

Summary
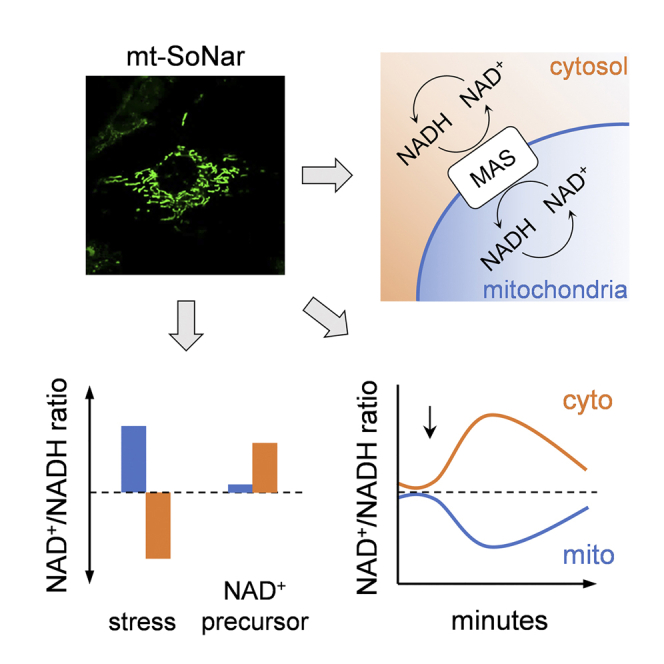
The ratio of oxidized to reduced NAD (NAD+/NADH) sets intracellular redox balance and antioxidant capacity. Intracellular NAD is compartmentalized and the mitochondrial NAD+/NADH ratio is intricately linked to cellular function. Here, we report the monitoring of the NAD+/NADH ratio in mitochondrial and cytosolic compartments in live cells by using a modified genetic biosensor (SoNar). The fluorescence signal of SoNar targeted to mitochondria (mt-SoNar) or cytosol (ct-SoNar) responded linearly to physiological NAD+/NADH ratios in situ. NAD+/NADH ratios in cytosol versus mitochondria responded rapidly, but differently, to acute metabolic perturbations, indicating distinct NAD pools. Subcellular NAD redox balance regained homeostasis via communications through malate-aspartate shuttle. Mitochondrial and cytosolic NAD+/NADH ratios are influenced by NAD+ precursor levels and are distinctly regulated under pathophysiological conditions. Compartment-targeted biosensors and real-time imaging allow assessment of subcellular NAD+/NADH redox signaling in live cells, enabling future mechanistic research of NAD redox in cell biology and disease development.
Keywords: biosensors, NAD+/NADH ratio, mitochondria, live-cell imaging
Graphical abstract
Highlights
-
•
The fluorescent biosensor for NAD+/NADH ratio, SoNar, is targeted to mitochondria
-
•
mt-SoNar and ct-SoNar monitor compartmentalized NAD+/NADH ratios in live cells
-
•
Mitochondrial NAD+/NADH ratio is separately maintained and distinctly regulated
-
•
Subcellular NAD redox regains homeostasis via malate-aspartate shuttle
Motivation
Nicotinamide adenine dinucleotide (NAD) exists in an oxidized (NAD+) or reduced (NADH) form and in different subcellular compartments, including mitochondria and cytosol. The NAD+/NADH ratio sets intracellular redox status and modulates vital biological processes. Mitochondria maintain a distinct NAD pool that is separated from the other parts of the cell. Traditional enzymatic cycling assays and mass spectrometry analysis do not have the spatiotemporal resolution to determine mitochondrial NAD+/NADH ratio. New approaches specifically monitoring NAD+/NADH redox status inside mitochondria of living cells are needed to define the mechanistic roles of mitochondrial NAD in cell biology and pathology.
Hu et al. expressed a fluorescent biosensor for NAD+/NADH ratio inside mitochondria. They show that mitochondrial NAD+/NADH redox ratio is separately maintained though connected with cytosolic NAD pools and responds differently to pathophysiological perturbations. The study presents an approach to evaluate NAD+/NADH ratios in subcellular compartments of live cells.
Introduction
Nicotinamide adenine dinucleotide (NAD), existing in oxidized (NAD+) and reduced (NADH) forms, is an electron carrier and signaling molecule participating in a broad spectrum of biological processes in live cells. NAD is present in multiple subcellular locations, e.g. mitochondria, cytosol, and nucleus, where it regulates important and distinct functions, from energy metabolism, gene expression, and DNA repair to protein modification (Goodman et al., 2020; Katsyuba and Auwerx, 2017; Verdin, 2015; Xiao and Loscalzo, 2020; Zhao et al., 2018). Changes in NAD+ level and/or NAD+/NADH ratio are reported in aging and diseases such as diabetes, cancer, neurodegeneration, and heart failure (Berthiaume et al., 2019; Horton et al., 2016; Hu et al., 2020; Lee et al., 2016; Lu et al., 2009; Neinast et al., 2019; Tateishi et al., 2015; Zou et al., 2020). Preclinical studies suggest that NAD metabolism and/or NAD+/NADH redox balance are potential therapeutic targets (Diguet et al., 2018; Katsyuba and Auwerx, 2017; Lee et al., 2016; Wang et al., 2016; Yoshino et al., 2018). Future mechanistic studies would require information regarding the regulation and roles of compartmentalized NAD pools and NAD+/NADH redox status in vivo. A major challenge in understanding intracellular NAD dynamics is the lack of tools that can reveal the real-time and site-specific changes of NAD pools and/or NAD+/NADH redox status in live cells.
Mitochondria are double-membrane organelles, and the majority of biological molecules including NAD cannot cross the inner membrane by diffusion. Therefore, mitochondria are thought to maintain a distinct NAD pool that is separated from the other parts of the cell. Although traditional enzymatic cycling assays and mass spectrometry analysis have been used to monitor whole-cell NAD pool and NAD+/NADH ratios, they require homogenization of the cell or tissue (Zhao et al., 2011; Zou et al., 2018), cannot differentiate subcellular NAD pools, and are endpoint rather than real-time measurements. Optical imaging in live cells has been used to evaluate the autofluorescence of NADH, which is predominantly from the mitochondria but indistinguishable from the signal of reduced NAD phosphate (NADPH) (Huang et al., 2002). Autofluorescence imaging of NADH and flavin adenine dinucleotide has been used to determine the redox ratio in live cells, which correlated with the NAD+/NADH ratio (Chance et al., 1979; Varone et al., 2014). The autofluorescence from cytosol and mitochondria can be separated by image segmentation (Bianchetti et al., 2021; Wallrabe et al., 2018). Nevertheless, autofluorescence measurement has critical limitations such as not being specific for mitochondria or NADH, not being a direct measurement of NAD+/NADH ratio, low contrast and spatial resolution even under confocal or two-photon microscopy, and requiring complicated image processing and analysis. New approaches that can specifically monitor NAD dynamics and NAD+/NADH redox status are needed to define the mechanistic roles of mitochondrial NAD in cell biology and pathology.
Recently, the circularly permuted yellow fluorescent protein (cpYFP) was inserted into the surface loops of T-Rex protein (Thermus aquaticus) to form a fluorescent biosensor, named SoNar, which has two excitation peaks (410 and 480 nm) and one emission peak (520 nm) (Zhao et al., 2015). Non-targeted SoNar has been characterized and used to monitor the cytosolic NAD+/NADH ratio in live cells and in a real-time manner (Chen et al., 2021; Gu et al., 2020; Hao et al., 2019; Oldham et al., 2015; Titov et al., 2016; Zhao et al., 2016). We modified SoNar and targeted it into the mitochondrial matrix. Using live-cell confocal imaging, we revealed that NAD+/NADH ratios in mitochondria and cytosol of cardiomyocytes were separately maintained. Metabolic substrates, cell contraction, and pathological stresses during ischemia reperfusion or chronic adrenergic stress led to differential changes of NAD+/NADH ratios in mitochondria and cytosol. Moreover, NAD+ precursors, nicotinamide riboside (NR) and nicotinamide mononucleotide (NMN), not only expanded the total intracellular NAD (NAD+ plus NADH) pool but also affected the NAD+/NADH ratio in a compartment-specific manner.
Results
Real-time monitoring of NAD+/NADH redox state in mitochondria and cytosol by modified SoNar in cultured cells
We generated mitochondria-targeted SoNar (mt-SoNar) by subcloning SoNar downstream of a 2× mitochondria-targeting sequence (2× COXVIII, Figure 1A). mt-SoNar exhibited a clear pattern of mitochondrial localization in cultured fibroblasts, epithelial cells, H9C2 cells, and adult rat cardiomyocytes (ACMs) (Figures 1B–1D and S1A). In ACMs, mt-SoNar fluorescence overlapped with the fluorescence of mitochondrial membrane potential dye TMRM while non-targeted SoNar (ct-SoNar) showed diffuse cytosolic localization (Figure 1D). SoNar contains a cpYFP element, which is sensitive to pH (Wang et al., 2008; Zhao et al., 2016). Therefore, we generated mitochondria-targeted cpYFP (mt-cpYFP) and non-targeted cpYFP (ct-cpYFP) to monitor pH changes in parallel (Figures 1D and S1A). The NAD+/NADH ratio was derived from SoNar fluorescence after normalization to the cpYFP fluorescence, thus eliminating the interference by potential changes of pH under given experimental conditions.
Figure 1.

Real-time monitoring of compartmentalized NAD+/NADH redox ratios in live cells
(A) Scheme showing the domains of SoNar and the sequence of 2× mitochondrial targeting sequence.
