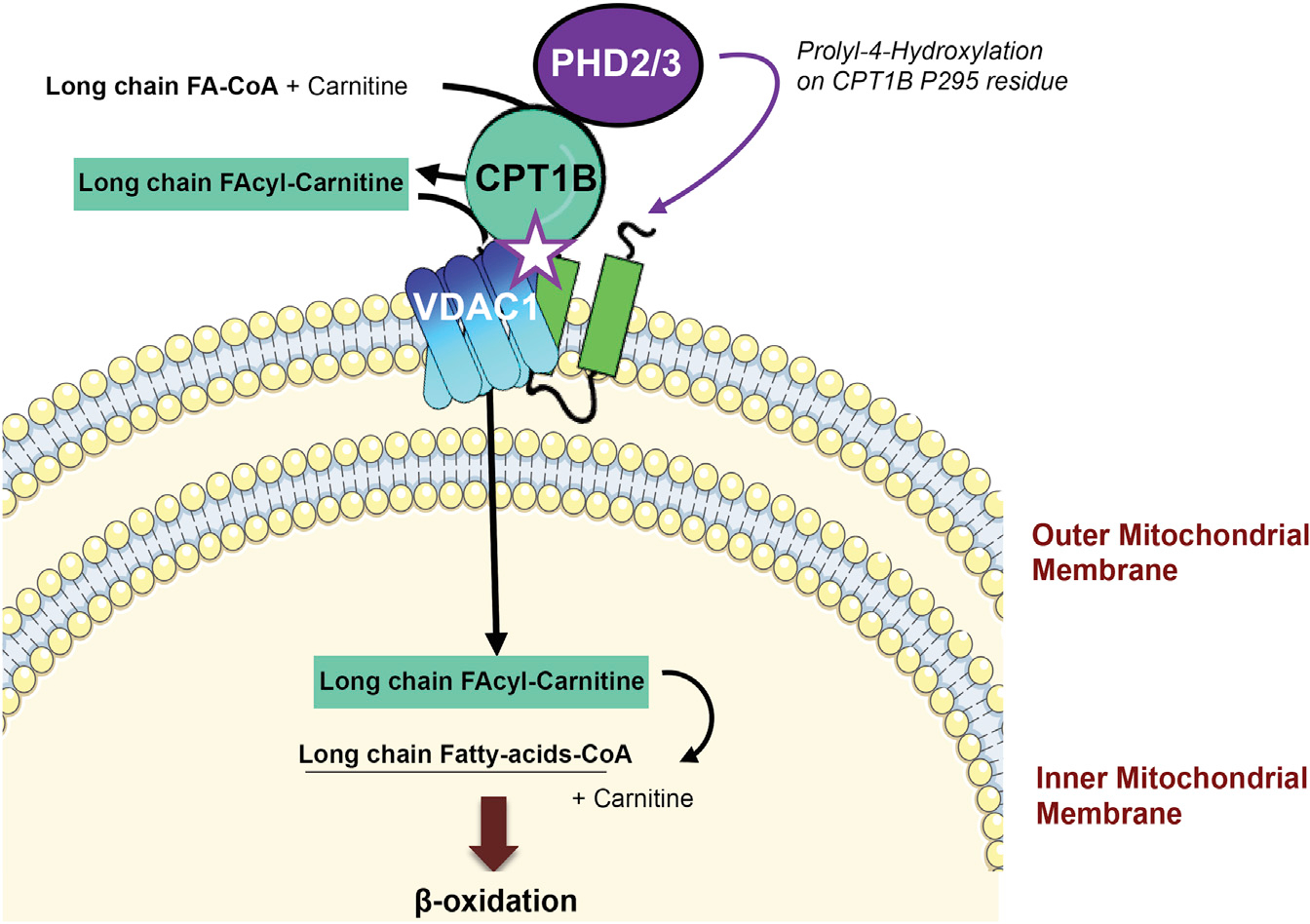
PHD2/3 catalyze prolyl-4-hydroxylation on CPT1B P295 residue, which promotes CPT1B/VDAC1 complex formation and facilitates LFCA mitochondrial uptake. Hypoxia inhibits PHD2/3 enzymatic activity, resulting in the disruption of CPT1B/VDAC1 complex and inhibition of LCFA mitochondrial uptake and metabolism.
Cardiac metabolism switch capacity is tightly based on oxygen availability. In fact, adult cardiomyocytes are high oxygen consumers, showing a net preference for LCFA as a fuel source that is the highest energetic in terms of ATP (Angelini et al., 2017; Brookes and Taegtmeyer, 2017; Pascual and Coleman, 2016). But, conversely, a high sensitivity to hypoxia is the price to pay, and therefore hypoxic condition (such as ischemia and myocardial infarction) used to lead to irreversible and extremely rapid loss of cardiomyocytes (Abozguia et al., 2009). By contrast, fetal immature cardiomyocytes possess a hypoxia resistance and a more glycolytic metabolism, which are both lost during the perinatal period, as the oxygen level rises up in the blood (Patterson and Zhang, 2010; Piquereau and Ventura-Clapier, 2018; Walejko et al., 2018). Therefore, the cardiac metabolism switch is a double-edged sword as it is beneficial during perinatal period, but it seems to be detrimental in failing cardiomyocytes. The clear understanding of this oxygen-dependent metabolic balance is foreseen as a key point of cardiac pathophysiological processes at the scope of therapeutics, yet still challenging to assess (Brookes and Taegtmeyer, 2017; Karlstaedt et al., 2018; Taegtmeyer, 2002).
Recent papers pointed out the importance of hypoxia-related genes, notably evocating a panel of hypoxia-specific MiRNAs or some hypoxia-response transcriptomic changes to explain this specific metabolism switch (Brahimi-Horn et al., 2007; Brahimi-Horn and Pouysségur, 2007b; Guan et al., 2019; Hadj-Moussa et al., 2018). Yet, although this approach was indeed helpful to establish a link between metabolic and hypoxia response, the cell response to such a drastic stress cannot be only a matter of post-transcriptional regulation. Here, we found that the loss of CPT1B prolyl-4-hydroxylation (thus leading to VDAC1/CPT1B complex dissociation) is one of the very first events following the loss of PHDs expression or activity, even forerunning alteration of the cardiac gene program, which also resumed FAO in days.
Mitochondrial integrity is another promising therapeutic candidate to prevent the heart from harmful effects of hypoxia, as well as to avoid a disastrous metabolism switch-back (Bayeva et al., 2013; Gustafsson and Gottlieb, 2008; Leung et al., 2014; Shen et al., 2018). In fact, mitochondria represent the bioenergetic core of the cells, supplying the TCA cycle with both glycolytic and FAO byproducts (Lemieux and Hoppel, 2009; Piquereau et al., 2013). In addition, the mitochondrial respiratory chain complex is not only the ATP generator of the cell but also the main site of oxygen consumption. In cardiomyocytes, the mitochondrial network represents 30% of the total cell volume, and it is highly concentrated between the sarcomere units, so at the closest position to the ATP consumption sites, suggesting a critical role in cell function (Piquereau et al., 2013). In addition, drastic changes affecting mitochondria morphology, recycling, and network maintenance are commonly described in striated muscle diseases, including cardiomyopathy and HF (Ferrari, 1996; Hoppel et al., 2009; Lesnefsky et al., 2001). Oxidative stress also contributes to irreversibly affect mitochondrial double-membrane integrity and matrix enzymes catalytic activity (Ferrari, 1996; Giordano, 2005; Kuroda et al., 2010; Lemasters, 2005; Lesnefsky et al., 2001). Recent publications notably demonstrated the crucial role of some mitochondrial complexes, such as ATP transporter or NOX4 in cardiac substrate preference and oxidative stress adaptation (Nabeebaccus et al., 2017; Springett et al., 2017), which tends to reinforce the involvement of mitochondrial integrity and/or mitochondrial membrane proteins complex formation in cardiac metabolism switch. In accordance with this concept, we demonstrated the importance of CPT1B/VDAC1 complex in LCFA metabolism in cardiomyocytes. We also found that inducing the expression of a less-hypoxia-sensitive form of CPT1B preserves its binding to VDAC1, and so did LCFA β-oxidation in PHD2/3-defecient cardiomyocytes. However, the consequences of such an overexpression remain unclear in vivo.
