

Abstract
2-azido-1H-benzo[d]imidazole derivatives 1a,b were reacted with a β-ketoester such as acetylacetone in the presence of sodium ethoxide to obtain the desired molecules 2a,b. The latter acted as a key molecule for the synthesis of new carbazone derivatives 4a,b that were submitted to react with 2-oxo-N-phenyl-2-(phenylamino)acetohydrazonoyl chloride to obtain the target thiadiazole derivatives 6a,b. The structures of the newly synthesized compounds were inferred from correct spectral and microanalytical data. Moreover, the newly prepared compounds were subjected to molecular docking studies with DNA gyrase B and exhibited binding energy that extended from −9.8 to −6.4 kcal/mol, which confirmed their excellent potency. The compounds 6a,b were found to be with the minimum binding energy (−9.7 and −9.8 kcal/mol) as compared to the standard drug ciprofloxacin (−7.4 kcal/mol) against the target enzyme DNA gyrase B. In addition, the newly synthesized compounds were also examined and screened for their in vitro antimicrobial activity against pathogenic microorganisms Staphylococcus aureus, E. coli, Pseudomonas aeruginosa, Aspergillus niger, and Candida albicans. Among the newly synthesized molecules, significant antimicrobial activity against all the tested microorganisms was obtained for the compounds 6a,b. The in silico and in vitro findings showed that compounds 6a,b were the most active against bacterial strains, and could serve as potential antimicrobial agents.
Keywords: benzimidazole; 1,3,4-thiadiazole; 1,2,3-triazoles; antimicrobial activity; docking study
1. Introduction
Nitrogen-containing heterocyclic analogues have received great interest in drug discovery because of their well-known activity in pharmaceutical and medicinal fields [1,2,3,4]. Benzimidazole derivatives represent an important heterocyclic class of active therapeutic agents because of their wide spectrum of biological and pharmaceutical applications including antibacterial, antifungal, antiviral, anticancer, antidiabetic, anticonvulsant, and anti-HIV agents [5,6]. In addition, 1,2,3-triazoles have attracted a great deal of interest from medicinal chemists in the design and development of potential drug candidates due to their high pharmacological properties such as antimicrobial, antitubercular, CNS depressant, and antihypertensive activities [7,8,9,10,11,12]. They have diverse applications in drug development for the treatment of some diseases including cancer, inflammation, malaria, and tuberculosis [13,14,15].
A wide range of antibacterial properties have been gained in hybrid molecules containing benzimidazole and triazole moieties, as shown in Figure 1.
Figure 1.

Hybrid molecules bearing benzimidazole and triazole moieties I–III as antibacterial agents.
On the other hand, DNA gyrase is considered an important bacterial enzyme that is involved in the control of topological transitions of DNA [16,17]. In addition, it is the intercellular target for a number of antibacterial agents as a paradigm for other DNA topoisomerases [18]. Therefore, DNA gyrase B has been selected as a therapeutic target for the identification and development of antimicrobial agents [19,20,21].
In light of the potential therapeutic properties of these heterocyclic compounds and with the contribution of our research work [18,22,23,24,25,26,27,28,29,30,31] to identify a new class of antimicrobial agents, a novel set of hybrid derivatives bearing benzimidazole and triazole moieties was synthesized. Their antimicrobial activities were performed in vitro. Moreover, the molecular docking study [32,33,34] was performed both to support the antibacterial activity and to understand the binding mode of the interactions of the screened compounds against the binding site of the target enzyme DNA gyrase B. Furthermore, the in silico absorption, distribution, metabolic, excretion, and toxicity (ADMET) and drug-likeness of the prepared compounds were also calculated to identify their bioavailability and toxicity.
2. Results and Discussion
2.1. Chemistry
In the present study, a preparation of the starting materials 1-(5-methyl-1-(7-aryl-1H-benzo[d]imidazol-2-yl)-1H-1,2,3-triazol-4-yl) ethan-1-one 2a,b, were obtained by the reaction of benzo[d]imidazoles 1a and/or 1b with β-ketoester acetylacetone in the presence of sodium ethoxide in ethanol under reflux (Scheme 1). Their structures were confirmed using spectral data and elemental analysis. The FT-IR spectrum of 2a and 2b exhibited significant strong absorption bands at v 1715 and 1705 for the carbonyl group, respectively, indicated the formation of the acetyl group. In addition, the 1H-NMR spectrum of 2a and 2b showed characteristic singlet signals at δ 2.10 and 2.42 ppm for compound 2a and at δ 2.16 and 2.47 ppm for compound 2b for the two methyl groups formed in each compound. The 13C-NMR of 2a exhibited two signals at δ 9.52 and 27.42 ppm for the two methyl groups and at 195.9 ppm for the carbonyl group. Additionally, the 13C-NMR of 2b exhibited two signals at δ 9.58 and 27.45 ppm for the two methyl groups and at 194.82 ppm for the carbonyl group. Moreover, the structures were also supported by their mass spectrum, which agree with their molecular formula.
Scheme 1.

Synthesis of acetyl triazole derivatives 2a,b.
The two acetyl triazole derivatives were stirred with methyl hydrazinecarbodithioate 3 in isopropyl alcohol for 2h at RT to afford the desired carbodithioate derivatives 4a,b (Scheme 2). The structures of 4a and 4b were confirmed on the basis of their spectral and microanalytical data. The FT-IR spectra of the two compounds were devoid of any signals for carbonyl groups. The 1H-NMR spectrum of 4a showed three singlet signals at δ 2.16, 2.36, and 2.47 ppm for the three methyl groups and two singlet signals at δ 8.95 and 11.58 ppm for the NH groups. The 1H-NMR spectrum of 4b exhibited three singlet signals at δ 2.18, 2.32, and 2.42 for the methyl groups and two singlet signals at δ 8.98 and 11.47 ppm for the NH groups. Their mass spectra agreed with their molecular formulas.