(B and C) Representative images of mt-SoNar expression in fibroblasts (B) and epithelial cells (C).
(D) Representative images of mt-SoNar, mt-cpYFP, ct-SoNar, and ct-cpYFP in adult rat cardiomyocytes 3 days after adenovirus-mediated gene expression. Scale bar: 20 μm.
(E and F) Responses of the fluorescence ratio (R488/405) of mt-SoNar and mt-cpYFP (E) and ct-SoNar and ct-cpYFP (F) to various NAD+/NADH ratios in permeabilized H9C2 cardiomyoblasts. n = 3.
(G) Summarized data showing time-dependent changes in mitochondrial NAD+/NADH ratio in response to acute addition of ketone bodies acetoacetic acid (AcAc, 1 mM) or β-hydroxybutyrate (β-OHB, 1 mM). n = 5 rats.
(H) Representative examples showing time-dependent changes of mitochondrial or cytosolic NAD+/NADH ratio in individual adult cardiomyocyte during ischemia (indicated by the gray area) and reperfusion.
(I and J) Summarized data showing the changes of mitochondrial (I) or cytosolic (J) NAD+/NADH ratio during simulated ischemia reperfusion in adult cardiomyocytes. n = 4–5 rats. ∗p < 0.05 versus control (value is set as 1.0).
Data are presented as mean ± SEM. See also Figure S1.
To test the response of mt-SoNar and ct-SoNar in situ, we used 0.001% or 0.3% digitonin to permeabilize cell membrane or mitochondrial inner membrane in H9C2 cardiomyoblasts (Figure S1B) and exposed permeabilized H9C2 cardiomyoblasts to NAD+/NADH ratios varying from 1,000:1 to 1:1, and monitored the fluorescence of mt-SoNar and ct-SoNar. When the NAD+/NADH ratio decreased, so did the fluorescence of mt-SoNar or ct-SoNar (Figures 1E and 1F). The fluorescence of mt-cpYFP or ct-cpYFP remained unchanged, indicating that changes of the NAD+/NADH ratio itself would not affect intracellular pH. The results showed that mt-SoNar and ct-SoNar responded linearly to a wide range of NAD+/NADH ratios, supporting the application of SoNar in monitoring physiological changes of the NAD+/NADH ratio in live cells. Moreover, overexpressing mt-SoNar had no impact on cell viability and proliferation (Figures S1C and S1D).
In light of this, we next determined whether SoNar can accurately and promptly detect changes of NAD+/NADH redox state in specific subcellular compartments of live cells. We found that 1 mM β-hydroxybutyrate (β-OHB) decreased while 1 mM acetoacetic acid (AcAc) increased the mitochondrial NAD+/NADH ratio detected by mt-SoNar (Figure 1G), consistent with the predicted change based on the equilibrium of NAD-dependent β-hydroxybutyrate dehydrogenase (BDH) reaction inside mitochondria: β-OHB drives the reaction to generate NADH (lowering the ratio) while AcAc drives the reaction to consume NADH (elevating the ratio) (Williamson et al., 1967). Moreover, SoNar detected a rapid decline of NAD+/NADH ratio in mitochondria and cytosol upon simulated ischemia (perfusion with no substrates and no oxygen solutions) and a quick rebound of NAD+/NADH ratios upon reoxygenation (Figures 1H–1J). The overshot of mt-SoNar signal upon reperfusion faithfully captured the excessive oxidation of NADH in the mitochondria (Ceconi et al., 2000; Varadarajan et al., 2001). These observations confirmed the ability of SoNar to monitor reversible changes of the NAD+/NADH ratio in subcellular compartments during perturbations of oxidative phosphorylation and NADH consumption.
Regulation of mitochondrial versus cytosolic NAD redox during metabolic challenge in adult cardiomyocytes
We next proceeded to determine the NAD redox state in mitochondria and cytosol during metabolic stress in ACMs. We found that addition of 1 mM pyruvate rapidly and dramatically increased the cytosolic NAD+/NADH ratio but decreased the mitochondrial NAD+/NADH ratio, providing a real-time visualization of pyruvate-induced metabolic changes in the two compartments (Figure 2A). The divergent change of NAD+/NADH ratio reflected pyruvate-driven NADH consumption via lactate dehydrogenase reaction in the cytosol (Patgiri et al., 2020) versus NADH production in the mitochondria through pyruvate oxidation and the tricarboxylic acid (TCA) cycle. On the other hand, incubation of ACMs with high concentrations of glucose (20 mM) decreased significantly the NAD+/NADH ratio in both cytosol and mitochondria (Figure 2B). This is in line with glucose feeding glycolysis in the cytosol and TCA cycle in mitochondria concomitantly. Interestingly, when the mitochondrial compartment was specifically perturbed by 5 μM rotenone, a compound that blocks the oxidation of NADH by complex I in the mitochondrial electron transport chain (ETC), the NAD+/NADH ratio decreased in both mitochondria and cytosol (Figure 2C), suggesting a robust communication between the two compartments. These observations collectively show that compartment-specific SoNar offers a reliable and sensitive tool for investigating the NAD+/NADH redox ratio in live cells.
Figure 2.

Mitochondrial and cytosolic NAD pools communicate via MAS
(A) Summarized data showing time-dependent changes of mitochondrial (mt-SoNar, blue) or cytosolic (ct-SoNar, red) NAD+/NADH ratio in response to acute addition of 1 mM pyruvate. n = 5 rats.
(B) Mitochondrial or cytosolic NAD+/NADH ratio in response to acute addition of 20 mM glucose. n = 4–5 rats. ∗p < 0.05 versus control (value is set as 1.0).
(C) Changes of mitochondrial or cytosolic NAD+/NADH ratio in response to acute addition of 5 μM rotenone. n = 4-5 rats. ∗p < 0.05 versus control (value is set as 1.0).
(D) Schematic model showing the regulation of mitochondrial and cytosolic NAD pools and their connection via MAS.
(E and F) Effects of 400 μM AOA and/or 1 μM FCCP preincubation on pyruvate-induced changes of NAD+/NADH ratio in the mitochondria (E) or cytosol (F) of adult cardiomyocytes. n = 4–6 rats. ∗p < 0.05 versus control (value is set as 1.0); #p < 0.05 versus pyruvate + FCCP.
(G) Effects of AOA on cytosolic or mitochondrial NAD+/NADH ratios in adult cardiomyocytes (ACM) or A549 cells. n = 4–5. ∗p < 0.05 versus control (value is set as 1.0).
(H) Summarized data showing time-dependent changes of cytosolic NAD+/NADH ratio in adult cardiomyocytes or A549 cells by 1 mM pyruvate with or without 400 μM AOA preincubation. n = 4–5.
(I) Effects of AOA on the recovery rate of cytosolic NAD+/NADH ratio after the pyruvate-induced peak changes in adult cardiomyocytes or A549 cells. n = 4–5. #p < 0.05 versus −AOA.
Data are presented as mean ± SEM.
Communications between mitochondria and cytosol via malate-aspartate shuttle
Metabolic shuttles that reside in the inner mitochondrial membrane, e.g., the glycerol-phosphate shuttle and malate-aspartate shuttle (MAS), which transport electrons between mitochondria and cytosol (Houtkooper et al., 2010; Lee et al., 2016), have been considered critical players in regulating cellular redox homeostasis (Figure 2D). Previous studies of their function largely relied on destructive biochemical analysis after chemical inhibition. Using ct-SoNar and mt-SoNar we sought to monitor the dynamics of NAD redox in mitochondria and cytosol in real time. First, we treated ACMs with pyruvate in the presence or absence of 1 μM carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone (FCCP). FCCP is known to uncouple mitochondrial respiration from ATP production and to stimulate maximal ETC activity. This causes robust mitochondrial NADH consumption through complex I. As shown in Figure 2A, pyruvate in the absence of FCCP caused a decrease in the mitochondrial redox as it stimulated NADH production in the TCA cycle. In the presence of FCCP, addition of pyruvate to ACMs no longer decreased the mitochondrial NAD+/NADH ratio (Figure 2E). Unexpectedly, FCCP also led to a robust increase in the NAD+/NADH ratio in cytosol, suggesting that maximal mitochondrial respiration utilized electrons carried by cytosolic NADH and not just NADH generated from pyruvate oxidation in mitochondria (Figure 2F). Treatment with an MAS inhibitor, amino-oxyacetic acid (AOA; 400 μM) blocked the rise of cytosolic NAD+/NADH ratio and increased mitochondrial redox during FCCP challenge, indicating MAS-mediated transportation of cytosolic NADH into mitochondria under this condition (Figures 2E and 2F). These results collectively suggest that when mitochondrial NADH consumption is accelerated, MAS plays a key role in supplying NADH to maintain mitochondrial redox by rebalancing the NAD+/NADH ratio in the direction of cytosol to mitochondria.