In mitochondria isolated from PHD_HWT and PHD_HKO mouse hearts, we performed lipid metabolomics, and we took as evidence an increase in many carnitine-conjugated long-, medium-, and short-chain fatty acids (C3–C16) (Figure S4). Similarly, in a mouse model of myocardial ischemia/reperfusion it was previously demonstrated that oxygen scarcity provokes a rapid compensatory increase in CPT1B level (as we showed 1 week after PHD2/3 disruption in Figures 4A and 4B) and a decreased protein level of CPT2, leading to the storage of carnitine conjugated long-chain fatty Acyls in the mitochondrial intermembrane space (Liepinsh et al., 2016).
The Km of the reaction leading to CPT1B prolyl-hydroxylation in vivo is currently unknown, but, given our experience using a proteomics approach, we can assume this rate to be extremely low. Of note, we also found that CPT1B activity is unchanged in the PHD_HKO mouse heart (Figure 4E). Therefore, the biochemical conditions may remain favorable to prolyl-hydroxylated CPT1B/VDAC1 complex formation, preserving LCFA conversion and shuttling during the first days following PHD2/3 disruption. This transient accumulation may also contribute to the generation of a positive gradient (outside > matrix) facilitating the entrance that will lately enhance the accumulation of carnitine-LCFA, while mitochondria swell in response to long-term hypoxia. This hypothesis is also in accordance with the transient higher rate of glucosamine metabolism found in PHD2/3_HKO NMVMs, which usually intends to upkeep the LC-FAO. In addition, not only do carnitine-conjugated long-chain fatty acyls accumulate but also medium- and short-chain fatty acyls. Since the transport of this shorter FA is mainly CPT/carnitine independent, this accumulation is thus more likely the result of the interruption of LC-FAO within the mitochondria. In addition, we can notice a lower level of LC-FA in mitochondrial fractions isolated from the heart of PHD_HKO mice, which can also be linked with our results (both in vivo and in vitro) showing lipid-droplet formation outside mitochondria in NRVMs in the absence of PHD2/3 activity or oxygen.
In conclusion, although at first counterintuitive with our primary hypothesis, the metabolomics performed on mitochondrial fractions from PHD_HKO mouse hearts is in fact in accordance with the literature regarding the short-term adaptation of metabolism to oxygen scarcity (short-term increase CPT1B) and long-term maladaptive damage-causing effects. It also demonstrates the critical role of CPT1B/VDAC1 complex while suggesting the importance of neighboring partners such as CPT2. To go further, it would be interesting to investigate whether the CPT1B/VDAC1 complex works alongside other mitochondrial complexes as integrity keepers, or even as a part of a bigger protein hetero-complex at the OMM.
For the last few decades, due to its top yet rate-limited position in the LCFA catabolic process, CPT1B has been thought of as a “tailoring” metabolic target in cardiac diseases (Arumugam et al., 2016; Chong et al., 2016; Kerr et al., 2017). However, some contradictory results trended to keep the real underlying functioning of CPT1B toughly debated. In fact, pharmacological inhibition of CPT1B is associated with some cardioprotective effects in both pre-clinical and clinical trials, hence reinforcing its therapeutics interest (Beadle and Frenneaux, 2015; Beadle et al., 2015; Kennedy and Horowitz, 1998; Kennedy et al., 1996; Singh et al., 2014). In a similar fashion, such CPT1 inhibition was able to replenish OXPHOS in fibroblasts deficient for tri-functional protein, a complex involved in downstream steps of LCFA catabolism (Lefort et al., 2017). In mice harboring AAV-driven cardiac-specific knockout, triggering the loss of CPT1B in this model prevented the adverse effects of HFD-induced type II diabetes (Zhang et al., 2016). In our case, PHD2/3 loss improved glucose metabolism, while HFD exacerbated and accelerated the deleterious cardiac outcome in PHD2/3 cardiomyocyte-specific knockout mice, notably due to higher range of lipid droplets accumulation within cardiac cells. Consequently, there should be two timelines involving CPT1B activity and the downstream fate of LCFA β-oxidation, when cardiac mitochondria cannot ensure a complete lipid catabolism in response to oxygen scarcity: a short-term inhibition of LCFA uptake must be beneficial, whereas a long-term blockage favors LCFA cytotoxic accumulation, compromising cardiomyocyte function. And, in fact, the lack of CPT1B in both skeletal and cardiac muscles led to a severe cardiomyopathy in mouse, due to lipotoxicity (Haynie et al., 2014). Therefore, targeting CPT1B activity should be done with caution, due to adverse side effects that are difficult to manage. Our work has shed some light on CPT1B regulation at the very first step of LCFA β-oxidation. It notably demonstrates the possibility to target its prolyl-4-hydroxylation-dependent binding to VDAC1 in order to limit the LCFA β-oxidation. Hence, prolyl-hydroxylation dependence of CPT1B/VDAC1 complex formation works like a safeguard for preserving cardiomyocyte integrity and metabolism.
The next step shall be to determine the main “mirror” effectors that may favor and reinforce the switch to glycolysis, following the dissociation of CPT1B and VDAC1 within hypoxic/failing cardiomyocytes. A recent dataset from the literature trends to strongly suggest a specific role of VDAC1 in the resistance to hypoxia and cell-metabolism switch, notably in cancer cells (Mazure, 2016). In rat brain, VDAC1 expression level is positively correlated to acetyl-carnitine level, which is an important bystander of mitochondrial OXPHOS but also commonly described as neuroprotective (Traina et al., 2006). Yet, the metabolic role of VDAC1 has been canonically linked to its binding to Hexokinase-2, a key enzyme of glycolysis (Krasnov et al., 2013). Conversely, here we described that VDAC1 can be involved in LCFA β-oxidation through its binding to CPT1B. Notably, an increase in the Hexokinase 2 protein level was found in the heart lysate at 2 weeks following the loss of PHD2/3, while CPT1B proteins remain back to normal values. Consequently, it may make sense to foresee VDAC1 as a metabolic gauge and metabolite gatekeeper in cardiomyocytes, by its ability to crosstalk between CPT1B and Hexokinase 2. A recent publication has described VDAC1 de-phosphorylation as a contribution to the cardio-protective effects of resveratrol in ischemia/reperfusion (Tian et al., 2019). Consequently, it is possible to emphasize that VDAC1/CPT1B complex formation/dissociation may be involved in the pathogenesis of HF and/or in the cardioprotective virtues of some pharmacological strategies that remain unclear for now. Post-translational modifications (including prolyl-4-hydroxylation events) impacting the VDAC1 protein may be also worth investigating during hypoxia response.
Our current study does not fully exclude a potential impact of CPT1B prolyl-hydroxylation independently from VDAC1. However, since the loss of CPT1B prolyl-hydroxylation does not impair the enzyme activity, while the CPT1B/VDAC complex dissociation is contemporary to the impairment of the LC-FAO, if not VDAC1 dependent, the loss of this CPT1B prolyl-hydroxylation would thus more likely impact a protein complex formation at the outer mitochondrial membrane. Although, as noted in this paper, the CPT1B/VDAC1 binding dynamics is particularly strong, we cannot exclude the potential interplaying of indirect or secondary other partners that remain unknown for now. In this case, VDAC1 binding would just act as an OOM anchoring/docking protein. Noteworthy, very recent data from the literature (Haloi et al., 2021) suggest that VDAC1/Hexokinase-II binding promotes HK-II anchorage to the OOM, and more interestingly that this binary complex formation decreases VDAC1 conductance. In other words, favoring glycolysis decreases VDAC1 opening range, which might be in accordance with our working model suggesting VDAC1 as a part of LCFA mitochondrial uptake.