Scheme 2.

Synthesis of carbodithioate derivatives 4a,b.
Compounds 4a and 4b were treated with 2-oxo-N-phenyl-2-(phenylamino)aceto hydrazonoyl chloride in ethanol containing a catalytic amount of TEA (2–3 drops) to obtain the corresponding 1,3,4-thiadiazole derivatives 6a,b (Scheme 3). Their chemical structures were inferred from their data, while the FT-IR spectrum of compound 6a showed a strong absorption band at 1685 for the carbamide group. Additionally, compound 6b exhibited a strong absorption band at 1692 for the carbamide group. Their 1H-NMR and 13C-NMR spectra showed characteristic signals; the 1H-NMR spectrum of compound 6a, for example, showed significant two signals at δ 2.38 and 2.46 ppm for the two methyl groups in addition to the two singlet signals at δ 9.81 and 11.52 ppm for the two NH groups. Its 13C-NMR spectrum showed two significant signals at 10.1 and 20.5 ppm for the two methyl groups. Their mass spectra supported their molecular formula.
Scheme 3.

Synthesis of thiadiazole derivatives 6a,b.
2.2. Computational Approach
2.2.1. Molecular Docking Study
Based on the bacterial activity results, the compounds (1a–6b) were docked to the target enzyme DNA gyrase subunit b to check their binding profile. The docking results exhibited that the compounds (1a–6b) were fitted well in the active site pockets of the target and possessed good binding energy values ΔG (−6.4 to −9.8 kcal/mol), as summarized in Table 1 and Figure 2. It has been observed that the derivative 1a and 1b formed arene- σ with the target enzyme through the amino acid residue Asn46 at distances of 3.91 and 3.93 Å, respectively. Moreover, 1a docked to the target through one HB with the residue Val71 at 1.87 Å. Compounds 2a and 2b showed the same interactions with the target enzyme as they formed three HB and one arene–cation interactions with the residues Ala47, Thr165, and Arg76. Compound 4a formed two HB interactions with Thr34 at 2.98 and 3.00 Å. On the other hand, derivative 4b exhibited one HB interaction with Asp49 at 2.20 Å. Compound 6a showed one HB with the target through Thr164 at 2.45. Finally, compound 6b, with the highest binding affinity of −9.8 kcal/mol, docked to the target DNA gyrase B through one HB with the residue Asp49 at 2.40 Å. The 2D and 3D representations of the intermolecular interactions of the best docked compounds 6a and 6b with the target enzyme are shown in Figure 2. The interactions of the other compounds and the reference drug are shown in Figure S1 in the Supplementary File Section.
Table 1.
Energy-based interactions and intermolecular bindings between the prepared compounds and the target enzyme.
| 2D Structure | Binding Energy kcal/mol |
Docked Complex (Amino Acid–Ligand) Interactions |
Distance (Å) |
|
|---|---|---|---|---|
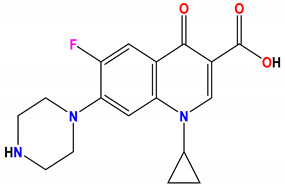
| Ciprofloxacin |
|
−7.4 |
H-bond Arg76:NH1—ciprofloxacin Thr165:OG1—ciprofloxacin Val43:O—ciprofloxacin |
3.00 2.74 2.04 |
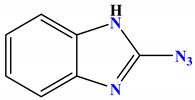
| 1a |
|
−6.4 |
H-bond Val71:O—compound 1a arene-σ Asn46:CB—compound 1a |
1.87 3.91 |
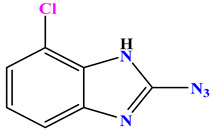
| 1b |
|
−6.5 |
arene-σ Asn46:CB—compound 1b |
3.93 |
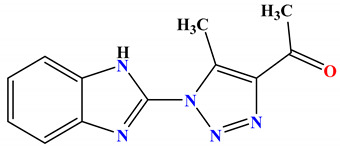
| 2a |
|
−7.9 |
H-bond Ala47:N—compound 2a Thr165:OG1—compound 2a Thr165:OG1—compound 2a arene-cation Arg76:NH1—compound 2a |
2.99 2.98 2.95 4.06 |
| 2b |
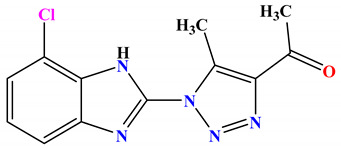
|
−8.1 |
H-bond Ala47:N—compound 2b Thr165:OG1—compound 2b Thr165:OG1—compound 2b arene-cation Arg76:NH1—compound 2b |
2.99 2.95 2.94 4.02 |
| 4a |
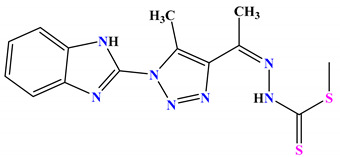
|
−7.0 |
H-bond Thr34:OG1—compound 4a Thr34:OG1—compound 4a |
2.98 3.00 |
| 4b |
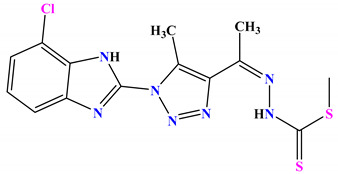
|
−7.1 |
H-bond Asp49:OD2—compound 4b |
2.20 |
| 6a |
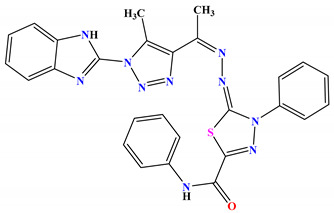
|
−9.7 |
H-bond Thr165:OG1—compound 6a |
2.45 |
| 6b |
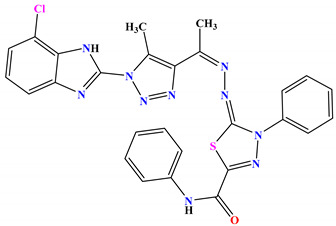
|
−9.8 |
H-bond Asp49:OD1—compound 6b |
2.40 |
Figure 2.