Since the relative sizes of NAD (NAD+ plus NADH) pools in intracellular compartments vary among different cell types, we sought to determine whether the shuttle flux affected the NAD redox states in a pool-size-dependent manner. We compared the effects of MAS inhibition by AOA on mitochondrial versus cytosolic NAD+/NADH ratios in two cell types with different subcellular NAD pool size. In ACMs, which had a predominant mitochondrial NAD pool (estimated to be 70% mitochondrial), AOA decreased the cytosolic NAD+/NADH ratio but had no effect on the mitochondrial NAD+/NADH ratio (Figure 2G). In contrast, in lung epithelial cell line A549 cells, which had a predominant cytosolic NAD pool, AOA had no effect on the cytosolic NAD+/NADH ratio but dramatically increased the mitochondrial NAD+/NADH ratio (Figure 2G). In ACMs treated with pyruvate, MAS played a critical role in restoring cytosolic NAD redox. AOA completely blocked the recovery of cytosolic NAD+/NADH ratio in ACMs (Figures 2H and 2I). These results suggested that ACMs rebalanced cellular redox via MAS-mediated transfer of NADH from mitochondria to cytosol under this condition. A549 cells, however, did not depend on MAS to recover cytosolic NAD+/NADH ratios after pyruvate treatment. AOA did not affect the amplitude of pyruvate-induced change or the recovery rate of cytosolic NAD+/NADH ratio in A549 cells (Figures 2H and 2I). Taken together, we found that the MAS flux played an important role in balancing the redox state of mitochondria and cytosol at rest and during metabolic stress. The impact of MAS on each compartment was negatively correlated with the relative size of the pool. The mitochondrial NAD pool size in ACMs is significantly larger, making it insensitive to MAS inhibition. However, the NAD+/NADH ratios in the smaller cytosolic NAD pool of ACMs are more vulnerable to inhibition. The opposite holds true for A549 cells where mitochondrial NAD pool size is smaller, thus showing greater sensitivity to MAS inhibition. The redox state in the larger cytosolic pool is less dependent on MAS. Thus, the communication mechanism may render the redox state of the smaller NAD pool in a given cell type more vulnerable to stress.
Subcellular NAD+/NADH ratios in beating cardiomyocytes were dependent on substrate metabolism
The NAD redox state is closely coupled with energy metabolism such that substrate metabolism turns NAD+ to NADH while oxidative phosphorylation oxidizes NADH to regenerate NAD+. Cardiac contraction consumes a significant amount of energy and thus would potentially affect the NAD+/NADH ratio. Whether and how contraction-associated energy demand alters NAD redox state in subcellular compartments has not been examined directly. Using SoNar and real-time confocal imaging, we found that electric pacing led to a drastic increase in the mitochondrial NAD+/NADH ratio accompanied by a decrease in cytosolic NAD+/NADH ratio in ACMs cultured with glucose as the sole substrate (Figure 3). These changes reflected increases in mitochondrial respiration and glycolysis, respectively, as they were abolished by inhibiting mitochondrial complex I activity with 5 μM rotenone (Figures 3A and 3C) or inhibiting glycolysis with the non-metabolizable glucose analog 2-deoxyglucose (2-DG; 2 mM, Figures 3B and 3D). To determine whether the changes were specific to cardiomyocytes using glucose as the substrate, we added β-OHB to the culture medium and found that the pacing-induced increase in mitochondrial NAD+/NADH ratio was significantly blunted while no change in cytosolic NAD+/NADH ratio was observed (Figure 3). A similar effect was also seen when long-chain fatty acids were added as an additional energy source (Figure 3). These results indicate that glucose metabolism, despite increased glycolysis, is insufficient to sustain bioenergetics and maintain the NAD+/NADH ratio in mitochondria during cardiomyocyte contraction. Additional substrates, e.g., ketone bodies and fatty acids, which directly fuel mitochondrial oxidative metabolism, are needed for maintaining NAD redox homeostasis during increased energy demand. These observations highlight the need for more physiologically relevant culture medium for metabolic studies. Furthermore, the results argue against using culture medium containing glucose only for bioenergetics studies in cardiomyocytes because the results and interpretation might be influenced by substrate deprivation.
Figure 3.

Modulation of subcellular NAD+/NADH ratios in beating cardiomyocytes
(A and B) Representative examples showing time-dependent changes of mitochondrial (A) or cytosolic (B) NAD+/NADH ratio in individual adult cardiomyocytes before and during electric pacing (indicated by the gray area). The cells were incubated in no glucose (No glu, gray circle), glucose only (Glu, 11 mM, blue circle), glucose plus inhibitor (rotenone [+Rot], 5 μM or 2-deoxyglucose [+2-DG], 2 mM, blue triangle), glucose plus β-OHB (1 mM, orange square), or glucose plus mixed fatty acids (+FA, 1 mM, red rhombus) solutions.
(C and D) Summarized data showing the changes of mitochondrial or cytosolic NAD+/NADH ratio after electric pacing for 7 min and in solutions containing the indicated substrates. n = 4–6 rats. ∗p < 0.05 versus before pacing (value is set as 1.0); #p < 0.05 versus pacing in glucose-only solution.
Data are presented as mean ± SEM.
Deranged mitochondrial NAD+/NADH ratio contributed to the detrimental effects of chronic adrenergic stimulation in cardiomyocytes
Pathological hypertrophy of cardiomyocytes is associated with significant changes of cell metabolism and function (Kolwicz et al., 2013). Using compartmentalized SoNar, we found that 10 μM PE increased the NAD+/NADH ratio in mitochondria but not in the cytosol of ACMs after 4 h (Figures 4A–4C). These results were confirmed by biochemical assessment of the NAD+/NADH ratio in mitochondria (AcAc/β-OHB ratio) or cytosol (pyruvate/lactate ratio) (Figures S2A and S2B). PE is known to stimulate glucose uptake and glycolysis in cardiomyocytes, which we hypothesize would reduce the cytosolic NAD+/NADH ratio. Consistently, a robust decrease in NAD+/NADH ratio in the cytosol was detected by ct-SoNar at 24 h after PE incubation, and this decrease was blocked by inhibition of glycolysis using 2-DG (Figure 4C). However, the change in mitochondrial NAD+/NADH ratio occurred earlier (at 4 h) than that in the cytosol and persisted through 24 h after PE treatment, and the increase was significantly reduced by adding β-OHB to the culture medium (Figure 4B). This observation suggested that PE-triggered increased mitochondrial NADH consumption was not met by glucose metabolism. PE-induced elevation in mitochondrial NAD+/NADH ratio was also partially blocked by the mitochondrial Ca2+ uniporter (MCU) inhibitor, Ru360 (Figure 4B), suggesting that increased mitochondrial Ca2+ uptake contributes to the redox imbalance. In support of this hypothesis, caffeine, which acutely increases the cytosolic Ca2+ level in ACMs, elevated the mitochondrial NAD+/NADH ratio, and the MCU inhibitor attenuated this increase (Figure 4D). Caffeine also elevated cytosolic NAD+/NADH ratio, which was not affected by Ru360 (Figure 4E), suggesting that Ca2+ uptake by mitochondria under this condition plays a negligible role in affecting the cytosolic redox state. As the mitochondrial membrane potential was maintained during caffeine stimulation (Figure S2C), the increase in NAD+/NADH ratio is unlikely due to damage caused by Ca2+ overload. We further explored whether the changes in mitochondrial or cytosolic NAD+/NADH ratio played a role in PE-induced cardiomyocyte damage. Sustained PE incubation for 24 h induced cell death and compromised cardiomyocyte contraction (Figures 4F–4G and S2D). Correcting the mitochondrial NAD+/NADH ratio by β-OHB attenuated these detrimental effects of PE (Figures 4F and 4G). Inhibiting glycolysis by 2-DG did not accomplish rescue. These results revealed that the mitochondrial redox ratio responded early to pathological stimulus and played an important role in cell death and dysfunction.
Figure 4.

Subcellular NAD+/NADH ratios and pathological stress
(A) Summarized data showing time-dependent changes of cytosolic or mitochondrial NAD+/NADH ratio in adult cardiomyocytes during acute phenylephrine (PE, 10 μM) treatment. n = 3–4.
(B and C) Mitochondrial or cytosolic NAD+/NADH ratio measured by mt-SoNar or ct-SoNar, respectively, before and after 4 or 24 h PE (10 μM) incubation with or without 1 mM β-OHB, 10 μM Ru360, or 2 mM 2-DG. n = 3–5 rats. ∗p < 0.05 versus control (value is set as 1.0); #p < 0.05 versus PE.
(D and E) Mitochondrial (D) or cytosolic (E) NAD+/NADH ratio in response to acute addition of 10 mM caffeine, which significantly increases cytosolic Ca2+, and the effects of 10 μM Ru360. n = 4–5 rats. ∗p < 0.05 versus control (value is set as 1.0); #p < 0.05 versus caffeine.
(F and G) Effects of PE (10 μM for 24 h) on cell death (F) and contraction amplitude (G) of adult cardiomyocytes. n = 3 rats. ∗p < 0.05 versus control (value is set as 1.0); #p < 0.05 versus PE.
Data are presented as mean ± SEM. See also Figure S2.