Finally, here we describe a regulatory axis in cardiomyocytes involving the oxygen sensors PHD2/3, the key enzyme of LCFA mitochondrial uptake CPT1B and the main OMM gatekeeper VDAC1. This oxygen-sensitive regulatory axis may play a crucial role in the cardiac metabolism switch and can thus be foreseen as a potential therapeutic target that may be actionable in the future.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for reagents may be directed to the corresponding author Liang Xie (Baylor College of Medicine, liangx@bcm.edu).
Materials availability
All unique reagents generated in this study are available from the Lead Contact, but we may require a payment and/or a completed Materials Transfer Agreement if there is potential for commercial application.
Data and code availability
Data reported in this paper will be shared by the Lead Contact upon request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the Lead Contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mouse models
All mice were housed in microisolator cages under pathogen-free conditions and subjected to 12-hour light/dark cycles, with food and water ad libitum. All studies were performed according to the National Institutes of Health Guide for the Care and Use of Laboratory Animals and approved by the Baylor College of Medicine Institutional Animal Care and Use Committee. Since we observed similar result in both female and male mice, both age-matched genders were used in this study.
Both PHD2/3flox/flox, CAG-CreER ± (PHD2/3_KO and PHD2/3_WT) and PHD2/3flox/flox, Myh6-CreER ± mice (PHD2/3_HKO and PHD2/3_HWT) were C57BL/6 background. Genotype of these mice were determined by PCR, according to the protocol provided by The Jackson Laboratory. Tamoxifen (Sigma, Cat# T5648) was dissolved in sterile corn oil (Sigma, Cat# C8267) at 10 mg/mL and stored at −80°C prior to use. In order to induce PHD2/3 disruption, 6~8 weeks old-mice were I.P. injected with tamoxifen (20 mg/kg/day) for four consecutive days, regardless their genotypes. After tamoxifen injection time shift, the mice (both PHD2/3_HWT and PHD2/3_HKO) were divided in two groups, and fed else with high fat diet (60% calories from fat, Research Diet #D12492) or with normal chow (provided by the mouse care facility). Body weight and 4 hours-fasting blood glucose level were monitored each week until the end point of the experiment (8 or 12 weeks after tamoxifen infusion, as needed). When samples must be collected, isoflurane (2%) inhalation anesthesia was first provided to the mice and euthanasia was then ensured by cervical dislocation on unconscious animals.
Cell lines and primary cells
HEK293 cells were grown in DMEM supplemented with 10% FBS and antibiotics (100 U/ml penicillin, 68.6 mol/L streptomycin). Primary neonatal rat ventricular cardiomyocytes (NRVMs) were isolated from 1–2 days old Sprague-Dawley rat pups (Charles Rivers) by using the Worthington Neonatal Cardiomyocyte Isolation System kit and following the manufacturer instructions (# LK003300, Worthington Biochemical Corporation). NRVMs were maintained in MEM (GIBCO by Life Technologics) supplemented with 10% Horse Serum (GIBCO by Life Technologics), 5% Fetal Bovine Serum (FBS, GIBCO by Life Technologics) and antibiotics (100 U/mL penicillin, 68.6 mmol/mL streptomycin). Two days after plating, NRVMs were transduced with lentiviral or adenoviral particles containing the constructs as described. When required, cells were treated with 1mM Dimethyloxalylglycine (DMOG) (Milipore Sigma CAS 89464–63-1) for 6 hours, or with 200 μM Etomoxir (Sigma, # E1905). Oligothiophosphorate 18-randomer control or targeting VDACs were manually designed and produced by TriLinks Biotechnologies. They were used directly on NRVMs at 1 μM, combined with Lipofectamine 2000 (1:10 ratio). Primary neonatal mouse cardiomyocytes (NMCMs) were isolated from P1-P3 PHD2/3_HWT and PHD2/3_HKO, following the same kit and instructions that used for NRVMs. Two days after plating, 4-OH Tamoxifen (100nM) was added to complete culture medium for 4 consecutive days, in order to trigger PHD2/3 knock-down.
METHODS DETAILS
Immunoblotting, immunoprecipitation
Primary neonatal rat ventricular cardiomyocytes (NRVMs) were isolated from 1–2 day old Sprague-Dawley rat pups (Charles Rivers) by using the Worthington Neonatal Cardiomyocyte Isolation System kit and following the manufacturer instructions (# LK003300, Worthington Biochemical Corporation). NRVMs were maintained in MEM (GIBCO by Life Technologics) supplemented with 10% Horse Serum (GIBCO by Life Technologics), 5% Fetal Bovine Serum (FBS, GIBCO by Life Technologics) and antibiotics (100 U/mL penicillin, 68.6 mmol/mL streptomycin). Two days after plating, NRVMs were transduced with lentiviral or adenoviral particles containing the constructs mentioned previously as needed. When required, cells were treated with 1mM Dimethyloxalylglycine (DMOG) (Milipore Sigma CAS 89464–63-1) for 6 hours, or with 200 μM Etomoxir (Sigma, # E1905). Oligothiophosphorate 18-randomer control or targeting VDACs were manually designed and produced by TriLinks Biotechnologies (sequence available upon request). They were used directly on NRVMs at 1 μM, combined with Lipofectamine 2000 (1:10 ratio). Primary neonatal mouse cardiomyocytes (NMCMs) were isolated from P1-P3 PHD2/3_HWT and PHD2/3_HKO, following the same kit and instructions that used for NRVMs. Two days after plating, 4-OH Tamoxifen (100nM) was added to complete culture medium for 4 consecutive days, in order to trigger PHD2/3 knock-down.
For immunoblotting and immunoprecipitation, cells were washed twice with cold PBS and harvested in cold lysis buffer (1% Triton X-100, 50 mM Tris, pH 7.4, 150 mM NaCl, 1mM EGTA, 1mM EDTA, protease and phosphatase inhibitors) on ice. Cell lysates were then clarified by centrifugation at 14,000 g for 10 minutes. Equal amounts of protein were used for western blotting. Immunoblotting was performed with antibodies directed against CPT1B (Abcam), PHD2 (Cell Signaling), PHD3 (Novus Biologicals), VDAC1 (Abcam), HK2 (Cell signaling), β-actin (Santa-Cruz Biotechnologies), anti-Flag-HRP (Sigma-Aldrich) and GAPDH (Santa-Cruz Biotechnologies). They were all diluted in 5% blocking bovine serum albumin (Bio-Rad) in TBS-T at the manufacturers recommended concentration. Immunoprecipitations and co-immunoprecipitations were carried out by adding anti-Flag resin (Sigma-Aldrich) to cell lysates, then the reactions were incubated with rotation overnight at 4°C. Beads were then washed 5 times with lysis buffer. Elution was performed by using 3XFlag peptide, and final proteins were analyzed by Western-blot Assay.
Lentiviral construct generation
Lentiviral vector encoding Flag-tagged CPT1B was purchased from Origene. Site-directed mutagenesis was achieved using Q5 mutagenesis kit (New England Biolabs), by using the respective primers. Then, resulting lentiviral plasmids encoding else full-length Flag-CPT1B-WT, shorter Flag-CPT1B (CPT1B1–616, CPT1B1–350, CPT1B1–219), or Flag-CPT1B-P295A, or Flag-CPT1B-P295R, or plasmid encoding FLAG-VDAC1 or carboxy-terminal truncated VDAC1 (VDAC1-ΔC) were co-transfected as needed with lentiviral packaging plasmids psPAX2 (#12260, Addgene) and pMD2.G (#12259, Addgene) in HEK293T cells to ensure lentiviral particles production.