The molecular interactions of the best docked compounds 6a and 6b with the target enzyme DNA gyrase B. Left side (2D): the residues are represented in 3 letter codes. Hydrogen bonds are represented by green and blue lines and pi-interactions are represented by orange lines. Right side (3D): the docked compounds are represented by gray stick models, and the active site pockets are shown by blue stick models. H-bond interactions are shown in green dashed lines. π-interactions are shown in orange lines.
On the other hand, the standard drug ciprofloxacin docked to the target enzyme with binding energy ΔG (−7.4 kcal/mol), and showed three HB interactions with the residues Arg76, Thr165, and Val43 at 3.00, 2.74, and 2.04 Å, respectively (Figure 2).
The results justified that the compounds 6a,b firmly docked to the active site pockets of the target enzyme of DNA gyrase subunit B, which functionally participates in DNA inhibitions.
2.2.2. Predictive ADMAT and Drug-Likeness of the Compounds
With the help of free accessible servers such as admetSAR, mol inspiration, and SwissADME, the pharmacokinetics, physicochemical, and drug-likeness of the newly prepared compounds are summarized in Table 2. ADMET analysis of the screened compounds exhibited that they have good absorption properties (%HIA) ranging from 99.57 to 100%. For distribution, the compounds do not permeate the blood–brain barrier (BBB) except compound 1a. Moreover, the molecules were negative in the AMES toxicity and carcinogenicity test, which suggests that they are non-mutagenic. In addition, the physiochemical properties results exhibited acceptable values as the compounds had M.W (molecular weight) (<500 g/mol), except compounds 6a,b. The partition coefficients of the compounds were <5. The TPSA (topological surface areas) were found to be in the favorable range (<140). Further, the HBA (H-bond acceptors) and HBD (donors) in the compounds comply with Lipinski′s rule of five and were found to be in the acceptable range. Overall, we can conclude that they showed good absorption, distribution, and oral bioavailability within the body by not violating Lipinski′s rule of five more than once.
Table 2.
ADMET and drug-likeness prediction of newly prepared molecules.
| Ciprofloxacin | Compound 1a | Compound 1b | Compound 2a | Compound 2b | Compound 4a | Compound 4b | Compound 6a | Compound 6b | |
|---|---|---|---|---|---|---|---|---|---|
| Molecular Weight (g/mol) | 331.13 | 159.15 | 193.60 | 241.25 | 275.70 | 345.46 | 379.90 | 534.61 | 569.05 |
| BBB permeant | No | No | Yes | No | Yes | No | No | No | No |
| %Human Intestinal Absorption (HIA+) | 97.95 | 99.51 | 99.57 | 100.00 | 100.00 | 99.20 | 99.32 | 100.00 | 100.00 |
| logp | −0.7 | 2.80 | 3.43 | 1.72 | 2.35 | 2.80 | 3.43 | 3.91 | 4.38 |
| TPSA A2 | 74.57 | 78.44 | 78.44 | 76.47 | 76.47 | 83.79 | 83.79 | 131.05 | 131.05 |
| HBA | 6 | 5 | 5 | 6 | 6 | 7 | 7 | 11 | 2 |
| HBD | 2 | 1 | 1 | 1 | 1 | 2 | 2 | 2 | 6 |
| N rotatable | 3 | 1 | 1 | 2 | 2 | 5 | 5 | 6 | 1 |
| N violations | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 1 |
| Volume A3 | 285.46 | 133.76 | 147.29 | 209.43 | 222.97 | 288.66 | 302.20 | 454.50 | 468.04 |
| Carcinogenicity | Noncarcinogenic | Noncarcinogenic | Noncarcinogenic | Noncarcinogenic | Noncarcinogenic | Noncarcinogenic | Noncarcinogenic | Noncarcinogenic | Noncarcinogenic |
| GI absorption | High | High | High | High | High | Low | Low | Low | Low |
| Bioavailability score | 0.55 | 0.55 | 0.55 | 0.55 | 0.55 | 0.55 | 0.55 | 0.17 | 0.17 |
2.3. Antimicrobial Activity
All the newly synthesized molecules were screened for their antimicrobial activity against the pathogenic microorganisms Aspergillus niger, candida albicans, Pseudomonas aeruginosa, Staphylococcus aureus, and E coli using the agar diffusion method. The inhibition zone diameters were measured and compared with that of the standard drugs, as tabulated in Table 3. Ciprofloxacin (5µg/disc) and nystatin (100 units/disc) were used as standard antibacterial and antifungal drugs, respectively. The results of the antimicrobial screening showed that compounds 6a and 6b had strong activity against all the tested pathogenic microbes. In addition, compounds 2a and 4a only showed effects against the Gram-negative and Gram-positive bacteria and had no effect on the fungi tested. On the other hand, compound 2b showed strong effects against the fungi tested and no effect against the bacteria tested. Finally, compound 4b revealed moderate activity against all the microorganisms tested, except Staphylococcus aureus.