NAD+ precursor supplementation selectively altered cytosolic NAD+/NADH ratio in cardiomyocytes
Recent studies suggest that depletion of intracellular NAD+ contributes to aging and diseases; boosting cellular NAD level with NAD+ precursors, e.g., nicotinamide mononucleotide (NMN) and NR, has emerged as a promising therapeutic approach (Diguet et al., 2018; Katsyuba and Auwerx, 2017; Lee et al., 2016; Wang et al., 2016; Yoshino et al., 2018). While NMN and NR are effective in raising cellular NAD+, their effect on NAD+/NADH ratios has rarely been assessed and their subcellular compartment-specific effect is unknown. We thus evaluated the impact of NAD+ precursors on the subcellular redox state in ACMs. We found the NAD+/NADH ratio in both cytosol and mitochondria to increase in the first few minutes of NMN treatment (0.5 mM and 1 mM, Figures 5A–5C). However, there was no change at lower NMN concentration (0.1 mM), suggesting that cellular uptake of NMN in cardiac cells could be concentration dependent. A recent report described Slc12a8 as a selective NMN transporter and suggested a KM value of 34.1 ± 8.3 μM for NMN (Grozio et al., 2019). Slc12a8 has minimal expression in cardiac cells and may not be relevant for ACM uptake of NMN. Considering that neither ct-SoNar nor mt-SoNar registered an increase or decrease in signal with 100 μM extracellular NMN concentrations, we speculate that, if an NMN-specific transporter does exist in cardiac cells, it must have a higher KM for NMN than Slc12a8. Alternatively, NMN could be first converted into NR by CD73 in the extracellular environment (Ratajczak et al., 2016). It has been argued that the hourly kinetics of extracellular NMN to NR conversion cannot explain the minute-order cellular uptake of NMN (Grozio et al., 2019). In our experiments, we observed minute-order cellular changes of NAD+/NADH ratio at 0.5 mM and 1 mM extracellular NMN concentrations. Furthermore, at equimolar concentration (1 mM), NMN increased the cellular NAD+ level at a faster rate, reaching the peak level in 24 h while the same level was reached in cells treated with NR at 72 h (Figures 5D and S3A), suggesting that NMN entered the cells without converting to NR.
Figure 5.

Mitochondrial NAD+/NADH redox is maintained despite significantly enlarged NAD pool
(A–C) Time-dependent responses of mitochondrial (mito, blue) or cytosolic (cyto, red) NAD+/NADH ratio to vehicle (A) or different doses of NAD+ precursor NMN (B and C). n = 3–5 rats. ∗p < 0.05 versus 0.1 mM NMN by two-way ANOVA.
(D–F) Total intracellular NAD+ level, NADH level, and NAD+/NADH ratio (D), and mitochondrial (E) or cytosolic (F) NAD+/NADH ratio after NMN (1 mM) incubation for 0, 4, 24, or 72 h. n = 4–8 rats. ∗p < 0.05 versus control (0 h, value is set as 1.0).
(G–I) Total intracellular NAD+ level, NADH level, and NAD+/NADH ratio (G), and mitochondrial (H) or cytosolic (I) NAD+/NADH ratio after long-term treatment of NAD+ precursor NR in vivo (500 mg/kg/day intraperitoneally for 10 days in rats) followed by NR or vehicle in cell culture (1 mM NR for 3 days on cardiomyocytes isolated from the rats after 10 days of in vivo NR treatment). n = 5–9 rats. ∗p < 0.05 versus no NR (value is set as 1.0).
Data are presented as mean ± SEM. See also Figure S3.
During 72-h incubation of NMN or NR whole-cell NAD+ levels increased significantly, reaching more than double the baseline value. The NADH level was also elevated, resulting in no change of NAD+/NADH ratio (Figures 5D and S3A). Compartment-specific measurement by SoNar showed that NMN caused an early increase in mitochondrial NAD+/NADH ratio at 15 min and 4 h, but the ratio returned to normal at 24 and 72 h (Figure 5E). NR had little effect on the mitochondrial NAD+/NADH ratio at 4–72 h (Figure S3B). The cytosolic NAD+/NADH ratio, however, was increased by both NMN and NR, and the increase was sustained over the treatment period of 72 h (Figures 5F and S3C). These data highlight the advantages of monitoring subcellular redox states using mt-SoNar and ct-SoNar over traditional enzymatic assays which were unable to detect changes in the NAD+/NADH ratio. In cardiomyocytes, the NAD pool is smaller and the NAD+/NADH ratio is higher in the cytosolic compartment compared with the mitochondrial compartment (Zhao et al., 2018); the sustained mitochondrial redox state likely masked the changes in the cytosolic pool in the whole-cell lysate assay.
To study the long-term effects of NAD precursors in vivo, we treated rats with NR for 10 days and then isolated the cardiomyocytes. The cells were infected with adenoviruses containing SoNar/cpYFP indicators and cultured for 3 days to allow their proper expression. Biochemical measurements using whole-cell lysates showed that NR in vivo (10 days in rat) plus NR in vitro (3 days on cells) significantly elevated the total cellular NAD pool as well as NAD+ and NADH levels, resulting in no change of NAD+/NADH ratio (Figure 5G). It should be noted that, without supplementing NR during the 3-day culture, NAD+ concentration declined to basal level (Figure 5G), which supported the dynamic regulation of NAD+ metabolism in cultured cells. Measurements using SoNar showed that the NAD+/NADH ratio in cytosol was moderately increased but the mitochondrial NAD+/NADH ratio was unchanged (Figures 5H and 5I).
These results suggest that supplementation of NAD+ precursors in ACM causes a transient increase in the mitochondrial NAD+/NADH ratio, which is restored by increasing NADH production. On the contrary, increasing NAD+ has a sustained effect on the cytosolic NAD+/NADH ratio, suggesting that a prolonged increase in the NAD+ level can be achieved in the cytosol. Furthermore, these effects are readily reversible after the withdrawal of precursors, consistent with rapid cellular NAD metabolism. The lack of increase in the mitochondrial NAD+/NADH ratio at 4 h of NR treatment could be due to a slower NAD+ synthesis. NR must first be converted to intracellular NMN via the ATP-dependent nicotinamide riboside kinase (Nrk) reaction. It has been shown that Nrk1 and/or Nrk2 are rate limiting in NR utilization for intracellular NAD+ synthesis (Ratajczak et al., 2016). Since the reaction is ATP dependent it could also be heavily regulated by the energetic status of the cell (ATP/ADP ratio), thereby limiting cytosolic NMN concentrations.
Discussion
In this study, we developed a mitochondrial-targeted genetic sensor (mt-SoNar) to assess the dynamic changes of NAD+/NADH redox state in mitochondria under stress conditions. The subcellular compartment-targeted SoNar reveals previously unrecognized changes of NAD redox in mitochondria versus that in the cytosol, thus filling an important methodological gap for monitoring the compartmentalized NAD+/NADH redox ratio in live cells and tissues, and in vivo. The study first tested mt-SoNar in cardiomyocytes because these cells are abundant in mitochondria, have a predominant mitochondrial NAD pool, and exhibit metabolic flexibility known to affect the mitochondrial redox state. We validated the results of mt-SoNar by using a well-accepted biochemical method for the mitochondrial NAD+/NADH ratio based on the BDH reaction equilibrium (Figure S2). The opposite responses of mt-SoNar to AcAc or β-OHB are also consistent with the BDH reaction. Moreover, mt-SoNar detected biphasic changes in mitochondrial NAD+/NADH ratio during simulated ischemia reperfusion (Figure 1H), which is consistent with the results of NAD(P)H autofluorescence (Zhang et al., 2018). In situ titration in permeabilized cells determined the direct response of mt-SoNar to a wide range of physiologically relevant NAD+/NADH ratios (Chen et al., 2016). We further demonstrated that SoNar is capable of reporting NAD+/NADH redox ratio in non-cardiomyocytes with a relatively small mitochondrial NAD pool, suggesting that compartment-targeted SoNar technology is broadly applicable to other cell types.
NAD+ and NADH are the most important redox pairs in the cell. Their ratio sets the intracellular redox environment, controls antioxidant capacity, and regulates cell signaling (Jones and Sies, 2015). The mitochondrial NAD+/NADH ratio connects energy metabolism with crucial mechanisms for cell survival and function. For instance, NADH generated by oxidative metabolism provides reduction equivalent to NADPH for its antioxidant activity (Nickel et al., 2015). Compromised mitochondrial metabolism can decrease NADH generation, increase the NAD+/NADH ratio, and impair antioxidant capacity, leading to oxidative stress, a mechanism underlying the contractile dysfunction of the failing heart (O'Rourke et al., 2021). On the other hand, a decrease in NAD+/NADH ratio could reduce the availability of NAD+ for proteins requiring NAD+ as co-substrate, such as sirtuin deacetylase, and hence affect cell signaling (Karamanlidis et al., 2013; Lee et al., 2016). Recently it has been shown that serine can generate substantial NADH during mitochondrial respiration inhibition to further tilt the redox balance, leading to suppression of cell growth (Yang et al., 2020). It should be noted that in these studies, NAD+ and NADH are measured either indirectly via enzymatic assays or in whole-cell/tissue samples, and thus do not provide accurate information regarding the specific changes in subcellular compartments. The compartment-targeted SoNar technology, which possesses subcellular spatiotemporal resolution in live cells and without the liability of shifting redox state during destructive sample processing, is expected to provide a powerful tool for discovery.
The results of this study show that mitochondria of cardiomyocytes maintain a separate NAD pool with an NAD+/NADH redox ratio distinct from that in the cytosol. The mitochondrial NAD+/NADH ratio responded promptly to acute metabolic, physiological, or pathological perturbations as well as chronic adrenergic stress. However, normal mitochondria have the ability to sustain the NAD+/NADH ratio under physiological stress conditions even during large increases of intracellular NAD+ level, due to a relatively larger NAD pool and a lower NAD+/NADH ratio (Zhao et al., 2018). The mitochondrial NAD pool, although independently maintained, communicated with the cytosolic NAD pool via MAS. Specifically, in cells that rely primarily on mitochondrial metabolism, MAS may set the NAD+/NADH homeostasis at resting conditions and shuttle NADH in or out of mitochondria to support elevated ETC activity or rebalance redox homeostasis. During acute and chronic pathological stress, the mitochondrial NAD+/NADH ratio is affected more dramatically or much earlier in the disease progression than the cytosolic ratio, further supporting a predominant role of the mitochondrial NAD pool and redox status in defining the cellular responses to stress. Importantly, altered mitochondrial NAD+/NADH ratio is not only a consequence of stress-induced metabolic responses but also a potential mechanism to mediate stress-induced cell damage and dysfunction. As such, the use of compartmentalized SoNar indicators, combined with manipulations of subcellular NAD+/NADH ratios and functional assays, will be able to uncover novel targets and pathways by which the NAD+/NADH redox ratio bridges a stress signal and a particular defect in cell function.