Measurement of oxidative phosphorylation (OXPHOS) and changes of fuel source capacity in isolated cardiomyocytes
A Seahorse Bioscience XF96 instrument (Seahorse Bioscience, North Billerica, MA) was used to measure oxygen consumption rate (OCR) or extracellular acidification rate (ECAR) from isolated NRVM or NMCM. Each well was seeded at a density of 25,000 cells per each. Cells were infected with lentiviral particles or treated with drugs as needed. The measurement was performed according to Mitostress (OCR), and following the manufacturer’s recommendation. Both parameters were then standardized with the cell number and protein concentration (that was determined at the end of each experiment). For changes of fuel source capacity, measurements were performed by XFp SeaHorse assay using the Mito Fuel Flex Test kit, after 4 days following the treatment of PHD_HWT or PHD_HKO neonate mouse cardiomyocytes with 4-OH tamoxifen.
Echocardiography
Transthoracic echocardiography was performed on mice anesthetized with 2% isoflurane inhalation by using a high-resolution system, equipped with a high-frequency ultrasound probe (RMV-707B), designed for small animals (Vevo 770, Visual Sonics, Canada). Parasternal long-axis and short-axis views were acquired. LV dimensions and wall thicknesses were determined from parasternal short axis M-mode images. The heart rate (HR) of the anesthetized animal was recorded. Ejection fraction, fraction shortening, and left ventricular mass were calculated by Vevo770 software simultaneously. Data represent averaged values of 12 cardiac cycles.
Glucose tolerance and insulin tolerance test
Overnight fasted mice were I.P injected with Glucose (2 g/Kg of body weight) diluted in sterile saline solution. Blood glucose level was monitored at time 0, 15, 30, 60 and 120 minutes after injection by using a glucometer (Freestyle Lite, Abbott Diabetes Care). Insulin sensitivity was tested on 4 hours-fasted mice that were I.P injected with insulin diluted in sterile saline solution (0.75U/kg of body weight). Blood glucose level was monitored following the same time course. Local analgesic cream was applied to the tail before blood collection and StyptStix (Henry Schein) was used on the cut at the end of the experiment.
Hyperinsulinemic euglycemic clamp in conscious mice
The studies were performed in unrestrained mice using the insulin clamp technique (using constant insulin doses) in combination with [3H] glucose and [14C] 2-deoxyglucose. In summary, mice were cannulated as described previously and allowed to recover for 4 to 7 days before the clamp. After an overnight fasting, mice received a primed dose of [3H] glucose (10 μCi) and then a constant rate intravenous infusion (0.1 μCi/min) of [3H] glucose using a syringe infusion pump for 90 minutes. Blood samples were collected for the determination of basal glucose production. After 90 minutes, mice were infused with insulin for 2 hours (4 milliunits/kg/min). Simultaneously, 25% glucose was infused at an adjusted rate to maintain the blood glucose level at 100–140 mg/dL. Blood glucose concentration was determined every 10 minutes by a glucometer. At the end of a 120-minute period, blood was collected for the measurements of hepatic glucose production and peripheral glucose disposal rates. For tissue specific uptake, we inject 2-deoxy-D-[1,−14C] glucose (10 μCi) into bolus during hyperinsulinemic-euglycemic clamp at 45 min before the end of the clamps and collect blood sample at 5, 10, 15, 25, 35 and 45 minutes. At the end of the clamp, mouse tissues were harvested for the evaluation of glucose uptake.
Histology and staining
Samples collection was performed following terminal euthanasia of the mice. For paraffin slides preparation, mice were perfused from transversal aorta with PBS and then with 4% formalin (vol/vol) in PBS. Tissues (Heart, liver, skeletal muscles) were left in 4% formalin bath overnight at 4°C and dehydration was performed with growing concentration of sucrose (15% then 30%) within the next days. Paraffin-embedding and sectioning was done at the Pathology and Histology Core Facility at Baylor College of Medicine. Final tissue sections were between 5–8 μm thick.
For frozen slides preparation, the heart was cut in half at the mid-part of myocardium, and the upper part was immediately embedded in Tissue-Tek Cryomold (Sakura, Electron Microscopy Sciences) containing Optimal Cutting Temperature compound (Fisher Scientific). The cryomold was then frozen at the surface of a methyl-butane bath (#M0167, TCI America) previously cooled at −80°C by using liquid nitrogen. Frozen sections were prepared on a Leica cryostat at an average thickness of 5–7 μm for routine histological staining (Eosin Hematoxylin). For lipid staining, Oil Red O staining was performed on 10–15 μm-thick tissue sections (heart and liver).
Mitochondria isolation and respiration
Mouse heart mitochondria isolation protocol was designed following the online protocol provided by Oroboros Instruments with modifications. Briefly, mice were euthanized as previously described. Half of the heart was minced in ice-cold mitochondrial isolation buffer (MIB, Mannitol 225 mM, Sucrose 70mM, HEPES 5mM, EGTA 1mM, and FA-free BSA 2.5 mg/mL, pH 7.2) and homogenized by using a 10mL glass dounce. Tissue homogenate was centrifuged at 800 g for 10 minutes at 4°C, and the collected supernatant was re-centrifuged at 10,000 g for 10 minutes at 4°C. Mitochondrial pellet was re-suspended in BSA-free MIB containing 10mM KH2PO4 and 5mM MgCl2 (pH 7.2). For respiration assessment, crude mitochondrial extract was directly seeded on XFP 96 plates containing MIB buffer by 10,000 g centrifugation for 10 minutes at +4°C.
CPT1 enzymatic activity assay
For enzymatic assessment, total CPT (CPT I and CPT II) activity was measured from 50 μg of crude mitochondrial suspension, and assayed in 250 μL reaction buffer (20mM HEPES, 1mM EGTA, 220mM sucrose, 40mM KCl, 0.1mM 5,5′-dithio-bis (2-nitrobenzoic acid) (DTNB), 1.3 mg/ml free-FA BSA, and 40 μM palmitoyl-CoA, pH 7.4) in 96-well microplate (Corning). Densitometry is read once at 412 nm by using a Tecan Ultraspec 3000 spectrophotometer. Then the reaction was started with the addition of L-carnitine (Sigma) at 1 mM simultaneously in each well. Optical density at 420 nm was monitored in 4 minutes immediately following the addition of L-Carnitine. In order to distinguish CPT1B to CPT1A activity, the experiments were performed in parallel with simultaneous addition of 1 mM L-Carnitine and 6 uM MalonylCoA. For in vitro CPT1 activity estimation, cell lysate was directly diluted at a 1:4 ratio in reaction buffer.
In vitro hydroxylation assay
Approximately 1 μg recombinant GST-CPT1B220–451 was incubated with 0.25 μg recombinant PHD2 in a reaction buffer containing 10 μM FeSO4, 100 μM 2-oxo-glutarate, 1 mM ascorbate, 2 mg BSA, and 50 μM Tris-HCl buffer, adjusted to pH 7.8. The enzyme reaction was carried out at 37°C for 30 minutes.