Table 3.
In vitro antimicrobial screening of the newly synthesized derivatives using the agar diffusion method.
| Compounds | Microorganism Inhibition Zone Diameters Using the Agar Diffusion Method (mm) | ||||
|---|---|---|---|---|---|
| Gram (+ve) Bacteria | Gram (−ve) Bacteria | Fungi | |||
| Staphylococcus aureus |
Escherichia
coli |
Pseudomonas aeruginosa | Aspergillus niger | Candida albicans | |
| 2a | 15 ± 0.14 | 12 ± 1.08 | 22 ± 1.01 | −ve | −ve |
| 2b | −ve | 5 ± 0.2 | −ve | 30 ± 1.16 | 27 ± 1.1 |
| 4a | 23 ± 0.8 | −ve | 13 ± 0.65 | −ve | −ve |
| 4b | −ve | −ve | 12 ± 08 | 14 ± 0.15 | 19 ± 1.04 |
| 6a | 24 ± 0.6 | 25 ± 0.9 | 17 ± 0.75 | 20 ± 0.9 | 16 ± 0.89 |
| 6b | 29 ± 1.2 | 21 ± 1.14 | 19 ± 0.79 | 18 ± 0.12 | 14 ± 0.58 |
| Ciprofloxacin | 20 ± 0.9 | 23 ± 1.02 | 21 ± 0.9 | −ve | −ve |
| Nystatin | −ve | −ve | −ve | 22 ± 0.18 | 23 ± 1.15 |
Values are expressed as mean ± standard deviation.
3. Experimental
3.1. Chemistry
3.1.1. Experimental Instrumentation
All melting points were determined on an electrothermal apparatus and are uncorrected. IR spectra were recorded (KBr discs) on a Shimadzu FT-IR 8201 PC spectrophotometer. 1H-NMR and 13C-NMR spectra were recorded in (CD3)2SO solutions on a BRUKER500 FT-NMR system spectrometer, and chemical shifts are expressed in ppm units using TMS as an internal reference. Mass spectra were recorded on a GC-MS QP1000 EX Shimadzu. Elemental analyses were carried out at the Microanalytical Center of Cairo University.
3.1.2. General Procedures for Synthesis of Derivatives 2a,b
2-azido-1H-benzo[d]imidazole 1a and/or 2-azido-7-chloro-1H-benzo[d]imidazole 1b (10 mmol) were reacted with acetylacetone (2 mL, 20 mmol) under reflux in ethanol containing sodium ethoxide (0.5 g, 10 mmol) for 5 h. The resulting solid that formed after cooling was collected and recrystallized from ethanol to afford 2a and 2b, respectively.
1-(1-(1H-benzo[d]imidazol-2-yl)-5-methyl-1H-1,2,3-triazol-4-yl)ethan-1-one derivatives 2a
White crystals, m.p. 218–220 °C, yield: 58%; FT-IR (KBr, cm−1): v 3461(NH), 2919, 2852 (CH), 1715(C=O), 1618 (C=N); 1600(C=C); 1H-NMR (500 MHz, DMSO-d6): δ 2.10 (s, 3H, CH3), 2.42 (s, 3H, CH3), 7.21 (d, 2H, J = 10Hz, ArH), 7.54 (d, 2H, J = 10Hz, ArH), 11.52 (s, 1H, NH); 13C-NMR (100 MHz, DMSO-d6): δ 9.5 (CH3), 27.4 (CH3), 115.2, 123.4, 133.1, 138.3, 138.7, 141.5, 195.9 (C=O); ESI-MS: m/z(%): 241 [M+] (11), 225 (12), 212 (13), 177 (50), 166 (18), 77 (91), 65 (100). Anal. Calcd. for C12H11N5O (241): C, 59.74; H, 4.60; N, 29.03% found: C, 59.78; H, 4.52; N, 29.01%.
1-(1-(7-chloro-1H-benzo[d]imidazol-2-yl)-5-methyl-1H-1,2,3-triazol-4-yl) ethan-1-one 2b
White crystals, m.p. 228–230 °C, yield: 52%; FT-IR (KBr, cm−1): v 3450 (NH), 2921, 2858 (CH), 1705 (C=O), 1622 (C=N); 1600 (C=C); 1H-NMR (500 MHz, DMSO-d6): δ 2.16 (s, 3H, CH3), 2.47 (s, 3H, CH3), 7.21–7.25 (m, 3H, ArH), 11.58 (s, 1H, NH); 13C-NMR (100 MHz, DMSO-d6): δ 9.5 (CH3), 27.4 (CH3), 113.5, 120.2, 124.4, 133.1, 138.3, 140.7, 141.5, 194.8 (C=O); ESI-MS: m/z(%): 275 [M+] (15), 273 (17), 265 (18), 238 (12), 222 (14), 170 (100), 166 (28), 65 (50). Anal. Calcd. for C12H10ClN5O (275): C, 52.28; H, 3.66; N, 25.40% found: C, 52.34; H, 3.62; N, 25.37%.
3.1.3. General Procedures for Synthesis of 4a,b
To a solution of 2a and/or 2b (10 mmol) in 2-propanol (20 mL), methyl hydrazine carbodithioate 3 (1.22 g, 10 mmol) was added. The mixture was stirred at RT for 2 h. The solid product was filtered off, recrystallized from Acetic acid to afford the target molecules 4a and 4b, respectively.