With the emerging interest in using NAD+ precursor supplementation as a therapy, the biological consequences of perturbing the intracellular NAD+ pool become a pressing question. We here reveal that the NAD+/NADH redox ratio in mitochondria is affected only transiently in normal cardiomyocytes despite a large (>2-fold) increase in intracellular NAD+ by precursor supplementation. This finding is consistent with the notion that mitochondria redox is tightly controlled. While the ability of maintaining the mitochondrial NAD+/NADH redox ratio is reassuring, it does not mean that increasing NAD+ would have no effect on mitochondria. Rather, it shows that mitochondria are able to respond and re-establish redox homeostasis, likely via increased conversion of NAD+ to NADH via oxidative metabolism. The cytosolic NAD+/NADH redox ratio, however, remains elevated during NAD+ precursor supplementation even after the increase of cellular NAD+ plateaus. Thus, increased NAD+ level outside mitochondria could be an important mediator of the biological effects of NAD+ precursor supplementation (Canto et al., 2015; Yoshino et al., 2018). Further studies are needed to distinguish the mechanisms mediated by increased NAD+ level from that by altered NAD+/NADH ratio.
In summary, we have developed a modified version of SoNar (mt-SoNar) that is genetically encoded and targeted into the mitochondrial matrix. Confocal imaging with mt-SoNar provides a specific signal of the mitochondrial NAD+/NADH ratio and can be used in conjunction with non-targeted SoNar to distinguish NAD redox in mitochondrial versus cytosolic compartments. Our findings demonstrate the power of monitoring subcellular NAD redox in live cells. Deciphering the NAD redox regulation in mitochondria versus cytosol in cardiomyocytes provides novel insight into stress response, which whole-cell study fails to resolve.
Limitations of the study
Although high optical resolution is not critical for SoNar, confocal imaging of mt-SoNar has advantages of obtaining high-intensity fluorescence exclusively from mitochondria and free of interference by motion artifact and contaminating signals. Due to the normalization process and the fact that GFPs are pH and temperature sensitive, SoNar is suitable for qualitative, but not quantitative, monitoring of time-dependent changes of NAD+/NADH ratios in live cells. It is also recommended that mt-SoNar is used in parallel with mt-cpYFP for correcting any potential influences of pH. To achieve reliable comparison and evaluation of SoNar fluorescence, the expression level of SoNar indicators, the setting of confocal imaging (e.g., temperature and laser intensity), and the baseline condition of cultured cells should be kept identical among the groups. This unique approach may have broad applications in studying the physiological regulation, disease implications, and therapeutic potential of manipulating subcellular NAD+/NADH redox in cultured cells, intact organs, or in vivo.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and virus strains | ||
| Turbo competent E. coli (high efficiency) | New England Biolabs | Cat# C2984H |
| Biological samples | ||
| Adult rat cardiomyocytes | This paper | N/A |
| Adult rat cardiac fibroblasts | This paper | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| 2-Deoxyglucose | Sigma-Aldrich | Cat# D8375 |
| Acetoacetic acid | Sigma-Aldrich | Cat# A8509 |
| Aminooxiacetic acid | Sigma-Aldrich | Cat# C13408 |
| β-hydroxybutyrate | Sigma-Aldrich | Cat# H6501 |
| Bovine serum albumin (BSA) | Sigma-Aldrich | Cat# A3803 |
| BrdU Labeling Reagent | Invitrogen | Cat# 00-0103 |
| Caffeine | Alfa Aesar | Cat# AAA1043122 |
| Calcium chloride (CaCl2) | Sigma-Aldrich | Cat# C4901 |
| Carbonyl cyanide 4-(trifluoromethoxy) phenylhydrazone (FCCP) | Sigma-Aldrich | Cat# C2920 |
| Collagenase type 2 | Worthington Corp. | Cat# LS004177 |
| DMEM media | Sigma-Aldrich | Cat# D6429 |
| Digitonin | Sigma-Aldrich | Cat# D141 |
| Fatty acid mixture | (Ritterhoff et al., 2020) | N/A |
| Fetal bovine serum (FBS) | Sigma-Aldrich | Cat# F8687 |
| Glucose | Sigma-Aldrich | Cat# G8270 |
| Glutathione | Sigma-Aldrich | Cat# G6013 |
| Hyaluronidase | Sigma-Aldrich | Cat# H3506 |
| Laminin | Life Technologies | Cat# 23017015 |
| Lipofectamine 2000 | Invitrogen | Cat# 11668019 |
| M199 medium | Sigma-Aldrich | Cat# M2520 |
| Magnesium sulfate (MgSO4) | Sigma-Aldrich | Cat# M7506 |
| N-(2-Hydroxyethyl)piperazine-N′- (2-ethanesulfonic acid), 4-(2-Hydroxyethyl) piperazine-1-ethanesulfonic acid (HEPES) |
Sigma-Aldrich | Cat# H3375 |
| NAD+ | Sigma-Aldrich | Cat# 481911 |
| NADH | Sigma-Aldrich | Cat# 481913 |
| Nicotinamide mononucleotide | Sigma-Aldrich | Cat# N3501 |
| Nicotinamide riboside | ChromaDex | Cat# NIAGEN |
| Opti-MEM | Invitrogen | Cat# 31985070 |
| Penicillin/Streptomycin (100×) | Thermo Fisher | Cat# 10378016 |
| Phenylephrine | Sigma-Aldrich | Cat# P1240000 |
| Potassium chloride (KCl) | Sigma-Aldrich | Cat# P3911 |
| Potassium phosphate monobasic (KH2PO4) | Sigma-Aldrich | Cat# P5655 |
| Pyruvate | Sigma-Aldrich | Cat# P2256 |
| Rotenone | Sigma-Aldrich | Cat# R8875 |
| Ru360 | Millipore | Cat# 557440 |
| Sodium bicarbonate (NaHCO3) | Sigma-Aldrich | Cat# S5761 |
| Sodium chloride (NaCl) | Sigma-Aldrich | Cat# S9888 |
| Sodium dithionite (Na2S2O4) | Sigma-Aldrich | Cat# 71699 |
| Tetramethylrhodamine, methyl ester (TMRM) | Sigma-Aldrich | Cat# T5428 |
| Trypan blue | Sigma-Aldrich | Cat# T6146 |
| Trypsin-EDTA | Gibco | Cat# 25200072 |
| Critical commercial assays | ||
| Ketone body assay kit | BioAssay Systems | Cat# EKBD-100 |
| L-Lactate assay kit | Cayman | Cat# 700510 |
| NAD+/NADH cell-based assay kit | BioAssay Systems | Cat# E2ND002 |
| Pyruvate assay kit | Cayman | Cat# 700470 |
| Experimental models: Cell lines | ||
| 293T cells | ATCC | Cat# CRL-3216 |
| A549 cells | ATCC | Cat# CCL-185 |
| H9C2 cardiomyoblasts | ATCC | Cat# CRL-1446 |
| Experimental models: Organisms/strains | ||
| Sprague Dawley | Envigo | Cat# 002 |
| Oligonucleotides | ||
| ct-SoNar-F: 5’-GCGAATTCATGAACC GGAAGTGGGGCCTGTG-3’ |
This paper | N/A |
| ct-SoNar-R: 5’-GAGAAGCTTCTAGCC CATCATCTCCTC-3’ |
This paper | N/A |
| ct-cpYFP-F: 5’-GCGAATTCATGTACA ACAGCGACAACGTCTATATC-3’ |
This paper | N/A |
| ct-cpYFP-R: 5’-GAGAAGCTTCTAGTT GTACTCCAGCTTGTGCCCC-3’ |
This paper | N/A |
| mt-COXVIII-F: 5’-GCAGATCTGCCACC ATGTCTGTTCTGACTCCTCTGCTGC TCCG-3’ |
This paper | N/A |
| mt-COXVIII-R: 5’-GCGGATCCCTTGG GATCGCCGAGACTGTGGATCTTGG CCCTGGGG-3’ |
This paper | N/A |
| SoNar-F: 5’-GCGGATCCATGAACCGGAAGTGGG GCCTGTGCATCGTGGGCATGGG-3’ |
This paper | N/A |
| SoNar-R: 5’-GCAAGCTTCTAGCCCAT CATCTCCTCCCGCCACTTGGGGTT-3’ |
This paper | N/A |
| Recombinant DNA | ||
| pCD316-mt-SoNar | This paper | N/A |
| pCD316-ct-cpYFP | This paper | N/A |
| pcDNA3.1-ct-SoNar | (Zhao et al., 2015) | N/A |
| pcDNA3.1-mt-cpYFP | (Wang et al., 2008) | N/A |
| Software and algorithms | ||
| Gen5 1.05 | Biotek | https://www.biotek.com/products/software-robotics |
| Graphpad Prism V8.1 | Graphpad software | https://download.cnet.com/s/graphpad-prism |
| ImageJ | NIH | https://imagej.nih.gov/ij/download.html |
| IonWizard | IonOptix | https://www.ionoptix.com/products/software/ |
| Leica Application Suite X | Leica | https://www.leica-microsystems.com/products/microscope-software/p/leica-las-x-ls |
| Other | ||
| Glass coverslip | Warner instruments | Cat# 640705 |
| High vacuum grease | Dow corning | Cat# HVG |
| Quick change chamber w/stimulation | Warner instruments | Cat# 640374 |
| Quick exchange platform (QE-1) | Warner instruments | Cat# 640375 |
| SA-20LZ stage adapter | Warner instruments | Cat# 642413 |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Dr. Wang Wang (wangwang@uw.edu).