LC-MS/MS analysis
Flag-CPT1B immunoprecipitated from NRVMs or in vitro hydroxylated GST-CPT1B220–451 was separated with SDS-PAGE and visualized with Coomassie Brilliant blue-stain. The target proteins were excised from the gel followed by enzyme digestion using Trypsin (GenDepot T9600) and Chymotrypsin (Promega V1061) separately. The peptides were resuspended in 10 μL of loading solution (5% methanol containing 0.1% formic acid) and subjected to nanoflow LC-MS/MS analysis with a nano-LC 1200 system (Thermo Fisher Scientific, San Jose, CA) coupled to Orbitrap Fusion Lumos ETD mass spectrometer (Thermo Fisher Scientific, San Jose, CA). The peptides were loaded on a two-column setup using a pre-column trap of 2cm × 100μm size (Reprosil-Pur Basic C18 1.9 μm, Dr.Maisch GmbH, Germany) and a 5cm × 150μm analytical column (Reprosil-Pur Basic C18 1.9 μm, Dr.Maisch GmbH, Germany). The peptides were eluted using a 75min discontinuous gradient of 5%–30% acetonitrile/0.1% formic acid at a flow rate of 750nl/min. The eluted peptides were directly electro-sprayed into mass spectrometer operated in the data-dependent acquisition (DDA) mode along with Parallel Reaction monitoring (PRM) mode. For DDA mode, the full MS scan was acquired in Orbitrap in the range of 350–1500 m/z at 120,000 resolution followed by MS2 in Ion Trap (CID 30% collision energy) with 15sec dynamic exclusion time. For PRM mode, the target precursor ions for P295 containing peptides were isolated in quadrupole with isolation width 1.6 m/z for entire scan duration. The MS2 was carried out in Orbitrap (60,000 resolution, AGC 1×105, max injection time 100ms) using CID fragmentation.
Database search and data validation
Obtained MS/MS spectra were searched against target-decoy Rattus Norvegicus NCBI refseq database (updated 2016_0126) in Proteome Discoverer 1.4 interface (Thermo Fisher) with Mascot algorithm (Mascot 2.4, Matrix Science). The precursor mass tolerance was confined within 20 ppm with fragment mass tolerance of 0.5 dalton and a maximum of two missed cleavage allowed. Dynamic modification of Oxidation on Methionine and Proline and protein N-terminal Acetylation was allowed. The peptides identified from mascot result file were validated with 5% false discover rate (FDR) and subject to manual verifications for correct assignment.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical information for each experiment, including the total n number analyzed and the specific tests performed, is reported in the figure legends. In general, data are shown as mean ± SEM for at least 4 independent experiments. Differences between two groups were analyzed with two-tailed Student’s t test. When experiments involved more than two groups, ANOVA single factor was conducted, followed by a post hoc analysis. All data with p < 0.05 were considered statistically significant and reported in the figure legends.
Supplementary Material
KEY RESOURCES TABLE.
| Reagent or resource | Source | Identifier |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| Anti-Cpt1b | Proteintech | Cat#22170-1-AP; RRID:AB_2713959 |
| Anti-PHD2 | Cell signaling | Cat#4835; RRID:AB_10561316 |
| Anti-Hexokinase II | Cell signaling | Cat#2867; RRID:AB_2232946 |
| Anti-Vdac1 | Abcam | Cat#Ab14734; RRID:AB_443084 |
| Anti-β-actin | Santa-Cruz Biotechnologies | Cat#sc-47778; RRID:AB_626632 |
| Anti-PHD3 | Novus Biologicals | Cat#NB100-303; RRID:AB_10003302 |
| Anti-Flag | Sigma-Aldrich | Cat#2426; AB_2616449 |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| Corn oil | Millipore Sigma | Cat#C8267 |
| Tamoxifen | Millipore Sigma | Cat#T5648 |
| High Fat diet 60% calories from fat | Research diet | Cat#D12492 |
| Dimethyloxalylglycine (DMOG) | Millipore Sigma | Cat#CAS 89464-63-1 |
| Etomoxir | Millipore Sigma | Cat#E1905 |
| L-Cartinine | Millipore Sigma | Cat#11242008001 |
| Bovine Serum Albumin Fraction V, heat shock, fatty acid free (FA-free BSA) | Millipore Sigma | Cat#3117057001 |
| Oligomycin | Millipore Sigma | Cat#495455-10MG |
| Carbonyl cyanide 4-(trifluoromethoxy) phenylhydrazone (FCCP) | Millipore Sigma | Cat#C2920-10MG |
| Rotenone | Millipore Sigma | Cat#557368-1GM |
| Antimycin A | Millipore Sigma | Cat#A8674-25MG |
| Sodium Palmitate | Millipore Sigma | Cat#P9767-5G |
| Palmitoyl-CoA lithium salt | Millipore Sigma | Cat#P9716-5MG |
| Malonyl coenzyme A lithium salt | Millipore Sigma | Cat#M4263-5MG |
| Lipofectamine 2000 | Thermo Scientific | Cat#11668019 |
|
| ||
| Critical commercial assays | ||
|
| ||
| Worthington Neonatal Biochemical Isolation System kit |
Worthington Biochemical Corporation |
Cat#LK003300 |
| RNA purification kit | QIAGEN | Cat#74104 |
| MitoFuel Flex kit | Agilent | Cat#103270-100 |
| iScript™ cDNA synthesis kit | Bio-Rad Laboratories | Cat#1708891 |
| iTaq SYBR Green supermix | Bio-Rad Laboratories | Cat#1725121 |
| TaqMan Universal PCR Master Mix | Thermo Scientific | Cat#4304437 |
| Seahorse XF HS Mini Analyzer|Seahorse XFp Analyzer - XFp 96well PDL miniplate |
Agilent | Cat#103722-100 |
|
| ||
| Experimental models: Cell lines | ||
|
| ||
| 293T cells | ATCC | Cat#CRL-3216 |
| Primary neonatal rat ventricular cardiomyocytes | This paper | N/A |
| Primary neonatal mouse ventricular cardiomyocytes | This paper | N/A |
|
| ||
| Experimental Models: Organisms/Strains | ||
|
| ||
| Sprague Dawley Rat | Charles River | Cat#24100570 |
| B6.FVB (129)-Tg (Myh6-cre/Esr1)1Jmk/J | Jackson Laboratory | Cat#005657 |
|
| ||
| Oligonucleotides | ||
|
| ||
| Oligothiophosphorate 18-randomer control | TriLinks Biotechnologies | Cat#O-30040-50 |
| Oligothiophosphorate targeting VDACs | TriLinks Biotechnologies | Cat#O-30030-50 |
|
| ||
| Recombinant DNA | ||
|
| ||
| Mouse Vdac1 | Origene | Cat#MR203842 |
| Mouse Cpt1b | Origene | Cat#MR210564 |
| psPAX2 | A gift from Didier Trono at EPFL | Addgene plasmid Cat#12260 |
| pMD2.G | A gift from Didier Trono at EPFL | Addgene plasmid Cat#12259 |
|
| ||
| Software and algorithms | ||
|
| ||
| ImageJ | NIH | https://imagej.nih.gov/ij/ |
| Vevo770 | FUJIFILM VisualSonics | https://www.visualsonics.com/product/imaging-systems/vevo-770 |
| Illustrator | Adobe Suite | https://www.adobe.com/fr/products/illustrator.html |
| Prism9 | Graphpad | https://www.graphpad.com/ |
| Excel | Microsoft office | https://www.microsoft.com/enus/microsoft-365/excel?rtc=1 |
|
| ||
| Other | ||
|
| ||
| Oxygen Control Glove Box | Coy Lab Products | Cat#8375065 |
| StyptStix Wood Applicator Sticks 6” 100/Vl | Henry Schein Inc. | Cat#112-6994 |
Highlights.
Loss of PHD2/3 decreases LCFA uptake and β-oxidation in cardiomyocytes
PHD2/3 bind and prolyl-hydroxylate CPT1B
CPT1B binds to VDAC1, which regulates LCFA metabolism
Hypoxia-insensible CPT1B rescues LCFA metabolism in PHD2/3-deficient cardiomyocytes
ACKNOWLEDGMENTS
This work is supported by NIH grants R01 HL122736 (L.X.), R01 HL061656 and HL112890 (X.P.), MMPC, R01DK114356 and UM1HG006348 (P.K.S.), and CPRIT Core Facility Award RP170005 and P30 Cancer Center support Grant NCI-CA125123 (S.Y.J.). We thank Cecilia Ljungberg (RNA In Situ Hybridization Core at the Jan and Dan Duncan Neurological Research Institute) for providing access to the cryostat used for the preparation of the cardiac frozen sections.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2021.109767.