Methyl-2-(1-(1-(1H-benzo[d]imidazol-2-yl)-5-methyl-1H-1,2,3-triazol-4-yl) ethylidene) hydrazine-1-carbodithioate 4a
Yellow crystals, m.p. 240–242 °C, yield: 75%; FT-IR (KBr, cm−1): v 3417 (broad, NH), 1622 (C=N); 1600 (C=C); 1H-NMR (500 MHz, DMSO-d6): δ 2.16 (s, 3H, CH3), 2.36 (s,3H, CH3), 2.47 (s, 3H, CH3), 7.35–7.85 (m, 4H, ArH), 8.95 (s,1H, NH), 11.58 (s, 1H, NH); 13C-NMR (100 MHz, DMSO-d6): δ 10.0 (CH3), 16.9 (CH3), 18.7 (CH3), 115.2, 123.2, 138.7, 141.5, 145.6 (C=N), 200.0 (C=S); ESI-MS: m/z(%): 345 [M+] (42), 325 (18), 293 (10), 271 (8), 240 (12), 222 (14), 197 (15), 181 (20), 166 (28), 77 (14), 50 (10). Anal. Calcd. for C14H15N7S2 (345): C, 48.68; H, 4.38; N, 28.38% found: C, 48.57; H, 4.32; N, 28.31%.
Methyl-2-(1-(1-(7-chloro-1H-benzo[d]imidazol-2-yl)-5-methyl-1H-1,2,3-triazol-4-yl) ethylidene) hydrazine-1-carbodithioate 4b
Yellow crystals, m.p. 263–265 °C, yield: 75%; FT-IR (KBr, cm−1): v 3419 (broad, NH), 1628 (C=N); 1612 (C=C); 1H-NMR (500 MHz, DMSO-d6): δ 2.18 (s, 3H, CH3), 2.32 (s,3H, CH3), 2.42 (s, 3H, CH3), 7.14–7.42 (m, 3H, ArH), 8.98 (s,1H, NH), 11.47 (s, 1H, NH); 13C-NMR (100 MHz, DMSO-d6): δ 9.1 (CH3), 16.9 (CH3), 19.7 (CH3), 117.2, 124.2, 139.7, 142.5, 142.6, 143.5 (C=N), 197.1 (C=S); ESI-MS: m/z(%): 379 [M+] (12), 352 (17), 287 (51), 252 (18), 250 (12), 227 (14), 170 (15), 165 (20), 70 (14), 65 (100). Anal. Calcd. for C14H14ClN7S2 (379): C, 44.26; H, 3.71; N, 25.81% found: C, 44.32; H, 3.65; N, 25.73%.
3.1.4. General Procedures for Synthesis of Compounds 6a,b
To a mixture of methyl carbodithioate 4a and/or 4b (1 mmol) and the appropriate 2-oxo-N-phenyl-2-(phenylamino) aceto hydrazonoyl chloride (0.27 gm, 1 mmol) in ethanol (20 mL), TEA (0.5 mL) was added; the mixture was stirred at RT for 2–4 h (monitored with TLC). The resulting solid was collected and recrystallized from the proper solvent to obtain the desired 1,3,4-thiadiazolines 6a and 6b, respectively.
5-(1-(1-(1-H-benzo[d]imidazol-2-yl)-5-methyl-1H-1,2,3-triazol-4-yl)ethylidene) hydrazono)-N,4-diphenyl-4,5-dihydro-1,3,4-thiadiazole-2-carboxamide 6a
Yellow crystals, m.p. 251–253 °C, yield: 67%; FT-IR (KBr, cm−1): v 3435 (broad, NH), 1685 (C=O), 1600 (C=N), 1590 (C=C); 1H-NMR (500 MHz, DMSO-d6): δ 2.38 (s,3H, CH3), 2.46 (s, 3H, CH3), 7.32–7.91 (m, 14H, ArH), 9.81 (s,1H, NH), 11.52 (s, 1H, NH); 13C-NMR (100 MHz, DMSO-d6): δ 10.1 (CH3), 20.5 (CH3), 115.20, 122.40, 123.06, 129.60, 133.7, 138.30, 138.90, 141.50 (C=N), 158.20 (C=N), 160.00 (C=O), 164.60 (C=N); ESI-MS: m/z(%): 534 [M+] (12), 520 (16), 492 (26), 478 (14), 425 (17), 357 (17), 312 (10), 287 (51%), 252 (18), 251 (12), 236 (14), 172 (15), 167 (21), 77 (12), 50 (100). Anal. Calcd. for C27H22N10OS (534): C, 60.66; H, 4.15; N, 26.20% found: C, 60.72; H, 4.11; N, 26.15%.
5-((1-(1-(7-chloro-1H-benzo[d]imidazol-2-yl)-5-methyl-1H-1,2,3-triazol-4-yl) ethylidene)hydrazono)-N,4-diphenyl-4,5-dihydro-1,3,4-thiadiazole-2-carboxamide 6b
Yellow crystals, m.p. 282–284 °C, yield: 65; FT-IR (KBr, cm−1): v 3447 (broad, NH), 1692 (C=O), 1611 (C=N), 1595 (C=C); 1H-NMR (500 MHz, DMSO-d6): δ 2.32 (s,3H, CH3), 2.48 (s, 3H, CH3), 7.37–7.81 (m, 13H, ArH), 9.82 (s,1H, NH), 11.57 (s, 1H, NH); 13C-NMR (100 MHz, DMSO-d6): δ 10.1 (CH3), 20.5 (CH3), 115.2, 122.4, 123.0, 129.6, 133.7, 138.3, 138.9, 148.5 (C=N), 156.2 (C=N), 162.0 (C=O), 165.6 (C=N); ESI-MS: m/z(%): 570 [M + 2] (15) 568 [M+] (14), 530 (12), 467 (30), 420 (16), 397 (12), 342 (18), 278 (61), 251 (11), 245 (17), 226 (14), 167 (15), 165 (22), 77 (17), 50 (100). Anal. Calcd. for C27H21ClN10OS (568): C, 56.99; H, 3.72; N, 24.61% found: C, 56.92; H, 3.65; N, 24.58%.