Materials availability
Most of the materials used in this study are commercially available. The mt-SoNar and mt-cpYFP plasmids are available upon request.
Experimental model and subject details
Animals
All the animal procedures were performed in accordance with the approved protocol by the Institutional Animal Care and Use Committee (IACUC) of the University of Washington. Rats (Sprague Dawley, male, 8 weeks old) were kept on regular rodent chow and water ad libitum in a vivarium with a 12 hr light/dark cycle at 22°C until use. Nicotinamide riboside chloride provided by ChromaDex was delivered intraperitoneally to rats at 500 mg/kg/day for ten days. Saline was delivered intraperitoneally as vehicle control.
Adult rat cardiomyocyte isolation, culture, and gene transfer
Adult cardiomyocytes were isolated from the heart of Sprague Dawley rats (200-250 g) by using standard enzymatic technique as we reported previously (Wang et al., 2008). Briefly, rat was anaesthetized by intraperitoneal injection of pentobarbital (50 mg/kg). The heart was quickly removed, cannulated via the ascending aorta, and mounted on a modified Langendorff perfusion system. The heart was perfused with oxygenated Krebs-Henseleit Buffer (KHB) containing NaCl (118 mM), KCl (4.8 mM), HEPES (25 mM), KH2PO4 (1.2 mM), MgSO4 (1.2 mM), and glucose (11 mM), and supplemented with collagenase II (Worthington, USA) and hyaluronidase (Sigma, USA) at 37°C. Rod shaped adult cardiomyocytes were collected and plated at a density of ∼2 × 104 cells per coverslip pre-coated with 20 μg/ml laminin (Life Technologies). Attached cardiomyocytes were infected with Ad-ct-SoNar, Ad-ct-cpYFP, Ad-mt-SoNar, or Ad-mt-cpYFP at a multiplicity of infection of 50-100. The cells were cultured in serum-free M199 medium (Sigma), which contains 5.5 mM glucose as fuel, and supplemented with 10 mM glutathione, 26.2 mM sodium bicarbonate, 0.02% bovine serum albumin and 50 U/ml penicillin–streptomycin for 72 hr to allow adequate gene expression.
Method details
Plasmids and recombinant adenoviral vectors
The cDNA of SoNar gene is a kind gift from Dr. Yi Yang (East China University of Science and Technology, China). We added the following mitochondria-targeting sequence (2× COXVIII):
ATGTCTGTTCTGACTCCTCTGCTGCTCCGGGGTCTCACAGGTTCCGCAAGAAGACTCCCCGTGCCTAGGGCCAAAATTCATTCACTGGGGGACCCCATGAGCGTGCTCACCCCACTCCTGCTGCGGGGGCTGACCGGCAGCGCTAGGCGGCTGCCAGTCCCCAGGGCCAAGATCCACAGTCTCGGCGATCCCAAG
The primers used for the subcloning of cytosolic SoNar (ct-SoNar) or cpYFP (ct-cpYFP), or mitochondria-targeted SoNar (mt-SoNar) are shown in Oligonucleotides list. Construction of recombinant adenoviral vectors containing ct-SoNar (Ad-ct-SoNar) or mt-SoNar (Ad-mt-SoNar) used protocols previously described (Hu et al., 2020). Ad-mt-cpYFP was reported before (Wang et al., 2008). Ad-ct-cpYFP was generated by Vector Biolabs. Concentrations of the viruses were estimated to be ∼1 × 1011 viral particles per ml. The viruses were aliquoted and stored at −80°C.
In situ calibration of SoNar indicators in permeabilized cells
Calibration of the SoNar indicators and their cpYFP controls followed a previously published protocol (Zhao et al., 2011). Briefly, to calibrate ct-SoNar or ct-cpYFP, indicator expressing H9C2 cells were trypsinized, suspended in PBS (pH = 7.4), and permeabilized by 0.001% digitonin. To calibrate mt-SoNar or mt-cpYFP, indicator expressing H9C2 cells were suspended in PBS buffer (pH = 8.0) and permeabilized by 0.3% digitonin. NAD+ and NADH were added at various ratios with the same total NAD concentration (400 μM). After incubation at room temperature for 15 min, the fluorescence was monitored by using a microplate fluorometer (excitation 488/405 nm and emission 520 nm).
Confocal imaging
Cells were mounted on the confocal microscope (Leica SP8) stage in a custom-made chamber as previously reported (Hu et al., 2020). Cells were immersed in Tyrode's solution containing NaCl (138 mM), KCl (3.7 mM), HEPES (20 mM), MgSO4 (1.2 mM), KH2PO4 (1.2 mM), CaCl2 (1 mM), glucose (5 mM) (pH = 7.4). The ct-SoNar and mt-SoNar were monitored in cells on separate coverslips. The confocal microscope is equipped with a 40×, 1.3 NA oil immersion objective. Dual-excitation images of SoNar or cpYFP were taken by alternating excitation at 405 and 488 nm and collecting emissions at >505 nm. For time-lapse confocal imaging, we first confirmed the appropriate mitochondrial or cytosolic localization pattern and adequate fluorescent signal. A group of cells within a randomly selected region of interest were scanned at an interval of 30 sec for ∼25 min, during which time different reagents were added. The metabolic substrates (1 mM acetoacetic acid, 1 mM β-hydroxybutyrate, 20 mM glucose, or 1 mM pyruvate) or chemicals (400 μM AOA, 1 μM FCCP, 10 mM caffeine, 10 μM Ru360, 10 μM phenylephrine, 0.1-1 mM nicotinamide mononucleotide, or 0.1-1 mM nicotinamide riboside chloride) were added as indicated in the Figures or figure legends. For some experiments, AOA (400 μM), FCCP (1 μM), 2-deoxyglucose (2-DG, 2 mM), or Ru360 (10 μM) were added 20 min before pyruvate (1 mM), caffeine (10 mM), PE (10 μM), or electric pacing. For PE experiments, fluorescence from the same cell was collected before and after the 4 hr PE incubation. Image processing used ImageJ. The relative changes in NAD+/NADH ratio was calculated as follows: (1) the ratios of fluorescence emission at 488 nm excitation over that of 405 nm excitation for SoNar (R488/405 for SoNar) or for cpYFP (R488/405 for cpYFP) were calculated; (2) R488/405 for SoNar was normalized by R488/405 for cpYFP to calculate NAD+/NADH ratio (Zhao et al., 2016); (3) the NAD+/NADH ratio at baseline condition (Control or before the addition of reagents) was used to further normalize the NAD+/NADH ratios after treatment.
For the ischemia and reperfusion mimetic experiments in cultured cardiomyocytes, cells were perfused with Tyrode's solution (pH = 7.4) without glucose and with Na2S2O4 (1 M) for 10 min and switched back to normal Krebs-Henseleit buffer (pH = 7.4) for 5 min (Zhang et al., 2018). For electric pacing experiment, cardiomyocytes were field stimulated at 1 Hz (50-60 V) for 7 min.
For mitochondrial potential analysis, adult cardiomyocytes were incubated with 20 nM TMRM for 15 min, and then caffeine (10 mM) was added. Images were obtained by a confocal microscopy (Leica, SP8). TMRM was excited at 543 nm and emission collected at >568 nm.
Cell viability assay
HEK293 cells were plated on 96-well plates (2 × 104/well) in 100 μl culture medium and transfected with 50 ng empty plasmids (Mock), mt-cpYFP plasmids, or mt-SoNar plasmids using lipofectamin 2000. Cells were cultured for 72 hr and MTT labeling reagent (10 μl, final concentration 0.5 mg/ml) was added and incubated for 4 hr at 37°C. DMSO (100 μl) were added and incubated at 37°C overnight. Absorbance at 590 nm were measured by using a microplate reader.
Cell proliferation assay
HEK293 cells were plated on 6-well plates (8 × 104/well) and transfected with 2 μg of the indicated plasmids using lipofectamin 2000 and cultured for 72 hr. BrdU Labeling Reagents (1 μM BrdU) were added and incubated for 2 hr at 37°C. BrdU positive cells were counted in 5 × 104 cells by a FACS machine.
Cell contractility
To monitor contraction of adult rat cardiomyocytes, the cells were cultured on glass coverslips with respective reagents treatment for 24 hr. The coverslip was then mounted in a chamber, filled with Tyrode's solution, and positioned on the microscope stage. Cardiomyocytes were paced at 1 Hz at 37°C and cell shortening was monitored by the IonOptix system. Parameters of cell contractility including cell shortening (%) were calculated by using the IonWizard.
Biochemical assays
Total intracellular NAD+ and NADH levels were measured by using a kit and following its instruction (BioAssay Systems, Cat. No. E2ND002). Briefly, ∼105 adult cardiomyocytes were homogenized in 100 μl NAD+ or NADH extraction buffer and heated to 60°C for 5 min. Then, 20 μl assay buffer and 100 μl opposite extraction buffer were added to neutralize the sample. After gentle mixing, samples were centrifuged at 14,000 rpm for 5 min and the supernatant collected. Solutions for standard curve contained 0, 3, 6, or 10 μM NAD(H) and were diluted from 500 μl 10 μM NAD(H) stock solution. Standards or samples (80 μl) were transferred into 96-well plate and freshly prepared working reagent (60 μl assay buffer, 1 μl enzyme A, 1 μl enzyme B, 14 μl lactate, and 14 μl MTT) was added and mixed quickly. Optical density (OD) was detected at 565 nm by using a microplate reader. NAD(H) concentration was calculated as follows:
ΔOD was calculated by subtracting OD value at 0 min from OD value at 15 min. Slope was calculated from the standard curve. n was dilution factor.