REFERENCES
- Abdurrachim D, Luiken JJ, Nicolay K, Glatz JF, Prompers JJ, and Nabben M (2015). Good and bad consequences of altered fatty acid metabolism in heart failure: evidence from mouse models. Cardiovasc. Res. 106, 194–205. [DOI] [PubMed] [Google Scholar]
- Abozguia K, Shivu GN, Ahmed I, Phan TT, and Frenneaux MP (2009). The heart metabolism: pathophysiological aspects in ischaemia and heart failure. Curr. Pharm. Des. 15, 827–835. [DOI] [PubMed] [Google Scholar]
- Angelini A, Pi X, and Xie L (2017). Dioxygen and Metabolism; Dangerous Liaisons in Cardiac Function and Disease. Front. Physiol. 8, 1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arumugam S, Sreedhar R, Thandavarayan RA, Karuppagounder V, and Watanabe K (2016). Targeting fatty acid metabolism in heart failure: is it a suitable therapeutic approach? Drug Discov. Today 21, 1003–1008. [DOI] [PubMed] [Google Scholar]
- Ashrafian H, and Frenneaux MP (2007). Metabolic modulation in heart failure: the coming of age. Cardiovasc. Drugs Ther. 21, 5–7. [DOI] [PubMed] [Google Scholar]
- Ashrafian H, Frenneaux MP, and Opie LH (2007). Metabolic mechanisms in heart failure. Circulation 116, 434–448. [DOI] [PubMed] [Google Scholar]
- Awan MM, and Saggerson ED (1993). Malonyl-CoA metabolism in cardiac myocytes and its relevance to the control of fatty acid oxidation. Biochem. J. 295, 61–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayeva M, Gheorghiade M, and Ardehali H (2013). Mitochondria as a therapeutic target in heart failure. J. Am. Coll. Cardiol. 61, 599–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beadle RM, and Frenneaux MP (2015). Reply: Perhexiline, Cardiac Energetics, and Heart Failure: Lessons From the First Law of Thermodynamics. JACC Heart Fail. 3, 660. [DOI] [PubMed] [Google Scholar]
- Beadle RM, Williams LK, Kuehl M, Bowater S, Abozguia K, Leyva F, Yousef Z, Wagenmakers AJ, Thies F, Horowitz J, and Frenneaux MP (2015). Improvement in cardiac energetics by perhexiline in heart failure due to dilated cardiomyopathy. JACC Heart Fail. 3, 202–211. [DOI] [PubMed] [Google Scholar]
- Beraud N, Pelloux S, Usson Y, Kuznetsov AV, Ronot X, Tourneur Y, and Saks V (2009). Mitochondrial dynamics in heart cells: very low amplitude high frequency fluctuations in adult cardiomyocytes and flow motion in non beating Hl-1 cells. J. Bioenerg. Biomembr. 41, 195–214. [DOI] [PubMed] [Google Scholar]
- Bohr HG, Shim I, Stein C, Ørum H, Hansen HF, and Koch T (2017). Electronic Structures of LNA Phosphorothioate Oligonucleotides. Mol. Ther. Nucleic Acids 8, 428–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brahimi-Horn MC, and Pouysségur J (2007a). Harnessing the hypoxia-inducible factor in cancer and ischemic disease. Biochem. Pharmacol. 73, 450–457. [DOI] [PubMed] [Google Scholar]
- Brahimi-Horn MC, and Pouysségur J (2007b). Hypoxia in cancer cell metabolism and pH regulation. Essays Biochem. 43, 165–178. [DOI] [PubMed] [Google Scholar]
- Brahimi-Horn MC, Chiche J, and Pouysségur J (2007). Hypoxia signalling controls metabolic demand. Curr. Opin. Cell Biol. 19, 223–229. [DOI] [PubMed] [Google Scholar]
- Brookes PS, and Taegtmeyer H (2017). Metabolism: A Direct Link Between Cardiac Structure and Function. Circulation 136, 2158–2161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho SW, Park JS, Heo HJ, Park SW, Song S, Kim I, Han YM, Yamashita JK, Youm JB, Han J, and Koh GY (2014). Dual modulation of the mitochondrial permeability transition pore and redox signaling synergistically promotes cardiomyocyte differentiation from pluripotent stem cells. J. Am. Heart Assoc. 3, e000693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong CR, Sallustio B, and Horowitz JD (2016). Drugs that Affect Cardiac Metabolism: Focus on Perhexiline. Cardiovasc. Drugs Ther. 30, 399–405. [DOI] [PubMed] [Google Scholar]
- Declercq PE, Falck JR, Kuwajima M, Tyminski H, Foster DW, and McGarry JD (1987). Characterization of the mitochondrial carnitine palmitoyltransferase enzyme system. I. Use of inhibitors. J. Biol. Chem. 262, 9812–9821. [PubMed] [Google Scholar]
- Doenst T, Pytel G, Schrepper A, Amorim P, Färber G, Shingu Y, Mohr FW, and Schwarzer M (2010). Decreased rates of substrate oxidation ex vivo predict the onset of heart failure and contractile dysfunction in rats with pressure overload. Cardiovasc. Res. 86, 461–470. [DOI] [PubMed] [Google Scholar]
- Dyck JR, and Lopaschuk GD (2002). Malonyl CoA control of fatty acid oxidation in the ischemic heart. J. Mol. Cell. Cardiol. 34, 1099–1109. [DOI] [PubMed] [Google Scholar]
- Ferrari R (1996). The role of mitochondria in ischemic heart disease. J. Cardiovasc. Pharmacol. 28 (Suppl 1), S1–S10. [DOI] [PubMed] [Google Scholar]
- Fraisl P, Aragonés J, and Carmeliet P (2009). Inhibition of oxygen sensors as a therapeutic strategy for ischaemic and inflammatory disease. Nat. Rev. Drug Discov. 8, 139–152. [DOI] [PubMed] [Google Scholar]
- Fraser F, Corstorphine CG, and Zammit VA (1997). Topology of carnitine palmitoyltransferase I in the mitochondrial outer membrane. Biochem. J. 323, 711–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeman RS, Hasbani DM, Lipscomb EA, Straub JA, and Xie L (2003). SM-20, EGL-9, and the EGLN family of hypoxia-inducible factor prolyl hydroxylases. Mol. Cells 16, 1–12. [PubMed] [Google Scholar]
- Giordano FJ (2005). Oxygen, oxidative stress, hypoxia, and heart failure. J. Clin. Invest. 115, 500–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg IJ, Trent CM, and Schulze PC (2012). Lipid metabolism and toxicity in the heart. Cell Metab. 15, 805–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan Y, Song X, Sun W, Wang Y, and Liu B (2019). Effect of Hypoxia-Induced MicroRNA-210 Expression on Cardiovascular Disease and the Underlying Mechanism. Oxid. Med. Cell. Longev. 2019, 4727283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gustafsson AB, and Gottlieb RA (2008). Heart mitochondria: gates of life and death. Cardiovasc. Res. 77, 334–343. [DOI] [PubMed] [Google Scholar]
- Hada T, Kato Y, Obana E, Yamamoto A, Yamazaki N, Hashimoto M, Yamamoto T, and Shinohara Y (2012). Comparison of two expression systems using COS7 cells and yeast cells for expression of heart/muscle-type carnitine palmitoyltransferase 1. Protein Expr. Purif. 82, 192–196. [DOI] [PubMed] [Google Scholar]
- Hadj-Moussa H, Logan SM, Seibel BA, and Storey KB (2018). Potential role for microRNA in regulating hypoxia-induced metabolic suppression in jumbo squids. Biochim. Biophys. Acta. Gene Regul. Mech. 1861, 586–593. [DOI] [PubMed] [Google Scholar]