3.2. Docking Study
In the present study, an in silico docking study was carried out to evaluate the binding geometries of the ligand molecules with the target enzyme. To understand the interactions of all the newly prepared compounds with DNA gyrase B, the crystallographic structure of the enzyme was downloaded from the Protein Data Bank (PDB: 1KZN) with resolution 2.3 Å [35]. In addition, the 2D structures of the ligand molecules were drawn using ChemDraw 16 (CambridgeSoft, Massachusetts, MA, USA), then converted to SDF format using Open Babel GUI [36]. The docking-based virtual screening approach was achieved using PyRx-tool [37]. To analyze the docking results, the discovery studio 3.5 was employed. In silico ADMET and drug-likeness predictions of the molecules were performed using free accessible servers such as admetSAR, mol inspiration, and SwissADME.
3.3. Antimicrobial Activity
3.3.1. Antimicrobial Assay
The antimicrobial activities of the tested compounds were tested against the pathogenic microorganisms Aspergillus niger and candida albicans (NRRL Y-477) using sabouraud dextrose agar medium and Pseudomonas aeruginosa (NRRL B-23, Gram-negative bacteria), E. coli (E. coli O157), and Staphylococcus aureus (NRRL B-313, Gram-positive bacteria) using nutrient agar medium.
3.3.2. Agar Diffusion Medium
The synthesized compounds were tested for their antimicrobial activity using the agar diffusion method (Cruickshank et al. 1975) [38].
4. Conclusions
Herein, in the present study, we reported the synthesis of a novel set of benzimidazole-triazole hybrid analogues, and the compounds were characterized by means of FT-IR, 1H-NMR, 13C-NMR, ESI-MS, and elemental analysis. All the compounds were evaluated for their antimicrobial activity against different strains of bacteria and fungi. Looking at the biological activity, we can conclude that all the compounds showed good to excellent results. In addition, the in vitro findings of the compounds were validated with the help of the molecular docking study with the target enzyme DNA gyrase B by investigating its binding modes of interaction. The molecular docking study revealed that all the synthesized compounds exhibited good binding energy toward the target enzyme. The in silico and in vitro findings showed that compounds 6a,b were the most active against bacterial strains. The results suggested that the compounds 6a,b could be used as promising scaffolds for the design and development of potential antimicrobial inhibitors.
Supplementary Materials
The following are available online. Figure S1. The molecular interactions of the other docked compounds and standard drug with the target enzyme DNA gyrase B. Left side (2D): the residues are represented in 3 letter codes. Hydrogen bonds are represented by green and blue lines and pi-interactions are represented by orange lines. Right side (3D): the docked compounds are represented by gray stick models, and the active site pockets are shown by blue stick models. H-bond interactions are shown in green dashed lines. π-interactions are shown in orange lines.
Author Contributions
Formal analysis, H.R.M.R.; Funding acquisition, M.M.A.-K. and T.A.Y.; Investigation, A.H.A. All authors designed the study, contributed to the revision of the drafts, and agreed on the final version to be submitted. All authors have read and agreed to the published version of the manuscript.
Funding
The authors extend their appreciation to the Deanship of Scientific Research at Imam Mohammad Ibn Saud Islamic University for funding this work through Research Group no. RG-21-09-72.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
Authors have no conflict of interest.
Sample Availability
Samples of the newly synthesized compounds are available from the authors.
Footnotes
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Hassan M.A., Seleem M.A., Younes A.M.M., Taha M.M., Abdel-Monsef A.-B.H. Synthesis and spectral characterization of some heterocyclic nitrogen compounds. Eur. J. Chem. 2013;4:121–123. doi: 10.5155/eurjchem.4.2.121-123.740. [DOI] [Google Scholar]
- 2.El-Sheshtawy H.S., Abdelmonsef A.H., Abboudy S.M., Younes A.M.M., Taha M.M., Hassan M.A. Synthesis, Structural, and Theoretical Studies of Quinazoline-2,4-dione Derivatives. Polycycl. Aromat. Compd. 2017;39:1–8. doi: 10.1080/10406638.2016.1255234. [DOI] [Google Scholar]
- 3.Hassan M.A., Mohamed A., Younes M., Taha M.M., Haredy A.-B., Monsef A. Synthesis and reactions of 3-aminotetrachloroquinazolin-2,4-dione. Eur. J. Chem. 2011;2:514–518. doi: 10.5155/eurjchem.2.4.514-518.479. [DOI] [Google Scholar]
- 4.Abdelmonsef A.H., Abdelhakeem M.A., Mosallam A.M., Temairk H., El-Naggar M., Okasha H., Rashdan H.R.M. A Search for Anti-inflammatory Therapies: Synthesis, In silico Investigation of the Mode of Action and In vitro Analyses of New Quinazolin-2, 4-dione Derivatives Targeting Phosphodiesterase-4 Enzyme. J. Heterocycl. Chem. 2021 doi: 10.1002/jhet.4395. [DOI] [Google Scholar]