Pyruvate and lactate were measured by using commercial kits (Cayman, Cat. No. 700510 and 700470, respectively). Briefly, cardiomyocytes (1 × 106) were washed by 1 ml PBS, collected, and centrifuged at 10,000 g for 5 min at 4°C. The supernatant was discarded, 0.5 ml of 0.25 M MPA was added to the cell pellet. The tube was vortexed and placed on ice for 5 min. After centrifugation at 10,000 g for 5 min at 4°C, the supernatant was removed and the pellet was resuspended in 25 μl of K2CO3 to neutralize the acid. After centrifugation again at 10,000 g for 5 min at 4°C, the supernatant was collected as samples for assay. The sample was diluted 1:2 with diluted assay buffer. Solutions for pyruvate standard curve contained 0, 2.5, 5, 10 or 20 μM pyruvate and solutions for lactate standard curve contained 0, 0.25, 0.5, 1 or 2 mM lactate. Standards or samples (20 μl) were transferred into 96-well plate and freshly prepared working reagent (50 μl assay buffer, 50 μl cofactor mixture, 10 μl fluorometric detector and 20 μl enzyme mixture) was added and mixed quickly. The plate was incubated at room temperature for 20 min and the fluorescence measured in a microplate reader (Biotech). The excitation wavelength was set at 530 nm and emission wavelength at 585 nm.
AcAc and β-OHB were measured by using a commercial kit (Bioassay systems, Cat. No. EKBD-100). Briefly, The 8 mM standard was prepared by mixing 5 μl AcAc or β-OHB standard with 45 μl distilled H2O. The standard (5 μl), H2O (5 μl) or samples (5 μl each in two wells, Sample well and Blank well) were transferred to separate wells of a 96-well plate. The working reagent, containing 195 μl AcAc or β-OHB buffer, 8 μl reconstituted AcAc or β-OHB reagent and 0.5 μl HBDH enzyme, was added to the standard, H2O, and Sample wells. The blank reagent, containing 195 μl AcAc or β-OHB buffer and 8 μl reconstituted AcAc or β-OHB reagent, was added to the Blank wells. The plate was gently tapped for mixing, incubated at room temperature for 5 min, and put in a microplate reader (Biotech) to measure OD340nm. The concentrations of AcAc or β-OHB were calculated using the equations:
To monitor cell death we incubated adult cardiomyocytes with 0.4% trypan blue for 3 min at room temperature. Trypan blue was washed out and then images were taken by a light microscope. Cells with blue color were defined damaged. Five images were taken randomly in each sample and >90 cells were counted in each field.
Quantification and statistical analysis
All experimental results are expressed as Mean ± SEM. The number of experiments using rat cardiomyocytes indicates the number of rats used. Data collected from multiple cardiomyocytes from the same rat were averaged to N = 1. When multiple experiments using different numbers of animals were pooled for statistical analysis, the range of number of animals was indicated in figure legend. Data comparisons among the groups were performed by using unpaired Student’s t-test, one-way ANOVA followed by Tukey's post hoc analysis, or two-way ANOVA when appropriate. A p value < 0.05 was considered statistically significant.
Acknowledgments
We thank Dr. Yi Yang for providing the SoNar construct and for conceptual advice. We thank Drs. Chi Fung Lee, Arianne Caudal, and Bo Zhou for technical support and helpful advice. This work was supported by the National Institutes of Health (NIH HL114760 and HL137266 to W.W., HL110349 and HL149695 to R.T., and 2T32DK007247-41 to M.W.) and the American Heart Association (18EIA33900041 to W.W.).
Author contributions
Q.H., R.T., and W.W. designed the experiments. Q.H. and D.W. performed the experiments. P.W. and Q.H. constructed plasmids and adenoviruses containing mt-SoNar, ct-SoNar, and ct-cpYFP. Q.H., D.W., M.W., R.T., and W.W. analyzed the data and wrote the manuscript.
Declaration of interests
The authors declare no competing interests.
Published: November 15, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.crmeth.2021.100116.
Contributor Information
Rong Tian, Email: rongtian@uw.edu.
Wang Wang, Email: wangwang@uw.edu.
Supplemental information
Data and code availability
All data reported in this paper will be shared by the lead contact upon request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- Berthiaume J.M., Kurdys J.G., Muntean D.M., Rosca M.G. Mitochondrial NAD(+)/NADH redox state and diabetic cardiomyopathy. Antioxid. Redox Signal. 2019;30:375–398. doi: 10.1089/ars.2017.7415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bianchetti G., Ciccarone F., Ciriolo M.R., De Spirito M., Pani G., Maulucci G. Label-free metabolic clustering through unsupervised pixel classification of multiparametric fluorescent images. Anal. Chim. Acta. 2021;1148:238173. doi: 10.1016/j.aca.2020.12.048. [DOI] [PubMed] [Google Scholar]
- Canto C., Menzies K.J., Auwerx J. NAD(+) metabolism and the control of energy homeostasis: a balancing act between mitochondria and the nucleus. Cell Metab. 2015;22:31–53. doi: 10.1016/j.cmet.2015.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceconi C., Bernocchi P., Boraso A., Cargnoni A., Pepi P., Curello S., Ferrari R. New insights on myocardial pyridine nucleotides and thiol redox state in ischemia and reperfusion damage. Cardiovasc. Res. 2000;47:586–594. doi: 10.1016/s0008-6363(00)00104-8. [DOI] [PubMed] [Google Scholar]
- Chance B., Schoener B., Oshino R., Itshak F., Nakase Y. Oxidation-reduction ratio studies of mitochondria in freeze-trapped samples. NADH and flavoprotein fluorescence signals. J. Biol. Chem. 1979;254:4764–4771. [PubMed] [Google Scholar]
- Chen C., Hao X., Lai X., Liu L., Zhu J., Shao H., Huang D., Gu H., Zhang T., Yu Z., et al. Oxidative phosphorylation enhances the leukemogenic capacity and resistance to chemotherapy of B cell acute lymphoblastic leukemia. Sci. Adv. 2021;7:eabd6280. doi: 10.1126/sciadv.abd6280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W.W., Freinkman E., Wang T., Birsoy K., Sabatini D.M. Absolute quantification of matrix metabolites reveals the dynamics of mitochondrial metabolism. Cell. 2016;166:1324–1337 e1311. doi: 10.1016/j.cell.2016.07.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diguet N., Trammell S.A.J., Tannous C., Deloux R., Piquereau J., Mougenot N., Gouge A., Gressette M., Manoury B., Blanc J., et al. Nicotinamide riboside preserves cardiac function in a mouse model of dilated cardiomyopathy. Circulation. 2018;137:2256–2273. doi: 10.1161/CIRCULATIONAHA.116.026099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodman R.P., Markhard A.L., Shah H., Sharma R., Skinner O.S., Clish C.B., Deik A., Patgiri A., Hsu Y.H., Masia R., et al. Hepatic NADH reductive stress underlies common variation in metabolic traits. Nature. 2020;583:122–126. doi: 10.1038/s41586-020-2337-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grozio A., Mills K.F., Yoshino J., Bruzzone S., Sociali G., Tokizane K., Lei H.C., Cunningham R., Sasaki Y., Migaud M.E., et al. Slc12a8 is a nicotinamide mononucleotide transporter. Nat. Metab. 2019;1:47–57. doi: 10.1038/s42255-018-0009-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu H., Chen C., Hao X., Su N., Huang D., Zou Y., Lin S.H., Chen X., Zheng D., Liu L., et al. MDH1-mediated malate-aspartate NADH shuttle maintains the activity levels of fetal liver hematopoietic stem cells. Blood. 2020;136:553–571. doi: 10.1182/blood.2019003940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao X., Gu H., Chen C., Huang D., Zhao Y., Xie L., Zou Y., Shu H.S., Zhang Y., He X., et al. Metabolic imaging reveals a unique preference of symmetric cell division and homing of leukemia-initiating cells in an endosteal niche. Cell Metab. 2019;29:950–965.e956. doi: 10.1016/j.cmet.2018.11.013. [DOI] [PubMed] [Google Scholar]