- Haloi N, Wen PC, Cheng Q, Yang M, Natarajan G, Camara AKS, Kwok WM, and Tajkhorshid E (2021). Structural basis of complex formation between mitochondrial anion channel VDAC1 and Hexokinase-II. Commun. Biol. 4, 667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haynie KR, Vandanmagsar B, Wicks SE, Zhang J, and Mynatt RL (2014). Inhibition of carnitine palymitoyltransferase1b induces cardiac hypertrophy and mortality in mice. Diabetes Obes. Metab. 16, 757–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He L, Kim T, Long Q, Liu J, Wang P, Zhou Y, Ding Y, Prasain J, Wood PA, and Yang Q (2012). Carnitine palmitoyltransferase-1b deficiency aggravates pressure overload-induced cardiac hypertrophy caused by lipotoxicity. Circulation 126, 1705–1716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiller S, Garces RG, Malia TJ, Orekhov VY, Colombini M, and Wagner G (2008). Solution structure of the integral human membrane protein VDAC-1 in detergent micelles. Science 321, 1206–1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hölscher M, Silter M, Krull S, von Ahlen M, Hesse A, Schwartz P, Wielockx B, Breier G, Katschinski DM, and Zieseniss A (2011). Cardiomyocyte-specific prolyl-4-hydroxylase domain 2 knock out protects from acute myocardial ischemic injury. J. Biol. Chem. 286, 11185–11194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoppel CL, Tandler B, Fujioka H, and Riva A (2009). Dynamic organization of mitochondria in human heart and in myocardial disease. Int. J. Biochem. Cell Biol. 41, 1949–1956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaasik A, Safiulina D, Zharkovsky A, and Veksler V (2007). Regulation of mitochondrial matrix volume. Am. J. Physiol. Cell Physiol. 292, C157–C163. [DOI] [PubMed] [Google Scholar]
- Karlstaedt A, Schiffer W, and Taegtmeyer H (2018). Actionable Metabolic Pathways in Heart Failure and Cancer-Lessons From Cancer Cell Metabolism. Front. Cardiovasc. Med. 5, 71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy JA, and Horowitz JD (1998). Effect of trimetazidine on carnitine palmitoyltransferase-1 in the rat heart. Cardiovasc. Drugs Ther. 12, 359–363. [DOI] [PubMed] [Google Scholar]
- Kennedy JA, Unger SA, and Horowitz JD (1996). Inhibition of carnitine palmitoyltransferase-1 in rat heart and liver by perhexiline and amiodarone. Biochem. Pharmacol. 52, 273–280. [DOI] [PubMed] [Google Scholar]
- Kerr M, Dodd MS, and Heather LC (2017). The ‘Goldilocks zone’ of fatty acid metabolism; to ensure that the relationship with cardiac function is just right. Clin. Sci. (Lond.) 131, 2079–2094. [DOI] [PubMed] [Google Scholar]
- Kolwicz SC Jr., Olson DP, Marney LC, Garcia-Menendez L, Synovec RE, and Tian R (2012). Cardiac-specific deletion of acetyl CoA carboxylase 2 prevents metabolic remodeling during pressure-overload hypertrophy. Circ. Res. 111, 728–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krasnov GS, Dmitriev AA, Lakunina VA, Kirpiy AA, and Kudryavtseva AV (2013). Targeting VDAC-bound hexokinase II: a promising approach for concomitant anti-cancer therapy. Expert Opin. Ther. Targets 17, 1221–1233. [DOI] [PubMed] [Google Scholar]
- Kuroda J, Ago T, Matsushima S, Zhai P, Schneider MD, and Sadoshima J (2010). NADPH oxidase 4 (Nox4) is a major source of oxidative stress in the failing heart. Proc. Natl. Acad. Sci. USA 107, 15565–15570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laczy B, Fuülöp N, Onay-Besikci A, Des Rosiers C, and Chatham JC (2011). Acute regulation of cardiac metabolism by the hexosamine biosynthesis pathway and protein O-GlcNAcylation. PLoS ONE 6, e18417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefort B, Gouache E, Acquaviva C, Tardieu M, Benoist JF, Dumas JF, Servais S, Chevalier S, Vianey-Saban C, and Labarthe F (2017). Pharmacological inhibition of carnitine palmitoyltransferase 1 restores mitochondrial oxidative phosphorylation in human trifunctional protein deficient fibroblasts. Biochim. Biophys. Acta Mol. Basis Dis. 1863, 1292–1299. [DOI] [PubMed] [Google Scholar]
- Lemasters JJ (2005). Selective mitochondrial autophagy, or mitophagy, as a targeted defense against oxidative stress, mitochondrial dysfunction, and aging. Rejuvenation Res. 8, 3–5. [DOI] [PubMed] [Google Scholar]
- Lemasters JJ, and Holmuhamedov E (2006). Voltage-dependent anion channel (VDAC) as mitochondrial governator–thinking outside the box. Biochim. Biophys. Acta 1762, 181–190. [DOI] [PubMed] [Google Scholar]
- Lemieux H, and Hoppel CL (2009). Mitochondria in the human heart. J. Bioenerg. Biomembr. 41, 99–106. [DOI] [PubMed] [Google Scholar]
- Lesnefsky EJ, Moghaddas S, Tandler B, Kerner J, and Hoppel CL (2001). Mitochondrial dysfunction in cardiac disease: ischemia–reperfusion, aging, and heart failure. J. Mol. Cell. Cardiol. 33, 1065–1089. [DOI] [PubMed] [Google Scholar]
- Leung CH, Wang L, Nielsen JM, Tropak MB, Fu YY, Kato H, Callahan J, Redington AN, and Caldarone CA (2014). Remote cardioprotection by transfer of coronary effluent from ischemic preconditioned rabbit heart preserves mitochondrial integrity and function via adenosine receptor activation. Cardiovasc. Drugs Ther. 28, 7–17. [DOI] [PubMed] [Google Scholar]
- Liepinsh E, Makrecka-Kuka M, Volska K, Kuka J, Makarova E, Antone U, Sevostjanovs E, Vilskersts R, Strods A, Tars K, and Dambrova M (2016). Long-chain acylcarnitines determine ischaemia/reperfusion-induced damage in heart mitochondria. Biochem. J. 473, 1191–1202. [DOI] [PubMed] [Google Scholar]
- Lopaschuk GD, Ussher JR, Folmes CD, Jaswal JS, and Stanley WC (2010). Myocardial fatty acid metabolism in health and disease. Physiol. Rev. 90, 207–258. [DOI] [PubMed] [Google Scholar]
- Martin-Puig S, Tello D, and Aragonés J (2015). Novel perspectives on the PHD-HIF oxygen sensing pathway in cardioprotection mediated by IPC and RIPC. Front. Physiol. 6, 137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marxsen JH, Stengel P, Doege K, Heikkinen P, Jokilehto T, Wagner T, Jelkmann W, Jaakkola P, and Metzen E (2004). Hypoxia-inducible factor-1 (HIF-1) promotes its degradation by induction of HIF-alpha-prolyl-4-hydroxylases. Biochem. J. 381, 761–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazure NM (2016). News about VDAC1 in Hypoxia. Front. Oncol. 6, 193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minamishima YA, Moslehi J, Padera RF, Bronson RT, Liao R, and Kaelin WG Jr. (2009). A feedback loop involving the Phd3 prolyl hydroxylase tunes the mammalian hypoxic response in vivo. Mol. Cell. Biol. 29, 5729–5741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nabeebaccus AA, Zoccarato A, Hafstad AD, Santos CX, Aasum E, Brewer AC, Zhang M, Beretta M, Yin X, West JA, et al. (2017). Nox4 reprograms cardiac substrate metabolism via protein O-GlcNAcylation to enhance stress adaptation. JCI Insight 2, e96184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagoshi T, Yoshimura M, Rosano GM, Lopaschuk GD, and Mochizuki S (2011). Optimization of cardiac metabolism in heart failure. Curr. Pharm. Des. 17, 3846–3853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogawa A, Yamamoto S, Kanazawa M, Takayanagi M, Hasegawa S, and Kohno Y (2000). Identification of two novel mutations of the carnitine/acylcarnitine translocase (CACT) gene in a patient with CACT deficiency. J. Hum. Genet. 45, 52–55. [DOI] [PubMed] [Google Scholar]