- 5.Ahmad N., Azad M.I., Khan A.R., Azad I. Benzimidazole as a Promising Antiviral Heterocyclic Scaffold: A Review. J. Sci. Arts. 2021;21:273–284. doi: 10.46939/J.Sci.Arts-21.1-b05. [DOI] [Google Scholar]
- 6.Ogurtsov V.A., Rakitin O.A., Rees C.W., Smolentsev A.A. 4,5-Dichloro-1,2-dithiole-3-thione in the synthesis of benzimidazole, benzoxazole and benzothiazole derivatives of 1,3-dithioles. Mendeleev Commun. 2003;13:50–51. doi: 10.1070/MC2003v013n02ABEH001750. [DOI] [Google Scholar]
- 7.Rashdan H.R.M., Abdelmonsef A.H., Shehadi I.A., Gomha S.M., Soliman A.M.M., Mahmoud H.K. Synthesis, Molecular Docking Screening and Anti-Proliferative Potency Evaluation of Some New Imidazo[2,1-b]Thiazole Linked Thiadiazole Conjugates. Molecules. 2020;25:4997. doi: 10.3390/molecules25214997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rashdan H.R.M., Shehadi I.A., Abdelmonsef A.H. Synthesis, anticancer evaluation, computer-aided docking studies, and ADMET prediction of 1,2,3-triazolyl-pyridine hybrids as human aurora B kinase inhibitors. ACS Omega. 2021;6:1445–1455. doi: 10.1021/acsomega.0c05116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rashdan H.R.M., Farag M.M., El-Gendey M.S., Mounier M.M. Toward rational design of novel anti-cancer drugs based on targeting, solubility, and bioavailability exemplified by 1,3,4-thiadiazole derivatives synthesized under solvent-free conditions. Molecules. 2019;24:2371. doi: 10.3390/molecules24132371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rashdan H.R.M., Shehadi I.A., Abdelrahman M.T., Hemdan B.A. Antibacterial Activities and Molecular Docking of Novel Sulfone Biscompound Containing Bioactive 1, 2, 3-Triazole Moiety. Molecules. 2021;26:4817. doi: 10.3390/molecules26164817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.El-Naggar M., Abd El-All A.S., El-Naem S.I.A., Abdalla M.M., Rashdan H.R.M. New potent 5α- Reductase and aromatase inhibitors derived from 1,2,3-triazole derivative. Molecules. 2020;25:672. doi: 10.3390/molecules25030672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rashdan H.R.M., El-Naggar M., Abdelmonsef A.H. Synthesis, Molecular Docking Studies and In Silico ADMET Screening of New Heterocycles Linked Thiazole Conjugates as Potent Anti-Hepatic Cancer Agents. Molecules. 2021;26:1705. doi: 10.3390/molecules26061705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Aouad M.R., Khan D.J.O., Said M.A., Al-Kaff N.S., Rezki N., Ali A.A., Bouqellah N., Hagar M. Novel 1, 2, 3-Triazole Derivatives as Potential Inhibitors against Covid-19 Main Protease: Synthesis, Characterization, Molecular Docking and DFT Studies. Chem. Sel. 2021;6:3468. doi: 10.1002/slct.202100522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li Y., Geng J., Liu Y., Yu S., Zhao G. Thiadiazole-a Promising Structure in Medicinal Chemistry. ChemMedChem. 2013;8:27–41. doi: 10.1002/cmdc.201200355. [DOI] [PubMed] [Google Scholar]
- 15.Mehta D., Taya P. Neetu. A review on the various biological activities of thiadiazole. Int. J. Pharm. Pharm. Sci. 2015;7:39–47. [Google Scholar]
- 16.Samadpour A.N., Merrikh H. DNA gyrase activity regulates DnaA-dependent replication initiation in Bacillus subtilis. Mol. Microbiol. 2018;108:115–127. doi: 10.1111/mmi.13920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Reece R.J., Maxwell A., Wang J.C. DNA gyrase: Structure and function. Crit. Rev. Biochem. Mol. Biol. 1991;26:335–375. doi: 10.3109/10409239109114072. [DOI] [PubMed] [Google Scholar]
- 18.El-Saghier A.M., El-Naggar M., Hussein A.H.M., El-Adasy A.-B.A., Olish M., Abdelmonsef A.H. Eco-Friendly Synthesis, Biological Evaluation, and In Silico Molecular Docking Approach of Some New Quinoline Derivatives as Potential Antioxidant and Antibacterial Agents. Front. Chem. 2021;9:1–14. doi: 10.3389/fchem.2021.679967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chopra B., Dhingra A.K., Prasad D.N., Bhardwaj S., Dubey S. Synthesis and in silico molecular docking studies on substituted piperic acid derivatives as inhibitors of bacterial DNA gyrase. Curr. Comput. Aided Drug Des. 2020;16:281–294. doi: 10.2174/1573409915666190710092032. [DOI] [PubMed] [Google Scholar]
- 20.Halim S.A., Sikandari A.G., Khan A., Wadood A., Fatmi M.Q., Csuk R., Al-Harrasi A. Structure-Based Virtual Screening of Tumor Necrosis Factor-α Inhibitors by Cheminformatics Approaches and Bio-Molecular Simulation. Biomolecules. 2021;11:329. doi: 10.3390/biom11020329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Halim S.A., Waqas M., Khan A., Al-Harrasi A. In Silico Prediction of Novel Inhibitors of SARS-CoV-2 Main Protease through Structure-Based Virtual Screening and Molecular Dynamic Simulation. Pharmaceuticals. 2021;14:896. doi: 10.3390/ph14090896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Abo-Bakr A.M., Alsoghier H.M., Abdelmonsef A.H. Molecular docking, modeling, semiempirical calculations studies and in vitro evaluation of new synthesized pyrimidin-imide derivatives. J. Mol. Struct. 2022;1249:131548. doi: 10.1016/j.molstruc.2021.131548. [DOI] [Google Scholar]