- Horton J.L., Martin O.J., Lai L., Riley N.M., Richards A.L., Vega R.B., Leone T.C., Pagliarini D.J., Muoio D.M., Bedi K.C., Jr., et al. Mitochondrial protein hyperacetylation in the failing heart. JCI Insight. 2016;2:e84897. doi: 10.1172/jci.insight.84897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houtkooper R.H., Canto C., Wanders R.J., Auwerx J. The secret life of NAD+: an old metabolite controlling new metabolic signaling pathways. Endocr. Rev. 2010;31:194–223. doi: 10.1210/er.2009-0026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Q., Zhang H., Gutierrez Cortes N., Wu D., Wang P., Zhang J., Mattison J.A., Smith E., Bettcher L.F., Wang M., et al. Increased Drp1 acetylation by lipid overload induces cardiomyocyte death and heart dysfunction. Circ. Res. 2020;126:456–470. doi: 10.1161/CIRCRESAHA.119.315252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang S., Heikal A.A., Webb W.W. Two-photon fluorescence spectroscopy and microscopy of NAD(P)H and flavoprotein. Biophys. J. 2002;82:2811–2825. doi: 10.1016/S0006-3495(02)75621-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones D.P., Sies H. The redox code. Antioxid. Redox Signal. 2015;23:734–746. doi: 10.1089/ars.2015.6247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karamanlidis G., Lee C.F., Garcia-Menendez L., Kolwicz S.C., Jr., Suthammarak W., Gong G., Sedensky M.M., Morgan P.G., Wang W., Tian R. Mitochondrial complex I deficiency increases protein acetylation and accelerates heart failure. Cell Metab. 2013;18:239–250. doi: 10.1016/j.cmet.2013.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katsyuba E., Auwerx J. Modulating NAD(+) metabolism, from bench to bedside. EMBO J. 2017;36:2670–2683. doi: 10.15252/embj.201797135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolwicz S.C., Jr., Purohit S., Tian R. Cardiac metabolism and its interactions with contraction, growth, and survival of cardiomyocytes. Circ. Res. 2013;113:603–616. doi: 10.1161/CIRCRESAHA.113.302095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee C.F., Chavez J.D., Garcia-Menendez L., Choi Y., Roe N.D., Chiao Y.A., Edgar J.S., Goo Y.A., Goodlett D.R., Bruce J.E., et al. Normalization of NAD+ redox balance as a therapy for heart failure. Circulation. 2016;134:883–894. doi: 10.1161/CIRCULATIONAHA.116.022495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Z., Scott I., Webster B.R., Sack M.N. The emerging characterization of lysine residue deacetylation on the modulation of mitochondrial function and cardiovascular biology. Circ. Res. 2009;105:830–841. doi: 10.1161/CIRCRESAHA.109.204974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neinast M., Murashige D., Arany Z. Branched chain amino acids. Annu. Rev. Physiol. 2019;81:139–164. doi: 10.1146/annurev-physiol-020518-114455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nickel A.G., von Hardenberg A., Hohl M., Loffler J.R., Kohlhaas M., Becker J., Reil J.C., Kazakov A., Bonnekoh J., Stadelmaier M., et al. Reversal of mitochondrial transhydrogenase causes oxidative stress in heart failure. Cell Metab. 2015;22:472–484. doi: 10.1016/j.cmet.2015.07.008. [DOI] [PubMed] [Google Scholar]
- O'Rourke B., Ashok D., Liu T. Mitochondrial Ca(2+) in heart failure: not enough or too much? J. Mol. Cell Cardiol. 2021;151:126–134. doi: 10.1016/j.yjmcc.2020.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oldham W.M., Clish C.B., Yang Y., Loscalzo J. Hypoxia-mediated increases in L-2-hydroxyglutarate coordinate the metabolic response to reductive stress. Cell Metab. 2015;22:291–303. doi: 10.1016/j.cmet.2015.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patgiri A., Skinner O.S., Miyazaki Y., Schleifer G., Marutani E., Shah H., Sharma R., Goodman R.P., To T.L., Robert Bao X., et al. An engineered enzyme that targets circulating lactate to alleviate intracellular NADH:NAD(+) imbalance. Nat. Biotechnol. 2020;38:309–313. doi: 10.1038/s41587-019-0377-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratajczak J., Joffraud M., Trammell S.A., Ras R., Canela N., Boutant M., Kulkarni S.S., Rodrigues M., Redpath P., Migaud M.E., et al. NRK1 controls nicotinamide mononucleotide and nicotinamide riboside metabolism in mammalian cells. Nat. Commun. 2016;7:13103. doi: 10.1038/ncomms13103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritterhoff J., Young S., Villet O., Shao D., Neto F.C., Bettcher L.F., Hsu Y.A., Kolwicz S.C., Jr., Raftery D., Tian R. Metabolic remodeling promotes cardiac hypertrophy by directing glucose to aspartate biosynthesis. Circ. Res. 2020;126:182–196. doi: 10.1161/CIRCRESAHA.119.315483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tateishi K., Wakimoto H., Iafrate A.J., Tanaka S., Loebel F., Lelic N., Wiederschain D., Bedel O., Deng G., Zhang B., et al. Extreme vulnerability of IDH1 mutant cancers to NAD+ depletion. Cancer Cell. 2015;28:773–784. doi: 10.1016/j.ccell.2015.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Titov D.V., Cracan V., Goodman R.P., Peng J., Grabarek Z., Mootha V.K. Complementation of mitochondrial electron transport chain by manipulation of the NAD+/NADH ratio. Science. 2016;352:231–235. doi: 10.1126/science.aad4017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varadarajan S.G., An J., Novalija E., Smart S.C., Stowe D.F. Changes in [Na(+)](i), compartmental [Ca(2+)], and NADH with dysfunction after global ischemia in intact hearts. Am. J. Physiol. Heart Circ. Physiol. 2001;280:H280–H293. doi: 10.1152/ajpheart.2001.280.1.H280. [DOI] [PubMed] [Google Scholar]
- Varone A., Xylas J., Quinn K.P., Pouli D., Sridharan G., McLaughlin-Drubin M.E., Alonzo C., Lee K., Munger K., Georgakoudi I. Endogenous two-photon fluorescence imaging elucidates metabolic changes related to enhanced glycolysis and glutamine consumption in precancerous epithelial tissues. Cancer Res. 2014;74:3067–3075. doi: 10.1158/0008-5472.CAN-13-2713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verdin E. NAD(+) in aging, metabolism, and neurodegeneration. Science. 2015;350:1208–1213. doi: 10.1126/science.aac4854. [DOI] [PubMed] [Google Scholar]
- Wallrabe H., Svindrych Z., Alam S.R., Siller K.H., Wang T., Kashatus D., Hu S., Periasamy A. Segmented cell analyses to measure redox states of autofluorescent NAD(P)H, FAD & Trp in cancer cells by FLIM. Sci. Rep. 2018;8:79. doi: 10.1038/s41598-017-18634-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W., Fang H., Groom L., Cheng A., Zhang W., Liu J., Wang X., Li K., Han P., Zheng M., et al. Superoxide flashes in single mitochondria. Cell. 2008;134:279–290. doi: 10.1016/j.cell.2008.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W., Karamanlidis G., Tian R. Novel targets for mitochondrial medicine. Sci. Transl. Med. 2016;8:326rv323. doi: 10.1126/scitranslmed.aac7410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williamson D.H., Lund P., Krebs H.A. The redox state of free nicotinamide-adenine dinucleotide in the cytoplasm and mitochondria of rat liver. Biochem. J. 1967;103:514–527. doi: 10.1042/bj1030514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao W., Loscalzo J. Metabolic responses to reductive stress. Antioxid. Redox Signal. 2020;32:1330–1347. doi: 10.1089/ars.2019.7803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L., Garcia Canaveras J.C., Chen Z., Wang L., Liang L., Jang C., Mayr J.A., Zhang Z., Ghergurovich J.M., Zhan L., et al. Serine catabolism feeds NADH when respiration is impaired. Cell Metab. 2020;31:809–821.e806. doi: 10.1016/j.cmet.2020.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshino J., Baur J.A., Imai S.I. NAD(+) intermediates: the biology and therapeutic potential of NMN and NR. Cell Metab. 2018;27:513–528. doi: 10.1016/j.cmet.2017.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H., Gong G., Wang P., Zhang Z., Kolwicz S.C., Rabinovitch P.S., Tian R., Wang W. Heart specific knockout of Ndufs4 ameliorates ischemia reperfusion injury. J. Mol. Cell Cardiol. 2018;123:38–45. doi: 10.1016/j.yjmcc.2018.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y., Hu Q., Cheng F., Su N., Wang A., Zou Y., Hu H., Chen X., Zhou H.M., Huang X., et al. SoNar, a highly responsive NAD+/NADH sensor, allows high-throughput metabolic screening of anti-tumor agents. Cell Metab. 2015;21:777–789. doi: 10.1016/j.cmet.2015.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y., Jin J., Hu Q., Zhou H.M., Yi J., Yu Z., Xu L., Wang X., Yang Y., Loscalzo J. Genetically encoded fluorescent sensors for intracellular NADH detection. Cell Metab. 2011;14:555–566. doi: 10.1016/j.cmet.2011.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y., Wang A., Zou Y., Su N., Loscalzo J., Yang Y. In vivo monitoring of cellular energy metabolism using SoNar, a highly responsive sensor for NAD(+)/NADH redox state. Nat. Protoc. 2016;11:1345–1359. doi: 10.1038/nprot.2016.074. [DOI] [PubMed] [Google Scholar]
- Zhao Y., Zhang Z., Zou Y., Yang Y. Visualization of nicotine adenine dinucleotide redox homeostasis with genetically encoded fluorescent sensors. Antioxid. Redox Signal. 2018;28:213–229. doi: 10.1089/ars.2017.7226. [DOI] [PubMed] [Google Scholar]
- Zou Y., Wang A., Huang L., Zhu X., Hu Q., Zhang Y., Chen X., Li F., Wang Q., Wang H., et al. Illuminating NAD(+) metabolism in live cells and in vivo using a genetically encoded fluorescent sensor. Dev. Cell. 2020;53:240–252.e247. doi: 10.1016/j.devcel.2020.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou Y., Wang A., Shi M., Chen X., Liu R., Li T., Zhang C., Zhang Z., Zhu L., Ju Z., et al. Analysis of redox landscapes and dynamics in living cells and in vivo using genetically encoded fluorescent sensors. Nat. Protoc. 2018;13:2362–2386. doi: 10.1038/s41596-018-0042-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data reported in this paper will be shared by the lead contact upon request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.