- Onay-Besikci A, Campbell FM, Hopkins TA, Dyck JR, and Lopaschuk GD (2003). Relative importance of malonyl CoA and carnitine in maturation of fatty acid oxidation in newborn rabbit heart. Am. J. Physiol. Heart Circ. Physiol. 284, H283–H289. [DOI] [PubMed] [Google Scholar]
- Ong SG, and Hausenloy DJ (2012). Hypoxia-inducible factor as a therapeutic target for cardioprotection. Pharmacol. Ther. 136, 69–81. [DOI] [PubMed] [Google Scholar]
- Pascual F, and Coleman RA (2016). Fuel availability and fate in cardiac metabolism: A tale of two substrates. Biochim. Biophys. Acta 1861, 1425–1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson AJ, and Zhang L (2010). Hypoxia and fetal heart development. Curr. Mol. Med. 10, 653–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedersen PL (2008). Voltage dependent anion channels (VDACs): a brief introduction with a focus on the outer mitochondrial compartment’s roles together with hexokinase-2 in the “Warburg effect” in cancer. J. Bioenerg. Biomembr. 40, 123–126. [DOI] [PubMed] [Google Scholar]
- Piquereau J, and Ventura-Clapier R (2018). Maturation of Cardiac Energy Metabolism During Perinatal Development. Front. Physiol. 9, 959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piquereau J, Caffin F, Novotova M, Lemaire C, Veksler V, Garnier A, Ventura-Clapier R, and Joubert F (2013). Mitochondrial dynamics in the adult cardiomyocytes: which roles for a highly specialized cell? Front. Physiol. 4, 102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rambold AS, Cohen S, and Lippincott-Schwartz J (2015). Fatty acid trafficking in starved cells: regulation by lipid droplet lipolysis, autophagy, and mitochondrial fusion dynamics. Dev. Cell 32, 678–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reszko AE, Kasumov T, David F, Thomas KR, Jobbins KA, Cheng JF, Lopaschuk GD, Dyck JR, Diaz M, Des Rosiers C, et al. (2004). Regulation of malonyl-CoA concentration and turnover in the normal heart. J. Biol. Chem. 279, 34298–34301. [DOI] [PubMed] [Google Scholar]
- Schreiber T, Salhöfer L, Quinting T, and Fandrey J (2019). Things get broken: the hypoxia-inducible factor prolyl hydroxylases in ischemic heart disease. Basic Res. Cardiol. 114, 16. [DOI] [PubMed] [Google Scholar]
- Sepa-Kishi DM, Wu MV, Uthayakumar A, Mohasses A, and Ceddia RB (2016). Antilipolytic and antilipogenic effects of the CPT-1b inhibitor oxfenicine in the white adipose tissue of rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 311, R779–R787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen J, Zhang JH, Xiao H, Wu JM, He KM, Lv ZZ, Li ZJ, Xu M, and Zhang YY (2018). Mitochondria are transported along microtubules in membrane nanotubes to rescue distressed cardiomyocytes from apoptosis. Cell Death Dis. 9, 81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoshan-Barmatz V, and Ben-Hail D (2012). VDAC, a multi-functional mitochondrial protein as a pharmacological target. Mitochondrion 12, 24–34. [DOI] [PubMed] [Google Scholar]
- Shoshan-Barmatz V, Israelson A, Brdiczka D, and Sheu SS (2006). The voltage-dependent anion channel (VDAC): function in intracellular signalling, cell life and cell death. Curr. Pharm. Des. 12, 2249–2270. [DOI] [PubMed] [Google Scholar]
- Singh S, Beadle R, Cameron D, Rudd A, Bruce M, Jagpal B, Schwarz K, Brindley G, Mckiddie F, Lang C, et al. (2014). Randomized double-blind placebo-controlled trial of perhexiline in heart failure with preserved ejection fraction syndrome. Future Cardiol. 10, 693–698. [DOI] [PubMed] [Google Scholar]
- Springett R, King MS, Crichton PG, and Kunji ERS (2017). Modelling the free energy profile of the mitochondrial ADP/ATP carrier. Biochim. Biophys. Acta Bioenerg. 1858, 906–914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein CA, and Colombini M (2008). Specific VDAC inhibitors: phosphorothioate oligonucleotides. J. Bioenerg. Biomembr. 40, 157–162. [DOI] [PubMed] [Google Scholar]
- Taegtmeyer H (2002). Switching metabolic genes to build a better heart. Circulation 106, 2043–2045. [DOI] [PubMed] [Google Scholar]
- Takeda K, Cowan A, and Fong GH (2007). Essential role for prolyl hydroxylase domain protein 2 in oxygen homeostasis of the adult vascular system. Circulation 116, 774–781. [DOI] [PubMed] [Google Scholar]
- Tan W (2012). VDAC blockage by phosphorothioate oligonucleotides and its implication in apoptosis. Biochim. Biophys. Acta 1818, 1555–1561. [DOI] [PubMed] [Google Scholar]
- Tian M, Xie Y, Meng Y, Ma W, Tong Z, Yang X, Lai S, Zhou Y, He M, and Liao Z (2019). Resveratrol protects cardiomyocytes against anoxia/reoxygenation via dephosphorylation of VDAC1 by Akt-GSK3 β pathway. Eur. J. Pharmacol. 843, 80–87. [DOI] [PubMed] [Google Scholar]
- Traina G, Bernardi R, Rizzo M, Calvani M, Durante M, and Brunelli M (2006). Acetyl-L-carnitine up-regulates expression of voltage-dependent anion channel in the rat brain. Neurochem. Int. 48, 673–678. [DOI] [PubMed] [Google Scholar]
- Turer AT, Malloy CR, Newgard CB, and Podgoreanu MV (2010). Energetics and metabolism in the failing heart: important but poorly understood. Curr. Opin. Clin. Nutr. Metab. Care 13, 458–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Leij FR, Cox KB, Jackson VN, Huijkman NC, Bartelds B, Kuipers JR, Dijkhuizen T, Terpstra P, Wood PA, Zammit VA, and Price NT (2002). Structural and functional genomics of the CPT1B gene for muscle-type carnitine palmitoyltransferase I in mammals. J. Biol. Chem. 277, 26994–27005. [DOI] [PubMed] [Google Scholar]
- Walejko JM, Koelmel JP, Garrett TJ, Edison AS, and Keller-Wood M (2018). Multiomics approach reveals metabolic changes in the heart at birth. Am. J. Physiol. Endocrinol. Metab. 315, E1212–E1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie L, Pi X, Townley-Tilson WH, Li N, Wehrens XH, Entman ML, Taffet GE, Mishra A, Peng J, Schisler JC, et al. (2015a). PHD2/3-dependent hydroxylation tunes cardiac response to β-adrenergic stress via phospholamban. J. Clin. Invest. 125, 2759–2771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie L, Pi X, Wang Z, He J, Willis MS, and Patterson C (2015b). Depletion of PHD3 protects heart from ischemia/reperfusion injury by inhibiting cardiomyocyte apoptosis. J. Mol. Cell. Cardiol. 80, 156–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Fang X, Dai M, Cao Q, Tan T, He W, Huang Y, Chu L, and Bao M (2016). Cardiac-specific down-regulation of carnitine palmitoyltransferase-1b (CPT-1b) prevents cardiac remodeling in obese mice. Obesity (Silver Spring) 24, 2533–2543. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data reported in this paper will be shared by the Lead Contact upon request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the Lead Contact upon request.