- 23.Abdelmonsef A.H., Mosallam A.M. Synthesis, in vitro biological evaluation and in silico docking studies of new quinazolin-2,4-dione analogues as possible anticarcinoma agents. J. Heterocycl. Chem. 2020;57:1637–1654. doi: 10.1002/jhet.3889. [DOI] [Google Scholar]
- 24.Noser A.A., El-Naggar M., Donia T., Abdelmonsef A.H. Synthesis, In Silico and In Vitro Assessment of New Quinazolinones as Anticancer Agents via Potential AKT Inhibition. Molecules. 2020;25:4780. doi: 10.3390/molecules25204780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Noser A.A., Abdelmonsef A.H., El-naggar M., Salem M.M. New Amino Acid Schiff Bases as Anticancer Agents via Potential Mitochondrial Complex I-Associated Hexokinase Inhibition and Targeting AMP-Protein Kinases/mTOR Signaling Pathway. Molecules. 2021;26:5332. doi: 10.3390/molecules26175332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Haredi Abdelmonsef A., Eldeeb Mohamed M., El-Naggar M., Temairk H., Mohamed Mosallam A. Novel Quinazolin-2,4-Dione Hybrid Molecules as Possible Inhibitors Against Malaria: Synthesis and in silico Molecular Docking Studies. Front. Mol. Biosci. 2020;7:105. doi: 10.3389/fmolb.2020.00105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sobhi M.G., Abdelhady H.A., Doaa Z.H., Abdelmonsef A.H., El-Naggar M., Elaasser M.M., Mahmoud H.K. Thiazole-Based Thiosemicarbazones: Synthesis, Cytotoxicity Evaluation and Molecular Docking Study. Drug Des. Devel. Ther. 2021;2021:659–677. doi: 10.2147/DDDT.S291579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shehadi I.A., Rashdan H.R.M., Abdelmonsef A.H. Homology Modeling and Virtual Screening Studies of Antigen MLAA-42 Protein: Identification of Novel Drug Candidates against Leukemia—An In Silico Approach. Comput. Math. Methods Med. 2020;2020:8196147. doi: 10.1155/2020/8196147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Haredi Abdelmonsef A. Computer-aided identification of lung cancer inhibitors through homology modeling and virtual screening. Egypt. J. Med. Hum. Genet. 2019;20:1–14. doi: 10.1186/s43042-019-0008-3. [DOI] [Google Scholar]
- 30.El-Maghraby A.M., Abdelmonsef A.H. Synthesis, characterization and in silico molecular docking studies of novel chromene derivatives as Rab23 inhibitors. Egypt. J. Chem. 2020;63:1341–1358. doi: 10.21608/ejchem.2019.15013.1911. [DOI] [Google Scholar]
- 31.HA A., SP L. Human Rab8b Protein as a Cancer Target—An In Silico Study. J. Comput. Sci. Syst. Biol. 2016;9:132–149. doi: 10.4172/jcsb.1000231. [DOI] [Google Scholar]
- 32.Rondla R., PadmaRao L.S., Ramatenki V., Haredi-Abdel-Monsef A., Potlapally S.R., Vuruputuri U. Selective ATP competitive leads of CDK4: Discovery by 3D-QSAR pharmacophore mapping and molecular docking approach. Comput. Biol. Chem. 2017;71:224–229. doi: 10.1016/j.compbiolchem.2017.11.005. [DOI] [PubMed] [Google Scholar]
- 33.Abdelmonsef A.H., Dulapalli R., Dasari T., Padmarao L.S., Mukkera T., Vuruputuri U. Identification of Novel Antagonists for Rab38 Protein by Homology Modeling and Virtual Screening. Comb. Chem. High Throughput Screen. 2016;19:875–892. doi: 10.2174/1386207319666161026153237. [DOI] [PubMed] [Google Scholar]
- 34.Dasari T., Kondagari B., Dulapalli R., Abdelmonsef A.H., Mukkera T., Padmarao L.S., Malkhed V., Vuruputuri U. Design of novel lead molecules against RhoG protein as cancer target–a computational study. J. Biomol. Struct. Dyn. 2017;35:3119–3139. doi: 10.1080/07391102.2016.1244492. [DOI] [PubMed] [Google Scholar]
- 35.Berman H.M., Battistuz T., Bhat T.N., Bluhm W.F., Bourne P.E., Burkhardt K., Feng Z., Gilliland G.L., Iype L., Jain S., et al. The protein data bank. Acta Crystallogr. Sect. D Biol. Crystallogr. 2002;58:899–907. doi: 10.1107/S0907444902003451. [DOI] [PubMed] [Google Scholar]
- 36.O’Boyle N.M., Banck M., James C.A., Morley C., Vandermeersch T., Hutchison G.R. Open Babel: An Open chemical toolbox. J. Cheminform. 2011;3:33. doi: 10.1186/1758-2946-3-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dallakyan S., Olson A.J. Chemical Biology. Volume 1263. Springer; Berlin/Heidelberg, Germany: 2015. Small-Molecule Library Screening by Docking with PyRx; pp. 243–250. [DOI] [PubMed] [Google Scholar]
- 38.Metzger B.E., Contreras M., Sacks D.A., Watson W., Dooley S.L., Foderaro M., Niznik C., Bjaloncik J., Catalano P.M., Dierker L. Hyperglycemia and adverse pregnancy outcomes. N. Engl. J. Med. 2008;358:1991–2002. doi: 10.1056/NEJMoa0707943. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Not applicable.